

Abstract
Background
Stem rot caused by Sclerotium rolfsii is a very important soil-borne disease of peanut. S. rolfsii is a necrotrophic plant pathogenic fungus with an extensive host range and worldwide distribution. It can infect peanut stems, roots, pegs and pods, leading to varied yield losses. S. rolfsii strains GP3 and ZY collected from peanut in different provinces of China exhibited a significant difference in aggressiveness on peanut plants by artificial inoculation test. In this study, de-novo genome sequencing of these two distinct strains was performed aiming to reveal the genomic basis of difference in aggressiveness.
Results
Scleotium rolfsii strains GP3 and ZY, with weak and high aggressiveness on peanut plants, exhibited similar growth rate and oxalic acid production in laboratory. The genomes of S. rolfsii strains GP3 and ZY were sequenced by Pacbio long read technology and exhibited 70.51 Mb and 70.61 Mb, with contigs of 27 and 23, and encoded 17,097 and 16,743 gene models, respectively. Comparative genomic analysis revealed that the pathogenicity-related gene repertoires, which might be associated with aggressiveness, differed between GP3 and ZY. There were 58 and 45 unique pathogen-host interaction (PHI) genes in GP3 and ZY, respectively. The ZY strain had more carbohydrate-active enzymes (CAZymes) in its secretome than GP3, especially in the glycoside hydrolase family (GH), the carbohydrate esterase family (CBM), and the polysaccharide lyase family (PL). GP3 and ZY also had different effector candidates and putative secondary metabolite synthetic gene clusters. These results indicated that differences in PHI, secreted CAZymes, effectors and secondary metabolites may play important roles in aggressive difference between these two strains.
Conclusions
The data provided a further understanding of the S. rolfsii genome. Genomic comparison provided clues to the difference in aggressiveness of S. rolfsii strains.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-021-07534-0.
Keywords: Comparative genomic analysis, PacBio sequel sequencing, Pathogenesis-related genes, Sclerotium rolfsii
Background
Sclerotium rolfsii is a destructive soil-borne fungal pathogen. Its sexual stage, Athelia rolfsii, belongs to Basidiomycota and rarely occurs in nature; thus, its role in the life cycle of the fungus is unknown [1]. S. rolfsii infects more than 600 plant species, especially economically important agricultural and horticultural crops including peanut, soybean, wheat, cotton, tomato, potato, cucurbit, and onion [2, 3], therefore a pathogen of wide host range. Moreover, S. rolfsii produces sclerotia, which plays a key role in the disease cycle and can survive in soil for long periods [4]. S. rolfsii can infect stems, roots, pegs, and pods of peanut and causes branches wilting, and even whole plant wilting. Peanut stem rot caused by S. roflsii is also known as southern stem rot, southern blight, white mold, and Sclerotium rot [5]. This fungal disease has been reported in many peanut producing regions of the world. Loss caused by peanut stem rot was estimated at 41 million US dollars in Georgia in 2011 [6]. Up to 30% yield loss was recorded in India [7]. Peanut stem rot has been epidemic in China recently, caused up to 50% yield loss in hotspots, and is the most serious peanut soil-borne disease in China [8].
Control of peanut stem rot disease is difficult because of wide range of hosts, profuse mycelium, abundant persistent sclerotia, and genetic variability of S. rolfsii populations [4]. Currently, there are only a few resistant commercial peanut cultivars available for use [9–11]. Limited success was achieved in developing resistant varieties to peanut stem rot in China [12]. Normally, approaches to control peanut stem rot include the application of fungicides and agronomic measures such as rotation with non-host crops or coverage of infected crop debris with deep plowing [13]. But these methods are still not effective to control this disease.
In order to implement effective integrated practices to control peanut stem rot, knowledge about the genetic basis of differently aggressive strains of S. rolfsii is a key component, as it is essential for host resistance assessment in a given region [14]. Earlier investigators observed differences in aggressiveness among isolates of S. rolfsii in the USA and India [15–18]. They were classified as highly, moderately, and weakly aggressive strains [16]. Until now, differences in aggressiveness have not been reported among S. rolfsii strains in China. In previous research, aggressiveness of S. rolfsii strains were found to be highly correlated with endo-PG production and growth rate [16], but the genetic basis of aggressiveness is still unknown.
The genetic variability of S. rolfsii stains has not been documented. Correlations between pathogenic traits and genetic patterns have rarely been identified. To gain the relevant insights, we sequenced two S. rolfsii strains GP3 and ZY, GP3 isolated from Guangxi province and ZY isolated from Henan province, China, by combing the Single Molecule Real-Time (SMRT) sequencing and Illumina technology. The two strains were in different mycelial compatibility groups (MCG) [19], possessed similar cultural morphology and growth rate on PDA media, produced similar amount of oxalic acid in vitro, but demonstrated different levels of aggressiveness on peanut plants in inoculation tests. The ZY strain was highly aggressive, and the GP3 strain was weakly aggressive. In comparison with GP3 strain, ZY strain had a slightly larger genomes size. The genomes annotation of GP3 and ZY revealed that many pathogenesis- related genes differed between them, including pathogen host interaction (PHI) genes, CAZymes, secreted proteins and secondary metabolites. This study will be meaningful for further identifying determinants of pathogenicity as well as deeply understanding of S. rolfsii infection mechanisms.
Results
Aggressiveness, growth rate and OA production
The typical symptoms caused by S. rolfsii strains ZY and GP3 on the peanut stems included unrestricted lesions at the infection sites followed by tissue maceration, finally partial plant even whole plant wilting. Disease severity was scored at 14 days past inoculation (dpi) and disease index showed a significant difference between these two strains. The disease index of ZY was 82.34, which was classified as highly aggressive. The disease index of GP3 was 32.2, which was regarded as weakly aggressive (Fig. 1a, b). The growth rate of these two strains was similar on PDA plate and showed no significant difference (Fig. 1c, d). There was no significant difference in the amount of oxalic acid (OA) produced by these two strains either by haloes revealing on the PDA plate containing bromophenol blue, or by OA amount in the culture filtrate as analyzed by KMnO4 titration (Fig. 1e, f).
Fig. 1.

Pathogenicity, mycelial growth and oxalic acid production of S. rolfsii GP3 and ZY. a Symptom of peanut plants caused by GP3 and ZY; b Disease index of peanut plants infected by GP3 and ZY; c Mycelial growth of GP3 and ZY on potato dextrose agar (PDA) plates; d Growth rate of GP3 and ZY on PDA plates, e Mycelial growth of GP3 and ZY on PDA plates containing bromophenol blue; f Oxalic acid content of GP3 and ZY in PDB medium
Genome sequence and assembly
A total of 9.97 Gb subreads with 8.80 kb average length was generated for ZY and 6.34 Gb subreads with 10.68 kb average length for GP3 by SMRT sequencing. After polishing with Illumina data, the assembled genomes of GP3 and ZY were 70.51 Mb and 70.61 Mb, respectively, containing 27 contigs with an N50 of 3.67 Mb for GP3, and 23 contigs with an N50 of 3.71 Mb for ZY (Table 1). The two strains had genome assemblies of a similar size, both slightly smaller than that of S. rolfsii strain MR10 (73.18 Mb) [20]. The completeness of the genome assemblies was assessed using BUSCO [21]. About 97.5% (1301/1335) and 97.2% (1298/1305) of gene groups required for the correct assembly of Basidiomycota were present in GP3 and ZY, respectively (Fig. S1). The average GC contents of the resulting S. rolfsii genomes of GP3 (46.27%) and ZY (46.29%) were comparable to S. rolfsii MR10 (46.16%) (Table 1). Gene candidates in the S. rolfsii GP3 and ZY genomes were predicted by a combined approach, and 17,097 and 16,743 genes with an average gene length of 2013.91 bp and 2039.76 bp were identified (Table 1). Approximately 93.27% (15,947) of GP3 genes and 93.93% (15,727) of ZY genes could be annotated by non-redundant nucleotide and protein sequences in the Cluster of Orthologous Groups (KOG), Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Non-redundant Protein (NR), and Swiss-Prot databases (Fig. S2, 3, 4, 5). The number of genes predicted in S. rolfsii strains GP3 and ZY was similar with that in S. rolfsii strain MR10 (16,830 genes) (Table 1). In this study, we identified 356 tRNAs, 48 rRNAs and 32 snRNAs in the genome of GP3, and 415 tRNAs, 55 rRNAs and 32 snRNAs in the genome of ZY (Table S1). Comparison of gene orthologous with nine Agricomycetes fungi by OrthoMCL [22], GP3 and ZY shared a similarly low number of unique genes with 75 for GP3 and 37 for ZY distributed in 62 and 19 gene families (Table S2), respectively. Sequence comparison between contigs of whole-genome assemblies indicated a good macrosynteny between GP3 and ZY. Especially, contig 3, 7, 10, 15, 16, and 17 of GP3 corresponded well with contig 1, 6, 14, 18, 15, and 16 of ZY (Fig. 2).
Table 1.
Genome characteristic and assemblies feature of S. rolfsii strains. S. rolfsii MR10 was the first sequenced S. rolfsii strain isolated from India. GP3 and ZY were isolated from China and sequenced in this study.
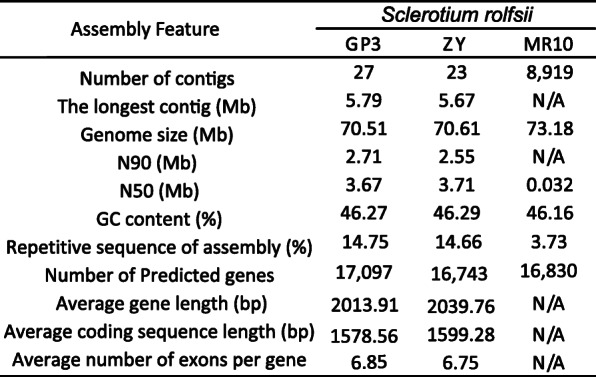
Fig. 2.
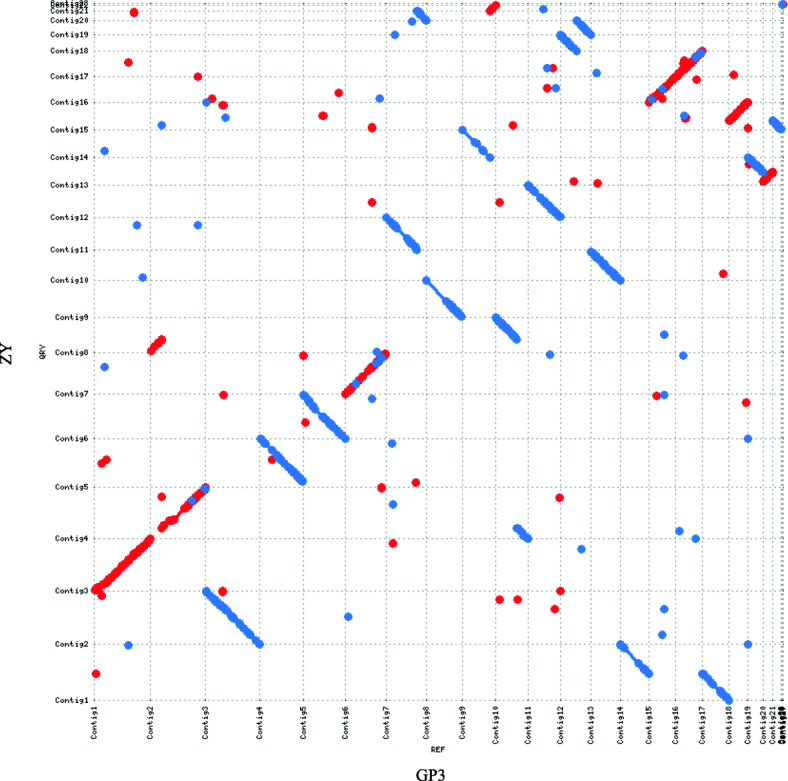
Genome synteny analysis between S. rolfsii strains GP3 and ZY. Dot-plots depicted nucleotide sequence matches detected via MUMer between all contigs of S. rolfsii GP3 and ZY. Contigs of ZY along the Y- axes, while contigs of GP3 along the X- axes. Sequence alignments exhibited a good macrosyntenic configuration
Repetitive element analysis
De novo and homology approaches were combined to identify repetitive sequences in the genomes of S. rolfsii GP3 and ZY. A total of 14.75% and 14.66% repetitive sequences were generated for GP3 and ZY, respectively (Table 1, Table S3). The abundance of repetitive sequences was similar between the two strains and much more than that of S. rolfsii strain MR10, which had a repetitive sequence content of 3.73% (Table 1). GP3 and ZY contained repetitive elements including DNA transposons, retroelements, and satellites. Retroelements were abundant in the studied genomes, accounting for 10.28% and 10.79% in GP3 and ZY. LTR was abundant in the retroelements, accounting for 9.85% and 10.38% in GP3 and ZY (Fig. 3, Table S4). Both abundance of LTR elements and retroelements in repetitive sequences were also found in S. rolfsii MR10 genome (Table S4).
Fig. 3.

Distribution of repetitive sequences in S. rolfsii strains GP3 and ZY genomes. The left circle plot shows repetitive sequences distribution in S. rolfsii strain GP3, the right circle plot shows repetitive sequence distribution in S. rolfsii strain ZY. Repetitive sequence were classified as retroelement (LTR, long terminal repeat; LINE, long interspersed repeat element; SINE short interspersed repeat element), DNA transposon element, satellite, others, and non-repetitive element of genome
Orthology analysis and phylogenetic analysis
The entire sets of predicted proteins of S. rolfsii GP3 and ZY were clustered with the OrthoMCL program [22] to identify gene families. Comparative analysis of the genomes of related species of Agaricomycetes, Basidiomycota showed that S. rolfsii strains had larger genomes but fewer total genes in comparison with most of the other species (Fig. S6). Of gene families, the unclustered genes number of GP3 and ZY were the least among fungi in Agaricomycetes. A Venn diagram of the OrthoMCL revealed that S. rolfsii strains shared 4813 genes with other four Agaricomycetes species (Fig. 4a).
Fig. 4.

Phylogenetic and comparative genomic study of S. rolfsii GP3 and ZY. a Venn diagram showing an overlap of gene families among S. rolfsii GP3, ZY, G. luxurians, P. croceum, and H. pinastri; b Maximum likelihood phylogenetic tree of GP3, ZY and nine fungi species in Agaricomycetes based on single-copy orthologous genes, with P. cyanescens used as an outgroup species
To understand the genetic relationship of GP3 and ZY to the related Agaricomycetes species, we generated a phylogenetic tree of single-copy genes based the orthologous gene family analysis of the two S. rolfsii strains and other Agaricomycetes fungi, including Armillaria gallica, Auricularia subglabra [23], Exidia glandulosa [24], Galerina marginata, Gymnopus luxurians [25], Hydnomerulius pinastri [25], Psilocybe cyanescens [25], Scleroderma citrinum [25], and Piloderma croceum [25]. The phylogenetic tree indicated that S. rolfsii strains were more closely related to E. glandulosa and A. subglabra, which belonged to Auriculariales, than to P. croceum, which belonged to Atheliaceae, the same as S. rolfsii (Fig. 4b).
Genes involved in pathogenicity
Homologs in PHI base
In total, we identified 4600 and 4603 potential pathogen-host interaction (PHI) genes by searching the PHI base (Fig. 5). Among them, 24 genes were predicted as effector category and 172 genes were identified as “increased virulence” in GP3, while ZY had 25 effectors and 138 genes related to “increased virulence”. Compared with S. rolfsii GP3, a total of 45 genes were unique in ZY, two of which were predicted as effector and one was predicted as “increased virulence”. We also found 58 genes of GP3 were not present in the ZY genome, 12 and 18 of which were predicted as “loss of pathogenicity” and “reduced virulence”, respectively (Table S5).
Fig. 5.
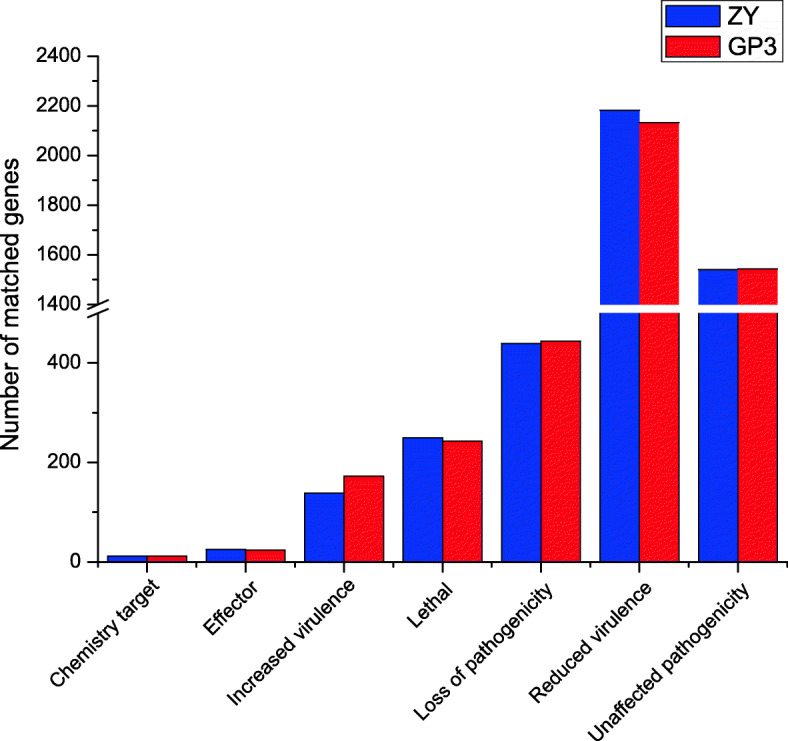
Pathogen-host interaction (PHI) genes of S. rolfsii GP3 and ZY. Distribution of S. rolfsii PHI genes in different phenotypes including chemistry target, effector, increased virulence, reduced virulence, lethal, loss of pathogenicity, and unaffected pathogenicity
CAZymes
The genomes of S. rolfsii GP3 and ZY contained 957 and 925 genes encoding putative CAZymes, distributed in 118 and 119 CAZyme families. Glycoside hydrolases (GHs) were dominant in the GP3 and ZY genomes (51.62 and 52.54%), followed by carbohydrate-binding modules (CBMs) and glycosyltransferases (GTs) (Fig. 6a). The CAZyme content of GP3 was slightly larger than that of ZY, and CAZyme content of both GP3 and ZY was more than that of S. rolfsii MR10 (902) (Fig. 6b).
Fig. 6.

Distribution of CAZymes, secreted CAZymes, and CAZymes involved in plant cell wall degradation of S. rolfsii GP3 and ZY. a Distribution of CAZymes in S. rolfsii GP3 and ZY; b Comparison of CAZymes of S. rolfsii strains with other 12 plant pathogens; c Comparison of CAZymes and secreted CAZymes in GP3 and ZY; d Comparison of CAzymes and secreted CAZymes involved in plant cell wall degradation in GP3 and ZY. Abbreviations: GH, Glycoside hydrolase; CBM, Carbohydrate-binding module; GT, Glycosyltransferase; AA, Auxiliary activity; CE, Carbohydrate esterase; PL, Polysaccharide lyase
Comparison of CAZyme content of S. rolfsii strains with other plant pathogens including six necrotrophic fungi (Aspergillus niger, Botrytis cinerea, Penicillium digitatum, Sclerotinia sclerotiorum, Rhizoctonia solani, and Verticillium dahliae), three hemibiotrophic fungi (Colletotrichum higginsianum, Fusarium graminearum, and Magnaporthe oryzae), and three biotrophic fungi (Puccinia graminis, Peronospora effusa, and Ustilago maydis) showed that the CAZyme content of S. rolfsii genome was the highest among above analyzed pathogens (Fig. 6b). Necrotrophic fungi had more CAZymes than biotrophic and hemibiotrophic fungi. In comparison with those necrotrophic plant pathogens of a broad host range, such as S. sclerotiorum, B. cinerea, and V. dahliae, the CAZyme content of S. rolfsii was much more than these fungi. Compared to Basidiomycota plant pathogens, CAZyme content of S. rolfsii was three times as much as R. solani and P. graminis, and four times as much as U. maydis (Fig. 6b). Besides differences in CAZyme content, the number of CAZymes involved in cellulose, hemicellulose, and pectin degradation of S. rolfsii strains GP3 and ZY was noticeably larger than that of those analyzed pathogens (Tables S6–S8), especially in the pectin degrading capacity.
Glycoside hydrolases are known to catalyze the hydrolysis of glycosidic bonds in carbohydrate molecules. S. rolfsii was rich in one glycosyl hydrolase family, GH28, a class of polygalacturonases involved in pectin degradation. The amount of GH28 was the same in GP3 and ZY (62 vs 62) and was larger than that in the other analyzed pathogens (Table S8). The expansion of GH28 was not found in the biotrophic and hemibiotrophic pathogens, such as U. maydis, P. graminis, and M. oryzae. In comparison with other analyzed necrotrophic pathogenic fungi, S. rolfsii strains had three times more GH28. Besides GH28, some other glycoside hydrolases involved in pectin degradation in S. rolfsii, such as GH35, GH51, and GH78, also had higher number in comparison with those pathogens (Table S8).
Secretome and effector
The putative secreted proteins of S. rolfsii GP3 and ZY were identified based on a comprehensive pipeline (Fig. S7). The genomes of GP3 and ZY were predicted to encode 536 (3.14%) and 551(3.29%) secreted proteins, respectively. Among the secreted protein candidates, there were 151 and 30 secreted CAZyme genes for ZY and GP3, including 113 GH, 20 CE, 15 CBM, and 3 PL genes for ZY, while 22 GH, 6 CE, and 2 CBM genes for GP3 (Fig. 6c, Table S9). In comparison with secreted CAZymes involved in cellulose, hemicellulose and pectin degradation, ZY had more of these genes than GP3 (Fig. 6d).
A total of 50 and 46 putative effector candidates for GP3 and ZY, respectively, were predicted by Effector P.1. After manual inspection with the criteria of 50 ≤ molecular weight ≤ 300 kDa, 0–1 predicted trans-membrane domain, and ≥ 4 cysteine residues, a total of 30 and 27 effector candidates for GP3 and ZY were identified (Table 2). Most of the putative effector candidates were small (average length of 146 and 152 amino acids, ranging from 52 to 278, and 58 to 291 amino acids for GP3 and ZY). These candidates were rich in cysteines (the average cysteine composition was 8.5% for GP3 and 8.6% for ZY). The functions of most effector candidates (73.33% and 44.44% of GP3 and ZY) were unknown. Comparison of putative effectors with PHI and CAZymes candidate genes showed that the number of genes, for “functional effector”, “loss of pathogenicity”, “reduced virulence”, GH, and CBM, differed between these two strains. ZY had two effectors and five GH genes, while GP3 had one GH gene and no effector overlapping with PHI and CAZyme candidate genes (Table S10). The function of these predicted effectors needs to be further verified in future research.
Table 2.
Putative effectors of S. rolfsii GP3 and ZY. Putative effectors of GP3 and ZY were functionally annotated in NCBI-NR and PHI base.

Secondary metabolites
The antiSMASH 4.0 software was used to identify the secondary metabolite gene clusters in the genome of S. rolfsii ZY and GP3. A total of 46 and 31 gene clusters were predicted to be related to secondary metabolism in ZY and GP3, respectively (Fig. 7). In ZY, two clusters were identified as non-ribosomal peptide synthase (NPRS). Three, one, and 12 clusters were predicted as Type I polyketide synthase (T1 PKS), NPRS/ T1 PKS, and terpene, respectively. Besides, 28 clusters were predicted as others. Compared to ZY, GP3 contained no NPRS cluster, the same number of NPRS/ T1 PKS clusters, two fewer T1 PKS clusters, three fewer terpene clusters, and 8 fewer other clusters (Fig. 7).
Fig. 7.

Putative secondary metabolite gene clusters in S. rolfsii ZY and GP3. Abbreviations: PKS, polyketide synthase; NRPS, nonribosomal peptides synthase; T1PKS, type 1 PKS; PKS-NRPS: hybrid NRPS-PKS enzymes
Discussion
Sclerotium rolfsii is a very important plant pathogen with a broad host range. To date, the genome of one strain MR10 with little information on its aggressiveness had been sequenced [20]. In the present study, we discovered two S. rolfsii strains that differed in aggressiveness on peanut plants. Meanwhile, the two strains did not show a significant difference in growth rate and oxalic acid production. Thus, we conducted genome sequencing of the two S. rolfsii strains and produced gapless high-quality genomes aiming to unravel the genomic basis underlying the difference in aggressiveness between the two strains.
During pathogenesis, S. rolfsii may produce cell wall degrading enzymes such as endo-polygalacturonase (endo-PG) [26, 27], cellulase [28], and polymethylagalacturonase [16] in conjunction with oxalic acid (OA) [16, 26]. Bateman and Beer [26] suggested that OA, pectinase, and cellulase acted synergistically in the destruction of host tissue by S. rolfsii. Secretion of OA and endo-PG concomitantly with rapid mycelial growth appeared to be the key requirements for establishing infection [16]. Earlier investigators observed differences in aggressiveness among isolates of S. rolfsii [16, 29, 30] and found that aggressiveness was highly correlated with endo-PG production and growth rate, provided a base level of OA [16]. OA was reported to have a positive correlation to the aggressiveness of S. roflsii isolates [30]. In contrast, OA was not correlated with the aggressiveness of S. rolfsii by Punja (1985) [16], who found that highly, moderately, and weakly aggressive strains produced similar amounts of oxalic acid. In the present study, we tested GP3 and ZY on the PDA plate containing bromophenol blue and measured the amount of OA produced in a liquid PDB medium. The results indicated that there was no significant difference in oxalic acid production between the weakly aggressive strain GP3 and the highly aggressive strain ZY, although oxalic acid is an essential aggressiveness factor for S. sclerotiorum [31]. S. rolfsii produced a basic level of oxalic acid to acidic environment that facilitates the optimal activity of certain sets of cell wall degrading enzymes and peptidases. However, OA was not the essential factor for difference in aggressiveness between S.rolfsii GP3 and ZY.
Plant cell walls are an important barrier that plants used to protect themselves from attacking by a range of organisms. Plant cell wall carbohydrates form a complex network of different polysaccharides that can be subdivided into three categories: cellulose, hemicellulose, and pectin. Plant pathogenic fungi employ diverse gene repertoires, including carbohydrate-active enzymes (CAZymes), to invade host plants and subvert host immune systems [32, 33]. CAZymes are known to play an important role in host-pathogen interactions and, along with effectors, are prime targets for studying aggressive factors in fungi [34, 35]. CAZyme families with potential roles in aggressiveness were examined in S. rolfsii strains GP3 and ZY. In our study, the CAZyme content in weakly aggressive strain GP3 was found to be slightly more than that in highly aggressive strain ZY. GP3 also had a noticeably higher number of enzymes in the AA, CBM, and CE families. GP3 and ZY had a similar number of enzymes involved in cellulose, hemicellulose, and pectin degradation, these results indicated that CAZyme content was not related to the difference in aggressiveness between S. rolfsii GP3 and ZY. We then undertook further analysis of the secreted CAZymes, which were involved in plant cell wall degradation that played an important role in phytopathogenic penetration of their hosts [36]. There was a significant difference in the levels of secreted CAZymes between GP3 and ZY. Highly aggressive strain ZY possessed three times more secreted CAZymes (105) than weakly aggressive strain GP3 (30). ZY also possessed more enzymes involved in pectin degradation, such as GH28. These results indicated that secreted CAZymes, especially polygalacutronases, may play an important role in different aggressiveness between S. rolfsii strains ZY and GP3. It was in accordance with the results of Punja (1985) [16], who reported that the aggressiveness of S. rolfsii was highly correlated with endo-PG production.
To establish infection, fungal plant pathogens secrete effector molecules that manipulate host physiology, including immune responses that are triggered when plant hosts sense invading pathogens [37–39]. Effectors have been discovered in multiple plant pathogenic fungi and exhibit numerous different functions depending on the fungal lifestyle. Necrotrophic fungi, which feed on dead tissue, often produce effectors that promote cell death, whereas biotrophic fungi, which require living tissue, produce effectors that prevent cell death [40–43]. In some soil-borne vascular necrotrophic pathogens with a broad range of host plants, effectors involved in aggressiveness have been identified. In S. sclerotiorum, about 70 effectors have been identified [44], a small, cysteine-rich secreted protein with a cyanoviron-N homology (CVNH) domain, attenuated aggressiveness when deleted [45]. A total of 127 putative effectors were identified in another broad host range necrotrophic pathogen V. dahliae [46]. Among them VdCP1 contributed to aggressiveness and triggered the host plant’s immune system [47]. Up to now, little experimental evidence for the existence of similar effector proteins was available for S. rolfsii. To identify putative effectors involved in aggressiveness, we searched the whole proteome of S. rolfsii and found that the effectors of GP3 and ZY were completely different. S. rolfsii existed as a multi-nuclear heterokaryon, in which individual cells may carry multiple nuclei [6]. The method for the stable transformation of S. rolfsii has not been available yet, and thus functional testing of pathogenic candidate genes in further studies will be challenging.
Despite the variety of pathogenicity-related mechanisms involved, accumulating evidence indicates that necrotrophic plant pathogens interact with their hosts in a manner much more subtle than originally considered and that signaling between them plays a significant role in the lifestyle of these pathogens [48]. The mechanism of differences in aggressiveness is complicated in plant pathogens. Besides secreted CAZymes and effectors, other factors may also be involved in aggressiveness on host plants. It was reported that the genomic islands might contribute to the expanded genetic diversity and aggressiveness of V. dahliae [49]. Aggressiveness-associated effectors were often found to be affected by both repeat activity and repeat-induced point mutations (RIP) in Leptosphaeria maculans and S. sclerotiorum [50, 51].
To understand the difference in aggressiveness of these two strains in genome, we presented here the gapless genome sequences of S. rolfsii GP3 and ZY. This work has provided important clues to factors involved in aggressive difference between these two S. rolfsii strains. The data will provide a useful foundation for further studies to understand the mechanism of S. rolfsii infection.
Conclusions
We generated long-read PacBio reads and gapless genome assemblies of two S. rolfsii strains with different levels of aggressiveness on peanut plants and then implemented a comparative genomic analysis of these two strains. The genome of S. rolfsii ZY and GP3 encoded different pathogen related gene repertoires. The obtained GP3 and ZY genome assemblies and annotation represent the few available Sclerotium genome resources for studying the pathogenic mechanism of this fungus toward peanut.
Methods
Isolates and oxalic acid production
Scleotium rolfsii strain ZY was originally collected from Zhengyang county (114.34 °E, 32.38 °N) in Henan province, and strain GP3 was collected from Guiping city (101.49 °E, 36.34 °N) in Guangxi province of China. The two strains were in different mycelial compatibility group (MCG) and exhibited similar growth rate on potato dextrose agar (PDA medium: 200 g peeled and sliced potatoes boiled for 20mins, 20 g dextrose, adjusted to pH 7.0, 20 g agar, to make the final volume to 1000 ml with distilled water [19]. Oxalic acid was detected by two methods. PDA containing bromophenol blue was used to test oxalic acid in PDA plate. Mycelium discs of each strain were placed at the center of PDA medium containing 0.5 g/l of bromophenol blue and kept at 30 °C in the dark, four petri dishes for each strain. The diameter of yellow halo was measured after three days. KMnO4 titration was used to detect oxalic acid in liquid PDB. The strains grew in liquid PDB medium, three replicates of 150 ml flasks containing 30 ml of medium were included for each isolate, three discs were added to each flask, and the flasks were incubated without shaking at 30 °C in the dark. The culture of each strain was filtered through a Whatman No.1 filter paper after 5 days incubation. Oxalic acid (OA) content in 5 ml filtrate was determined using a KMnO4 titration method following the procedure of Kritzman’s [52].
Pathogenicity test
The experiment was conducted as a completely randomized design in three replications. Each plot consisted of three rows with 15 susceptible peanut plants per row. The plants were inoculated 50–55 days after sowing. S. rolfsii inoculum was prepared just before inoculation. Oat grains were soaked in water for 4 h, sterilized at 121 °C for 30 mins twice after water removed. The fresh mycelium discs of S. rolfsii GP3 and ZY were transferred to the flasks containing sterilized oat grains, respectively. The oat grains culture maintained in the dark at 30 °C until surface of grains covered by S. rolfsii mycelium. The oat grains inoculum was mixed with equal amount of sterilized sand to ensure uniform delivery of inoculum. Each plant was inoculated with 2 g of S. rolfsii oat inoculum and sand mixture. The plots were watered to field capacity after inoculation. Disease symptoms were investigated 14 days after inoculation. A 1–5 scale for the severity of wilting according to Shokes’ method [53], where 1 = no symptom, 2 = lesions on stem only, 3 = up to 25% of the plant wilting, 4 = 26–50% of the plant wilting, and 5 = > 50% of the plant wilting. Disease index was calculated by using the following formula. DI = {[Σ (number of plants × corresponding diseases scale)] / (total number of plants × the maximum disease scale)} × 100. Different level of aggressiveness was determined according to Punja (1985) [16], high aggressiveness with DI more than 66.7, and weak aggressiveness showing DI less than 33.3.
DNA and RNA purification
To prepare genomic DNA and RNA for sequencing, GP3 and ZY were cultured on PDA plates overlaid by cellophane films and maintained in the dark at 30 °C for 3–4 days. Mycelia were collected and grounded for DNA and RNA extraction. High-molecular-weight genomic DNA for single-molecule real-time (SMRT) was extracted using the SMRTbellTM Templated Prep Kit 1.0 (PACBIO). The genomic DNA for Illumina sequencing was extracted using a CTAB method as previously described [54]. Total RNA was extracted from mycelia using the TRIZOL Kit (Invitrogen, Carlsbad, CA, USA) following the manufacture’s protocol.
Genome sequencing and assembly
For PacBio Sequel genome sequencing, high molecular weight genomic DNA (20 μg) was random sheared with Covaris- g-Tube with a goal of DNA fragments of approximately 20 kb and end-repaired according to the manufacturer’s instructions. A blunt-end ligation reaction followed by exonuclease treatment was performed to generate the SMRT Bell template, then the library was qualified and quantified using an Agilent Bioanalyzer 12 kb DNA Chip (Agilent Technologies, Santa Clara, CA, USA) and a Qubit fluorimeter (Invitrogen, Carlsbad, CA, USA). SMRT Bell cells were sequenced using the PacBio Sequel sequencing platform (Nextomics Biosciences, Co., Ltd., Wuhan, China). After adaptor removed and low quality reads filtered out, a total of 594,166 and 1,124,070 high quality reads covering 6,343,564,369 and 9,972,706,733 base pairs were generated for S. rolfsii strains GP3 and ZY, respectively.
For Illumina sequencing, about 100 μg of genomic DNA were sheared to ~ 180 bp using a Covaris LE instrument and adapted for Illumina sequencing on Illumina Hiseq Xten platform (San Diego, CA, USA) by NextOmics Biosciences. Illumina short reads were trimmed using Trimmomatic version 0.36 [55], read length for Illumina sequencing procedure for genomic DNA was 150 bp, a total of 6.42 Gb and 7.03 Gb clean data were yielded for GP3 and ZY, respectively.
The cDNA libraries were prepared by Illumina TreSeq RNA Sample Preparation Kit (Illumina, Inc., San Diego, CA, USA) and validated according to Illumina’s low-throughput protocol. The RNA-seq was conducted on an Illumina HiSeq 2500 Platform with 150 bp paired-end strategy.
A de novo genome assemblies of ZY and GP3 were generated with the PacBio Sequel reads using CANU pipeline (v1.5) [56] with default setting. The assemblies were adjusted using Arrow program, and polished using Illumina reads by Pilon [20]. Finally, the completeness of assemblies was evaluated using BUSCO [21].
Repetitive elements analysis
Repetitive elements were identified by using different methods. Transposable elements were analyzed using four programs, two programs for de novo prediction, including Repeat Moldeler (https://www.repeatmasker.org/RepeatModeler) and LTR finder [57], and the database based programs Repeat Masker (https://www.repeatmasker.org/) and Repeat-ProteinMasker (submodule in Repeatmasker) with default parameters to search Repbase [58]. Tandem Repeats Finder (TRF) was used to identify tandem repeat sequences [59]. MicroSAtellites (MISA) (https://www.plob.org/tag/misa) was used to identify simple sequence repeats (SSR) with default setting.
Genome annotation
Gene predication was performed by using a combination of ab initio-based and homology-based methods. To aid gene annotation, we generated transcript assemblies based on RNA of GP3 and ZY, respectively. For ab initio-based prediction, Augustus v2.4 [60] and Genscan (version 1.0) [61] were used to de novo predict protein coding genes with the default setting. Exonerate [62] was used to predict the gene structure with RNA-seq data. For homology-based predication, GeneWise [63] was used to predict protein coding genes by homology analysis with known protein sequences from six related species of Basidiomycota, including Galerina marginata, Gymnopus luxurians, Hydnomerulius pinastri, Jaapia argillacea, Piloderma croceum, and Plicaturopsis crispa. EvidenceModler (EVM) was used to compute the weighed consensus gene structure annotation [64]. The final gene sets were obtained after removed TE transposable elements by Tranposon PSI [65].
The predicted gene sets of S. rolfsii GP3 and ZY were functionally annotated based on similarity comparison with homologous in public databases. BLASTP was used to align the protein sequences by automated searches in NCBI-NR, Swiss-Prot (https://www.expasy.org/sprot/), KEGG, GO and KOG database with E-values≤1e-5. Gene function domain annotation was conducted by InterProScan program [66]. The pathway analyses were conducted by KAAS-KEGG Automatic Annotation Serve [67]. The candidate non-coding RNA (ncRNA) was annotated by two approaches, BLAST was used to align the S. rolfsii genome against the Rfam database [68], and tRNA scan-SE [69] and RNAmmer [70] were used to predict tRNAs and rRNAs, respectively.
Analysis of orthologous gene families in Agaricomycetes
Orthology comparison was conducted by OrthoMCL [22] (http://va.orthomcl.org) with e-value less than le-5 among protein sets of S. rolfsii GP3, ZY and nine related species of Agaricomycetes, including Armillaria gallica (GenBank: GCA_002307695.1), Auricularia subglabra (GenBank: GCA_000265015.1), Exidia glandulosa (GenBank: GCA_001632375.1), Galerina marginata (GenBank: GCA_000697645.1), Gymnopus luxurians (GenBank: GCA_000827265.1), Hydnomerulius pinastri (GenBank: GCA_000827185.1), Psilocybe cyanescens (GenBank: GCA_002938375.1), Scleroderma citrinum (GenBank: GCA_000827425.1), and Piloderma croceum (GenBank: GCA_000827315.1).
Phylogenetic analysis and synteny analysis
The phylogenetic tree of S. rolfsii GP3, ZY and the above related nine species of Agaricomycetes was constructed by single copy gene based on the orthologous gene family analysis. Mafft [71] software was conducted to align the protein sequence of the single copy gene, converted to coding sequence alignment. Gblocks [72] was used to extract the well-aligned regions of each coding sequence alignments. RAxML 8.2.12 [73] was carried out to generate the maximum-likelihood tree with 100 bootstrap replicates with P. cyanescens as an outgroup. The whole genome aligner Murmer 3.06 [74] was used for comparative analysis of the assemblies of GP3 and ZY. Dot plots between contigs of GP3 and ZY were created by MuMerplot programs from the MuMmer package.
Identification of the pathogenicity related genes
The S. rolfsii GP3 and ZY protein sets were used to conduct a BLASTP search against PHI base (a database of Pathogen Host Interactions) with e-value less than 1e-5 to identify pathogenicity genes. Putative carbohydrate active enzymes (CAZymes) of S. roflsii GP3 and ZY were annotation using dbCAN (dbCAN HMMs 5.0) [75] servers, with an e-value of less than 1e-5 and more than 70% coverage. CAZymes were classified by following modules: Glycoside Hydrolases (GHs), Polysaccharide Lyases (PLs), Carbohydrate Esterases (CEs), Glycosyl Transferase (GTs), Carbohydrate-Binding Modules (CBMs), and Auxillary Activities (AAs) as described in CAZyme database classification (http://www.cazy.org) [76].
Secretome and effector predication
The predicted secretome of S. rolfsii strains GP3 and ZY was conducted based on the following pipeline. SignalP version 4.0 [77] was used to analyze signal peptide and cleavage sites of S. rolfsii GP3 and ZY proteins. Candidate proteins with signal peptide were identified by Protcomp 9.0 (http://www.softberry.com/berry.phtml) using the LocDB and PotLoc DB databases and proteins predicted as extracellular or unknown were kept for next analysis. The candidate proteins were conducted by TMHMM version 2.0 [78] to identify protein with transmembrane domains, and all proteins with 0 TM or 1 TM, if located in the predicted N- terminal signal peptide, were kept. The candidate proteins that harbored a putative glycophosphatidylinositol membrane-anchoring domain were identified by GPIsom (https://gpi.unibe.ch/) [79]. The remaining proteins without GPI-anchor were predicted with Target P [80], proteins with a Target P Loc = S or – were kept in the final secretome databases. The candidate secretory proteins were blasted in NR database and PHI database to annotate the protein function and searched against CAZyme database for function of CAZymes. The candidate effectors were identified by passing the secretome through the program Effector P 1.0 [81]. Putative effectors were screened for those candidates with molecular weight ranged from 50 to 300 amino acids, and at least 4 cysteine amino acids in their sequences [82–84].
Secondary metabolites synthetic gene cluster predication
Secondary metabolites synthetic gene clusters were predicted by the web-based software antiSMASH (antibiotics and Secondary Metabolite Analysis 4.0) [85].
Supplementary Information
Additional file 1: Figure S1. Statistics of BUSCO assessment of S. rolfsii GP3 and ZY genome assemblies. a Total searched BUSCOs of S. rolfsii GP3 and ZY; b Distribution of different BUSCOs in GP3 and ZY. S, Complete and single-copy BUSCOs; D, Complete and duplicated BUSCOs; F, Fragmented BUSCOs; M, Missing BUSCOs
Additional file 2: Figure S2. Genome annotation statistics of S. rolfsii GP3 and ZY by blasting against five databases including GO, KOG, KEGG, NR and Swiss_pro
Additional file 3: Figure S3. KOG distribution of predicted proteins of S. roflsii GP3 and ZY
Additional file 4: Figure S4. Go annotation enrichment analysis of genes of S. rolfsii GP3 and ZY. a Number of genes in biological process, cellular component, and molecular function of S. rolfsii GP3 and ZY; b Percent of genes involved in molecular function, biological process, and cellular component of S. rolfsii GP3; c Percentage of genes involved in molecular function, biological process, and cellular component of S. rolfsii ZY
Additional file 5: Figure S5. KEGG classification of genes of S. rolfsii GP3 and ZY. a Distribution of genes among processes, metabolism, and organismal systems of S. rolfsii GP3; b Distribution of genes among processes, metabolism, and organismal systems of S. rolfsii ZY
Additional file 6: Figure S6. Analysis of orthologs of two S. rolfsii strains and other species in Agaricomycetes
Additional file 7: Figure S7. The pipeline for putative secretomes and effectors analysis of S. roflsii GP3 and ZY
Additional file 8: Table S1. List of ncRNAs in genomes of S. rolfsii GP3 and ZY
Additional file 9: Table S2. Comparison of gene families of GP3, ZY, and other species in Agaricomycetes
Additional file 10: Table S3. Identification of repetitive elements of S. rolfsii GP3 and ZY by five programs
Additional file 11: Table S4. List of repetitive elements of each category in the genomes of S. rolfsii GP3, ZY, and MR10
Additional file 12: Table S5. Unique Pathogen Host Interaction (PHI) genes in the genome of S. rolfsii GP3 and ZY
Additional file 13: Table S6. Comparison of CAZymes involved in cellulose degradation in the genomes of plant pathogens. The values reflect the total numbers in each family of CAZymes.
Additional file 14: Table S7. Comparison of CAZymes involved in hemicellulose degradation in the gnomes of plant pathogens. The values reflect the total numbers in each family of CAZymes.
Additional file 15: Table S8. Comparison of CAZymes involved in pectin degradation in the genomes of plant pathogens. The values reflect the total numbers in each family of CAZymes.
Additional file 16: Table S9. List of secreted CAZymes in the genome of S. rolfsii GP3 and ZY
Additional file 17: Table S10. Overlapping of putative effectors with PHI and CAZymes genes
Acknowledgments
We thank Dr. Huizhang Zhao for analyzing CAZymes of S. rolfsii ZY and GP3.
Abbreviations
- AA
Auxiliary activity
- A. subglabra
Auricularia subglabra
- B. cinerea
Botrytis cinerea
- CAZyme
Carbohydrate-active Enzyme
- CBM
Carbohydrate-binding module
- CE
Carbohydrate esterase
- endo-PG
Endo-polygalacturonase
- E. glandulosa
Exidia glandulosa
- GH
Glycoside hydrolase
- G. luxurians
Gymnopus luxurians
- GPI
Glycosylphosphatidylinositol anchor
- GT
Glycosyltransferase
- P. croceum
Piloderma croceum
- P. cyanescens
Psilocybe cyanescens
- P. tinctorius
Pisolithus tinctorius
- PL
Polysaccharide lyase
- PHI
Pathogen- host interaction
- P. graminis
Puccinia graminis
- R. solani
Rhizoctonia solani
- S. rolfsii
Sclerotium rolfsii
- S. sclerotiorum
Sclerotinia sclerotiorum
- U. maydis
Ustilago maydis
- V. dahliae
Verticillium dahliae
Authors’ contributions
The research was designed and written by LYY, bioinformatics analysis was performed by ZHW, the experiments were performed by WDS, PMF, LYW, and YPK, manuscript was critically revised by YL, DXH, YNC, XW, HS, and BSL. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31971981).
Availability of data and materials
The genomes of S. rolfsii strains GP3 and ZY were deposited in GenBank under BioProject numbers: PRJNA635225, PRJNA635226, and BioSample numbers: SAMN15029893, SAMN15029894, respectively.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Liying Yan, Email: yanliying@caas.com.
Zhihui Wang, Email: wangzhihui0229@126.com.
Wanduo Song, Email: songwanduo@163.com.
Pengmin Fan, Email: 907459930@qq.com.
Yanping Kang, Email: xiaohaozi412@163.com.
Yong Lei, Email: leiyong@caas.cn.
Liyun Wan, Email: susun19846@163.com.
Dongxin Huai, Email: dxhuai@163.com.
Yuning Chen, Email: ynchen126@126.com.
Xin Wang, Email: wangxin456_2000@163.com.
Hari Sudini, Email: h.sudini@cgiar.org.
Boshou Liao, Email: liaoboshou@163.com.
References
- 1.Nalim FA, Starr JL, Woodard KE, Segner S, Keller NP. Mycelial compatibility groups in Texas peanut field populations of Sclerotium rolfsii. Phytopathology. 1995;85(12):1507–1512. doi: 10.1094/Phyto-85-1507. [DOI] [Google Scholar]
- 2.Harlton CE, Uvesque CA, Punja ZK. Genetic diversity in Sclerotium (Athelia) rolfsii and related species. Phytopathology. 1995;85(10):1269–1281. doi: 10.1094/Phyto-85-1269. [DOI] [Google Scholar]
- 3.Cilliers A, Herselman L, Pretorius Z. Genetic variability within and among mycelia compatibility groups of Sclerotium rolfsii in South Africa. Phytopathology. 2000;90(9):1026–1031. doi: 10.1094/PHYTO.2000.90.9.1026. [DOI] [PubMed] [Google Scholar]
- 4.Punja ZK. The biology, ecology, and control of Sclerotium rolfsii. Annu Rev Phytopathol. 1985;23(1):97–127. doi: 10.1146/annurev.py.23.090185.000525. [DOI] [Google Scholar]
- 5.Backman PA, Brenneman TB. Peanut stem rot. In: Kakalis-Burelle PDM, Rodriguez-kabana R, Smith DH, Subrahmanyam P, editors. Compendium of Peanut Diseases. 2. MN: American Phytopathological Society; 1997. pp. 36–37. [Google Scholar]
- 6.Kemerait R. Georgia plant disease loss estimates. In: Woodward JWW. editor. CAES Publications UGA Cooperative Extension. 2011;P 102–4:13.
- 7.Mayee CD, Datur VV. Diseases of groundnut in the tropics. Rev Tropic Plant Pathol. 1998;5:85–118. [Google Scholar]
- 8.Chen KR, Ren L, Xu L, Chen W, Liu F, Fang XP. Research progress on peanut southern stem rot caused by Sclerotium rolfsii. Chin J Oil Crop Sci. 2018;40(2):302–308. [Google Scholar]
- 9.Branch WD, Brenneman TB. Stem rot disease evaluation of mass-selected peanut populations. Crop Prot. 1999;18(2):127–130. doi: 10.1016/S0261-2194(98)00103-3. [DOI] [Google Scholar]
- 10.Branch WD, Brenneman TB. Field evaluation for the combination of white mold and tomato spotted wilt disease resistance among peanut genotypes. Crop Prot. 2009;28(7):595–598. doi: 10.1016/j.cropro.2009.03.008. [DOI] [Google Scholar]
- 11.Woodward JE, Brenneman TB, Kemerait RC, Smith NB, Culbreath AK, Stevenson KL. Use of resistant cultivars and reduced fungicide programs to manage peanut diseases in irrigated and non-irrigated fields. Plant Dis. 2008;92(6):896–902. doi: 10.1094/PDIS-92-6-0896. [DOI] [PubMed] [Google Scholar]
- 12.Yan LY, Song WD, Lei Y, Wan LY, Huai DX, Kang YP, Chen YN, Liao BS. Evaluation of peanut accessions for resistance to Sclerotium stem rot. Chin J Oil Crop Sci. 2019;41(5):1–7. [Google Scholar]
- 13.Li JT, Fan HF, Wang JM, Liu F. Toxicity and field control efficacy of four fungicides against Sclerotium rolfsii. Chin J Oil Crop Sci. 2013;35(6):686–691. [Google Scholar]
- 14.Remesal E, Jordan-Ramirez R, Jimenez-Diaz RM, Navas-Cortes JA. Mycelial compatibility groups and pathogenic diversity in Sclerotium rolfsii populations from sugar beet crops in Mediterranean-type climate regions. Plant Pathol. 2012;61(4):739–753. doi: 10.1111/j.1365-3059.2011.02552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandra Sekhar Y, Khayum Ahammed S, TNVKV P, Sarada Jayalakshmi Devi R. Morphological and pathogenic variability of Sclerotium rolfsii isolates causing stem rot in groundnut. Int J Pure App Biosci. 2017;5(5):478–487. doi: 10.18782/2320-7051.3003. [DOI] [Google Scholar]
- 16.Punja ZK, Huang JS, Jenkins SF. Relationship of mycelial growth and production of oxalic acid and cell wall degrading enzymes to virulence in Sclerotium rolfsii. Can J Plant Pathol. 1985;7(2):109–216. doi: 10.1080/07060668509501485. [DOI] [Google Scholar]
- 17.Jebaraj MD, Aiyanathan KEA, Nakkeeran S. Virulence and genetic diversity of Sclerotium rolfsii Sacc. Infecting groundnut using nuclear (RAPD & ISSR) markers. J Environ Biol. 2017;38(1):147–159. doi: 10.22438/jeb/38/1/ms-274. [DOI] [Google Scholar]
- 18.Cooper WE. Strains of, resistance to, and antagonists of Sclerotium rolfsii. Phytopathology. 1961;51:113–116. [Google Scholar]
- 19.Song WD, Yan LY, Lei Y, Wan LY, Huai DX, Kang YP, Ren XP, Jiang HF, Liao BS. Analysis of genetic variation among Sclerotium rolfsii isolates from China based on mycelial compatibility groups, ITS sequence and biological characteristics. Acta Phytopathol Sin. 2018;48(3):305–312. [Google Scholar]
- 20.Iquebal MA, Rukam S, Parakhia TMV, Deepak S, Sarika J, Rathod VM, Padhiyar SM, Kumar N, Rai A, Kumar D. Draft whole genome sequence of groundnut stem rot fungus Athelia rolfsii revealing genetic architect of its pathogenicity and virulence. Sci Rep. 2017;7(1):5299. doi: 10.1038/s41598-017-05478-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 22.Li L, CJJr S, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Floiudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, Martinez AT, Otillar T, Spatafora JW, Yadav JS, et al. The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336(6089):1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 24.Nagy LG, Riley R, Tritt A, Adam C, Daum C, Daum C, Flouda D, Sun H, Yadav JS, Tanilinan J, et al. Comparative genomics of early- diverging mushroom-forming fungi provides insights into the origins of lignocellulose decay capabilities. Mol Biol Evol. 2016;33(4):959–970. doi: 10.1093/molbev/msv337. [DOI] [PubMed] [Google Scholar]
- 25.Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canbäck B, Choi C, Cichocki N, Clum A, et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet. 2015;47(4):410–415. doi: 10.1038/ng.3223. [DOI] [PubMed] [Google Scholar]
- 26.Bateman DF, Beer SV. Simultaneous production and synergistic action of oxalic acid and polygalacturonase during pathogenicity by Sclerotium rolfsii. Phytopathology. 1965;55:204–211. [PubMed] [Google Scholar]
- 27.Bateman DF. The polygalacturonase complex produced by Sclerotium rolfsii. Physiol Plant Pathol. 1972;2(2):175–184. doi: 10.1016/0048-4059(72)90025-2. [DOI] [Google Scholar]
- 28.Bateman DF. Some characteristics of the cellulase system produced by Sclerotium rolfsii Sacc. Phytopathology. 1969;59:37–42. [PubMed] [Google Scholar]
- 29.Xie CZ, Huang CH, Vallad GE. Mycelial compatibility and pathogenic diversity among Sclerotium rolfsii isolates in the southern United States. Plant Dis. 2014;98(12):1685–1694. doi: 10.1094/PDIS-08-13-0861-RE. [DOI] [PubMed] [Google Scholar]
- 30.Saraswathi M, Jaya MR. Variation in oxalic acid production by groundnut isolates of Sclerotium rolfsii. Biores Bull. 2014;3(2):1–3. [Google Scholar]
- 31.Amselem J, Cuomo CA, van Kan JAL, Viaud M, Benito EP, Couloux A, Coutinho PM, de Vries RP, Dyer PS, Fillinger S, Fournier E, Gout L, Hahn M, Kohn L, Lapalu N, Plummer KM, Pradier JM, Quévillon E, Sharon A, Simon A, ten Have A, Tudzynski B, Tudzynski P, Wincker P, Andrew M, Anthouard V, Beever RE, Beffa R, Benoit I, Bouzid O, Brault B, Chen Z, Choquer M, Collémare J, Cotton P, Danchin EG, da Silva C, Gautier A, Giraud C, Giraud T, Gonzalez C, Grossetete S, Güldener U, Henrissat B, Howlett BJ, Kodira C, Kretschmer M, Lappartient A, Leroch M, Levis C, Mauceli E, Neuvéglise C, Oeser B, Pearson M, Poulain J, Poussereau N, Quesneville H, Rascle C, Schumacher J, Ségurens B, Sexton A, Silva E, Sirven C, Soanes DM, Talbot NJ, Templeton M, Yandava C, Yarden O, Zeng Q, Rollins JA, Lebrun MH, Dickman M, et al. Genomic analysis of the necrotophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011;7(8):e1002230. doi: 10.1371/journal.pgen.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiapello H, Mallet L, Guerin C, Aguileta G, Amselem J, Kroj T, Ortega-Abboud E, Lebrun MH, Henrissat B, Gendrault A, et al. Deciphering genome content and evolutionary relationships of isolates from the fungus Magnaporthe oryzae attacking different host plants. Genome Biol Evol. 2015;7(10):2896–2912. doi: 10.1093/gbe/evv187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubicek CP, Starr TL, Glass NL. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu Rev Phytopathol. 2014;52(1):427–451. doi: 10.1146/annurev-phyto-102313-045831. [DOI] [PubMed] [Google Scholar]
- 34.Guo M, ChenY DY, DongY GW, Zhai S, Dong SM, Zhang ZG, Wang YC, Wang P, Zheng XB. The bZIP transcription factor MoAP1 mediates the oxidative stress response and is critical for pathogenicity of the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 2011;7(2):e1001302. doi: 10.1371/journal.ppat.1001302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malinovsky FG, Batoux M, Schwessinger B, Youn JH, Stransfeld L, Win J, Kim SK, Zipfel C. Antagonistic regulation of growth and immunity by the Arabidopsis basic helix-loop-helix transcription factor homolog of brassinosteroid enhanced expression interacting with increased leaf inclination binding bHLH1. Plant Physiol. 2014;164(3):1443–1455. doi: 10.1104/pp.113.234625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Liu XB, Cai JM, Chen YP, Li BX, Guo ZK, Huang GX. Genomic characteristics and comparative genomics analysis of the endophytic fungus Sarocladium brachiariae. BMC Genomics. 2019;20(1):782. doi: 10.1186/s12864-019-6095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cook D, Mesarich CH, Thomma BPHJ. Understanding plant immunity as a surveillance system to detect invasion. Annu Rev Phytopathol. 2015;53(1):541–563. doi: 10.1146/annurev-phyto-080614-120114. [DOI] [PubMed] [Google Scholar]
- 38.Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444(7117):323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 39.Rovenich H, Boshoven JC, Thomma BPHJ. Filamentous pathogen effector functions: of pathogens, hosts and microbiomes. Curr Opin Plant Biol. 2014;20:96–103. doi: 10.1016/j.pbi.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Friesen TL, Faris JD, Solomon PS, Oliver RP. Host-specific toxins: effectors of necrotrophic pathogenicity. Cell Microbiol. 2008;10(7):1421–1428. doi: 10.1111/j.1462-5822.2008.01153.x. [DOI] [PubMed] [Google Scholar]
- 41.Lu S, Gillian Turgeon B, Edwards MC. A ToxA-like protein from Cochliobolus heterostrophus induces light-dependent leaf necrosis and acts as a virulence factor with host selectivity on maize. Fungal Genet Biol. 2015;81:12–24. doi: 10.1016/j.fgb.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 42.Whigham E, Qi S, Mistry D, Surana P, Xu R, Fuerst G, Pliego C, Biindschedler LV, Spanu PD, Dickerson JA, et al. Broadly conserved fungal effector BEC1019 suppresses host cell death and enhances pathogen virulence in powdery mildew of barley (Hordeum vulgare L.) Mol Plant-Microbe Interact. 2015;28(9):968–983. doi: 10.1094/MPMI-02-15-0027-FI. [DOI] [PubMed] [Google Scholar]
- 43.Lyu X, Shen C, Fu Y, Xie J, Jiang D, Li G, Cheng J. A small secreted virulence-related protein is essential for the necrotrophic interactions of Sclerotinia sclerotiorum with its host plants. PLoS Pathog. 2016;12(2):e1005435. doi: 10.1371/journal.ppat.1005435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang Y, Yajima W, Davis MR, Kav NNV, Strelkov SE. Disruption of a gene encoding a hypothetical secreted protein from Sclerotinia sclerotiorum reduces its virulence on canola (Brassica napus) Can J Plant Pathol. 2013;35(1):46–55. doi: 10.1080/07060661.2012.745904. [DOI] [Google Scholar]
- 45.Lyu X, Shen C, Fu Y, Xie J, Jiang D, Li G, Cheng J. Comparative genomic and transcriptional analyses of the carbohydrate-active enzymes and secretomes of phytopathogenic fungi reveal their Agaricomycetes roles during infection and development. Sci Rep. 2015;5(1):15565. doi: 10.1038/srep15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kombrink A, Sanchez-Vallet A, Thomma BPHJ. The role of chitin detection in plant-pathogen interactions. Microbes Infect. 2011;13(14-15):1168–1176. doi: 10.1016/j.micinf.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Z, Fradin EF, de Jonge R, van Esse HP, Smit P, Liu CM, Thomma BPHJ. Optimized agroinfiltration and virus-induced gene silencing to study Ve1-mediated Verticillium resistance in tobacco. Mol Plant-Microbe Interact. 2013;26(2):182–190. doi: 10.1094/MPMI-06-12-0161-R. [DOI] [PubMed] [Google Scholar]
- 48.van Kan JA. Licensed to kill: the lifestyle of a necrotrophic plant pathogen. Trends Plant Sci. 2006;11(5):247–253. doi: 10.1016/j.tplants.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 49.Kombrink A, Thomma BPHJ. LysM effectors: secreted proteins supporting fungal life. PLoS Pathog. 2013;9(12):e1003769. doi: 10.1371/journal.ppat.1003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rouxel T, Grandaubert J, Hane JK, Hoede C, van de Wouw AP, Couloux A, Dominguez V, Anthouard V, Bally P, Bourras S, Cozijnsen AJ, Ciuffetti LM, Degrave A, Dilmaghani A, Duret L, Fudal I, Goodwin SB, Gout L, Glaser N, Linglin J, Kema GHJ, Lapalu N, Lawrence CB, May K, Meyer M, Ollivier B, Poulain J, Schoch CL, Simon A, Spatafora JW, Stachowiak A, Turgeon BG, Tyler BM, Vincent D, Weissenbach J, Amselem J, Quesneville H, Oliver RP, Wincker P, Balesdent MH, Howlett BJ, et al. Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat Commun. 2011;2(1):202. doi: 10.1038/ncomms1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derbyshire M, Denton-Giles M, Hegedus D, Seifbarghi S, Rollins J, van Kan J, Seidl MF, Faino L, Mbengue M, Navaud O, et al. The complete genome sequence of the phytopathogenic fungus Sclerotinia slcerotiorum reveals insights into the genome architecture of broad host range pathogens. Genome Biol Evol. 2017;9(3):593–618. doi: 10.1093/gbe/evx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kritzman G, Chet I, Kenis Y. The role of oxalic acid in the pathogenic behavior of Sclerotium rolfsii Sacc. Expt Mycol. 1977;1(4):280–285. doi: 10.1016/S0147-5975(77)80003-0. [DOI] [Google Scholar]
- 53.Shokes FM, Weber Z, Gorbet DW, Pudelko HA, Taczanowski M. Evaluation of peanut genotypes for resistance to southern stem rot using an agar disk technique. Peanut Sci. 1998;25(1):12–17. doi: 10.3146/i0095-3679-25-1-4. [DOI] [Google Scholar]
- 54.Wu ZH, Wang TH, Huang W, Qu YB. A simplified method for chromosome DNA preparation from filamentous Fungi. Mycosystema. 2001;20:575–577. [Google Scholar]
- 55.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;3(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao X, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:265–268. doi: 10.1093/nar/gkm456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet genome Res. 2005;110(14):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 59.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:465–467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Bio. 1997;268(1):78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 62.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005;6(1):31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Birney E, Durbin R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000;10(4):547–548. doi: 10.1101/gr.10.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, Whte O, Buell CR, Wortman JR. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9(1):R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yagi M, Kosugi S, Hirakawa H, Ohimiya A, Tanase K, Harada T, Kishimoto K, Nakayama M, Ichimura K, Onozaki T, et al. Sequence analysis of the genome of carnation (Dianthus caryophyllus L.) DNA Res. 2014;21(3):231–241. doi: 10.1093/dnares/dst053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jones P, Binns D, Chang HY, Fraser M, Li WZ, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 2007; 35:W182-W185, Web Server, DOI: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed]
- 68.Jones DA, Bertazzoni S, Turo CJ, Syme RA, Hane JK. Bioinformatic prediction of plant-pathogenicity effector proteins of fungi. Curr Opin Microbiol. 2018;46:43–49. doi: 10.1016/j.mib.2018.01.017. [DOI] [PubMed] [Google Scholar]
- 69.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lagesen K, Hallin P, Rodland EA, Starfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 73.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2014;5:12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yin YB, Mao XZ, Yang JC, Chen X, Mao FL, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acid Res. 2012;40:445–451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:490–495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 78.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 79.Fankhauser N, Maser P. Identification of GPI anchor attachment signals by a Kohonen self-organizing map. Bioinformatics. 2005;21(9):1846–1852. doi: 10.1093/bioinformatics/bti299. [DOI] [PubMed] [Google Scholar]
- 80.Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S. Prediction of twin-arginine signal peptides. BMC Bioinform. 2005;6(1):167. doi: 10.1186/1471-2105-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sperschneider J, Gardiner DM, Dodds PN, Tini F, Covarelli L, Singh KB, Manners JM, Taylor JM. EffectorP: predicting fungal effector proteins from secretomes using machine learning. New Phytol. 2015;210(2):743–761. doi: 10.1111/nph.13794. [DOI] [PubMed] [Google Scholar]
- 82.de Wit PJ, Mehrabi R, van den Burg HA, Stergiopoulos I. Fungal effector proteins: past, present and future. Mol Plant Pathol. 2009;10(6):735–747. doi: 10.1111/j.1364-3703.2009.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Franceschetti M, Maqbool A, Jimenez-Dalmaroni MJ, Pennington HG, Kamoun S, Banfield MJ. Effectors of filamentous plant pathogens: commonalities amid diversity. Microbiol Mol Biol Rev. 2017;81:e00066–e00016. doi: 10.1128/MMBR.00066-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sonah H, Deshmukh RK, Belanger RR. Computational prediction of effector proteins in fungi: opportunities and challenges. Front Plant Sci. 2016;7:126. doi: 10.3389/fpls.2016.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blin K, Wolf T, Chevrette MG, Lu ZW, Schwalen CJ, Kautsar SA, Suarez Duran HG, los Santos ELC d, Kim HU, Nave M, et al. AntiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017;45(W1):W36–W41. doi: 10.1093/nar/gkx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Statistics of BUSCO assessment of S. rolfsii GP3 and ZY genome assemblies. a Total searched BUSCOs of S. rolfsii GP3 and ZY; b Distribution of different BUSCOs in GP3 and ZY. S, Complete and single-copy BUSCOs; D, Complete and duplicated BUSCOs; F, Fragmented BUSCOs; M, Missing BUSCOs
Additional file 2: Figure S2. Genome annotation statistics of S. rolfsii GP3 and ZY by blasting against five databases including GO, KOG, KEGG, NR and Swiss_pro
Additional file 3: Figure S3. KOG distribution of predicted proteins of S. roflsii GP3 and ZY
Additional file 4: Figure S4. Go annotation enrichment analysis of genes of S. rolfsii GP3 and ZY. a Number of genes in biological process, cellular component, and molecular function of S. rolfsii GP3 and ZY; b Percent of genes involved in molecular function, biological process, and cellular component of S. rolfsii GP3; c Percentage of genes involved in molecular function, biological process, and cellular component of S. rolfsii ZY
Additional file 5: Figure S5. KEGG classification of genes of S. rolfsii GP3 and ZY. a Distribution of genes among processes, metabolism, and organismal systems of S. rolfsii GP3; b Distribution of genes among processes, metabolism, and organismal systems of S. rolfsii ZY
Additional file 6: Figure S6. Analysis of orthologs of two S. rolfsii strains and other species in Agaricomycetes
Additional file 7: Figure S7. The pipeline for putative secretomes and effectors analysis of S. roflsii GP3 and ZY
Additional file 8: Table S1. List of ncRNAs in genomes of S. rolfsii GP3 and ZY
Additional file 9: Table S2. Comparison of gene families of GP3, ZY, and other species in Agaricomycetes
Additional file 10: Table S3. Identification of repetitive elements of S. rolfsii GP3 and ZY by five programs
Additional file 11: Table S4. List of repetitive elements of each category in the genomes of S. rolfsii GP3, ZY, and MR10
Additional file 12: Table S5. Unique Pathogen Host Interaction (PHI) genes in the genome of S. rolfsii GP3 and ZY
Additional file 13: Table S6. Comparison of CAZymes involved in cellulose degradation in the genomes of plant pathogens. The values reflect the total numbers in each family of CAZymes.
Additional file 14: Table S7. Comparison of CAZymes involved in hemicellulose degradation in the gnomes of plant pathogens. The values reflect the total numbers in each family of CAZymes.
Additional file 15: Table S8. Comparison of CAZymes involved in pectin degradation in the genomes of plant pathogens. The values reflect the total numbers in each family of CAZymes.
Additional file 16: Table S9. List of secreted CAZymes in the genome of S. rolfsii GP3 and ZY
Additional file 17: Table S10. Overlapping of putative effectors with PHI and CAZymes genes
Data Availability Statement
The genomes of S. rolfsii strains GP3 and ZY were deposited in GenBank under BioProject numbers: PRJNA635225, PRJNA635226, and BioSample numbers: SAMN15029893, SAMN15029894, respectively.