

Abstract
T cell therapies utilizing engineered T cells show great promise for cancer immunotherapy, as illustrated by the CD19 paradigm. Much of the excitement about this approach, and second generation CARs in particular, is due to the dramatic clinical results recently reported by a few centers, especially in acute lymphoblastic leukemia, and the applicability of this approach, in principle, to a wide range of cancers. Extending the use of CAR therapies to cancers other than B cell malignancies will require selective tumor targeting with minimal or acceptable “on-target, off-tumor” effects. The identification of new CAR target antigens is thus one of the next big challenges to address. Recognizing the paucity of currently available tumor-specific targets, we have developed broadly applicable approaches to enhance the tumor selectivity and safety of engineered T cells. Here we review two promising concepts. One is to improve tumor targeting based on combinatorial antigen recognition. The other utilizes receptors that provide antigen-specific inhibition, which we named iCARs, to divert T cells from the normal tissues one wants to protect.
Introduction
Engineered adoptive T cell therapies that utilize exogenous receptors to redirect the specificity and function of T lymphocytes are emerging as a promising cancer therapy1. The targeting of tumor cells (or tumor stroma) may be mediated by expressing either a T cell receptor (TCR) or a chimeric antigen receptor (CAR)2,3. In addition to mediating antigen recognition, CARs that encode costimulatory domains have the capacity to augment T cell function and persistence, extending the function of the transduced antigen receptor far beyond what a TCR or first generation CAR can achieve3. Dual-signaling CARs not only redirect T cell specificity but also exert a broad range of immune-modulatory functions4,5. Such CARs, which we named “second generation CARs”, have recently induced remarkable responses in patients with B cell malignancies6–10, most strikingly in patients with acute lymphoblastic leukemia11–13.
In principle, any cell surface molecule can be targeted through a CAR, thus over-riding tolerance to self-antigens and filling any gaps in the T cell repertoire. CARs are not HLA-restricted, and may bind to any cell surface structure, whether a protein, carbohydrate or glycolipid. Identifying appropriate targets for CAR therapy is critical as it is challenging to identify targets that allow for complete tumor eradication, while avoiding damage to normal tissues that express the same target antigen (“on-target, off-tumor effect”) complications from reacting to target on normal cells. Over a decade ago, we provided proof-of-principle of CAR T cell function utilizing human peripheral blood T cells targeted to CD19 in order to eradicate a broad range of B cell malignancies14. CD19 is a cell-surface antigen found on most B lineage lymphomas and leukemias15. We chose CD19 because it is highly expressed in these tumors, uncommonly lost and, most importantly, only found amongst normal tissues in the B cell lineage. Thus, a successful therapy would be expected to induce a B cell aplasia, as is indeed observed in murine models16,17 and in patients treated with CD19 CAR-targeted T cells6–13. Most importantly, no cytotoxicity against another tissue or organ is expected, and indeed none has been reported to date.
Other instances of “on-target, off-tumor” responses have been observed, sometimes with serious consequences. Undesirable but manageable toxicities have been observed upon targeting carbonic anhydrase IX (jaundice) or MART-1 (ophthalmic and auditory effects)18,19. The targeting of HER2 with a CAR and MAGE-A3 antigens with TCRs has led to fatal complications caused by unanticipated reactivity in the cardiovascular or central nervous systems20–23.
One therapeutic solution is to utilize suicide genes in the T cell therapy to be able to abort a threatening undesirable response24–26. The downsides to this effective approach are that it is reactive, rather than preventive, and further results in abrogating therapeutic benefits as all infused T cells undergo ablation irrespective of their function or location. Approaches that prevent rather than treat adverse effects would be much preferable. The simplest scenario is to have an absolutely tumor-specific target. This is, however, seldom the case amongst currently known candidates5,27. Here we review two promising alternative approaches. One is to improve tumor targeting based on combinatorial antigen recognition28. The other is to utilize receptors that provide antigen-specific inhibition, which we named iCARs, to divert T cells from the normal tissues one wants to protect29.
Combinatorial antigen recognition
In order for CAR and TCR therapies to become a reality for the treatment of a spectrum of cancers, approaches to finely control the targeting of the therapies are needed. This is a challenge in the absence of an identified tumor-unique antigen for most tumors. We discuss here one approach to achieve tumor selectivity despite not having access to a tumor-restricted target. Thus, T cells may be rendered more selective through combinatorial antigen recognition, i.e. the requirement for recognizing a set combination of antigens in order to undergo full activation. We recently described a strategy that integrates combinatorial antigen recognition, split signaling, and balanced strength of T cell activation and costimulations20.
This approach requires the coexpression of two types of receptors, each one recognizing a different antigen and providing a different signal. T cell activation is provided through TCR or CAR-mediated recognition of one antigen, while costimulation is independently mediated by a chimeric costimulatory receptor (CCR)30 that is triggered by the presence of a second antigen. Such dual-targeting and division of labor is required but not sufficient to achieve the desired effect. Indeed, we found that T cells receiving both activating and costimulatory signals at one tumor site acquire enhanced function resulting in greater activity against tissues that only express the “activating antigen”28. Functionally, the dual-antigen expressing tissue becomes equivalent to a secondary lymphoid organ, from where “recharged” T cells that have received both activating and costimulatory signals may recirculate and thereafter display enhanced activity at other sites without requiring further costimulation. This pattern mirrors physiological responses wherein priming by professional antigen presenting cells presenting antigen and providing costimulation is followed by effective peripheral reactivity where costimulation is no longer required31. This outcome is contrary to the purpose of enhancing tumor selectivity and therefore needs to be prevented. Thus, for the dual-targeting, split-signaling approach to succeed, one further needs to abolish this abscopal effect and alter the activating signal. (Figure 1). To enforce tumor selectivity, we therefore diminished the efficiency of T cell activation to a level where it is ineffective without rescue provided by simultaneous CCR recognition of the second antigen.
Figure 1. Combinatorial Antigen Recognition Allows For Selective Tumor Eradication.
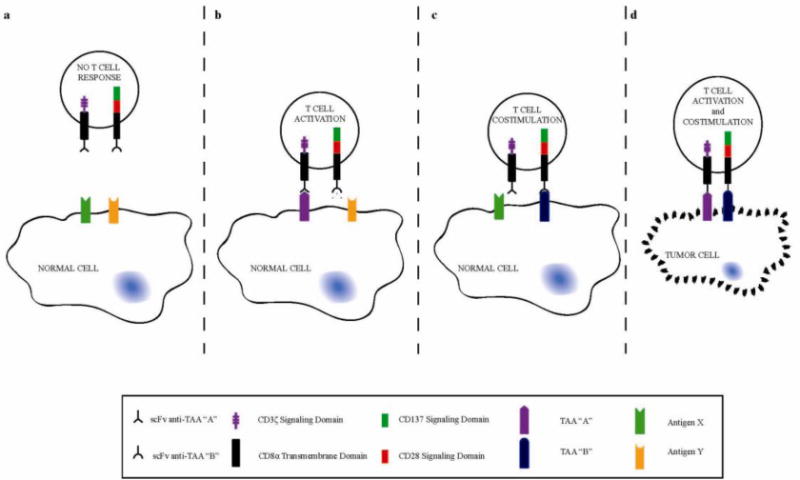
By using a CAR to supply a CD3ζ signal (purple) upon binding respective antigen (“A”) and a CCR to supply CD28 and CD137 signals (green and red) upon binding respective antigen (“B”), selective tumor eradication can be accomplished. Engineered T cells will remain unresponsive to cells not expressing either antigen specific to the CAR or CCR (A). Upon binding of the CAR alone (B), T cells can receive only T cell activation that can result in short-term cell lysis depending on the affinity or efficacy of the CAR binding alone. By reducing the affinity or efficacy of the CAR, activity to single positive cells can be avoided. Having only the CCR bind cells single positive cells (C) will engage T cell costimulation, but without activation no response of CCR binding alone can be measured. Only when T cells encounter tumor cells identified to be double positive for CAR and CCR respective antigens (D) can both T cell activation and costimulation occur, resulting in complete eradication of double positive tumor cells.
In a prostate cancer model, we engineered CARs that provide an activation signal upon recognition of prostate stem cell antigen (PSCA). Rather than providing costimulatory support through the CAR itself, as is the case in second generation CARs3–5, we engineered chimeric costimulatory receptors (CCRs), which do not signal in response to natural costimulatory ligands but rather to an antigen, in this case prostate-specific membrane antigen (PSMA). The CCR we used in this instance provided combined CD28 and CD137 signals, to provide costimulation when PSMA is coexpressed with PSCA. In isolation, the binding of the CCR, which lacks a T cell activating domain and is therefore not a CAR, does not result in an observable immune response. On the other hand, a first generation CAR can direct significant cytotoxicity, albeit for short-term responses and without the potential to support robust T cell proliferation and cytokine secretion. Thus, while the response against target cells that only express the CCR-targeted antigen (PSMA) is unmeasurable, the CAR-mediated response to PSCA can be significant when utilizing a standard Car. To abolish reactivity against PSCA+, PSMA− tumors, representing a sum agate for normal tissue expressing PSCA, we designed suboptimal PSCA-specific CARs and found we could eliminate PSCA+ PSMA+ while sparing PSCA+ PSMA− and PSCA− PSMA+ tumors.
Several approaches to reducing the activity of the activating receptor may be considered. These include epitope selection, binding affinity and activating motifs. Having too high a function of the CAR or too high an affinity of a TCR will indeed predispose to higher activity against tissues expressing the target antigen, including in this example normal tissues that express PSCA. In order to avoid this undesirable outcome, proper function and affinity of the activating receptor must be identified.
In the case of the TCR, receptor affinity is a key determinant of the strength of T cell activation, proliferation, cell surface expression of markers, polarity, asymmetric division, differentiation, and ability to infiltrate tissues32. The weaker strength of activation requires stronger costimulation, a paradigm we exploit in combinatorial antigen recognition by rescuing a poor T cell activating receptor with a CCR specific for another antigen known to be coexpressed in tumor cells.
In conclusion, the two-receptor CAR+CCR approach allows for targeting a combination of antigens that individually have expression on different normal tissues, but are coexpressed by cancerous cells. This control allows for rescuing the potential utility of target antigens that on their own are inadequate targets because of the on-target, off-tumor toxicities they may cause. Based on these principles, we can engineer T cell activation and costimulation to selectively eradicate tumor cells, while avoiding destruction to normal tissues.
Dynamic switches through the iCAR approach
Recent clinical experiences indicate that specific targeting does not necessarily avert the “on-target, off-tumor” toxicities of certain engineered receptors33. The observation that a MAGE-A3- targeting TCR cross-reacted with an identical epitope in MAGE-A9 and a non-identical epitope in MAGE-A12 leading to significant neurological toxicity underscores two critical problems in using targeting to control toxicities34. It was previously unrecognized that MAGE-A9/12 were expressed in human brain tissue, thus without reliable databases for expression “surfaceomes” of individual cell types, a priori predictions of such toxicities are impossible35,36. The ability of TCR/CARs to recognize nonidentical epitopes further complicates ruling out toxicities, as each combinatorial targeting domain will potentially increase this risk.
An alternative strategy is the use of a second receptor, the function of which is to control the toxicities of the TCR/CAR, rather than expecting to accomplish this solely through improvements in targeting of the TCR/CAR. This approach takes cues from the physiological control of T cell autoimmunity, which requires lifelong positive/negative thymic selection and immune regulation through a multitude of receptors to stop inappropriate and toxic T cell responses37. This would be especially attractive for receptors that display potent tumor elimination with unfortunate concurrent toxicity against a critical tissue, such as the neurotoxicity described above. The use of suicide genes is limited in these situations, because of the potential for permanent or fatal damage from the toxicity and the kinetics of the toxicity, which would require elimination of the therapeutic T cells before the benefit of their antitumor effects. Using physiology as a guide, we can attempt to direct the actions of engineered T cells in terms of what needs to be done (eradication of tumor cells) and what should be avoided (toxicities).
A paradigm demonstration of this strategy is the use of inhibitory chimeric antigen receptors (iCARs)38. These synthetic receptors provided a potent antigen specific inhibitory signal to limit the functions of T cells, thus when engaged these receptors induce dominant negative signals over any activation cue the T cell experiences. When introduced into primary human T cells, PD-1 and CTLA-4 based iCARs are able to suppress cytotoxicity, cytokine release, and proliferation in T cells activated by endogenous TCRs or clinically relevant CARs. Importantly, this inhibition was only initiated if the iCAR engaged its antigen, as the reactivity of T cells against tissues not expressing the iCAR antigen was not affected.
If T cells were permanently inhibited by iCARs upon engagement of a normal tissue, this would result in a “suicide gene” effect induced at the site of the toxicity (although an improved one, since it would not requiring the appearance of symptoms before the toxicity could be controlled). However, the ideal function of the iCAR strategy relies on the reversible inhibition of the T cell function, so that T cells initially prevented by the iCAR from damaging a normal tissue can still recirculate and deliver therapeutic functionally against a tumor (Figure 2). This type of reversibly inhibited functionality in T cell has been found physiologically in a variety of circumstances including pregnancy, tumor microenvironments, and hypoxia39–41. Upon infusion into a patient, T cell will statistically redistribute to target and off-target sites. One potential goal is to limit T cell interaction at the off-target sites and encourage recruitment to on-target sites. PD-1 and CTLA-4 signaling domains used in the iCAR design have been mechanistically implicated in altering the motility “stop” signal of T cells, thus encouraging their migration to alternative targets42,43.
Figure 2. iCARs Allow Dynamic T Cell Behavior At Tumor and Normal Tissue Sites.
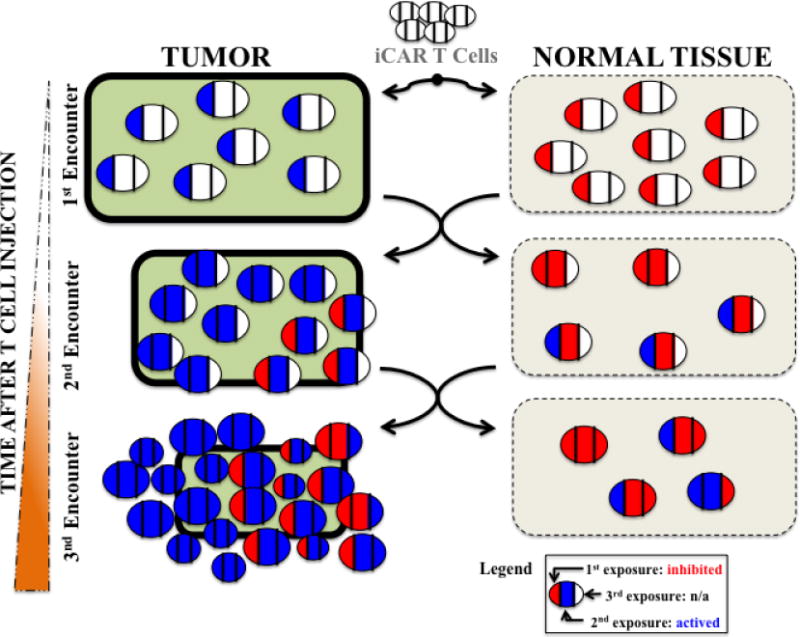
Each round of antigen exposure is schematically represented as blue for activation (driven by the activating receptor) and red for inhibition (delivered through the iCAR). The rounds of exposure are tracked in each individual cell by color changes (red->blue->red: inhibition->activation->inhibition). Initially, all the iCAR T cells seed both the tumor and off-target normal tissue equally. During the first exposure in the tumor tissue, the T cells are activated, while in the normal tissue they are repressed. The activation causes the T cells to expand, but there is also recirculation between the two sites, so that upon the second exposure redistribution has occurred. As the T cells continue to receive activating signals at the tumor site, they continue to proliferate and destroy the tumor aided by T cells that initially encountered the normal tissue. At the normal tissue, the iCAR continue to temporarily inhibit most T cells with some potentially permanently anergized.
We were able to demonstrate the reversible nature of the iCAR using two approaches. By sequentially exposing iCAR positive T cells to target cells followed by off-target cells (as well as the reverse sequence), we were able to demonstrate the continued function of both the positive signal (TCR/CAR killing) and the negative signal (iCAR temporary inhibition), independent of what cells the T cells had initially encountered. Our in vivo experiments used a mixture of hematological target and off-target cells that equally seeded the bone marrow of a mouse, which challenged the iCAR T cells to discern what not to kill in the same microenvironment as what to kill, and crucially as they were simultaneously killing target cells. By visualizing this choice with real time microscopy analysis of target and off target in the same well, the iCARs showed potent and specific control of the T-cell response with protection of off target cells at levels reaching 90%. Furthermore, we demonstrated that the level of inhibition depended on the stoichiometry of the iCAR, the positive signal receptor, and both respective ligands. Thus, clinical use of the iCARs could possess significant potential for a personalized medicine strategy, where the potency of inhibition is tailored to the specifics of each treatment.
The reversible nature of T cell regulation is sustained through receptor phosphorylation states in the T cell, thus allowing for rapid dynamic behavioral changes through the appropriate changes in phosphorylation states39,44. We analyzed the signaling induced by the PD-1 iCAR, showing increased levels of phosphorylation of both SHP1 and SHP2, which mediate reversible inhibition of T cell activation. Additionally, we have found several other ITAM/ITIM-associated immunoreceptors being phosphorylated by the engagement of the iCAR. Some of these are novel and we are following up to understand how iCARs can be used to interrogate inhibitory signaling pathways in primary human T-cells.
The majority of effector T cells co-express more than one immune inhibitory receptor45,46. The signaling cascades downstream of inhibitory receptors are likely to overlap, but their functions are not redundant37,47. The interplay among the inhibitory receptors plays diverse roles in the control of activated T cells. Therefore, iCARs could be designed that combine the signaling moieties of these molecules, much like the second generation CARs combined killing and costimulation signals. For example, recent work has shown that tumor-specific T cells from lymph nodes exhibited different reactivity due to differential expression of several immune inhibitory receptors (upregulation of TIM-3, LAG-3 and CTLA-4 and down-regulation of KLRG-1) as compared to cells with the same specificity in blood47. As more becomes known about inhibitory receptors, iCARs could be designed for situation specific needs, and these layers will combine to allow for rational uses of the next generation of iCARs.
The concept of universal targeting systems can also be applied to iCARs, potentially with greater efficacy than positive CARs. The use of biotin-binding immune receptor or anti-fluorescein isothiocyanate (FITC) CARs potentially allowing targeting of multiple antigens labeled with biotinylated or FITC tagged antibodies, but have shown poor in vivo tumor control potentially from the difficulty of dosing the right amount of antibody48,49. These problems are avoided in using a universal recognition domain with iCARs, since the aim is to protect a normal tissue. The toxicity associated with CARs and TCRs has been shown to be reactive against random tissues and therefore it is ideal to develop a “zip code” like system of surface antigen receptors marking each physiologically critical tissue (brain, liver, lung, heart, etc). A library of antibodies against each tissue could then offer protection of normal tissues from any T cell based therapy toxicity using a universal iCAR approach.
The goal of the iCARs is to provide engineered T cells with dynamic control over their activation and function, while accepting that they are bound to engage bystander tissues due to imperfections in the targeting. Rather than wait for the appearance of toxicities to implement a static control strategy through suicide genes, iCARs attempt to take advantage of T cell capacitates as a decision-making entity, utilizing a dynamic control strategy. For example, considering the cross-reactivity of anti-MAGE-A3 TCRs with brain tissue is inevitable, the use of an iCAR targeting a neuronal antigen absent on melanoma cells, would cue T cells that an off-targeting event had occurred potentially preventing neurotoxicity.
Conclusion
Very few candidate targets identified to date are truly tumor-specific5,27. It is therefore necessary to discover new targets, as well as to devise operational strategies that address the risk of intolerable “on-target, off-tumor” effects. While tumor cell specific antigen identification will improve with new proteomic and analytical technologies, current CAR/TCR based T cell therapies will require systems to control and direct their reactivates beyond the inherent targeting of their scFv. Greater understanding of physiological T cell functionalities combined with advances in synthetic biology allow for new found capabilities to explore how to best shape engineered T cell-mediated eradication of specific cell populations based on cell surface phenotypes. Using a multiple logic gate approach through novel synthetic receptor can facilitate or suppress T cell responses, and thus provides the power to unleash a robust immune response only when a particular combination of antigens are identified by the T cells. We discussed two recent cell engineering methods to balance T cell signaling to better control the robust immune responses to only cancerous cells. We imagine that incorporation of additional receptors to further fine-tune this delicate balance of signaling is necessary to advance engineered T cell therapies. These methods allow for unlocking the true potential of immunotherapy to eradicate multiple types of cancers.
Acknowledgments
This research has been supported for the past two decades by the ACGT, the CRI, the DOD, the NCI, Stand-Up to Cancer/AACR, the V Foundation and philanthropic gifts from Mr. and Mrs. Goodwin and the Commonwealth Foundation for Cancer Research, the Lake Road, Majors Family, Mr. and Mrs. Mallah and Mr. L. Sanders foundations.
V.D.F. was supported by a training grant from the National Heart, Lung, and Blood Institute (NHLBI; T32 GM007739-31S1) to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD/PhD program. The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. C.K. was supported by DOD PCTA PC101964.
References
- 1.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 2.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer cell. 2003;3:431–437. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 3.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 4.Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer discovery. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kochenderfer JN, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalos M, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brentjens RJ, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brentjens RJ, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grupp SA, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davila ML, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014 doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brentjens RJ, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 15.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112:1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pegram HJ, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012 doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PloS one. 2013;8:e61338. doi: 10.1371/journal.pone.0061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamers CH, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 19.Johnson LA, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan RA, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan RA, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cameron BJ, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linette GP, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lupo-Stanghellini MT, et al. Clinical impact of suicide gene therapy in allogeneic hematopoietic stem cell transplantation. Hum Gene Ther. 2010;21:241–250. doi: 10.1089/hum.2010.014. [DOI] [PubMed] [Google Scholar]
- 25.Sadelain M. Eliminating cells gone astray. N Engl J Med. 2011;365:1735–1737. doi: 10.1056/NEJMe1109971. [DOI] [PubMed] [Google Scholar]
- 26.Morgan RA. Live and let die: a new suicide gene therapy moves to the clinic. Mol Ther. 2012;20:11–13. doi: 10.1038/mt.2011.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanada K, Restifo NP. Double or nothing on cancer immunotherapy. Nat Biotechnol. 2013;31:33–34. doi: 10.1038/nbt.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Science translational medicine. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krause A, et al. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. The Journal of experimental medicine. 1998;188:619–626. doi: 10.1084/jem.188.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liechtenstein T, Dufait I, Lanna A, Breckpot K, Escors D. Modulating Co-Stimulation during Antigen Presentation to Enhance Cancer Immunotherapy. Immunology, endocrine & metabolic agents in medicinal chemistry. 2012;12:224–235. doi: 10.2174/187152212802001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King CG, et al. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity. 2012;37:709–720. doi: 10.1016/j.immuni.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. doi: 10.1038/nbt.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgan RA, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zammatteo N, et al. DNA microarray to monitor the expression of MAGE-A genes. Clin Chem. 2002;48:25–34. [PubMed] [Google Scholar]
- 36.da Cunha JP, et al. Bioinformatics construction of the human cell surfaceome. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16752–16757. doi: 10.1073/pnas.0907939106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.June CH, et al. Inhibition of tyrosine phosphorylation prevents T-cell receptor-mediated signal transduction. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:7722–7726. doi: 10.1073/pnas.87.19.7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohta A, et al. Hypoxia-induced and A2A adenosine receptor-independent T-cell suppression is short lived and easily reversible. International immunology. 2013 doi: 10.1093/intimm/dxt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simpson-Abelson MR, et al. Human ovarian tumor ascites fluids rapidly and reversibly inhibit T cell receptor-induced NF-kappaB and NFAT signaling in tumor-associated T cells. Cancer Immun. 2013;13:14. [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider H, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972–1975. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 43.Fife BT, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nature immunology. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parry RV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and cellular biology. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nature reviews. Immunology. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory Receptor Expression Depends More Dominantly on Differentiation and Activation than “Exhaustion” of Human CD8 T Cells. Front Immunol. 2013;4:455. doi: 10.3389/fimmu.2013.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baitsch L, et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PloS one. 2012;7:e30852. doi: 10.1371/journal.pone.0030852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urbanska K, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72:1844–1852. doi: 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamada K, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:6436–6445. doi: 10.1158/1078-0432.CCR-12-1449. [DOI] [PubMed] [Google Scholar]