

SUMMARY
Neurons can maintain stable synaptic connections across adult life. However, the signals that regulate expression of synaptic proteins in the mature brain are incompletely understood. Here we describe a transcriptional feedback loop between the biosynthesis and repertoire of specific phospholipids and the synaptic vesicle pool in adult Drosophila photoreceptors. Mutations that disrupt biosynthesis of a subset of phospholipids cause degeneration of the axon terminal and loss of synaptic vesicles. While degeneration of the axon terminal is dependent on neural activity, activation of Sterol Regulatory Element Binding Protein (SREBP) is both necessary and sufficient to cause synaptic vesicle loss. Our studies demonstrate that SREBP regulates synaptic vesicle levels by interacting with tetraspanins, critical organizers of membranous organelles. SREBP is an evolutionarily conserved regulator of lipid biosynthesis in non-neuronal cells; our studies reveal a surprising role for this feedback loop in maintaining synaptic vesicle pools in the adult brain.
eTOC blurb
Tsai et al. describe how the biosynthesis of specific phospholipids is linked to neurodegeneration and synaptic vesicle loss in adult Drosophila photoreceptors. Neurodegeneration is dependent on activity, while vesicle depletion engages a transcriptional feedback loop that includes SREBP and tetraspanins.
INTRODUCTION
The structural stability of synaptic connections is central to maintaining brain function throughout adult life. As neural activity can be highly dynamic, this structural stability requires feedback mechanisms that can act across a range of timescales to maintain appropriate levels of synaptic proteins and lipids (Bezprozvanny and Hiesinger, 2013). However, the molecular mechanisms that regulate synaptic components and vesicle pools over long timescales are incompletely understood. In addition, synapse loss is a common feature of many neurodegenerative diseases, and can be both a critical component of disease onset and a strong predictor of cognitive impairment (Davies et al., 1987; DeKosky and Scheff, 1990; Terry et al., 1991; Masliah et al., 1991; Yoshiyama et al., 2007; Nemani et al., 2010). Thus, identifying the molecular mechanisms by which adult neurons maintain synapses is essential to understanding both fundamental biological processes and disease states.
Photoreceptors are highly dynamic, with vesicle release rates that can vary across two orders of magnitude over short time scales. In cone photoreceptors, rapid synaptic vesicle recycling results in plasma membrane turnover equivalent to the entire surface area of the cell within an hour (Choi et al., 2005). Similarly, photoreceptors in flies maintain extremely high rates of vesicle fusion, up to hundreds of vesicles per second per release site (van Steveninck and Laughlin, 1996), requiring rapid replenishment (Stuart et al., 2007). As photoreceptors must maintain their response properties across the lifespan of the animal, this highly dynamic character suggests specialized mechanisms for maintaining synaptic vesicle pools. Here we used a forward genetic screen in Drosophila photoreceptors to uncover a mechanism for maintaining synaptic vesicles.
Studies in a variety of systems have implicated specific membrane phospholipids in the regulation of neuronal development and function. Phosphatidylethanolamine (PE) is highly enriched in synaptic vesicle membranes, and is found at high levels in the mammalian retina (Anderson, 1970; Breckenridge et al., 1973). One pathway for PE biosynthesis utilizes three enzymes, ethanolamine kinase (easily shocked in Drosophila), phosphatidylethanolamine cytidylyltransferase (pect), and ethanolamine phosphotransferase (CG33116, CG7149). Intriguingly, knock-down of PE biosynthetic genes in Drosophila results in light-dependent retinal degeneration (Midorikawa et al., 2010), and easily shocked (eas) mutants display neuronal hyper-excitability, and defects in dendrite development that emerge after the dendritic field is established in sensory neurons (Pavlidis et al., 1994; Meltzer et al., 2017). However, how mutations in PE biosynthetic enzymes would affect the levels of specific lipids in individual neuron types is unknown. In non-neuronal cells, PE negatively regulates SREBP, an evolutionarily conserved transcription factor, to control lipid biosynthesis (Dobrosotskaya et al., 2002; Seegmiler et al., 2002), and while many downstream targets of SREBP are known, SREBP targets in adult neurons are only incompletely understood (Shao and Espenshade, 2012). Taken together, these data demonstrate that PE is required for the maintenance of neuronal structure and function, and typically affects SREBP activity, but the mechanistic links between PE biosynthesis and neuronal structure, particularly at synapses, are unclear.
RESULTS
A forward genetic screen identifies mutations in pect
To identify genes required for photoreceptor function, we performed a forward genetic screen based on a visual behavioral assay using somatic mosaic animals that depends on functional synaptic connections between photoreceptors and their postsynaptic targets (Clandinin et al., 2001). From this screen, we obtained two recessive lethal mutations, omb362 and omb593, affecting conserved domains of the gene encoding Pect (omb362 D26N; omb593 H55Y; See STAR Methods). Further complementation testing identified an additional point mutation, designated 38K (A247V), as well as a molecular null allele, LL06325 (Schuldiner et al., 2008 and data not shown).
Pect mutant photoreceptors undergo progressive degeneration and lose synaptic markers
The Drosophila retina is composed of 800 facets, ommatidia, each of which contains eight photoreceptors (R cells), R1-R8. The six outer photoreceptors, R1-R6, extend axons into the lamina, the first optic neuropil, forming stereotyped columnar elements, cartridges, that contain six presynaptic axon terminals and their postsynaptic targets (Meinertzhagen and Hanson, 1993; Figure 1A). Antibody markers label the Drosophila glycoprotein Chaoptin, specific to R cell axon terminals (Fujita et al., 1982), the vesicle-associated Cysteine String Protein CSP (Zinsmaier et al., 1994; Hamanaka and Meinertzhagen, 2010), the SNARE complex member Syntaxin (Schulze et al., 1995), and the active zone marker Bruchpilot (Wagh et al., 2006; Hamanaka and Meinertzhagen, 2010). On the day of eclosion (Day 0), axon terminals in pect mutant R cells were morphologically similar to controls and displayed robust expression of CSP, Syntaxin and Bruchpilot (Figure 1B–D, F–H; Figure S1A–D, H–J). Moreover, the morphology and arrangement of R cell bodies in the retina was also normal (Figure 1E, I). However, over the first five days of adult life, while control R cell axon terminals were unchanged, mutant axon terminals developed irregular, swollen morphologies, and progressively lost both CSP and Syntaxin labeling, but retained Bruchpilot labeling (Figure 1N–P, R–T; Figure S1A–S). In parallel, the retina degenerated, consistent with previous observations (Figure 1Q, U; Midorikawa et al., 2010). To verify that these phenotypes were caused by mutations in pect, we expressed a UAS-pect rescue construct in mutant animals under the control of the photoreceptor-specific driver GMR-Gal4. In control flies, expression UAS-pect had no effect on R cell axon terminals, while in somatic mosaic mutants, expression of this construct had no effect at eclosion, but strongly rescued all R cell phenotypes five days later (Figure 1J–M, V–Y; S1A, T–Y; data not shown). Somatic mosaic animals in which R cells were homozygous mutant for any of the four pect alleles, including the putative molecular null allele LL06325, displayed indistinguishable phenotypes, and were rescued UAS-pect (Figure S1N–Y, and data not shown). Finally, pect mutant photoreceptors were morphologically normal during mid-pupal development (data not shown). Thus, strong reduction of function alleles of pect cause degeneration and loss of synaptic marker labeling in mature photoreceptors.
Figure 1. Pect mutants display adult-onset synaptic vesicle loss, R cell axon terminal degeneration, and R cell body degeneration.

(A) Schematic of Drosophila visual system. Photoreceptors (R cells) project axons from the retina directly into the lamina. Cross sections of the retina reveal a regular arrangement of seven R cell rhabdomeres, while cross sections of the lamina reveal R cell axon terminals. (B-M) Day 0. (B-E) Control flies. (F-I) pectomb593 somatic mosaic animals. (J-M) pectomb593 somatic mosaic animals expressing eye-specific full-length Pect. (N-Y) Day 5. (N-Q) Control flies. (R-U) pectomb593 somatic mosaic animals. (V-Y) pectomb593 somatic mosaic animals expressing eye-specific full-length Pect. (B, F, J, N, R, V) Synaptic vesicle labeling (CSP in magenta). (C, G, K, O, S, W) Photoreceptor axon terminals (Chaoptin in green). (D, H, L, P, T, X) A marker of the active zone (Bruchpilot in white). Inset panels display high magnification views of single cartridges. (E, I, M, Q, U, Y) Plastic sections of the retina. Scale bar: 15 µm. n = 10 for each genotype. See also Figure S1.
To examine the specificity of the synaptic changes seen in pect mutants, we used five additional markers associated with synaptic terminals in Drosophila (Südhof et al., 1989; Lahey et al., 1994; Klagges et al., 1996; Harden et al., 1996; Godenschwege et al., 2004; Hofbauer et al., 2009; Hamanaka and Meinertzhagen, 2010). Aa2/2 labels the epidermal growth factor receptor pathway substrate clone 15 (EPS-15), which localizes to the synaptic vesicle pool surrounding the heads of capitate projections. Anti-DPAK recognizes Drosophila p21 activated kinase, while anti-synapsin recognizes several Synapsin isoforms. Discs-large (DLG) labels both pre- and postsynaptic structures, and capitate projections. Of these, expression of A2/2, DLG and synapsin are reduced in pect mutant R cell axon terminals five days after eclosion, while expression of Dynamin and DPAK were unaffected (Figure S1A). Thus, some, but not all, synaptic markers are affected by this manipulation.
We next examined eye-specific somatic mosaic animals using electron microscopy (Figure 2; Figure S2). In pect mutants observed within 8 hours of eclosion, cartridge organization was typically normal (Figure 2A, B) with only a modest increase in the variance in the number of R1-R6 terminals observed in mutant cartridge cross sections relative to controls (Figure 2P). Within each cartridge, the number of presynaptic release sites at T-bar ribbons between R cells and their postsynaptic targets was the same in pect mutant terminals as in controls (Figure 2Q). These observations argue strongly that R cells in pect mutant animals make synapses only with the correct post-synaptic targets. However, while individual R cell axon terminals were of normal size (Figure 2A–D, R), synaptic vesicles were reduced in number, lowering the density of synaptic vesicles to approximately 55% of that in controls (Figure 2C, D, J, S). These remaining vesicles were evenly spaced, and distributed in small patches (Figure 2I), a phenotype previously observed in endocytic mutants (Fabian-Fine et al., 2003; Verstreken et al., 2003; Dickman et al., 2005). To quantify this “patchy” distribution, we compared Ra, the measured nearest-neighbor distance between synaptic vesicle profiles to Re, the same distance based on the calculated density of synaptic vesicles assuming a random distribution (Figure S2A). If vesicles were distributed at random, this ratio would be 1 but, consistent with the presence of vesicle clusters, was significantly less than 1 in pect mutant terminals (Figure S2B). A further phenotype, also found in other endocytic mutant R cell terminals, was an approximately 70% increase in the number of shallow profiles of capitate projection organelles, sites of vesicle endocytosis (Figure S2C; Fabian-Fine et al., 2003). Finally, pect mutant terminals also contained rare profiles of dense-core vesicles at eclosion (Figure 2H, S2D), in addition to the small, clear synaptic vesicles, which contain histamine (Borycz et al., 2005), suggesting that pect mutant terminals may accumulate neuropeptides. In sum, 8 hours after eclosion, pect mutant terminals have normal synapses but display defects consistent with changes in vesicle membrane trafficking and endocytosis.
Figure 2. Ultrastructural analysis of pect mutants reveals synaptic vesicle loss and progressive degeneration.
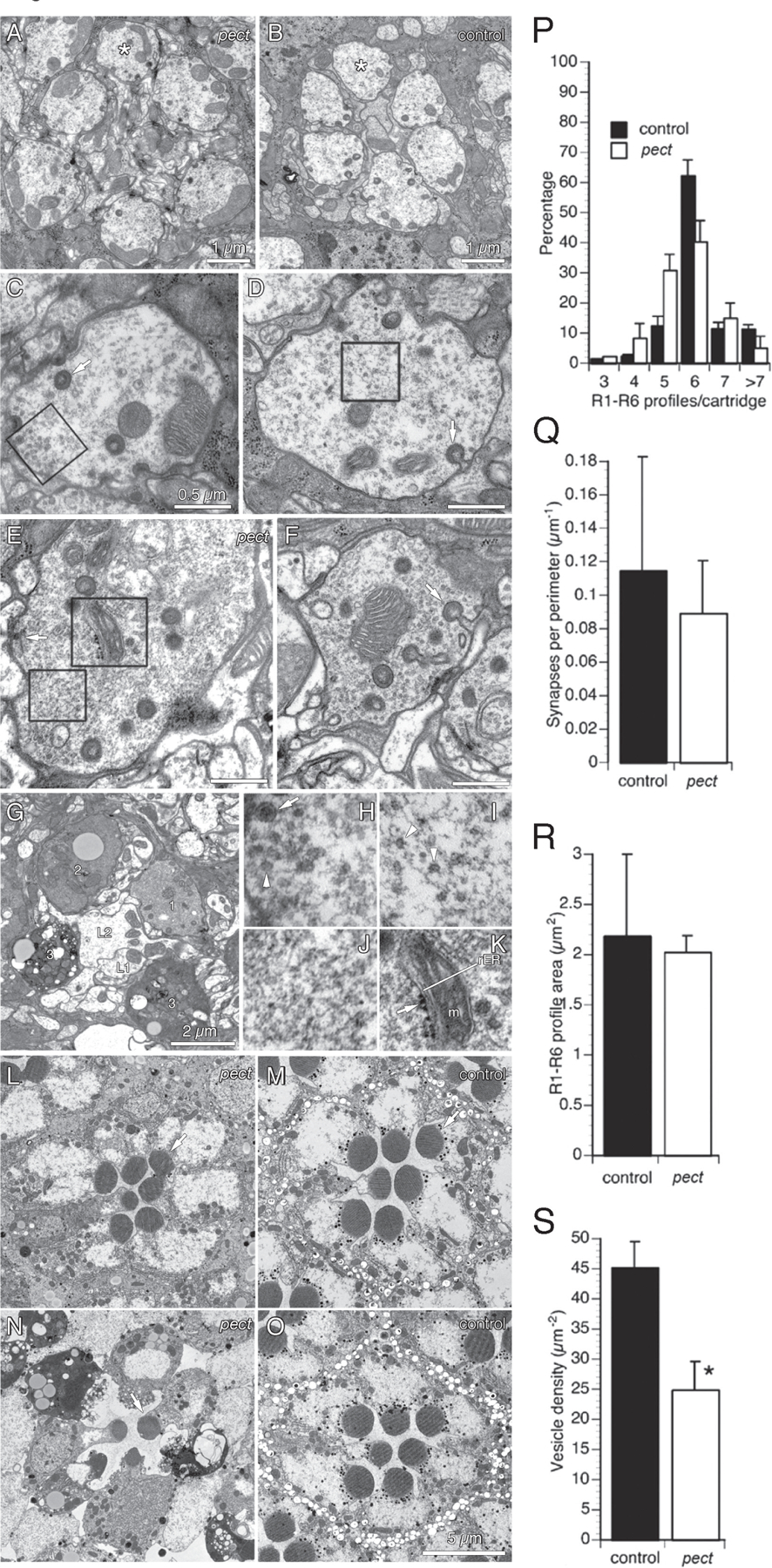
(A, B) Pect mutant and control lamina cartridges 8 hours after eclosion. (C, D) Single pect mutant and control R1-R6 axon terminals 8 hours after eclosion. (C) Box encloses a patch of vesicles in a pect mutant R cell axon terminal, enlarged in (H). (D) Box encloses a patch of vesicles in a control R cell axon terminal, enlarged in (I). Arrow indicates an invaginating capitate projection. (E) Pect mutant R cell axon terminal. Upper box encloses structure with the morphological character of rough endoplasmic reticulum, enlarged in (K). Lower box encloses a vesicle patch, enlarged in (J). The arrow points to the pre-synaptic ribbon of a tetrad synapse. (F) Pect mutant R cell axon terminal. Arrow indicates head of an invaginating capitate projection. (G) Lamina cartridge from day 5 pect mutant. Four R1-R6 terminals surround the axons of two target interneurons, L1 and L2. The four R cell axon terminals are undergoing different stages of degeneration (labeled 1–3). (H-K) Enlargements of pect mutant R cell cytoplasm boxed in C-E. (H) ~30 nm clear synaptic vesicles (arrowhead) and ~100 nm dense core vesicle (arrow). (I) Remaining clear synaptic vesicles cluster in small patches with normal spacing (arrowheads). (J) Clear synaptic vesicles are absent in this section of a pect terminal. (K) Profiles with the morphological character of endoplasmic reticulum (rER) with ribosomes (arrow), smooth ER and mitochondrion labeled m. (L, M) Retinas of day 0 pect mutant and control flies. (N) Retina of day 5 pect mutant. Only a single R1-R6 rhabdomere (arrow) still has well preserved structure. (O) Retina of day 5 control with normal rhabdomeres. (P-U) Quantification of pect mutant R cell axon terminal defects 8 hours post-eclosion. (P) Distribution of the number of R1-R6 axon terminals per cartridge in control and pect animals. (Q) Tetrad synapse profiles per µm of R1-R6 membrane perimeter. (R) R1-R6 terminal profile areas in control and pect mutants. (S) Synaptic vesicle density in pect mutant terminals relative to control terminals. Data represented as mean ± SEM. Scale bars indicated in images. Differences significant at p < 0.01 (S) (unweighted means ANOVA, Tukey HSD test). See also Figure S2.
Ultrastructural analysis of pect mutants revealed additional neurodegenerative changes in older flies. At five days of age, R1-R6 terminals in control flies appeared relatively unchanged from eclosion (data not shown). However, many pect mutant R1-R6 terminals lacked most vesicles and displayed electron-dense cytoplasm, consistent with degeneration (Figure 2G; Brandstätter et al., 1991). This degeneration was progressive, with most terminals being normal soon after eclosion, transitioning smoothly to most terminals displaying severe degeneration at 5 days of age (Figure S2E). Associated with these changes, there was also a general loss of capitate projections and synaptic T-bar ribbons, as well as the presence of structures with the morphological character of endoplasmic reticulum (Figure 2K; Figure S2F and data not shown). Finally, while pect mutant photoreceptor cell bodies appeared normal 8 hours after eclosion, the retinas of 5 day old animals also underwent degeneration, displaying both electron dense cytoplasm and loss of rhabdomere structure (Figure 2L–O). Thus, Pect is required to maintain the structural organization of R cell bodies and axon terminals.
Pect is required for the biosynthesis of specific phospholipid species
We next examined how mutations in pect altered phospholipid biosynthesis by performing mass spectrometric (MS) analysis of retinas dissected from control and pect mutant somatic mosaic flies on the day of eclosion. As phosphatidylcholine (PC) and phosphatidylethanolamine (PE) are the most common constituents of membranes in Drosophila (Jones et al., 1992), we compared the relative levels of PE and PC molecular species containing between 16 and 24 carbons in fatty acid chain length. In addition to being classified by their headgroups (PE or PC in this case), phospholipid structures are described by the lengths of the carbon chains, and by the number of carbon-carbon double bonds within these chains, summed over both fatty acid groups. By convention, phospholipids with 36 or more carbons and 6 or more double bonds are classified as long chain, polyunsaturated fatty acids (LC-PUFAs). These studies revealed that in control retinas, 76% of membrane glycerophospholipids comprised PC species, while PE species accounted for the remaining 24% (Supplemental Table 1). Moreover, photoreceptor membranes include a diverse repertoire of PC and PE species, with 97.9% of lipid species containing fewer than 6 double bonds and 36 or fewer carbons, and 2.1% comprising LC-PUFA species with between 6 and 12 double bonds (Figure 3A). In pect mutant retinas, the overall proportions of PC and PE species were not significantly changed (81% PC species, 19% PE species). However, loss of Pect activity caused a significant shift in the relative levels of a specific subset of PC and PE species (Figure 3B). Amongst PC species, loss of Pect activity lowered the levels of PC 32:0, while increasing the levels of PC 32:2 (Figure 3B, Supplemental Table 1). All of these changes were substantially reversed by expression of a rescuing Pect transgene. Amongst PE species, loss of Pect activity significantly lowered the levels of PE 34:1 and PE 36:2, changes that were also significantly reversed by the rescuing Pect transgene. Finally, aggregate levels of PC and PE LC-PUFA species were not affected by Pect manipulation. These data argue that Pect is not essential for the synthesis of either PC or PE per se, but that Pect activity is required to maintain the appropriate levels of specific phospholipid species, with particular effects on the relative proportions of specific PC and PE species. Thus, the degenerative phenotypes we observed in pect mutant retinas are not the result of generalized deficits in membrane phospholipid composition, but rather reflect changes in the biosynthesis of a small number of specific phospholipids.
Figure 3. Pect mutants have specific changes in PC and PE phospholipid species.
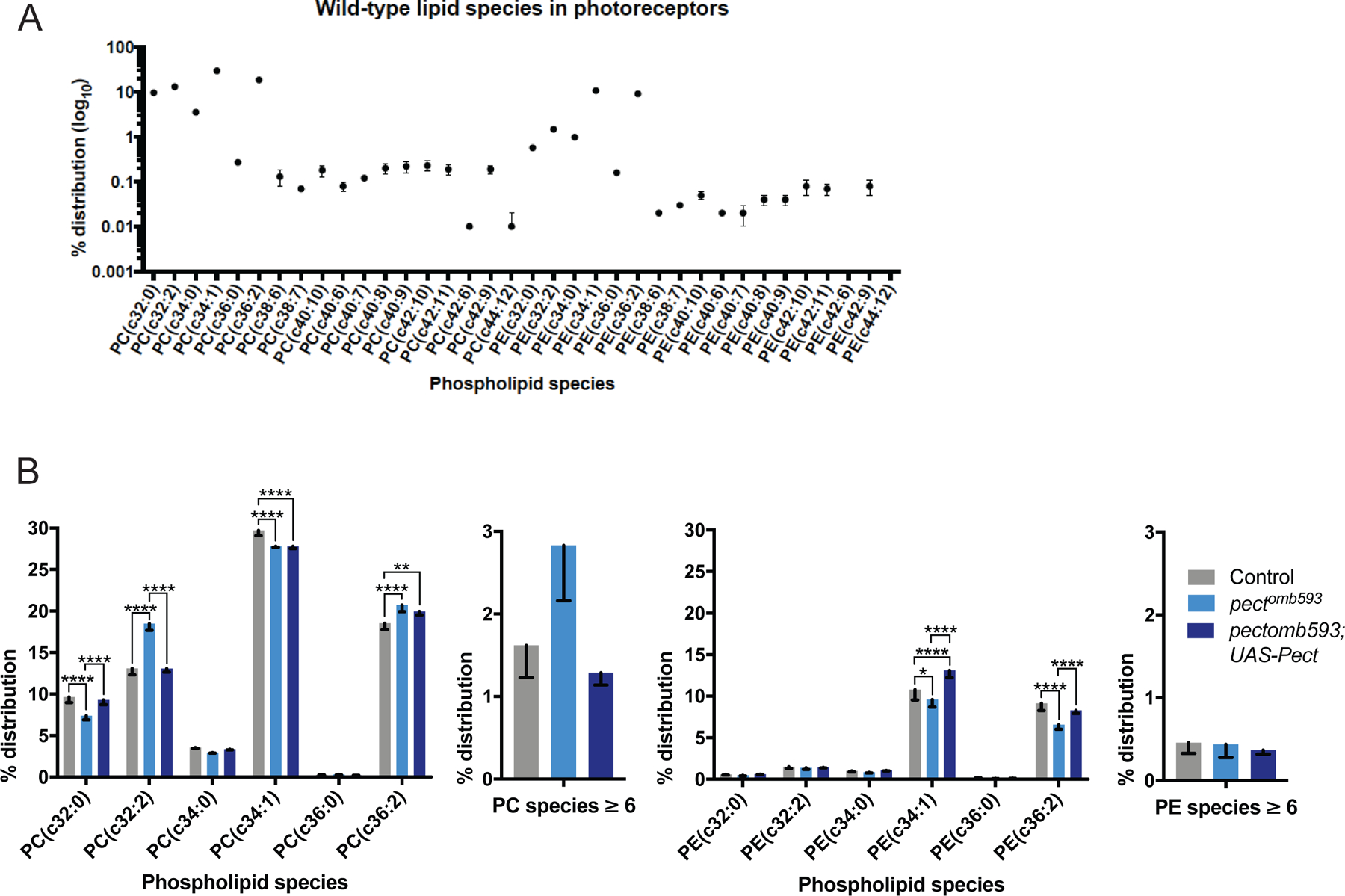
(A) Quantitative measurements of PE and PC species in control retinas, displaying the relative percent contribution of each species to the total distribution of PE and PC phospholipids in the membrane. (B) Quantitative comparisons of PE and PC species between control (gray), pectomb593 somatic mosaic animals (light blue) and pectomb593 somatic mosaic animals expressing eye-specific full-length Pect (dark blue), displaying the relative percent contribution of each species. Left graphs display a subset of PC species as well as an aggregate sum of all PC species with six or more double bonds; right graphs display the same subsets of PE species as well as an aggregate sum of all PE species with six or more double bonds. Data represented as mean ± SEM. Differences significant at p < 0.01 (**), p < 0.0001 (*****) (one-way ANOVA; t-test). n = 4 replicates of 20 retinas per genotype. See also Figure S3 and Supplemental Table 1.
Pect is expressed in R cells
To examine Pect expression, we used an antibody against Pcyt2, the human homolog of Pect, which shares 59% identity with Drosophila Pect. In control flies, on the day of eclosion, this antibody specifically labeled R1-R6 axon terminals (Figure S3A–C), a staining pattern that disappeared when R cells were homozygous mutant for the molecular null allele of pect, pectLL06325 (Figure S3D–F). Photoreceptor-specific expression of UAS-pect in pectLL06325 somatic mosaic animals restored robust staining of axon terminals (Figure S3G–J). Thus, Pect protein is expressed by adult photoreceptors.
Axon terminal degeneration in pect mutant R cells is activity-dependent
Deficits in PE biosynthesis cause light-dependent degeneration of R cell rhabdomeres (Midorikawa et al., 2010). To determine whether the axonal phenotypes in pect mutant R cells were activity-dependent, pect somatic mosaic animals were reared in the dark to reduce R cell activity (Niven et al., 2007). Under these conditions, pect mutant axon terminals were more strongly labeled by Chaoptin, and nearly normal in morphology, compared with pect mutants reared under standard lighting (Figure 4A, C, D, F, G, I, N). However, in spite of strong suppression of the degeneration of the axon terminal, synaptic marker expression was not increased by dark rearing (Figure 4B, E, H, M). We next examined whether this suppression was directly due to reductions in light exposure via effects on phototransduction or reflected changes in light-evoked synaptic activity. To do this, we took advantage of a null allele in synaptotagmin 1 (syt) that blocks activity-dependent vesicle release at the neuromuscular junction and in R cells (DiAntonio et al., 1993; Stowers and Schwarz, 1999). Importantly, syt homozygous mutant R cells displayed no detectable changes in axon terminal morphology or synaptic staining (Stowers and Schwarz, 1999; data not shown). Strikingly, as in dark-reared flies, pect, syt double mutant R cells displayed significantly reduced degeneration of axon terminals but continued to display the characteristic loss of synaptic markers associated with pect mutations (Figure 4J–N). Finally, both dark-rearing of pect mutants and pect, syt double mutants displayed suppression of cell body degeneration in the retina (Figure S4A–D). Taken together, these data demonstrate that degeneration of the R cell axon terminal and cell body is activity-dependent and requires synaptic vesicle exocytosis. These studies also show that the loss of synaptic protein expression seen in pect mutants was unaffected by reductions in activity, suggesting a distinct underlying mechanism.
Figure 4. Pect photoreceptor axon terminal degeneration is independent of synaptic vesicle loss.
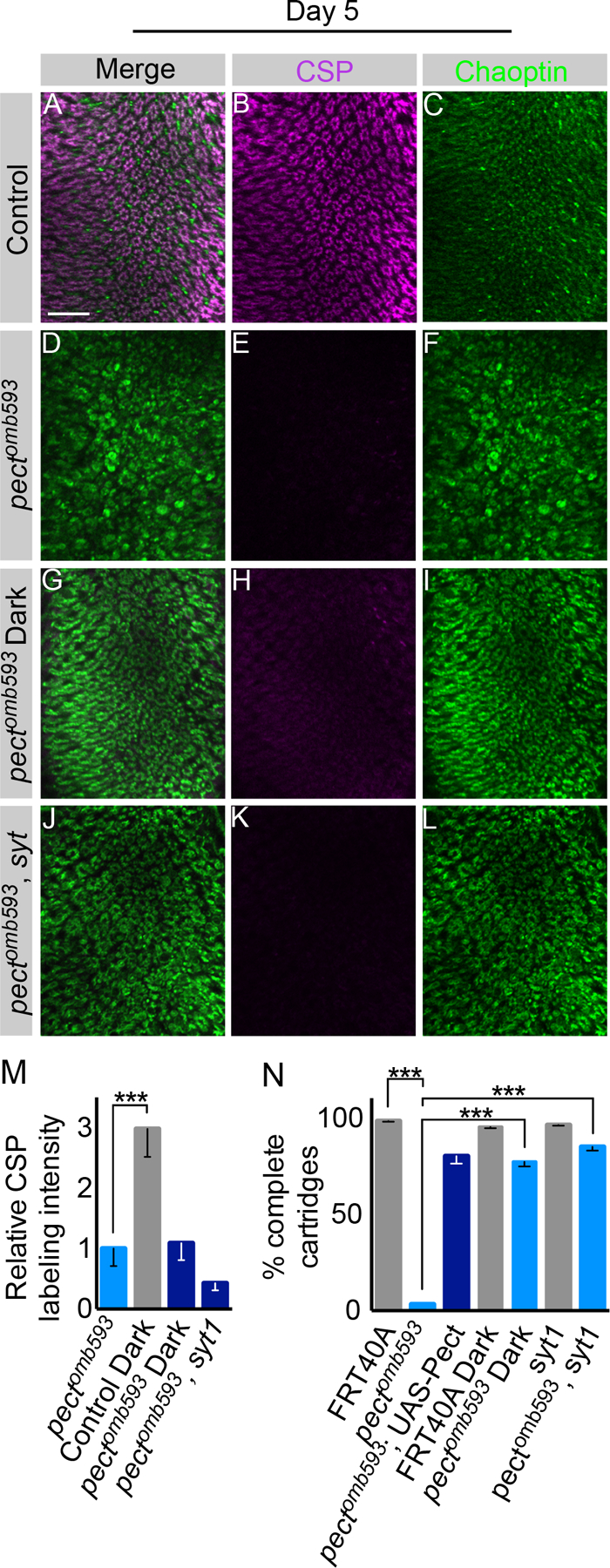
Day 5 flies. (A-C) Control. (D-F) Pectomb593 somatic mosaic animals. (G-I) Pectomb593 somatic mosaic animals reared in the dark. (J-L) Pectomb593, syt double mutant somatic mosaic animals. (M) Relative CSP labeling intensity in dark-reared control, dark-reared pectomb593, and pectomb593, syt double mutants compared to pectomb593 mutants. (N) Cartridge quantification using Chaoptin labeling for pectomb593 mutants, dark-reared control, dark-reared pectomb593, pectomb593, and pectomb593, syt double mutants. Scale bar: 15 µm. Data represented as mean ± SEM. Differences significant at p < 0.001 (***) (one-way ANOVA; t-test). n = 9 for each genotype. See also Figure S4.
Loss of synaptic marker expression in pect mutant R cells requires SREBP
In non-neuronal cells, reductions in PE levels activate the transcription factor SREBP, inducing expression of metabolic regulatory factors, including genes involved in lipid synthesis. As pect mutants disrupt the synthesis of specific PE species, we hypothesized that increased activation of SREBP might be functionally relevant. To test this, we first expressed an RNA interference construct directed against SREBP in pect somatic mosaic mutants. Western blotting of dissected retinas revealed that this construct reduced SREBP expression by approximately 83% (Figure S5A). Control flies expressing either GMR-Gal4 alone or GMR-Gal4 driving SREBP-IR displayed normal synaptic vesicle staining and stereotyped cartridge structures (Figure 5A–C; data not shown). Strikingly, pect mutants expressing SREBP-IR displayed increased expression of CSP relative to pect mutants alone (Figure 5D–I, Q). However, knock-down of SREBP failed to suppress degeneration of the axon terminal or the retina (Figure 5P and data not shown). Next, we confirmed these RNAi studies by reducing the activity of SREBP by 50% using animals that were heterozygous for a molecular null allele, SREBP∆189. Indeed, reducing SREBP dosage produced suppressed the loss of CSP expression (Figure 5Q; Figure S5B–J). Thus SREBP is required for the loss of CSP expression seen in pect mutant R cells, a process that is mechanistically distinct from degeneration of the axon terminal.
Figure 5. Pect synaptic vesicle loss requires SREBP.
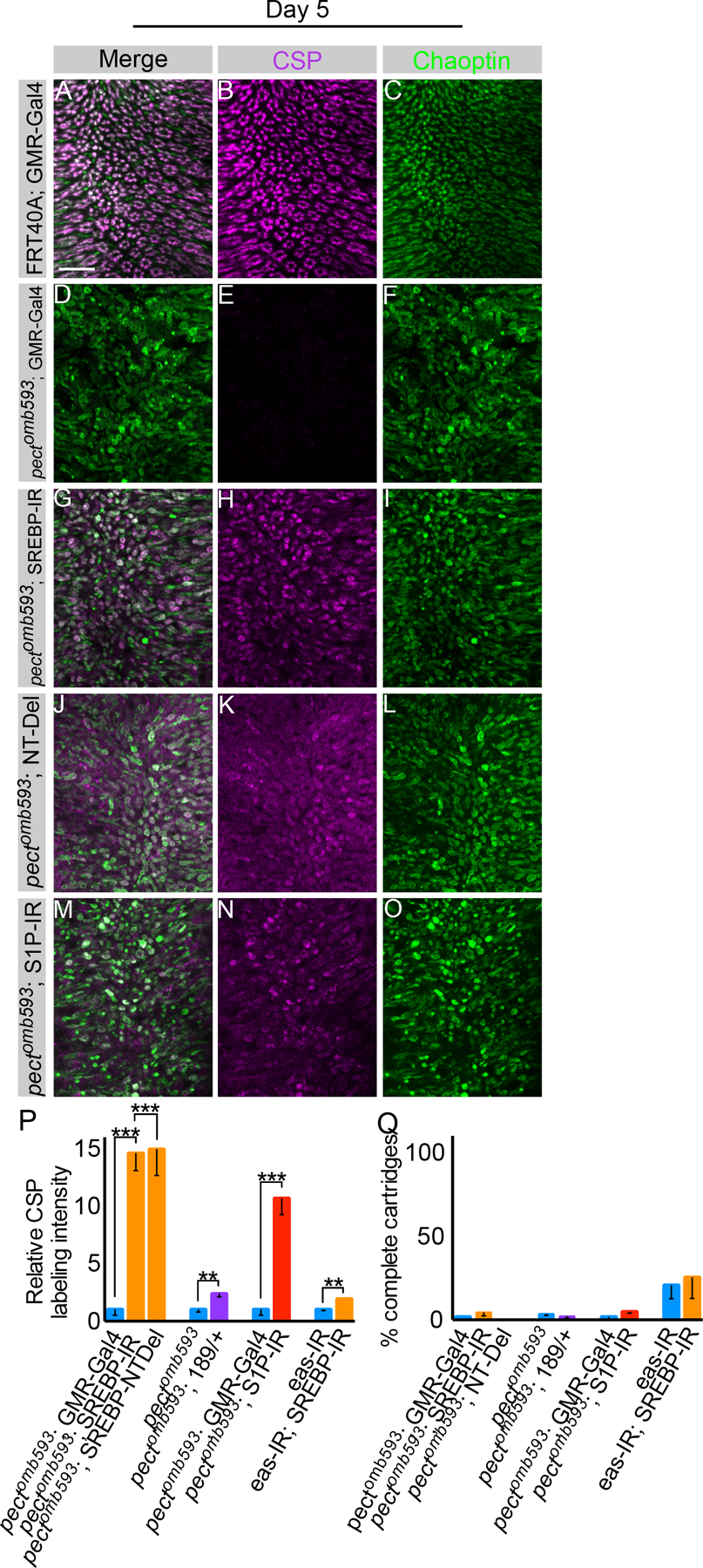
Day 5 flies. (A-C) Control flies expressing GMR-Gal4. (D-F) Pectomb593 somatic mosaic animals expressing GMR-Gal4. (G-I) Pectomb593 somatic mosaic animals expressing UAS-SREBP-IR driven by GMR-Gal4. (J-L) Pectomb593 somatic mosaic animals expressing UAS-SREBP-NT-Del using GMR-Gal4. (M-O) Pectomb593 somatic mosaic animals expressing S1P-IR. (P) Relative CSP labeling intensity in pectomb593 mutants expressing UAS-SREBP-IR and UAS-SREBP-NTDel relative to pectomb593 mutants expressing GMR-Gal4 alone, pectomb593 mutants containing SREBP∆189/+ compared to pectomb593 mutants alone, pectomb593 mutants expressing SIP-IR using GMR-Gal4 relative to pectomb593 mutants expressing GMR-Gal4 alone, and eye-specific eas-IR with SREBP-IR relative to flies expressing eas-IR alone. (Q) Cartridge quantification using Chaoptin labeling for pectomb593 mutants expressing pectomb593 mutants expressing UAS-SREBP-IR and UAS-SREBP-NTDel relative to pectomb593 mutants expressing GMR-Gal4 alone, pectomb593 mutants containing SREBP∆189/+ compared to pectomb593 mutants alone, pectomb593 mutants expressing SIP-IR using GMR-Gal4 relative to pectomb593 mutants expressing GMR-Gal4 alone, and eye-specific eas-IR with SREBP-IR relative to flies expressing eas-IR. Scale bar: 15 µm. Data represented as mean ± SEM. Differences significant at p < 0.01 (**), p < 0.001 (***) (one-way ANOVA; t-test). n = 10 for each genotype. See also Figure S5.
We next tested whether this epistatic genetic interaction between pect and SREBP was specific to Pect, or more general to PE biosynthesis. The CDP-ethanolamine pathway that includes Pect converts ethanolamine to PE in three steps (Figure S5K). If the CDP-ethanolamine pathway was specifically required in adult R cells, inhibiting either the first or last enzymatic step should produce a degenerative phenotype similar to that in pect mutants. However, three genes have been identified as ethanolamine phosphotransferases, making genetic dissection of the third enzymatic step complex. We therefore used eye-specific expression of an RNA interference construct directed against eas, the upstream gene, and successfully phenocopied the degenerative phenotype, producing bloated R cell axon terminals and a decrease in CSP staining (Figure 5P, Q; Figure S5L–Q). Moreover, knock-down of SREBP in the background of reduced Eas modestly suppressed the loss of CSP staining (Figure 5P, Q; Figure S5R–T). Thus, the degenerative phenotypes we observed in pect mutants, and the genetic epistasis with SREBP, reflects deficits in this biosynthesis pathway.
We next examined the mechanism underlying this genetic interaction. SREBP is a transmembrane protein that is normally localized to the ER and is activated by proteolytic cleavage to release an N-terminal fragment that contains a helix-loop-helix DNA binding domain (Wang et al., 1994; Hua et al., 1995). To test whether the genetic interaction between SREBP and pect mutants reflected increased transcriptional activity, we expressed a dominant negative SREBP transgene in pect somatic mosaic animals. UAS-SREBP-NTDel, contains a deletion in the highly acidic region of the N-terminal domain that is required for transcriptional activation (reviewed in Goldstein et al., 2002). This dominant negative protein retains the transcription factor in the endoplasmic reticulum. Eye-specific expression of this dominant negative SREBP transgene in pect mutant R cells suppressed the loss of CSP staining but did not ameliorate the degeneration of the axon terminal (Figure 5J–L, P, Q). Finally, we knocked-down S1P, the enzyme required for the first proteolytic cleavage in the SREBP activation cascade, in pect mutant R cells (Amarneh et al., 2009). This manipulation strongly suppressed the synaptic vesicle loss (Figure 5M–Q). Thus, activation of SREBP by its canonical pathway is required for the loss of CSP staining seen in pect mutants.
Activated SREBP is up-regulated in pect mutant R cells
These genetic interactions predict that SREBP should be activated in pect mutants. We therefore determined whether pect mutant R cells contain elevated levels of the proteolytically-cleaved, nuclear form of SREBP (Seegmiller et al., 2002). In dissected retinas from control flies, we detected both the full-length SREBP precursor (p-SREBP) as well as the shorter, nuclear form (n-SREBP) (Figure 6A). Importantly, both the full-length precursor and the activated, nuclear forms of SREBP were elevated in pect mutant retinas (Figure 6A). As these retinal dissections do not include the photoreceptor axon terminals, these changes in protein levels reflect changes in the cell body and nucleus. Finally, that both the full-length and activated forms of SREBP are elevated in these mutants is consistent with previous observations that vertebrate SREBP is positively auto-regulated (Sato et al., 1996).
Figure 6. Nuclear SREBP is up-regulated in pect mutants.
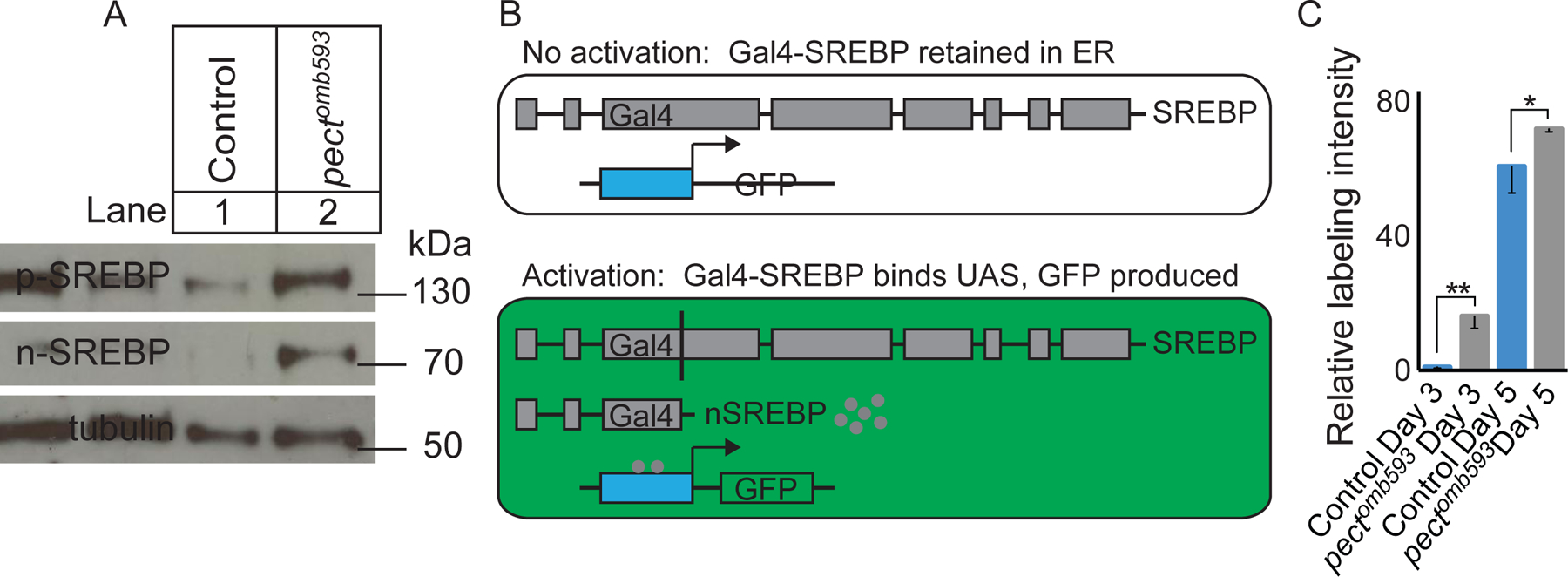
(A) Western blot of control and pectomb593 retinas using anti-SREBP antibody directed against the N-terminal domain of SREBP, with an anti-tubulin loading control. Lane 1: Control. Lane 2: pectomb593. (B) Diagram of the Gal4-VP16-SREBP construct and its activation. (C) Quantification of relative GFP labeling intensity. Day 3 control, day 3 pectomb593 mutants, day 5 control, and day 5 pectomb593 relative to day 3 control. Data represented as mean ± SEM. Differences significant at p < 0.05 (*), p < 0.01 (**) (one-way ANOVA; t-test). n = 10 for each genotype. See also Figure S6.
To examine whether increased levels of activated SREBP were associated with increased transcriptional activity, we took advantage of a Gal4-SREBP, UAS-GFP strain that acts as a transcriptional reporter of SREBP activation (Kunte et al., 2006). In this reporter, the DNA binding domain of SREBP is replaced with Gal4-VP16, but retains all of the domains that target SREBP to the ER for processing. Thus, this Gal4-VP16-SREBP derivative is inactive until it has been subject to the normal proteolytic processing cascade associated with SREBP activation (Figure 6B). Higher levels of GFP expression thus reflect increased activated, nuclear SREBP. Control animals expressing this reporter displayed significantly lower levels of GFP fluorescence than pect mutant somatic mosaic animals (Figure 6C). This increase in GFP expression paralleled the progressive loss of synaptic marker expression seen in pect mutants, as reporter activity could not be detected on the day of eclosion, rose to modest levels by day 3, and was markedly higher on day 5 after eclosion (data not shown, and Figure S6A–H).
SREBP activation is sufficient to cause synaptic vesicle loss
To test whether activation of SREBP alone was sufficient to induce synaptic vesicle loss in an otherwise wild-type animal, we took advantage of SREBP1-452, a truncated fragment of the N-terminus that comprises the first 452 amino acids and contains the active transcription factor domain. This construct produces a constitutively active form of SREBP (Yang et al., 1994; Yang et al., 1995). We used Rh1-Gal4 to restrict expression of this construct to R1-R6 cells late in development, and observed that expression of SREBP1-452 was sufficient to produce significant loss of synaptic vesicle labeling but had no effect on active zones, a phenotype qualitatively similar to the synaptic changes observed in pect mutants (Figure 7A–G). Conversely, expression of full-length SREBP had no effect (Figure S7A). Consistent with this difference, Western blot analysis revealed that increased expression of the activated form of SREBP were only observed using the UAS-SREBP1-452 transgene, not the full-length transgene (Figure S7B). Notably when we over-expressed nuclear SREBP, we also observed increased levels of full-length SREBP, consistent with a positive auto-regulatory feedback loop, as we also observed in pect mutants (Figure 6A).
Figure 7. Activation of SREBP is sufficient to induce synaptic vesicle loss.
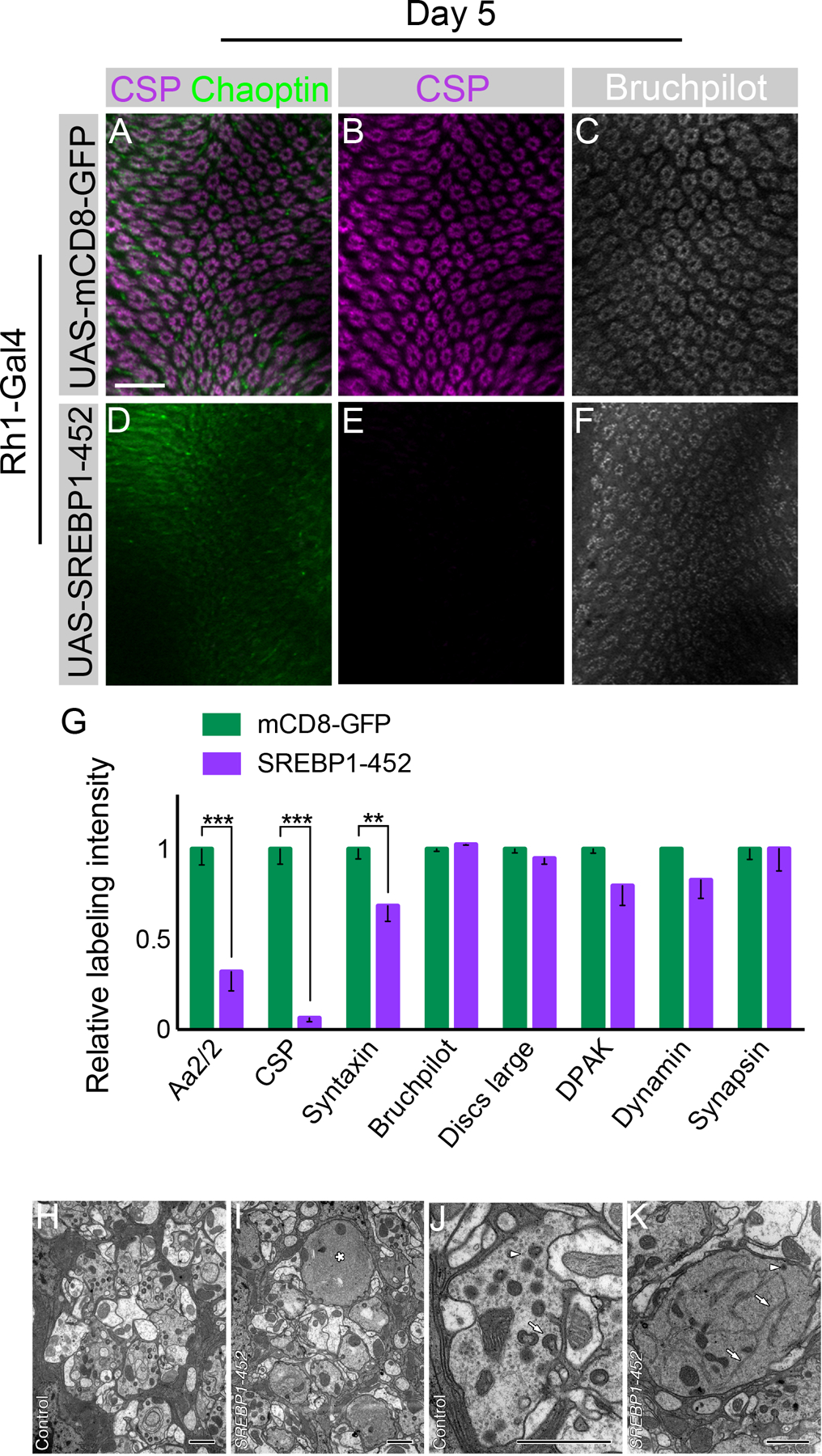
Day 5 flies. (A-C) Flies expressing UAS-mCD8-GFP driven by Rh1-Gal4 with CSP (magenta), Chaoptin (green), and Bruchpilot (white) labeling. (D-F) Flies expressing UAS-SREBP1-452 driven by Rh1-Gal4. (G) Quantification of relative synaptic labeling observed using several markers (Aa2/2, CSP, Syntaxin, Discs large, DPAK, Dynamin, Bruchpilot, and Synapsin) in Rh1-Gal4 >> UAS-SREBP1-452 animals relative to Rh1-Gal4 >> UAS-mCD8-GFP control. Differences significant at p < 0.01 (**), and p < 0.001 (***) (one-way ANOVA; multiple t-tests). (H, I) Single control and SREBP1-452 mutant lamina cartridges. The mutant (I) has a single, large R1-R6 terminal (*) which lacks obvious synaptic organelles. Other terminals appear largely normal. (J, K) Single control and SREBP1-452 mutant (K) R1-R6 terminals. Note the enlarged mutant terminal with extensive smooth, branched structures with the morphological character of endoplasmic reticulum profiles (arrows), single DCV (arrowhead), and capitate projections, lacking in this profile, but which are abundant in the control terminal (arrow, J). Note also the presence of numerous synaptic vesicle profiles (arrowhead) in the control terminal (J) that are lacking in the mutant (K). These form no clear clusters but are distributed throughout the profile of the terminal. Data represented as mean ± SEM. Scale bar: 15 µm (A-F), 1 µm (H-K). n = 10 for each genotype. See also Figure S7.
To better compare the synaptic changes seen in pect mutants to those associated with expression of SREBP1-452, we compared expression of eight synaptic markers in pect mutant R cell terminals, and R cell terminals that were wild-type for pect, but which expressed SREBP1-452. Of these eight markers, expression of five, namely CSP, Syntaxin, DLG, Synapsin and Aa2/2, was reduced in pect mutants, while expression of three others, namely DPAK, Dynamin, and Bruchpilot was unaffected (Figure S1A). Expression of SREBP1-452 caused significant reductions in three of the five markers affected in pect mutants, namely CSP, Syntaxin and Aa2/2, leaving the remaining five markers unchanged (Figure 7G). A common feature of those markers with reduced expression is that all label synaptic vesicles. Thus, activation of SREBP causes a qualitatively similar, but more specific pattern of synaptic marker loss than that seen in pect mutants.
R cell axon terminals in flies expressing SREBP1-452 exhibited many of the same synaptic phenotypes displayed in pect mutant terminals, but lacked the degenerative changes in the axon terminal and cell body. As in pect mutants, expression of SREBP1-452 caused a reduction in the packing density of synaptic vesicles and resulted in the increased presence of structures with the morphological character of endoplasmic reticulum (Figure 7H–K, S7C). SREBP1-452 mutant cartridges at 2–3 days after eclosion exhibited a range of R1-R6 terminal phenotypes, some with enlarged profiles that lacked most capitate projections and had extensive structures with the morphological character of smooth endoplasmic reticulum and occasional profiles of dense core vesicles neighboring other, more normal terminals (Figure 7H–K). The density of T-bar ribbon profiles around the perimeter of the R-cell terminal was also lower than in control terminals (Figure S7D). Finally, the structure of R cell bodies was also slightly perturbed in R cells expressing SREBP1-452 compared with those in control ommatidia (Figure S7E, F). Thus, activation of SREBP in R1-R6 cells is sufficient to cause synaptic vesicle loss.
We next examined whether activation of SREBP affects either the transcription of synaptic genes, or the levels of synaptic proteins, or both. To do this, we performed reverse transcription qPCR on cDNA isolated from dissected retinas comparing flies expressing SREBP1-452 to those expressing UAS-mCD8-GFP, and examined two of the most prominent synaptic proteins, CSP and Syntaxin. These studies revealed that expression of SREBP1-452 changed the mRNA expression levels for each of these two genes less than two-fold (1.61-fold for CSP, 0.78-fold for Syntaxin, comparing SREBP1-452 to mCD8-GFP). We also performed Western blot analysis on dissected retinas of the same genotypes, and examined CSP and Syntaxin protein levels. We observed that expression of SREBP1-452 caused a 11.9-fold decrease in CSP protein levels and a 2.2-fold decrease in Syntaxin protein levels (Figure S7G). Thus, for both of these genes, the changes in transcription were insufficient to account for the observed changes in protein levels arguing that SREBP activation likely has indirect effects on these genes.
RNA profiling identifies nuclear SREBP-induced transcriptional changes
To identify SREBP target genes that might mediate this regulatory effect, we performed RNA-seq analysis of dissected adult retinal tissue 5 days after eclosion to identify potential causal targets. We compared gene expression profiles in which Rh1-Gal4 was used to express mCD8-GFP versus SREBP1-452. We detected the transcripts of 8924 genes in the two control replicates, as well as 8885 genes in the two SREBP1-452 replicates (Supplemental Table 2). Both of these datasets included 20/22 (91%) of the genes previously known or implicated in retinal degeneration or phototransduction, as well as 37/93 (40%) of the genes identified as differentially expressed between wild-type heads and heads derived from eyeless flies (Xu et al., 2004). Thus, this dataset represents a significant fraction of the adult retinal transcriptome. In our dataset, only 219 genes (2.46% of the SREBP 1-452 transcriptome, p<0.0011) changed expression when SREBP was activated (Figure 8A). 128 of these genes increased expression between 2 and 24 fold; 91 of these genes decreased expression between 2 and 62 fold (Figure 8B). To validate these changes, we selected 10 genes for qPCR, eight of which had large decreases in expression according to our RNA-seq analysis, and two of which displayed no significant difference. These qPCR results were entirely consistent with our RNA-seq studies (Figure S8A).
Figure 8. Tetraspanins are functionally critical SREBP effectors.
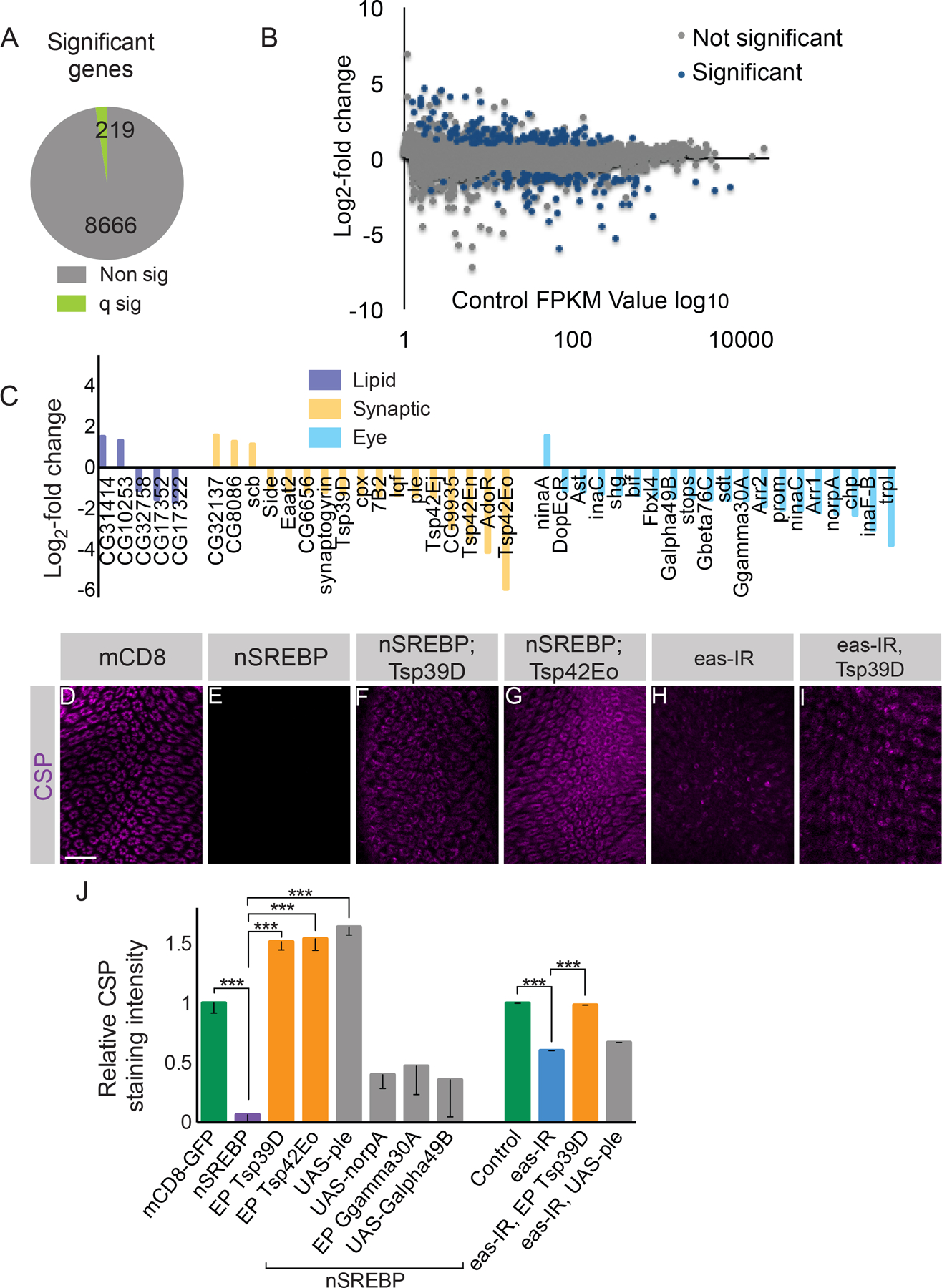
(A) A small group of genes display significantly (q sig) altered expression between flies expressing Rh1-Gal4 >> UAS-mCD8-GFP versus Rh1-Gal4 >> UAS-SREBP1-452. (B) Control log10 FPKM values versus log2-fold change in expression. (C) Log2-fold change in FPKM values from RNA-seq in the SREBP1-452 condition relative to control for lipid biosynthetic genes, synaptic genes, and eye and phototransduction-related genes. (D-G) Lamina cross sections stained with an antibody directed against CSP. (D) Control flies in which UAS-mCD8-GFP is driven by Rh1-Gal4. (E) Flies expressing UAS-SREBP1-452 driven by Rh1-Gal4. (F) Flies co-expressing UAS-SREBP1-452 with EP:Tsp39D driven by Rh1-Gal4. (G) Flies expressing UAS-SREBP1-452 and EP:Tsp42Eo driven by Rh1-Gal4. (H) Flies expressing eye-specific eas-IR. (I) Flies co-expressing eye-specific eas-IR and EP Tsp39D. (J) Quantification of relative CSP labeling. Data represented as mean ± SEM. Differences significant at p < 0.001 (***) (one-way ANOVA). Scale bar: 15 µm. n = 9 for each genotype. See also Figure S8 and Supplemental Table 2.
Expression of 27 genes was strongly affected by SREBP activation, causing changes in expression of approximately 8 fold or greater (Supplemental Table 2). Of these, 14 genes were significantly up-regulated, all of which were only weakly expressed in wild-type photoreceptors, and remained expressed at low levels when SREBP was activated (Supplemental Table 2). Conversely, the 13 genes that were down-regulated by activation of SREBP were expressed at a variety of levels in wild-type R cells. As we will discuss further below, this subset of genes included two members of the tetraspanin family, Tsp42Eo and Tsp42En, located at adjacent chromosomal loci. More broadly, SREBP activation affected the expression of 5 genes involved in lipid biosynthesis, 20 genes that had previously been described as playing roles in phototransduction or retinal structure, and 17 additional genes with synaptic functions (Figure 8C). Strikingly, 19 of 20 phototransduction genes as well as 14 of 17 synaptic genes were down-regulated by SREBP activation (Figure 8C). Taken together, these data demonstrate that activation of SREBP regulates a handful of lipid biosynthetic genes, and reduces expression of many genes involved in phototransduction, synaptic transmission and R cell structure.
Tetraspanins are functionally critical targets of SREBP regulation
This RNA-seq dataset provided a list of candidate SREBP target genes, a subset of which could be functionally critical. We noted that four members of the tetraspanin family, Tsp42Eo, Tsp42En, Tsp42Ej and Tsp39D were downregulated by SREBP1-452. Tetraspanins are transmembrane proteins that regulate synaptic development in flies, and vertebrate photoreceptor morphology (Kopczynski et al., 1996; Travis et al., 1991; Bascom et al., 1992). We therefore hypothesized that this gene family might represent functionally important SREBP effectors. As genetic tools existed to allow photoreceptor-specific over-expression of Tsp39D and Tsp42Eo, we compared the effects of co-expressing these two genes with SREBP1-452 to the effects of co-activating four other genes that were also downregulated by SREBP activation, namely pale, no receptor potential A, Ggamma30A and Galpha49B. Remarkably, co-expression of either Tsp39D or Tsp42Eo significantly suppressed the effects of SREBP 1-452 on CSP expression (Figure 8D–G, J). However, while three of the four control genes had no effect, we also observed phenotypic suppression using a ple transgene. To determine whether these genetic interactions reflected the activities of bona fide downstream effectors of SREBP, versus artifactual effects on expression of the SREBP1-452 transgene (caused by the presence of additional Gal4 binding sites associated with the interacting transgene), we performed Western blots on dissected retinas and stained for SREBP. These studies revealed that while Tsp39D expression had no effect on the level of SREBP1-452, expression of Ple reduced SREBP1-452 levels (Figure S8B). Thus, Tsp39D but not Ple, acts as a downstream effector of SREBP.
If down-regulation of tetraspanins causes synaptic vesicle loss in response to SREBP activation, upregulation of tetraspanins should suppress the synaptic phenotypes associated with mutations in pect. However, the tetraspanin loci we identified, Tsp39D and the Tsp42E complex lies extremely close to the centromere, meaning that they cannot be used in mosaic clones with pect null alleles. As an alternate approach, we took advantage of the EAS knock-down manipulation that causes phenotypes similar to those seen in pect mutants (Figure S5). Remarkably, over-expression of Tsp39D in the Eas knock-down background suppressed the loss of CSP staining (Figure 8H–J). Thus, at least some tetraspanins are functional downstream effectors of SREBP.
DISCUSSION
Summary
These studies demonstrate that disrupting the biosynthesis of specific membrane phospholipids causes adult onset degeneration of R cells and loss of synaptic vesicles. These two phenotypes arise via distinct molecular mechanisms that can be doubly dissociated using genetic and physiological manipulations (Figure S9A–E). Degeneration of the axon terminal is an activity-dependent process that requires calcium-mediated vesicle fusion. Conversely, loss of synaptic vesicles is driven by activation of the transcription factor SREBP. Thus, in these cells, SREBP is activated by alterations in the levels of specific phospholipids. Here, SREBP affects the expression of a specific subset of genes that are largely not directly involved in lipid regulation, thus defining a previously unknown SREBP function. Rather, SREBP activation leads to reduced expression of four tetraspanins. Restoring expression of either of two of these tetraspanins suppresses the effects of SREBP activation, demonstrating that tetraspanins are functional effectors of SREBP in photoreceptors. Thus, a specialized feedback loop from the synaptic terminal to the nucleus links the levels of specific phospholipids to photoreceptor function and synaptic vesicle number. We propose that this feedback loop matches the vesicular demand for phospholipids to their production. As SREBP is evolutionarily conserved, and recent studies have linked SREBP to neuronal damage in several contexts, we speculate that this feedback loop plays a central role in maintaining synaptic vesicle pools in the healthy aging brain.
The relative levels of specific phospholipids are critical to neuronal structure and function
In Drosophila, mutations that disrupt phospholipid biosynthesis cause broad defects in brain function, including increased seizure activity and photoreceptor degeneration (Pavlidis et al., 1994; Midorikawa et al., 2010). However, these and other studies examining phospholipid composition in flies have either not quantified phospholipid levels, or have not differentiated different phospholipid species (Pavlidis et al., 1994, Midorikawa et al., 2010; Lim et al., 2011). Our studies using a high-resolution lipidomic approach demonstrate that biosynthesis of specific PE and PC species is required for maintaining synaptic vesicle pools and the axon terminal in adult photoreceptors. Moreover, the biosynthetic enzyme Pect is found at the axon terminal. We speculate that the production of specific phospholipids can occur locally, coupling precise levels of phospholipids to the cellular processes that require them in the axonal compartment. Finally, recent work has demonstrated that derivatives of very long chain (VLC) PC species are neuroprotective in vertebrate photoreceptors and neurons (Jun et al., 2017; Bhattacharjee et al., 2017). While we detected only one VLC PC precursor, PC c44:12, representing 0.01% of the total PC species in the fly retina (Supplementary Table 1), future studies will determine the extent to which derivatives of this or other PC or PE species play roles in maintaining adult photoreceptor axons and synapses in Drosophila.
Our work suggests the following model (Figure S9B). Our ultrastructural analysis of pect mutants reveals phenotypes in the axon terminal that are strongly reminiscent of those in endocytic mutants (Fabian-Fine et al., 2003; Verstreken et al., 2003; Dickman et al., 2005). Consistent with this, blocking exocytosis in pect mutants by either reducing light exposure, or by genetic means, suppresses axon terminal degeneration. We infer, then, that the inability to retrieve synaptic vesicles from the plasma membrane is sufficient to cause neuronal degeneration, and that the availability of specific phospholipids can be rate-limiting for endocytosis. These results are consistent with previous studies in C. elegans that demonstrated that a phospholipid desaturase causes defects in endocytosis through effects on synaptojanin, a critical component in endocytosis (Marza et al., 2008). At the same time, altering phospholipid production may also impair vesicle biogenesis, in which case blocking synaptic transmission could suppress neuronal degeneration by removing the demand for vesicle biogenesis via an as-yet unknown mechanism.
A previously undescribed function for SREBP in adult R cells
SREBP is a central regulator of genes involved in lipid biosynthesis in many cell types (Rawson, 2003). Our data support the notion that SREBP plays an additional role in Drosophila photoreceptors. As the levels of only a few phospholipids are altered in pect mutants, SREBP activation appears linked to the detection of changes in levels in these PE and PC species. Moreover, while activation of SREBP does up-regulate a small number of genes involved in lipid biosynthesis, it also down-regulates many genes involved in phototransduction and synaptic function. Amongst these, our genetic interaction studies demonstrate that tetraspanins are functionally critical SREBP effectors. Tetraspanins are transmembrane proteins that have been linked to synapse development (Kopczynski et al., 1996), lysosomal function in R cells (Xu et al., 2004) and to outer segment structure and function in the vertebrate retina (Travis et al., 1991; Bascom et al., 1992). Moreover, recent work has demonstrated that they can serve as cholesterol binding proteins, further implicating this family in the regulation of membrane function (Zimmerman et al., 2016). While unraveling the specific molecular mechanisms that link tetraspanin function to synaptic vesicle pools remains a challenge for future studies, our model for this role of SREBP represents an extension of SREBP’s longstanding role in regulating lipid biosynthesis. In particular, a central role for phospholipids that is unique to neurons is as a critical component of synaptic vesicles. We hypothesize that when SREBP is activated, and tetraspanin expression is reduced, either the biogenesis of synaptic vesicles is downregulated, or their turnover and degradation is increased, shrinking the synaptic vesicle pool in an activity-independent manner. As a result, the cellular demand for the specific phospholipids found in synaptic vesicles is reduced. More broadly, these studies suggest that SREBP might complement its longstanding role in lipid biosynthesis with an additional role in controlling phospholipid utilization. Finally, by combining the high resolution lipidomic approach we have developed to work with small populations of labelled cells, with the powerful genetic tools available in this system, future work may shed further light on the regulation of SREBP activity and phospholipid levels.
A role for SREBP in neurological disease
SREBP has been linked to both neurodegenerative disease and stroke (Taghibiglou et al., 2009, Do et al., 201, Barbero-Camps et al., 2013). Recent studies in flies have demonstrated that reactive oxygen species can activate SREBP to cause lipid droplet formation in glia (Liu et al., 2015). However, the molecular mechanisms by which SREBP might act in these contexts are unknown. In addition, mutations in a human tetraspanins have been linked to intellectual disability (Bassani et al., 2012, Verpelli et al., 2013). Our demonstration that SREBP acts through tetraspanins to regulate synaptic vesicle pools and negatively regulates other genes required for synaptic function suggests a unifying mechanism for these seemingly disparate observations. Taken together, these studies argue that SREBP plays an evolutionarily conserved role in regulating neuronal and synaptic function, suggesting a link between the neuronal phospholipid repertoire and synapse maintenance in the adult brain.
EXPERIMENTAL PROCEDURES
Chemical mutagenesis, mosaic analysis, rescue and histological methods were as described in (Clandinin et al., 2001; Lee et al., 2001). Adult eyes were prepared for dissection, fixation, and embedding in epoxy following (Gaengel and Mlodzik, 2008). Ultrastructural studies were performed as previously described (Fabian-Fine et al., 2003). RNA was isolated from pools of dissected eyes and used for sequencing library preparation and analysis using an Illumina Genome Analyzer IIX, with two biologic replicates per genotype. Phospholipids from 10 dissected retinas per biological replicate were Folch extracted, and subjected to normal phase chromatography followed by MS (Rivas-Serna et al. 2017). Western blots from dissected retinas were performed following standard methods, with at least two biological replicates. See STAR methods for further details.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Thomas R. Clandinin (trclandinin@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Husbandry and Genetics
Fly stocks were maintained at 25°C on standard medium. Chemical mutagenesis for the forward genetic screen was performed using EMS on standard conditions (Clandinin et al., 2001). Somatic mosaic animals were produced using the FLP/FRT system. We used a dominant eye-specific cell lethal, gmr-hid, to reduce the size of the twin spot. The GAL4/UAS system was employed to drive transgene expression in specific cell types (Brand and Perrimon, 1993; reviewed in Duffy, 2002).
METHOD DETAILS
Fly stocks
The following alleles were used: pectomb362; pectomb593; pect38K; pectLL06325; synaptotagminAD4 (Dr. Tom Schwarz, Boston Children’s Hospital). The following drivers were used: Rh1-Gal4; GMR-Gal4; and ey-Gal4, GMR-Gal4. The SREBP reporter Gal4-SREBP, UAS-GFP was obtained from Dr. Robert Rawson (UTSW). All RNAi lines were received from the Vienna Drosophila RNAi Center (Dietzl et al, 2007). UAS-Pect was made using cDNA clone RE62261 from the Drosophila Genomics Resource Center. The following UAS transgenes were obtained from the Bloomington Drosophila Stock Center at Indiana University and Dr. Robert Rawson (UTSW): UAS-SREBP1-452; UAS-SREBP; UAS-SREBP-NTDel; UAS-mCD8-GFP. The following EP lines and UAS transgenes were obtained from the Bloomington Drosophila Stock Center at Indiana University: EP Tsp39D; EP Tsp42Eo; UAS-ple; UAS-norpA; EP Galpha49B; EP Ggamma30A.
Animal Husbandry and Genetics
Fly stocks were maintained at 25°C on standard medium. Chemical mutagenesis for the forward genetic screen was performed using EMS on standard conditions (Clandinin et al., 2001). Somatic mosaic animals were produced using the FLP/FRT system. We used a dominant eye-specific cell lethal, gmr-hid, to reduce the size of the twin spot. The GAL4/UAS system was employed to drive transgene expression in specific cell types (Brand and Perrimon, 1993; reviewed in Duffy, 2002).
Semithin Sections
Adult eyes were prepared for dissection, fixation, and embedding in epoxy (Gaengel and Mlodzik, 2008). Semithin sections were mounted on slides using DPX mounting medium. Images were acquired on a Zeiss Axio Imager.Z1 microscope using a 100X objective lens with an AxioCam HR microscope camera.
Electron Microscopy
Organelle counts from mutant and control R1-R6 photoreceptor terminals were made from the means of the means for 3 flies, with approximately 15 profiles per fly. We counted the numbers of profiles of synaptic organelles: synaptic vesicles, T-bar ribbons at synapses, and capitate projections, sites of invagination into the photoreceptor terminal by surrounding epithelial glial cells (Stark and Carlson, 1986). These were counted as one of three related forms of EM profile (Pyza and Meinertzhagen, 1998): shallow (with a head invaginating not more than half its diameter); single (penetrating, with a single head profile); or multiple-headed (penetrating, with more than one head). To express these profile counts with respect to the size of the R1-R6 terminal profile, we normalized the raw counts to the length of the R1-R6 profile perimeter and cross-sectional area, measured using software (ImageJ, NIH). We tested organelle counts for statistical significances first by an unweighted means ANOVA, followed by a Tukey HSD test, using software (Systat 5.2.1). We also marked the x,y coordinates for the centers of synaptic vesicle profiles, to determine spatial dispersion, using previously reported methods (Dickman et al., 2005) and spatial statistics (Clark and Evans, 1954).
Immunohistochemistry
Adult brains were dissected in 2% PLP under previously described conditions (Lee et al., 2001). The following antibodies were used: mouse anti-Chaoptin mAb24B10 (1:20, Developmental Hybridoma Bank), mouse anti-CSP (1:10), mouse anti-Syntaxin (1:10), mouse anti-Bruchpilot (1:10), chicken anti-GFP (1:2000), mouse anti-Discs large (1:20), mouse anti-EPS-15 (1:20); mouse anti-Synapsin (1:20); rabbit anti-DPAK (1:100); rabbit anti-Dynamin (1:100), goat anti-mouse AlexaFluor488 IgG1 (1:200), goat anti-rabbit AlexaFluor488 (1:200), goat anti-chicken 488 (1:200), goat anti-mouse AlexaFluor546 IgG1 (1:200), goat anti-mouse AlexaFluor594 IgG2a (1:200), goat anti-mouse AlexaFluor594 IgG2b (1:200), goat anti-rabbit 594 (1:200), and goat anti-mouse Alexa 633 IgG2a (1:200).
Confocal Imaging
Images were acquired on a Leica TCS SP2 AOBS confocal microscope, using a 40x NA 1.25 lens. Images were rendered using Imaris (Bitplane), and figures were prepared using Photoshop and Illustrator (Adobe).
Quantification of Immunolabeling
Anti-Chaoptin and anti-CSP were used as reference immunolabels in order to delineate the area of interest for each lamina section. First, reference staining images were filtered (using a 2D averaging filter with fixed window size). Subsequently, images were thresholded (by the mean pixel intensity in the filtered image), eroded (using a fixed size square structuring element), and holes, if present, were filled. Pixel intensities from raw images were analyzed within these defined areas of interest using a fixed threshold value, and the total fraction of pixels above the fixed threshold value was determined. Quantification analysis was performed using MatLab. For the degeneration phenotype, cartridges were counted manually, and the number of complete cartridges containing 6 R cell terminals was divided by the total number of cartridges scored. Statistical analysis was performed by one-way ANOVA using GraphPad (Prism).
Western Blot and Immunoblotting
Aliquots were boiled for 5 minutes and subjected to SDS-PAGE on an 8% polyacrylamide gel, before transfer to nitrocellulose. Blots were then incubated in 7% milk in TBS-Tween. After one hour, blots were washed in TBS-Tween, then incubated overnight in mouse anti-dSREBP-N-terminus (3B2, a generous gift from Dr. Robert Rawson). Subsequently, blots were incubated in goat anti-mouse IgG-HRP (Santa Cruz Biotechnology) in 7% milk for one hour. Blots were incubated with Thermo Scientific SuperSignal West Dura Chemiluminescent substrate and then exposed to film. After film was processed, blots were stripped and re-probed overnight with rat anti-tubulin (Abcam 6161) overnight, incubated in goat anti-rat IgG-HRP (Santa Cruz Biotechnology), and processed as described above. Blots were quantified using ImageJ (NIH).
Western Blot Quantification
Western blot quantification was performed using ImageJ analysis. Lanes were identified using ImageJ with calculation of area under the curve. For each band, the relative density, of each band to the control condition, was calculated. Subsequently, the adjusted density, the relative density of each sample divided by the relative density of the loading control, was then determined.
RNA profiling
RNA was isolated from eyes as specified above and measured using a Bioanalyzer RNA 6000 Nano kit (Agilent Technologies), NanoDrop 2000 (Thermo Scientific), and Qubit Fluorometer (Invitrogen). Fragmented double-stranded cDNA were prepared using TruSeq RNA Sample Preparation Kit (Illumina) and used for sequencing library preparation automatically with an Illumina TruSeq adapter on the SPRIWorks System I (Beckman-Coulter) by using cartridges and method cards specific to the Illumina sequencing system, followed by specific PCR enrichment. The sequencing reactions were performed using the Illumina Genome Analyzer IIX (SR36x1) sequencing platform according to manufacturer’s instructions, with two biologic replicates per genotype. High-throughput sequencing was performed using the Stanford Functional Genomics Facility.
RNA profiling analysis
Initially, all reads were analyzed for quality using Galaxy (Blankenberg et al., 2010a; Blankenberg et al., 2010b). Reads were subsequently mapped to the Drosophila melanogaster reference genome using TopHat (Trapnell et al., 2009). Mapped reads were then analyzed using Cufflinks to generate a FPKM score (Trapnell et al., 2010; Trapnell et al., 2012). CuffDiff was used to compare Cufflink files from all conditions. This enabled the generation of both p and q values after significance testing between groups, taking biologic replicates into account.
Phospholipid Extraction
Phosholipids from disected retinas were extracted by a Folch extraction. Approximately 10 dissected retinas were used for each biological replicate. Chloroform:methanol (2:1, v/v) was added to sonication for 15 s in ice. This homogenate was ground with a dounce homogenizer on ice and CaCl2 (0.025%) was added. After 10 min shaking, this mixture was centrifuged and bottom layer removed. The upper layer was washed with chloroform:methanol and centrifuged. The bottom layers were combined and dried under N2. Phospholipid extracts were resuspended in 75% acetonitrile 25% water/methanol solution prior to LC/triple quad MS analysis.
Mass Spectrometry
Phospholipid extracts were analysed using LC/triple quad MS (Rivas-Serna et al. 2017). Phospholipid separation was subjected to normal phase chromatography using an Agilent 1260 Infinity LC system followed by MS analysis using an Agilent 6430 Triple-Quad LC/MS system (Santa Clara, CA) to obtain the relative abundance of PC and PE phospholipids. The MS was operated in multiple reaction monitoring mode (MRM) and positive ion mode.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed in Graphpad Prism 7. For both one- and two-way ANOVA analyses, Bonferonni corrected two-tailed tests were performed. Error bars represent standard error of the mean.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Chaoptin | Developmental Studies Hybridoma Bank | DSHB Cat# 24B10, RRID:AB_528161 |
| Mouse anti-CSP | Developmental Studies Hybridoma Bank | DSHB Cat# CSP2, RRID:AB_10805296 |
| Mouse anti-Syntaxin | Developmental Studies Hybridoma Bank | DSHB Cat# 8c3, RRID:AB_528484 |
| Mouse anti-Bruchpilot | Developmental Studies Hybridoma Bank | DSHB Cat# nc82, RRID:AB_2314866 |
| Mouse anti-Discs large | Developmental Studies Hybridoma Bank | DSHB Cat# 4F3 anti-discs large, RRID:AB_528203 |
| Mouse anti-EPS-15 | Developmental Studies Hybridoma Bank | Halder P; PLoS One. 2011 Cat# Eps-15, RRID:AB_2567895 |
| Mouse anti-Synapsin | Developmental Studies Hybridoma Bank | DSHB Cat# 3C11, RRID:AB_2313867 |
| Rabbit anti-DPAK | Developmental Studies Hybridoma Bank | Harden et al., 1996 Cat# DPAK, RRID:AB_2314339 |
| Rabbin anti-Dynamin | Developmental Studies Hybridoma Bank | Estes et al., 1996 Cat# Dynamin, RRID:AB_2314348 |
| Goat anti-mouse AlexaFluor 488 IgG1 | Invitrogen | Invitrogen Cat# A-21121, RRID:AB_141514 |
| Goat anti-rabbit AlexaFluor488 | Invitrogen | Invitrogen Cat# A-11034, RRID:AB_2576217 |
| Goat anti-chicken 488 | Invitrogen | Invitrogen Cat# A-11039, RRID:AB_2534096 |
| Goat anti-mouse AlexaFluor546 IgG1 | Invitrogen | Invitrogen Cat# A-21123, RRID:AB_2535765 |
| Goat anti-mouse AlexaFluor594 IgG2a | Invitrogen | Invitrogen Cat# A-21135, RRID:AB_2535774 |
| Goat anti-mouse AlexaFluor594 IgG2b | Invitrogen | Invitrogen Cat# A-21145, RRID:AB_2535781 |
| Goat anti-rabbit AlexaFluor594 | Invitrogen | Invitrogen Cat# A-11012, RRID:AB_141359 |
| Goat anti-mouse AlexaFluor633 IgG2a | Invitrogen | Invitrogen Cat# A-21136, RRID:AB_2535775 |
| Goat anti-rat IgG-HRP | Santa Cruz Biotechnology | Santa Cruz Biotechnology Cat# sc-2006, RRID:AB_1125219) |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | Santa Cruz Biotechnology Cat# sc-2055, RRID:AB_631738 |
| Mouse anti-dSREBP-N-terminus | Gift from Dr. Robert Rawson | N/A |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: SREBP-IR | Vienna Drosophila RNAi Center (VDRC) | FlyBase: FBst0462097 |
| D. melanogaster: S1P-IR | Vienna Drosophila RNAi Center (VDRC) | FlyBase: FBst0465869 |
| D. melanogaster: eas-IR | Vienna Drosophila RNAi Center (VDRC) | FlyBase: FBst0475642å |
| D. melanogaster: pectomb362 | Clandinin Lab | N/A |
| D. melanogaster: pectomb593 | Clandinin Lab | N/A |
| D. melanogaster: pect38K | Clandinin Lab | N/A |
| D. melanogaster: pectLL06325 | Kyoto Stock Center | FlyBase: FBst0320403 |
| D. melanogaster: synaptotagminAD4 | Gift from Dr. Tom Schwarz | FlyBase: FBal0032847 |
| D. melanogaster: Rh1-Gal4 | Bloomington Drosophila Stock Center | BDSC: 8688 |
| D. melanogaster: GMR-Gal4 | Bloomington Drosophila Stock Center | FlyBase: FBst0009146 |
| D. melanogaster: Gal4-SREBP, UAS-GFP | Gift from Dr. Robert Rawson | FlyBase: FBst0039612 |
| D. melanogaster: UAS-SREBP1-452 | Gift from Dr. Robert Rawson | FlyBase: FBst0041018 |
| D. melanogaster: UAS-SREBP | Gift from Dr. Robert Rawson | FlyBase: FBst0008236 |
| D. melanogaster: UAS-SREBP-NTDel | Gift from Dr. Robert Rawson | FlyBase: FBal0156413 |
| D. melanogaster: UAS-mCD8-GFP | Bloomington Drosophila Stock Center | FlyBase: FBst0056172 |
| D. melanogaster: Tsp39D | Bloomington Drosophila Stock Center | FlyBase: FBst0022366 |
| D. melanogaster: Tsp42Eo | Bloomington Drosophila Stock Center | FlyBase: FBst0028424 |
| D. melanogaster: UAS-ple | Bloomington Drosophila Stock Center | FlyBase: FBst0037539 |
| D. melanogaster: UAS-norpA | Bloomington Drosophila Stock Center | FlyBase: FBst0035529 |
| D. melanogaster: Galpha49B | Bloomington Drosophila Stock Center | FlyBase: FBst0030734 |
| Clones | ||
| cDNA clone RE62261 | Drosophila Genomics Resource Center | FlyBase: FBcl0202946 |
| Software and Algorithms | ||
| ImageJ | ImageJ | RRID:SCR_003070 |
| MatLab | MatLab | RRID:SCR_001622) |
| Galaxy | Galaxy Project | RRID:SCR_006281 |
| TopHat | TopHat | RRID:SCR_013035 |
| Cufflinks | Cufflinks | RRID:SCR_014597 |
| Cuffdiff | Cuffdiff | RRID:SCR_001647 |
| Graphpad Prism 7 | GraphPad | RRID:SCR_002798 |
Highlights.
Disrupting phospholipid synthesis causes degeneration of Drosophila photoreceptors
Neurodegeneration in these cells is activity dependent, and requires vesicle fusion
The transcription factor SREBP regulates the synaptic vesicle pool
Tetraspanins interact genetically with SREBP to control vesicle numbers
ACKNOWLEDGMENTS
The authors are grateful to Dr. Robert Rawson (UT Southwestern), Dr. Tom Schwarz (Boston Children’s Hospital), the Developmental Hybridoma Bank, the Drosophila Genome Center (Kyoto Stock Center), the Bloomington Drosophila Stock Center, and the Vienna Drosophila RNAi Center (VDRC) for providing fly stocks and reagents. We also thank Dr. Richard J. Reimer (Stanford University), Dr. Xinnan Wang (Stanford University), and members of the Clandinin Lab for providing helpful comments. This work was supported by NIH R01 EY015231 (TRC) and by NIH R01 EY03592 (IAM), and by grants from the Canadian Institute for Health Research (MTC). JWT was supported by NIH T32 GM007365 and the Stanford School of Medicine Medical Scientist Training Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Please see Supplemental Table 2.
The RNA-sequencing data have been deposited in the Sequence Read Archive (SRA) with BioProject ID PRJNA512933 and the following URL links:
http://www.ncbi.nlm.nih.gov/bioproject/512933
https://www.ncbi.nlm.nih.gov/biosample/10690876
https://www.ncbi.nlm.nih.gov/biosample/10690877
KEY RESOURCES TABLE
Please see Key Resources Table.
REFERENCES
- Amarneh B, Matthews KA, and Rawson RB (2009). Activation of Sterol Regulatory Element-binding Protein by the caspase Drice in Drosophila larvae. The Journal of biological chemistry 284, 9674–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RE (1970). Lipids of ocular tissues. IV. A comparison of the phospholipids from the retina of six mammalian species. Experimental eye research 10, 339–344. [DOI] [PubMed] [Google Scholar]
- Barbero-Camps E, Fernandez A, Martinez L, Fernandez-Checa JC, and Colell A (2013). APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer’s disease. Human molecular genetics 22, 3460–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bascom RA, Manara S, Collins L, Molday RS, Kalnins VI, and McInnes RR (1992). Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron 8, 1171–1184. [DOI] [PubMed] [Google Scholar]
- Bassani S, Cingolani LA, Valnegri P, Folci A, Zapata J, Gianfelice A, Sala C, Goda Y, and Passafaro M (2012). The X-linked intellectual disability protein TSPAN7 regulates excitatory synapse development and AMPAR trafficking. Neuron 73, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, and Hiesinger PR (2013). The synaptic maintenance problem: membrane recycling, Ca2+ homeostasis and late onset degeneration. Molecular neurodegeneration 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee S, Jun B, Belayev L, Heap J, Kautzmann MA, Obenaus A, Menghani H, Marcell SJ, Khoutorova L, Yang R, Petasis NA, and Bazan NG, (2017). Elovanoids are a novel class of homeostatic lipid mediators that protect neural cell integrity upon injury. Sci. Adv 3(9):e1700735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko A, and Galaxy T (2010a). Manipulation of FASTQ data with Galaxy. Bioinformatics 26, 1783–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, and Taylor J (2010b). Galaxy: a web-based genome analysis tool for experimentalists. Current protocols in molecular biology / edited by Ausubel Frederick M [et al. ] Chapter 19, Unit 19 10 11–21. [DOI] [PMC free article] [PubMed]
- Borycz JA, Borycz J, Kubow A, Kostyleva R, and Meinertzhagen IA (2005). Histamine compartments of the Drosophila brain with an estimate of the quantum content at the photoreceptor synapse. Journal of neurophysiology 93, 1611–1619. [DOI] [PubMed] [Google Scholar]
- Brand AH, and Perrimon N (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Brandstätter JH, Shaw SR, and Meinertzhagen IA (1991). Terminal degeneration and synaptic disassembly following receptor photoablation in the retina of the fly’s compound eye. The Journal of neuroscience : the official journal of the Society for Neuroscience 11, 1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge WC, Morgan IG, Zanetta JP, and V incendon G (1973). Adult rat brain synaptic vesicles. II. Lipid composition. Biochimica et biophysica acta 320, 681–686. [DOI] [PubMed] [Google Scholar]
- Choi SY, Borghuis BG, Rea R, Levitan ES, Sterling P, and Kramer RH (2005). Encoding light intensity by the cone photoreceptor synapse. Neuron 48, 555–562. [DOI] [PubMed] [Google Scholar]
- Clandinin TR, Lee CH, Herman T, Lee RC, Yang AY, Ovasapyan S, and Zipursky SL (2001). Drosophila LAR regulates R1-R6 and R7 target specificity in the visual system. Neuron 32, 237–248. [DOI] [PubMed] [Google Scholar]
- Clark PJ, and Evans FC (1954). Distance to Nearest Neighbor as a Measure of Spatial Relationships in Populations. Ecology 35, 445–453. [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, and Yates PO (1987). A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. Journal of the neurological sciences 78, 151–164. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, and Scheff SW (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Annals of neurology 27, 457–464. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Parfitt KD, and Schwarz TL (1993). Synaptic transmission persists in synaptotagmin mutants of Drosophila. Cell 73, 1281–1290. [DOI] [PubMed] [Google Scholar]
- Dickman DK, Horne JA, Meinertzhagen IA, and Schwarz TL (2005). A slowed classical pathway rather than kiss-and-run mediates endocytosis at synapses lacking synaptojanin and endophilin. Cell 123, 521–533. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156. [DOI] [PubMed] [Google Scholar]
- Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW, et al. (2011). Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS genetics 7, e1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrosotskaya IY, Seegmiller AC, Brown MS, Goldstein JL, and Rawson RB (2002). Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science 296, 879–883. [DOI] [PubMed] [Google Scholar]
- Duffy JB (2002). GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis 34, 1–15. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Verstreken P, Hiesinger PR, Horne JA, Kostyleva R, Zhou Y, Bellen HJ, and Meinertzhagen IA (2003). Endophilin promotes a late step in endocytosis at glial invaginations in Drosophila photoreceptor terminals. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 10732–10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita SC, Zipursky SL, Benzer S, Ferrus A, and Shotwell SL (1982). Monoclonal antibodies against the Drosophila nervous system. Proceedings of the National Academy of Sciences of the United States of America 79, 7929–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaengel K, and Mlodzik M (2008). Microscopic analysis of the adult Drosophila retina using semithin plastic sections. Methods in molecular biology 420, 277–287. [DOI] [PubMed] [Google Scholar]
- Godenschwege TA, Reisch D, Diegelmann S, Eberle K, Funk N, Heisenberg M, Hoppe V, Hoppe J, Klagges BR, Martin JR, et al. (2004). Flies lacking all synapsins are unexpectedly healthy but are impaired in complex behaviour. The European journal of neuroscience 20, 611–622. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Rawson RB, and Brown MS (2002). Mutant mammalian cells as tools to delineate the Sterol Regulatory Element-Binding Protein pathway for feedback regulation of lipid synthesis. Archives of Biochemistry and Biophysics 397, 139–148. [DOI] [PubMed] [Google Scholar]
- Hamanaka Y, and Meinertzhagen IA (2010). Immunocytochemical Localization of Synaptic Proteins to Photoreceptor Synapses of Drosophila melanogaster. Journal of Comparative Neurology 518, 1133–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden N, Lee J, Loh HY, Ong YM, Tan I, Leung T, Manser E, and Lim L (1996). A Drosophila homolog of the Rac- and Cdc42-activated serine/threonine kinase PAK is a potential focal adhesion and focal complex protein that colocalizes with dynamic actin structures. Molecular and cellular biology 16, 1896–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr DR, Fyrst H, Phan V, Heinecke K, Georges R, Harris GL, and Saba JD (2003). Sply regulation of sphingolipid signaling molecules is essential for Drosophila development. Development 130, 2443–2453. [DOI] [PubMed] [Google Scholar]
- Hofbauer A, Ebel T, Waltenspiel B, Oswald P, Chen YC, Halder P, Biskup S, Lewandrowski U, Winkler C, Sickmann A, et al. (2009). The Wuerzburg hybridoma library against Drosophila brain. Journal of neurogenetics 23, 78–91. [DOI] [PubMed] [Google Scholar]
- Hua X, Sakai J, Ho YK, Goldstein JL, and Brown MS (1995). Hairpin orientation of sterol regulatory element-binding protein-2 in cell membranes as determined by protease protection. The Journal of biological chemistry 270, 29422–29427. [DOI] [PubMed] [Google Scholar]
- Jones HE, Harwood JL, Bowen ID, and Griffiths G (1992). Lipid composition of subcellular membranes from larvae and prepupae of Drosophila melanogaster. Lipids 27, 984–987. [DOI] [PubMed] [Google Scholar]
- Jun B, Mukherjee PK, Asatryan A, Kautzmann MA, Heap J, Gordon WC, Bhattacharjee S, Yang R, Petasis NA, Bazan NG (2017). Elovanoids are novel cell-specific lipid mediators necessary for neuroprotective signaling for photoreceptor integrity. Sci. Rep 7(1):5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klagges BR, Heimbeck G, Godenschwege TA, Hofbauer A, Pflugfelder GO, Reifegerste R, Reisch D, Schaupp M, Buchner S, and Buchner E (1996). Invertebrate synapsins: a single gene codes for several isoforms in Drosophila. The Journal of neuroscience 16, 3154–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopczynski CC, Davis GW, and Goodman CS (1996). A neural tetraspanin, encoded by late bloomer, that facilitates synapse formation. Science 271, 1867–1870. [DOI] [PubMed] [Google Scholar]
- Kunte AS, Matthews KA, and Rawson RB (2006). Fatty acid auxotrophy in Drosophila larvae lacking SREBP. Cell metabolism 3, 439–448. [DOI] [PubMed] [Google Scholar]
- Lahey T, Gorczyca M, Jia XX, and Budnik V (1994). The Drosophila tumor suppressor gene dlg is required for normal synaptic bouton structure. Neuron 13, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HY, Wang W, Wessells RJ, Ocorr K, and Bodmer R (2011). Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes & development 25, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marza E, Long T, Saiardi A, Sumakovic M, Eimer S, Hall DH, and Lesa GM (2008). Polyunsaturated fatty acids influence synaptojanin localization to regulate synaptic vesicle recycling. Molecular biology of the cell 19, 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Albright T, Mallory M, and Terry RD (1991). Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta neuropathologica 81, 428–433. [DOI] [PubMed] [Google Scholar]
- Meinertzhagen IA, and Hanson TE (1993). The development of the optic lobe. In The Development of Drosophila melanogaster (Plainview, NY: Cold Spring Harbor Laboratory Press; ), pp. 1363–1491. [Google Scholar]
- Meltzer S, Bagley JA, Perez GL, O’Brien CE, DeVault L, Guo Y, Jan LY, Jan Y (2017). Phospholipid homeostasis regulates dendrite morphogenesis in Drosophila sensory neurons. Cell Reports 21, 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midorikawa R, Yamamoto-Hino M, Awano W, Hinohara Y, Suzuki E, Ueda R, and Goto S (2010). Autophagy-dependent rhodopsin degradation prevents retinal degeneration in Drosophila. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 10703–10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, and Edwards RH (2010). Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niven JE, Anderson JC, and Laughlin SB (2007). Fly photoreceptors demonstrate energy-information trade-offs in neural coding. PLoS biology 5, e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Ramaswami M, and Tanouye MA (1994). The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell 79, 23–33. [DOI] [PubMed] [Google Scholar]
- Pyza E, and Meinertzhagen IA (1998). Neurotransmitters alter the numbers of synapses and organelles in photoreceptor terminals in the lamina of the housefly, Musca domestica. Journal of comparative physiology A, Sensory, neural, and behavioral physiology 183, 719–727. [DOI] [PubMed] [Google Scholar]
- Rawson RB (2003). The SREBP pathway--insights from Insigs and insects. Nature reviews Molecular cell biology 4, 631–640. [DOI] [PubMed] [Google Scholar]
- Rivas-Serna IM, Keelan M, Mazurak V, and Clandinin MT (2017). Modification of ganglioside content of human gastric epithelial cell membrane decreases Helicobacter pylori adhesion. Journal of Pediatric Gastroenterology and Nutrition. 65 (4): 456–461. [DOI] [PubMed] [Google Scholar]
- Sato R, Inoue J, Kawabe Y, Kodama T, Takano T, and Maeda M (1996). Sterol-dependent transcriptional regulation of sterol regulatory element-binding protein-2. The Journal of biological chemistry 271, 26461–26464. [DOI] [PubMed] [Google Scholar]
- Schuldiner O, Berdnik D, Levy JM, Wu JS, Luginbuhl D, Gontang AC, and Luo L (2008). piggyBac-based mosaic screen identifies a postmitotic function for cohesin in regulating developmental axon pruning. Developmental cell 14, 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze KL, Broadie K, Perin MS, and Bellen HJ (1995). Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell 80, 311–320. [DOI] [PubMed] [Google Scholar]
- Seegmiller AC, Dobrosotskaya I, Goldstein JL, Ho YK, Brown MS, and Rawson RB (2002). The SREBP pathway in Drosophila: regulation by palmitate, not sterols. Developmental cell 2, 229–238. [DOI] [PubMed] [Google Scholar]
- Stark WS, and Carlson SD (1986). Ultrastructure of capitate projections in the optic neuropil of Diptera. Cell and tissue research 246, 481–486. [DOI] [PubMed] [Google Scholar]
- Stowers RS, and Schwarz TL (1999). A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics 152, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart AE, Borycz J, and Meinertzhagen IA (2007). The dynamics of singaling at the histaminergic photoreceptor synapse of arthropods. Progress in neurobiology 82, 202–227. [DOI] [PubMed] [Google Scholar]
- Sudhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, et al. (1989). Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science 245, 1474–1480. [DOI] [PubMed] [Google Scholar]
- Taghibiglou C, Martin HG, Lai TW, Cho T, Prasad S, Kojic L, Lu J, Liu Y, Lo E, Zhang S, et al. (2009). Role of NMDA receptor-dependent activation of SREBP1 in excitotoxic and ischemic neuronal injuries. Nature medicine 15, 1399–1406. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, and Katzman R (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Annals of neurology 30, 572–580. [DOI] [PubMed] [Google Scholar]
- Shao W and Espenshade PJ (2012). Expanding roles for SREBP in metabolism. Cell Metabolism 16, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis GH, Sutcliffe JG, and Bok D (1991). The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron 6, 61–70. [DOI] [PubMed] [Google Scholar]
- van Steveninck RRD, and Laughlin SB (1996). The rate of information transfer at graded-potential synapses. Nature 379, 642–645. [Google Scholar]
- Verpelli C, Montani C, Vicidomini C, Heise C, and Sala C (2013). Mutations of the synapse genes and intellectual disability syndromes. European journal of pharmacology. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, and Bellen HJ (2003). Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 40, 733–748. [DOI] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Durrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, et al. (2006). Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron 49, 833–844. [DOI] [PubMed] [Google Scholar]
- Wang X, Sato R, Brown MS, Hua X, and Goldstein JL (1994). SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 77, 53–62. [DOI] [PubMed] [Google Scholar]
- Xu H, Lee S, Suzuki E, Dugan KD, Stoddard A, Li H, Chodosh LA, and Montell C (2004). A lysosomal tetraspanin associated with retinal degeneration identified via a genome-wide screen. The EMBO Journal 23, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Sato R, Goldstein JL, and Brown MS (1994). Sterol-resistant transcription in CHO cells caused by gene rearrangement that truncates SREBP-2. Genes & development 8, 1910–1919. [DOI] [PubMed] [Google Scholar]
- Yang J, Brown MS, Ho YK, and Goldstein JL (1995). Three different rearrangements in a single intron truncate Sterol Regulatory Element Binding Protein-2 and produce sterol-resistant phenotype in three cell lines. The Journal of biological chemistry 270, 12152–12616. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, and Lee VM (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]
- Zimmerman B, Kelly B, McMillan BJ, Seegar TC, Dror RO, Kruse AC, and Blacklow SC (2016). Crystal structure of a full-length human tetraspanin revelas a cholesterol-binding pocket. Cell 167, 1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, and Benzer S (1994). Paralysis and early death in cysteine string protein mutants of Drosophila. Science 263, 977–980. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.