


Abstract
Generation of conditional knockout (cKO) and various gene-modified cells is laborious and time-consuming. Here, we established an all-in-one cKO system, which enables highly efficient generation of cKO cells and simultaneous gene modifications, including epitope tagging and reporter gene knock-in. We applied this system to mouse embryonic stem cells (ESCs) and generated RNA helicase Ddx1 cKO ESCs. The targeted cells displayed endogenous promoter-driven EGFP and FLAG-tagged DDX1 expression, and they were converted to Ddx1 KO via FLP recombinase. We further established TetFE ESCs, which carried a reverse tetracycline transactivator (rtTA) expression cassette and a tetracycline response element (TRE)-regulated FLPERT2 cassette in the Gt(ROSA26)Sor locus for instant and tightly regulated induction of gene KO. By utilizing TetFE Ddx1F/F ESCs, we isolated highly pure Ddx1F/F and Ddx1−/− ESCs and found that loss of Ddx1 caused rRNA processing defects, thereby activating the ribosome stress–p53 pathway. We also demonstrated cKO of various genes in ESCs and homologous recombination-non-proficient human HT1080 cells. The frequency of cKO clones was remarkably high for both cell types and reached up to 96% when EGFP-positive clones were analyzed. This all-in-one cKO system will be a powerful tool for rapid and precise analyses of gene functions.
INTRODUCTION
Gene knockout (KO) in cells or mice has made remarkable contributions to the understanding of molecular functions of mammalian genes. When we analyze essential genes, a conditional knockout (cKO) strategy is often required to elucidate their functions. The conventional cKO methodology, however, demands at least several months to prepare the cKO cells. First, loxP or FRT sequences must be introduced upstream and downstream of target coding exon(s) via homologous recombination. After deletion of the selection marker, the other allele must be targeted in the same way. To shorten these labor-intensive and time-consuming processes, researchers have employed genome editing technologies to generate cKO cells. Flemr et al. generated cKO embryonic stem cells (ESCs) by TALEN-assisted simultaneous integration of loxP sequences flanking the target exon in both alleles (1). Chen et al. developed a similar approach to generate cKO ESCs using the CRISPR/Cas9 system (2). Andersson-Rolf et al. reported one-step generation of cKO ESCs via CRISPR/Cas9-assisted homologous recombination of a Cre-regulated invertible gene-trap cassette (3). However, these methods still require large-scale screening to isolate correct cKO clones even though homologous recombination-proficient ESCs were utilized in these studies. Moreover, none of these methods allow simultaneous modification of target genes for monitoring and tagging. Another issue with the cKO methodology is how to delete the floxed or FRTed (i.e. FRT-flanked) genes. High expression of Cre is reportedly toxic, and potential toxicity of FLP is also a concern (4–7). Inducible expression systems or 4-hydroxytamoxifen (4OHT)-regulated recombinases are often utilized to circumvent this risk. These systems are also advantageous for preparing large amounts of cells with KO of an essential gene because they allow expansion of cKO cells before deletion. However, incomplete deletion and/or leaky activity of the recombinases can render the experimental results complicated and unreliable (8–10).
In this study, we developed a novel all-in-one cKO system, which enables purification of KO cells and thus fine analysis of gene functions. To demonstrate the use of this approach, we generated cells with cKO of an essential gene, Ddx1, as well as other genes, including some DDX1-binding protein genes, using ESCs and homologous recombination-non-proficient human HT1080 cells.
The 37 members of the DEAD box RNA helicase family are defined by nine well-conserved motifs (Q, I, Ia, Ib and II to IV), which are considered to be essential to their enzymatic activity (11,12). DEAD box 1 (DDX1), a member of the DEAD box family, was found to be co-amplified with MYCN in neuroblastoma (13,14). Our previous studies revealed the importance of DDX1 in testicular and colorectal tumor development (15,16). However, since the consequences of DDX1 amplification in neuroblastoma are controversial (17,18), it is not yet clear how DDX1 is involved in tumor development. In addition to tumorigenesis, DDX1 also plays important roles in various biological processes, including tRNA splicing, immunoglobulin class switching, viral replication and early embryogenesis (19–23). Although Ddx1 KO is lethal in mouse embryos at the pre-implantation stage, the underlying molecular mechanism(s) remains to be clarified. To elucidate the essential functions of Ddx1, an efficient cKO system has been sought, and the all-in-one cKO system described in this study meets this demand.
The mammalian ribosome is a large molecular complex comprising 79 ribosomal proteins and four rRNAs (28S, 18S, 5.8S and 5S) (24). Of the four rRNAs, the 28S, 18S and 5.8S rRNAs are transcribed by RNA polymerase I as a polycistronic 47S precursor rRNA. The 47S rRNA passes through a complex endo- and exoribonuclease-mediated processing cascade that sequentially trims the precursor rRNAs to form smaller precursors and eventually generates the mature rRNAs (25,26). Since ribosomes are generated at the expense of notable cellular resources, cells possess a ribosome surveillance system (27). When ribosome biogenesis is disturbed, ribosomal proteins, including RPL11, are released from the nucleoli and bind to MDM2. This binding inhibits the ubiquitinase activity of MDM2, thereby upregulating the level of its substrate protein p53 (28–30).
Here, we show that conditional loss of Ddx1 in ESCs resulted in aberrant rRNA maturation, which induced p53 upregulation and apoptosis via the ribosome stress pathway.
MATERIALS AND METHODS
Cell culture
129/Ola-derived E14tg2a ESCs (provided by H. Niwa, RIKEN) and C57BL/6N-derived RENKA ESCs (provided by K. Sakimura, Niigata University) were maintained on mitomycin C-treated mouse embryonic fibroblast (MtCMEF) cells in 2i medium composed of KnockOut DMEM (Thermo Fisher Scientific), 15% KnockOut Serum Replacement (Thermo Fisher Scientific), 2 mM l-alanyl-l-glutamine solution (Wako), 5 μg/ml insulin (Sigma), 1 μM PD0325901 (Wako), 3 μM CHIR99021 (Chemscene), mouse LIF (prepared in-house with a final concentration higher than 1000 U/ml), 1 mM sodium pyruvate (Sigma), 0.1 mM non-essential amino acids (Sigma), 0.1 mM 2-mercaptoethanol (Sigma), and 50 U/ml penicillin/streptomycin (PS) (Sigma) (31). When E14tg2a-derived cells were used for experiments, the cells were cultured on gelatin-coated plates with LIF medium composed of DMEM (Thermo Fisher Scientific), containing 10% fetal bovine serum (FBS) (Biosera), mouse LIF, 1 mM Na pyruvate, 0.1 mM non-essential amino acids, 0.1 mM 2-mercaptoethanol and PS. All other cells were cultured in DMEM supplemented with 10% FBS and PS. The recombinant lentiviruses were prepared as previously described (32).
All-in-one conditional gene knockout
Left and right homologous arms in the targeting vectors for Ddx1, Hnrnpk, Rtraf and Ddx39 were designed using the following genomic sequences: Ddx1, chr12: 13246592–13246048 and 13246047–13245146; Hnrnpk, chr13: 58401298–58400342 and 58400341–58399742; Rtraf, chr14: 19823109–19822571 and 19822570–19821634; Ddx39, chr8: 83718848–83719452 and 83719453–83720358 in GRCm38.p4. The following genomic sequences were used as homologous arms for targeting of human DDX1 and HNRNPK: DDX1, Chr2: 15594671−15595174 and 15595175–15596194; and HNRNPK, Chr9: 83979255–83978204 and 83978203–83977627 in GRCh38.p12. Potential splicing donor sites in the EGFP and FLAG-tag sequences derived from the 3× FLAG-tag system (Sigma) were removed via codon conversion to avoid unintended splicing. The protein-coding sequence upstream of the CRISPR/Cas9 cleavage site was inserted between the FLAG sequence and the right arm. Silent mutations were introduced in the coding sequence to protect the targeting vector from CRISPR/Cas9 digestion. The CRISPR/Cas9 targeting sequences for Hnrnpk, Rtraf, p53, DDX1, and HNRNPK were 5′-CTTCTCACCAAATTCACCAT-3′, 5′-CTGGCCCAAGTTCTTTGAAA-3′, 5′-GAAGTCACAGCACATGACGG-3′, 5′-GCCTGAGATTGCACAAGCTG-3′, and 5′-CTTCTCACCAAATTCACCAT-3′, respectively, and other sequences are described in each figure. PX458a was constructed by replacing the GFP sequence in pSpCas9(BB)-2A-GFP (PX458), which was a gift from Feng Zhang (Addgene plasmid #48138; http://n2t.net/addgene:48138; RRID: Addgene_48138), with Ametrine, and this construct was used for the expression of Cas9 and sgRNA (33). To isolate all-in-one cKO ESC clones, a targeting vector and a CRISPR/Cas9 vector for each gene were co-transfected using Lipofectamine 3000 (Thermo Fisher Scientific) (day 0). Ametrine-positive transfected cells were sorted by FACSAria III (BD Biosciences) on day 1. Two days after sorting, cells were replated on feeder cells and cultured for 4–7 days. Alternatively, single-cell sorting of Ametrine/EGFP-double-positive cells was performed on day 3. The genotypes of the isolated clones were analyzed via genomic PCR using the primers described in Supplementary Table S1. Indels of non-targeted alleles were analyzed by sequencing of PCR products generated with primers for the non-targeted allele. Southern blotting was performed using digoxigenin-labeled probes. The primers used to prepare labeled probes are described in Supplementary Table S1. For all-in-one cKO in HT1080 cells, cells transfected with a targeting vector and a CRISPR/Cas9 vector were cultured in the presence of 2 μM M3814 (LKT Laboratories), and Ametrine-positive cells were sorted on day 1. Sorted cells were cultured with 1–2 × 104 MtCMEF cells/cm2 to support proliferation. M3814 was omitted on day 3, and single-cell sorting of EGFP-positive cells was performed on day 6. Sorted cells were further cultured with MtCMEF cells.
Cloning of TetFE cells
The gene-loading vectors (GLVs) used to generate TetFE cells were as follows: GLV1 encoded TRE-regulated Ametrine linked to FLPERT2 via the P2A peptide sequence. This expression cassette was encompassed by the HS4 insulator element. GLV2 encoded CAG-promoter-driven mRuby linked to rtTA via the P2A peptide sequence. GLVs were introduced into the Gt(ROSA26)Sor locus via NHEJ-dependent knock-in using the CRISPR/Cas9 vectors TetFE CR1 and CR2, which cleaved Gt(ROSA26)Sor intron 1 and the backbone sequence of GLV1, respectively (34). The target sequences of TetFE CR1 and CR2 are 5′-ACTGGAGTTGCAGATCACGA-3′ and 5′-AGGTCATCACGGCCATAGCT-3′, respectively. To generate TetFE cells, GLV1, GLV2, TetFE CR1, TetFE CR2, and Bxb1 integrase-expressing vectors (provided by T. Ohbayashi, Tottori University) were co-transfected into E14tg2a cells using Lipofectamine 3000, and Ametrine-positive cells were sorted the next day. Two days after sorting, cells were replated on G418-resistant feeder cells and selected using G418 (350 μg/ml, Wako). G418-resistant clones were isolated, and the targeting of the GLVs was confirmed via genomic PCR using primers described in Supplementary Table S1.
Preparation and analysis of Ddx1−/− ESCs
TetFE Ddx1F/F and Ddx1−/− cells were prepared as follows, unless otherwise stated: TetFE Ddx1F/F cells were cultured in LIF medium in the presence of 2 μg/ml Dox (Sigma) for 1 day to induce FLPERT2 expression, followed by treatment with Dox and 1 μM 4OHT (LKT Laboratories) for 2 days. 4OHT was omitted in the preparation of TetFE Ddx1F/F cells. Cells were further cultured for 1 day in LIF medium without Dox and 4OHT to silence FLPERT2. EGFP-positive and -negative cells were then sorted to isolate Ddx1F/F and Ddx1−/− cells, respectively. For the cell growth assay, 1 × 105 sorted cells were inoculated in 12-well plates, and the numbers of cells were counted on days 2, 3, and 4 after sorting. Cells were replated on day 2 to prepare samples for day 4. Total RNA was purified using ISOGEN (Nippon Gene), and cDNA was prepared using PrimeScript RT Master Mix (Perfect Real Time, Takara) according to the manufacturer's protocol. qPCR was performed using THUNDERBIRD SYBR qPCR Mix (Toyobo) and the Light Cycler 480 System (Roche). The primers used for the qRT-PCR are listed in Supplementary Table S1. Standard curves were generated for each primer set, and the expression levels were normalized to the level of the Gapdh housekeeping gene. The TUNEL assays were performed using the in situ Apoptosis Detection Kit (Takara) according to the manufacturer's protocol. Predesigned Dicer-Substrate Short Interfering RNAs to Rpl11 or negative control DsiRNA (Integrated DNA Technologies) were transfected using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) for the knockdown experiments. Western blotting was performed using the following primary antibodies: anti-DDX1 (A300–521A, Bethyl Laboratories), anti-p53 (2524S, Cell Signaling Technology), anti-ACTB-HRP (017–24573, Wako), anti-FLAG M2-HRP (A8592, Sigma), anti-γH2A.X (613401, Biolegend), anti-p19ARF (NB200–174, Novus Biological), anti-HNRNPK (ab23644, Abcam), anti-RTRAF (19848-1-AP, Proteintech) and anti-DDX39/39B (11723–1-AP, Proteintech).
Pulse-chase labeling
Pulse-chase labeling was performed as previously described (35–37). Briefly, 2 days after sorting, Ddx1F/F and Ddx1−/− cells were labeled with 5 μCi [5,6-3H] uridine for 30 min, followed by incubation in LIF medium containing 0.5 mM uridine for the indicated times. Total RNA was harvested with ISOGEN, and RNA samples corresponding to 10 000 cpm were loaded in tricine/triethanolamine buffer-based gels to separate the long rRNA precursors. The separated RNA was transferred to Zeta-Probe membrane (Bio-Rad) and visualized with FLA-7000 using a Storage Phospor Screen for tritium (GE Healthcare). Incorporation of [5,6-3H] uridine into the total RNA was quantitated using an LSC-6100 instrument (Aloka).
Identification of DDX1-associated proteins
To identify DDX1-associated proteins, FLAG-tagged DDX1 was immunoprecipitated from Ddx1F/F RENKA cells cultured in 2i medium on gelatin-coated dishes. Briefly, a cell lysate of Ddx1F/F RENKA cells was prepared using lysis buffer (20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EGTA, 50 μM ZnSO4, 1 mM MgCl2, 5% glycerol and 1× cOmplete EDTA-free protease inhibitor Cocktail (Roche)), and the lysate was incubated with anti-FLAG M2 Affinity Gel for 2.5 h at 4°C. The beads were washed four times with lysis buffer, followed by a wash with PBS/5% glycerol. The beads were then treated with 10 and 200 μg/ml 1× FLAG peptide to wash out the cellular proteins directly bound to the anti-FLAG M2 antibody with weak affinities. After washing, FLAG-DDX1 was eluted with 1 mg/ml 3× FLAG peptide. The sample was loaded on 10−20% SuperSep Ace gel (Wako) for SDS-PAGE, and the gel was stained using the Silver Stain MS Kit (Wako) to dissect the bands of DDX1-binding proteins for LC–MS. The stained bands were excised and digested with trypsin. Control samples were also dissected at the corresponding sizes and processed in the same way to validate the non-specifically bound proteins. The tryptic peptide samples were analyzed using a nanoLC Ultra 1D plus nano liquid chromatography system Eksigent Technologies (SCIEX) coupled to a 5600 Triple TOF mass spectrometer (SCIEX), which was operated in data-dependent acquisition mode. The raw mass spectrometric files were analyzed with ProteinPilot version 4.5beta software (SCIEX) using the Paragon™ algorithm against the UniProtKB-mouse database (24 027 entries) supplemented with 262 frequently observed contaminants, including human keratins, bovine serum proteins, and proteases. The total protein score was calculated from the scores of the individual peptides, where the peptide score = –log(1 – confidence) (i.e. a peptide with 95% confidence would have a score of 1.3, and a peptide with 99% confidence would have a score of 2.0).
RESULTS
Disruption of Ddx1 via CRISPR/Cas9
To knockout mouse Ddx1 in cells, we first designed an sgRNA, CR1, which targets exon 2 of Ddx1 (Supplementary Figure S1A and B). Exon 2 encodes the Q-motif required for ATP binding and hydrolysis (38) (Supplementary Figure S1C). The Q-motif is highly conserved among species (Supplementary Figure S1C). We first confirmed the efficient digestion of substrate DNA using CR1 (Supplementary Figure S1D). Subsequently, the RENKA (ESC), NIH3T3 (fibroblast), and B16F10 (melanoma) cell lines were subjected to this gene-disruption experiment. Consequently, we could not obtain any Ddx1-disrupted clones from any cell lines (Supplementary Figure S1E and F and Table S2). Note that the ESC clone #3 had mutations in both alleles, but one of these was a G-to-A point mutation (Ala16Ser) (Supplementary Figure S1E and F). This mutation does not induce a frame-shift or destroy the local domain structure by deleting/inserting amino acids; therefore, we did not consider Ddx1 to be functionally disrupted in this clone. Furthermore, we did not obtain any Ddx1-disrupted cells using another sgRNA, CR2, which targeted exon 3 of Ddx1 (Supplementary Figure S1B, C and G and Table S2), while disruption of a non-lethal gene (Slc20a2) (39) was highly efficient with this procedure (Supplementary Figure S1H and I and Table S2). These results indicated that Ddx1 disruption is lethal in mouse cells. Of note, we did not obtain any clones with in-frame mutations in both Ddx1 alleles. We assume that no indels can acceptably maintain the domain structure and function of DDX1. Indeed, alignment of the Q-motif sequences from various species showed no gaps (Supplementary Figure S1C).
All-in-one conditional knockout system
To elucidate the essential functions of Ddx1, we needed to generate cKO cells. Since existing cKO methods are time-consuming and less efficient, we developed a new method, designated as the ‘all-in-one conditional gene knockout system’, which enables the conditional knockout of a target gene as well as the introduction of an epitope tag and a fluorescent reporter system in one step (Figure 1 and Supplementary Figure S2). This method utilizes CRISPR/Cas9-assisted homologous recombination with a targeting vector that encodes an FRT-flanked promoterless EGFP gene followed by a P2A peptide sequence, a FLAG-tag sequence, and the ORF sequence of the target gene upstream of the CRISPR/Cas9 cleavage site (Figure 1A and Supplementary Figure S2). Knock-in of the targeting vector introduces an endogenous promoter-driven copy of the EGFP gene and a FLAG-tagged version of the target gene linked via the P2A peptide sequence. EGFP and the FLAG-tagged target protein are separately expressed via the function of the P2A peptide. The FRT-EGFP-FRT cassette does not contain a stop codon and is translated in frame. Introduction of FLP recombinase induces EGFP deletion and the formation of a frame-shift mutation in the target gene. The CRISPR/Cas9 target site is designed in a protein-coding region; therefore, CRISPR/Cas9-assisted targeting generates either homozygous (F/F) or heterozygous (F/mut) cell clones (Figure 1B). F/mut cells are generated when one allele is disrupted via CRISPR/Cas9-mediated non-homologous end joining.
Figure 1.

Overview of the all-in-one conditional gene knockout method. (A) Strategy to generate all-in-one Ddx1 cKO ESCs. The CR1 target sequence is underlined, and the cleavage site is indicated by an arrowhead. Black box, UTR; yellow box, ORF of exon 1; blue box, exon 2 upstream of the cleavage site of CR1; red box, downstream of the cleavage site of CR1; white box, exon 3; white triangle, FRT sequence. (B) Expression of DDX1, EGFP, and FLAG-DDX1 driven by the endogenous Ddx1 promoter in Ddx1F/mut, Ddx1F/F or FLP-induced cells. (C) Various applications of all-in-one cKO ESCs for functional analysis of target genes.
We applied this system to generate Ddx1 cKO ESCs (Figure 2A). ESC clones were randomly picked without analyzing EGFP expression. Genomic PCR was performed using primers spanning the homology arm to check the genotype. We found that 18 out of 68 clones had a targeted allele and 85% of targeted clones were Ddx1 cKO (i.e. Ddx1F/F or Ddx1F/mut with a frame-shift mutation, exon/intron disruption, an in-frame stop codon, or a large indel) (Table 1, Supplementary Table S3, and Figure 2B and C). Genomic DNA of clones #1 and #2 was further analyzed by Southern blotting, and homozygous targeting in clone #2 was confirmed (Figure 2D). Note the PCR-verified non-targeted allele-negative clones were not necessarily F/F (Supplementary Figure S3). They could be F/mut with a large indel in the mutant allele, which cannot be detected by PCR. Therefore, Southern blotting is essential to identify F/F clones. Nevertheless, any clones that test negative by non-targeted allele PCR (i.e. F/F or F/mut with a large indel) are available for cKO because a large indel destroys the exon/intron structure and/or introduces a stop codon into the reading frame. The genotypes of all F/F clones utilized in this study were verified by both genomic PCR and Southern blotting. We confirmed expression of FLAG-tagged DDX1 in a Ddx1F/F ESC clone via western blotting (Figure 2E). As expected, the size of FLAG-tagged DDX1 was slightly larger than that of endogenous DDX1. We did not detect the EGFP and FLAG-DDX1 fusion protein, which should be ∼30 kDa larger than the FLAG-DDX1 protein. The high cleaving efficiency may be due to the use of P2A peptide, which has the highest cleaving efficiency among commonly utilized 2A peptides in vivo (40). The protein and mRNA expression levels of DDX1 were comparable to those in parental ESCs (Figure 2E and F). Furthermore, Ddx1 promoter-driven expression of EGFP was detected via fluorescence microscopy and flow cytometry (Figure 2G and H). To enable instant large-scale preparation of Ddx1KO ESCs, we constructed a lentivirus vector (Lv-a2FE) that encodes the fluorescent protein Ametrine and a 4OHT-regulated codon-optimized FLP (FLPERT2) recombinase (Supplementary Figure S4A). 4OHT treatment of Lv-a2FE-transduced Ddx1 cKO ESCs converted most of them to EGFP-negative cells. DDX1 expression was almost entirely depleted in these EGFP-negative cells (Supplementary Figure S4B). Furthermore, we confirmed that Ddx1−/− ESCs did not express any appreciable partial DDX1 proteins, which could be generated from an alternative promoter or downstream ATG codon after KO induction (Supplementary Figure S4C). By taking advantage of the FLAG-tagged DDX1 in the Ddx1F/F ESCs, DDX1-associated proteins were pulled down with FLAG antibody and were then subjected to liquid chromatography–tandem mass spectrometric (LC–MS/MS) analysis (Supplementary Figure S4D). As a result, various DDX1-interacting proteins were identified, including the previously reported DDX1-binding proteins RTCB, FAM98B and RTRAF (Supplementary Table S4). These results demonstrated that our all-in-one conditional gene knockout system functioned as expected.
Figure 2.

Characterization of Ddx1 cKO ESCs. (A) Scheme of Ddx1 targeting. Arrows and arrowheads represent primers used for genomic PCR. The sizes of PCR products generated with these primer sets are shown. The probe position for Southern blotting is depicted in the figure. B, BsrGI recognition site. (B) Thirteen targeted allele-positive clones were analyzed by three-primer PCR. Primers for the 5′ junction of the targeted site were used. The upper band represents the targeted allele, while the lower band represents the non-targeted allele. C, wild-type control. (C) The 3′ junction of the targeted site was analyzed by three-primer PCR. The upper band represents the non-targeted allele, while the lower band represents the targeted allele. C, wild-type control. (D) BsrGI-digested genomic DNA of clones #1 and #2 was analyzed by Southern blotting. C, wild-type control. Clone #2 was used as Ddx1F/F ESCs hereafter. (E) DDX1 expression in the indicated cells was analyzed via western blotting. (F) Expression of Ddx1 mRNA in parental ESCs and Ddx1F/F cells was analyzed by quantitative RT-PCR (n = 3; NS, not significant). (G) EGFP expression in Ddx1F/F cells was analyzed via fluorescence microscopy. (H) Expression of EGFP was analyzed via flow cytometry. Gray, parental ESCs; Blue, Ddx1F/F ESCs. (I) Dox-inducible FLPERT2 expression system of TetFE ESC. (J) Expression of fluorescent proteins under the indicated conditions was analyzed via flow cytometry. (K) DDX1 expression in sorted EGFP+ (F/F) and EGFP− (–/–) cells was analyzed via western blotting. Cells were harvested on day 2.
Table 1.
Generation of All-in-one cKO ES cells
| Classification | ||||||||
|---|---|---|---|---|---|---|---|---|
| Screening | Target locus | Clones analyzed | Targeted allele positive clones | cKOa | F/ mut (in-frame indel) | F/wt | Misc. | |
| Transfected cells | No selection | Ddx1 | 68 | 18/68 | 11/13b | 2/13 | 0/13 | 0/13 |
| (26%) | (85%) | (15%) | (0%) | (0%) | ||||
| Rtraf | 23 | 6/23 | 5/6 | 1/6 | 0/6 | 0/6 | ||
| (26%) | (83%) | (17%) | (0%) | (0%) | ||||
| Ddx39 | 24 | 3/24 | 0/3 | 2/3 | 0/3 | 1/3c | ||
| (13%) | (0%) | (67%) | (0%) | (33%) | ||||
| Hnrnpk | 23 | 1/23 | 0/1 | 1/1 | 0/1 | 0/1 | ||
| (4%) | (0%) | (100%) | (0%) | (0%) | ||||
| GFP+ cells | Ddx1 | 18 | 18/18 | 15/18 | 3/18 | 0/18 | 0/18 | |
| (100%) | (83%) | (17%) | (0%) | (0%) | ||||
| Hnrnpk | 8 | 8/8 | 5/8 | 3/8 | 0/8 | 0/8 | ||
| (100%) | (63%) | (38%) | (0%) | (0%) | ||||
acKO includes F/F and F/mut with a frame-shift mutation, exon/intron disruption, an in-frame stop codon, or a large indel.
bThirteen out of 18 targeted allele positive clones were analyzed by three primer PCR and sequencing.
cMixture of multiple clones.
Generation of Dox/4OHT-inducible Ddx1 cKO ESCs
Although Lv-a2FE allowed efficient knockout of the target gene, we noticed that the population of Lv-a2FE-transduced Ddx1 cKO ESCs always contained 10–30% of EGFP-negative cells even without 4OHT treatment (Supplementary Figure S4A), presumably due to leaky FLPERT2 activity. To solve this problem, we established TetFE ESCs, which carried a reverse tetracycline transactivator (rtTA) expression cassette and a tetracycline response element (TRE)-regulated FLPERT2 cassette (Figure 2I and Supplementary Figure S5). We inserted these cassettes into the Gt(ROSA26)Sor locus by using modified simultaneous or sequential integration of multiple gene-loading vectors system (SIM system) (41) (Supplementary Figure S5A and B). Constitutive rtTA expression and Dox-inducible FLPERT2 expression were verified via the fluorescence of surrogate markers, i.e. mRuby or Ametrine, respectively (Supplementary Figure S5C). We then targeted Ddx1 in TetFE ESCs to establish TetFE Ddx1 cKO ESCs, and found that there was no background Ddx1 deletion (Figure 2J). Highly pure Ddx1F/F and Ddx1−/− cell populations could be prepared by sorting cells on the basis of their EGFP expression levels (Figure 2J and K). These results indicated that conditional gene knockout was strictly regulated in the TetFE ESCs; thus we used TetFE ESCs for the following all-in-one cKO experiments. To demonstrate the applicability of this all-in-one cKO system for various genes, we attempted to target Hnrnpk, Rtraf and Ddx39 in TetFE ESCs, and successfully obtained cKO ESCs of these genes (Supplementary Figure S6). When isolating clones without analyzing EGFP expression, the frequency of targeted clones varied from 4% to 26% depending on the target (Table 1). In the case of Hnrnpk, only 1 out of 23 clones had a targeted allele. However, we realized that targeted ESCs were easily obtained by isolating EGFP-positive clones (Supplementary Figure S6). To verify the frequency of targeted cells among EGFP-positive cells, we performed single-cell sorting of EGFP-positive cells for isolation of Ddx1 and Hnrnpk cKO clones. This modified procedure dramatically improved the targeting efficiency, and all analyzed clones were targeted allele-positive in both cases (Table 1). Flow cytometric analysis of the cKO clones revealed that the EGFP expression level depended on the strength of the target gene promoter (Supplementary Figure S6F, M and S). In the case of Ddx39 cKO screening, we obtained Ddx39F/mut but not Ddx39F/F ESC clones (Supplementary Figure S6P). We analyzed the genomic sequence around the CRISPR/Cas9 cleavage site and found a Ddx39F/mut ESC clone with a frame-shift mutation in the non-targeted allele (Supplementary Figure S6Q). We confirmed that the Ddx39−/mut ESCs lacked DDX39 protein expression (Supplementary Figure S6R–T). Hnrnpk−/− and Rtraf−/− ESCs exhibited severe growth defects and cell death, whereas the Ddx39−/mut ESCs proliferated normally (data not shown). These results indicate that the all-in-one cKO system is applicable for various genes including non-essential genes. We further applied the system to the homologous recombination-non-proficient, pseudo-diploid human fibrosarcoma cell line HT1080 (Supplementary Figure S7). We attempted to generate DDX1 or HNRNPK cKO HT1080 cells to address the applicability of the system. Although we did not obtain any cKO clones without EGFP sorting, targeting in the presence of the DNA-PK inhibitor M3814 followed by single-cell sorting of EGFP-positive cells resulted in a cKO efficiency of almost 100% (Supplementary Tables S3 and S5). These results indicate that the all-in-one cKO system is a useful method not only for ESCs but also for various cell lines.
p53 upregulation in Ddx1−/− ESCs
We first compared the proliferation of Ddx1F/F and Ddx1−/− ESCs, and found that loss of Ddx1 resulted in a severe growth defect (Figure 3A). Furthermore, the number of TUNEL-positive apoptotic cells was significantly increased in the Ddx1−/− ESCs (Figure 3B). Consistently, we could not establish a Ddx1−/− ESC clone as in Ddx1 disruption experiments. Since p53 plays an important role in the regulation of cell growth and apoptosis, we analyzed the expression level of p53 protein. Ddx1−/− cells showed clear upregulation of p53 protein from day 2, when cells displayed a significant proliferation defect (Figure 3A and C). Consistently, expression levels of several p53-regulated genes, including Cdkn1a, Perp, Fas, Pmaip1, Plk2 and Mdm2, were significantly increased in Ddx1−/− ESCs (Figure 3D). These results indicated that loss of Ddx1 rapidly induces the p53 signaling pathway.
Figure 3.
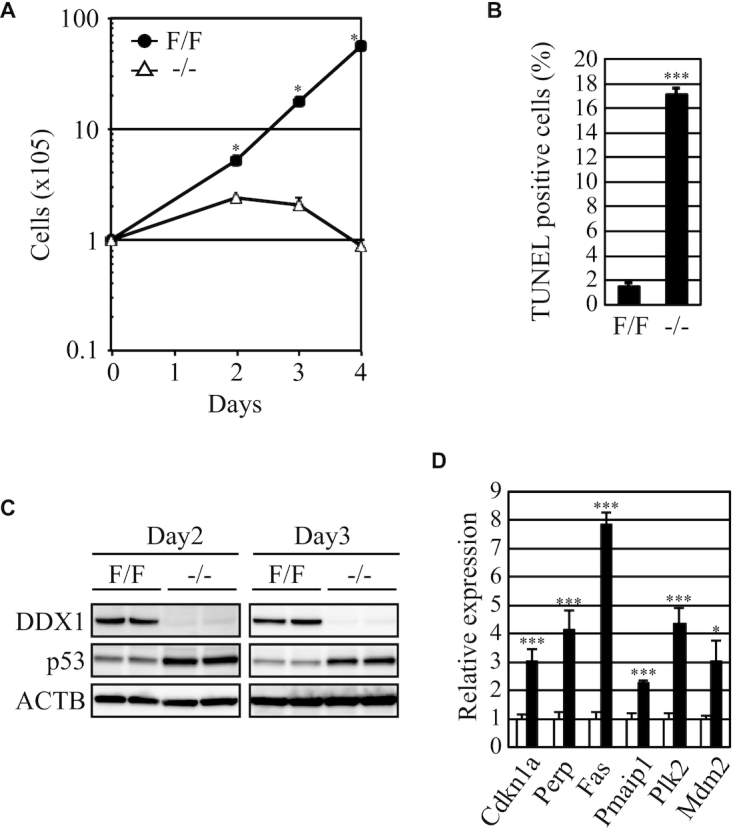
Proliferation of Ddx1−/− ESCs. (A) The numbers of Ddx1F/F or Ddx1−/− cells were counted on the indicated days after sorting (n = 3; *P < 0.05). (B) Apoptosis of Ddx1F/F and Ddx1−/− cells was analyzed on day 3 (n = 3; ***P < 0.005). (C) The p53 expression levels in Ddx1F/F or Ddx1−/− cells were analyzed via western blotting. (D) The expression levels of p53-regulated genes were analyzed via qRT-PCR (n = 3; *P < 0.05; ***P < 0.005).
rRNA processing defect in Ddx1−/− ESCs
We next investigated how the p53 pathway was activated in Ddx1−/− ESCs. The levels of phosphorylated Ser139 in H2AX (γH2AX) and p19 ARF protein were comparable between Ddx1F/F and Ddx1−/− ESCs, suggesting that neither massive DNA damage nor p19ARF induction was responsible for the p53 upregulation (Figure 4A).
Figure 4.

Activation of the ribosome stress pathway in Ddx1−/− ESCs. (A) The levels of γH2A.X and p19ARF were analyzed via western blotting. Cells were harvested on day 2. (B) Knockdown of Rpl11 mRNA was confirmed via qRT-PCR. NC, negative control. Cells were harvested on day 2. (C) The effect of Rpl11 knockdown on p53 expression was analyzed via western blotting. Cells were harvested on day 2. (D) Cells pulse-labeled with [3H]uridine were harvested at the indicated time points, and their total RNA was separated on a formaldehyde gel to analyze rRNA processing. (E) The ratios of 18S or 28S RNA and rRNA precursors were quantified at 120 min (n = 3; *P < 0.05). (F) [3H]uridine incorporation into the total RNA (n = 3; NS, not significant).
Upregulation of p53 is also induced by defects in ribosome biogenesis. When ribosome biogenesis is disturbed, ribosomal proteins, including RPL11, are released from the nucleoli, after which they bind to MDM2. This binding inhibits the ubiquitinase activity of MDM2, thereby upregulating the level of its substrate protein p53 (28–30). Previous reports demonstrated that Rpl11 depletion is sufficient to prevent p53 upregulation caused by ribosome biogenesis defects (30,42).
To determine whether ribosome stress is involved in the p53 upregulation in Ddx1−/− ESCs, Rpl11 expression was depleted using two different siRNAs (Figure 4B). Both siRNAs inhibited p53 upregulation in Ddx1−/− ESCs (Figure 4C). Furthermore, pulse-chase labeling with [3H]uridine revealed that rRNA processing from 47S-45S early precursors (primary transcript plus (PTP)) to 28S and 18S mature rRNAs was significantly impaired in Ddx1−/− ESCs (Figure 4D and E). We confirmed that the absolute amounts of [3H]uridine incorporated into the total RNA did not differ significantly between Ddx1F/F and Ddx1−/− ESCs (Figure 4F). These results demonstrated that rRNA processing defects triggered p53 upregulation in Ddx1−/− ESCs.
p53KO notably rescued the proliferation and survival defects of Ddx1−/− ESCs
To verify the essential role of p53 in the growth defect of Ddx1−/− ESCs, we disrupted the p53 gene in Ddx1 cKO ESCs (Figure 5A). Two Ddx1cKOp53KO ESC clones (#1 and #2) were chosen for further experiments, and we confirmed the lack of DDX1 expression in Dox/4OHT-treated GFP-negative cells (Figure 5B). We then compared the growth rate of Ddx1−/−p53KO ESCs with those of Ddx1−/−, Ddx1F/F and Ddx1F/Fp53KO ESCs. While the number of Ddx1−/− cells was severely diminished by day 4, the phenotype was significantly rescued by p53 disruption (Figure 5C and D). These results indicated that p53 plays an important role in the growth and survival defects of Ddx1−/− ESCs.
Figure 5.
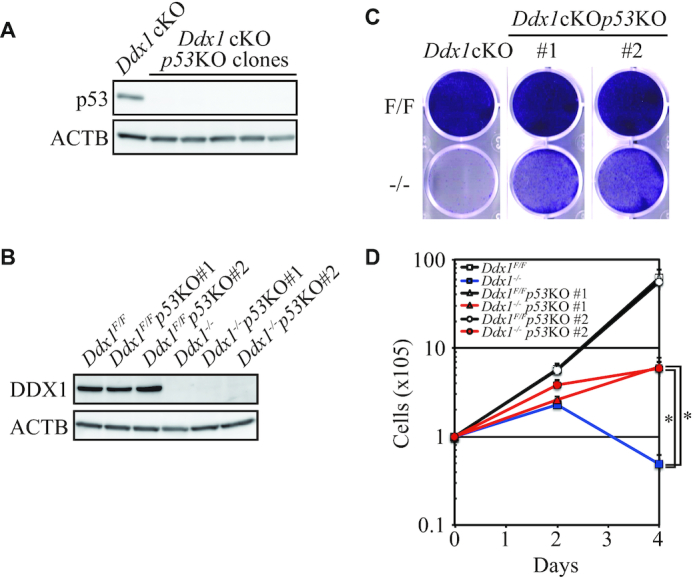
p53 KO partially rescued the proliferation and survival of Ddx1−/− ESCs. (A) p53 expression in Ddx1cKOp53KO clones was analyzed via western blotting. (B) Expression of DDX1 was analyzed via western blotting. (C) Sorted cells were cultured for 4 days without passaging and then stained with 0.05% crystal violet. (D) Proliferation of the sorted cells (n = 3; *P < 0.05).
DISCUSSION
In this study, we developed an all-in-one cKO system that enabled not only conditional knockout generation but also epitope tagging and fluorescent marking of the target gene in one step. Therefore, this system is useful for analyzing the expression and functions of various genes (Figure 1C). We detected statistical differences in target gene expression between parental cells and Rtraf cKO ESCs or DDX1 cKO HT1080 cells via qRT-PCR (Supplementary Figures S6L and S7E). This may be due to the inserted sequence and/or clonal variation in target gene expression. However, the slight differences would not adversely affect analyses of the molecular functions of the target genes in most cases. The all-in-one cKO system requires a recombinase recognition sequence that has two reading frames without a stop codon (Supplementary Figure S2). FRT, but not loxP, fulfills this requirement; therefore, we employed FLP recombinase for the system. However, other recombinases or even Cre with use of the loxP mutant may also be applicable. Combinatorial use of other recombinases may help to establish multi-cKO cells. We applied the all-in-one cKO method to Ddx1 and found that 26% of ESC clones acquired the targeted allele without any selection, and that 85% of them were cKO clones (Table 1). Although the frequency of targeted clones varied depending on the target site, single-cell sorting of EGFP-positive cells yielded a frequency of cKO clones from 63% to 96% in ESCs and HT1080 cells. Interestingly, all analyzed HT1080 clones, which tested negative for a non-targeted allele by PCR, were homozygously targeted clones (Supplementary Figure S7C and J). On the other hand, some ESC clones, which tested negative for a non-targeted allele by PCR, were F/mut with a large indel (Supplementary Figures S3 and S6J). The high frequency of homozygous targeting may be due to use of M3814, which potently blocks NHEJ by inhibiting DNA-PK and thus enhances homologous recombination-dependent DNA repair (43). M3814 may also enable efficient isolation of F/F clones in ESCs and other cell lines. The efficiency of previously reported cKO methods using genome editing technology was ∼5%, and they were applied only to homologous recombination-proficient ESCs (1–3). Thus, our method is remarkably advantageous for minimizing the time and labor required to obtain cKO clones with various cell types. The relatively smaller insert size may contribute to the high efficiency. In the case of targeting essential genes such as Ddx1, selective pressure for targeted insertion to preserve gene expression could also contribute to the cKO efficiency. Nevertheless, this mechanism is not a prerequisite for the all-in-one cKO system because we could also efficiently generate cKO clones for the non-essential gene Ddx39. This all-in-one cKO system might also be applicable to fertilized eggs for generating cKO mice, and we plan to explore this possibility. On the other hand, when utilizing this system, caution is advised because a mutant allele or FLPed allele of cKO cells may express part of the target gene due to use of an alternative start codon, second promoter, or neo-exon as these are found in indel-based gene-disrupted cells (44–47). Therefore, cKO cells must be carefully characterized before utilization. In the case of Ddx1 cKO, we did not find any appreciable partial DDX1 proteins in Ddx1−/− ESCs (Supplementary Figure S4C). Moreover, we could not establish a Ddx1 KO clone by CRISPR/Cas9-induced gene disruption or knockout induction from Ddx1F/F cells, indicating that functionally compensatory DDX1 fragments were not expressed from the mutant allele or KO-induced FRTed allele. Careful design of the targeting site may help to minimize potential problems. To generate Ddx1 cKO cells, we targeted the ATP-binding region, which is crucial for helicase activity. Therefore, any partial proteins expressed from the mutant allele would lack a major function of DDX1. Leaky CreERT2 or FLPERT2 activity has been an important technical problem for keeping conditional alleles intact. We overcame this issue by generating TetFE ESCs, which did not show background FLP activity. We can now fully maintain an EGFP-positive population even after long-term culture and can easily obtain KO cells via Dox/4OHT treatment (Figure 2I–K and Supplementary Figure S6). Furthermore, removal of Dox/4OHT instantly eliminated the recombinase and allowed fine analysis of the molecular functions, as presented in this study. An issue when using TetFE ESCs is the incomplete deletion of conditional alleles when the knockout is induced (Figure 2J). The deletion efficiency was 50–60% against any target tested in this study. Thus, cell sorting based on EGFP expression driven by the endogenous promoter was useful to eliminate remaining undeleted cells and to prepare highly pure F/F and −/− cells. This purification method will not work for genes whose expression is not detectable via the endogenous promoter-driven EGFP. Brighter fluorescent proteins, such as mNeonGreen or multimerized EGFP, could improve detection sensitivity (48,49). Increasing the expression level of inducible FLPERT2 or utilizing inducible FLP, which shows stronger recombination activity than FLPERT2, might be helpful to obtain a higher yield of −/− cells without EGFP sorting, although leaky activity could compromise the purity to some extent. The modified SIM system that allows further integration of multiple FLPERT2 expression units into the Gt(ROSA26)Sor locus would be useful for investigating this possibility.
In this study, we found that Ddx1 disruption was lethal in ESCs. Since we also could not isolate Ddx1-disrupted NIH3T3 and B16F10 clones, Ddx1 appears to be an essential gene in various cell types. These results were consistent with the pre-implantation lethality of Ddx1 KO mice (22). By contrast, previous reports showed that certain cell types tolerated loss of DDX1 (16,23). Other DDX family proteins could compensate for the loss of DDX1 in these cells.
We revealed that DDX1 has an important function in rRNA processing. Interestingly, Ddx1 is an evolutionally young gene that is absent in yeast. This feature implies that DDX1 is involved in an rRNA maturation step specific to higher eukaryotes, such as the processing of external transcribed spacers (5′- and 3′-ETS) and internal transcribed spacers 1 (ITS1) in pre-rRNAs (25,26). The DEAD box RNA helicase Ddx51 is an example of such a gene. A previous report showed that Ddx51, which is also absent in yeast, is essential for higher eukaryote-specific 3′-ETS processing (50). We showed that loss of Ddx1 induced p53 upregulation via the ribosome stress pathway. However, considering the versatile molecular functions of DDX1 and the complex p53 regulation mechanisms, Ddx1 KO may also activate other pathways that lead to p53 activation at a later stage. Further investigation is necessary to clarify this point.
Disruption of p53 partially rescued the growth defect of Ddx1−/− ESCs. However, although Ddx1−/−p53 KO ESCs survived for more than 6 days after sorting, Ddx1−/−p53 KO ESCs showed a slower growth rate compared with that of the parental ESCs, and these cells did not proliferate indefinitely. These results suggested that the p53-independent growth inhibitory pathway(s) was activated via Ddx1 disruption in ESCs. Ddx1−/−p53 KO ESCs could be an important tool for discovering the novel DDX1 functions.
DATA AVAILABILITY
The mass spectrometry data have been deposited to the ProteomeXchange Consortium via jPOST (51). The accession numbers are PXD023575 for ProteomeXchange and JPST001033 for jPOST.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Professor Shigeru Chaen (Nihon University) for the educational support of S.T.
Contributor Information
Teruhiko Suzuki, Stem Cell Project, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan.
Eiji Katada, Stem Cell Project, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan; Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8510, Japan.
Yuki Mizuoka, Stem Cell Project, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan; Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8510, Japan.
Satoko Takagi, Stem Cell Project, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan; Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8510, Japan.
Yasuhiro Kazuki, Chromosome Engineering Research Center (CERC), Tottori University, 86 Nishicho, Yonago 683-8503, Japan; Division of Genome and Cellular Functions, Department of Molecular and Cellular Biology, School of Life Science, Faculty of Medicine, Tottori University, Yonago 683-8503, Japan.
Mitsuo Oshimura, Chromosome Engineering Research Center (CERC), Tottori University, 86 Nishicho, Yonago 683-8503, Japan.
Mayumi Shindo, Center for Basic Technology Research, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan.
Takahiko Hara, Stem Cell Project, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan; Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8510, Japan; Graduate School of Science, Department of Biological Science, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji-shi, Tokyo 192-0397, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science (JSPS) KAKENHI [JP18K06047 to T.S., JP23390256 to T.H., in part]; Core Research for Evolutionary Science and Technology (CREST) program of the Japanese Science and Technology Agency (JST) [JPMJCR18S4 to Y.K., T.S.]. Funding for open access charge: CREST program of the JST [JPMJCR18S4 to Y.K., T.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Flemr M., Bühler M.. Single-step generation of conditional knockout mouse embryonic stem cells. Cell Rep. 2015; 12:709–716. [DOI] [PubMed] [Google Scholar]
- 2. Chen L., Ye Y., Dai H., Zhang H., Zhang X., Wu Q., Zhu Z., Spalinskas R., Ren W., Zhang W.. User-friendly genetic conditional knockout strategies by CRISPR/Cas9. Stem Cells Int. 2018; 2018:9576959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andersson-Rolf A., Mustata R.C., Merenda A., Kim J., Perera S., Grego T., Andrews K., Tremble K., Silva J.C., Fink J.et al.. One-step generation of conditional and reversible gene knockouts. Nat. Methods. 2017; 14:287–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Loonstra A., Vooijs M., Beverloo H.B., Allak B.A., van Drunen E., Kanaar R., Berns A., Jonkers J.. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:9209–9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Silver D.P., Livingston D.M.. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol. Cell. 2001; 8:233–243. [DOI] [PubMed] [Google Scholar]
- 6. Kondo S., Takahashi Y., Shiozawa S., Ichise H., Yoshida N., Kanegae Y., Saito I.. Efficient sequential gene regulation via FLP-and Cre-recombinase using adenovirus vector in mammalian cells including mouse ES cells. Microbiol. Immunol. 2006; 50:831–843. [DOI] [PubMed] [Google Scholar]
- 7. Galla M., Schambach A., Falk C.S., Maetzig T., Kuehle J., Lange K., Zychlinski D., Heinz N., Brugman M.H., Göhring G.et al.. Avoiding cytotoxicity of transposases by dose-controlled mRNA delivery. Nucleic Acids Res. 2011; 39:7147–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kristianto J., Johnson M.G., Zastrow R.K., Radcliff A.B., Blank R.D.. Spontaneous recombinase activity of Cre-ERT2 in vivo. Transgenic Res. 2017; 26:411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Y., Suckale J., Masjkur J., Magro M.G., Steffen A., Anastassiadis K., Solimena M.. Tamoxifen-independent recombination in the RIP-CreER mouse. PLoS One. 2010; 5:e13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vooijs M., Jonkers J., Berns A.. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. 2001; 2:292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jarmoskaite I., Russell R.. RNA helicase proteins as chaperones and remodelers. Annu. Rev. Biochem. 2014; 83:697–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Linder P., Jankowsky E.. From unwinding to clamping—the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011; 12:505–516. [DOI] [PubMed] [Google Scholar]
- 13. Godbout R., Squire J.. Amplification of a DEAD box protein gene in retinoblastoma cell lines. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:7578–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Godbout R., Li L., Liu R.Z., Roy K.. Role of DEAD box 1 in retinoblastoma and neuroblastoma. Future Oncol. 2007; 3:575–587. [DOI] [PubMed] [Google Scholar]
- 15. Tanaka K., Okamoto S., Ishikawa Y., Tamura H., Hara T.. DDX1 is required for testicular tumorigenesis, partially through the transcriptional activation of 12p stem cell genes. Oncogene. 2009; 28:2142–2151. [DOI] [PubMed] [Google Scholar]
- 16. Tanaka K., Ikeda N., Miyashita K., Nuriya H., Hara T.. DEAD box protein DDX1 promotes colorectal tumorigenesis through transcriptional activation of the LGR5 gene. Cancer Sci. 2018; 109:2479–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Squire J.A., Thorner P.S., Weitzman S., Maggi J.D., Dirks P., Doyle J., Hale M., Godbout R.. Co-amplification of MYCN and a DEAD box gene (DDX1) in primary neuroblastoma. Oncogene. 1995; 10:1417–1422. [PubMed] [Google Scholar]
- 18. Weber A., Imisch P., Bergmann E., Christiansen H.. Coamplification of DDX1 correlates with an improved survival probability in children with MYCN-amplified human neuroblastoma. J. Clin. Oncol. 2004; 22:2681–2690. [DOI] [PubMed] [Google Scholar]
- 19. Fang J., Kubota S., Yang B., Zhou N., Zhang H., Godbout R., Pomerantz R.J.. A DEAD box protein facilitates HIV-1 replication as a cellular co-factor of Rev. Virology. 2004; 330:471–480. [DOI] [PubMed] [Google Scholar]
- 20. Xu L., Khadijah S., Fang S., Wang L., Tay F.P., Liu D.X.. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J. Virol. 2010; 84:8571–8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Popow J., Jurkin J., Schleiffer A., Martinez J.. Analysis of orthologous groups reveals archease and DDX1 as tRNA splicing factors. Nature. 2014; 511:104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hildebrandt M.R., Germain D.R., Monckton E.A., Brun M., Godbout R.. Ddx1 knockout results in transgenerational wild-type lethality in mice. Sci. Rep. 2015; 5:9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ribeiro de Almeida C., Dhir S., Dhir A., Moghaddam A.E., Sattentau Q., Meinhart A., Proudfoot N.J.. RNA helicase DDX1 converts RNA G-quadruplex structures into R-loops to promote IgH class switch recombination. Mol. Cell. 2018; 70:650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomson E., Ferreira-Cerca S., Hurt E.. Eukaryotic ribosome biogenesis at a glance. J. Cell Sci. 2013; 126:4815–4821. [DOI] [PubMed] [Google Scholar]
- 25. Tomecki R., Sikorski P.J., Zakrzewska-Placzek M.. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells - focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 2017; 591:1801–1850. [DOI] [PubMed] [Google Scholar]
- 26. Henras A.K., Plisson-Chastang C., O’Donohue M.F., Chakraborty A., Gleizes P.E.. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA. 2015; 6:225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Warner J.R., Vilardell J., Sohn J.H.. Economics of ribosome biosynthesis. Cold Spring Harb. Symp. Quant. Biol. 2001; 66:567–574. [DOI] [PubMed] [Google Scholar]
- 28. Liu Y., Deisenroth C., Zhang Y.. RP-MDM2-p53 pathway: linking ribosomal biogenesis and tumor surveillance. Trends Cancer. 2016; 2:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morgado-Palacin L., Varetti G., Llanos S., Gómez-López G., Martinez D., Serrano M.. Partial loss of Rpl11 in adult mice recapitulates diamond-blackfan anemia and promotes lymphomagenesis. Cell Rep. 2015; 13:712–722. [DOI] [PubMed] [Google Scholar]
- 30. Bursac S., Brdovcak M.C., Donati G., Volarevic S.. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta. 2014; 1842:817–830. [DOI] [PubMed] [Google Scholar]
- 31. Gertsenstein M., Nutter L.M., Reid T., Pereira M., Stanford W.L., Rossant J., Nagy A.. Efficient generation of germ line transmitting chimeras from C57BL/6N ES cells by aggregation with outbred host embryos. PLoS One. 2010; 5:e11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki T., Kazuki Y., Oshimura M., Hara T.. Highly efficient transfer of chromosomes to a broad range of target cells using chinese hamster ovary cells expressing murine leukemia virus-derived envelope proteins. PLoS One. 2016; 11:e0157187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He X., Tan C., Wang F., Wang Y., Zhou R., Cui D., You W., Zhao H., Ren J., Feng B.. Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016; 44:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mansour F.H., Pestov D.G.. Separation of long RNA by agarose-formaldehyde gel electrophoresis. Anal. Biochem. 2013; 441:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stefanovsky V.Y., Moss T.. Metabolic labeling in the study of mammalian ribosomal RNA synthesis. Methods Mol. Biol. 2016; 1455:133–145. [DOI] [PubMed] [Google Scholar]
- 37. Yoshikawa H., Komatsu W., Hayano T., Miura Y., Homma K., Izumikawa K., Ishikawa H., Miyazawa N., Tachikawa H., Yamauchi Y.et al.. Splicing factor 2-associated protein p32 participates in ribosome biogenesis by regulating the binding of Nop52 and fibrillarin to preribosome particles. Mol. Cell. Proteomics. 2011; 10:M110.006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanner N.K., Cordin O., Banroques J., Doère M., Linder P.. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol. Cell. 2003; 11:127–138. [DOI] [PubMed] [Google Scholar]
- 39. Jensen N., Schrøder H.D., Hejbøl E.K., Füchtbauer E.M., de Oliveira J.R., Pedersen L.. Loss of function of Slc20a2 associated with familial idiopathic basal ganglia calcification in humans causes brain calcifications in mice. J. Mol. Neurosci. 2013; 51:994–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim J.H., Lee S.R., Li L.H., Park H.J., Park J.H., Lee K.Y., Kim M.K., Shin B.A., Choi S.Y.. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One. 2011; 6:e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suzuki T., Kazuki Y., Oshimura M., Hara T.. A novel system for simultaneous or sequential integration of multiple gene-loading vectors into a defined site of a human artificial chromosome. PLoS One. 2014; 9:e110404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bursać S., Brdovčak M.C., Pfannkuchen M., Orsolić I., Golomb L., Zhu Y., Katz C., Daftuar L., Grabušić K., Vukelić I.et al.. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:20467–20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Riesenberg S., Chintalapati M., Macak D., Kanis P., Maricic T., Pääbo S.. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 2019; 47:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smits A.H., Ziebell F., Joberty G., Zinn N., Mueller W.F., Clauder-Münster S., Eberhard D., Fälth Savitski M., Grandi P., Jakob P.et al.. Biological plasticity rescues target activity in CRISPR knock outs. Nat. Methods. 2019; 16:1087–1093. [DOI] [PubMed] [Google Scholar]
- 45. Tuladhar R., Yeu Y., Tyler Piazza J., Tan Z., Rene Clemenceau J., Wu X., Barrett Q., Herbert J., Mathews D.H., Kim J.et al.. CRISPR-Cas9-based mutagenesis frequently provokes on-target mRNA misregulation. Nat. Commun. 2019; 10:4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sharpe J.J., Cooper T.A.. Unexpected consequences: exon skipping caused by CRISPR-generated mutations. Genome Biol. 2017; 18:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Makino S., Fukumura R., Gondo Y.. Illegitimate translation causes unexpected gene expression from on-target out-of-frame alleles created by CRISPR-Cas9. Sci. Rep. 2016; 6:39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shaner N.C., Lambert G.G., Chammas A., Ni Y., Cranfill P.J., Baird M.A., Sell B.R., Allen J.R., Day R.N., Israelsson M.et al.. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods. 2013; 10:407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Genové G., Glick B.S., Barth A.L.. Brighter reporter genes from multimerized fluorescent proteins. BioTechniques. 2005; 39:doi:10.2144/000112056. [DOI] [PubMed] [Google Scholar]
- 50. Srivastava L., Lapik Y.R., Wang M., Pestov D.G.. Mammalian DEAD box protein Ddx51 acts in 3′ end maturation of 28S rRNA by promoting the release of U8 snoRNA. Mol. Cell. Biol. 2010; 30:2947–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Okuda S., Watanabe Y., Moriya Y., Kawano S., Yamamoto T., Matsumoto M., Takami T., Kobayashi D., Araki N., Yoshizawa A.C.et al.. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. 2017; 45:D1107–D1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data have been deposited to the ProteomeXchange Consortium via jPOST (51). The accession numbers are PXD023575 for ProteomeXchange and JPST001033 for jPOST.