


Abstract
RNA helicases play important roles in diverse aspects of RNA metabolism through their functions in remodelling ribonucleoprotein complexes (RNPs), such as pre-ribosomes. Here, we show that the DEAD box helicase Dbp3 is required for efficient processing of the U18 and U24 intron-encoded snoRNAs and 2′-O-methylation of various sites within the 25S ribosomal RNA (rRNA) sequence. Furthermore, numerous box C/D snoRNPs accumulate on pre-ribosomes in the absence of Dbp3. Many snoRNAs guiding Dbp3-dependent rRNA modifications have overlapping pre-rRNA basepairing sites and therefore form mutually exclusive interactions with pre-ribosomes. Analysis of the distribution of these snoRNAs between pre-ribosome-associated and ‘free’ pools demonstrated that many are almost exclusively associated with pre-ribosomal complexes. Our data suggest that retention of such snoRNPs on pre-ribosomes when Dbp3 is lacking may impede rRNA 2′-O-methylation by reducing the recycling efficiency of snoRNPs and by inhibiting snoRNP access to proximal target sites. The observation of substoichiometric rRNA modification at adjacent sites suggests that the snoRNPs guiding such modifications likely interact stochastically rather than hierarchically with their pre-rRNA target sites. Together, our data provide new insights into the dynamics of snoRNPs on pre-ribosomal complexes and the remodelling events occurring during the early stages of ribosome assembly.
INTRODUCTION
Production of eukaryotic ribosomes, which involves the assembly of four ribosomal RNAs (rRNAs) and 79 (Saccharomyces cerevisiae; yeast)/80 (human) ribosomal proteins, is one of the most energy-consuming cellular processes (1–3). In yeast, the best characterised model system for analysing ribosome assembly, this process requires the co-ordinated action of over 200 assembly factors that transiently associate with pre-ribosomal particles to facilitate correct assembly of the ribosomal subunits (4,5).
A nascent pre-rRNA transcript (35S in yeast) containing three of the four rRNAs (18S, 5.8S and 25S) separated by internal transcribed spacers (ITS1 and ITS2) and flanked by external transcribed spacers (5′ ETS and 3′ ETS) is synthesised by RNA polymerase I (Pol I) in the nucleolus (6). The pre-rRNA transcripts undergo various site-specific cleavages to release the mature rRNAs, several of which occur already co-transcriptionally, and an array of rRNA modifications are introduced (7,8). The vast majority of rRNA modifications are 2′-O-methylations (Nm) and pseudouridylations (Ψ), which are largely introduced by small nucleolar RNPs (snoRNPs) composed of a small nucleolar RNA (snoRNA) and associated proteins (9,10). SnoRNAs basepair with pre-rRNA sequences to direct the modification of specific target rRNA nucleotides by their associated methyltransferase (Nop1 (fibrillarin in humans)) or pseudouridine synthetase [Cbf5 (Dyskerin in humans)] (9,11). Their extensive pre-rRNA interactions mean that snoRNPs likely also make important contributions to the early stages of ribosome assembly by regulating the order and dynamics of pre-rRNA folding, although how this takes place is currently poorly understood. Together with the various base methylations introduced by stand-alone modification enzymes, Nm and Ψ cluster in functionally important regions of the ribosome, such as the peptidyl transferase centre, decoding site and intersubunit interface (12–14). Globally, rRNA modifications are suggested to contribute to the stability and conformational flexibility of ribosome structure and thereby influence translation efficiency and fidelity. The recent discovery of substoichiometric rRNA modifications in various species (8,15–17) has revealed rRNA modifications as a source of ribosome heterogeneity and highlighted their potential roles in the translational control of gene expression (10).
During pre-rRNA maturation, various ribosomal proteins and biogenesis factors are recruited to the nascent pre-rRNA transcript giving rise to the small subunit (SSU) processome and early 90S pre-ribosomal particles (18). Structural rearrangements and pre-rRNA cleavages separate the pre-40S (SSU) and pre-60S (large subunit, LSU) particles, which follow independent assembly pathways in the nucleolus and nucleoplasm (4). Maturation of both the pre-40S and pre-60S particles requires the dynamic association and dissociation of numerous assembly factors. Pre-40S particles containing the 20S pre-rRNA are rapidly exported to the cytoplasm where final maturation and quality control steps take place. In contrast, nucleolar and nucleoplasmic maturation of pre-60S complexes involves numerous intermediate steps before the particles achieve export competence and can translocate to the cytoplasm (19,20).
During their maturation, pre-ribosomal complexes undergo extensive remodelling, involving the establishment of rRNA folds present in mature ribosomes, a process which is often closely coupled with the recruitment and release of ribosomal proteins, assembly factors and snoRNPs. These structural rearrangements can serve as important checkpoints during the assembly pathway and energy-driven enzymes, such as AAA-ATPases, GTPases, kinases and ATP-dependent RNA helicases, have emerged as key regulators of these transitions (21–23). RNA helicases are characterised by the presence of two RecA-like domains containing conserved sequence motifs involved in RNA binding, ATP binding and hydrolysis, and the coupling of these to achieve unwinding (24). Of the >20 RNA helicases implicated in ribosome biogenesis in yeast (23), the majority function during the early stages of ribosome assembly, likely due to the more open conformation of pre-ribosomal particles at this stage, which allows greater access to their target sites for remodelling. While examples of helicases that are necessary for the association or dissociation of specific ribosomal proteins and assembly factors have been described (25–27), several RNA helicases have been implicated in modulating the dynamics of particular snoRNPs on pre-ribosomes. For example, a specific subset of snoRNAs accumulate on early pre-LSU particles when Prp43 is lacking and the helicase is suggested to remodel such particles to facilitate the access of other snoRNPs (28,29). Similarly, Dhr1, Has1, and Rok1 have all been linked to release of individual snoRNAs (U3, U14 and snR30 respectively) from pre-SSU complexes (28,30–33). Nevertheless, the functions of other RNA helicases that act on pre-ribosomal complexes have remained largely unexplored.
Although Dbp3 has been implicated in pre-60S biogenesis, little is known about its role(s) in ribosomal subunit assembly. Here we show that the catalytic activity of the RNA helicase Dbp3 is required for efficient conversion of the 27SA pre-rRNA to 27SB. This pre-rRNA processing defect is rescued by reduced expression of the box C/D snoRNP component Nop56, implying that the function of Dbp3 in pre-LSU maturation is linked to snoRNPs. Consistent with this, using northern blotting, RiboMeth-seq (RMS) and reverse transcription, quantitative PCR (RT-qPCR) analysis of snoRNA levels on pre-ribosomes, we observe defects in snoRNA maturation, rRNA 2′-O-methylation and the pre-ribosomal accumulation of a subset of box C/D snoRNPs in cells lacking Dbp3. We propose that Dbp3 is indirectly involved in promoting release of specific snoRNPs from pre-ribosomes, which is necessary for efficient snoRNP recycling and snoRNP access to proximal sites to enable stoichiometric rRNA modification. Our data implicate another RNA helicase, Prp43, in directly resolving the pre-rRNA basepairing of several snoRNAs guiding Dbp3-dependent rRNA 2′-O-methylations.
MATERIALS AND METHODS
Molecular cloning
The coding sequence of Dbp3, amplified from yeast genomic DNA (Supplementary Table S1), was cloned into a pQE-80-based vector for the expression of proteins with an N-terminal His10-ZZ tag in Escherichia coli. To express mutant versions of Dbp3 from the same vector, site-directed mutagenesis using oligonucleotides listed in Supplementary Table S1 was performed to convert glutamate 263 to glutamine in the expressed protein (Dbp3E263Q) or to delete amino acids 22–48 (Dbp3Δ22–48). For exogenous expression of Dbp3 in yeast, the coding sequence of Dbp3 and 500 basepairs upstream and downstream of it were cloned into pRS415 (Supplementary Tables S1 and S2). The Dbp3E263Q mutant was also created in this vector using site-directed mutagenesis. Overexpression of snR67 was achieved by cloning the sequence encoding the snoRNA and flanking regions into a dedicated snoRNA expression construct for expression of the snoRNA from within the intron of the actin gene under the control of a pGAL1 promoter (Supplementary Tables S1 and S2) (34).
Expression of recombinant proteins in E. coli and purification
Expression of His10-ZZ-Dbp3 or His10-ZZ-Dbp3E263Q was induced in BL21 codon plus cells by addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 16 h at 18°C. Cells were pelleted and resuspended in Lysis buffer (50 mM Tris–HCl pH 7.0, 500 mM NaCl, 1 mM MgCl2, 10% glycerol, 10 mM imidazole and 1 mM phenylmethylsulfonyl fluoride (PMSF)). After disruption by sonication, the cell lysate was cleared by centrifugation at 20 000 × g for 30 min at 4°C. Polyethylamine (PEI) was added to the soluble fraction to a concentration of 0.05% and the lysate was incubated at 4°C for 15 min before centrifuging at 33 000 × g for 30 min at 4°C. The cleared lysate was incubated with cOmplete His-tag purification resin (Roche) for 2 h at 4°C. After thorough washing steps with a buffer composed of 50 mM Tris–HCl pH 7.0, 500 mM NaCl, 1 mM MgCl2, 10% glycerol and 30 mM imidazole, bound proteins were eluted with a buffer containing 50 mM Tris–HCl pH 7.0, 500 mM NaCl, 1 mM MgCl2, 10% glycerol and 300 mM imidazole. The eluate was dialysed against a buffer containing 50 mM Tris–HCl pH 7.0, 120 mM NaCl, 2 mM MgCl2 and 20% glycerol.
In vitro ATPase assays
NADH-coupled assays were used to monitor the hydrolysis of ATP (35,36). Reactions containing 45 mM Tris–HCl pH 7.4, 25 mM NaCl, 2 mM MgCl2, 1 mM phosphoenolpyruvate, 300 μM NADH, 20 U/ml pyruvate kinase/lactic dehydrogenase and 4 mM ATP were supplemented with 250 nM recombinant His10-ZZ-tagged Dbp3/Dbp3E263Q and 0–1 μM RNA (5′-GUAAUGAAAGUGAACGUAAAACAAAACAAAAC-3′). The absorbance at 340 nm was monitored using a BioTEK Synergy plate reader and the rate of ATP hydrolysis was calculated using the following equation where Kpath is the molar absorption co-efficient for a defined optical path length, which is defined as reaction volume (150 μl/well) and background NADH decomposition.
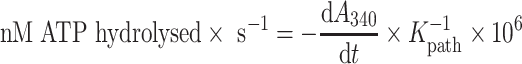 |
Yeast strains and growth conditions
Yeast strains used in this study are listed in Supplementary Table S3 and were grown in YPD/G (1% yeast extract, 2% peptone, 2% glucose/galactose) or synthetic media lacking leucine where appropriate. To generate the Dbp3 complementation system, the pRS415-based constructs for the expression of DBP3 from its endogenous promoter, or the empty pRS415 plasmid, were used to transform either wild type yeast or a Δdbp3 strain. Yeast strains lacking individual snoRNA genes, or the SNR72–78 or SNR67–53 clusters, were generated by substitution of the relevant gene with a marker cassette. Homologous recombination was similarly used to insert a truncated pGAL promoter (pGALS) and the sequence encoding an HA tag immediately upstream of the coding sequence of PRP43 to generate the pGALS-HA-Prp43 strain. To deplete Prp43, cells of the pGALS-HA-Prp43 strain were grown in exponential phase in media containing galactose as the carbon source before switching to growth in media containing glucose for 8 h.
Protein extraction and western blotting
Yeast cells were lysed by vortexing with glass beads and proteins were precipitated using 15% trichloroacetic acid (TCA). Proteins were separated by SDS-PAGE and analysed by western blotting using the following primary antibodies [anti-Prp43 (kindly provided by Yves Henry); anti-HA (Sigma-Aldrich #H3663]; anti-Pgk1 [ThermoFisher Scientific #459250)].
RNA extraction, northern blotting and 4sU metabolic labelling
Total RNA was extracted from exponentially growing yeast cells using acidic phenol and chloroform extracts as previously described (27). For analysis of long RNAs (>400 nt) by northern blotting, 6 μg total RNA was separated by denaturing (glyoxal) agarose gel electrophoresis and transferred to a nylon membrane by vacuum blotting. Alternatively, to detect RNAs <400 nt, total RNA was separated by denaturing (7 M urea) polyacrylamide gel electrophoresis (PAGE) and transferred to a nylon membrane by electro-wet blotting. Membranes were then pre-hybridized in 0.25 M sodium phosphate pH 7.0, 7% SDS (w/v), 1 mM EDTA before addition of 5′ [32P]-labelled DNA oligonucleotides (Supplementary Table S1) and incubation overnight at 37°C. After washing steps, membranes were exposed to phosphorimager screens and signals detected using a Typhoon FLA9500 phosphorimager. For detection of nascent RNAs (37), exponentially growing yeast cells were grown in the presence of 200 μM 4-thiouracil (4sU) for 20 min at 30°C. Total RNA was extracted and 50 μg 4sU labelled total RNA was incubated with 5 μg MTSEA Biotin-XX (Biotium) in 10 mM Tris–HCl pH 7.4, 1 mM EDTA pH 8 in the dark at 4°C for 30 min. RNAs were re-extracted and 12 μg RNA was separated by denaturing agarose gel electrophoresis and transferred to a nylon membrane. Biotinylated RNAs were detected using IRDye-conjugated streptavidin (LICOR) using a LICOR Odyssey CLx.
Sucrose density gradient centrifugation
Sucrose density gradient centrifugation was essentially performed as previously described (38,39). Whole cell extracts prepared from exponentially growing yeast cells by grinding in liquid nitrogen were clarified by centrifugation at 20 000 × g for 10 min. Cleared lysates were separated on 10–45% sucrose gradients in an SW-40Ti rotor for 16 h at 23 500 rpm. Fractions of 530 μl were taken and RNA was extracted as described above for analysis by northern blotting or RT-qPCR.
Analysis of snoRNA levels on pre-ribosomes by RT-qPCR
Analysis of the relative proportions of 75 yeast snoRNAs between pre-ribosomal and non-ribosomal complexes was performed as in (31). In brief, whole cell extracts from wild type yeast and the Ddbp3 strain were separated by sucrose density gradient centrifugation and RNA was extracted from gradient fractions using phenol:chloroform:isoamylalcohol (25:24:1). RNAs from fractions containing (pre-)ribosomal and non-ribosomal complexes were combined. 5–10 μg RNA was polyadenylated for 2 h at 37°C using 8 U E. coli poly(A) polymerase (NEB) in the presence of 5 mM ATP and 2.5 mM MnCl2. Small polyadenylated RNAs were isolated using the MirVana miRNA isolation kit (Life Technologies) according to the manufacturer's instructions. Polyadenylated small RNAs were quantified and quality controlled (260 nm/280 nm ratio) using a Nanodrop and 12.5 μg (free pool) or 7.5 μg (pre-ribosome-associated) RNA was used for reverse transcription. Reactions were carried out in 1× Superscript III reaction buffer supplemented with RNasin and 5 mM DTT using an anchored oligo d(T) primer (31), Superscript III reverse transcriptase (1000 U; Thermo Fisher Scientific) and 1 mM dNTPs (Roche). RNA was digested using 25 U RNase H (NEB) and 10 U RNase A for 2 h at 37°C. The cDNA was purified using the QIAquick PCR purification kit (Qiagen) according to the manufacturer's instructions. cDNA concentrations were determined using a Nanodrop and equal amounts from each sample were used for qPCR analyses. snoRNA-specific forward qPCR primers (Sigma) (31) were designed to generate amplicons of 80–100 bp and have equal melting temperatures, similar to that of the common reverse primer. Primer pairs were tested for amplification of a single amplicon using a product melting curve. The amplification efficiency and linearity of cDNA amplification was determined and only primer pairs with an amplification efficiency of >90% were used. qPCR was performed using SYBR® Green Jumpstart Taq Readymix (Sigma), 0.3 μM primers and 1× ROX (Reference of qPCR; Sigma) in a Mx3000P qPCR machine (Agilent Technologies). Cycling conditions were 95°C for 5 min, 40 cycles of 95°C for 10 sec, 55°C for 10 s and 72°C for 15 s, followed by 95°C for 10 s, 55°C for 10 s and 95°C for 30 s. qPCR data were analysed using MxPro software. Technical triplicate reactions were performed and values differing by >0.5 Cq were excluded. The values obtained in samples from the Δdbp3 strain were normalised to those from the wild type (WT) samples and ratios of pre-ribosomal versus unbound levels for each snoRNA were calculated using the formula 2−(Ct(WT) – Ct(Δdbp3)unbound)/2−(Ct(WT) – Ct(Δdbp3) pre-ribosomal). Three independent experiments were performed and statistical analyses for significance thresholds were determined as previously described (31).
RiboMeth-seq
RiboMeth-seq was essentially performed as previously reported (40). In brief, 5 μg total RNA was fragmented under denaturing conditions using an alkaline buffer (pH 9.9). Subsequently, the RNA was separated on a denaturing (urea) polyacrylamide gel, fragments in the size of 20–40nt were excised and ligated to adapters using a modified tRNA ligase. cDNA was generated using Superscript III (Thermo Fisher Scientific) and sequenced on a PI Chip v3 using the Ion Proton platform. Reads were mapped to the yeast rDNA and snoRNA sequences, and the RMS score (fraction methylated) was calculated as ‘score C’ in Birkedal et al. (8). In a few cases, a barcode correction was applied when calculating the RMS score as described previously (41). The numbers of sequencing reads mapping to specific RNAs was used as a measure of their levels and is presented as reads per kilobase of transcript, per million mapped reads (RPKM) ± SEM. Statistical significance was determined using Student's t-test (P < 0.05).
RNase H-based cleavage assays for monitoring RNA 2′-O-methylation
The methylation status of specific rRNA nucleotides was monitored using RNase H-based cleavage assays (42). Chimeric 2′-O-methylated RNA-DNA oligonucleotides (Supplementary Table S4) were annealed to total RNA before treatment with RNase H (NEB) for 30 min at 37°C. Reactions were stopped by addition of 240 mM NaAc pH 5.2 and 1 mM EDTA before RNA extraction using phenol:chloroform:isoamylalcohol (25:24:1). Samples were separated by denaturing agarose gel electrophoresis and analysed by northern blotting.
CLASH analysis of CRAC data
Identification of snoRNA-rRNA hybrids in the CRAC data was performed using a bioinformatics pipeline developed for crosslinking, ligation and analysis of sequence hybrids (43).
RESULTS
The catalytic activity of Dbp3 is required for efficient production of the 27SB pre-rRNA
Dbp3 has been shown to associate with early pre-LSU particles in vivo and to possess RNA duplex unwinding activity in vitro (44,45), however the requirement of this catalytic activity for ribosome assembly has not been addressed. To establish a catalytically inactive mutant of Dbp3, wild type Dbp3 (Dbp3WT) and a mutant version in which glutamate 263 within the conserved DEAD motif responsible for ATP binding and hydrolysis was substituted for glutamine (Dbp3E263Q) were recombinantly expressed with N-terminal His10-ZZ tags in Escherichia coli (E. coli) and purified by nickel affinity chromatography (Figure 1A and B). In vitro NADH-coupled ATPase assays were then used to monitor the rates of ATP hydrolysis by these two proteins in the presence of different amounts of RNA. Compared to a control sample without protein, Dbp3WT, but not Dbp3E263Q, showed minimal ATPase activity in the absence of RNA (Figure 1C). Addition of increasing concentrations of RNA stimulated the rate of ATP hydrolysis by Dbp3WT, but Dbp3E263Q never hydrolysed ATP above the background level (Figure 1C), confirming that this amino acid substitution abolishes ATP hydrolysis by the protein.
Figure 1.
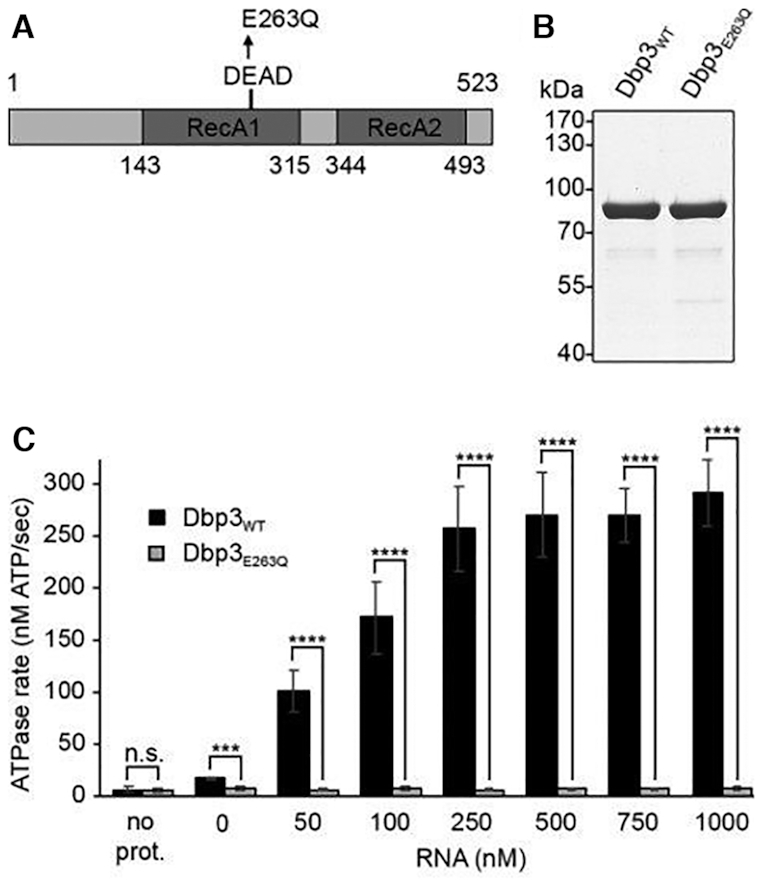
The putative RNA helicase Dbp3 is an RNA-dependent ATPase. (A) Schematic view of Dbp3 showing the amino acid boundaries of the RecA1 and RecA2 domains as well as the amino acid substitution made within the evolutionarily conserved DEAD motif. (B) N-terminally His10-ZZ tagged wild type Dbp3 (Dbp3WT) and an equivalent protein carrying a glutamate to glutamine substitution at position 263 (Dbp3E263Q) were recombinantly expressed in E. coli and purified by Ni2+-affinity chromatography. Purified proteins were separated by SDS-PAGE and visualised by Coomassie staining. (C) In vitro NADH-coupled ATPase assays were used to monitor ATP hydrolysis by Dbp3WT and Dbp3E263Q in the presence of increasing amounts of RNA. A sample containing no protein was included as a control for background hydrolysis of ATP. The data represent the mean of three independent experiments ± standard deviation. ***P< 0.001, ****P< 0.0001, n.s. = non-significant.
Next, to explore the requirement of this catalytic activity for ribosome biogenesis, a yeast complementation system was generated. Wild type BY4741 yeast or a strain where DBP3, which is not essential, had been deleted from the genome (Δdbp3; Supplementary Figure S1A and B) were transformed with pRS415-based constructs for the expression of untagged Dbp3WT or Dbp3E263Q from the endogenous DBP3 promoter, or as a control, the empty plasmid. Growth analyses of these strains revealed that the Δdbp3 strains containing either an empty plasmid or the plasmid for expression of Dbp3WT grew as wild type yeast and cells expressing Dbp3E263Q had only a very mild growth impairment (Figure 2A). Pre-rRNA processing intermediates from these strains were analysed by northern blotting using a mixture of probes hybridising within the ITS1 or ITS2 regions of the pre-rRNA transcript (Figure 2B). Compared to the wild type strain carrying an empty pRS415 vector, cells lacking Dbp3 showed elevated levels of the 35S pre-rRNA and 33S/32S and 27SA processing intermediates as well as a markedly reduced level of 27SB (Figure 2C and D). The significantly increased 27SA-27SB ratio in the Δdbp3 strain indicates that Dbp3 is required for the efficient conversion of 27SA to 27SB, which occurs by processing at the A3, B1 and B2 sites. This result is consistent with a previous analysis indicating the requirement for Dbp3 for processing at the A3 site (44). Importantly, these pre-rRNA processing defects were rescued by expression of plasmid-derived Dbp3WT, confirming that they are caused by the lack of Dbp3. In contrast, expression of exogenous Dbp3E263Q in the Δdbp3 background showed increased levels of the 35S and 27SA pre-rRNAs and loss of the 27SB intermediate (Figure 2C and D), demonstrating that the catalytic activity of Dbp3 is necessary for its function in LSU biogenesis. To determine whether these pre-rRNA processing defects affect production of the mature 25S rRNA, the production of nascent 25S rRNA in the strains of the complementation system was determined by metabolic labelling (37). Strains were grown in the presence of 4-sU, newly synthesised 4-thiouridine-containing RNAs were labelled with biotin and, after separation by denaturing agarose gel electrophoresis, biotinylated RNAs were detected using fluorescently labelled streptavidin. Lack of Dbp3 or expression of Dbp3E263Q did not affect production of the 25S RNA (Figure 2E and F), suggesting that the pre-rRNA processing defects observed reflect subtle kinetic differences in the processing pathway.
Figure 2.

The role of Dbp3 in processing of the 27SA–27SB pre-rRNAs is linked to box C/D snoRNPs. (A) Wild type yeast (WT) or a strain lacking Dbp3 (Δdbp3) were transformed with an empty pRS415 vector (EV) or plasmids for the expression of Dbp3WT or Dbp3E263Q from the endogenous DBP3 promoter. Growth of these strains in exponential phase was monitored by measuring the OD of cultures at 600 nm every 2 h. The data represent the mean of three independent experiments ± standard deviation. (B) Schematic view of the major pre-rRNA processing intermediates present in yeast. Black rectangles indicate mature rRNA sequences and black lines represent internal transcribed spacers (ITS1 and ITS2) and external transcribed spacers (5′ ETS and 3′ ETS). The positions of selected pre-rRNA cleavage sites are marked on the 35S pre-rRNA transcript. A magnified view of the ITS1 and ITS2 regions with major pre-rRNA cleavage sites and the hybridisation positions of probes used for northern blotting (indicated by grey lines) is shown below the 35S transcript. (C) Pre-rRNA levels in the strains described in (A) were analysed by northern blotting using a mixture of probes hybridising within ITS1 or ITS2. Mature rRNAs were visualised by methylene blue staining. (D) The ratio of 27SA/27SB in the samples shown in (C) was calculated in three independent experiments and is shown as mean ± standard deviation. Significance was calculated using a Student's t-test (**P< 0.01, ***P< 0.001, n.s. = non-significant). (E) The strains described in (A) were grown in the presence of 4sU and nascent RNAs were labelled with biotin, separated by denaturing agarose gel electrophoresis and detected with fluorescently labelled streptavidin. (F) The ratio of 25S/18S in the samples shown in (E) was calculated in three independent experiments and is shown as mean ± standard deviation. Significance was calculated using a Student's t-test (n.s. = non-significant). (G) Pre-rRNA processing in wild type yeast (WT), cells lacking Dbp3 (Δdbp3), cells expressing reduced levels of Nop56 (NOP56DAmP) or both (Δdbp3-NOP56DAmP) was analysed as in (C). (H) The ratio of 27SA/27SB in the samples shown in (G) was calculated in three independent experiments and is shown as mean ± standard deviation. Significance was calculated using a Student's t-test (**P< 0.01, n.s. = non-significant).
The role of Dbp3 in LSU biogenesis is linked to box C/D snoRNPs
Interestingly, a high-throughput screen based on expression of an artificial snoRNA guiding an rRNA 2′-O-methylation that impairs cellular growth (46), identified Dbp3 as a potential regulator of rRNA 2′-O-methylation (Nicholas Watkins and Robert van Nues, unpublished). This suggested that the pre-rRNA processing defects observed in the absence of Dbp3 and its function in ribosome assembly may be linked to rRNA 2′-O-methylation-guiding box C/D snoRNPs. To test this hypothesis, yeast strains were generated in the wild type and Δdbp3 backgrounds where expression of the essential core box C/D snoRNP component Nop56 is reduced by introduction of an antibiotic resistance cassette into the 3′ untranslated region (UTR) of the NOP56 gene, leading to destabilisation of the transcribed mRNA (NOP56DAmP) (Supplementary Figure S1A and B) (47). In the Nop56DAmP strain, the levels of a subset of snoRNAs examined were observed to be increased (Supplementary Figure S1C) (48). Analysis of pre-rRNA processing confirmed the pre-rRNA processing defects previously observed in cells lacking Dbp3 (Figure 2B and C) and revealed that a reduced level of Nop56 leads to defects in early pre-rRNA processing, characterised by accumulation of the 35S pre-rRNA and aberrant 23S intermediate as well as reduced levels of the 20S and 27SA/B processing intermediates (Figure 2G). In the Δdbp3+NOP56DAmP strain, the 35S pre-rRNA and aberrant 23S species were observed to accumulate and the amount of the 20S processing intermediate was reduced, consistent with the lack of Nop56 in this strain. However, the amount of 27SA and 27SB, and the ratio between these processing intermediates was comparable to that in wild type cells (Figure 2G and H). The finding that co-depletion of Nop56 rescues the pre-rRNA processing phenotype caused by lack of Dbp3 supports the notion that Dbp3 may be functionally linked to rRNA methylation.
Dbp3 is required for efficient 2′-O-methylation of specific nucleotides within the 25S rRNA sequence
To explore the link between Dbp3 and rRNA 2′-O-methylation in more detail, RiboMeth-Seq (RMS) analysis (8) was performed on RNAs from wild type cells and those lacking Dbp3. RNAs were subjected to partial alkaline hydrolysis, which does not cleave 2′-O-methylated nucleotides, and the resultant RNA fragments were copied into a cDNA library that was subjected to Ion Torrent sequencing (40). After mapping of the obtained sequencing reads to the S. cerevisiae transcriptome, the normalised numbers of reads mapping to the 25S and 18S rRNAs was determined, confirming that lack of Dbp3 does not alter mature rRNA levels (Supplementary Figure S2A). The number of read ends mapping to each nucleotide of the 18S and 25S rRNAs was then determined and used to calculate an RMS score for each 2′-O-methylated nucleotide. Consistent with previous analyses (8), in wild type yeast, the majority of 2′-O-methylated sites were almost fully modified, with only few exceptions (Figure 3A and B). While the extent of modification at only one site within the 18S rRNA of the SSU, Um1269 guided by snR55, was affected by lack of Dbp3 (Figure 3A), strikingly, lack of Dbp3 caused clear reductions in the extent of 2′-O-methylation at various (18/37) sites within the 25S rRNA (Figure 3B). Dbp3-dependent 2′-O-methylations are present along the length of the 25S rRNA sequence and are interspersed with non-Dbp3-dependent modifications. As no structural information on pre-ribosomal complexes bound by Dbp3 is currently available, the affected 2′-O-methylations were mapped on the tertiary structure of the 25S rRNA in the earliest pre-LSU complex where the complete rRNA sequence is visible, which is a pre-60S particle purified via the nucleolar/nucleoplasmic GTPase Nog2 (49). This showed that while these modifications are all close to the peptidyltransferase centre and tRNA binding sites of the ribosome, they do not strictly cluster in one specific region (Figure 3C).
Figure 3.

Dbp3 is required for efficient 2′-O-methylation of specific rRNA nucleotides. (A, B) RiboMeth-seq analysis was performed on wild type yeast (WT) or a strain lacking Dbp3 (Δdbp3). RiboMeth-seq scores, indicating the fraction of methylation, are plotted for each 2′-O-methylated nucleotide in the 18S rRNA (A) and the 25S rRNA (B). The data shown are the mean of two biological replicates and error bars represent standard deviation. The snoRNAs that guide each modification are indicated in blue and modifications for which the RMS score is decreased by >10% in the absence of Dbp3 are highlighted in red. (C) Dbp3-dependent (red) and Dbp3-independent (black) 2′-O-methylations are mapped onto the tertiary structure of a pre-60S complex purified via Nog2 (PBD: 3JCT) (49). (D–F) Wild type yeast cells or a strain lacking Dbp3 were transformed with an empty pRS415 vector (EV) or plasmids for the expression of Dbp3WT or Dbp3E263Q from the endogenous DBP3 promoter. RNA was isolated from these strains and RNase H (RH)-mediated cleavage assays targeting Am876 (snR72), Um898 (snR40) or Um2724 (snR67) were performed, and full length 25S rRNA and specific cleavage products (indicated by arrow heads) were detected by northern blotting using probes hybridising up- and down-stream of the cleavage sites (D and E) or only downstream of the cleavage site (F). Asterisks indicate non-specific cleavage products – note that the extent of non-specific RNase H-mediated cleavage is reduced upon cleavage at a specific cleavage site.
We next addressed the question of whether the catalytic activity of Dbp3, or merely its presence, is required for efficient rRNA 2′-O-methylation. While RNase H efficiently cleaves RNA in RNA–DNA hybrids, its cleavage activity is inhibited by the presence of RNA 2′-O-methylation. The extent of modification at specific sites can therefore be monitored using RNase H together with chimeric RNA-DNA oligonucleotides targeting different modified nucleotides in the rRNA (42). Cleavage assays monitoring three Dbp3-dependent 2′-O-methylation sites (25S-Am876, 25S-Um898 and 25S-Um2724) were first performed on RNAs derived from wild type yeast and cells lacking the snR72 (snR72–78 cluster), snR40 or snR67 snoRNAs that guide these modifications to verify the effectiveness of the method. Northern blotting analysis of the reaction products using probes hybridising upstream and/or downstream of the cleavage sites revealed no specific cleavage of the 25S rRNA in the absence of RNase H or in RNA derived from wild type yeast, consistent with the RMS data showing that these sites are normally almost fully modified. However, reduced levels of the full length 25S rRNA and the generation of products of sizes corresponding to the expected cleavage fragments was observed for RNAs lacking the snoRNA guiding each modification under investigation (Supplementary Figure S2B–E). Cleavage assays were then performed on RNAs purified from wild type yeast or Δdbp3 cells complemented with an empty vector (EV) or plasmids for the expression of wild type Dbp3 (Dbp3WT) or catalytically inactive Dbp3 (Dbp3E263Q). Again, no specific cleavage activity was observed in the absence of RNase H and in the presence of RNase H, the 25S rRNA remained uncleaved in the wild type strain carrying an empty vector (Figure 3D-F). In contrast, the 25S rRNA derived from Δdbp3 cells complemented with either the empty vector or the plasmid for expression of Dbp3E263Q was partially cleaved by RNase H leading to reduced levels of the full length 25S rRNA and generation of the expected cleavage fragments (Figure 3D–F). rRNA 2′-O-methylation of these sites was largely restored by expression of plasmid-derived Dbp3WT in the Δdbp3 strain as only minimal cleavage was observed. The reduced 2′-O-methylation of 25S-Am876, 25S-Um898 and 25S-Um2724 upon expression of Dbp3E263Q supports a role for the catalytic activity of Dbp3 in rRNA 2′-O-methylation.
The intron-derived U18 and U24 snoRNAs are incompletely processed in the absence of Dbp3
The substoichiometric 2′-O-methylation of numerous nucleotides within the 25S rRNA in the absence of Dbp3 raised the possibility that the cellular levels of the snoRNAs that guide these modifications are reduced when Dbp3 is lacking. The numbers of sequencing reads from the RMS analysis of wild type and Δdbp3 cells mapping to each yeast snoRNA were used to compare snoRNA levels in these two strains. Lack of Dbp3 was not observed to cause any significant differences in snoRNA levels and northern blotting analysis of the subset of snoRNAs guiding Dbp3-dependent rRNA 2′-O-methylations confirmed this finding (Supplementary Figure S3A and B). This demonstrates that altered snoRNA levels are not the basis of the reduced rRNA 2′-O-methylation detected in Δdbp3. However, while the overall levels of the U18 and U24 snoRNAs were not affected by lack of Dbp3, northern blotting for the U18 snoRNA revealed the presence of two species and deletion of DBP3 caused a shift towards the longer form (Supplementary Figure S3B and Figure 4A). Similarly, it appeared that a longer form of U24 accumulated upon deletion of DBP3. In contrast to the majority (73/79) of yeast snoRNAs that are synthesised as mono- or poly-cistronic transcripts, U18 and U24 are encoded within the introns of protein coding pre-mRNAs from which they are released by debranching and/or endonucleolytic cleavage, followed by exonucleolytic processing (50,51). Notably, maturation of all other intron-encoded snoRNAs (snR38, snR39, snR44, snR54, snR59 and snR191) was not affected by lack of Dbp3 (Supplementary Figures S3B and S4A). To investigate the nature of the longer U18 and U24 versions, northern blotting was performed on total RNA extracted from wild type yeast and cells lacking either Dbp3 or the 3′-5′ exonuclease Rrp6, which is implicated in 3′ end processing of snoRNAs (52). Only the longer version of U18, enriched in Δdbp3, was present in the Δrrp6 strain, suggesting that Dbp3 contributes to 3′ maturation of the U18 snoRNA (Figure 4A). To verify this, the RMS sequencing reads from wild type and Δdbp3 datasets were mapped to the annotated U18 sequence ±50 nt and the number of reads mapping to each nucleotide was determined. Interestingly, for U18 from wild type cells, this revealed high sequence coverage from the 5′ G, which forms the first basepair of the terminal stem, to 7 nt beyond the annotated 3′ end of the snoRNA. Sequence coverage was also observed for a further 13 nt (Figure 4B and C). Strikingly, the numbers of sequencing reads mapping to nucleotides beyond the annotated 3′ end of the U18 snoRNA were notably higher in the Δdbp3 strain than in wild type yeast (Figure 4B and C), supporting the involvement of Dbp3 in 3′ end processing of this snoRNA.
Figure 4.

Maturation of the intron-encoded U18 and U24 snoRNAs requires Dbp3. (A) Total RNA from wild type yeast (WT), and Δdbp3 and Δrrp6 strains was separated by denaturing PAGE and analysed by northern blotting using probes hybridising to the U18 and U24 snoRNAs. (B and C) RMS sequencing reads derived from wild type yeast or the Δdbp3 strain were mapped to the annotated U18 sequence ±50 nt, and after normalization for expression level, the numbers of reads mapping to each nucleotide were determined. Profiles for the 5′ end (B) and 3′ end (C) are shown. Nucleotides of box C (B) and box D (C) are underlined and the annotated 5′ and 3′ ends (61) are indicated. (D) Schematic view of the secondary structure of the pre-U18 snoRNA with key features indicated. Basepairing is shown according to (62). (E) Total RNA from wild type yeast (WT) or a strain lacking Dbp3 (Δdbp3) carrying an empty pRS415 vector (EV) or plasmids for the expression of Dbp3WT, Dbp3E263Q or Dbp3Δ22–48 from the endogenous DBP3 promoter was analysed as in (A). (F) Extracts from yeast expressing Nop1-His-Tev protease cleavage site-ProtA (HTP) in the wild type or Δdbp3 backgrounds were used for pulldown assays on IgG sepharose. Co-purified RNAs were analysed as in (A). (G) Whole cell extracts prepared from wild type yeast or the Δdbp3 strain were separated by sucrose density gradient centrifugation. RNA from individual fractions was separated by denaturing PAGE and analysed by northern blotting using probes to the snoRNAs indicated to the right. The fractions containing ‘free’ snoRNAs and pre-ribosome-associated snoRNAs are indicated.
Northern blotting analysis revealed that an extended form of U24 accumulated when Dbp3 is lacking was of an intermediate length between that detected in the wild type and Δrrp6 strains (Figure 4A). This suggests that a 3′ extended U24 precursor is only partially processed in the absence of Dbp3, however, this could not be supported by analysis of RMS sequencing reads (Supplementary Figure S4B and C). In contrast to pre-U18, where the 5′ and 3′ ends form a relatively weak terminal stem structure, pre-U24 forms a strong 7–8 bp terminal stem (Figure 4D and Supplementary Figure S4D). As alkaline hydrolysis, which is used to generate RNA fragments in the RMS procedure, is impaired by nucleotide basepairing, it is possible that the presence of the terminal stem structure of pre-U24 reduces read coverage of the precursor sequences, preventing detection of the effect of Dbp3 on 3′ end processing. Interestingly, expression of Dbp3E263Q also impaired processing of both U18 and U24 (Figure 4E), indicating that the catalytic activity of Dbp3 contributes to its function in U18 and U24 snoRNA biogenesis. The G-patch protein Gno1 has also been implicated in facilitating U18 and U24 3′ processing and a KK(E/D) motif, present in several nucleolar proteins including Nop56 and Nop58, was found to be important for this function. As Dbp3 also contains a KK(E/D) motif, we tested the requirement of this for efficient U18 and U24 maturation. Expression of Dbp3 lacking its KK(E/D) motif (Dbp3Δ22–48) also led to accumulation of the longer forms of these snoRNAs, but the 3′ processing defect observed was not as strong as in cells lacking Dbp3 or its catalytic activity (Figure 4E).
Given these findings, it is possible that the reduced 2′-O-methylation of the rRNA nucleotides guided by these snoRNAs (U18 – 25S-Am649, 25S-Am650, U24 – 25S-Cm1437, 25S-Am1449, 25S-Gm1450) arises due to the defects in snoRNA biogenesis. However, although lack of Dbp3 strongly impairs production of the mature U18 and U24 snoRNAs (Figure 4A), the RMS data implies that partial 2′-O-methylation of the U18/U24-guided sites can still occur, and that the extent of modification at the 2/3 sites guided by each snoRNA differ (Figure 3B). Therefore, to investigate whether the 3′ extended forms of U18 and U24 may be functional, their interactions with the 2′-O-methyltransferase Nop1 and their associations with pre-ribosomes were examined. Nop1-containing complexes were isolated from cell extracts prepared from wild type yeast expressing His-TEV protease cleavage site-Protein A (HTP)-tagged Nop1 or the equivalent strain lacking Dbp3. This revealed a similar enrichment of both processed and unprocessed U18 and U24 with Nop1 from the two strains, implying that both snoRNA versions are incorporated into snoRNPs and that incorporation is likely not dependent on Dbp3 (Figure 4F). The distribution of (pre-)U18 and (pre-)U24 between pre-ribosome-associated and non-pre-ribosome-associated pools was then examined. Whole cell extracts prepared from wild type yeast or cells lacking Dbp3 were subjected to sucrose density centrifugation and RNAs present in each fraction were examined by northern blotting (Figure 4G). This revealed that both the mature and precursor forms of U18 and U24 associate with pre-ribosomes. It is not possible to exclude that although the immature versions of the U18 and U24 snoRNPs associate with pre-ribosomes, they are not functional. However, it has previously been shown that the U24 snoRNA within its intron lariat is capable of directing rRNA modification (53), suggesting that the short 3′ snoRNA extension observed when Dbp3 is lacking probably does not influence the ability of these snoRNPs to mediate rRNA 2′-O-methylation. It is likely, therefore, that there is an alternative mechanistic basis for the rRNA modification defects observed at the U18 and U24-guided sites when Dbp3 is absent.
A subset of box C/D snoRNAs accumulate on pre-ribosomes when Dbp3 is lacking
As snoRNA levels and the processing of most snoRNAs are not affected by lack of Dbp3, an explanation for the reduced rRNA 2′-O-methylations observed in Δdbp3 could be that access of these snoRNAs to pre-ribosomes is impaired when DBP3 is deleted. Therefore, to quantitatively monitor snoRNA levels on pre-ribosomes, whole cell extracts prepared from wild type yeast or cells lacking Dbp3 were subjected to sucrose density centrifugation. Fractions containing (pre-)ribosomal complexes and those containing non-ribosome associated proteins, RNAs and small complexes were identified based on the absorbance at 260 nm and those belonging to each group were combined. RNA extracted from these two pools was reverse transcribed to produce cDNA, which served as a template for qPCR to determine the levels of each of the 75 snoRNAs present in yeast (27,31). The ratio of pre-ribosome-associated to non-ribosome-associated RNA was determined for each snoRNA and compared between the wild type and Δdbp3 strains. A value of 1 reflects no variation between the relative proportions of non-ribosome-associated and pre-ribosomal snoRNA in the two strains while higher values reflect snoRNA accumulation on pre-ribosomes when Dbp3 is lacking and lower values indicate reduced amounts of pre-ribosomal snoRNA in Δdbp3. The values 2 and 0.5 were calculated as upper and lower statistical significance thresholds for accumulation on and exclusion from pre-ribosomes respectively and the values obtained for most snoRNAs varied within these limits. However, several box C/D snoRNAs (U18, U24, snR39, snR39b, snR50, snR55, snR59, snR60, snR61, snR67, snR69, snR74 and snR79) significantly accumulated on pre-ribosomes in the absence of Dbp3, whereas no specific changes in the levels of the H/ACA box snoRNAs were detected (Figure 5 and Supplementary Figure S5). Intriguingly, 10 of the 13 snoRNAs (U18, U24, snR39, snR39b, snR50, snR55, snR59, snR60, snR67 and snR69) that accumulate on pre-ribosomes when Dbp3 is lacking guide Dbp3-dependent 2′-O-methylations, implying that alterations in snoRNA levels on pre-ribosomes may indeed underlie the defects in rRNA 2′-O-methylation observed in Δdbp3. These results do not, however, support the initial hypothesis that lack of Dbp3 affects rRNA 2′-O-methylation by directly impairing access of the snoRNAs guiding these modifications to pre-ribosomes as the amount of these snoRNPs on pre-ribosomes was increased rather than reduced.
Figure 5.

A subset of box C/D snoRNAs accumulate on pre-ribosomes when Dbp3 is lacking. Extracts were separated by sucrose density gradient centrifugation as in (Figure 4G). Fractions containing either (pre-)ribosomal complexes or non-ribosome-associated snoRNPs were pooled and RNA was extracted. Polyadenylation and reverse transcription were performed and the levels of each of the 75 yeast snoRNAs in each sample was determined by RT-qPCR. The relative distribution of each box C/D snoRNA between (pre-)ribosome-bound and non-ribosome-associated fractions was calculated and differences in this ratio between the wild type and Δdbp3 strains are shown graphically. Three independent experiments were performed and the data are presented as mean ± standard deviation. Dashed red lines indicate the upper and low thresholds for significant accumulation and exclusion of snoRNAs on/from pre-ribosomes respectively.
Alterations in snoRNA dynamics on pre-ribosomes when Dbp3 is lacking affect rRNA 2′-O-methylation at specific sites
The extensive interactions snoRNAs form with their rRNA substrates involve both the snoRNA guide sequence as well as extra basepairing regions that stabilise snoRNA–pre-rRNA interactions to promote rRNA modification (11,54). Mapping of the snoRNA–rRNA basepairing of the Dbp3-dependent snoRNAs onto the secondary structure of the 25S rRNA sequence (55) revealed that many of these snoRNAs have partially overlapping basepairing sites (Figure 6A-C and Supplementary Figure S6). Pre-rRNA basepairing by these snoRNAs will therefore be mutually exclusive and failure to release one snoRNP after modification would be expected to impede access of the snoRNP with an overlapping basepairing site, leading to reduced 2′-O-methylation of the adjacent site. Assuming stochastic recruitment of such snoRNAs to pre-ribosomes, this would result in both snoRNA accumulation on pre-ribosomes and substoichoimetric rRNA 2′-O-methlyation. Notably, the U18 snoRNP is proposed to exist in two alternative conformations to target the adjacent sites (34) (Figure 6C) and if so, failure to release the snoRNP from one target site would impede modification of the other site by preventing access of the snoRNP in its alternative conformation. It is possible, therefore, that the model of snoRNA retention impairing modification at proximal sites is not only applicable for modifications installed by different snoRNPs, but that competition could also occur between differently assembled forms of a single snoRNP.
Figure 6.

Many snoRNAs that accumulate on pre-ribosomes in the absence of Dbp3 and guide Dbp3-dependent rRNA 2′-O-methylations have overlapping pre-rRNA basepairing sites. (A–E) 2′-O-methylations reduced in Δdbp3 and the snoRNAs that guide them are highlighted on the secondary structure of the 25S rRNA (55) in red and the pre-rRNA nucleotides involved in basepairing interactions with the guiding snoRNAs are indicated in green. The basepairing of relevant snoRNAs guiding non-Dbp3-dependent rRNA 2′-O-methylations is indicated by thick black lines. Extra basepairing of snoRNAs guiding Dbp3-dependent 2′-O-methylations is shown in blue and that of non-Dbp3 dependent snoRNAs is in grey. Magnified views of selected areas of interest from domain I (A), domain V (B), domain 0 (C), domain IV (D) and domain V (E) are shown. (F) Whole cell extracts prepared from wild type yeast were separated by sucrose density gradient centrifugation. RNA from individual fractions was separated by denaturing PAGE and analysed by northern blotting using probes to the snoRNAs indicated to the right. The fractions containing ‘free’ snoRNAs and pre-ribosome-associated snoRNAs are indicated.
Retention of a snoRNP impairing modification at a proximal site can explain many of the effects on pre-ribosomal snoRNA levels and rRNA 2′-O-methylation observed by RT-qPCR and RMS, but several of the Dbp3-dependent modifications are introduced by snoRNPs that do not have overlapping basepairing sites on the pre-rRNA and/or do not accumulate on pre-ribosomes in the absence of Dbp3. Therefore, other mechanisms likely also contribute to regulation of rRNA modification by Dbp3. For example, the pre-rRNA basepairing sites of snR76 and snR67 do not overlap with those of any other snoRNA affected by Δdbp3 (Figure 6D and E), and the snR40, snR72, snR73, snR76 and snR78 snoRNPs guide Dbp3-dependent rRNA 2′-O-methylations but do not accumulate on pre-ribosomes in the absence of Dbp3 (Figures 3B and 5). Pre-rRNA modification by snoRNPs is a dynamic process in which snoRNPs are transiently recruited to pre-ribosomes but after modification, they are released and recycled to other pre-ribosomal particles to install further modifications. It is therefore possible that retention of snoRNPs on particular pre-ribosomal particles when Dbp3 is lacking reduces the amount of ‘free’ snoRNP available for (re-)recruitment to other pre-ribosomes thus leading to impaired rRNA 2′-O-methylation. To explore this possibility further, we examined the proportions of snoRNAs guiding Dbp3-dependent rRNA modifications that are pre-ribosome-associated in wild type cells using sucrose density gradient separation followed by northern blotting. This revealed that some of these snoRNAs (e.g. U18, snR39, snR39b, snR50, snR59, snR69 and snR78) are present in both pre-ribosome-associated and non-ribosome-associated pools, while others (e.g. U24, snR40, snR60, snR67, snR72, snR73 and snR76) are almost exclusively present on pre-ribosomes (Figure 6F). On the one hand, this result may explain why not all snoRNAs guiding Dbp3-dependent modifications are observed to accumulate on pre-ribosomes in Δdbp3, as the absence of a free pool of snoRNA even in wild type cells may prevent significant increases in the amount of pre-ribosomal snoRNA when Dbp3 is lacking. On the other hand, these data support the model that inefficient recycling of snoRNAs that are largely pre-ribosome-associated could reduce the amount of available snoRNP below the critical threshold required for stoichiometric rRNA modification of nascent particles. In the case of snoRNPs such as U24, snR50, snR72, snR40 and snR60 that have both overlapping basepairing sites on the pre-rRNA and are almost exclusively pre-ribosome-associated, it is likely that both impaired snoRNP access caused by retention of a snoRNP targeting a proximal site as well as limiting amounts of available snoRNP due to compromised recycling contribute to the reduced rRNA 2′-O-methylation observed in Δdbp3.
Overexpression of a snoRNA rescues the methylation defect caused by lack of Dbp3
To explore how changes in the available amount of a snoRNA that accumulates on pre-ribosomes in the absence of Dbp3 influence rRNA 2′-O-methylation, we focused on snR67. This snoRNP installs two modifications within the 25S rRNA sequence that are distant on the linear rRNA sequence (snR67 – 25S-Gm2619 and 25S-Um2724). The basepairing snR67 forms to guide 25S-Gm2619 is spatially distinct from the basepairing regions of any other snoRNA and the basepairing snR67 forms to introduce the 25S-Um2724 modification only weakly overlaps with that of snR51, a snoRNA unaffected by Dbp3. Notably, snR67 is largely pre-ribosome-associated in wild type cells and is almost exclusively associated with these complexes in the absence of Dbp3 (Figure 6F and Supplementary Figure S7A). This suggests that alterations in the dynamics of the available snR67 between its two modification sites would likely affect the extent of 2′-O-methylation of these positions. A plasmid for the overexpression of snR67 from within the intron of the actin pre-mRNA, under the control of a pGAL1 promoter was generated and used to produce wild type and Δdbp3 yeast strains capable of overexpressing snR67 (Supplementary Figure S7B) (34). RNase H-based cleavage assays were then performed to monitor the extent of 2′-O-methylation of 25S-Gm2619 and 25S-Um2724. The 25S rRNA remained intact in the absence of RNase H or when assays were performed on total RNA derived from wild type cells carrying an empty plasmid or overexpressing snR67 (Figure 7A and B). As previously, the 25S rRNA derived from cells lacking Dbp3 with normal snR67 levels was cleaved by RNase H indicating reduced 2′-O-methylation of 25S-Gm2619 and 25S-Um2724 (Figure 7A and B). Upon overexpression of snR67, however, the rRNA modification defects caused by lack of Dbp3 were rescued (Figure 7A and B), suggesting that failure to release snR67 from one of its pre-rRNA basepairing sites in the absence of Dbp3 limits the amount of snoRNP available to basepair with and modify its other target site.
Figure 7.
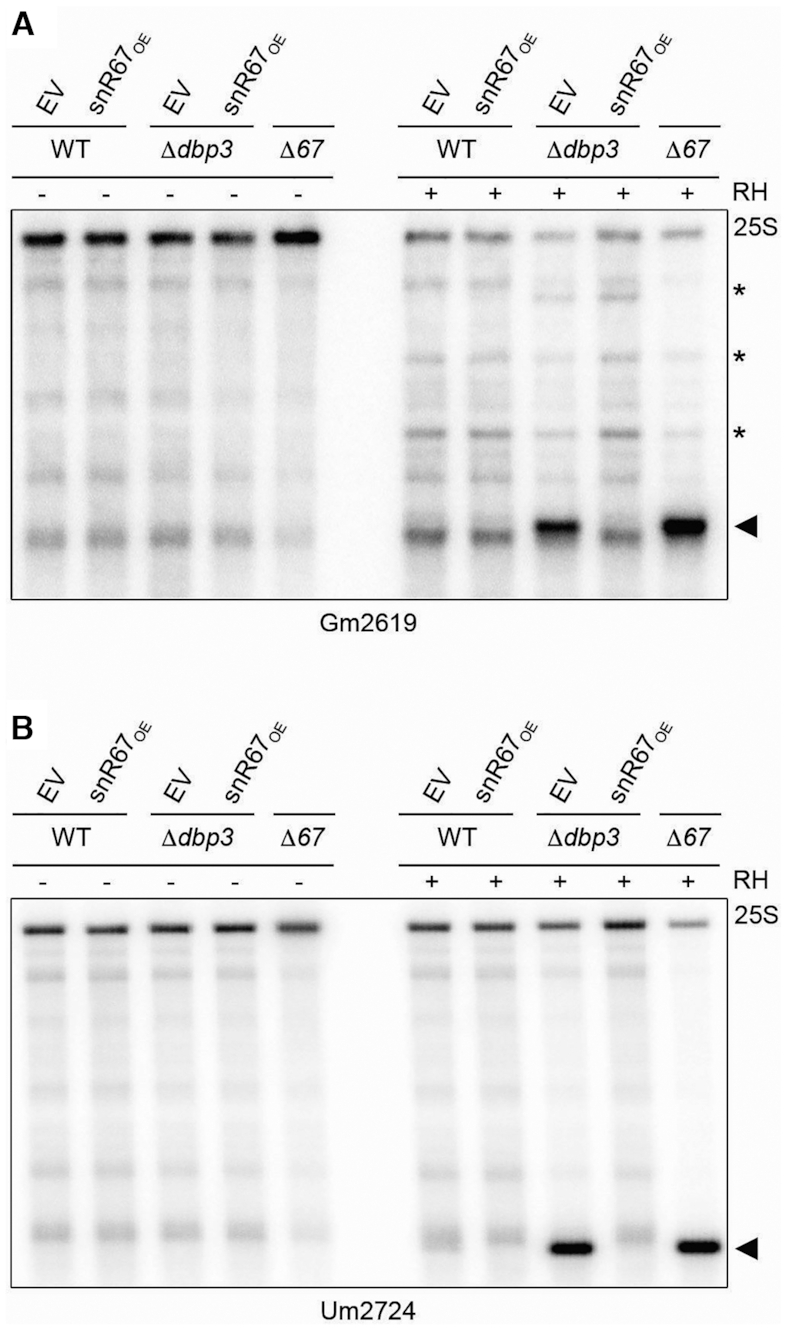
Overexpression of snR67 rescues 2′-O-methylation of Gm2619 and Um2724 when Dbp3 is lacking. (A, B) RNA from wild type yeast or strains lacking Dbp3 transformed with an empty pRS416 vector (EV) or a plasmid for the overexpression of snR67 from a pGAL1 promoter (snR67OE), or a strain in which SNR67 was deleted (Δ67) was subjected to RNase H (RH)-mediated cleavage targeting 25S-Gm2619 (A) or 25S-Um2724 (B). Full length 25S rRNA and cleavage products (indicated by arrow head) were detected by northern blotting using probes hybridising downstream of the cleavage sites. Asterisks indicate non-specific cleavage products – note that the extent of non-specific RNase H-mediated cleavage is reduced upon cleavage at a specific cleavage site.
The DEAH box RNA helicase Prp43 likely directly unwinds the pre-rRNA basepairing of a subset of snoRNAs guiding Dbp3-dependent rRNA 2′-O-methylations
Intriguingly, several of the snoRNAs that are retained on pre-ribosomes when Dbp3 is absent have previously been shown to accumulate on pre-ribosomes when the RNA helicase Prp43 is lacking (snR59, snr50, snR39, snR39b and U18) (28), suggesting that these helicases both contribute to facilitating release of a subset of snoRNAs from pre-ribosomes. We therefore investigated whether, similar to Dbp3, lack of Prp43 causes reduced 2′-O-methylation of sites proximal to those guided by the retained snoRNAs. A yeast strain was constructed in which expression of Prp43 is under the control of the galactose-inducible/glucose-repressible pGALS promoter. Importantly, in the presence of galactose, Prp43 is expressed close to its endogenous level and growth in glucose-containing media leads to efficient depletion of the protein after 6–8 h (Figure 8A and B). RMS analysis of the wild type and pGALS-HA-Prp43 strains grown in glucose-containing media revealed extensive effects on rRNA 2′-O-methylation with the RMS scores for 8 of the 18 sites in the 18S rRNA and 25 of the 37 sites in the 25S rRNA reduced >10% upon depletion of Prp43 (Figure 8C and D). Analysis of snoRNA levels in these datasets showed that with the exception of snR128 and snR64, which were up-regulated and guide non-Dbp3-dependent rRNA 2′-O-methylations, snoRNA levels are not significantly affected by lack of Prp43 (Supplementary Figure 8A). Furthermore, depletion of Prp43 was observed to significantly reduce mature 18S rRNA but not 25S rRNA levels (Supplementary Figure S8B). Notably, eleven 2′-O-methylations within the 25S rRNA affected by depletion of Prp43, were also reduced in the absence of Dbp3, further supporting the model linking snoRNA retention to impaired rRNA methylation.
Figure 8.

Prp43 is required for efficient rRNA 2′-O-methylation and crosslinks to snoRNA–pre-rRNA duplexes. (A) Proteins from wild type yeast (WT) and the pGALS-HA-Prp43 strain grown in galactose-containing media were separated by SDS-PAGE. Western blotting was performed using antibodies against Prp43 and, as a loading control, Pgk1. (B) The pGALS-HA-Prp43 strain was grown in exponential phase in media containing galactose (Gal) or glucose (Glu) for 12 h. Cells were harvested at the indicated time points, and proteins were extracted, separated by SDS-PAGE and analysed by western blotting using antibodies against the HA tag and Pgk1. (C, D) RiboMeth-seq analysis was performed on RNA derived from wild type yeast and the pGALS-HA-Prp43 strain grown exponentially in media containing glucose for 8 h. RiboMeth-seq scores, indicating the fraction of methylation, are plotted for each 2′-O-methylated nucleotide in the 18S rRNA (C) and the 25S rRNA (D). The data shown are the mean of two biological replicates and error bars represent standard deviation. The snoRNAs that guide each modification are indicated in blue and modifications for which the RMS score is decreased by >10% when Prp43 is depleted are highlighted in red. (E) The positions of predicted snoRNA basepairing sites (light blue) and chimeric reads identified in the Prp43 (upper) and WT (lower) CRAC datasets (26) by Hyb (dark blue) are mapped according to their positions on the 25S rRNA sequence. Sites of overlap between snoRNA basepairing sites and CLASH hybrids are shown in red. The profile of Prp43 crosslinking on the 25S rRNA sequence (28) is shown in grey with the peak heights given on the right.
The finding that lack of either Dbp3 or Prp43 individually leads to snoRNA retention on pre-ribosomes and impaired rRNA methylation implies that these proteins do not act redundantly, but rather suggests that they regulate snoRNA dynamics on pre-ribosomes in different ways. Transcriptome-wide identification of the RNA binding sites of Prp43 showed that this helicase crosslinks to many of the snoRNAs that accumulate on pre-ribosomes when the Prp43 or Dbp3 are lacking, as well as to their pre-rRNA target sites (28), suggesting that Prp43 may play a direct role in releasing these snoRNAs by resolving their basepairing with the pre-rRNA. This model necessitates that Prp43 simultaneously contacts both the snoRNA and pre-rRNA sequences involved in the basepairing interaction. To determine if this is the case, further bioinformatic analysis of the Prp43 CRAC dataset using the Hyb bioinformatic pipeline (43) was performed to identify chimeric sequencing reads generated by the ligation of physically proximal sequences during the CRAC procedure. This revealed numerous such chimeric reads containing the sequences of Dbp3/Prp43-dependent snoRNAs and their pre-rRNA target sites within the Prp43 CRAC dataset (Figure 8E). These findings strongly suggest that Prp43 binds to these RNA sequences when they are basepaired, thereby supporting a direct role for the helicase in unwinding of these snoRNA–pre-rRNA duplexes.
DISCUSSION
Here, we demonstrate the requirement of Dbp3 catalytic activity for efficient processing of the 27SA pre-rRNA and uncover a role for the helicase in regulating rRNA modification. Our data show that approximately half of the 2′-O-methylations in the 25S rRNA are introduced substoichiometrically in the absence of Dbp3. The extensive reduction in rRNA 2′-O-methylation observed in the absence of Dbp3 coupled with the fact that lack of Dbp3 does not affect cell growth or production of the mature 25S and 18S rRNA makes the Δdbp3 strain a potential tool with which to study the effect of rRNA hypomethylation on translation efficiency and fidelity.
A subset of box C/D snoRNAs, many of which guide Dbp3-dependent rRNA 2′-O-methylations, accumulate on pre-ribosomes when the helicase is lacking. Close inspection of the interactions snoRNAs guiding Dbp3-dependent modifications make with the pre-rRNA and analysis of the distribution of these snoRNAs between pre-ribosome-associated and non-ribosomal pools revealed that most of these snoRNAs either have overlapping basepairing sites on the pre-rRNA and/or are predominantly pre-ribosome-associated (Table 1). We therefore propose that Dbp3 facilitates the release of a subset of snoRNAs from pre-ribosomes and that retention of these snoRNAs in the absence of Dbp3 can impede the access of other snoRNPs to proximal sites and/or impair snoRNP recycling such that snoRNAs become limiting for rRNA modification. Interestingly, snR69 and snR78, which guide Dbp3-dependent 25S rRNA 2′-O-methylations have overlapping basepairing sites with other box C/D snoRNAs (snR71 and snR66 respectively). However, the 2′-O-methylations guided by the overlapping snoRNAs are not affected by lack of Dbp3. While it is possible that the effects on 2′-O-methylation of 25S-Um2421 and 25S-Cm2948 arise due to alternative mechanisms, it is also possible that the differential effects on rRNA 2′-O-methylation reflect differences in the affinities of the snoRNAs for their pre-rRNA basepairing sites. snR69 and snR71 both form canonical and extra-basepairing interactions with the 25S rRNA, which overlap (Figure 6B and Supplementary Figure S6). snR71 forms more extensive rRNA basepairing interactions than snR69 and has a unique interaction site, raising the possibility that snR71 outcompetes snR69 from pre-ribosome interaction leading to stoichiometric 2′-O-methylation of 25S-Am2946 (snR71) and substoichiometric methylation of 25S-Cm2948 (snR69). Alternatively, the rRNA remodelling event induced by Dbp3 may favour snR71 basepairing or methylation by other means or render release of this snoRNA, but not snR69, dependent on Dbp3. A similar phenomenon is observed in the case of snR55, which guides the only Dbp3-dependent 2′-O-methylation in the 18S rRNA. Notably, the 18S rRNA basepairing site of snR55 overlaps with that of snR40, a snoRNA that guides methylations in both the 18S and 25S rRNAs. The 25S rRNA 2′-O-methylation guided by snR40 is affected by lack of Dbp3 and it is possible therefore that the effect of lack of Dbp3 on 18S-Um1269 reflects complex inter-subunit interactions of snR40. As these modifications take place co-transcriptionally in the context of 90S pre-ribosomal particles such effects are conceivable, and it is important to note that the analysis of snoRNA levels on pre-ribosomes does not differentiate the amount of snoRNA present on pre-40S and pre-60S complexes. Given the highly dynamic and interconnected nature of early ribosome assembly events as well as the potential for long-range consequences of individual remodelling steps, dissecting the molecular mechanisms underlying all observations remains challenging.
Table 1.
Overview of rRNA 2′-O-methylations and box C/D snoRNAs affected by lack of Dbp3
| rRNA 2′-O-methylation | Guiding snoRNA | Overlapping box C/D snoRNA | snoRNA accumulation on pre-ribosomes in Δdbp3 vs WT | Non-ribosome-associated snoRNA | Comment |
|---|---|---|---|---|---|
| 18S-Um1269 | snR55 | snR40 | + | + | Overlapping snoRNA basepairing but proximal modification not affected |
| 25S-Am649 | U18 | U18 | + | +++ | Overlapping snoRNA basepairing |
| 25S-Cm650 | U18 | U18 | + | +++ | Overlapping snoRNA basepairing |
| 25S-Gm805 | snR39b | snR39/snR59 | + | ++ | Overlapping snoRNA basepairing |
| 25S-Am807 | snR39/snR59 | snR39b/snR60 | +/+ | ++/+ | Overlapping snoRNA basepairing |
| 25S-Am817 | snR60 | snR39/snR59 | + | - | Overlapping snoRNA basepairing & limiting amount of snoRNA available |
| 25S-Gm867 | snR50 | snR72 | + | + | Overlapping snoRNA basepairing |
| 25S-Am876 | snR72 | snR50 | - | + | Overlapping snoRNA basepairing |
| 25S-Um898 | snR40 | snR60 | - | - | Overlapping snoRNA basepairing |
| 25S-Gm908 | snR60 | snR40 | + | - | Overlapping snoRNA basepairing & limiting amount of snoRNA available |
| 25S-Cm1437 | U24 | U24 | + | - | Overlapping snoRNA basepairing |
| 25S-Am1449 | U24 | U24 | + | - | Overlapping snoRNA basepairing |
| 25S-Gm1450 | U24 | U24 | + | - | Overlapping snoRNA basepairing |
| 25S-Cm2197 | snR76 | - | - | - | Limiting amount of snoRNA available |
| 25S-Um2421 | snR78 | snR66 | - | + | Overlapping snoRNA basepairing but proximal modification not affected |
| 25S-Gm2619 | snR67 | - | + | - | Limiting amount of snoRNA available |
| 25S-Um2724 | snR67 | snR51 | + | - | Limiting amount of snoRNA available |
| 25S-Cm2948 | snR69 | snR71 | + | +++ | Overlapping snoRNA basepairing but proximal modification not affected |
| 25S-Cm2959 | snR73 | - | - | - | Limiting amount of snoRNA available |
Sites of reduced rRNA 2′-O-methylation in Ddbp3 compared to wild type are listed alongside the snoRNAs guiding each modification. snoRNAs that have overlapping basepairing sites with those guiding the target modification are listed. Accumulation (+) or lack of accumulation (–) of the guiding snoRNA on pre-ribosomes in Ddbp3 compared to wild type is indicated in the fourth column. The amount of non-ribosome-associated guiding snoRNA is given (– = none/very little, +,++,+++ = low, medium, high amounts). Comment reflects the proposed mechanism for the reduction in rRNA 2′-O-methylation observed in Ddbp3 compared to wild type alongside any additional relevant information.
A role for Dbp3 in the biogenesis of the intron-encoded snoRNAs U18 and U24 was also uncovered. However, the findings that pre-U18 and pre-U24 associated with Nop1 and pre-ribosomes, together with partial modification of the U18 and U24 target sites in the absence of Dbp3, implies that this additional function of Dbp3 is not the sole basis of the methylation defects observed at these positions. The 3′-5′ exonuclease Rrp6 is suggested to process the final 1–3 nucleotides of snoRNAs and our northern blot analyses suggest a similar length extension of U18 and U24 when Dbp3 is lacking. Analysis of RMS sequencing reads generated by alkaline hydrolysis of total RNA suggests also the presence of longer 3′ extended forms of the U18 snoRNA. It is intriguing that extended forms of the U18 and U24 snoRNAs accumulate in the absence of Dbp3, while processing of the other intron-encoded snoRNAs seems unaffected. This suggests that the biogenesis pathway of the intron-encoded snoRNAs may differ. Mechanistically, it remains unclear what role Dbp3 plays in facilitating 3′ processing of the U18 and U24 snoRNAs. 3′ extended forms of U18 and U24 are also observed when the G-patch domain of Gno1, which mediates interactions with Prp43 that regulate the activity of the helicase, is deleted (56,57). Together with the observation that the catalytic activity of Dbp3 contributes to U18 and U24 maturation, this suggests that helicase action, perhaps to resolve pre-snoRNA secondary structures, may be involved in efficient 3′ end processing. Notably, both Dbp3 and Gno1 contain KK(E/D) motifs, which are also present in the core box C/D snoRNP proteins Nop56 and Nop58. The finding that like Gno1, lack of the KK(E/D) motif of Dbp3 impairs processing of U18 and U24 further suggests that an interplay between factors containing this motif, possibly together with an unknown protein binding the KK(E/D) motif, may underlie the observed processing defects.
Interestingly, many of the effects of DBP3 deletion on snoRNA levels on pre-ribosomes, especially those that guide 2′-O-methylations within domain I, are also observed when the RNA helicase Prp43 is depleted. Similarly, although depletion of Prp43 has a broader effect on rRNA 2′-O-methylation than lack of Dbp3, perhaps due to its functions in pre-mRNA splicing as well as ribosomes biogenesis, reduced levels of either of these helicases impacts a largely overlapping set of modifications. It is possible that these common phenotypes occur due to the action of Prp43 and Dbp3 on pre-LSU complexes at the same stage of maturation and reflect general effects caused by impairing the assembly pathway at this point. However, these effects on pre-ribosomal snoRNA levels are not observed upon depletion of other LSU biogenesis factors whose depletion causes pre-rRNA processing defects similar to lack of Dbp3 or Prp43, e.g. Mak5 (27). The data therefore rather suggest that Dbp3 and Prp43 both contribute to regulating the dynamics of a subset of snoRNAs on pre-ribosomes, thereby influencing rRNA 2′-O-methylation. Dbp3 and Prp43 belong to the DEAD and DEAH box families of RNA helicases respectively. Mechanistically, DEAH box helicases, including Prp43, act in a processive manner and are able to translocate along RNA strands (58) making them highly suitable enzymes for directly unwinding multiple snoRNA-pre-RNA duplexes. Indeed, mining available Prp43 CRAC datasets (28) indicated the crosslinking of Prp43 to snoRNAs when they are basepaired to their pre-rRNA target sites, supporting a direct role for Prp43 in resolving these duplexes. In contrast to DEAH box helicases, DEAD box proteins generally remodel their substrates by inducing local strand unwinding (59). While several DEAD box helicases are implicated in snoRNA release from pre-ribosomes, in these cases, each helicase is only linked to an individual snoRNA, e.g. Has1 and U14, and Rok1 and snR30 (26,29,32). Consistent with action by local strand unwinding, Dbp3 has been shown to be capable of unwinding an RNA duplex of 10 nucleotides (nt) in vitro (45). Although this duplex is similar in length to the basepairing interactions box C/D snoRNAs form with their pre-rRNA targets (10–19 nt, in the case of Dbp3-dependent snoRNAs), a direct role of Dbp3 in releasing the 13 snoRNAs that accumulate on pre-ribosomes in its absence would necessitate numerous and extensive conformational changes of Dbp3 on the pre-ribosomes to enable its catalytic site to directly contact each of the snoRNA–pre-rRNA duplexes. Even within the highly dynamic process of ribosome assembly, this extent of re-positioning or dissociation/re-association of a single protein is improbable, implying that, in contrast to Prp43, Dbp3 likely affects snoRNA dynamics on pre-ribosomes indirectly. We anticipate that rearrangement of a particular pre-rRNA region by Dbp3 induces long-range effects on pre-ribosome structure that impede the release of specific snoRNPs that basepair with spatially distinct pre-rRNA sequences. It is important to note that the early pre-LSU particles bound by Dbp3 are highly dynamic structures and therefore lack of this helicase could influence the dynamics of any of the numerous pre-ribosome maturation events that are occurring simultaneously on such particles. Accordingly, it is likely that the impaired processing of the 27SA pre-rRNA observed upon deletion of DBP3 is a downstream consequence of failure of release of specific snoRNPs during preceding pre-LSU maturation steps. Despite extensive efforts to identify the pre-rRNA binding site of Dbp3 using the UV crosslinking and analysis of cDNA (CRAC (60)) approach and its derivatives, this has so far not been possible. On the one hand, this may be due to the insensitivity of the interacting Dbp3 amino acids and pre-RNA sequences to crosslinking but, on the other hand, it may reflect a very transient interaction between Dbp3 and the pre-ribosome. Structural analysis of pre-ribosomal particles containing Dbp3 that allow visualisation of the helicase active site on its target RNA sequence(s) will therefore likely be required to gain further mechanistic insight into the precise pre-ribosome remodelling event(s) regulated by Dbp3.
Analyses of the timing of rRNA 2′-O-methylation demonstrate that the majority of these modifications are introduced co-transcriptionally (7,8). As these modifications cluster within the rRNA sequences that form functionally important regions of the pre-ribosome and box C/D snoRNAs form extensive basepairing with their pre-rRNA substrates, it is intuitive that snoRNPs mediating proximal modifications must form mutually exclusive interactions with pre-ribosomal particles. However, it has remained unclear whether such ‘overlapping’ snoRNPs act hierarchically or stochastically or whether examples of both exist. Our detection of correlated mild accumulation on pre-ribosomes and fractional rRNA 2′-O-methylation by such snoRNA/Ps suggest that most do not normally act in a strictly defined order. A growing body of evidence supports the step-wise assembly and compaction of different pre-ribosomal regions in eukaryotes (4), analogous to the hierarchical model of bacterial ribosome assembly. While the stochastic pre-ribosome-association of snoRNAs with overlapping basepairing sites in the same domain is accommodated within this framework, it is likely that snoRNPs are broadly recruited to pre-ribosomes in the order in which their target sites are transcribed and that snoRNAs guiding modifications in different domains of the ribosome that compact at different stages instead act sequentially.
DATA AVAILABILITY
GEO database: identifier GSE155720.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Anne-Caren Zeiss and Nidhi Sharma for molecular cloning and preparation of yeast strains, and Dr Grzegorz Kudla for help with CLASH analysis.
Contributor Information
Gerald Ryan R Aquino, Department of Molecular Biology, University Medical Centre Göttingen, Humboldtallee 23, 37073 Göttingen, Germany.
Nicolai Krogh, Department of Cellular and Molecular Medicine, University of Copenhagen, 3B Blegdamsvej, 2200N Copenhagen, Denmark.
Philipp Hackert, Department of Molecular Biology, University Medical Centre Göttingen, Humboldtallee 23, 37073 Göttingen, Germany.
Roman Martin, Department of Molecular Biology, University Medical Centre Göttingen, Humboldtallee 23, 37073 Göttingen, Germany.
Jimena Davila Gallesio, Department of Molecular Biology, University Medical Centre Göttingen, Humboldtallee 23, 37073 Göttingen, Germany.
Robert W van Nues, Biosciences Institute, Newcastle University, Framlington Place, Newcastle upon Tyne NE2 4HH, United Kingdom.
Claudia Schneider, Biosciences Institute, Newcastle University, Framlington Place, Newcastle upon Tyne NE2 4HH, United Kingdom.
Nicholas J Watkins, Biosciences Institute, Newcastle University, Framlington Place, Newcastle upon Tyne NE2 4HH, United Kingdom.
Henrik Nielsen, Department of Cellular and Molecular Medicine, University of Copenhagen, 3B Blegdamsvej, 2200N Copenhagen, Denmark; Genomics group, Faculty of Biosciences and Aquaculture, Nord University, 8049, Bodø, Norway.
Katherine E Bohnsack, Department of Molecular Biology, University Medical Centre Göttingen, Humboldtallee 23, 37073 Göttingen, Germany.
Markus T Bohnsack, Department of Molecular Biology, University Medical Centre Göttingen, Humboldtallee 23, 37073 Göttingen, Germany; Göttingen Center for Molecular Biosciences, Georg-August University Göttingen, Justus-von-Liebig-Weg 11, 37077 Göttingen, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [SFB860 to M.T.B. and K.E.B., and BO3442/2-2 (SPP1784) to M.T.B.]; University Medical Centre Göttingen [to M.T.B. and K.E.B.]; Biotechnology and Biological Sciences Research Council/Medical Research Council (UK) [BB/R00143X/1 to N.J.W. and C.S.]; Wellcome Trust [092076 to N.J.W.]; Royal Society (UK) [UF150691 to C.S.]; EU COST Action CA16120 [STSM41493 to G.R.R.A. and M.T.B.]; Danish Research Council for Independent Research [DFF-4183-00486 to H.N.]; Lundbeck Foundation [R198-2015-174 to H.N.]; Danish Cancer Society [R167-A10943-17-S2 to N.K.]. Funding for open access charge: University Medical Center Goettingen.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ben-Shem A., Garreau de Loubresse N., Melnikov S., Jenner L., Yusupova G., Yusupov M.. SOM: the structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011; 334:1524–1529. [DOI] [PubMed] [Google Scholar]
- 2. Warner J.R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 1999; 24:437–440. [DOI] [PubMed] [Google Scholar]
- 3. Anger A.M., Armache J.-P., Berninghausen O., Habeck M., Subklewe M., Wilson D.N., Beckmann R.. Structures of the human and Drosophila 80S ribosome. Nature. 2013; 497:80–85. [DOI] [PubMed] [Google Scholar]
- 4. Klinge S., Woolford J.L.J.. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 2019; 20:116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bohnsack K.E., Bohnsack M.T.. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 2019; 38:e100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henras A.K., Plisson-Chastang C., O’Donohue M.-F., Chakraborty A., Gleizes P.-E.. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA. 2015; 6:225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koš M., Tollervey D.. Yeast Pre-rRNA processing and modification occur cotranscriptionally. Mol. Cell. 2010; 37:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Birkedal U., Christensen-Dalsgaard M., Krogh N., Sabarinathan R., Gorodkin J., Nielsen H.. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chemie - Int. Ed. 2015; 54:451–455. [DOI] [PubMed] [Google Scholar]
- 9. Watkins N.J., Bohnsack M.T.. The box C/D and H/ACA snoRNPs: Key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip. Rev. RNA. 2012; 3:397–414. [DOI] [PubMed] [Google Scholar]
- 10. Sloan K.E., Warda A.S., Sharma S., Entian K.-D., Lafontaine D.L.J., Bohnsack M.T.. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017; 14:1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Nues R.W., Granneman S., Kudla G., Sloan K.E., Chicken M., Tollervey D., Watkins N.J.. Box C/D snoRNP catalysed methylation is aided by additional pre-rRNA base-pairing. EMBO J. 2011; 30:2420–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Decatur W.A., Fournier M.J.. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002; 27:344–351. [DOI] [PubMed] [Google Scholar]
- 13. Sharma S., Lafontaine D.L.J.. View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem. Sci. 2015; 40:560–575. [DOI] [PubMed] [Google Scholar]
- 14. Bachellerie J.P., Cavaillé J.. Guiding ribose methylation of rRNA. Trends Biochem. Sci. 1997; 22:257–261. [DOI] [PubMed] [Google Scholar]
- 15. Krogh N., Jansson M.D., Hafner S.J., Tehler D., Birkedal U., Christensen-Dalsgaard M., Lund A.H., Nielsen H.. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic. Acids. Res. 2016; 44:7884–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marchand V., Blanloeil-Oillo F., Helm M., Motorin Y.. Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucleic. Acids. Res. 2016; 44:e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hebras J., Krogh N., Marty V., Nielsen H., Cavaille J.. Developmental changes of rRNA ribose methylations in the mouse. RNA Biol. 2020; 17:150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barandun J., Chaker-Margot M., Hunziker M., Molloy K.R., Chait B.T., Klinge S.. The complete structure of the small-subunit processome. Nat. Struct. Mol. Biol. 2017; 24:944–953. [DOI] [PubMed] [Google Scholar]
- 19. Sloan K.E., Gleizes P.-E., Bohnsack M.T.. Nucleocytoplasmic transport of RNAs and RNA-protein complexes. J. Mol. Biol. 2016; 428:2040–2059. [DOI] [PubMed] [Google Scholar]
- 20. Gerhardy S., Menet A.M., Pena C., Petkowski J.J., Panse V.G.. Assembly and nuclear export of pre-ribosomal particles in budding yeast. Chromosoma. 2014; 123:327–344. [DOI] [PubMed] [Google Scholar]
- 21. Kressler D., Hurt E., Bassler J.. Driving ribosome assembly. Biochim. Biophys. Acta. 2010; 1803:673–683. [DOI] [PubMed] [Google Scholar]
- 22. Prattes M., Lo Y.-H., Bergler H., Stanley R.E.. Shaping the nascent ribosome: AAA-ATPases in eukaryotic ribosome biogenesis. Biomolecules. 2019; 9:715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin R., Straub A.U., Doebele C., Bohnsack M.T.. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 2013; 10:4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sloan K.E., Bohnsack M.T.. Unravelling the mechanisms of RNA helicase regulation. Trends Biochem. Sci. 2018; 43:237–250. [DOI] [PubMed] [Google Scholar]
- 25. Dembowski J.A., Kuo B., Woolford J.L.. Has1 regulates consecutive maturation and processing steps for assembly of 60S ribosomal subunits. Nucleic. Acids. Res. 2013; 41:7889–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khoshnevis S., Askenasy I., Johnson M.C., Dattolo M.D., Young-Erdos C.L., Stroupe M.E., Karbstein K.. The DEAD-box protein Rok1 orchestrates 40S and 60S ribosome assembly by promoting the release of Rrp5 from Pre-40S ribosomes to allow for 60S maturation. PLoS Biol. 2016; 14:e1002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bruning L., Hackert P., Martin R., Davila Gallesio J., Aquino G.R.R., Urlaub H., Sloan K.E., Bohnsack M.T.. RNA helicases mediate structural transitions and compositional changes in pre-ribosomal complexes. Nat. Commun. 2018; 9:5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bohnsack M.T., Martin R., Granneman S., Ruprecht M., Schleiff E., Tollervey D.. Prp43 bound at different sites on the Pre-rRNA performs distinct functions in ribosome synthesis. Mol. Cell. 2009; 36:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leeds N.B., Small E.C., Hiley S.L., Hughes T.R., Staley J.P.. The splicing factor Prp43p, a DEAH box ATPase, functions in ribosome biogenesis. Mol. Cell. Biol. 2006; 26:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang X.-H., Fournier M.J.. The helicase Has1p is required for snoRNA release from pre-rRNA. Mol. Cell. Biol. 2006; 26:7437–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bohnsack M.T., Kos M., Tollervey D. Quantitative analysis of snoRNA association with pre-ribosomes and release of snR30 by Rok1 helicase. EMBO Rep. 2008; 9:1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin R., Hackert P., Ruprecht M., Simm S., Bruning L., Mirus O., Sloan K.E., Kudla G., Schleiff E., Bohnsack M.T.. A pre-ribosomal RNA interaction network involving snoRNAs and the Rok1 helicase. RNA. 2014; 20:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sardana R., Liu X., Granneman S., Zhu J., Gill M., Papoulas O., Marcotte E.M., Tollervey D., Correll C.C., Johnson A.W.. The DEAH-box helicase Dhr1 dissociates U3 from the pre-rRNA to promote formation of the central pseudoknot. PLoS Biol. 2015; 13:e1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Nues R.W., Watkins N.J.. Unusual C’/D’ motifs enable box C/D snoRNPs to modify multiple sites in the same rRNA target region. Nucleic Acids Res. 2016; 45:2016–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kiianitsa K., Solinger J.A., Heyer W.-D.. NADH-coupled microplate photometric assay for kinetic studies of ATP-hydrolyzing enzymes with low and high specific activities. Anal. Biochem. 2003; 321:266–271. [DOI] [PubMed] [Google Scholar]
- 36. Choudhury P., Hackert P., Memet I., Sloan K.E., Bohnsack M.T.. The human RNA helicase DHX37 is required for release of the U3 snoRNP from pre-ribosomal particles. RNA Biol. 2019; 16:54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Braun C.M., Hackert P., Schmid C.E., Bohnsack M.T., Bohnsack K.E., Perez-Fernandez J.. Pol5 is required for recycling of small subunit biogenesis factors and for formation of the peptide exit tunnel of the large ribosomal subunit. Nucleic Acids Res. 2019; 48:405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sloan K.E., Leisegang M.S., Doebele C., Ramírez A.S., Simm S., Safferthal C., Kretschmer J., Schorge T., Markoutsa S., Haag S.et al.. The association of late-acting snoRNPs with human pre-ribosomal complexes requires the RNA helicase DDX21. Nucleic Acids Res. 2015; 43:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Memet I., Doebele C., Sloan K.E., Bohnsack M.T.. The G-patch protein NF-kappaB-repressing factor mediates the recruitment of the exonuclease XRN2 and activation of the RNA helicase DHX15 in human ribosome biogenesis. Nucleic Acids. Res. 2017; 45:5359–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krogh N., Birkedal U., Nielsen H.. RiboMeth-seq: Profiling of 2′-O-Me in RNA. Methods Mol. Biol. 2017; 1562:189–209. [DOI] [PubMed] [Google Scholar]
- 41. Krogh N., Kongsbak-Wismann M., Geisler C., Nielsen H.. Substoichiometric ribose methylations in spliceosomal snRNAs. Org. Biomol. Chem. 2017; 15:8872–8876. [DOI] [PubMed] [Google Scholar]
- 42. Yu Y.T., Shu M.D., Steitz J.A.. A new method for detecting sites of 2′-O-methylation in RNA molecules. RNA. 1997; 3:324–331. [PMC free article] [PubMed] [Google Scholar]
- 43. Travis A.J., Moody J., Helwak A., Tollervey D., Kudla G.. Hyb: a bioinformatics pipeline for the analysis of CLASH (crosslinking, ligation and sequencing of hybrids) data. Methods. 2014; 65:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weaver P.L., Sun C., Chang T.H.. Dbp3p, a putative RNA helicase in Saccharomyces cerevisiae, is required for efficient pre-rRNA processing predominantly at site A3. Mol. Cell. Biol. 1997; 17:1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garcia I., Albring M.J., Uhlenbeck O.C.. Duplex destabilization by four ribosomal DEAD-box proteins. Biochemistry. 2012; 51:10109–10118. [DOI] [PubMed] [Google Scholar]
- 46. Liu B., Fournier M.J.. Interference probing of rRNA with snoRNPs: a novel approach for functional mapping of RNA in vivo. RNA. 2004; 10:1130–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Breslow D.K., Cameron D.M., Collins S.R., Schuldiner M., Stewart-Ornstein J., Newman H.W., Braun S., Madhani H.D., Krogan N.J., Weissman J.S.. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods. 2008; 5:711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lafontaine D.L., Tollervey D.. Synthesis and assembly of the box C+D small nucleolar RNPs. Mol. Cell. Biol. 2000; 20:2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu S., Tutuncuoglu B., Yan K., Brown H., Zhang Y., Tan D., Gamalinda M., Yuan Y., Li Z., Jakovljevic J.et al.. Diverse roles of assembly factors revealed by structures of late nuclear pre-60S ribosomes. Nature. 2016; 534:133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petfalski E., Dandekar T., Henry Y., Tollervey D.. Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol. Cell. Biol. 1998; 18:1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Giorgi C., Fatica A., Nagel R., Bozzoni I.. Release of U18 snoRNA from its host intron requires interaction of Nop1p with the Rnt1p endonuclease. EMBO J. 2001; 20:6856–6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Allmang C., Kufel J., Chanfreau G., Mitchell P., Petfalski E., Tollervey D.. Functions of the exosome in rRNA, snoRNA and snRNA synthesis. EMBO J. 1999; 18:5399–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ooi S.L., Samarsky D.A., Fournier M.J., Boeke J.D.. Intronic snoRNA biosynthesis in Saccharomyces cerevisiae depends on the lariat-debranching enzyme: intron length effects and activity of a precursor snoRNA. RNA. 1998; 4:1096–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kiss-László Z., Henry Y., Bachellerie J.P., Caizergues-Ferrer M., Kiss T.. Site-specific ribose methylation of preribosomal RNA: A novel function for small nucleolar RNAs. Cell. 1996; 85:1077–1088. [DOI] [PubMed] [Google Scholar]
- 55. Petrov A.S., Bernier C.R., Gulen B., Waterbury C.C., Hershkovits E., Hsiao C., Harvey S.C., Hud N.V, Fox G.E., Wartell R.M.et al.. Secondary structures of rRNAs from all three domains of life. PLoS One. 2014; 9:e88222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bohnsack K.E., Ficner R., Jonas S., Bohnsack M.T. Regulation of DEAH-box RNA helicases by G-patch proteins. Biol. Chem. doi:10.1515/hsz-2020-0338. [DOI] [PubMed] [Google Scholar]
- 57. Guglielmi B., Werner M.. The yeast homolog of human PinX1 is involved in rRNA and small nucleolar RNA maturation, not in telomere elongation inhibition. J. Biol. Chem. 2002; 277:35712–35719. [DOI] [PubMed] [Google Scholar]
- 58. Hamann F., Enders M., Ficner R.. Structural basis for RNA translocation by DEAH-box ATPases. Nucleic Acids Res. 2019; 47:4349–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ozgur S., Buchwald G., Falk S., Chakrabarti S., Prabu J.R., Conti E.. The conformational plasticity of eukaryotic RNA-dependent ATPases. FEBS J. 2015; 282:850–863. [DOI] [PubMed] [Google Scholar]
- 60. Bohnsack M.T., Tollervey D., Granneman S.. Identification of RNA Helicase Target Sites by UV Cross-linking and Analysis of cDNA. 2012; 1st edn.Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- 61. Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.Ret al.. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic. Acids. Res. 2012; 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leontis N.B., Westhof E.. Geometric nomenclature and classification of RNA base pairs. RNA. 2001; 7:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GEO database: identifier GSE155720.