


Abstract
The compact CRISPR/Cas9 system, which can be delivered with their gRNA and a full-length promoter for expression by a single adeno-associated virus (AAV), is a promising platform for therapeutic applications. We previously identified a compact SauriCas9 that displays high activity and requires a simple NNGG PAM, but the specificity is moderate. Here, we identified three compact Cas9 orthologs, Staphylococcus lugdunensis Cas9 (SlugCas9), Staphylococcus lutrae Cas9 (SlutrCas9) and Staphylococcus haemolyticus Cas9 (ShaCas9), for mammalian genome editing. Of these three Cas9 orthologs, SlugCas9 recognizes a simple NNGG PAM and displays comparable activity to SaCas9. Importantly, we generated a SlugCas9-SaCas9 chimeric nuclease, which has both high specificity and high activity. We finally engineered SlugCas9 with mutations to generate a high-fidelity variant that maintains high specificity without compromising on-target editing efficiency. Our study offers important minimal Cas9 tools that are ideal for both basic research and clinical applications.
INTRODUCTION
Compact CRISPR/Cas9 nucleases (<1100 aa) can be delivered with their gRNA and a full-length promoter for their expression by a single adeno-associated virus (AAV) (1–4) for in vivo genome editing, and thus hold great promise for gene therapy. The active complex is composed of a Cas9 nuclease and a guide RNA (gRNA), which together recognize a target DNA that is complementary to the 20 bp-protospacer sequence in the gRNA. Upon recognition, this complex generates a site-specific double-strand break (DSB) (5–9). Target site recognition also requires a specific protospacer adjacent motif (PAM) (5,10–12) unique to the Cas9 protein, which limits the targeting scope of Cas9. To expand targeting scope, several compact Cas9 nucleases have been developed for mammalian genome editing (2,7,13–15). We recently developed a compact SauriCas9 recognizing simple NNGG PAM, but the specificity is moderate (16).
The ideal compact Cas9 tools should maintain the properties of broadened PAM targeting range, high activity and high specificity. In this study, we identified three compact SaCas9 orthologs, Staphylococcus lugdunensis Cas9 (SlugCas9), Staphylococcus lutrae Cas9 (SlutrCas9) and Staphylococcus haemolyticus (ShaCas9), for mammalian genome editing. Initial SaCas9 proteins require a longer PAM than the commonly used SpCas9 for targeting (NNGRRT for SaCas9, NNNRRT for SaCas9-KKH, NGG for SpCas9) (2,5,17), which limits their widespread usage. The new SlugCas9 contains a compact NNGG PAM, analogous to the NGG PAM of SpCas9, and is ∼1 kp smaller in size. To reduce the propensity of off-target cleavage, we generated a SlugCas9-based chimeric Cas9 nuclease with high specificity and activity. Importantly, we also engineered a variant of SlugCas9 to be of high-fidelity, hereby referred to as SlugCas9-HF. This new Cas protein encompasses all desired properties such as recognizing an NNGG PAM, displays high specificity and maintains high activity.
MATERIALS AND METHODS
Cell culture and transfection
HEK293T, HeLa and Human foreskin fibroblast (HFF) cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS (Gibco). N2a cells were maintained in 45% DMEM and 45% Opti-MEM supplemented with 10% FBS (Gibco). HCT116 cells were maintained in McCoy’s 5A supplemented with 10% FBS (Gibco). A375 cells were maintained in RPMI-1640 supplemented with 10% FBS (Gibco). All the cells were supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin, and cultured at 37°C and 5% CO2. For SlugCas9, Sa-SlugCas9, ShaCas9 and SlutrCas9 PAM sequence screening, HEK293T cells were plated into 10 cm dishes and transfected at 50–60% confluency with Cas9-gRNA-expressing plasmid (15 μg) using 30 μl of Lipofectamine 2000 (Life Technologies) in Opti-MEM. For genome editing comparisons of SaCas9, SlugCas9, SlugCas9-HF, Sa-SlugCas9, ShaCas9 and SlutrCas9, cells were seeded on 48-well plates and transfected with Cas9 plasmid (300 ng) and gRNA plasmid (200 ng) using 1 μl of Lipofectamine 2000 (Life Technologies) in Opti-MEM according to the manufacturer’s instructions. For base editing capability of SlugCas9, HEK293T cells were seeded on 48-well plates and transfected with SlugABEmax/SlugBE4max plasmid (300 ng) and gRNA plasmid (200 ng) using 1 μl of Lipofectamine 2000 (Life Technologies) in Opti-MEM(Gibco), according to the manufacturer’s instructions.
Plasmid construction
Cas9 expression plasmid construction: the plasmid pX601 (addgene#61591) was amplified by primers px601-F/px601-R to remove SaCas9. The human codon-optimized Cas9 genes (Supplementary Table S1) were synthesized by HuaGene (Shanghai, China) and cloned into the pX601 backbone by NEBuilder assembly tool (NEB). The sequence of each Cas9 was confirmed by Sanger sequencing (GENEWIZ, Suzhou, China).
SlugBE4max and SlugABEmax plasmid construction: The primers ABEmax-F/ABEmax-R and AncBE4max-F/ AncBE4max-R were used to amplify pCMV_ABEmax_P2A_GFP (Addgene #112101) and pCMV_AncBE4max (Addgene #112094), respectively, to remove SpCas9n. SlugCas9n was amplified from SlugCas9 plasmid by primers ABE-SlugCas9(D10A)-F/ABE-SlugCas9(D10A)-R and BE4-SlugCas9(D10A)-F/BE4-SlugCas9 (D10A)-R, respectively. The PCR products were cloned into pCMV-ABEmax_P2A_GFP and pCMV_AncBE4max backbone to obtain SlugABEmax and SlugBE4max, respectively. All the primer sequences are listed in Supplementary Table S2.
gRNA expression plasmids construction: The plasmid hU6-Sa_tracr was digested with the restriction endonuclease BsaI. gRNAs were inserted into plasmid between two BsaI restriction sites, respectively. All target sequences are listed in Supplementary Table S3.
PAM sequence analysis
Twenty base-pair sequences (AAGCCTTGTTTGCCACCATG/ GTGAGCAAGGGCGAGGAGCT) flanking the target sequence (GAACGGCTCGGAGATCATCATTGCG NNNNNNN) were used to fix the target sequence. GCG and GTGAGCAAGGGCG AGGAGCT were used to fix 7-bp random sequence. Target sequences with in-frame mutations were used for PAM analysis. The 7-bp random sequence was extracted and visualized by WebLog3 (18) and PAM wheel chart to demonstrate PAMs (10).
Genome editing for endogenous sites
HEK293T cells were seeded into a 48-well plate and transfected with Cas9 plasmids (300 ng) and gRNA plasmids (200 ng) by Lipofectamine 2000 (1 μl) (Life Technologies). Cells were collected 3–5 days after transfection. For HeLa, A375, HCT116, HFF and N2a cells, we transfected SlugCas9 (300 ng) and gRNA plasmids (200 ng) by Lipofectamine 3000 (1 μl) (Life Technologies). Cells were collected 5–7 days after transfection. Genomic DNA was isolated, and the target sites were PCR-amplified by nested PCR amplification and purified by a Gel Extraction Kit (QIAGEN) for deep sequencing.
Genome editing for stimulating HDR
The donor DNA (single-stranded oligonucleotides) was synthesized by GENEWIZ (Shanghai, China). A total of 2 × 105 HEK293T cells were transfected with 1 μg of Cas9-expressing plasmid DNA, 0.5 μg of gRNA-expressing plasmids, and 100 pmol of donor oligonucleotides by electroporation and then seeded into 12-wells. Electroporation voltage, width and number of pulses were 1150 V, 30 ms and 1 pulse, respectively. Genomic DNA was isolated by DNeasy Blood and Tissue Kit (QIAGEN), and the target sites were PCR-amplified, and then purified by a Gel Extraction Kit (QIAGEN). Then, 400 ng of PCR products was digested by EcoRI restriction enzyme, and samples were run on a 2% agarose gel.
Base editing with SlugABEmax/SlugBE4max
HEK293T cells were seeded into 48-well at a density of 20 000 cells/250 μl and transfected with SlugABEmax/ SlugBE4max (300 ng) and gRNA plasmids (200 ng). Cells were collected and the genomic DNA was isolated 3–5 days after transfection. Genomic DNA was isolated, and the target sites were PCR amplified by nested PCR amplification and purified by a Gel Extraction Kit (QIAGEN) for deep sequencing.
Test of Cas9 specificity
To test the specificity of SlugCas9, SlugCas9-HF, Sa-SlugCas9, ShaCas9 and SlutrCas9, we generated a GFP-reporter cell line with NNGGA (CTGGA) PAM. The cells were seeded into 48-well and transfected with Cas9 plasmids (300 ng) and gRNA plasmids (200 ng) by 1 μl of Lipofectamine 2000. Three days after editing, the GFP-positive cells were analyzed on the Calibur instrument (BD). Data were analyzed using FlowJo. For SaCas9, specificity, we re-analyzed the previously generated data (16).
AAV production
HEK293T cells were seeded at approximately 60–70% confluency in a 10 cm dish the day before transfection. For each well, 4 μg of Cas9-gRNA expressing plasmid, 4 μg of pAAV-RC (Gene-Bank: AF369963), and 8 μg of pAAV-helper were transfected using 160 μL of PEI (0.1% m/v, Poly-sciences, Cat# 23966 [pH 4.5]). Media were changed 6–8 h after transfection. After 72 h, cells are scrapped and poured into a 15-ml conical centrifuge tube. Spin at 4000 g at 4°C for 5 min, and transfer supernatant into a new 15-ml tube. Resuspend the cell pellet in 1 ml of PBS Buffer. Transfer to a new 15-ml conical tube. Freeze in liquid nitrogen for 1–2 min, thaw at 37°C for 3–4 min, and repeat three times. Spin at 4000 g at 4°C for 10 min. Mix the two supernatants together and filter with a 0.45-μm polyvinylidene fluoride filter. Add one-half volume of the mixed solution (1 M NaCl + 10% PEG8000), and incubate at 4°C overnight. After centrifugation at 4°C for 2 h at 12 000 g, discard the flow-through and add 500 μl of chilled PBS. The quantitative PCR reveals that AAV titration is 1.0 × 1012 copies/ml. About 100 μl of the virus was added into a 12-well plate with 60–80% confluency of cells, resulting in a multiplicity of infection (MOI) of ∼2 × 105 viral genomes/cell.
GUIDE-seq
GUIDE-seq experiments were performed as described previously (19), with minor modifications. Briefly, 2 × 105 HEK293T cells were transfected with 1 μg of SlugCas9/ SlugCas9-HF, 0.5 μg of gRNA plasmids, and 100 pmol of annealed GUIDE-seq oligonucleotides by electroporation and then seeded into 12 wells. Electroporation voltage, width and number of pulses were 1150 V, 30 ms and 1 pulse, respectively. Genomic DNA was extracted with a DNeasy Blood and Tissue Kit (QIAGEN) 5–7 days after transfection according to the manufacturer’s protocol. Prepare the genome library and deep sequencing.
Western blotting
HEK293T cells were seeded into a six-well plate and transfected with Cas9-expressing plasmids (2 μg) by Lipofectamine 2000 (1 μl) (Life Technologies). Three days after transfection, cell samples were collected and were lysed with NP-40 buffer (Beyotime) in the presence of 1 mM phenylmethanesulfonyl fluoride (PMSF) (Beyotime). Then, centrifugation at 12 000 rpm for 10 min at 4°C, the supernatant was collected for western blotting. The protein was separated by 8% SDS-PAGE gel and then transferred to polyvinylidene fluoride (PVDF) (Thermo) membrane. After blocking with 5% (wt./vol.) BSA (Sigma) in TBS-T (0.1% Tween 20 in 1× TBS) buffer for 1 h at room temperature, the membrane was incubated in the primary antibody (Anti-HA tag (1:1000; ab236632, Abcam) and anti GAPDH (1:2000; 5174s, Cell Signaling)) at 4°C overnight. TBS-T was used to wash three times for 5 min each time. The second antibody (1:10 000; ab6721, Abcam) was incubated for 1 h at room temperature, and then washed three times and imaged.
Test of gRNA activity in mouse cells
Six gRNAs targeting mouse Myh6 Exon 3 were selected and each of them was cloned into BsaI-digested Sa-SlugCas9 expression vector. Each gRNA activity was tested in the N2a cells. Transfection was performed with Lipofectamine 2000 according to the manufacturer’s instructions (Thermo Fisher Scientific). In brief, N2a cells were seeded onto a 48-well plate one day before transfection, and transfection was performed at about 70–80% confluency. Sa-SlugCas9 with gRNA plasmid (500 ng) were transfected.
Preparing of Cas9 and gRNA mRNA
Sa-SlugCas9 and gRNA DNA templates containing a T7 promoter were obtained by PCR-amplification on Sa-SlugCas9 plasmid using NEB Q5® High-Fidelity DNA Polymerase. Purified PCR products were used as templates for in vitro transcription using HiScribe™ T7 High Yield RNA Synthesis Kit (NEB, New England Biolabs). Cas9 mRNA and gRNAs were then purified using RNAClean XP Beckman Coulter, and resuspended in RNase-free water. Both of them were stored in -80°C. Oligonucleotides used for cloning and amplification are listed in Supplementary Table S2.
Microinjection of mouse zygotes
All the experiments and animal care procedures were approved by the Animal Ethical and Welfare Committee of Fudan University, Shanghai, China. C57BL/6 female mice for oocyte generation and male mice for mating were purchased from Shanghai SLAC Laboratory Animal. C57BL/6 mice (4 weeks old) were used for zygote collection.
Mouse zygotes were obtained from super-ovulated 3-week-old C57BL/6 females submitted to intra-peritoneal injection of pregnant mare serum gonadotropin (PMSG 5 units/mouse) followed by human chorionic gonadotropin (hCG, 5 units/mouse) 42–48 h later and then mated overnight with C57BL/6 males. After 2 h of incubation, the fertilized eggs were microinjected with a mixture of Sa-SlugCas9 mRNA (100 ng/μl) and individual gRNA (50 ng/μl). We performed pronuclear injections and cultured the zygotes to the morula stage in KSOM, CO2 incubator at 37°C for 3 days. Embryos were collected into 10 μl QuickExtract™ DNA Extraction Solution (Epicentre) and incubated at 65°C for 6 min then incubated at 98°C for 2 min. The target sites were PCR-amplified and extracted by Gel Extraction Kit (QIAGEN). The PCR products were sequenced by Sanger sequencing.
Quantification and statistical analysis
All the data are shown as mean ± SD. Statistical analyses were conducted using Microsoft Excel. Two-tailed, paired Student’s t-tests were used to determine statistical significance when comparing two groups, whereas analyses of variance (ANOVA) is used for comparisons between for three or more groups. A value of P < 0.05 was considered to be statistically significant. (*P < 0.05, **P < 0.01, ***P < 0.001). All the data are listed in Supplementary Data S1.
RESULTS
Identification of SaCas9 orthologs for genome editing
To identify novel compact Cas9 nucleases for genome editing, we used Staphylococcus aureus Cas9 (SaCas9) as a reference and searched for related orthologs from UniProt (20). We focused on Cas9 nucleases with relatively high identity (at least 50%) to SaCas9 and subsequently selected five orthologs for characterization, including Staphylococcus equorum Cas9 (SeqCas9, 1053 aa, 97.1% identity), Staphylococcus lugdunensis Cas9 (SlugCas9, 1054 aa, 63.2% identity), Staphylococcus epidermidis Cas9 (SepCas9, 1099 aa, 64.2% identity), Staphylococcus haemolyticus Cas9 (ShaCas9, 1055 aa, 63.2% identity) and Staphylococcus lutrae Cas9 (SlutrCas9, 1054 aa, 59.1% identity). Importantly, these Cas9 orthologs differ in at least one residue located at the protein domain that is important for PAM recognition, corresponding to residues N986 and R991 in SaCas9 (Figure 1A) (21), suggesting that these orthologs may recognize different PAMs.
Figure 1.

A GFP-activation assay reveals that SlugCas9, ShaCas9 and SlutrCas9 enable genome editing. (A) PAM interaction (PI) domain sequence alignment of SaCas9 orthologs. Amino acids important for PAM recognition are indicated by box. (B) Schematic of the GFP-activation assay for Cas9 activity testing. A GFP reporter is disrupted by a protospacer followed by a 7-bp random sequence between ATG and GFP coding sequence. The reporter library is stably integrated into HEK293T cells. Genome editing will induce GFP expression for a portion of cells. (C) Transfection of SlugCas9, ShaCas9 and SlutrCas9 with gRNAs induce GFP expression.
We first used a previously reported GFP-activation PAM screening assay (Figure 1B) (16) to evaluate active genome editing and to identify the Cas9s’ respective PAMs. In this assay, a protospacer followed by a 7-bp random sequence is inserted between the translation initiation codon (ATG) and GFP coding sequence, inducing frameshift mutation. The reporter library is stably integrated into HEK293T cells. Genome editing can induce in-frame mutations in a portion of cells, resulting in GFP-positive cells. Each Cas9 ortholog was human-codon optimized, synthesized and cloned into a mammalian SaCas9 expression plasmid construct. The canonical SaCas9 gRNA scaffold was used for gRNA expression (2). Transfection of SlugCas9, ShaCas9 or SlutrCas9 with gRNAs could induce GFP expression (Figure 1C, upper panel), suggesting that they indeed enabled active genome editing in mammalian cells. Transfection of SeqCas9 or SepCas9 with gRNAs could not induce GFP expression (Figure 1C, lower panel). GFP-positive cells were subsequently isolated by flow cytometry and the target DNA was PCR-amplified for deep-sequencing. Sequencing results revealed that insertions and deletions (indels) occurred at the target sites for these three Cas9 nucleases at sites encoded in their respective protospacers (Figure 2A). WebLogo revealed that SlugCas9 preferred G-rich PAM (Figure 2B), and a PAM wheel revealed that SlugCas9 preferred an NNGG PAM (Figure 2C). Both WebLogo and the PAM wheel revealed that ShaCas9 and SlutrCas9 preferred an NNGGV (V = A or C or G) and NNGRR (R = A or G) PAM, respectively (Figure 2D–G).
Figure 2.
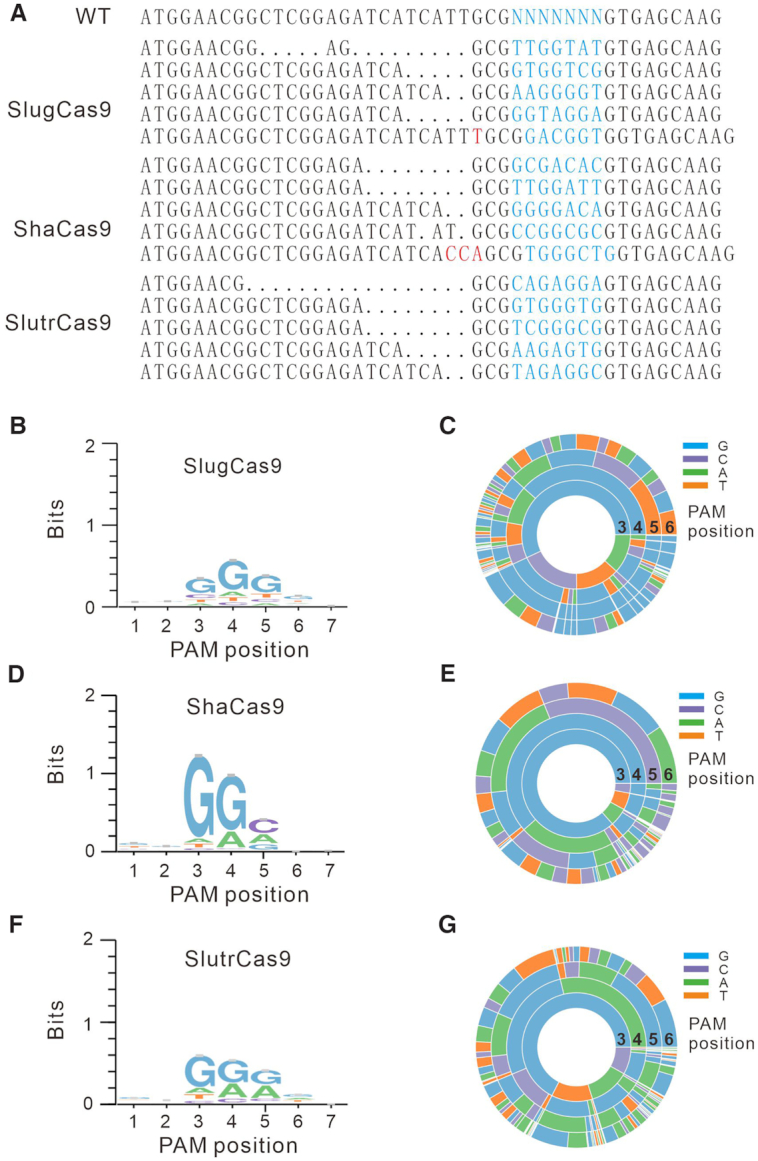
PAM sequence analysis. (A) Deep sequencing reveals that SlugCas9, ShaCas9 and SlutrCas9 generate indels on the targets. (B, D, F) WebLogos for SlugCas9, ShaCas9 and SlutrCas9 are generated based on deep sequencing data. (C, E, G) PAM wheels for SlugCas9, ShaCas9 and SlutrCas9 are generated based on deep sequencing data.
SaCas9 orthologs enable genome editing for endogenous loci
In this study, we used the SaCas9 construct that developed by Ran et al. (2) to express all Cas9 nucleases so that the expression level is comparable (Figure 3A). Similar expression level of SaCas9 and SlugCas9 was confirmed by western blot (Figure 3B). To test the genome editing capability of these three Cas9 nucleases, we selected a panel of endogenous targets loci. All three Cas9 nucleases efficiently generated indels in HEK293T cells (Figure 3C, Supplementary Figure S1A and B) at the respective sites. We focused further analysis on SlugCas9 due to its short and simple PAM. Selected target sites in Figure 3C contained an NNGRRT PAM that can be edited by SaCas9, allowing for a side-by-side comparison of editing efficiency at the same site. The editing results revealed that SlugCas9 and SaCas9 displayed similar efficiency at 14 endogenous loci (Figure 3C and D).
Figure 3.

Genome editing for endogenous sites. (A) Schematic of the Cas9 expression constructs. All Cas9 expression constructs have the same backbone as SaCas9 construct. (B) Protein expression level of SaCas9, SlugCas9 and SlugCas9-HF was measured by western blot. NC: negative control cells without Cas9 transfection. (C) Comparison of SaCas9, SlugCas9 and SlugCas9-HF efficiency for genome editing at 14 endogenous loci. Additional ‘g’ is added for U6 promoter transcription (n = 3). (D) Quantification of editing efficiency for SaCas9, SlugCas9 and SlugCas9-HF.
We further tested the genome editing capacity of SlugCas9 in additional cell types, including A375, HCT116, HeLa, human foreskin fibroblast (HFF, primary cells) cells and N2a (mouse neuroblastoma cell line) cells. SlugCas9 generated indels in all these cell types with varying efficacies (Supplementary Figure S2A–E). To facilitate delivery efforts, we packaged SlugCas9 together with its gRNA into AAV2 and infected HEK293T, A375, HCT116, HeLa and HFF cells. Indels were detected in all cell types, but frequencies varied depending on the loci and cell types (Supplementary Figure S3A–E).
We further identified the SlugCas9 trans-activating CRISPR RNA (tracrRNA) and established a single guide RNA (sgRNA). The secondary structure of SlugCas9 sgRNA was similar to that of SaCas9 sgRNA except that the forth thymine cannot pair with guanine (Supplementary Figure S4A). We changed the forth thymine to cytosine so that it can pair with guanine. SlugCas9 was transfected together with SaCas9 sgRNA, wild-type SlugCas9 sgRNA or engineered SlugCas9 sgRNA into HEK293T cells for genome editing. These three sgRNAs displayed comparable activity (Supplementary Figure S4B). In the following experiments, we used SaCas9 sgRNA for SlugCas9. We further identified SlutrCas9 and ShaCas9 tracrRNAs and established sgRNAs whose secondary structures were similar to that of SaCas9 sgRNA (Supplementary Figure S5A). When ShaCas9 and SlutrCas9 were used with SaCas9 sgRNA or their own sgRNA, the editing efficiencies were comparable (Supplementary Figure S5B and C). Taken together, we recommend SlugCas9, ShaCas9 and SlutrCas9 as minimal and efficient enzymes for genome editing.
Base editing with SlugCas9
Base editing is a powerful technology that enables the programmable conversion of an A:T base pair to G:C or a C:G base pair to T:A in the mammalian genome (22–25). This technology utilizes the fusion of a catalytically disabled Cas9 nuclease to a nucleobase deaminase enzyme. To decrease protein size and facilitate delivery efforts, SaCas9, which is ∼1 kb smaller than SpCas9, has been used for base editing (26). To test whether SlugCas9 can be employed for base editing, we generated a nickase form of SlugCas9 (SlugCas9n) by introducing a D10A mutation. We replaced the nickase form of SpCas9 with SlugCas9n in BE4max (27) to generate APOBEC1–SlugCas9n–UGI (SlugBE4max, Supplementary Figure S6A). We tested the editing capability of SlugBE4max at a panel of 34 endogenous sites. SlugBE4max and the respective gRNA plasmid DNA was transfected into HEK293T cells. Five days after transfection, targeted deep sequencing revealed C to T base editing ranging from 2.5% to 43.8% (Supplementary Figure S6A). We also replaced the nickase form of SpCas9 with SlugCas9n in the ABEmax (27) plasmid to generate TadA-TadA*-SlugCas9n (SlugABEmax) (Supplementary Figure S6B). SlugABEmax and the respective gRNA plasmid DNA were transfected into HEK293T cells. Five days after transfection, targeted deep sequencing revealed that A to G base editing ranging from 2.1% to 42.4% (Supplementary Figure S6B). Collectively, these data demonstrated that SlugCas9 can be used for base editing at varying efficiencies.
Engineering of SlugCas9 for improved specificity
Next, we evaluated the specificity of SlugCas9 by using a previously developed GFP-activation approach (16). We generated a panel of gRNAs with dinucleotide mutations along the protospacer to detect the specificity of SlugCas9. SlugCas9 showed robust activity for both on-target and off-target cleavage (Figure 4A), indicating that it has strong off-target effects. We also evaluated the specificity of ShaCas9 and SlutrCas9, both of which displayed substantial off-target effects (Supplementary Figure S7). Recently, Tan et al. introduced quadruple mutations into SaCas9 to generate a highly specific SaCas9 variant (SaCas9-HF) (28). To improve the specificity of SlugCas9, we used pairwise alignment and introduced the corresponding quadruple mutations (R247A, N415A, T421A and R656A) into SlugCas9 to generate SlugCas9-HF (Figure 4B). Interestingly, SlugCas9-HF dramatically improved specificity (Figure 4A).
Figure 4.

Analysis of SlugCas9 and SlugCas9-HF specificity. (A) Schematic of the GFP-activation assay for specificity analysis is shown on the top. A panel of gRNAs with dinucleotide mutations is shown below. Each gRNA activity for SlugCas9 and SlugCas9-HF is analyzed based on GFP expression. Mismatches are shown (n = 3). (B) Schematic of the SlugCas9-HF. Mutations are shown below. (C) Off-targets for EMX1 locus are analyzed by GUIDE-seq. Read numbers for on- and off-targets are shown on the right. Mismatches compared with the on-target site are shown.
To compare genome-wide off-target effects of SlugCas9 and SlugCas9-HF, GUIDE-seq was performed (19). Following transfection of SlugCas9 variants, the gRNA plasmid, and the GUIDE-seq oligos, we prepared libraries for deep sequencing. Sequencing and analysis revealed that on-target cleavage occurred for both Cas9 nucleases, reflected by the high GUIDE-seq read counts (Figure 4C). Four off-target sites were identified for SlugCas9 but no off-target sites were identified for SlugCas9-HF with this particular protospacer. In summary, these data indicated that the specificity of SlugCas9-HF was significantly improved.
To ensure no other effects were the cause of increased specificity, we compared the SlugCas9-HF activity side-by-side to SlugCas9. The SlugCas9-HF expression construct was the same as SlugCas9 and their protein expression level was similar, revealed by western blot (Figure 3A and B). The editing results revealed that SlugCas9 and SlugCas9-HF displayed similar efficiency although efficiencies varied for specific loci (Figure 3C and D). These data demonstrated that SlugCas9-HF improved specificity without compromising activity.
Engineering of SlugCas9 for expanding targeting scope
A previous study has shown that a SaCas9 variant (SaCas9-KKH) with triple mutations (E782K/N968K/R1015H) relieved the PAM sequence requirement at position 3 of the 6 base pair PAM (17). SlugCas9 already contained a K967 residue, which correlates to position N968 of SaCas9. To broaden the targeting scope of SlugCas9, we introduced two mutations (Q782K/R1014H) that correspond to E782K and R1015H on SaCas9, resulting in SlugCas9-KH. We performed a PAM screening assay to reveal that SlugCas9-KH preferred an NNRG (R = A or G) PAM (Supplementary Figure S8A). Next, we introduced the corresponding triple mutations (Q783K/Y968K/R1015H) into ShaCas9, resulting in ShaCas9-KKH. The PAM screening assay also revealed that ShaCas9-KKH had relieved PAM preference and now preferred an NNRRC PAM (Supplementary Figure S8B). Next, we introduced triple mutations (Q782K/Y966K/R1013H) into SlutrCas9, resulting in SlutrCas9-KKH. Interestingly, SlutrCas9-KKH relieved PAM restriction at position 3. The PAM screening assay revealed that the SlutrCas9-KKH now preferred an NNNRR PAM (Supplementary Figure S8C). Since all these modified Cas9 nucleases are restricted by two or three nucleotides at PAM, we did not further test their activity.
A chimeric Cas9 nuclease for specific and efficient genome editing
We previously replaced the SaCas9 PAM interacting (PI) domain with the Staphylococcus Auricularis Cas9 (SauriCas9) PI domain to generate a chimeric Cas9 nuclease (Sa-SauriCas9, 1056 aa), which displayed high fidelity and broad targeting scope (16). In this study, we used the same approach to generate a chimeric Sa-SlugCas9 (1055 aa, Figure 5A). The GFP-activation PAM screening assay revealed that the Sa-SlugCas9 preferred an NNGG PAM (Figure 5B and C). The GFP-activation assay also revealed that the Sa-SlugCas9 specificity was similar to SaCas9 (Figure 5D). We tested the activity of Sa-SlugCas9 at a panel of endogenous loci in HEK293T cells and efficient editing was observed (Figure 5E). To compare the ability of Sa-SlugCas9, SlugCas9-HF, SlugCas9 and SaCas9 for stimulating homologous directed repair (HDR), we designed gRNAs targeting GRIN2B gene and single-stranded DNA oligonucleotides containing an EcoRI restriction site that serve as the donor fragments (Supplementary Figure S9A). The HDR products can be detected by restriction fragment length polymorphism (RFLP). Five days after transfection of Cas9/gRNA with oligonucleotides in HEK293T cells, genomic DNA was isolated and target sites were PCR-amplified. The digestion results revealed that these four Cas9 nucleases displayed similar ability for stimulating HDR (Supplementary Figure S9B).
Figure 5.

Characterization of Sa-SlugCas9 for genome editing. (A) Schematic diagram of Sa-SlugCas9. (B and C) WebLogo and PAM wheel of Sa-SlugCas9 are generated from deep-sequencing data. (D) Specificity of Sa-SlugCas9 and SaCas9 is measured by the GFP-activation assay. A panel of gRNAs with dinucleotide mismatches (red) is shown below (n = 3). (E) Sa-SlugCas9 generates indels for a panel of 28 endogenous loci (n = 3).
In order to get a whole picture of the editing activity for the Cas9 nucleases developed in this study (including SlugCas9, ShaCas9, SlutrCas9, SlugCas9-HF and Sa-SlugCas9) and in our previous study (including SauriCas9 and Sa-SauriCas9) (16), we calculated the endogenous editing efficiencies for each Cas9 and compared them to SaCas9 (Supplementary Figure S10A). Notably, the SauriCas9 and Sa-SauriCas9 data was generated in a previous study (16). SlugCas9, SlugCas9-HF, Sa-SlugCas9 and SauriCas9 displayed similar activity to SaCas9, whereas Sa-SauriCas9, ShaCas9 and SlutrCas9 displayed lower activity compared to SaCas9. We also calculated the relative editing efficiencies (off-target efficiency normalized by on-target efficiency) at off-targets obtained from GFP-based specificity assay for each Cas9 and compared them to SaCas9 (Supplementary Figure S10B). SlugCas9-HF and Sa-SauriCas9 displayed higher specificity (lower editing efficiency at off-targets) compared to SaCas9. Sa-SlugCas9 displayed similar specificity compared to SaCas9. Other Cas9 nucleases displayed lower specificity compared to SaCas9. We also showed all the Cas9 variant maps used in this study for easy of comparison (Supplementary Figure S11).
To test whether Sa-SlugCas9 enabled efficient editing in mouse zygotes, we targeted the myosin heavy chain 6 (Myh6) gene, a bi-allelic disruption of which induces a cardiac phenotype (29). We designed a list of six gRNAs targeting exon 3 and tested activity in mouse N2a cells (Supplementary Figure S12A). The T7EI digestion assay revealed that all gRNAs efficiently edited the gene (Supplementary Figure S12B) at an average of 18.3% indel rate. Next, we coinjected Sa-SlugCas9 mRNA with in vitro transcribed gRNA into mouse zygotes (Supplementary Figure S12C and D). Interestingly, we observed that 3 out of 13 the embryos were edited 3 days after injection (Supplementary Figure S12E). These data demonstrated that Sa-SlugCas9 enabled efficient genome editing in mouse zygotes.
DISCUSSION
Small Cas9 nucleases together with their gRNA and a full-length promoter can be packaged into the size-limited AAV vector and hold great promise for gene therapy. Although several small Cas9 nucleases have been developed for genome editing, many genomic sites cannot be edited due to low activity or PAM limitation. Three type II-C Cas9 orthologs have been used for mammalian genome editing, including Neisseria meningitidis Cas9 (NmeCas9) (7,13), Campylobacter jejuni Cas9 (CjeCas9) (14) and N. meningitidis Cas9 (Nme2Cas9) (15). However, type II-C Cas9 nucleases generally display low editing efficiency (30–32). In addition, NmeCas9 and CjeCas9 require longer PAMs, N4GAYW/N4GYTT/ N4GTCT and N4RYAC, respectively. SaCas9 belonging to type II-A ortholog displayed higher activity (31), but it has limited utility due to its long PAM sequence requirement (NNGRRT). Engineered SaCas9 variants have increased the targeting scope (recognizing NNNRRT PAM) (17,33), but it still requires a triple nucleotide PAM.
Our group have developed five small Cas9 nucleases, including SauriCas9 (16), Sa-SauriCas9 (16), SlugCas9, Sa-SlugCas9 and SlugCas9-HF, which recognize a simple NNGG PAM. To facilitate to select an optimal Cas9 nuclease for genome editing, we compare the activity and specificity of these Cas9 nuclease to SaCas9. SauriCas9, SlugCas9, Sa-SlugCas9, SlugCas9 and SaCas9 display similar activity, while Sa-SauriCas9 displays lower activity. SauriCas9 and SlugCas9 display lower specificity compared to SaCas9. Sa-SlugCas9 displays similar specificity compared to SaCas9. SlugCas9-HF and Sa-SauriCas9 display higher specificity compared to SaCas9. Considering both activity and specificity, we suggest that SlugCas9-HF is first choice among these five Cas9 nucleases for genome editing.
Supplementary Material
ACKNOWLEDGEMENTS
Z.H., S.W., S.G., J.W., M.L. H.M., Y.W. and L.H. performed experiments. C.Z. and T.Q. analyzed the data. J.L. revised the manuscript. H.L., D.L., F.L., D.L., H.W. and Y.W. provide experimental guidance. Y.W. wrote the manuscript.
Contributor Information
Ziying Hu, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Chengdong Zhang, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China; School of Public Health, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China.
Shuai Wang, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Siqi Gao, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Jingjing Wei, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Miaomiao Li, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Linghui Hou, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Huilin Mao, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Yanyan Wei, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Tao Qi, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Hongmao Liu, Shanghai Key Laboratory for Assisted Reproduction and Reproductive Genetics, Center for Reproductive Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200135,China.
Dong Liu, School of Life Sciences, Co-innovation Center of Neuroregeneration, Jiangsu Key Laboratory of Neuroregeneration, Nantong University, Nantong 226001, China.
Feng Lan, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China.
Daru Lu, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China; NHC Key Laboratory of Birth Defects and Reproductive Health, Chongqing Key Laboratory of Birth Defects and Reproductive Health, Chongqing Population and Family Planning, Science and Technology Research Institute, Chongqing, 400020, China.
Hongyan Wang, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Jixi Li, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China.
Yongming Wang, State Key Laboratory of Genetic Engineering, School of Life Sciences, Zhongshan Hospital, Obstetrics and Gynecology Hospital, Fudan University, Shanghai 200438, China; School of Life Sciences, Co-innovation Center of Neuroregeneration, Jiangsu Key Laboratory of Neuroregeneration, Nantong University, Nantong 226001, China; Shanghai Engineering Research Center of Industrial Microorganisms, Shanghai, 200438, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81870199, 82070258, 81630087]; Foundation for Innovative Research Groups of the National Natural Science Foundation of China [31521003]; State Key Laboratory Opening Program [SKLGE1809]; Science and Technology Research Program of Shanghai [19DZ2282100]; Project of Chongqing Science Committee [cstc2020jcyj-zdxm0180, cstc2020jcyj-msxm3584]. Funding for open access charge: National Natural Science Foundation of China [81870199].
Conflict of interest statement. The authors have applied for a patent related to this work.
REFERENCES
- 1. Wu Z., Yang H., Colosi P.. Effect of genome size on AAV vector packaging. Mol. Ther.: J. Am. Soc. Gene Ther. 2010; 18:80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S.et al.. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015; 520:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jo D.H., Koo T., Cho C.S., Kim J.H., Kim J.S., Kim J.H.. Long-term effects of in vivo genome editing in the mouse retina using campylobacter jejuni cas9 expressed via adeno-associated virus. Mol. Ther.: J. Am. Soc. Gene Ther. 2019; 27:130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koo T., Lu-Nguyen N.B., Malerba A., Kim E., Kim D., Cappellari O., Cho H.Y., Dickson G., Popplewell L., Kim J.S. Functional rescue of dystrophin deficiency in mice caused by frameshift mutations using campylobacter jejuni Cas9. Mol. Ther.: J. Am. Soc. Gene Ther. 2018; 26:1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A.et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M.. RNA-guided human genome engineering via Cas9. Science. 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xie Y., Wang D., Lan F., Wei G., Ni T., Chai R., Liu D., Hu S., Li M., Li D.et al.. An episomal vector-based CRISPR/Cas9 system for highly efficient gene knockout in human pluripotent stem cells. Sci. Rep. 2017; 7:2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J., Manghwar H., Sun L., Wang P., Wang G., Sheng H., Zhang J., Liu H., Qin L., Rui H.et al.. Whole genome sequencing reveals rare off-target mutations and considerable inherent genetic or/and somaclonal variations in CRISPR/Cas9-edited cotton plants. Plant Biotechnol. J. 2019; 17:858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leenay R.T., Maksimchuk K.R., Slotkowski R.A., Agrawal R.N., Gomaa A.A., Briner A.E., Barrangou R., Beisel C.L.. Identifying and Visualizing Functional PAM Diversity across CRISPR-Cas Systems. Mol. Cell. 2016; 62:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chatterjee P., Jakimo N., Jacobson J.M.. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 2018; 4:eaau0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chatterjee P., Lee J., Nip L., Koseki S.R.T., Tysinger E., Sontheimer E.J., Jacobson J.M., Jakimo N.. A Cas9 with PAM recognition for adenine dinucleotides. Nat. Commun. 2020; 11:2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hou Z., Zhang Y., Propson N.E., Howden S.E., Chu L.F., Sontheimer E.J., Thomson J.A.. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA. 2013; 110:15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim E., Koo T., Park S.W., Kim D., Kim K., Cho H.Y., Song D.W., Lee K.J., Jung M.H., Kim S.et al.. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017; 8:14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edraki A., Mir A., Ibraheim R., Gainetdinov I., Yoon Y., Song C.Q., Cao Y., Gallant J., Xue W., Rivera-Perez J.A.et al.. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol. Cell. 2019; 73:714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu Z., Wang S., Zhang C., Gao N., Li M., Wang D., Wang D., Liu D., Liu H., Ong S.G.et al.. A compact Cas9 ortholog from Staphylococcus Auricularis (SauriCas9) expands the DNA targeting scope. PLoS Biol. 2020; 18:e3000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kleinstiver B.P., Prew M.S., Tsai S.Q., Nguyen N.T., Topkar V.V., Zheng Z., Joung J.K.. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015; 33:1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: A sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P.et al.. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015; 33:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. The UniProt, C. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nishimasu H., Cong L., Yan W.X., Ran F.A., Zetsche B., Li Y., Kurabayashi A., Ishitani R., Zhang F., Nureki O.. Crystal Structure of Staphylococcus aureus Cas9. Cell. 2015; 162:1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R.. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017; 551:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R.. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thuronyi B.W., Koblan L.W., Levy J.M., Yeh W.H., Zheng C., Newby G.A., Wilson C., Bhaumik M., Shubina-Oleinik O., Holt J.R.et al.. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 2019; 37:1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E.et al.. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020; 38:883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim Y.B., Komor A.C., Levy J.M., Packer M.S., Zhao K.T., Liu D.R.. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017; 35:371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R.. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018; 36:843–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tan Y., Chu A.H.Y., Bao S., Hoang D.A., Kebede F.T., Xiong W., Ji M., Shi J., Zheng Z.. Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. PNAS. 2019; 116:20969–20976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johansen A.K., Molenaar B., Versteeg D., Leitoguinho A.R., Demkes C., Spanjaard B., de Ruiter H., Akbari Moqadam F., Kooijman L., Zentilin L.et al.. Postnatal Cardiac Gene Editing Using CRISPR/Cas9 With AAV9-Mediated Delivery of Short Guide RNAs Results in Mosaic Gene Disruption. Circ. Res. 2017; 121:1168–1181. [DOI] [PubMed] [Google Scholar]
- 30. Tsui T.K.M., Hand T.H., Duboy E.C., Li H. The Impact of DNA topology and guide length on target selection by a cytosine-specific Cas9. ACS Synth. Biol. 2017; 6:1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Y., Liu K.I., Sutrisnoh N.B., Srinivasan H., Zhang J., Li J., Zhang F., Lalith C.R.J., Xing H., Shanmugam R.et al.. Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biol. 2018; 19:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma E., Harrington L.B., O’Connell M.R., Zhou K., Doudna J.A.. Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol. Cell. 2015; 60:398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma D., Xu Z., Zhang Z., Chen X., Zeng X., Zhang Y., Deng T., Ren M., Sun Z., Jiang R.et al.. Engineer chimeric Cas9 to expand PAM recognition based on evolutionary information. Nat. Commun. 2019; 10:560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.