


Abstract
The hydrolytic deamination of adenosine-to-inosine (A-to-I) by RNA editing is a widespread post-transcriptional modification catalyzed by the adenosine deaminase acting on RNA (ADAR) family of proteins. ADAR-mediated RNA editing modulates cellular pathways involved in innate immunity, RNA splicing, RNA interference, and protein recoding, and has been investigated as a strategy for therapeutic intervention of genetic disorders. Despite advances in basic and translational research, the mechanisms regulating RNA editing are poorly understood. Though several trans-acting regulators of editing have been shown to modulate ADAR protein expression, previous studies have not identified factors that modulate ADAR catalytic activity. Here, we show that RNA editing increases upon intracellular acidification, and that these effects are predominantly explained by both enhanced ADAR base-flipping and deamination rate at acidic pH. We also show that the extent of RNA editing increases with the reduction in pH associated with conditions of cellular hypoxia.
Graphical Abstract
Graphical Abstract.
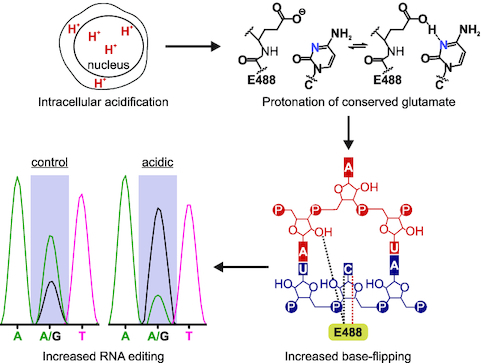
Intracellular acidification drives protonation of a conserved glutamate in the ADAR deaminase domain, which in turn enhances base-flipping and increases the extent of RNA editing.
INTRODUCTION
Dynamic and rapidly coordinated gene expression relies upon various post-transcriptional mechanisms that modify RNA sequence, structure, and stability (1). Prevalent among more than one hundred RNA processing events that shape the transcriptional landscape is the conversion of adenosine to inosine (A-to-I) by RNA editing (2,3). It has been predicted that the human transcriptome contains as many as one-hundred million A-to-I editing sites, comprising selectively edited adenosines in protein-coding regions as well as hyper-edited adenosine clusters in non-coding, repetitive sequences such as short interspersed nuclear elements (SINEs) (2,4,5). A-to-I editing is generally identified as adenosine to guanosine (A-to-G) discrepancies during comparisons of genomic and cDNA sequences due to the base-pairing of cytidine to inosine (like guanosine) during reverse transcriptase-mediated first-strand cDNA synthesis. Many cellular machines also recognize inosine as guanosine, indicating that A-to-I editing constitutes functional A-to-G substitutions that can modulate diverse pathways involved in innate immunity, RNA splicing, RNA interference, and protein recoding (6,7).
The specificity and frequency of A-to-I editing are dictated by both cis- and trans-acting regulatory elements. Cis-acting factors such as RNA sequence context influence the extent of site-specific A-to-I conversion, but the formation of an extended region of double-stranded RNA (dsRNA) by intramolecular base-pairing is paramount for editing (8–10). The major trans-regulatory factors are the editing enzymes themselves, referred to as adenosine deaminases acting on RNA (ADARs), which catalyze the deamination of adenosine residues within dsRNA substrates (7). Two active members of the vertebrate ADAR family, ADAR1 and ADAR2, each contain multiple copies of a dsRNA binding domain (dsRBD) and a carboxyl-terminal adenosine deaminase domain (11). The RNA editing reaction involves three main steps: (i) ADAR binding to the dsRNA substrate, (ii) flipping the targeted adenosine out of the RNA duplex into the enzyme active site and (iii) hydrolytic deamination at position 6 of the purine ring (12).
Expression of ADAR1 and ADAR2 is ubiquitous, but enriched in the brain, along with inosine-containing mRNAs (13,14). Interestingly, many editing-dependent recoding events in mRNAs occur within transcripts critical for nervous system function. The extent of editing for these RNAs varies spatiotemporally and carries functional consequences for encoded proteins including changes in calcium permeability through GluA2 subunit-containing AMPA receptors, alterations in inactivation dynamics for the Kv1.1-subtype of voltage-gated potassium channel, and modulation of G-protein coupling efficacy and constitutive activity for the 2C-subtype of serotonin receptor (5HT2C) (5,15–17). Furthermore, alterations in ADAR1 or ADAR2 expression have been shown to result in neurobehavioral phenotypes, as well as embryonic or early postnatal lethality, in animal models (18–22). Dysregulation of RNA editing in humans has been implicated in disorders of innate immunity and nervous system function including Aicardi-Goutières syndrome, epilepsy, suicide, amyotrophic lateral sclerosis and schizophrenia (23–27). Taken together, these data not only demonstrate an important role for A-to-I editing in numerous physiological systems, but also suggest that editing may be regulated to produce transcriptional plasticity that can endow biological systems with adaptive capacities in the face of changing environmental or physiologic conditions (28,29).
In mammals, several proteins regulate ADAR stability and subsequent RNA editing. The coordinate action of a positive regulator, Pin1, and a negative regulator, WWP2, modulate ADAR2 expression through post-translational interactions (30). AIMP2 also inhibits editing by decreasing ADAR protein levels (5). Although these trans-acting regulators provide a mechanism for editing regulation by modulating ADAR stability, studies spanning the past decade have consistently concluded that changes in ADAR protein expression do not fully account for differences in the extent of A-to-I conversion (5,31–36). Accordingly, it is likely that other factors modulate ADAR activity, rather than protein expression, to alter the extent of editing for ADAR substrates.
Recent studies of the structural basis for ADAR base-flipping have revealed the importance of a highly conserved glutamate (E1008 in ADAR1 and E488 in ADAR2) residing in the deaminase domains of each enzyme (37). This residue stabilizes the flipped-out conformation of the RNA duplex, presumably by occupying the space vacated by the flipped-out adenosine and hydrogen bonding with the complementary-strand orphaned base. Consistent with the idea that this glutamate requires protonation to stabilize the altered nucleic acid conformation, an enhancement in base-flipping and deamination rate is observed when this glutamate is mutated to a glutamine (E1008Q and E488Q in ADAR1 and ADAR2, respectively), which is fully protonated at physiologically relevant pH (12,38). These findings indicate that protonation of this glutamate may be critical for optimal catalytic activity and may alter the overall rate of ADAR catalysis during pH shifts in the cell.
To determine if editing is regulated by such changes in pH, we have employed cell culture model systems in which we analyzed either ADAR1 or ADAR2-mediated editing of transfected minigene-derived RNAs or the editing of endogenous RNA targets in response to acidic or more physiologically normal pH conditions. We also have performed in vitro studies to examine both editing kinetics and ADAR base-flipping in response to changes in pH. Finally, we have investigated how such changes in pH that occur in a cell culture model of hypoxia might affect the extent of site-selective A-to-I conversion. Our results show that RNA editing increases upon acidification, and that these effects are predominantly explained by enhanced base-flipping and deamination rate of ADARs at decreased pH.
MATERIALS AND METHODS
Plasmids for transfection
cDNAs encoding mouse ADAR1 (p110) or rat ADAR2 were subcloned into mammalian expression vectors as previously described (17). To generate ADAR-Q mutants, E1008Q and E488Q mutations were respectively introduced into wild-type ADAR1 and ADAR2 plasmids using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies), where the polymerase chain reaction (PCR) reactions were supplemented with 4% DMSO. Plasmids encoding human 5HT2C, mouse GluA2, and human Gli1 minigenes, containing the minimum sequences required for editing, were subcloned into either the pRC-CMV or pcDNA3 mammalian expression vectors (Thermo-Fisher) (17,39,40).
Cell culture
HEK293T and HeLa cells were maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% heat inactivated fetal bovine serum (Gibco) and 1% 10,000 μg/ml penicillin–streptomycin (Gibco) and incubated in a humidified chamber at 37°C and 5% CO2. Twenty-four hours prior to HEK293T cell transfection, cells were seeded in six-well plates (for RNA analysis) or 100 mm dishes (for protein analysis). HEK293T cells in six-well plates were transiently co-transfected with 1 μg ADAR1, ADAR1-E1008Q (ADAR1-Q), ADAR2 or ADAR2-E488Q (ADAR2-Q) as well as 1 μg of one of the substrate minigene-expressing plasmids (as described above) using Fugene 6 (Promega) according to manufacturer's instructions. HEK293T cells in 100 mm dishes were transiently co-transfected with 3 μg of ADAR1 or ADAR2 expression plasmids, as well as 3 μg of the 5HT2C minigene-containing plasmid using Fugene 6 according to manufacturer's instructions. Twenty-four hours after transfecting HEK293T cells or seeding HeLa cells, they were washed with 1× Dulbecco's phosphate-buffered saline (DPBS) and incubated with high-glucose DMEM containing 3.7 mg/ml NaHCO3 (for control samples) or high-glucose DMEM containing a reduced concentration of NaHCO3 (either 2, 1.5, 1, 0.5, 0.17 or 0 mg/ml NaHCO3 for experimental samples) for an additional 1, 3, 6, 12 or 24 h at 37°C and 5% CO2. Following this 24-h incubation, a pH meter was used to measure the pH of the cell culture medium immediately after removing dishes from the tissue culture incubator. The establishment of acidic culture medium via reduced NaHCO3 concentration is similar to that previously reported by Kondo and colleagues (41,42).
Niclosamide treatment
HEK293T and HeLa cells were seeded separately into 6-well plates. HEK293T cells were co-transfected with ADAR1 or ADAR2 and 5HT2C expression vectors as described above. Twenty-four hours after transfecting HEK293T cells or seeding HeLa cells, cells were washed with 1× DPBS and incubated with high-glucose DMEM containing 3.7 g/l NaHCO3 and treated for 6 h with either 5 μM niclosamide (diluted from a freshly prepared 10 mM stock solution in DMSO) or an equivalent volume of DMSO. Total RNA was extracted from cells immediately after this 6-h incubation to quantify RNA editing.
Cell culture hypoxia
HEK293T were seeded into six-well plates and co-transfected with ADAR1 or ADAR2 and 5HT2C expression vectors as described above. Twenty-four hours after transfecting HEK293T cells or seeding HeLa cells, cells were washed with 1× DPBS and incubated with high-glucose DMEM containing 3.7 g/l NaHCO3. Cells exposed to normoxic conditions were placed in the tissue culture chamber at 37°C and 5% CO2 for 24 h. Induction of hypoxia was achieved by subjecting cells to a 1% O2/5% CO2/94% N2 environment within a modular incubator chamber (STEMCELL Technologies) placed inside the tissue culture chamber for 24 h, as described previously (43). The pH of the cell culture medium was measured immediately following normoxic or hypoxic treatment.
Analysis of RNA editing from cultured cells
Total RNA was extracted from HEK293T and HeLa cells using 1 mL Trizol Reagent (Invitrogen) according to manufacturer's instructions. DNA was removed from RNA samples using the Turbo™ DNase kit (Invitrogen), and following the rigorous DNase treatment procedure, cDNA was prepared from 1 μg of DNase-treated RNA sample using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). When preparing cDNA, a control reaction lacking reverse transcriptase (–RT) was prepared in parallel for each RNA sample to ensure the absence of genomic DNA contamination. PCR amplicons were generated using the target-specific primers listed in Table 1 and then sequenced. RNA editing in HEK293T cells was quantified using a high-throughput sequencing-based strategy, as described previously (32). RNA editing levels in HeLa cells were determined by quantifying the relative peak heights at edited positions in Sanger sequencing-derived electropherograms (44).
Table 1.
Target-specific primers for quantitative analysis of RNA editing in cultured cells
| Transcript | Sense or antisense | Primer sequence |
|---|---|---|
| 5HT2C | Sense | 5′-ATT AAC CCT CAC TAA AGG GAC GCT GGA TCG GTA TGT AGC A-3′ |
| Antisense | 5′-TAA TAC GAC TCA CTA TAG GGC GTC TGT ACG TTG TTC ACA GTA C-3′ | |
| GluA2 (Q/R, +3, +4, +60 and +88 sites) | Sense | 5′-ATT AAC CCT CAC TAA AGG GAA TAG TCT CTG GTT TTC CTT GGG-3′ |
| Antisense | 5′-TAA TAC GAC TCA CTA TAG GGA GCT GGT GAC ATC TTT ATG GTG-3′ | |
| GluA2 (sites +241 through +271) | Sense | 5′-ATT AAC CCT CAC TAA AGG GAG TTG ATC AGG TGT TTC CCT GGT-3′ |
| Antisense | 5′-TAA TAC GAC TCA CTA TAG GGC GAT CTA AAA TCG CCC ATT TTC CC-3′ | |
| Gli1 | Sense | 5′-ATT AAC CCT CAC TAA AGG GAG GAC AGA ACT TTG ATC CTT ACC TCC-3′ |
| Antisense | 5′-TAA TAC GAC TCA CTA TAG GGC ATA TAG GGG TTC AGA CCA CTG-3′ | |
| Cog3 | Sense | 5′-ATT AAC CCT CAC TAA AGG GAC AGT CCT TAC TTG GAG CGT CA-3′ |
| Antisense | 5′-TAA TAC GAC TCA CTA TAG GGC TGA ATA AAC TGC TCA CAG GCC-3′ | |
| Eef2k | Sense | 5′-ATT AAC CCT CAC TAA AGG GAG TAA TTT ACA GCA GGA CGC TTT CA-3′ |
| Antisense | 5′-TAA TAC GAC TCA CTA TAG GGG TAG AGA CAG GGT CTC GC-3′ | |
| Blcap (Y/C site) | Sense | 5′-GAC AGC CAG AGA GCA CAG-3′ |
| Antisense | 5′-TGA GCA GGT AGA AGC CCA T-3′ |
Western blotting
HEK293T and HeLa cells were seeded separately into 100 mm plates. HEK293T cells were co-transfected with ADAR1 or ADAR2 and 5HT2C expression vectors as described above. Twenty-four hours after transfecting HEK293T cells or seeding HeLa cells, cells were washed with 1× DPBS and incubated with high-glucose DMEM containing 3.7 or 0 g/l NaHCO3 for 24 h. HEK293T and HeLa whole cell lysates were prepared using RIPA lysis and extraction buffer. 20 μg of each sample was resolved by electrophoresis on a 4–20% gradient SDS-PAGE gel and then transferred to a nitrocellulose membrane using a Trans-Blot® SD semi-dry transfer cell (Bio-Rad). After blocking the membrane with Intercept PBS blocking buffer (LI-COR) for 1 h, the membrane was probed using either a mouse ADAR1 monoclonal antibody (sc-73408, Santa Cruz; 1:1000 dilution) or an affinity-purified rat ADAR2 antiserum (Exalpha Biologicals; 1:375 dilution), as well as a polyclonal affinity-purified β-actin antibody (sc-1616-R, Santa Cruz; 1:1000 dilution). ADAR1 and β-actin signals were detected using IRDye secondary antibodies (LI-COR; 1:20,000 dilution), and ADAR2 signals were detected using Alexa Fluor® 790 light-chain specific anti-sheep IgG secondary antibody (Jackson ImmunoResearch; 1:50,000). All blots were imaged using an Odyssey CLx infrared imaging system (LI-COR) and quantified using Image Studio Lite (LI-COR).
Quantitative RT-PCR analysis
cDNA generated from HeLa cells was used to prepare reactions for duplex TaqMan® Gene Expression Assays (Applied Biosystems) according to manufacturer's instructions. Eef2k (Hs00179434_m1), Blcap (Hs00705669_s1), Cog3 (Hs00230134_m1) and Adar2 (Hs00953723_m1) expression were assessed using 20× FAM dye-labeled TaqMan® probes, and β-actin (Hs03023943_g1) expression was assessed using 20× VIC dye/MGB-labeled and a primer limited TaqMan® endogenous control. Expression of each target gene was assayed using three biological replicates, and each sample was run in technical triplicate. Relative changes in gene expression were determined using the ΔΔCt method (45).
pHrodo intracellular pH assessment
HEK293T and HeLa cells were seeded separately on poly-l-lysine- and laminin-coated 35-mm glass bottom dishes (MatTek Life Sciences). Twenty-four hours later, cells were washed with Live Cell Imaging Solution (LCIS) and incubated for 30 min with 2 μl of pHrodo Red AM Intracellular pH Indicator (Invitrogen) diluted in 20 μl of PowerLoad concentrate and added to 2 ml of pre-warmed LCIS. Cells were then washed in DPBS and incubated for 6 h at 37°C and 5% CO2 with high-glucose DMEM lacking phenol red and containing either 0 or 3.7 g/l NaHCO3. For niclosamide experiments, cells were incubated with high-glucose DMEM containing 3.7 g/l NaHCO3 and treated for 6 h with either 5 μM niclosamide or an equivalent volume of DMSO. Plates were then removed from the cell culture incubator and 2 μl of BioTracker™ 488 Nuclear Dye (Sigma-Aldrich) were added directly to the cell culture medium and incubated at 37°C for 10 min. Immediately following this incubation, z-stack images were captured using a Zeiss LSM 510 META Inverted confocal microscope. pHrodo Red and BioTracker™ 488 were excited at 543 and 488 nm, respectively. The average pHrodo Red fluorescence intensity per cell was quantified using Fiji image processing software.
General biochemical procedures
Unless otherwise stated, all regents were purchased from commercial sources (Fisher Scientific or Sigma Aldrich) and were used without further purification. Reagents for in vitro transcription, in vitro editing, and PCR amplification were purchased from: Promega: Access RT-PCR kit, RQ1 DNase free RNase; Qiagen: Gel Extraction kit; Zymo Research: DNA Clean & Concentrator kit; Syd Labs: Spin columns for PCR product clean up; New England BioLabs: Molecular-biology-grade bovine serum albumin (BSA), and RNase inhibitor. SDS-polyacrylamide gels were visualized with a Molecular Dynamics 9400 Typhoon phosphorimager. Data were analyzed with Molecular Dynamics ImageQuant 5.2 software. All MALDI analyses were performed at the University of California, Davis Mass Spectrometry Facilities using a Bruker ultrafleXtreme MALDI TOF/TOF mass spectrometer. Oligonucleotide masses were determined with Mongo Oligo Calculator v2.08. Unless otherwise noted, oligonucleotides were purchased from either Dharmacon or Integrated DNA Technologies.
Purification of oligonucleotides
All oligonucleotides for biochemical experiments were purified by denaturing polyacrylamide gel electrophoresis (PAGE) and visualized by UV shadowing. Oligonucleotides extracted from the gel using the crush and soak method for 16 h at 4°C into 0.5 M NH4OAc and 1 mM EDTA. Polyacrylamide fragments were removed using a 0.2 μm pore size cellulose acetate membrane filter (Corning). Oligonucleotides were precipitated from 75% ethanol containing 75 mM NaOAc at –70°C for 2 h. The resulting pellet was dried under vacuum and resuspended in nuclease free water.
In vitro transcription and preparation of editing target RNA
Target RNA was transcribed from a DNA template with the MEGAScript T7 Kit (ThermoFisher). DNA digestion was performed using RQ1 RNase-free DNase (Promega). The DNase-treated RNA product was purified by 4% PAGE as described above. Purified 5HT2c target RNA (180 nM) was added to 1× TE buffer and 100 mM NaCl, heated to 95°C for 5 min, and slowly cooled to room temperature.
Protein overexpression and purification of ADAR2 constructs
Human ADAR2 deaminase domain (ADAR2d), human ADAR2d-E488Q, wild-type human ADAR2, and human ADAR2-E488Q were expressed and purified as previously described (46). Purification was carried out by lysing cells in buffer containing 20 mM Tris–HCl, pH 8.0, 5% glycerol, 1 mM 2-mercaptoethanol, 750 mM NaCl, 35 mM imidazole, and 0.01% Nonidet P-40 using a French press. Cell lysate was clarified by centrifugation (39 000 × g for 1 h). Lysate was passed over a 3 ml Ni-NTA column, which was then washed in 3 steps with 20 mL lysis buffer, wash I buffer (20 mM Tris–HCl, pH 8.0, 5% glycerol, 1 mM 2-mercaptoethanol, 750 mM NaCl, 35 mM imidazole, 0.01% Nonidet P-40), wash II buffer (20 mM Tris–HCl, pH 8.0, 5% glycerol, 1 mM 2-mercaptoethanol, 35 mM imidazole, 500 mM NaCl), and eluted with 20 mM Tris–HCl, pH 8.0, 5% glycerol, 1 mM 2-mercaptoethanol, 400 mM imidazole, 100 mM NaCl. Fractions containing the target protein were pooled and concentrated to 30–80 μM for use in biochemical assays. Purified wild-type ADAR2 was stored in 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 20% glycerol and 1 mM 2-mercaptoethanol at –70°C. Protein concentrations were determined using BSA standards visualized by SYPRO orange staining of SDS-polyacrylamide gels.
Protein overexpression and purification of ADAR1 p110
MBP-tagged human ADAR1 p110 construct was cloned into a pSc vector using standard PCR techniques (47). The generated construct (yeast codon optimized) consisted of an N-terminal MBP-tag, a tobacco etch virus (TEV) protease cleavage site followed by the human ADAR1 p110 gene. The construct was transformed in Saccharomyces cerevisiae BCY123 cells and overexpressed as described previously (46). Purification was carried out by lysing cells in lysis/binding buffer containing 50 mM Tris–HCl, pH 8.0, 5% glycerol, 5 mM 2-mercaptoethanol, 1000 mM KCl, 0.05% NP-40 and 50 μM ZnCl2 using a microfluidizer. Cell lysate was clarified by centrifugation (39 000 × g for 50 min). Lysate was passed over a 2 ml NEB amylose column (pre-equilibrated with binding buffer), which was then washed in 2 steps with 50 ml binding buffer followed by 100 mL wash buffer (50 mM Tris–HCl, pH 8.0, 5% glycerol, 5 mM 2-mercaptoethanol, 500 mM KCl, 0.01% NP-40 and 50 μM ZnCl2) and eluted with buffer containing 50 mM Tris–HCl, pH 8.0, 10% glycerol, 5 mM 2-mercaptoethanol, 500 mM KCl, 0.01% NP-40, 50 μM ZnCl2 and 20 mM maltose. Fractions containing the target protein were pooled and dialyzed against a storage buffer containing 50 mM Tris–HCI, pH 8.0, 400 mM KCI, 0.5 mM EDTA, 0.01% NP-40, 10% glycerol and 1 mM tris(2-carboxyethyl)phosphine. Dialyzed protein was concentrated to 2–50 μM and stored as aliquots at –70°C until further use in biochemical assays. Protein concentrations were determined using BSA standards visualized by SYPRO orange staining of SDS-polyacrylamide gels.
Deamination assay with ADAR2d, ADAR2d-E488Q, ADAR2 and ADAR1 p110
Deamination assays were performed under single-turnover conditions in 15 mM Tris–HCl (pH 7.0 to 8.5) or 15 mM Bis–Tris–HCl (pH 6.0 to 7.0), 3% glycerol, 60 mM KCl, 1.5 mM EDTA, 0.003% Nonidet P-40, 3 mM MgCl2, 160 U/ml RNAsin, 1.0 μg/ml yeast tRNA, 10 nM RNA, and 75 nM ADAR2d, ADAR2d-E488Q, or wild-type ADAR2. Each reaction solution was incubated at 30°C for 30 min before the addition of enzyme. Reactions were then incubated at 30°C for varying times prior to quenching with 190 μl 95°C water and heating at 95°C for 5 min. Reaction products were used to generate cDNA using RT-PCR (Promega Access RT-PCR System). The target-specific primers used to generate these PCR amplicons are presented in Table 2. DNA was purified using a DNA Clean & Concentrator kit (Zymo) and subjected to Sanger Sequencing using an ABI Prism 3730 Genetic Analyzer at the UC Davis DNA Sequencing Facility. The sequencing peak heights were quantified in 4Peaks v1.8. Data were fit to the equation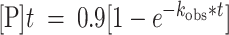
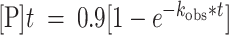 for ADAR2 and
for ADAR2 and 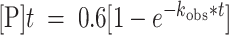 for ADAR1 p110 where
for ADAR1 p110 where 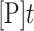 is percent edited at time
is percent edited at time 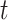 , and
, and 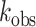 is the observed rate constant. Each experiment was carried out in triplicate where the
is the observed rate constant. Each experiment was carried out in triplicate where the 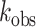 reported is the average of each replicate ± standard deviation (SD). Statistical significance between groups was determined by one-way ANOVA using Prism software (GraphPad). For the ADAR1 p110 enzyme, deamination reactions were performed as above with the following modifications: The final reaction solution for ADAR1 p110 contained 15 mM Tris–HCl (pH 7.0 to 8.5) or 15 mM Bis–Tris–HCl (pH 6.0 to 7.0), 4% glycerol, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% Nonidet P-40, 160 U/ml RNAsin, 1.0 μg/ml yeast tRNA and 10 nM RNA, and 150 nM ADAR1 p110.
reported is the average of each replicate ± standard deviation (SD). Statistical significance between groups was determined by one-way ANOVA using Prism software (GraphPad). For the ADAR1 p110 enzyme, deamination reactions were performed as above with the following modifications: The final reaction solution for ADAR1 p110 contained 15 mM Tris–HCl (pH 7.0 to 8.5) or 15 mM Bis–Tris–HCl (pH 6.0 to 7.0), 4% glycerol, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% Nonidet P-40, 160 U/ml RNAsin, 1.0 μg/ml yeast tRNA and 10 nM RNA, and 150 nM ADAR1 p110.
Table 2.
Target-specific primers for quantitative analysis of in vitro RNA editing
| Transcript | Sense or antisense | Primer sequence |
|---|---|---|
| 5HT2C | Sense | 5′-TGG GTA CGA ATT CCC ACT TAC GTA CAA GCT T-3′ |
| Antisense | 5′-AGA ACC CGA TCA AAC GCA AAT GTT AC- 3′ | |
| Gli1 | Sense | 5′-CAG AAC TTT GAT CCT TAC CTC C-3′ |
| Antisense | 5′-CAT ATA GGG GTT CAG ACC ACT G-3′ |
ThermoFluor melting temperature analysis of recombinant ADAR2, ADAR2d or ADAR2d-E488Q protein
Spectra were obtained using a Bio-Rad CFX Connect Real-Time PCR Detection System. Solutions contained 15 mM Tris–HCl (pH 7.0 to 8.5) or 15 mM Bis–Tris–HCl (pH 6.0 to 7.0), 3% glycerol, 60 mM KCl, 1.5 mM EDTA, 0.003% Nonidet P-40, 3 mM MgCl2 and 2X SYPRO orange dye, with or without protein. Samples containing protein included 3 μM wild-type ADAR2, or 4 μM of either deaminase domain protein (ADAR2d or ADAR2d E488Q). To a 96-well clear bottom PCR plate was added 20 μl of each solution. Wells were sealed with PCR plate sealing film. Fluorescence was measured as the solutions were heated from 5°C to 90°C at a rate of 2°C/min. The derivative of fluorescence signal as a function of temperature (–dF/dT) was exported, and the background values of the buffered solution without protein was subtracted from each sample. Melting temperature was determined as the temperature where the derivative of fluorescence signal was at a minimum. Measurements were performed in triplicate. Melting temperature values reported are the average of each replicate ± standard deviation (SD). Statistical significance between groups was determined by one-way ANOVA using Prism software (Graph Pad).
Preparation of duplex substrates for base-flipping analysis
Oligonucleotides previously described for use in ADAR2 base-flipping analyses were purchased from Dharmacon (12). RNAs were purified by 18% PAGE as previously described. PAGE purified top and bottom strands were annealed for a final concentration of 30 μM edited strand, 45 μM guide strand, 30 mM Tris–HCl, 6% glycerol, 120 mM KCl, 3 mM EDTA, 0.006% NP-40 and 0.6mM DTT. The mixture was heated to 95°C for 5 min, and slowly cooled to room temperature.
Base-flipping assay using a fluorescent RNA substrate
Fluorescence measurements were performed using a CLARIOstar microplate reader and a Nunc MaxiSorp 384-well black bottom plate. Excitation was at 320 nm and fluorescence emission was scanned from 340 to 430 nm with 0.2 nm resolution. Spectra were obtained for solutions containing 2.5 μM RNA, with or without 10 μM ADAR2, in 36 mM Tris–HCl (pH 7.0 to 8.5) or Bis–Tris–HCl (pH 6.0 to 7.0), 7% glycerol, 142 mM KCl, 3.6 mM EDTA, 0.007% NP-40 and 0.7 mM DTT at room temperature (48). The background fluorescence of the enzyme buffered at each pH was subtracted from the spectrum of the complex, and the background fluorescence of the buffer alone at each pH was subtracted from the RNA. Each spectrum is an average of three independent measurements that were LOWESS fit using Graphpad Prism software. The fluorescence intensity values at λmax were used to determine the fluorescence enhancement by ADAR in the formula FE = (FIADAR-RNA – FIRNA)/FIRNA where FE is fluorescence enhancement, and FI values are the fluorescence intensity of samples containing either RNA or RNA in the presence of ADAR2. Fluorescence enhancement values were normalized so that the value of FIADAR-RNA at pH 7.5 corresponds to 1.
RESULTS
Intracellular acidification increases RNA editing
Kinetic analyses of ADAR activity in which a conserved glutamate in the deaminase domain was mutated to a glutamine, which is fully protonated under normal conditions, revealed enzyme hyperactivity (12,37,38). These observations suggested that editing activity by wild-type ADARs could be enhanced when the intracellular proton concentration is increased beyond that normally observed under control conditions. To determine the effects of cellular acidification on RNA editing, minigene constructs encoding the minimum essential RNA duplexes required for editing of 5HT2C, GluA2 and Gli1 transcripts (Figure 1A) were transiently co-expressed with either ADAR1 or ADAR2 in a human embryonic kidney (HEK293T) cell line, followed by quantitative analysis of site-selective A-to-I conversion (17,39,40). For these studies, the pH of the cell culture medium was adjusted to either pH 7.4 (control) or pH 6.7 (acidic) by manipulation of the bicarbonate concentration, which affects the buffering capacity of the media in the 5% CO2 incubator environment. Acidification significantly increased ADAR1 and ADAR2-mediated editing of numerous sites within the examined transcripts (Figure 1B–D), although the magnitude of the effect was both site-dependent and dependent upon the specific ADAR acting upon it. Notably, sites preferentially recognized by a specific ADAR underwent robust increases in editing under acidic conditions (up to ∼40%) when acted upon by that ADAR. For example, ADAR2-mediated editing of the GluA2 +262 site increased from 29.4% ± 0.02 to 68.3% ± 0.8 under acidic conditions. Such large changes in editing levels are comparable to the largest changes in editing that have been observed, such as in response to ADAR1 induction by interferon treatment (49). However, the effect of acidification on editing appeared less pronounced on several 5HT2C and GluA2 sites that are not preferentially targeted by a particular ADAR (Figure 1D and Supplementary Figure S1). To further examine these effects using a more complex RNA target, we used a high-throughput sequencing-based strategy to quantify the 32 RNA permutations arising from combinatorial editing at up to five sites within 5HT2C transcripts (sites A, B, E, C and D). Results from this analysis showed that acidification significantly increased the expression of several more highly edited 5HT2C RNA isoforms at the expense of less edited RNA species (Supplementary Figure S2).
Figure 1.

Effects of acidification on A-to-I editing in transfected cells. (A) The predicted secondary structures of GluA2, Gli1 and 5HT2C minigene-derived RNA transcripts is presented; editing sites are indicated in inverse lettering; nt = nucleotides. (B–D) Quantification of site-selective ADAR1 or ADAR2-mediated editing in minigene-derived RNAs from HEK293T cells incubated with control media (pH 7.4) or under acidic conditions (pH 6.7). Results for 24-h editing assays for GluA2 and 5HT2C, and 6-h editing assays for Gli1 are shown. Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups for each site in GluA2 and 5HT2C transcripts was determined using the Holm–Sidak t-test for multiple comparisons; statistical significance between groups for the Gli1 R/G site was determined using an unpaired t-test with Welch's correction; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001; ns, not significant.
To verify that changing the bicarbonate concentration to manipulate the extracellular pH concomitantly produced changes in intracellular pH, we used a fluorescent pH indicator, pHrodo Red, whose fluorescence intensity is inversely correlated with intracellular pH. pHrodo fluorescence significantly increased upon incubation with the acidic cell culture medium (pH 6.7) (Figure 2A). This highly fluorescent pHrodo Red signal colocalized with a vital nuclear dye (BioTracker 488), indicating acidification of the nucleus, which has been shown to be the major site of ADAR localization and RNA editing (50,51).
Figure 2.

Time- and pH-dependent effects of intracellular acidification on the site-specific editing of 5HT2C transcripts. (A) Representative 20× confocal images of live HEK293T cells incubated with control media (pH 7.4; top) or acidic media (pH 6.7; bottom) and double labelled with pHrodo Red AM Intracellular pH Indicator and BioTracker 488 Green Nuclear Dye (left); normalized mean pHrodo Red fluorescence intensity per cell is presented (right); means ± SD (n = 115 control cells from three independent experiments and n = 106 acidic cells from three independent experiments) were compared by using the unpaired t-test with Welch's correction; ****P < 0.0001. (B) A comparison of the extent of ADAR1 or ADAR2-mediated editing for 5HT2C transcripts at control (pH 7.4, —•—) or acidic pH (pH 6.7, —○—) after 1, 3, 6, 12 or 24 h is presented. Plotted values represent the means of three biological replicates ± SD. Statistical significance between groups at a given time point was determined using Sidak's multiple comparisons test; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. (C) Quantification of ADAR1 or ADAR2-mediated 5HT2C editing across a pH range from 6.7 to 7.4. Plotted values are the means of three biological replicates (○) ± SD.
To examine pH-dependent increases in RNA editing as a function of time, we quantified A-to-I modification of 5HT2C transcripts by ADAR1 or ADAR2 over a time course from 1 to 24 h (Figure 2B and Supplementary Figure S3). We chose 5HT2C RNA for these studies because it contains multiple editing sites that are recognized by both ADARs, though previous studies have shown that ADAR1 preferentially edits the A- and B-sites, and ADAR2 preferentially edits the D-site (17). The extent of editing for all five 5HT2C sites was increased significantly under acidic conditions for most time points. For several sites, a significant increase in editing at pH 6.7 (acidic) compared to pH 7.4 (control) could be observed in as little as 1 h and continued to increase over control levels over the entire 24-h time period. Interestingly, ADAR1 editing of the A-site as well as ADAR2 editing of several sites appeared to increase logarithmically under acidic conditions, whereas the time-dependent increases in editing under control conditions appeared more linear. We also examined pH-dependent alterations in Gli1 editing at two timepoints, 6 and 24 h (Figure 1C and Supplementary Figure S4, respectively). Acidification-induced increases in editing at the R/G site were observed only at the earlier timepoint. Interestingly, the absence of increased R/G site editing at reduced pH after 24 h was concurrent with increased editing of additional adenosine residues within the Gli1 transcript, many of which exhibited increased editing upon acidification. These results indicate that editing of the R/G site reached its endpoint by 24 h and further suggest that the rate of catalysis for this site is more rapid than for other Gli1 sites.
To further examine how RNA editing is affected by alterations in pH, we analyzed ADAR1 or ADAR2-mediated editing of 5HT2C transcripts over a pH range from 6.7 to 7.4 that was established by varying the concentration of bicarbonate in the cell culture medium between 0 and 3.7 mg/ml NaHCO3 (Supplementary Figure S5A). ADAR1 editing at the A-, B- and C-sites, as well as ADAR2 editing at all five sites, showed incremental increases in editing with decreasing pH (Figure 2C; statistical analysis of Figure 2C shown in Supplementary Figure S5B; and Supplementary Figure S6). The largest stepwise increase in pH-dependent ADAR1 editing was observed between pH 7.4 and 7.3, whereas the largest stepwise increase in pH-dependent ADAR2 editing was observed between pH 6.9 and 6.8.
Although numerous studies have concluded that differences in steady-state ADAR protein levels do not fully account for differences in the cell- and region-specific RNA editing profiles, previous studies in cell culture model systems have shown a correlation between editing and ADAR expression (5,31–36,52,53). To determine whether the reductions in extracellular pH that promoted increased editing affected ADAR protein levels in transfected cells, ADAR expression from HEK293T cells co-transfected with ADAR1 or ADAR2 and the 5HT2C minigene were quantified after a 24-h incubation with cell culture medium under control (pH 7.4) or acidic (pH 6.7) conditions. Quantitative western blotting analyses revealed significant increases in both ADAR1 and ADAR2 protein expression upon cellular acidification (Figure 3), suggesting that pH-dependent increases in ADAR protein in this model system could be responsible for the increases in editing observed at reduced pH.
Figure 3.
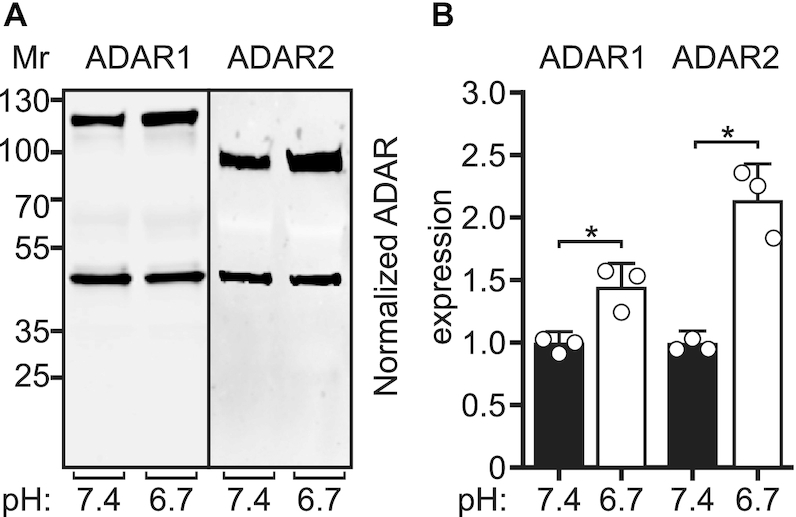
ADAR protein expression in response to acidification in HEK293T cells. (A) Representative Western blots for ADAR1 (110 kDa) and ADAR2 (80 kDa) protein expression from HEK293T cells incubated with control (pH 7.4) or acidic media (pH 6.7); β-actin (43 kDA) was used as a loading control. (B) Quantification of ADAR1 and ADAR2 protein expression normalized to the β-actin loading control is presented. Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups was determined using the unpaired t-test with Welch's correction; *P ≤ 0.05.
Because HEK293 cell lines are largely devoid of endogenous editing activity, they are well-suited to study the isolated, site-specific activities of recombinant ADAR1 or ADAR2 on various substrates via transient transfection (35). However, ADAR expression and RNA editing in a heterologous expression system may be subject to different regulatory mechanisms than those present in cell lines exhibiting endogenous editing activity. To address this possibility, we investigated the effect of acidification on RNA editing in HeLa cells, which show appreciable levels of endogenous editing (35). Relative to HeLa cells incubated with control medium, HeLa cells incubated with acidic culture medium displayed increased pHrodo fluorescence, demonstrating that this manipulation also produced intracellular acidification in this cell line (Figure 4A and Supplementary Figure S7). We then assessed the editing of three transcripts endogenously expressed in HeLa cells: Eef2k, Blcap and Cog3. Eef2k and Blcap previously were shown to be preferentially edited by ADAR1, while Cog3 was shown to be preferentially edited by ADAR2 (54–57). Acidification significantly increased editing of these transcripts without concomitant changes in endogenous ADAR1 protein expression (Figure 4B and C). Even though Cog3 editing was significantly increased at reduced pH, ADAR2 protein expression was not detectable under control or acidic conditions, though quantitative RT-PCR (qRT-PCR) analyses revealed a significant decrease in ADAR2 mRNA expression at reduced pH (Supplementary Figure S8). To determine whether changes in the expression levels of the RNA editing substrates could account for pH-dependent effects on editing, we also used qRT-PCR to examine the expression of each of the three editing substrates under control and acidic conditions. Blcap expression remained unchanged between conditions, while Eef2k and Cog3 expression decreased and increased at acidic pH, respectively, suggesting that acidification increases the extent of editing regardless of whether or not substrate levels change (Figure 4D). Taken together, these data indicate that acidification-induced increases in endogenous RNA editing cannot be explained by accompanying changes in the expression levels of ADARs or their RNA substrates in HeLa cells.
Figure 4.

Effects of cellular acidification on endogenous A-to-I editing in HeLa cells. (A) Quantification of normalized mean pHrodo Red fluorescence intensity per cell from HeLa cells incubated with control media (pH 7.4) or acidic media (pH 6.7) is presented; means ± SD (n = 124 control cells from three independent experiments and n = 115 acidic cells from three independent experiments) were compared using the unpaired t-test with Welch's correction; ****P < 0.0001. (B) Representative electropherogram traces from Sanger sequencing of Cog3, Eef2k or Blcap RT-PCR amplicons generated from HeLa cells incubated with control media (pH 7.4; top) or acidic media (pH 6.7; bottom). The edited position within each trace is highlighted in yellow (left); quantification of editing is presented (right) after 24 h of incubation with control (pH 7.4) or acidic (pH 6.7) media. Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups was determined using the unpaired t-test with Welch's correction; **P ≤ 0.01; ***P ≤ 0.001; ****P < 0.0001. (C) Representative Western blot for ADAR1 protein expression from HeLa cells incubated with control (pH 7.4) or acidic media (pH 6.7); β-actin (43 kDA) was used as a loading control (left). Quantification of ADAR1 protein expression normalized to the β-actin loading control is presented (right). Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups was determined using the unpaired t-test with Welch's correction; ns, not significant. (D) Quantification of relative mRNA expression for Cog3, Eef2k and Blcap from HeLa cells incubated with control (pH 7.4) or acidic media (pH 6.7). Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups was determined using unpaired t-test; *P ≤ 0.05; **P ≤ 0.01; ns, not significant. (E) Quantification of normalized mean pHrodo Red fluorescence intensity per cell from HeLa cells treated with DMSO or 5 μM niclosamide is presented; means ± SD (n = 69 DMSO-treated cells from three independent experiments and n = 70 niclosamide-treated cells from three independent experiments) were compared using unpaired t-test with Welch's correction; ****P < 0.0001. (F) Representative electropherogram traces from Sanger sequencing of Cog3, Eef2k or Blcap RT-PCR amplicons generated from HeLa cells treated with DMSO (top) or 5 μM niclosamide (bottom). The edited position within each trace is highlighted in yellow (left); quantification of editing is presented (right). Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups was determined using unpaired t-test with Welch's correction; *P ≤ 0.05; **P ≤ 0.01.
To verify that the observed increases in RNA editing resulted from decreased pH rather than cellular mechanisms associated with altered extracellular bicarbonate levels, we employed an alternative method to induce intracellular acidification by treating cells with niclosamide, a protonophore which has previously been shown to trigger intracellular acidification in the absence of extracellular acidification (58). Using pHrodo Red, we confirmed that niclosamide treatment induced intracellular acidification in HeLa and HEK293T cells, although the extent of increase in pHrodo fluorescence after niclosamide treatment was less than that observed after incubation with acidic medium (Figure 4E, Supplementary Figure S9, and Supplementary Figure S10A and B). Accordingly, smaller but significant increases in editing were observed upon 6-h treatment with 5 μM niclosamide compared to vehicle-treated controls (Figure 4F and Supplementary Figure S10C and D).
The RNA editing reaction is intrinsically pH-sensitive
While increases in RNA editing produced by acidification in HEK293T cells could result from increased ADAR protein expression, such a mechanism cannot account for the acidification-induced increases in editing in HeLa cells. As such, the observed increases in RNA editing at reduced pH also could be explained by other molecular mechanisms including activation of various pH-regulated signaling pathways or the effects of pH on RNA structure and the intrinsic pH sensitivity of ADAR activity. To directly examine this last possibility, in vitro editing assays were performed to quantify deamination rate constants (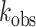 ) at varying pH (from pH 6.0 to pH 8.5) using purified, recombinant ADAR protein and an in vitro transcribed 5HT2C substrate. For most sites examined, an inverse correlation between deamination rate and pH between pH 6.5 and 8.5 was observed. For example, ADAR1 deamination of the A-site was most efficient at pH 6.5 (Figure 5A). Similarly, ADAR2 deamination of the A-, B- and C-sites also was most efficient at pH 6.5 and pH 7.0 (Figure 5B). The rate of editing for these sites is about 1.5-fold less efficient at pH 7.5 than at either pH 7.0 or pH 6.5. To assess whether the observed differences in catalytic rate resulted from changes in pH-dependent stability of ADAR proteins, the melting temperature of recombinant ADAR2 protein was quantified using SYPRO Orange, a dye that increases in fluorescence intensity upon thermal denaturation of the protein (59). Results from this ThermoFluor analysis revealed that ADAR2 was relatively stable from pH 6.5 to pH 8.5, with a melting temperature of ∼53°C across this range (Figure 5C). At pH 6.0 however, the melting temperature of the ADAR2 protein was significantly decreased, an instability that paralleled the observed reduction in catalytic rate for the 5HT2C editing sites (Figure 5A–C).
) at varying pH (from pH 6.0 to pH 8.5) using purified, recombinant ADAR protein and an in vitro transcribed 5HT2C substrate. For most sites examined, an inverse correlation between deamination rate and pH between pH 6.5 and 8.5 was observed. For example, ADAR1 deamination of the A-site was most efficient at pH 6.5 (Figure 5A). Similarly, ADAR2 deamination of the A-, B- and C-sites also was most efficient at pH 6.5 and pH 7.0 (Figure 5B). The rate of editing for these sites is about 1.5-fold less efficient at pH 7.5 than at either pH 7.0 or pH 6.5. To assess whether the observed differences in catalytic rate resulted from changes in pH-dependent stability of ADAR proteins, the melting temperature of recombinant ADAR2 protein was quantified using SYPRO Orange, a dye that increases in fluorescence intensity upon thermal denaturation of the protein (59). Results from this ThermoFluor analysis revealed that ADAR2 was relatively stable from pH 6.5 to pH 8.5, with a melting temperature of ∼53°C across this range (Figure 5C). At pH 6.0 however, the melting temperature of the ADAR2 protein was significantly decreased, an instability that paralleled the observed reduction in catalytic rate for the 5HT2C editing sites (Figure 5A–C).
Figure 5.

Effect of varying pH on in vitro A-to-I editing. (A) Rate of in vitro ADAR1- or (B) ADAR2-mediated 5HT2C editing at half-pH intervals from 6.0 to 8.5. Plotted values represent the means of three technical replicates (○) ± SD. Statistical significance between groups was determined using one-way ANOVA with Tukey's multiple comparisons test; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. (C) Quantification of ADAR2 melting temperature across the pH range used for in vitro editing experiments. Plotted values represent the means of three technical replicates (○) ± SD. Statistical significance between groups was determined using one-way ANOVA with Tukey's multiple comparisons test; **P ≤ 0.01; ns, not significant. (D) Rate of in vitro ADAR2d-mediated Gli1 R/G site editing. Plotted values represent the means of three technical replicates (○) ± SD. Statistical significance between groups was determined using one-way ANOVA with Tukey's multiple comparisons test; **P ≤ 0.01; ****P ≤ 0.0001.
Taken together, these in vitro analyses demonstrate that the RNA editing reaction is intrinsically pH-sensitive, a property that may arise through changes in ADAR-substrate binding or catalysis at reduced pH, but not by changes in ADAR protein expression. To exclude the possibility that the observed increase in editing rates resulted from enhanced ADAR2-substrate binding via the double-stranded RNA binding domains (dsRBDs), the pH-sensitivity of the ADAR catalytic domain alone was assessed by taking advantage of the efficient editing of Gli1 by the ADAR2 deaminase domain (ADAR2d) lacking dsRBDs (39). ADAR2d deamination of the Gli1 R/G site was 40- to 130-fold more efficient under acidic conditions than at pH 7.5, with 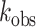 = 3.1 min–1 ± 0.2 at pH 6.5 and
= 3.1 min–1 ± 0.2 at pH 6.5 and 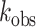 = 0.023 min–1 ± 0.006 at pH 7.5 (Figure 5D). These data indicate that RNA editing can be facilitated under acidic conditions independently of substrate interactions with the ADAR dsRBDs.
= 0.023 min–1 ± 0.006 at pH 7.5 (Figure 5D). These data indicate that RNA editing can be facilitated under acidic conditions independently of substrate interactions with the ADAR dsRBDs.
Protonation of a conserved glutamate residue in the ADAR base-flipping loop partially accounts for increases in RNA editing at acidic pH
A recent investigation of the structural basis for base-flipping by ADAR2 revealed the importance of a highly conserved glutamate, E488 (corresponding to E1008 in ADAR1), residing in the deaminase domain of the enzyme (37). This residue stabilizes the flipped-out conformation of the RNA duplex, presumably by occupying the space vacated by the flipped-out adenosine and hydrogen bonding with the complementary-strand orphaned base (Figure 6A and B and Supplementary Figure S11). Mutant ADAR proteins bearing a glutamate-to-glutamine substitution at this residue (ADAR1-Q and ADAR2-Q) exhibit increased catalytic activity via enhanced base-flipping (12,31,32). As this glutamine is fully protonated under normal physiologic conditions at pH 7.4, these observations are consistent with the idea that the corresponding glutamate residue in wild-type ADARs requires protonation to support RNA stabilization during the base-flipping step in catalysis (Figure 6A and B). To further examine how base-flipping is modulated by ADAR protonation, we compared base-flipping for ADAR2 and ADAR2-Q proteins as a function of pH using a 2-aminopurine (2-AP)-modified GluA2 transcript to measure 2-AP fluorescence intensity, which has been shown previously to correlate with base-flipping (12,48). The fluorescence intensity observed with the ADAR2-Q mutant was greater than that exhibited with the wild-type ADAR2 protein at each pH, confirming that ADAR2-Q has enhanced base-flipping abilities (Figure 6C). However, a differential pH dependence between wild-type ADAR2 and the ADAR2-Q mutant enzyme was observed where the fluorescence intensity with the wild-type enzyme increased with decreasing pH, whereas fluorescence intensity with the ADAR2-Q mutant was maximal at pH 7.0, but dropped off significantly with increasing or decreasing pH (Figure 6C). These data indicate that ADAR base-flipping is intrinsically pH-dependent and that base-flipping by the ADAR-Q mutant enzyme is less affected by acidification than the wild-type ADAR2 protein.
Figure 6.

Effects of a glutamate-to-glutamine substitution on the pH-dependence of ADAR base-flipping and editing. (A) The crystal structure of ADAR2d bound to dsRNA (PDB ID: 5HP3 & 5ED1) shows the base-flipped conformation stabilized by contacts between residue 488 and the orphan base. (B) An illustration of the hydrogen bonding contact between ADAR2 and the orphan base showing protonation-dependent hydrogen bonding for wild-type ADAR2. (C) Normalized fluorescence enhancement from a dsRNA substrate containing 2-aminopurine in the edited position, corresponding to base-flipping by the ADAR2 enzyme. Plotted values represent the means of three technical replicates ± SD. Statistical significance between groups was determined using one-way ANOVA with Tukey's multiple comparisons test; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001; ns, not significant. (D) Rates of in vitro ADAR2d- and ADAR2d-Q mediated Gli1 (+23 site) editing. Plotted values represent the means of three technical replicates (○) ± SD. Statistical significance between groups was determined using the Holm-Sidak t-test for multiple comparisons; *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001. (E) Quantification of ADAR2d and ADAR2d-Q melting temperatures across the pH range used for in vitro editing experiments. Plotted values represent the means of three technical replicates (○) ± SD. (F) Quantification of the extent of acidification-induced increases in 5HT2C editing mediated by wild-type ADARs or ADAR-Q mutants (Δ % editing = % site-selective editing at pH 6.7 - % site-selective editing at pH 7.4). Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups for each 5HT2C site was determined using the Holm–Sidak t-test for multiple comparisons; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
To further compare the relative pH sensitivity of wild-type and mutant (ADAR-Q) proteins, we measured the rate of Gli1 deamination (+23 site) by ADAR2d and ADAR2d-Q enzymes from pH 6.5-8.0. Interestingly, the deamination rates for the wild-type and mutant deaminase domains are similar at pH 6.5, with 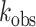 = 0.91 min–1 ± 0.2 and
= 0.91 min–1 ± 0.2 and 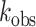 = 1.3 min–1 ± 0.09, respectively. However, the efficiency of wild-type ADAR2d deamination decreased over 450-fold with increasing pH, while the efficiency of ADAR2d-Q deamination only decreased between 2.5- and 5-fold with increasing pH (Figure 6D). Moreover, ADAR2d-Q deaminated Gli1 more efficiently than ADAR2d at each pH despite decreased stability of the mutant protein across the entire pH range (Figure 6E). These results show that the pH-dependence of deamination is much greater for wild-type ADAR2d than for ADAR2d-Q. Finally, the site-specific editing of 5HT2C transcripts was assessed using wild-type and ADAR-Q mutant enzymes in transfected HEK293T cells incubated under control (pH 7.4) or acidic (pH 6.7) conditions. Results from this analysis showed that while the extent of editing was increased for both ADAR1-Q and ADAR2-Q mutants under acidic conditions, acidification-induced increases in editing by ADAR1-Q and ADAR2-Q were attenuated relative to those exhibited by their wild-type ADAR counterparts (Figure 6F). Taken together, these data suggest that protonation of ADAR1 at E1008 or ADAR2 at E488 partially accounts for the increases in RNA editing observed at acidic pH.
= 1.3 min–1 ± 0.09, respectively. However, the efficiency of wild-type ADAR2d deamination decreased over 450-fold with increasing pH, while the efficiency of ADAR2d-Q deamination only decreased between 2.5- and 5-fold with increasing pH (Figure 6D). Moreover, ADAR2d-Q deaminated Gli1 more efficiently than ADAR2d at each pH despite decreased stability of the mutant protein across the entire pH range (Figure 6E). These results show that the pH-dependence of deamination is much greater for wild-type ADAR2d than for ADAR2d-Q. Finally, the site-specific editing of 5HT2C transcripts was assessed using wild-type and ADAR-Q mutant enzymes in transfected HEK293T cells incubated under control (pH 7.4) or acidic (pH 6.7) conditions. Results from this analysis showed that while the extent of editing was increased for both ADAR1-Q and ADAR2-Q mutants under acidic conditions, acidification-induced increases in editing by ADAR1-Q and ADAR2-Q were attenuated relative to those exhibited by their wild-type ADAR counterparts (Figure 6F). Taken together, these data suggest that protonation of ADAR1 at E1008 or ADAR2 at E488 partially accounts for the increases in RNA editing observed at acidic pH.
RNA editing increases during hypoxia
Various physiologic and pathophysiologic conditions have been shown to induce changes in intracellular or extracellular pH such as hypoxia, inflammatory signaling, tumorigenesis, and metabolic acidosis (60,61). Previous studies have shown that acid-base disturbances that occur during hypoxia induce both intra- and extracellular acidification (62). To determine whether physiologic manipulation of the cell culture environment could modulate the extent of RNA editing by ADAR1 and ADAR2, we used a cell culture model of hypoxia (43). Transfected HEK293T cells were incubated for 24 h under hypoxic (1% O2) or normoxic (20% O2) conditions, followed by quantification of 5HT2C editing profiles, determinations of the pH of the cell culture media, as well as quantification of ADAR protein expression. Results from these studies showed that hypoxia significantly increased ADAR1 and ADAR2-mediated editing of all five 5HT2C sites and concomitantly decreased the pH of the cell culture medium by 0.6 ± 0.01 pH units relative to normoxic conditions (Figure 7A–C). Furthermore, no significant differences in ADAR1 or ADAR2 protein expression were detected between hypoxic and normoxic conditions, indicating that the increased RNA editing produced by hypoxia cannot be explained by concomitant changes in ADAR expression (Figure 7D and E). Similar effects of hypoxia on endogenous RNA editing and ADAR expression were observed in HeLa cells (Supplementary Figure S12), indicating that that the extent of RNA editing is inversely correlated with pH changes produced by a physiologically relevant cell culture model of metabolic stress.
Figure 7.

A-to-I editing in response to hypoxia. (A) Quantification of the extent of ADAR1 or (B) ADAR2-mediated 5HT2C editing from HEK293T cells incubated under normoxic (20% O2) or hypoxic (1% O2) conditions. Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups for each 5HT2C site was determined using the Holm-Sidak t-test for multiple comparisons; **P ≤ 0.01; ***P ≤ 0.001. (C) pH of the cell culture medium following 24-h incubation of HEK293T cells under normoxic (20% O2) or hypoxic (1% O2) conditions. Means ± SD from three independent experiments (○) were compared using the unpaired t-test with Welch's correction; ***P ≤ 0.001. (D) Representative Western blots for ADAR1 (110 kDa) and ADAR2 (80 kDa) protein expression from HEK293T cells incubated under normoxic (20% O2) or hypoxic (1% O2) conditions; β-actin (43 kDA) was used as a loading control. (E) Quantification of ADAR1 and ADAR2 protein expression normalized to the β-actin loading control is presented. Plotted values represent the means of three biological replicates (○) ± SD. Statistical significance between groups was determined using the unpaired t-test with Welch's correction; ns, not significant.
DISCUSSION
The mechanisms underlying changes in RNA editing profiles in response to physiologic signals are not well-defined. Most of the known trans-acting regulators of editing modulate ADAR protein levels, yet steady-state ADAR protein expression fails to fully account for the observed spatiotemporal variations in A-to-I conversion (5,31–36). Though dynamic regulation of ADAR activity—rather than ADAR expression—could explain significant changes in RNA editing, the mechanisms regulating such activity remain elusive. Previous structural and biochemical characterization of ADARs and ADAR mutants has suggested a pH-sensitive deamination mediated by these enzymes (12,37,38). Therefore, our studies have focused on how changes in pH regulate RNA editing. Analysis of numerous editing sites from various RNA substrates in HEK293T and HeLa cells, as well as in vitro biochemical systems, revealed significant increases in ADAR1 and ADAR2-mediated editing under acidic conditions relative to editing at a physiologic pH of ∼7.4 (Figures 1–2 and Figures 4–5). These multiple lines of evidence from different model systems indicate that the results presented here are robust and reproducible. Moreover, several sites, including the A-, B- and E-sites of 5HT2C RNAs, as well as various adenosines within Gli1 transcripts, are virtually unrecognized or edited at very low levels by one or both ADARs at pH 7.4, but undergo robust editing as a result of acidification (Figure 1 and Supplementary Figure S4). Although these increases in editing were accompanied by increases in intracellular ADAR protein levels in HEK293T cells, results from experiments using HeLa cells and in vitro studies using recombinant ADARs indicated that increased ADAR expression is not required to observe acidification-mediated increases in editing (Figures 4–5). Indeed, these in vitro data demonstrate an intrinsic and robust pH-sensitivity of ADAR catalysis (Figure 5). While acidification could alter the rate of editing by affecting ADAR binding and/or catalytic efficiency, pH-sensitive editing of Gli1 by ADAR2d (lacking the dsRBDs) showed that increased ADAR activity did not result solely from increased dsRBD-mediated binding under acidic conditions (Figures 5D and 6D). Rather, our data show that the ADAR deaminase domain—and specifically the base-flipping loop—is fundamental to the intrinsic pH-sensitivity of ADAR catalysis (Figures 5D and 6).
During base-flipping, a highly conserved glutamate residue within the ADAR base-flipping loop invades the vacated space and hydrogen bonds with the base opposite the flipped-out adenosine to stabilize the strained nucleic acid conformation (Figure 6A and B) (37). Consistent with the hypothesis that this interaction may be sensitive to changes in proton concentration, mutation of this critical glutamate to a glutamine, which is fully protonated at pH 7.4, results in increased catalytic efficiency via enhanced base-flipping of the ADAR-Q mutant (12,38). Our analysis suggests that the ADAR2 reaction is accelerated by low pH regardless of the identity of the orphan base. Protonation of E488 enables this residue to donate a hydrogen-bond to contact an orphan cytidine (Figure 6A and B). In addition, protonation of this residue neutralizes the negative charge on the side chain, decreasing charge repulsion with RNA during the flipping step for either adenosine-uridine base pairs (Supplementary Figure S11) or adenosine-cytidine mismatches (Figure 6B). Our studies further show that base-flipping and editing activity for wild-type ADARs increase with decreasing pH, yet the pH-dependency of these properties is diminished for ADAR-Q mutants (Figure 6C–F). While the pKa of ADAR1 E1008 and ADAR2 E488 have not been determined, our data suggest that protonation of these single amino acid residues within the ADARs enhances base-flipping to increase RNA editing at acidic pH, and is the primary determinant that accounts for the pH‐dependence of deamination (Figure 6C–F). Though our studies using ADAR-Q mutants also suggest that these mutant enzymes are resistant but not completely insensitive to reductions in pH, the residual pH-sensitivity exhibited by ADAR-Q mutants in vitro may be attributed to protonation of other amino acid residues involved in other steps of the editing reaction (e.g. RNA binding, deprotonation of the reactive water molecule, etc.), as well as pH-dependent increases in the thermodynamic stability of targeted RNA duplexes. These factors, as well as increased ADAR expression under acidic conditions, may account for acidification-induced increases in editing by ADAR-Q mutants in HEK293T cells.
The pH optima for most enzymes coincide with the pH of the subcellular compartments in which they reside (63,64). For example, the pH optimum of many proteases involved in prohormone processing coincides with an intragranular pH of ∼5.5, whereas the pH optimum of many cytoplasmic enzymes is ∼7.4 (65,66). Surprisingly, ADARs function optimally between pH 6.5–7.0, well below the normal pH of the nucleus or cytoplasm (60). For many 5HT2C sites, large stepwise changes in editing were observed between pH 7.4 and 7.3 (for ADAR1 editing) or between pH 6.9 and 6.8 (for ADAR2 editing) in HEK293T cells (Figure 2C), and between pH 7.5 and 7.0 in vitro (Figure 5), suggesting that ADARs could serve as pH sensors to modulate RNA editing patterns in response to physiologically-driven pH shifts.
Cytoplasmic acidification often results from the accumulation of acid equivalents produced as metabolic byproducts (60). Although cells engage various regulatory mechanisms to maintain pH homeostasis, these systems can be overwhelmed during periods of unusually high metabolic activity, leading to intracellular acidification (60). Cells grown in a hypoxic environment demonstrated both acidification of their culture medium as well as a significant increase in site-selective A-to-I conversion without concomitant increases in ADAR expression (Figure 7 and Supplementary Figure S12). These observations are consistent with previous studies that have reported increases in editing under hypoxic conditions but lacked evidence to suggest that the observed increases in RNA editing are driven by parallel increases in ADAR protein levels or through modulation of other hypoxia-sensitive cellular pathways (67–69). The present studies support the hypothesis that metabolic stress during hypoxia triggers intracellular acidification, which in turn enhances base-flipping activity to increase the overall ADAR catalytic rate and editing. Similar pH-dependent regulatory mechanisms may exist in conditions such as inflammation and epilepsy, which also are associated with both intracellular acidification as well as increased RNA editing (23,70–76).
During hypoxia/ischemia, epilepsy, MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes), and other pathophysiologic conditions in which pH homeostasis is disrupted, electrically-active cells such as neurons and cardiomyocytes experience broad disturbances in ion dynamics, often resulting in increased intracellular Ca2+ concentration to induce cytotoxicity and membrane hyperexcitability (60). Though the present studies of pH-dependent increases in RNA editing were limited to several model transcripts, it is likely that editing of many sites in the transcriptome increases upon intracellular acidification since editing is favored under such conditions. While it is unknown how such global increases in editing might influence overall physiology, studies using Drosophila model systems have shown that ADAR overexpression or knockdown results in decreased or increased neuronal excitability, respectively (77). Since many editing-dependent recoding events affect the function of proteins involved in membrane excitability, it is intriguing to speculate that increased editing of various RNA targets may serve as a pH-dependent homeostatic mechanism to limit membrane hyperexcitability and protect against excitotoxic damage (77,78).
The observation that acidic pH enhances base-flipping and increases the rate of deamination by ADARs has implications beyond our understanding of mechanisms of natural regulation of A-to-I editing. Several recent reports have described efforts to direct ADAR reactions for therapeutic benefit (79,80). One approach uses a guide RNA to form a duplex at a target site that can recruit endogenous ADAR enzymes for deamination of a specific adenosine (49,81). Thus, this directed RNA editing approach can ‘repair’ G-to-A mutations associated with genetic disease. Optimization of ADAR guide RNAs requires a comprehensive understanding of factors that control RNA editing efficiency. The results described here are likely to stimulate future efforts to develop modifications to guide RNAs that mimic the effects of low pH.
DATA AVAILABILITY
The data that support the findings of this work are available from the corresponding author upon reasonable request. Structures referenced can be found in the Protein Data Bank under accession codes 5HP3 & 5ED1.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Kayla Shumate for critical reading of the manuscript. We also would like to thank the Vanderbilt Technologies for Advanced Genomics (VANTAGE), the Vanderbilt Cell Imaging Shared Resource (CISR), the Vanderbilt Creative Data Solutions Shared Resource (CDS), the University of California Davis DNA Technologies Core and the University of California Davis Campus Mass Spectrometry Facilities Core. VANTAGE is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126), and NIH/NCRR (G20 RR030956), while CISR is supported by NIH grant DK020593. We also are grateful to Henrietta Lacks and to her surviving family members for their contribution of HeLa cells and their impact upon biomedical research.
Contributor Information
Turnee N Malik, Training Program in Neuroscience, Vanderbilt University, Nashville, TN 37232, USA.
Erin E Doherty, Department of Chemistry, University of California, Davis, Davis, CA 95616, USA.
Vandana M Gaded, Department of Chemistry, University of California, Davis, Davis, CA 95616, USA.
Theodore M Hill, Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN 37232, USA.
Peter A Beal, Department of Chemistry, University of California, Davis, Davis, CA 95616, USA.
Ronald B Emeson, Training Program in Neuroscience, Vanderbilt University, Nashville, TN 37232, USA; Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN 37232, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Joel G. Hardman and Mary K. Parr Endowed Chair in Pharmacology and the National Institutes of Health [DK119508 to R.B.E. and GM061115 to P.A.B]; E.E.D. was supported by a training grant [GM113770] from the National Institutes of Health. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Funding for open access charge: National Institutes of Health [DK119508].
Conflict of interest statement. P.A.B. is a consultant for Agios Pharmaceuticals, ProQR Therapeutics and Beam Therapeutics. P.A.B. has ownership interest in Beam Therapeutics.
REFERENCES
- 1. Grosjean H. Fine-Tuning of RNA Functions by Modification and Editing. 2005; NY: Springer. [Google Scholar]
- 2. Walkley C.R., Li J.B.. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017; 18:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roundtree I.A., Evans M.E., Pan T., He C.. Dynamic RNA modifications in gene expression regulation. Cell. 2017; 169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bazak L., Haviv A., Barak M., Jacob-Hirsch J., Deng P., Zhang R., Isaacs F.J., Rechavi G., Li J.B., Eisenberg E.et al.. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014; 24:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tan M.H., Li Q., Shanmugam R., Piskol R., Kohler J., Young A.N., Liu K.I., Zhang R., Ramaswami G., Ariyoshi K.et al.. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017; 550:249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bass B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002; 71:817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010; 79:321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eggington J.M., Greene T., Bass B.L.. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011; 2:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nishikura K., Yoo C., Kim U., Murray J.M., Estes P.A., Cash F.E., Liebhaber S.A.. Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J. 1991; 10:3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Phelps K.J., Tran K., Eifler T., Erickson A.I., Fisher A.J., Beal P.A.. Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res. 2015; 43:1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goodman R.A., Macbeth M.R., Beal P.A.. ADAR proteins: structure and catalytic mechanism. Curr. Top. Microbiol. Immunol. 2012; 353:1–33. [DOI] [PubMed] [Google Scholar]
- 12. Kuttan A., Bass B.L.. Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E3295–E3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paul M.S., Bass B.L.. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 1998; 17:1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jacobs M.M., Fogg R.L., Emeson R.B., Stanwood G.D.. ADAR1 and ADAR2 expression and editing activity during forebrain development. Dev. Neurosci. 2009; 31:223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brusa R., Zimmermann F., Koh D.S., Feldmeyer D., Gass P., Seeburg P.H., Sprengel R.. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995; 270:1677–1680. [DOI] [PubMed] [Google Scholar]
- 16. Bhalla T., Rosenthal J.J., Holmgren M., Reenan R.. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat. Struct. Mol. Biol. 2004; 11:950–956. [DOI] [PubMed] [Google Scholar]
- 17. Burns C.M., Chu H., Rueter S.M., Hutchinson L.K., Canton H., Sanders-Bush E., Emeson R.B.. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997; 387:303–308. [DOI] [PubMed] [Google Scholar]
- 18. Higuchi M., Maas S., Single F.N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P.H.. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000; 406:78–81. [DOI] [PubMed] [Google Scholar]
- 19. Palladino M.J., Keegan L.P., O’Connell M.A., Reenan R.A.. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell. 2000; 102:437–449. [DOI] [PubMed] [Google Scholar]
- 20. Tonkin L.A., Saccomanno L., Morse D.P., Brodigan T., Krause M., Bass B.L.. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 2002; 21:6025–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hartner J.C., Schmittwolf C., Kispert A., Muller A.M., Higuchi M., Seeburg P.H.. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004; 279:4894–4902. [DOI] [PubMed] [Google Scholar]
- 22. Wang Q., Miyakoda M., Yang W., Khillan J., Stachura D.L., Weiss M.J., Nishikura K.. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 2004; 279:4952–4961. [DOI] [PubMed] [Google Scholar]
- 23. Vollmar W., Gloger J., Berger E., Kortenbruck G., Kohling R., Speckmann E.J., Musshoff U.. RNA editing (R/G site) and flip-flop splicing of the AMPA receptor subunit GluR2 in nervous tissue of epilepsy patients. Neurobiol. Dis. 2004; 15:371–379. [DOI] [PubMed] [Google Scholar]
- 24. Niswender C.M., Herrick-Davis K., Dilley G.E., Meltzer H.Y., Overholser J.C., Stockmeier C.A., Emeson R.B., Sanders-Bush E.. RNA editing of the human serotonin 5-HT2C receptor. alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology. 2001; 24:478–491. [DOI] [PubMed] [Google Scholar]
- 25. Hideyama T., Yamashita T., Aizawa H., Tsuji S., Kakita A., Takahashi H., Kwak S.. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol. Dis. 2012; 45:1121–1128. [DOI] [PubMed] [Google Scholar]
- 26. Breen M.S., Dobbyn A., Li Q., Roussos P., Hoffman G.E., Stahl E., Chess A., Sklar P., Li J.B., Devlin B.et al.. Global landscape and genetic regulation of RNA editing in cortical samples from individuals with schizophrenia. Nat. Neurosci. 2019; 22:1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rice G.I., Kasher P.R., Forte G.M., Mannion N.M., Greenwood S.M., Szynkiewicz M., Dickerson J.E., Bhaskar S.S., Zampini M., Briggs T.A.et al.. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012; 44:1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rosenthal J.J. The emerging role of RNA editing in plasticity. J. Exp. Biol. 2015; 218:1812–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garrett S., Rosenthal J.J.. RNA editing underlies temperature adaptation in K+ channels from polar octopuses. Science. 2012; 335:848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marcucci R., Brindle J., Paro S., Casadio A., Hempel S., Morrice N., Bisso A., Keegan L.P., Del Sal G., O’Connell M.A. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 2011; 30:4211–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wahlstedt H., Daniel C., Enstero M., Ohman M.. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009; 19:978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hood J.L., Morabito M.V., Martinez C.R. 3rd, Gilbert J.A., Ferrick E.A., Ayers G.D., Chappell J.D., Dermody T.S., Emeson R.B.. Reovirus-mediated induction of ADAR1 (p150) minimally alters RNA editing patterns in discrete brain regions. Mol. Cell. Neurosci. 2014; 61:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Porath H.T., Hazan E., Shpigler H., Cohen M., Band M., Ben-Shahar Y., Levanon E.Y., Eisenberg E., Bloch G.. RNA editing is abundant and correlates with task performance in a social bumblebee. Nat. Commun. 2019; 10:1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J.B., Church G.M.. Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat. Neurosci. 2013; 16:1518–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schaffer A.A., Kopel E., Hendel A., Picardi E., Levanon E.Y., Eisenberg E.. The cell line A-to-I RNA editing catalogue. Nucleic Acids Res. 2020; 48:5849–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sapiro A.L., Freund E.C., Restrepo L., Qiao H.H., Bhate A., Li Q., Ni J.Q., Mosca T.J., Li J.B.. Zinc finger RNA-binding protein Zn72D regulates ADAR-mediated RNA editing in neurons. Cell Rep. 2020; 31:107654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matthews M.M., Thomas J.M., Zheng Y., Tran K., Phelps K.J., Scott A.I., Havel J., Fisher A.J., Beal P.A.. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016; 23:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Y., Havel J., Beal P.A.. A phenotypic screen for functional mutants of human adenosine deaminase acting on RNA 1. ACS Chem. Biol. 2015; 10:2512–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eifler T., Pokharel S., Beal P.A.. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry. 2013; 52:7857–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rueter S.M., Dawson T.R., Emeson R.B.. Regulation of alternative splicing by RNA editing. Nature. 1999; 399:75–80. [DOI] [PubMed] [Google Scholar]
- 41. Kondo A., Osawa T.. Establishment of an extracellular acidic pH culture system. J. Vis. Exp. 2017; 129:56660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kondo A., Yamamoto S., Nakaki R., Shimamura T., Hamakubo T., Sakai J., Kodama T., Yoshida T., Aburatani H., Osawa T.. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. 2017; 18:2228–2242. [DOI] [PubMed] [Google Scholar]
- 43. Wu D., Yotnda P.. Induction and testing of hypoxia in cell culture. J. Vis. Exp. 2011; 54:2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Malik T.N., Cartailler J.P., Emeson R.B.. Quantitative analysis of adenosine-to-inosine RNA editing. Methods Mol. Biol. 2021; 2181:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 46. Mizrahi R.A., Phelps K.J., Ching A.Y., Beal P.A.. Nucleoside analog studies indicate mechanistic differences between RNA-editing adenosine deaminases. Nucleic Acids Res. 2012; 40:9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Macbeth M.R., Bass B.L.. Large-scale overexpression and purification of ADARs from Saccharomyces cerevisiae for biophysical and biochemical studies. Methods Enzymol. 2007; 424:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stephens O.M., Yi-Brunozzi H.Y., Beal P.A.. Analysis of the RNA-editing reaction of ADAR2 with structural and fluorescent analogues of the GluR-B R/G editing site. Biochemistry. 2000; 39:12243–12251. [DOI] [PubMed] [Google Scholar]
- 49. Merkle T., Merz S., Reautschnig P., Blaha A., Li Q., Vogel P., Wettengel J., Li J.B., Stafforst T.. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019; 37:133–138. [DOI] [PubMed] [Google Scholar]
- 50. Desterro J.M., Keegan L.P., Lafarga M., Berciano M.T., O’Connell M., Carmo-Fonseca M.. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003; 116:1805–1818. [DOI] [PubMed] [Google Scholar]
- 51. Maas S., Gommans W.M.. Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res. 2009; 37:5822–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Patterson J.B., Samuel C.E.. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol. Cell. Biol. 1995; 15:5376–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fritzell K., Xu L.D., Otrocka M., Andreasson C., Ohman M.. Sensitive ADAR editing reporter in cancer cells enables high-throughput screening of small molecule libraries. Nucleic Acids Res. 2019; 47:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tran S.S., Jun H.I., Bahn J.H., Azghadi A., Ramaswami G., Van Nostrand E.L., Nguyen T.B., Hsiao Y.E., Lee C., Pratt G.A.et al.. Widespread RNA editing dysregulation in brains from autistic individuals. Nat. Neurosci. 2019; 22:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rossetti C., Picardi E., Ye M., Camilli G., D’Erchia A.M., Cucina L., Locatelli F., Fianchi L., Teofili L., Pesole G.et al.. RNA editing signature during myeloid leukemia cell differentiation. Leukemia. 2017; 31:2824–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen W., He W., Cai H., Hu B., Zheng C., Ke X., Xie L., Zheng Z., Wu X., Wang H.. A-to-I RNA editing of BLCAP lost the inhibition to STAT3 activation in cervical cancer. Oncotarget. 2017; 8:39417–39429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Silvestris D.A., Picardi E., Cesarini V., Fosso B., Mangraviti N., Massimi L., Martini M., Pesole G., Locatelli F., Gallo A.. Dynamic inosinome profiles reveal novel patient stratification and gender-specific differences in glioblastoma. Genome Biol. 2019; 20:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tran Thi, Kitami T. Niclosamide activates the NLRP3 inflammasome by intracellular acidification and mitochondrial inhibition. Commun Biol. 2019; 2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huynh K., Partch C.L.. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 2015; 79:28 29 21–28 29 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Casey J.R., Grinstein S., Orlowski J.. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010; 11:50–61. [DOI] [PubMed] [Google Scholar]
- 61. Kamel K.S., Oh M.S., Halperin M.L.. L-lactic acidosis: pathophysiology, classification, and causes; emphasis on biochemical and metabolic basis. Kidney Int. 2020; 97:75–88. [DOI] [PubMed] [Google Scholar]
- 62. Yao H., Haddad G.G.. Calcium and pH homeostasis in neurons during hypoxia and ischemia. Cell Calcium. 2004; 36:247–255. [DOI] [PubMed] [Google Scholar]
- 63. Garcia-Moreno B. Adaptations of proteins to cellular and subcellular pH. J. Biol. 2009; 8:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Talley K., Alexov E.. On the pH-optimum of activity and stability of proteins. Proteins. 2010; 78:2699–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brett C.L., Donowitz M., Rao R.. Does the proteome encode organellar pH. FEBS Lett. 2006; 580:717–719. [DOI] [PubMed] [Google Scholar]
- 66. Davidson H.W., Peshavaria M., Hutton J.C.. Proteolytic conversion of proinsulin into insulin. Identification of a Ca2+-dependent acidic endopeptidase in isolated insulin-secretory granules. Biochem. J. 1987; 246:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nevo-Caspi Y., Amariglio N., Rechavi G., Paret G.. A-to-I RNA editing is induced upon hypoxia. Shock. 2011; 35:585–589. [DOI] [PubMed] [Google Scholar]
- 68. Ben-Zvi M., Amariglio N., Paret G., Nevo-Caspi Y.. F11R expression upon hypoxia is regulated by RNA editing. PLoS One. 2013; 8:e77702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nigita G., Acunzo M., Romano G., Veneziano D., Lagana A., Vitiello M., Wernicke D., Ferro A., Croce C.M.. microRNA editing in seed region aligns with cellular changes in hypoxic conditions. Nucleic Acids Res. 2016; 44:6298–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tyrtyshnaia A.A., Lysenko L.V., Madamba F., Manzhulo I.V., Khotimchenko M.Y., Kleschevnikov A.M.. Acute neuroinflammation provokes intracellular acidification in mouse hippocampus. J. Neuroinflamm. 2016; 13:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yang J.H., Luo X., Nie Y., Su Y., Zhao Q., Kabir K., Zhang D., Rabinovici R.. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology. 2003; 109:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang G., Wang H., Singh S., Zhou P., Yang S., Wang Y., Zhu Z., Zhang J., Chen A., Billiar T.et al.. ADAR1 Prevents Liver Injury from Inflammation and Suppresses Interferon Production in Hepatocytes. Am. J. Pathol. 2015; 185:3224–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Raimondo J.V., Burman R.J., Katz A.A., Akerman C.J.. Ion dynamics during seizures. Front. Cell Neurosci. 2015; 9:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Raimondo J.V., Irkle A., Wefelmeyer W., Newey S.E., Akerman C.J.. Genetically encoded proton sensors reveal activity-dependent pH changes in neurons. Front. Mol. Neurosci. 2012; 5:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Streit A.K., Derst C., Wegner S., Heinemann U., Zahn R.K., Decher N.. RNA editing of Kv1.1 channels may account for reduced ictogenic potential of 4-aminopyridine in chronic epileptic rats. Epilepsia. 2011; 52:645–648. [DOI] [PubMed] [Google Scholar]
- 76. Srivastava P.K., Bagnati M., Delahaye-Duriez A., Ko J.H., Rotival M., Langley S.R., Shkura K., Mazzuferi M., Danis B., van Eyll J.et al.. Genome-wide analysis of differential RNA editing in epilepsy. Genome Res. 2017; 27:440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li X., Overton I.M., Baines R.A., Keegan L.P., O’Connell M.A. The ADAR RNA editing enzyme controls neuronal excitability in Drosophila melanogaster. Nucleic Acids Res. 2014; 42:1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rosenthal J.J., Seeburg P.H.. A-to-I RNA editing: effects on proteins key to neural excitability. Neuron. 2012; 74:432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cox D.B.T., Gootenberg J.S., Abudayyeh O.O., Franklin B., Kellner M.J., Joung J., Zhang F. RNA editing with CRISPR-Cas13. Science. 2017; 358:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Montiel-Gonzalez M.F., Vallecillo-Viejo I.C., Rosenthal J.J.. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 2016; 44:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Qu L., Yi Z., Zhu S., Wang C., Cao Z., Zhou Z., Yuan P., Yu Y., Tian F., Liu Z.et al.. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019; 37:1059–1069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this work are available from the corresponding author upon reasonable request. Structures referenced can be found in the Protein Data Bank under accession codes 5HP3 & 5ED1.