


Abstract
N6-methyladenosine (m6A) is the most pervasive modification in eukaryotic mRNAs. Numerous biological processes are regulated by this critical post-transcriptional mark, such as gene expression, RNA stability, RNA structure and translation. Recently, various experimental techniques and computational methods have been developed to characterize the transcriptome-wide landscapes of m6A modification for understanding its underlying mechanisms and functions in mRNA regulation. However, the experimental techniques are generally costly and time-consuming, while the existing computational models are usually designed only for m6A site prediction in a single-species and have significant limitations in accuracy, interpretability and generalizability. Here, we propose a highly interpretable computational framework, called MASS, based on a multi-task curriculum learning strategy to capture m6A features across multiple species simultaneously. Extensive computational experiments demonstrate the superior performances of MASS when compared to the state-of-the-art prediction methods. Furthermore, the contextual sequence features of m6A captured by MASS can be explained by the known critical binding motifs of the related RNA-binding proteins, which also help elucidate the similarity and difference among m6A features across species. In addition, based on the predicted m6A profiles, we further delineate the relationships between m6A and various properties of gene regulation, including gene expression, RNA stability, translation, RNA structure and histone modification. In summary, MASS may serve as a useful tool for characterizing m6A modification and studying its regulatory code. The source code of MASS can be downloaded from https://github.com/mlcb-thu/MASS.
INTRODUCTION
RNA modification is an evolutionarily conserved modification closely associated with various biological processes and human diseases (1–5). There exist over 160 types of known RNA modifications to date, including the modifications in mRNAs, tRNAs, rRNAs and noncoding RNAs (6). Among them, N6-methyladenosine (m6A) is the most abundant mRNA modification and has emerged as a critical epitranscriptomic factor in diverse biological processes, such as RNA splicing, translation and RNA stability maintenance, immune response, DNA damage repair and development (7–10). N6-methyladenosine is generated under the catalyzation of a methyltransferase complex (also known as an m6A writer) composing of METTL3, METTL14, WTAP, RBM15, KIAA1429 and ZC3H13. To participate in various biological processes, m6A is recognized and bound by several RNA-binding proteins (also known as m6A readers), including YTHDC1, YTHDC2 and YTHDF3. An m6A modification can also be removed by several demethylases (also known as m6A erasers), including FTO and ALKBH5. A variety of m6A writers, readers and erasers determines the diversity of the functions of this modification. To identify transcriptome-wide m6A profiles, several experimental techniques combined with next-generation sequencing have been developed, such as MeRIP-seq, m6A-seq and miCLIP-seq, which can detect m6A sites at single-nucleotide resolution (11–13). Recently, several new methods based on the Oxford nanopore direct RNA sequencing reads, such as Tombo and Epinano, have also been used to detect m6A sites (14–17). Significant progress has been made in characterizing m6A functions with the aid of these experimental pipelines. However, these experimental techniques are generally too expensive and time-consuming to be applied to large genomes of eukaryotes. In addition, these high-throughput experiments may miss a certain amount of m6A sites due to biases caused by read mapping and the employed probing methods. On the other hand, computational methods provide a relatively cheap and effective tool complementary to current high-throughput experimental profiling techniques. These in silico methods are able to not only predict m6A sites but also capture the underlying sequence patterns related to this modification.
Recently, machine learning has been applied in solving many computational biology questions, such as the identification of RNA-binding protein (RBP) targets (18), the prediction of translation stalling (19) and the detection of single-cell DNA methylation states (20). A number of computational methods have also been proposed to characterize the sequence features of m6A modification. For example, Chen et al. proposed iRNA-Methyl, a machine learning based method that utilized a support vector machine (SVM) framework to identify m6A sites (21). Zhou et al. used a random forest based model to identify mammalian m6A sites, which achieved an improved performance using novel features extracted from the contextual sequences of the modification sites (22). Wei et al. proposed an ensemble learning based method that trained several SVMs with three different types of feature descriptors, including physico-chemical information, position-specific information and position-specific k-mer nucleotide propensities (23). Zhang et al. applied a convolutional neural network (CNN) model combined with a recurrent neural network (RNN) to capture both spatial and temporal features of RNA sequences (24), which can also provide useful clues into understanding the predicted m6A sites via the features captured by the employed CNN. Although these computational methods have been shown capable of identifying a number of novel m6A sites, they are quite limited in accuracy, interpretability and generalizability. Moreover, most of the existing prediction frameworks are only designed for single species, which means they cannot be directly applied to investigate the similarity and difference of m6A sites among multiple species. Previous studies have shown that the landscapes of m6A are highly conserved between human and mouse (12), and the evolution of m6A is processed in parallel with that of consensus RNA sequence motifs in primates (2). Therefore, identifying interpretable features among multiple species may provide useful insights into understanding the biological roles of m6A modification and discover potential regulatory factors that are shared among different species. In addition, although it has been observed that most of the detected m6A sites contain the consensus motif DRACH (where D = A, G or U, R = A or G, H = A, C, or U) (13,25,26), only a small fraction of the DRACH motifs in the transcriptome can be methylated. Hence, capturing the sequence patterns around the core DRACH motifs associated with m6A modification may offer novel insights into its regulatory mechanisms.
To overcome the shortcomings of the existing m6A prediction methods, further characterize multiple-species m6A sites and decipher the biological functions of m6A modification, we propose a multi-task curriculum learning based model, called MASS (M6A predictor for multiple SpecieS), to predict m6A sites across different species. Multi-task learning (27–29) is a learning strategy that trains a group of related tasks simultaneously. Such a training strategy enables the learning model to generalize better and share information among different tasks and has been successfully used in a variety of learning problems (30–32). Naturally, the identification of m6A sites across different species can be defined as a multi-task learning problem. However, the variation of genome sizes across different species generally makes it difficult to train multiple models simultaneously (33,34). To address this problem, we trained all the tasks sequentially following the strategy of curriculum learning which is a type of learning starting from relatively easy tasks and gradually increasing task difficulty (35,36). Such a curriculum learning strategy can be used here to alleviate the influences of the variances in genome sizes. Performance evaluation on data from the literature revealed that our model outperforms the state-of-the-art prediction methods, with at least 5% higher performance in terms of the area under the precision-recall curve (AUPRC) score. In addition, the sequence features captured by our model can be well-mapped to the known binding motifs of m6A-associated RBPs, which indicates that our model can help characterize the complex mechanisms and functions of m6A. Moreover, we screened for important conserved genes enriched with m6A sites across different species, which may provide useful molecular clues concerning m6A functions. Our downstream analyses also revealed that MASS can be applied to characterize properties of essential biological factors or processes associated with m6A, such as gene expression, RNA stability, RNA structure, translation, and histone modification. To our best knowledge, MASS is the first attempt to extract shared sequence features across multiple species in the prediction of m6A sites. Our results demonstrated that MASS can serve as a reliable tool to characterize m6A sites and offer novel insights into the underlying regulatory mechanisms of m6A.
MATERIALS AND METHODS
Datasets
Three benchmark datasets were used in our computational tests. The first one, denoted as sramp17, was a mammalian dataset downloaded from the Ensembl database (http://www.ensembl.org) according to the supplementary data of SRAMP (22). Human and mouse data from this dataset were used to evaluate our model on single species. The second dataset, denoted as nano20, was adopted from the nanopore direct RNA sequencing data collected in (14). Here, the original nanopore sequencing signals were 1D numeric vectors rather than modification marks on raw sequences. To make them suitable for our model, we processed them according to the protocol described in (16) and converted them into RNA modification data in the same format as in sramp17. The third dataset was downloaded from RMBase v2.0 (http://rna.sysu.edu.cn/rmbase/), which contains the m6A annotations of seven species, including zebrafish, human, mouse, rhesus, rat, chimpanzee and pig. These data covered a dozen different cell lines or tissues (including HepG2, brain, HEK293T, GM12878, HeLa, U2OS, H1A, H1B, NPC, ESC, LCL, CD8T, MT4, A549, MONO-MAC-6, Huh7, Jurkat, iSLK and PBT003) from the literature. In the above three datasets, only m6A sites containing the DRACH motifs were retained as positive samples. Those unmethylated adenosines in DRACH of the whole transcriptome were used as negative samples (non-m6A sites). Considering that non-m6A sites are much more distributed in the transcriptome than m6A sites, we also kept the positive-to-negative ratio as 1:10 as described in (22). To generate input sequences, we expanded each site by 50 nucleotides (nts) upstream and downstream, respectively. For each dataset, we randomly selected 80% of the input sequences as training data to train our model, and 20% as test data to assess our model performance. To further reduce the redundancy in sequence samples, we employed the CD-HIT-EST tool (37) to remove test samples that have a certain similarity (with a similarity score above 80%) to the training samples. The statistics of the two datasets used in our tests can be found in Supplementary Tables S5 and S6. For the downstream analyses, the gene expression data were collected from the GEO dataset with accession number GSM2072352, the mRNA half-lives and ribosome profiles were curated from (9) with GEO accession number GSE49339 and the histone modification (H3K36me3) data were downloaded from the GEO database with accession numbers GSM733685 and GSM733711.
Overview of MASS
Our multi-task curriculum learning based model consists of three main parts, including data organization, feature extractor and splitting classifiers (Figure 1). In data organization, we oversampled positive samples (with replacement) to construct a balanced dataset and packed the positive and negative samples into mini-batches with a ratio of 1:1. The input sequences were then encoded into binary vectors based on a one-hot encoding strategy (Figure 1A and B). The feature extractor designed for capturing features shared among different species is composed of convolution layers, bi-directional Long Short Term Memory (LSTM) units, and multi-head attention layers (i.e., multiple concatenated attention layers), which are used for local feature extraction, global feature extraction and learning of feature importance scores of individual positions in the input sequence, respectively (Figure 1A). A residual architecture that has been widely used in various deep learning tasks (38,39) to attenuate gradient vanishing is also adopted in our convolutional layers to build a deeper and more predictive neural network (40,41). The splitting classifiers are multi-layer fully connected neural networks for predicting the probabilities that the input sites of interest of the corresponding species are methylated. Each classifier of the splitting classifiers corresponds to a particular species and is independent of each other. In the training process, the input sequences are sampled and encoded in the data organization component and the encoded vectors are then fed to the feature extractor following the sequence of species defined according to the phylogenetic tree to generate feature maps. Finally, the m6A site prediction scores of different species are computed by the corresponding splitting classifiers based on the feature maps, individually.
Figure 1.

An overview of MASS. (A) Schematic illustration of the pipeline. Samples from multiple-species were first encoded by a one-hot encoder and then fed into our multi-task learning model sequentially. The sequence of species was determined by the evolutionary distances based on the phylogenetic tree. The multi-task learning model is composed of the shared feature extractor and several splitting classifiers, each corresponding to the prediction task of a species. BN: batch normalization layer, BLSTM: bi-directional long short term memory units, Conv: convolutional layer, Dense: fully connected layer. (B) Preparation of the training data. Since there are many more negative samples (unmethylated sites) than positive samples (methylated sites), we constructed the balanced sample set by oversampling positive samples. The balanced sample set was then divided into mini-batches to train the model.
Sequence encoding by dilated convolution
To fully exploit input sequences, we employ a dilated convolution technique that was originally proposed in the computer vision field (42–45) in the feature extractor of our model. The filters of the dilated convolution layers consider the combinations of long-range features, which thus increases the diversities of the higher-level features captured from the input sequences. More specifically, let  be a discrete function,
be a discrete function, 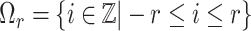 and
and 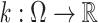 be a discrete filter of size (2r + 1). Then, the 1D dilated convolution operator can be defined as
be a discrete filter of size (2r + 1). Then, the 1D dilated convolution operator can be defined as
 |
(1) |
where i represents the index of the discrete filter, j represents the index of the given input, p represents the index of the filtered input, *l stands for an l-dilated convolution and l represents the dilation factor (1-dilated convolution represents the traditional discrete convolution operation). Here, the dilation factors for three concatenated dilated convolutional layers are set to one, three and five, respectively, according to the conventional gapped-k-mer strategy (22,46).
Multi-task curriculum learning with a shared representation
To integrate multi-species m6A data to improve the performance of the prediction model, we designed a feature extractor shared across all species following the hard parameter sharing strategy (27) and then trained an independent classifier for each species. For a given training set X consisting of samples for T species and their corresponding label set Y, which can be represented as
 |
(2) |
 |
(3) |
where Xt stands for the sample set of the t-th species, and Yt represents its corresponding label set. Then, the loss function 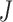 for our multi-task learning model can be defined as
for our multi-task learning model can be defined as
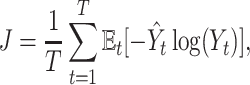 |
(4) |
where 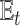 represents the expectation of the cross entropy for the t-th species,
represents the expectation of the cross entropy for the t-th species,  is calculated by
is calculated by
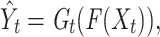 |
(5) |
where  represents the shared feature extractor, and
represents the shared feature extractor, and 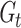 represents the splitting classifier of the t-th species. Several issues will arise if we apply multi-task learning directly to the multi-species m6A site prediction problem (see Supplementary Note S1). To address these issues, we further apply a curriculum learning strategy based on the known evolutionary relationships of all species in the training process of our multi-task model. More specifically, given a species set S (|S| = T) and their corresponding phylogenetic tree
represents the splitting classifier of the t-th species. Several issues will arise if we apply multi-task learning directly to the multi-species m6A site prediction problem (see Supplementary Note S1). To address these issues, we further apply a curriculum learning strategy based on the known evolutionary relationships of all species in the training process of our multi-task model. More specifically, given a species set S (|S| = T) and their corresponding phylogenetic tree 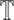 , the similarity between species si and sj can be defined as
, the similarity between species si and sj can be defined as 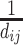 , where dij represents the shortest path from si to sj on
, where dij represents the shortest path from si to sj on 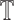 . More details on the training process of our multi-task curriculum learning model can be found in Algorithm 1.
. More details on the training process of our multi-task curriculum learning model can be found in Algorithm 1.

Our multi-task curriculum learning model can alleviate potential overfitting issues that are generally prone to occur in single-task models when the training set is not large enough and thus facilitate the training process (47). After training, common features across multiple species are captured by the feature extractor, which in principle should be able to enhance the generalization capability and interpretability of our model (47,48).
Multi-head attention mechanism for site weighting
Generally, not all features along the contextual sequence contribute to the final prediction in classification tasks. To enable our model to capture the importance scores of features from individual input positions in different tasks, we also adopt a special self-attention mechanism named multi-head attention in our deep learning framework (49). More specifically, the self-attention mechanism employed in our model can be represented by a weighted sum over the features of individual positions along the input sequence 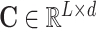 (L stands for the length of the input sequence, and d stands for the feature dimension of each site), that is
(L stands for the length of the input sequence, and d stands for the feature dimension of each site), that is
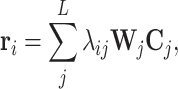 |
(6) |
where  stands for the response of the i-th site (dk stands for a hyperparameter representing the response dimension),
stands for the response of the i-th site (dk stands for a hyperparameter representing the response dimension), 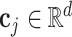 stands for the j-th site in the sequence C,
stands for the j-th site in the sequence C, 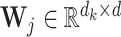 stands for the learnable weight matrix, and λij stands for a scalar value that assigns weights to individual sites along the sequence, which can be written as
stands for the learnable weight matrix, and λij stands for a scalar value that assigns weights to individual sites along the sequence, which can be written as
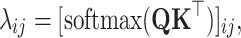 |
(7) |
where 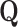 and
and 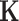 can be represented as
can be represented as
 |
(8) |
where 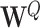 and
and 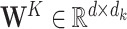 represent the learnable weight matrices. To provide a more powerful representation of feature subspace at different positions, we concatenate several self-attention layers to construct a so-called multi-head attention layer (49), that is,
represent the learnable weight matrices. To provide a more powerful representation of feature subspace at different positions, we concatenate several self-attention layers to construct a so-called multi-head attention layer (49), that is,
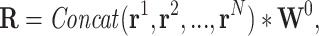 |
(9) |
where  represents a learnable weight matrix, N stands for the hyperparameter representing the number of heads,
represents a learnable weight matrix, N stands for the hyperparameter representing the number of heads, 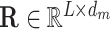 stands for the final output of the multi-head attention layer and Concat( · ) stands for the concatenate operation. In this work, N is set to 8, as suggested by the original paper in (49).
stands for the final output of the multi-head attention layer and Concat( · ) stands for the concatenate operation. In this work, N is set to 8, as suggested by the original paper in (49).
The multi-head attention layer in our model captures the connections between distant locations along the input sequence, which thus enhances the learning ability of the prediction model (49–51). In addition, the multi-head attention layer allows our model to capture coexisting sequence patterns associated with m6A modification, which thus can further increase the interpretability of the model.
Motif calling and global motif visualization
To interpret our model, we extracted and visualized the sequence motifs captured by MASS. More specifically, we scanned the entire input sequence to collect all subsequences that activate the convolutional filters (50,52,53), and then computed the positional weight matrix (PWM) of each motif based on the collected subsequences for the corresponding filter. For each species, we computed the PWMs by feeding the trained neural network with the sequences containing m6A sites from the test set. In particular, given an input sequence of length N, and a sliding window of size K and stride 1, we can obtain in total (N − K + 1) output values for each filter. Suppose that an output value is denoted by oi, and its corresponding subsequence is represented by 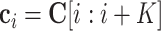 , where
, where 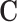 stands for the input sequence. If oi is larger than the average
stands for the input sequence. If oi is larger than the average 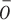 ,
,  is then collected to generate the PWM, which is also used as the local motif for our downstream analyses.
is then collected to generate the PWM, which is also used as the local motif for our downstream analyses.
Although motifs derived from the convolutional filters provide direct visualization of the important sequence patterns, they can only show the independent sequence features, such as protein binding sites. Further analyses of protein cooperation based on the motif co-occurrences, however, cannot be performed in this way. To address this problem, we employed a method called ‘class optimization’ visualization, which was originally proposed in the computer vision field (54,55) and had also been used in sequence analyses (53). More specifically, we maximized the prediction score P+(S) of an input sequence S (represented as a four-row matrix of scalars) while keeping the neural network model weights unchanged as follows:
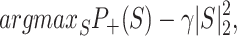 |
(10) |
where γ is the regularization parameter. With the above objective function, the sequence S is optimized through a gradient descent optimizer and adjusted at each training step to gain a higher output score. We then converted such a (locally) optimized sequence S into a PWM with a softmax function. In contrast to the local motifs, this optimized sequence depicts all possible sequence patterns that simultaneously determine a specific class (representing the methylated sites here).
Model training and model selection
To train our multi-task learning model, we split our dataset into three independent parts, i.e., training, validation and test sets. The mini-batch gradient descent algorithm was used in the training process. To make the training process more stable, we also applied the Adam algorithm (56) that can determine the learning rate automatically based on the current batch gradient. The ‘save best’ strategy, as described in (57,58), was adopted during the training procedure. To gain a better performance, we applied a line search strategy (59,60) to determine the setting of hyperparameters, including learning rate, the number of units, and the filter size of our model. In particular, we first randomly initialized all of the hyperparameters to train a basic model. Then we searched one hyperparameter with the others fixed in a reasonable range according to a 5-fold cross-validation procedure. This process was repeated until all hyperparameters were determined. The five-fold validation results and the final values of hyperparameters are shown in Supplementary Table S7. After all the hyperparameters were determined, they were used to train MASS again on the whole dataset.
Our implementation of MASS relied on the TensorLayer (61), a TensorFlow-based deep learning library. In addition, the NVIDIA TITAN X was used to speed up the training process.
Single species and multiple species training
To our best knowledge, there exists no multi-species model for m6A prediction that can capture both single species and cross-species features simultaneously. Here, we evaluated our model from both single-species and multi-species perspectives to demonstrate that it can predict m6A sites precisely. For our multi-task learning task, we considered several test settings with different configurations of species. We first trained individual single species models, and then gradually added a species according to its evolutionary distance to human. In particular, we considered different settings with two, three, five and seven species, respectively. We also compared the performance of MASS to that of the existing methods, including SRAMP and DeepM6ASeq. Since DeepM6ASeq (24) is not particularly designed for multi-species training, we evaluated its performance only on single species. For the SRAMP model (22), as the originated paper did not provide the source code for training, we did not include it in the performance comparison on the nanopore sequencing data and the multi-species prediction task, which would require the retraining of the model.
RESULTS
MASS accurately predicts m6A sites
We first assessed the prediction performance of MASS on single-species data from the sramp17 dataset from (22). In particular, we tested MASS on the data concerning human and mouse. We also compared its performance to that of two existing methods: SRAMP (22) and DeepM6ASeq (24). Since SRAMP processes long reads by cutting them into short reads with DRACH motifs and generates a prediction score for each short read, here we used its highest prediction score among all reads as the final score for a given sequence, as described in (24). As shown in Figure 2, MASS trained on single-species data (denoted by MASS-Single) showed the best performance in terms of the two classical metrics, i.e., area under the receiver operating characteristic (AUROC) and area under the precision-recall curve (AUPRC), compared to the two baseline classifiers. In particular, our model on the human data achieved AUROC and AUPRC scores of 0.858 and 0.394, respectively, which were 3.8% and 9.4% higher than those of DeepM6ASeq and 3.1%, and 6.4% higher than those of SRAMP, respectively (Figure 2A and Supplementary Figure S1a). For the mouse dataset, our model achieved AUROC and AUPRC scores of 0.841 and 0.357, respectively, which were 4.2% and 8.2% higher than those of DeepM6ASeq, and 3.2% and 4.4% higher than those of SRAMP, respectively. On average, the AUROC scores of MASS-Single were 4.9% and 3.8% higher than those of DeepM6ASeq and SRAMP, respectively, and AUPRC scores were 29.9% and 16.5% higher than those of DeepM6ASeq and SRAMP, respectively (Figure 2B and Supplementary Figure S1b). Although our model was only slightly better in terms of AUROC, it achieved much higher AUPRC scores than the baselines. Note that AUPRC is more critical and informative than AUROC for an imbalanced dataset (62,63). Thus, the test results on single-species data showed that our model can predict more accurate m6A sites than the state-of-the-art baseline methods.
Figure 2.

The performance of different models on single-species data from the sramp17 dataset and multi-species data from the RMBase 2.0. (A andB) The AUPRC (area under the corresponding PR curve) scores of MASS-Single, DeepM6ASeq and SRAMP on the sramp17 single-species data for human (a) and mouse (b), respectively. (C andD) The AUPRC scores of MASS-Single, MASS-2species, MASS-3species, MASS-5species, MASS and DeepM6ASeq on the RMBase 2.0 data in human (c) and chimpanzee (d), respectively. MASS-kspecies represents our model trained using samples from k species. MASS represents our model using the samples from all the species.
To further demonstrate the generalizability of our model to plants and data from other sequencing technologies, such as nanopore direct RNA sequencing, we also compared the performance of our model on a nanopore direct RNA sequencing data from Arabidopsis (called nano20) derived from (14) with DeepM6ASeq. As shown in Supplementary Figure S4, MASS trained on nano20 achieved an AUROC of 0.706 and an AUPRC of 0.442, which were 15% and 27.8% higher than those of DeeepM6ASeq, respectively. This simple but promising comparison result showed that our model can potentially be applied to data from a broad range of species and sequencing technologies to make a quality prediction of m6A sites.
Next, we evaluated the prediction performance of our multi-task learning model on multiple-species data derived from RMBase 2.0 (64), also using DeepM6ASeq as a baseline for comparison. When trained all seven species, MASS achieved an AUROC score of 0.911 and an AUPRC score of 0.647 on human, which were 2.2% and 11.9% higher than those of DeepM6ASeq, respectively (Figure 2C and Supplementary Figure S1c). Similar comparison results are given for chimpanzee in Figure 2D and Supplementary Figure S1d. We also assessed the performances of our model in different test settings, including models trained on single-species, two-species, three-species, five-species and seven-species data (Supplementary Table S1, and Supplementary Figures S2 and S3). These tests showed that our multi-task model trained on multi-species data can improve the prediction of m6A sites compared to the model trained on single-species data. Overall, the evaluation results demonstrated that integrating multi-species data through the multi-task learning framework employed in our model can improve the m6A site prediction results.
MASS depicts the landscapes of m6A across multiple species
Once our model was validated, we applied it to predict the m6A modification likelihood of each DRACH motif in all mRNA transcripts of different species, particularly human and mouse. The DRACH motifs with predicted probabilities >0.5 were collected as putative m6A sites. The densities of putative m6A sites along mRNAs (including 5′-UTR, CDS and 3′-UTR regions) were calculated by averaging the results from all mRNAs. We also compared the prediction results to those of MASS-Single and baseline methods, including DeepM6ASeq and SRAMP. We observed that the m6A sites predicted by all four models were mainly located in the 3′-UTR regions and significantly enriched near the stop codons and the tails of 3′-UTR regions (Figure 3). Although the distributions of most of the m6A sites predicted by the four different models were consistent with each other, there were several distinct regions between the prediction results of our model and the other models. For example, the densities of the m6A sites predicted by MASS and MASS-Single were slightly higher than those predicted by DeepM6ASeq and SRAMP in the 5′-UTR and CDS regions, while much lower in the 3′-UTR regions (Figure 3A and B). The overall distributions of predicted m6A sites were highly consistent with the previously known trend about the distributions of CLIP-seq peaks of the m6A reader YTHDF1 (65). We also applied MASS to illustrate the distributions of m6A along mRNAs in all the seven species and observed similar distributions among the six mammals, but different distributions among mammals and zebrafish (Figure 3C). These similarities and differences between the predicted landscapes of m6A sites of different species were also consistent with the previous known patterns concerning the distributions of m6A sites in the transcriptomes of the species obtained through the m6A binding proteins (including YTHDF1, YTHDF2 and WTAP) or m6A antibodies, respectively (12,65–69). These results suggested that our multi-species model can depict the landscapes of m6A in multiple species in a reasonably accurate manner.
Figure 3.

The distributions of the predicted m6A sites along mRNAs. (A andB) The distributions of the m6A sites on human mRNAs (a) and mouse mRNAs (b) predicted by different models, including MASS, MASS-Single, SRAMP and DeepM6ASeq. (C) The distributions of the m6A sites predicted by MASS for all the seven species, including zebrafish, human, mouse, chimpanzee, rhesus, rat and pig.
MASS provides a protein-binding perspective for understanding m6A functions
To interpret our deep learning model, we also visualized and analyzed the sequence features captured by our model from both local segments of the input or the entire input (see ‘Materials and Methods’ section). In particular, we first collected the local sequence features (i.e., within the local segments) captured by 300 convolution filters (each with a length of 18 nts) that were highly active in the first convolution layer of the feature extractor in our model. These sequences features were then overlaid together to generate position weight matrices (PWMs) that were regarded as local motifs. Next, we visualized these local motifs and mapped them to the known binding motifs of RBPs obtained from the CISBP-RNA database. As expected, the consensus motifs of m6A (‘GGACU’) and the binding motifs of several m6A-associated RBPs repeatedly occurred in our identified local motifs for each species (Figure 4 and Supplementary Figure S4). The analysis results suggested that our model is able to capture the local sequence features of m6A sites that can be aligned well with the site recognition patterns of m6A related binding proteins. For example, the binding motif of HNRNPA2B1, a mediator of an m6A associated process (70), well matches to the local motifs identified by our model in chimpanzee (Figure 4). Overall, an average of 21.94% of the 300 local motifs captured by MASS (see Supplementary Table S2) from seven species can be annotated by TOMTOM (71). Since a number of motifs from several species contained in CISBP-RNA of several species were not derived directly from experiments, in order to further verify that the sequence features captured by our model are valid, we also compared the local sequence patterns captured by our model to those motifs obtained from other databases that are fully verified by experiments, including Dominguez2018 (72), Ray2013 (73) and ATtRACT (74). The results showed that the sequence features captured by our model can also significantly match the motifs in these databases (see Supplementary Figure S6 for the details).
Figure 4.

Local sequence motifs (i.e., within short segments) captured by our model. For each species, we show three examples of the most significant mappings of the local sequence motifs captured by our model to the CIS-BP-RNA motif database. The motifs derived from the CIS-BP-RNA database and our model are shown on the top and bottom, respectively.
Though the local motifs are able to depict certain properties of m6A, the long-range co-occurrence motifs around m6A sites (i.e., along with the entire input sequences, which were also called the global motifs hereafter) can provide another angle to investigate the underlying mechanisms of m6A. Here, we applied a technique called class optimization (54) to capture such global sequence motifs of m6A. We found that for all seven species, there existed a ‘GGACA’ motif at the 5′ end of the input sequence (Figure 5A, dashed gray box) and a ‘GGUG’ motif near the center of the input sequence (Figure 5A, solid gray box). Previous studies indicated that the distribution of SRSF1-binding sites is highly correlated with m6A sites (75,76). In addition, we found that the ‘GGUG’ motif identified by MASS can be matched to the binding motif of TAF15, a binding partner of the m6A reader RBMX (77), which may also help explain the co-occurrence of ‘GGUG’ motif and m6A. To test whether the single-species model (i.e., trained using only single-species data) can capture these co-occurrence motifs or not, we also analyzed the optimized input sequences of MASS-Single using the same procedure as in the multi-species model. However, we did not observe any co-occurrence motifs in all the seven species (Figure 5B), which thus demonstrated the advantages of integrating multi-species data in our multi-task learning model to capture the global sequence features of m6A. These results suggested that the global motifs derived from our multi-species model can help characterize the complex mechanisms and functions of m6A modification.
Figure 5.

Global sequence motifs (i.e., within the entire input sequence) captured by our model. (A) The global sequence motifs of each species derived by the ‘class optimization’ of MASS, which was trained on multiple-species data. The ‘GGACA’ pattern in the dashed gray box can be matched to the binding motif of SRSF1, which has been reported to by closely related to the functions of m6A (75,76). The ‘GGUG’ motif in the solid gray boxes can be mapped to the binding motifs of TAF15, which is an interactor of the m6A reader RBMX. (B) The co-occurrence features were missed in the global sequence motifs of each species derived by the model trained on single-species data.
MASS detects important conserved genes enriched with m6A sites among different species
Based on the distributions of the predicted m6A sites in mRNAs, we defined a variable called the m6A capacity to assess the overall enrichment of m6A in an mRNA transcript (see Supplementary Note S2). We computed the m6A capacities of all mRNA transcripts in the seven species based on the prediction results of our model. The mRNAs with m6A capacities above the median were regarded as high m6A capacity transcripts. We then compared the numbers of high m6A capacity transcripts (including their orthologs) shared among different species (from two to seven species) between MASS and MASS-Single, which were trained using multiple-species and single-species data (see ‘Materials and Methods’ section), respectively. Our comparison results showed that MASS was able to identify more conservation across different species (from two to seven) than the single-species model (Figure 6), implying that our multi-species model can capture more common features of m6A among species than the single-species one.
Figure 6.

Comparison of the numbers of shared mRNAs with high m6A capacities derived by MASS and MASS-Single across different species.
Furthermore, we screened for mRNAs with high m6A capacities shared among all the seven species derived by MASS and MASS-Single, respectively. We found that most of the high-m6A-capacity mRNAs derived by our model were related to m6A functions (Table 1). For example, among all the identified genes, TUBB2A, which encodes a protein participating in mitosis and intracellular transport processes, had been found to interact with the general m6A writer METTL3 (78). We also found that there were more mRNAs in the MASS set that have support from the m6A literature than in the MASS-Single set (Table 1). For example, CSNK1D is one of the genes in the MASS set and it was reported previously that the deletion of the m6A locus in its 3′-UTR may elongate the circadian period of mouse cells (79). These observations indicated that high-m6A-capacity transcripts conserved among the species may be highly regulated by m6A for important consensus biological functions. These results also suggested that our model derived from multi-species data can provide new insights into understanding the regulatory functions of m6A in mRNAs.
Table 1.
The set of the mRNAs with high-m6A-capacities derived by MASS (which was trained on multiple-species data) had more support from the literature than that derived by MASS-Single (which was trained on single-species data)
| Intersection | MASS (multi-species only) | MASS-Single (single species only) | ||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Description | Reference | Gene | Description | Reference | Gene | Description | Reference |
| TUBB2A | METTL3 interactor | BioGRID | CSNK1D | elongate the circadian period in vivo | (79) | ACTC1 | KIAA1429 interactor | BioGRID |
| HLA-A | RA-associated m6A-SNPs | (90) | NME1-NME2 | miCLIP targets | (91) | TRIM10 | ||
| ACTC1 | METTL3 and METTL14 interactor | BioGRID | TUBA1A | m6A enriched gene | (92) | HBG1 | ||
| HLA-C | KIAA1429 interactor | BioGRID | GH2 | MIR181A2 | ||||
| DDX39A | METTL3 interactor | BioGRID | CCL3L3 | |||||
| PPAN-P2RY11 | m6A related gene | (91) | CSH2 | |||||
| ALDH16A1 | METTL14 interactor | (93) | CSH1 | |||||
| DHRS4L2 | High level m6A | (94) | NME2 | |||||
| ACTG2 | TUBA8 | |||||||
| RAB37 | CSHL1 | |||||||
| HLA-B | ||||||||
| ACTA1 | ||||||||
| HLA-G | ||||||||
| TRIM26 | ||||||||
| HLA-F | ||||||||
| HLA-E | ||||||||
| RAB26 | ||||||||
To further explore the molecular functions of the m6A methylated transcripts, we also selected the top 10% mRNAs with the highest m6A capacities for the gene ontology (GO) enrichment analysis in each species. GO molecular function terms that were significantly enriched (P < 0.05) in each species were collected (Figure 7). We found that several terms, including structural molecular activity, protein binding, and sequence-specific DNA binding were repeatedly enriched in different species (Supplementary Table S3; also see Supplementary Table S4 for complete results), which implied that m6A may play a critical role in protein formation and protein binding processes (80,81). We also analyzed the most significantly enriched molecular function terms in each species, and found that the highly enriched terms were similar among mouse, rat and human (Figure 7), which further verified the conservation of m6A profiles between human and mouse (12). Though more experiments are still needed, our prediction results may provide useful molecular hints for further studies of m6A regulation.
Figure 7.

The GO (gene ontology) enrichment analysis for the set of top genes with the highest m6A capacities (see Supplementary Note S2 for the detailed definition) in each species. Here, we only show GO terms that were among the top five significant ones in each species.
MASS can be applied to characterize the properties of gene regulation
In this section, we show that, based on the prediction results of MASS, we can further analyze the associations between m6A modification and the biological processes, such as gene expression regulation, RNA stability, RNA structure, translation and histone modification.
mRNAs with high m6A capacities are repressed in expression
It is well known that m6A can be widely involved in multiple biological processes including RNA stability, splicing, translation and pre-miRNA processing (9,82–84). Therefore, we speculated that m6A can also play an important role in gene expression regulation. To examine the relationship between the predicted m6A profiles and gene expression, we compared the expression levels of mRNAs with m6A capacities above the average to those with m6A capacities below the average. As a result, mRNAs with high m6A capacities were significantly down-regulated (Figure 8A, P = 2.78 × 10−27, Wilcoxon rank sum test), which can also be supported by the observations reported in previous studies (9,78).
Figure 8.

Downstream analyses of the relationships between m6A and other biological processes. (A) Comparison of the gene expressions between mRNAs with m6A capacities higher than the average (> Avg.) and those with m6A capacities less than the average (< Avg.). mRNAs with higher m6A capacities are significantly less expressed (P = 2.78 × 10−27, Wilcoxon rank sum test), ‘FPKM’: fragments per kilobase per million mapped fragments. (B) Comparison of the stability between mRNAs with m6A capacities higher than the average (> Avg.) and those with m6A capacities less than the average (< Avg.). mRNAs with higher m6A capacities were significantly more unstable (P = 9.35 × 10−95, Wilcoxon rank sum test). (C) Comparisons of translation rates (represented by the number of ribosome-bound fragments) between mRNAs with m6A capacities higher than the average (> Avg.) and those with m6A capacities less than the average (< Avg.), which shows that high m6A capacity RNAs translate faster (P = 5.95 × 10−30, Wilcoxon rank sum test). ‘RPKM’: reads per kilo base per million mapped reads. (D) Comparisons of m6A potentials in different RNA structure types. The predicted m6A potentials in single-stranded regions (SS) were higher that those in double-stranded (DS) regions (P = 2.11 × 10−12, Wilcoxon rank sum test) and poised regions (PRs, P = 5.41 × 10−4, Wilcoxon rank sum test). (E) The scatter plot of average m6A potentials normalized by peak lengths and H3K36me3 peak densities transformed by log2. The predicted m6A potentials were positively correlated to H3K36me3 densities (Pearson correlation = 0.55, P = 0.0, two-tailed t-test).
mRNAs with higher m6A capacities are more unstable
Though we observed a negative impact of m6A on gene expression, the regulation mechanisms of m6A in determining mRNA stability are not obvious. To further depict the relationship between mRNA stability and m6A, we collected mRNA half-lives from (9) and associated them with the corresponding m6A capacities derived from the m6A prediction results. In particular, we compared the half-lives of mRNAs with m6A capacities greater than the average (>Avg.) to those with m6A capacities lower than the average (<Avg.), and found that mRNAs with higher m6A capacities were generally more unstable (Figure 8B, P = 9.35 × 10−95, Wilcoxon rank sum test). This observation is also consistent with the previous reports indicating that m6A promotes mRNA decay by promoting the binding of m6A readers (9). This further verified that the m6A capacity derived by our model can represent the overall methylation states of the corresponding mRNAs.
mRNAs with higher m6A capacities show higher translation efficiency
We further investigated the association between mRNA translation efficiency and the corresponding m6A capacities derived by our model. We first collected two replicated data sets concerning mRNA translation efficiency from the literature (9). Then, we compared the translation rates of mRNAs with m6A capacities greater than the average (>Avg.) to those of mRNAs with m6A capacities lower than the average (<Avg.), and observed that mRNAs with higher m6A capacities were translated in slower rates (Figure 8C, P = 9.78 × 10−31, Wilcoxon rank sum test). Previous studies had demonstrated the functions of m6A in controlling mRNA translation efficiency (83,85,86), and our analysis results were consistent with the conclusions drawn from these studies, which thus also provided another implicit evidence to support the performance of our model.
m6A shows a preference for single-stranded regions of RNAs
Since the secondary structures of RNAs are also involved in the regulation of RNAs, we hypothesized that m6A sites were non-uniformly distributed in different types of RNA structural regions. To test this hypothesis, we first collected the parallel analysis of RNA structure (PARS) data from (87) and annotated three types of regions, including single-stranded regions (SSs), double-stranded regions (DSs) and poised regions (PRs, i.e., regions that cannot be defined as DSs or SSs) according to the descriptions in the original paper (87). Then we compared the probabilities of the predicted m6A sites located in the aforementioned three different types of RNA structural regions (Figure 8D). We found that the potentials of m6A sites in single-stranded regions were significantly greater than those in double-stranded regions (P = 2.11 × 10−12, Wilcoxon rank sum test) and poised regions (P = 5.41 × 10−4, Wilcoxon rank sum test; the effect sizes are given in Supplementary Table S1). The preference for m6A single-stranded regions in RNAs suggested that m6A may play a certain role in regulating RNA structures, which are also supported by the experimental results in previous studies (80,81).
Predicted m6A potentials are positively correlated with H3K36me3 densities
Recently, it has been shown that H3K36me3 can guide m6A deposition by recruiting METTL14, which is a component of an m6A writer complex (88). Thus, we hypothesized that the m6A profiles derived from MASS can be extensively correlated with the enrichment of H3K36me3 at the transcriptome scale. To verify the speculated correlation between H3K36me3 and m6A, we collected the H3k36me3 peaks and calculated the average m6A potentials in each H3K36me3 peak region according to the predicted results of MASS. We found that the average m6A potentials were positively correlated with the densities of H3k36me3 peaks (Figure 8E), which suggested a broad association between H3K36me3 and m6A. This result further demonstrated the reliability of our prediction results and implied that our model may be applied to investigate the relationships between m6A and other histone modifications.
DISCUSSION
In this paper, we propose a deep learning framework based on multi-task curriculum learning to predict the transcriptome-wide likelihoods of m6A sites across multiple species. To our best knowledge, this is the first attempt to model the m6A profiles of multiple species simultaneously. In our multi-task learning model, the consensus features of m6A shared among multiple species are learned in the shared feature extractor, which can reduce potential species-specific bias in the experimental data. Therefore, this strategy increases the accuracy and generalization capacity of our model in predicting novel m6A sites. In addition, characterizing the m6A profiles of different species simultaneously can shed light on the similarity and the difference of m6A patterns among different species. Though several deep learning based models have already been proposed for m6A site prediction in recent years (24,89), most of them are insufficient in model interpretability. Here, our model provides both local and global views of the sequence features, which can help interpret the deep learning model from a protein–RNA binding perspective. Moreover, since our multi-task learning model efficiently and logically integrates m6A data from multiple species, it can capture more consensus and specific features of m6A in different species than the single-species model or the existing methods.
In the downstream analyses, we further investigated the relationships between m6A and gene expression, RNA stability, translation efficiency, RNA structure stability and histone modification. The analyses based on our prediction results were highly consistent with the conclusions drawn in the previous studies, suggesting that the potential applications of our model in solving m6A-associated biological problems. Note that the data that we used here to calculate the associations between m6A and other variables were mainly collected from different cell lines or tissues. Thus, further experiments to control relevant variables are still needed. In addition, the current version of our multi-species model only considers seven vertebrate species (i.e., plants and prokaryotes are not included), which may prevent our model from uncovering the similarities and differences of m6A patterns either between animals and plants or between eukaryotes and prokaryotes. Therefore, integrating data from more species will be regarded as an important direction in our future work.
Contact for reagent and resource sharing
Further information and requests for resource sharing may be directed to and will be fulfilled by the corresponding authors Dr. Jianyang Zeng (zengjy321@tsinghua.edu.cn) and Dr. Tao Jiang (jiang@cs.ucr.edu).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr. Hailin Hu and Ms. Xiaotong Mo for helpful discussions and suggestions about the manuscript.
Contributor Information
Yuanpeng Xiong, Bioinformatics Division, BNRIST/Department of Computer Science and Technology, Tsinghua University, Beijing 100084, China.
Xuan He, Institute for Interdisciplinary Information Sciences, Tsinghua University, Beijing 100084, China.
Dan Zhao, Institute for Interdisciplinary Information Sciences, Tsinghua University, Beijing 100084, China.
Tingzhong Tian, Institute for Interdisciplinary Information Sciences, Tsinghua University, Beijing 100084, China.
Lixiang Hong, Institute for Interdisciplinary Information Sciences, Tsinghua University, Beijing 100084, China.
Tao Jiang, Department of Computer Science and Engineering, University of California, Riverside, CA 92521, USA; Bioinformatics Division, BNRIST/Department of Computer Science and Technology, Tsinghua University, Beijing 100084, China.
Jianyang Zeng, Institute for Interdisciplinary Information Sciences, Tsinghua University, Beijing 100084, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [61872216, 81630103, 61772197, 31900862]; Turing AI Institute of Nanjing; National Key Research and Development Program of China [2018YFC0910404]; Zhongguancun Haihua Institute for Frontier Information Technology. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lin S., Choe J., Du P., Triboulet R., Gregory R.I.. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell. 2016; 62:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ma L., Zhao B., Chen K., Thomas A., Tuteja J.H., He X., He C., White K.P.. Evolution of transcript modification by N6-methyladenosine in primates. Genome Res. 2017; 27:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Batista P.J. The RNA modification N6-methyladenosine and its implications in human disease. Genomics Proteom. Bioinform. 2017; 15:154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roignant J.-Y., Soller M.. m6A in mRNA: an ancient mechanism for fine-tuning gene expression. Trends Genet. 2017; 33:380–390. [DOI] [PubMed] [Google Scholar]
- 5. Deng X., Su R., Feng X., Wei M., Chen J.. Role of N6-methyladenosine modification in cancer. Curr. Opin. Genet. Dev. 2018; 48:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M.et al.. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 2012; 41:D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Motorin Y., Helm M.. RNA nucleotide methylation. Wiley Interdiscip. Rev. 2011; 2:611–631. [DOI] [PubMed] [Google Scholar]
- 8. Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.-G.et al.. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011; 7:885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G.et al.. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014; 505:117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roost C., Lynch S.R., Batista P.J., Qu K., Chang H.Y., Kool E.T.. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc. 2015; 137:2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R.. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012; 149:1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M.et al.. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012; 485:201. [DOI] [PubMed] [Google Scholar]
- 13. Linder B., Grozhik A.V., Olarerin-George A.O., Meydan C., Mason C.E., Jaffrey S.R.. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods. 2015; 12:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parker M.T., Knop K., Sherwood A.V., Schurch N.J., Mackinnon K., Gould P.D., Hall A.J., Barton G.J., Simpson G.G.. nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. Elife. 2020; 9:e49658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Q., Fang L., Yu G., Wang D., Xiao C.L., Wang K.. Detection of DNA base modifications by deep recurrent neural network on Oxford nanopore sequencing data. Nat. Commun. 2019; 10:2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu H., Begik O., Lucas M.C., Ramirez J.M., Mason C.E., Wiener D., Schwartz S., Mattick J.S., Smith M.A., Novoa E.M.. Accurate detection of m 6 A RNA modifications in native RNA sequences. Nat. Commun. 2019; 10:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Senol Cali D., Kim J.S., Ghose S., Alkan C., Mutlu O.. nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief. Bioinformatics. 2019; 20:1542–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831. [DOI] [PubMed] [Google Scholar]
- 19. Zhang S., Zhou J., Hu H., Gong H., Chen L., Cheng C., Zeng J.. A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res. 2015; 44:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Angermueller C., Lee H.J., Reik W., Stegle O.. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017; 18:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen W., Feng P., Ding H., Lin H., Chou K.-C.. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015; 490:26–33. [DOI] [PubMed] [Google Scholar]
- 22. Zhou Y., Zeng P., Li Y.-H., Zhang Z., Cui Q.. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016; 44:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei L., Chen H., Su R.. M6APred-EL: A sequence-based predictor for identifying N6-methyladenosine sites using ensemble learning. Mol. Ther.-Nucl. Acids. 2018; 12:635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y., Hamada M.. DeepM6ASeq: prediction and characterization of m6A-containing sequences using deep learning. BMC Bioinformatics. 2018; 19:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harper J.E., Miceli S.M., Roberts R.J., Manley J.L.. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990; 18:5735–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wei C.-M., Moss B.. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 1977; 16:1672–1676. [DOI] [PubMed] [Google Scholar]
- 27. Zhang Y., Qiang Y.. An overview of multi-task learning. Natl. Sci. Rev. 2018; 5:30–34. [Google Scholar]
- 28. Ruder12 S., Bingel J., Augenstein I., Søgaard A.. Sluice networks: Learning what to share between loosely related tasks. stat. 2017; 1050:23. [Google Scholar]
- 29. Misra I., Shrivastava A., Gupta A., Hebert M.. Cross-stitch networks for multi-task learning. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. 2016; Las Vegas, NV: IEEE; 3994–4003. [Google Scholar]
- 30. Domhan T., Hieber F.. Using target-side monolingual data for neural machine translation through multi-task learning. Proceedings of the 2017 Conference on Empirical Methods in Natural Language Processing. 2017; Copenhagen, DK: ACL; 1500–1505. [Google Scholar]
- 31. Tian B., Zhang Y., Wang J., Xing C.. Hierarchical Inter-Attention Network for Document Classification with Multi-Task Learning. IJCAI. 2019; Macao, CHN: Morgan Kaufmann Publishers Inc; 3569–3575. [Google Scholar]
- 32. Zhang W., Li R., Zeng T., Sun Q., Kumar S., Ye J., Ji S.. Deep model based transfer and multi-task learning for biological image analysis. EEE transactions on Big Data. 2016; 6:322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ng W., Dash M.. An evaluation of progressive sampling for imbalanced data sets. Sixth IEEE International Conference on Data Mining-Workshops (ICDMW’06). 2006; Hong Kong, CHN: IEEE; 657–661. [Google Scholar]
- 34. Ertekin S., Huang J., Bottou L., Giles L.. Learning on the border: active learning in imbalanced data classification. Proceedings of the sixteenth ACM conference on Conference on information and knowledge management. 2007; Lisbon, PT: ACM; 127–136. [Google Scholar]
- 35. Bengio Y., Louradour J., Collobert R., Weston J.. Curriculum learning. Proceedings of the 26th annual international conference on machine learning. 2009; Montreal, CAN: ACM; 41–48. [Google Scholar]
- 36. Pentina A., Sharmanska V., Lampert C.H.. Curriculum learning of multiple tasks. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. 2015; Boston, MA: IEEE; 5492–5500. [Google Scholar]
- 37. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chollet F. Xception: Deep learning with depthwise separable convolutions. Proceedings of the IEEE conference on computer vision and pattern recognition. 2017; Honolulu, HI: IEEE; 1251–1258. [Google Scholar]
- 39. Szegedy C., Ioffe S., Vanhoucke V., Alemi A.. Inception-v4, inception-resnet and the impact of residual connections on learning. Proceedings of the AAAI Conference on Artificial Intelligence. 2017; 31:San Francisco, CA: AAAI. [Google Scholar]
- 40. LeCun Y., Bengio Y., Hinton G.. Deep learning. Nature. 2015; 521:436–444. [DOI] [PubMed] [Google Scholar]
- 41. He K., Zhang X., Ren S., Sun J.. Deep residual learning for image recognition. Proceedings of the IEEE conference on computer vision and pattern recognition. 2016; Las Vegas, NV: IEEE; 770–778. [Google Scholar]
- 42. Yang Z., Hu Z., Salakhutdinov R., Berg-Kirkpatrick T.. Improved variational autoencoders for text modeling using dilated convolutions. International conference on machine learning. 2017; Syndey, AUS: ACM; 3881–3890. [Google Scholar]
- 43. Yu F., Koltun V., Funkhouser T.. Dilated residual networks. Proceedings of the IEEE conference on computer vision and pattern recognition. 2017; Honolulu, HI: IEEE; 472–480. [Google Scholar]
- 44. Yang Z., Hu Z., Salakhutdinov R., Berg-Kirkpatrick T.. Improved variational autoencoders for text modeling using dilated convolutions. Proceedings of the 34th International Conference on Machine Learning-Volume 70. 2017; Syndey, AUS: ACM; 3881–3890. [Google Scholar]
- 45. Chor B., Horn D., Goldman N., Levy Y., Massingham T.. Genomic DNA k-mer spectra: models and modalities. Genome Biol. 2009; 10:R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ghandi M., Lee D., Mohammad-Noori M., Beer M.A.. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 2014; 10:e1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y., Yang Q.. An overview of multi-task learning. Natl. Sci. Rev. 2018; 5:30–43. [Google Scholar]
- 48. Subramanian S., Trischler A., Bengio Y., Pal C.J.. Learning General Purpose Distributed Sentence Representations via Large Scale Multi-task Learning. International Conference on Learning Representations. 2018; Vancouver, CAN: OpenReview.net. [Google Scholar]
- 49. Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser Ł., Polosukhin I.. Attention is all you need. Advances in neural information processing systems. 2017; Red Hook, NY: Curran Associates Inc; 5998–6008. [Google Scholar]
- 50. Hu H., Xiao A., Zhang S., Li Y., Shi X., Jiang T., Zhang L., Zhang L., Zeng J.. DeepHINT: understanding HIV-1 integration via deep learning with attention. Bioinformatics. 2019; 35:1660–1667. [DOI] [PubMed] [Google Scholar]
- 51. Almagro Armenteros J.J., Sønderby C.K., Sønderby S.K., Nielsen H., Winther O.. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017; 33:3387–3395. [DOI] [PubMed] [Google Scholar]
- 52. Kelley D.R., Snoek J., Rinn J.L.. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016; 26:990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lanchantin J., Singh R., Wang B., Qi Y.. Deep motif dashboard: Visualizing and understanding genomic sequences using deep neural networks. Pacific Symposium on Biocomputing 2017. 2017; Kohala Coast, Hawaii: World Scientific; 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zeiler M.D., Fergus R.. Visualizingand understanding convolutional networks. European conference on computer vision. 2014; Cham: Springer; 818–833. [Google Scholar]
- 55. Nguyen A., Yosinski J., Clune J.. Understanding neural networks via feature visualization: A survey. Explainable AI: interpreting, explaining and visualizing deep learning. 2019; Cham.Springer; 55–76. [Google Scholar]
- 56. Jais I.K.M., Ismail A.R., Nisa S.Q.. Adam optimization algorithm for wide and deep neural network. Knowl. Eng. Data Sci. 2019; 2:41–46. [Google Scholar]
- 57. Lim L.A., Keles H.Y.. Foreground segmentation using convolutional neural networks for multiscale feature encoding. Pattern Recognition Letters. 2018; 112:256–262. [Google Scholar]
- 58. Moolayil J. An introduction to deep learning and keras. Learn Keras for Deep Neural Networks. 2019; Berkeley, CA: Apress; 1–16. [Google Scholar]
- 59. Nie D., Zhang H., Adeli E., Liu L., Shen D.. 3D deep learning for multi-modal imaging-guided survival time prediction of brain tumor patients. International Conference on Medical Image Computing and Computer-Assisted Intervention. 2016; Athens, Greece: Springer; 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bergstra J.S., Bardenet R., Bengio Y., Kégl B.. Algorithms for hyper-parameter optimization. Advances in neural information processing systems. 2011; Red Hook, NY: Curran Associates Inc; 2546–2554. [Google Scholar]
- 61. Dong H., Supratak A., Mai L., Liu F., Oehmichen A., Yu S., Guo Y.. Tensorlayer: a versatile library for efficient deep learning development. Proceedings of the 25th ACM international conference on Multimedia. 2017; NY: ACM; 1201–1204. [Google Scholar]
- 62. Wan F., Hong L., Xiao A., Jiang T., Zeng J.. NeoDTI: neural integration of neighbor information from a heterogeneous network for discovering new drug–target interactions. Bioinformatics. 2018; 35:104–111. [DOI] [PubMed] [Google Scholar]
- 63. Davis J., Goadrich M.. The relationship between Precision-Recall and ROC curves. Proceedings of the 23rd international conference on Machine learning. 2006; NY: ACM; 233–240. [Google Scholar]
- 64. Xuan J.-J., Sun W.-J., Lin P.-H., Zhou K.-R., Liu S., Zheng L.-L., Qu L.-H., Yang J.-H.. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2017; 46:D327–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi H., Zhang X., Weng Y.-L., Lu Z., Liu Y., Lu Z., Li J., Hao P., Zhang Y., Zhang F.et al.. m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature. 2018; 563:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schwartz S., Bernstein D.A., Mumbach M.R., Jovanovic M., Herbst R.H., León-Ricardo B.X., Engreitz J.M., Guttman M., Satija R., Lander E.S.et al.. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014; 159:148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhao B.S., Wang X., Beadell A.V., Lu Z., Shi H., Kuuspalu A., Ho R.K., He C.. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017; 542:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tan B., Liu H., Zhang S., da Silva S.R., Zhang L., Meng J., Cui X., Yuan H., Sorel O., Zhang S.-W.et al.. Viral and cellular N6-methyladenosine and N6-2’-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat. Microbiol. 2018; 3:108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shen H., Hong W., Rui L., Mengnan H., Tiandong C., Long J., Lamei D., Shilin T., Yan L., Hongfeng L., Xuewei L.et al.. mRNA N6-methyladenosine methylation of postnatal liver development in pig. PLoS One. 2017; 12:e0173421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alarcón C.R., Goodarzi H., Lee H., Liu X., Tavazoie S., Tavazoie S.F.. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell. 2015; 162:1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S.. Quantifying similarity between motifs. Genome Biol. 2007; 8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dominguez D., Freese P., Alexis M.S., Su A., Hochman M., Palden T., Bazile C., Lambert N.J., Nostrand E.L.V., Pratt G.A.A.. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins - ScienceDirect. Mol. Cell. 2018; 70:854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A.. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013; 499:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Giudice G., Sánchez-Cabo F., Torroja C., Lara-Pezzi E.. ATtRACT—a database of RNA-binding proteins and associated motifs. Database. 2016; 2016:baw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ben-Haim M.S., Moshitch-Moshkovitz S., Rechavi G.. FTO: linking m 6 A demethylation to adipogenesis. Cell Res. 2015; 25:3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang Y., Sun B.-F., Xiao W., Yang X., Sun H.-Y., Zhao Y.-L., Yang Y.-G.. Dynamic m 6 A modification and its emerging regulatory role in mRNA splicing. Sci. Bull. 2015; 60:21–32. [Google Scholar]
- 77. Jungmichel S., Rosenthal F., Altmeyer M., Lukas J., Hottiger M.O., Nielsen M.L.. Proteome-wide identification of poly (ADP-Ribosyl) ation targets in different genotoxic stress responses. Mol. Cell. 2013; 52:272–285. [DOI] [PubMed] [Google Scholar]
- 78. Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X.et al.. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014; 10:93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fustin J.-M., Kojima R., Itoh K., Chang H.-Y., Ye S., Zhuang B., Oji A., Gibo S., Narasimamurthy R., Virshup D.et al.. Two Ck1δ transcripts regulated by m6A methylation code for two antagonistic kinases in the control of the circadian clock. Proc. Natl. Acad. Sci. USA. 2018; 115:5980–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T.. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature. 2015; 518:560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu N., Zhou K.I., Parisien M., Dai Q., Diatchenko L., Pan T.. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017; 45:6051–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tang C., Klukovich R., Peng H., Wang Z., Yu T., Zhang Y., Zheng H., Klungland A., Yan W.. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA. 2018; 115:E325–E333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C.. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015; 161:1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Alarcón C.R., Lee H., Goodarzi H., Halberg N., Tavazoie S.F.. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015; 519:482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhuang M., Li X., Zhu J., Zhang J., Niu F., Liang F., Chen M., Li D., Han P., Ji S.-J.. The m6A reader YTHDF1 regulates axon guidance through translational control of Robo3.1 expression. Nucleic Acids Res. 2019; 47:4765–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Slobodin B., Han R., Calderone V., Vrielink J.A.F.O., Loayza-Puch F., Elkon R., Agami R.. Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation. Cell. 2017; 169:326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wan Y., Qu K., Zhang Q.C., Flynn R.A., Manor O., Ouyang Z., Zhang J., Spitale R.C., Snyder M.P., Segal E., Chang H.Y.. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014; 505:706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang H., Weng H., Zhou K., Wu T., Zhao B.S., Sun M., Chen Z., Deng X., Xiao G., Auer F.et al.. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature. 2019; 567:414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Huang Y., He N., Chen Y., Chen Z., Li L.. BERMP: a cross-species classifier for predicting m6A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci. 2018; 14:1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mo X.-B., Zhang Y.-H., Lei S.-F.. Genome-wide identification of N6-methyladenosine (m6A) SNPs associated with rheumatoid arthritis. Front. Genetics. 2018; 9:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Vu L.P., Pickering B.F., Cheng Y., Zaccara S., Nguyen D., Minuesa G., Chou T., Chow A., Saletore Y., MacKay M.et al.. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017; 23:1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Anders M., Chelysheva I., Goebel I., Trenkner T., Zhou J., Mao Y., Verzini S., Qian S.-B., Ignatova Z.. Dynamic m6A methylation facilitates mRNA triaging to stress granules. Life Sci. Allian. 2018; 1:e201800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schwartz S., Mumbach M.R., Jovanovic M., Wang T., Maciag K., Bushkin G.G., Mertins P., Ter-Ovanesyan D., Habib N., Cacchiarelli D.et al.. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014; 8:284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Molinie B., Wang J., Lim K.S., Hillebrand R., Lu Z.-X., Van Wittenberghe N., Howard B.D., Daneshvar K., Mullen A.C., Dedon P.et al.. m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat. Methods. 2016; 13:692–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.