

Abstract
Treatment against visceral leishmaniasis (VL) is mainly hampered by drug toxicity, long treatment regimens and/or high costs. Thus, the identification of novel and low-cost antileishmanial agents is urgent. Acarbose (ACA) is a specific inhibitor of glucosidase-like proteins, which has been used for treating diabetes. In the present study, we show that this molecule also presents in vitro and in vivo specific antileishmanial activity against Leishmania infantum. Results showed an in vitro direct action against L. infantum promastigotes and amastigotes, and low toxicity to mammalian cells. In addition, in vivo experiments performed using free ACA or incorporated in a Pluronic® F127-based polymeric micelle system called ACA/Mic proved effective for the treatment of L. infantum-infected BALB/c mice. Treated animals presented significant reductions in the parasite load in their spleens, livers, bone marrows and draining lymph nodes when compared to the controls, as well as the development of antileishmanial Th1-type humoral and cellular responses based on high levels of IFN-γ, IL-12, TNF-α, GM-CSF, nitrite and IgG2a isotype antibodies. In addition, ACA or ACA-treated animals suffered from low organ toxicity. Treatment with ACA/Mic outperformed treatments using either Miltefosine or free ACA based on parasitological and immunological evaluations performed one and 15 days post-therapy. In conclusion, data suggest that the ACA/Mic is a potential therapeutic agent against L. infantum and merits further consideration for VL treatment.
Keywords: Treatment, Drug repositioning, Acarbose, Visceral leishmaniasis, Miltefosine, Leishmania infantum
Introduction
Leishmaniasis is a disease complex found in 98 countries in the world, where approximately 380 million people are susceptible to the infection by the Leishmania parasites, which are obligate intracellular pathogens able to invade phagocytic cells of the mammalian hosts [56]. Approximately 20 parasite species cause disease in humans, and clinical manifestations include visceral and tegumentary leishmaniasis [7, 20]. Visceral leishmaniasis (VL) is caused by Leishmania infantum and L. donovani species, being fever, anemia, wasting, hepatosplenomegaly and patient’s immune suppression the clinical manifestations of active disease. VL is almost always fatal, if acute and left untreated [4].
Treatment against VL relies on the use of pentavalent antimonials. However, drug toxicity, parenteral administration and long therapy regimens represent significant hindrances to effective treatment. In addition, parasite resistance has been reported, being this a main cause for relapse of infections in affected patients [9, 50]. Alternatively to pentavalent antimony compounds, Amphotericin B (AmpB) has been extensively used to treat VL. However, although it is highly effective against parasites, it is also toxic to the patient, causing nephrotoxicity, hypokalemia and myocarditis, among others [31]. AmpB-based liposomal formulations have minimized the side effects caused by treatment with the free drug, but its high cost prevents it from becoming a widespread therapeutic agent [51]. Paromomycin has also been used against Leishmania infection; however, this drug targets a relatively restricted range of Leishmania species and parasite resistance has been reported [38]. Moreover, Miltefosine, which was originally described as an anti-tumor agent, presents antileishmanial potential against distinct parasite species by inhibiting the biosynthesis of the glycosyl-phosphatidyl-inositol receptor, a key molecule for Leishmania intracellular survival [27, 36, 58]. However, miltefosine causes teratogenicity and parasitic resistance to this drug has also been described, thus limiting its clinical application [50].
In this context, the identification of new antileishmanial agents is a challenge that remains to be overcome. Recently, a Leishmania proteome mining strategy was performed to select new drug targets against the parasites. Good drug candidates should be relevant for Leishmania survival within its mammalian hosts but should present low homology to human proteins [10]. In that study, a hypothetical protein, which was functionally annotated as a glucosidase-like protein and associated with Leishmania N-Glycan biosynthesis metabolic pathway, came up as a promising candidate. This parasite protein was also predicted to be potential target of two specific inhibitors, Acarbose (ACA) and Miglitol. The authors tested Miglitol against L. amazonensis and L. infantum, proving its effectiveness against both Leishmania species. Treatment with Miglitol significantly reduced the percentage of infection and number of recovered amastigotes from infected macrophages. Therefore, the authors recommended the use of Miglitol as a potential candidate to treat VL [10]. ACA is a specific inhibitor of glucosidase-like proteins. Administration of ACA causes a significant decrease in the plasma glucose levels of patients and thus, this drug has been used effectively to treat type-2-diabetes [21, 22]. In addition, to the best of our knowledge, there are no reports on the antileishmanial activity of ACA.
Poloxamer-based micelle systems have been explored to improve the efficacy of old drugs and/or reduce their toxicity [15, 28]. In addition, they have been also used to potentiate the properties of new antileishmanial agents [29, 42, 54]. Poloxamers are thermo-reversible and non-ionic surfactant co-polymers that present amphiphilic nature consisting of hydrophilic and hydrophobic segments. They are easily manufactured, present low production cost, good stability and efficient targeting ability [34, 54]. Regarding immunological aspects, Poloxamer-based micelles have shown adjuvant effect by stimulating the development of a Leishmania-specific Th1-type response, when antileishmanial targets are in vivo tested against infection by the parasites [15, 25, 53]. In this context, in the present study, a ACA-carrying delivery system based on polymeric micelles was evaluated to treat against L. infantum infection.
The immunity for VL has been studied and resistance against infection involves the induction of the T-helper 1 (Th1) cell response based on the activation of CD4+ and CD8+ T cells, with concomitant production of cytokines such as IFN-γ, IL-12 and GM-CSF, among others. On the other hand, susceptibility to infection is related to the production of Th2-type profile cytokines, such as IL-4, IL-5 and IL-10, among others [14, 24]. Based on this understanding about the immunological mechanisms of leishmaniasis, distinct agents have been evaluated regarding their antileishmanial immunity, however, little progress has been made beyond the experimental stage [39, 55, 59].
In the present study, ACA was evaluated in vitro and in vivo against L. infantum species. The free molecule or incorporated into a Poloxamer 407 (Pluronic® F127)-based polymeric micelle system (ACA/Mic) was used to treat L. infantum-infected mice, and Miltefosine was used as a control drug. Parasitological, immunological and toxicological evaluations were performed one and 15 days post-treatment. Results showed that treatment using ACA or ACA/Mic induced a polarized and specific antileishmanial Th1-type immune response, which was reflected by significant reductions in the parasite load in the spleen, liver, bone marrow (BM) and draining lymph nodes (dLNs), besides low organ toxicity in the treated animals.
Materials and methods
Chemicals
Miltefosine (C21H46NO4P), Acarbose (C25H43NO18), Amphotericin B (AmpB, C47H73NO17) and Poloxamer 407 (Pluronic® F127) were purchased from Sigma-Aldrich (catalog numbers: 58066-85-6, 56,180-94-0, 1397-89-3 and 16,758, respectively; St. Louis, USA).
Parasite and animals
L. infantum (MHOM/BR/1970/BH46) strain was used. Parasites were used until the 5th in vitro culture passage to maintain their infectivity. The stationary promastigotes were grown at 24 °C in complete Schneider's medium (Sigma-Aldrich, St. Louis, MO, USA), which was composed by medium plus 20% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich, USA) and 20 mM l-glutamine pH 7.4 at 24 °C [12]. Female BALB/c mice (8 weeks old) were purchased from the Institute of Biological Sciences of Federal University of Minas Gerais (UFMG, Belo Horizonte, Minas Gerais, Brazil), and kept under specific pathogen-free conditions. The study was approved by the Committee for the Ethical Handling of Research Animals of UFMG (protocol number 085/2017).
In vitro antileishmanial activity
The 50% Leishmania inhibitory concentration (IC50) was evaluated by incubating L. infantum stationary promastigotes in the absence or presence of ACA (0 to 100.0 µg/mL) or AmpB (0 to 10.0 µg/mL), which was used as a control, in 96-well culture plates (Nunc, Nunclon, Roskilde, Denmark) for 48 h at 24 °C. Cell viability was assessed by 3-(4.5-dimethylthiazol-2-yl)-2.5-diphenyl tetrazolium bromide (Sigma-Aldrich, USA) method. The optical density (OD) values were read in a microplate spectrophotometer (Molecular Devices, Spectra Max Plus, San Jose, CA, USA) at 570 nm. Results were entered into Microsoft Excel (version 10.0) spreadsheets and IC50 values were calculated by sigmoidal regression of the dose–response curves [52].
Cytotoxicity assay
ACA cytotoxicity to murine macrophages (CC50) and red blood cells (RBC50) was evaluated by determining the compound’s concentration required for 50% reduction of cell viability. To evaluate the CC50, macrophages derived from female BALB/c mice were obtained by peritoneal lavage with 5 mL cold PBS pH 7.4. Peritoneal exudate cells were centrifuged at 1000×g for 10 min and resuspended in RPMI 1640 medium. Cells (5 × 105 per well) were then incubated in the absence of presence of ACA (0 to 100.0 µg/mL) or AmpB (0 to 10.0 µg/mL) for 48 h at 37 °C in 5% CO2. Macrophage viability was assessed by MTT method. To evaluate the RBC50, a 5% (v/v) human red blood cells suspension was incubated in the absence of presence of ACA (0 to 100.0 µg/mL) or AmpB (0 to 10.0 µg/mL) for 1 h at 37 °C in 5% CO2 when the suspension was centrifuged for 10 min at 1000×g and lysis percentage was determined spectrophotometrically at 570 nm. The absence of (blank) or 100% hemolysis were determined by replacing ACA or AmpB with an equal volume of phosphate-buffered saline pH 7.4 (PBS) or distilled water, respectively. CC50 and RBC50 values were calculated by sigmoidal regression of dose–response curves with Microsoft Excel software (version 10.0) [29].
Treatment of infected macrophages
ACA efficacy for treatment of murine cells was evaluated in vitro by incubating macrophages (5 × 105) in RPMI 1640 medium supplemented with 20% FBS and 20 mM l-glutamine pH 7.4, for 24 h at 37 °C in 5% CO2. Parasites (5 × 106 cells) were then added to the wells and cultures were incubated for 48 h at 37 °C in 5% CO2. Free parasites were removed by extensive washing with medium. Infected macrophages were then left untreated or treated with ACA (0, 2.5, 5.0 and 10.0 µg/mL) or AmpB (0, 0.25, 0.5 and 1.0 µg/mL) for 48 h at 37 °C in 5% CO2. After fixation with 4% (w/v) paraformaldehyde, cells were washed and stained with Giemsa. Percentage of infected macrophages, infectiveness reduction and number of amastigotes per macrophage were determined by counting 200 cells, in triplicate, using an optical microscope [29].
Inhibition of infection by pre-treatment of L. infantum parasites
Stationary promastigotes (5 × 106 cells) were incubated in the absence or presence of ACA (0, 2.5, 5.0 and 10.0 µg/mL) or AmpB (0, 0.25, 0.5 and 1.0 µg/mL), for 4 h at 24 °C. Parasites were washed three times in RPMI 1640, quantified and used to infect macrophages at a ratio of 10 parasites per one macrophage for 24 h at 37 °C in 5% CO2. After fixation with 4% (w/v) paraformaldehyde, cells were washed and stained with Giemsa. Percentage of infected macrophages, reduction of infection and number of amastigotes per macrophage were determined by counting 200 cells, in triplicate, using an optical microscope [54].
Evaluation of mitochondrial membrane potential
Stationary promastigotes (107 cells) were cultured in the absence or presence of ACA (0.35 and 0.70 μg/mL, corresponding to one and two times the IC50 values, respectively) for 24 h at 25 °C. Parasites were washed in PBS and incubated with 500 nM MitoTracker Red CM-H2XROS (Invitrogen, USA) for 30 min in the dark and at room temperature. After washing twice with PBS, samples were added to a black 96-well plate and fluorescence intensity was measured using a fluorometer (FL×800, BioTek Instruments, Inc., Winooski, VT, USA) with excitation and emission wavelengths of 528 nm and 600 nm, respectively. Parasites incubated with carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazon (FCCP; 5.0 µM, Sigma-Aldrich, USA) for 10 min were used as control [54].
Production of reactive oxygen species (ROS)
Stationary promastigotes (107 cells) were cultured in the absence or presence of ACA (0.35 and 0.70 μg/mL, corresponding to one and two times the IC50 values, respectively) for 24 h at 25 °C. Parasites were incubated with 20 µM cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; Sigma-Aldrich, USA) for 30 min in the dark and at room temperature. Fluorescence intensity was measured in a spectrofluorometer (Varioskan® Flash, Thermo Scientific, USA) with excitation and emission wavelengths of 485 and 528 nm, respectively. H2O2-treated parasites (4.0 mM) were used as control [54].
Evaluation of lipid accumulation in the parasites
Stationary promastigotes (107 cells) were cultured in the absence or presence of ACA (0.35 and 0.70 μg/mL, corresponding to one and two times the IC50 values, respectively) for 24 h at 25 °C. Parasites were incubated with Nile Red (1.0 μg/mL; Sigma-Aldrich, USA) for 30 min in the dark and at room temperature. Fluorescence intensity was measured in a spectrofluorometer (Varioskan® Flash, Thermo Scientific, USA), with excitation and emission wavelengths of 485 and 528 nm, respectively [2].
Preparation of ACA-containing micelles
ACA-containing micelles (ACA/Mic) were prepared as described previously [29]. Briefly, Poloxamer 407 (18% w/w) was diluted in PBS under magnetic agitation for 18 h at 4 °C. 8 mg of ACA were added to 500 µL of dichloromethane and solubilized using a vortex. The mixture was then added to the Poloxamer solution under vigorous magnetic agitation in an ice bath until a viscous emulsion was obtained. The dichloromethane was evaporated using a rotary evaporator (Buchi, Flawil, Switzerland) and the ACA-containing micelles were obtained as a transparent yellow gel at room temperature. Empty micelles were similarly prepared using 18% w/w Poloxamer 407 without the addition of ACA.
Infection and treatment
Mice (n = 12 per group) were infected subcutaneously with 107 L. infantum stationary promastigotes injected into their right hind footpad according to previously described [28, 42, 54]. Sixty days post-infection, infected mice were administered with saline alone (50 μL, PBS); empty micelles (50 μL, B/Mic: 10 mg/kg body weight); Miltefosine (50 μL, 2 mg/kg body weight); free ACA (50 μL, 5 mg/kg body weight); or ACA-containing micelles (50 μL, ACA/Mic: 5 mg/kg body weight). Except for Miltefosine which was administered by oral route, all other compounds were administered subcutaneously, every 2 days for a period of 10 days. Half of the animals were euthanized one and 15 days post-treatment, after which parasitological and immunological evaluations were performed.
Evaluation of murine humoral response
Anti-parasite IgG1 and IgG2a isotype antibody levels were evaluated by ELISA assay in sera samples from infected and treated mice collected one and 15 days post-treatment. Briefly, L. infantum Soluble Leishmania Antigen (SLA) was used as the coating antigen (1.0 µg/well) and sera samples were diluted 1:100 in PBS-T (PBS plus 0.05% (v/v) Tween 20). Anti-mouse IgG1 and IgG2a horseradish-peroxidase conjugated antibodies (Sigma-Aldrich, USA) were used both at a 1:10,000 dilution in PBS-T. Reactions were developed in the presence of H2O2, ortho-phenylenediamine and citrate–phosphate buffer pH 5.0 for 30 min and in the dark. Next, reactions were stopped by addition of 2 N H2SO4, after which OD values were measured at 492 nm in an ELISA microplate spectrophotometer (Molecular Devices, Spectra Max Plus, Canada).
Analysis of the cellular profile
Spleens of infected and treated mice were collected one and 15 days post-treatment, when splenocytes were plated in 24-well plates (Nunc) and incubated in DMEM supplemented with 20% FBS and 20 mM l-glutamine at a pH 7.4. Cells (5 × 106/mL) were unstimulated (medium) or stimulated with L. infantum SLA (50 μg/mL) for 48 h at 37 °C in 5% CO2. IFN-γ, IL-4, IL-10, IL-12p70 and GM-CSF levels were measured in the culture supernatants by capture ELISA assay (BD Pharmingen®, San Diego, CA, USA), according to the manufacturer’s instructions. Nitrite production was also evaluated in the same cellular supernatants by Griess reaction. To investigate the participation of T cells in the IFN-γ production in the Miltefosine-, ACA- or ACA/Mic-treated mice, splenocytes (5 × 106/mL) were unstimulated (medium), stimulated with L. infantum SLA (50 μg/mL; control) or stimulated and incubated with anti-CD4 (GK 1.5) or anti-CD8 (53–6.7) monoclonal antibody (5 μg each; Pharmingen®, USA), for 48 h at 37 °C in 5% CO2. In all cases, cell supernatants were collected after 48 h incubation and used to quantify IFN-γ cytokine. Appropriate isotype-matched controls [rat IgG2a (R35-95) and rat IgG2b (95–1)] were used [29]. IFN-γ, TNF-α and IL-10-producing CD4+ and CD8+ T cell profiles were evaluated in Miltefosine-, ACA- or ACA/Mic-treated mice by flow cytometry technique using the animals’ spleens collected 15 days post-treatment. Briefly, spleen cells (5 × 106/mL) were cultured in RPMI 1640 medium and then either stimulated with SLA (50 μg/mL) or left untreated for 48 h at 37 °C in 5% CO2.
Immunophenotyping of spleen cell subsets and intracellular cytokines
IFN-γ, TNF-α and IL-10 cytokines were also evaluated in the spleen cells of the infected animals by flow cytometry technique. For this, splenocytes were incubated in the presence of 200 μL of RPMI medium in 96-well round-bottom culture plates at a concentration of 5 × 105 cells per well. Cells were unstimulated (medium) or stimulated with L. infantum SLA (20.0 μg/mL) for 48 h at 37 °C with 5% CO2. Cells were incubated with phorbol myristate acetate (5.0 ng/mL), ionomycin (1.0 μg/mL) or lipopolysaccharide (0.01 μg/uL), diluted in complete RPMI medium. Afterwards, cells were treated with Brefeldin A (10.0 μg/mL) for 4 h, and stained with Fixable Viability Stain 450 (BD Biosciences) for 15 min at room temperature, followed by anti-mouse CD3 FITC (clone 145.2C11), anti-mouse CD4 BV605 (clone RM4-5) or anti-mouse CD8α PerCP Cy5.5 (clone 53–6.7) (BD Biosciences Bioscience, USA), for 30 min at room temperature [6]. Next, fixation (FACS fixing solution), washing and permeabilization were performed. Permeabilized cells were then stained with anti-mouse IFN-γ AF700 (clone XMG1.2), anti-mouse TNF-α PE-Cy7 (clone LG.3A10) or anti-mouse IL-10 APC (JES5-16E) (BD Biosciences Bioscience, USA). Cells were acquired (100,000 events) on LSR Fortessa cytometer (BD Biosciences, USA) using FACSDiva software. For analysis in FlowJo software, dead cells were excluded after FVS450 stain and viable cells were gated for stained CD3+CD4+ and CD3+CD8+ cells and intracellular cytokine production. Results were expressed as percentage of CD4+ and CD8+ T cells in the SLA-stimulated cultures.
Estimation of parasite load
Parasite load was estimated in spleen, liver, bone marrow (BM) and draining lymph nodes (dLN) of infected and treated animals, one and 15 days post-treatment. Briefly, organs were macerated in a glass tissue grinder using sterile PBS, and tissue debris was removed by centrifugation at 150×g. Cells were then concentrated by centrifugation at 2000×g, pellets were resuspended in 1 mL of complete Schneider’s medium and serially diluted using the same medium (10–1 to 10–12 dilutions). Each sample was plated in triplicate and analyzed after 7 days of having set up the cultures, at 24 °C. Results were expressed as the negative log of the titer (the dilution corresponding to the last positive well) adjusted per milligram of the organ. The parasitism in the animals’ spleens was evaluated also by the quantitative PCR (qPCR) technique [3]. Briefly, spleen DNA was extracted using Wizard Genomic DNA Purification Kit (Promega Corporation) and resuspended in milli-Q water. The parasite load was estimated using specific primers to amplify L. infantum kDNA: Forward (5′-CCTATTTTACACCAACCCCCAGT-3′) and Reverse (5′-GGGTAGGGGCGTTCTGCGAAA-3′). Mouse gene encoding for β-actin (Forward: 5′-CAGAGCAAGAGAGGTATCC-3′; Reverse: 5′-TCATTGTAGAAGGTGTGGTGC-3′) was used as a control. Standard curves for kDNA and β-actin were obtained from DNA extracted from 1 × 108 parasites and 1 × 108 peritoneal macrophages, respectively, under the same experimental conditions used to extract the samples. Reactions were processed and analyzed in an ABI Prism 7500 Sequence Detection System (96 wells-plate; Applied Biosystems) using 2 × SYBR™ Select Master Mix (5 µL; Applied Biosystems), with 2 mM of each primer (1 µL) and 4 µL of DNA (25 ng/µL). Samples were incubated at 95 °C for 10 min, and submitted to 40 cycles of 95 °C for 15 s and 60 °C for 1 min. For each cycle, fluorescence data were collected. Results were calculated by interpolation from a calibration curve, which was run in parallel to the samples, performed in duplicate and expressed as the number of L. infantum organisms per total DNA.
In vivo toxicity
ACA toxicity was evaluated in vivo in the treated and infected mice, by means of dosage of aspartate aminotransferase (AST), alanine aminotransferase (ALT), direct bilirubin and total bilirubin, which were used as hepatic damage markers, and of creatine kinase muscle brain fraction (CK-MB), which was used as cardiac damage marker. The analyses were performed using commercial kits (Labtest Diagnostica®, Belo Horizonte) according to the manufacturer’s instructions.
Statistical analysis
IC50, CC50 and RBC50 values were calculated by dose–response curves using Microsoft Excel software (version 10.0) and plotted in GraphPad Prism 5.03. Results were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s post-test for comparison between the groups. Results were expressed as mean ± standard deviation for each group. Two independent experiments were performed and similar results were observed. Differences were considered significant when P < 0.05.
Results
In vitro biological assays
ACA antileishmanial activity was evaluated in vitro against L. infantum species. AmpB was used as a control drug. Results showed IC50 values of 0.47 ± 0.1 and 0.11 ± 0.03 µg/mL when ACA and AmpB were used, respectively (Table 1). CC50 and RBC50 values were 265.4 ± 16.5 µg/mL and 454.6 ± 13.4 µg/mL for ACA, and 0.92 ± 0.13 µg/mL and 12.2 ± 1.6 µg/mL for AmpB, respectively, with corresponding Selectivity Index values of 564.7 and 8.4. Treatment of infected macrophages showed infectiveness reduction of 87.4% and 67.5% when ACA and AmpB were used at concentrations of 10.0 and 1.0 µg/mL, respectively (Table 2). Inhibition of infection using pre-treated parasites showed similar infectiveness reduction levels of 87.7% (ACA) and 73.3% (AmpB) (Table 3).
Table 1.
In vitro biological activity
| Compound | IC50 (µg/mL) | CC50 (µg/mL) | SI | RBC50 (µg/mL) |
|---|---|---|---|---|
| Acarbose | 0.47 ± 0.1 | 265.4 ± 16.5 | 564.7 | 454.6 ± 13.4 |
| Amphotericin B | 0.11 ± 0.03 | 0.92 ± 0.13 | 8.4 | 12.2 ± 1.6 |
L. infantum stationary promastigotes were incubated with ACA (0–100 µg/mL) or AmpB (0–10.0 µg/mL) for 48 h at 24 °C. Cell viability was analyzed by MTT method and 50% Leishmania inhibitory concentration (IC50) was calculated by sigmoidal regression of the corresponding dose–response curve. Similarly, murine macrophages were incubated with ACA (0–100 µg/mL) or AmpB (0–10.0 µg/mL), and 50% macrophage inhibitory concentration (CC50) was determined by sigmoidal regression of the corresponding dose–response curve. Selectivity index (SI) was calculated as the ratio between CC50 and IC50 values. 50% inhibition of human red cells (RBC50) viability was calculated by incubating a 5% red cells suspension with ACA (0–100.0 µg/mL) or AmpB (0–10.0 µg/mL) for 1 h at 37 °C in 5% CO2. Lysis percentage was evaluated spectrophotometrically, and the absence (blank) or 100% of hemolysis were determined by replacing ACA for an equal volume of PBS or distilled water, respectively. Results are expressed as mean ± standard deviation
Table 2.
Treatment of infected macrophages
| Compound | Concentration (µg/mL) | Percentage of infected macrophages after treatment | Infectiveness reduction (%) | Number of amastigotes per macrophage |
|---|---|---|---|---|
| Acarbose | 10.0 | 9.4 ± 0.7 | 87.4 | 0.1 ± 0 |
| 5.0 | 19.8 ± 2.6 | 73.4 | 0.5 ± 0.2 | |
| 2.5 | 33.4 ± 5.4 | 55.1 | 1.4 ± 0.3 | |
| 0 | 74.4 ± 5.5 | (–) | 3.3 ± 0.6 | |
| Amphotericin B | 1.0 | 24.2 ± 3.0 | 67.5 | 1.1 ± 0.2 |
| 0.50 | 33.6 ± 4.0 | 54.8 | 1.8 ± 0.3 | |
| 0.25 | 49.9 ± 3.5 | 32.9 | 2.6 ± 0.6 | |
| 0 | 74.4 ± 5.5 | (–) | 3.3 ± 0.6 |
Murine macrophages (5 × 105 cells) were incubated in RPMI 1640 medium supplemented with 20% FBS and 20 mM l-glutamine at pH 7.4, for 24 h at 37 °C in 5% CO2. L. infantum stationary promastigotes were used to infect the macrophages (at a ratio of 10 parasites per one macrophage) for 48 h at 37 °C in 5% CO2. Free parasites were removed by extensive washing with medium and infected macrophages were treated with ACA (0, 2.5, 5.0 and 10.0 µg/mL) or AmpB (0, 0.25, 0.5 and 1.0 µg/mL) for 48 h at 24 °C in 5% CO2. Percentage of infected macrophages, infectiveness reduction and the number of recovered amastigotes per cell were determined by counting 200 macrophages, in triplicate. Results are expressed as mean ± standard deviation
Table 3.
Inhibition of infection by pre-treatment of parasites
| Compound | Concentration (µg/mL) | Infection percentage using pre-treated parasites | Infectiveness reduction (%) | Number of amastigotes per macrophage |
|---|---|---|---|---|
| Acarbose | 10.0 | 8.7 ± 1.0 | 87.7 | 0.2 ± 0 |
| 5.0 | 16.6 ± 3.0 | 76.6 | 0.6 ± 0.3 | |
| 2.5 | 30.1 ± 4.0 | 57.5 | 1.3 ± 0.4 | |
| 0 | 70.8 ± 4.2 | (–) | 3.4 ± 0.4 | |
| Amphotericin B | 1.0 | 18.9 ± 2.6 | 73.3 | 0.9 ± 0.4 |
| 0.50 | 28.9 ± 3.6 | 59.2 | 1.3 ± 0.4 | |
| 0.25 | 44.5 ± 4.0 | 37.1 | 2.3 ± 0.5 | |
| 0 | 70.8 ± 4.2 | (–) | 3.4 ± 0.4 |
L. infantum stationary promastigotes (5 × 106 cells) were pre-treated with ACA (0, 2.5, 5.0 and 10.0 µg/mL) or AmpB (0, 0.25, 0.5 and 1.0 µg/mL) for 4 h at 24 °C. Pre-treated parasites were washed in RPMI 1640 and used to infect murine macrophages (at a ratio of 10 parasites per one macrophage) for 24 h at 37 °C in 5% CO2. Percentage of infected macrophages, infectiveness reduction and the number of recovered amastigotes per cell were determined by counting 200 macrophages, in triplicate. Results are expressed as mean ± standard deviation
The proof of concept for the mechanism of action of ACA as an inhibitory drug was evaluated in L. infantum. Results showed that the molecule induced 36.9% and 33.6% reduction in the parasite’ mitochondrial membrane potential (ΔΨm) when used at concentrations of 0.35 and 0.70 µg/mL, respectively (Fig. 1a). FCCP-treated parasites used as control presented 47.7% reduction. Induction of oxidative stress was also observed in ACA-treated parasites, which was reflected by a 92.9% and 124.1% increase in reactive oxygen species (ROS) production when ACA was used at concentrations of 0.35 and 0.70 µg/mL, respectively (Fig. 1b). H2O2-treated parasites, used as a positive control, showed an increase in ROS production of 145.6%. Furthermore, treatment with ACA (0.35 and 0.70 µg/mL) induced a significant increase in the accumulation of lipid bodies in the order of 55.9% and 87.0%, respectively (Fig. 2).
Fig. 1.

Evaluation of Leishmania mitochondrial membrane potential (ΔΨm) and ROS production. L. infantum stationary promastigotes (107 cells) were cultured in the absence (control) or presence of ACA (0.35 μg/mL and 0.70 µg/mL, corresponding to one and two times the IC50 value, respectively) for 24 h at 25 °C. Cells were incubated for 30 min in the dark with 500 nM MitoTracker to evaluate ΔΨm or with 20 µM cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) to evaluate ROS production. In both cases, after having washed twice with PBS, treated promastigotes were transferred to a black 96-well plate and fluorescence intensity was measured using a fluorometer. FCCP (5.0 µM) and H2O2 (4.0 mM) were used as positive controls, respectively. In a percentage reduction of ΔΨm after treatment, compared to untreated parasites (control, considered as 100%). In b ROS production values after treatment, compared to untreated parasites as control. Results are expressed as arbitrary units (a.u.) of fluorescence intensity and correspond to the mean of three independent experiments. (*), (**) and (***) indicate statistically significant difference in relation to the untreated control (P < 0.05, P < 0.01 and P < 0.001, respectively)
Fig. 2.
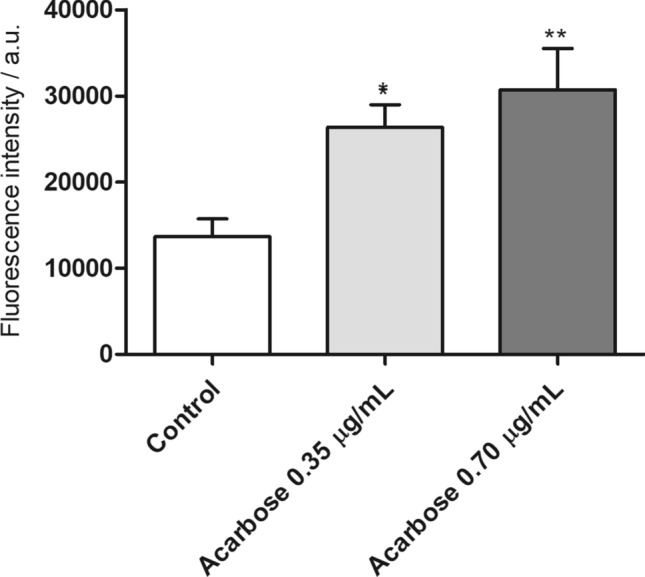
Accumulation of lipid bodies in L. infantum promastigotes. L. infantum stationary promastigotes (107 cells) were cultured in the absence or presence of ACA (0.35 and 0.70 μg/mL, corresponding to one and two times the IC50 values, respectively) for 24 h at 25 °C. Parasites were incubated with Nile Red (1.0 μg/mL) for 30 min in the dark and at room temperature. Fluorescence intensity was measured in a spectrofluorometer, with excitation and emission wavelengths of 485 and 528 nm, respectively. (*) and (**) indicate statistically significant difference in relation to the untreated control (P < 0.1 and P < 0.01, respectively)
Analysis of murine humoral responses developed after infection and treatment
Anti-Leishmania humoral response of infected and treated mice was evaluated one and 15 days post-therapy. Results showed that Miltefosine, ACA or ACA/Mic-treated mice presented significantly higher levels of anti-parasite IgG2a antibody when compared to IgG1 levels. On the contrary, saline- and B/Mic-treated groups produced significantly higher levels of antileishmanial IgG1 antibody (Fig. 3). Similar results were observed one and 15 days post-therapy, suggesting the development of a sustained humoral response profile after treatment of infection.
Fig. 3.
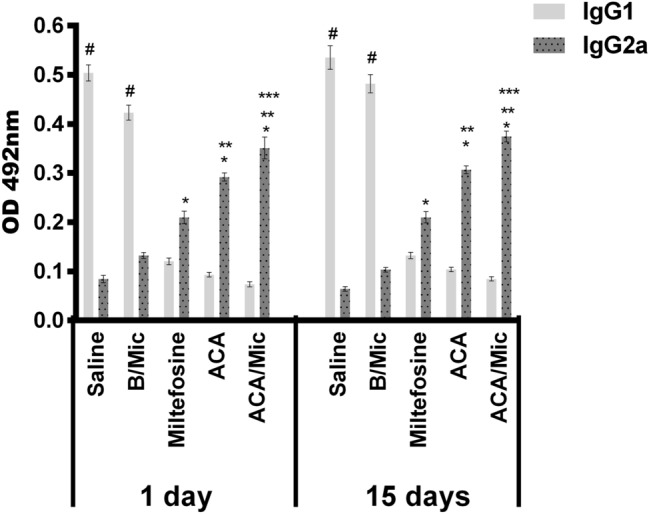
Evaluation of antibody response by indirect ELISA technique. Sera samples were collected from L. infantum-infected and treated mice, one and 15 days post-therapy. Levels of antileishmanial IgG1 and IgG2a isotype antibodies were measured by ELISA assay. Bars indicate the mean ± standard deviation of the groups. (*) indicate statistically significant difference in relation to the saline and B/Mic groups (P < 0.05). (**) indicate statistically significant difference in relation to the Miltefosine group (P < 0.05). (***) indicate statistically significant difference in relation to the ACA group (P < 0.05). (#) indicate statistically significant difference in relation to the Miltefosine, ACA and ACA/Mic groups (P < 0.05)
Analysis of cellular response after treatment
Cellular response of infected and treated mice was evaluated one and 15 days post-treatment by means of dosage of Th1 and Th2-type profile cytokines. Results obtained one day after treatment showed that spleen cells from Miltefosine, ACA or ACA/Mic-treated mice produced significantly higher levels of IFN-γ, IL-12 and GM-CSF, as well as low IL-4 and IL-10 levels. On the other hand, saline- and B/Mic-treated mice groups produced significantly higher levels of antileishmanial IL-4 and IL-10 cytokines (Fig. 4). Nitrite production was also evaluated in the cell supernatant, and results showed that treatment with Miltefosine, ACA or ACA/Mic induced increased levels of this molecule, when compared to the control groups (Fig. 5). To evaluate the participation of T cells in the IFN-γ production in the Miltefosine-, ACA- or ACA/Mic-treated mice, anti-CD4 and anti-CD8 monoclonal antibodies were separately added to the in vitro cultures when stimulus using SLA was performed. Results showed reductions in the levels of this cytokine in the evaluated animals, when compared to the IFN-γ values found in the cell supernatant of stimulated cultures but non-incubated with the monoclonal antibodies; suggesting then that both CD4+ and CD8+ T cells were important for the IFN-γ production in these treated animals (Fig. 6). A flow cytometry assay showed also that Miltefosine-, ACA- or ACA/Mic-treated mice presented significantly higher levels of IFN-γ- and TNF-α-producing CD4+ and CD8+ T cells, when compared to the controls. Conversely, mice groups receiving saline or B/Mic presented significantly higher IL-10-producing CD4+ T cell levels (Fig. 7).
Fig. 4.
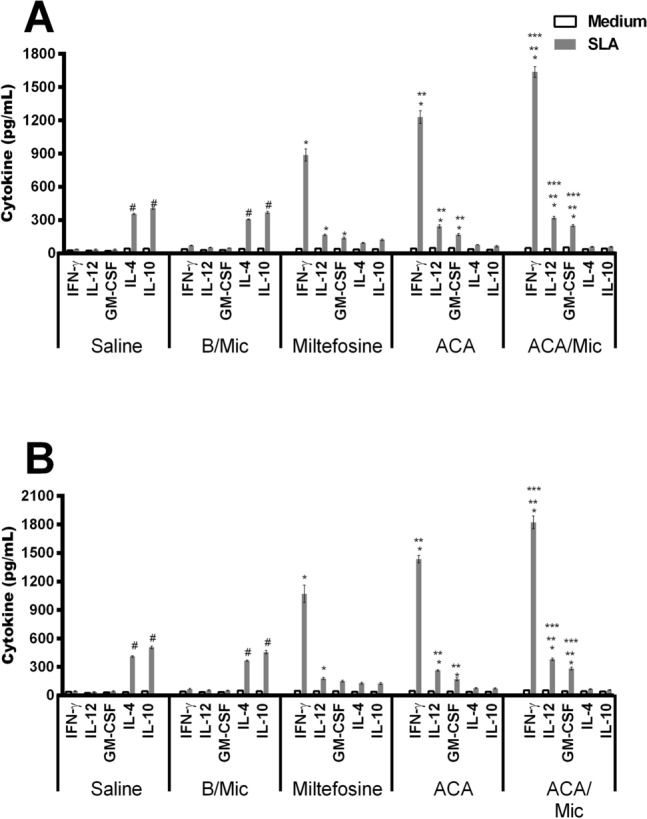
Analysis of cytokine production by capture ELISA technique. L. infantum-infected mice (n = 12 per group) were treated with saline, B/Mic, Miltefosine, ACA or ACA/Mic and their spleens removed, one and 15 days post-therapy. Spleen cells were unstimulated (medium) or stimulated with L. infantum SLA (50 μg/mL), for 48 h at 37 °C in 5% CO2. IFN-γ, IL-4, IL-10, IL-12p70 and GM-CSF levels were measured in the cell supernatants by capture ELISA, one (a) and 15 (b) days post-treatment. Bars indicate the mean ± standard deviation of the groups. (*) indicate statistically significant difference in relation to the saline and B/Mic groups (P < 0.05). (**) indicate statistically significant difference in relation to the Miltefosine group (P < 0.05). (***) indicate statistically significant difference in relation to the ACA group (P < 0.05). (#) indicate statistically significant difference in relation to the Miltefosine, ACA and ACA/Mic groups (P < 0.05)
Fig. 5.
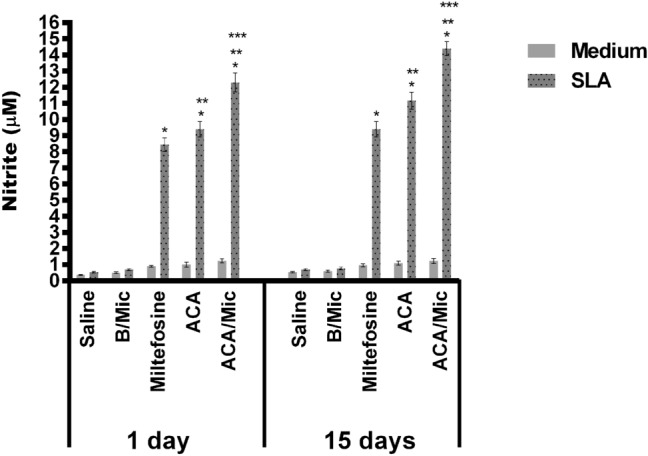
Evaluation of nitrite secretion by Griess reaction. The cellular supernatants used to quantify cytokines (Fig. 5) were also employed to evaluate nitrite production. Bars indicate the mean ± standard deviation of the groups. (*) indicate statistically significant difference in relation to the saline and B/Mic groups (P < 0.05). (**) indicate statistically significant difference in relation to the Miltefosine group (P < 0.05). (***) indicate statistically significant difference in relation to the ACA group (P < 0.05)
Fig. 6.
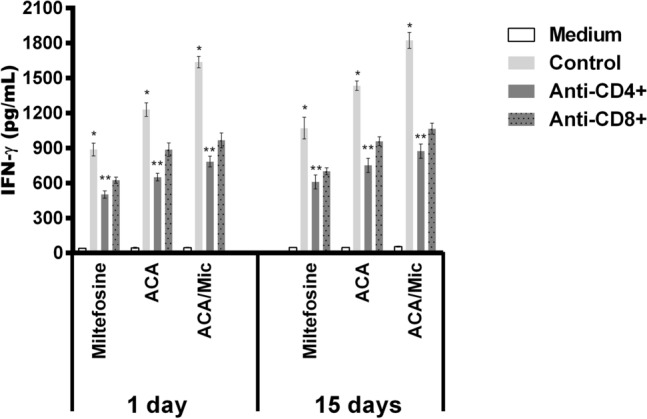
Evaluation of IFN-γ production by T-cell subtypes. Splenocytes (5 × 106/mL) of the Miltefosine-, ACA- or ACA/Mic-treated mice were unstimulated (Medium), stimulated with L. infantum SLA (50 μg/mL; Control) or stimulated and incubated with anti-CD4 (GK 1.5) or anti-CD8 (53–6.7) monoclonal antibody (5 μg each; Pharmingen®, USA), for 48 h at 37 °C in 5% CO2. In all cases, cell supernatants were collected after incubation and used to quantify IFN-γ by capture ELISA. (*) indicate statistically significant difference in comparison to the use of anti-CD4 and anti-CD8 monoclonal antibodies (P < 0.05). (**) indicate statistically significant difference in comparison to the use of anti-CD8 monoclonal antibody (P < 0.05)
Fig. 7.

Evaluation of intracytoplasmic cytokine-producing T-cells by flow cytometry technique. L. infantum-infected mice were treated, euthanized 15 days post-therapy and their spleen cells collected. Spleen cells were left unstimulated (medium) or stimulated with L. infantum SLA. IFN-γ, TNF-α and IL-10-producing T cell levels were evaluated in both CD4+ and CD8+ T cell subpopulations. Results were calculated as the ratio between CD4+ and CD8+ T-cell percentages in the stimulated and unstimulated cultures. Bars indicate the mean ± standard deviation of the groups. (*) indicate statistically significant difference in relation to the saline and B/Mic groups (P < 0.05). (**) indicate statistically significant difference in relation to the Miltefosine group (P < 0.05). (***) indicate statistically significant difference in relation to the ACA group (P < 0.05)
Evaluation of parasite load
To evaluate parasitological efficacy of therapeutics, the parasite load in infected and treated animals was analyzed in two different periods of time post-treatment. In both cases, results showed that Miltefosine-, ACA- or ACA/Mic-treated mice presented significant reductions in the parasite percentage in their livers, spleens, BMs and dLNs when compared to the untreated and infected group mice (Fig. 8). One day post-therapy, mice groups receiving Miltefosine, ACA or ACA/Mic presented reductions in the parasite percentage in the order of 60.0%, 65.0% and 80.0%, respectively, in their spleens; of 42.9%, 50.0% and 78.6%, respectively, in their livers; of 54.5%, 63.6% and 77.3%, respectively, in their dLNs; and of 41.7%, 50.0% and 75.0%, respectively, in their BMs, when compared to the saline group. Fifteen days post-treatment, reductions in the parasite percentage in Miltefosine-, ACA- and ACA/Mic-treated mice were of 54.5%, 63.6% and 77.3%, respectively, in their spleens; of 43.7%, 50.0% and 75.0%, respectively, in their livers; of 45.8%, 62.5% and 70.8%, respectively, in their dLNs; and of 42.9%, 50.0% and 64.3%, respectively, in their BMs, when compared to the saline group. The splenic parasite load was also evaluated by qPCR technique, and results showed reductions in the parasite percentage in Miltefosine-, ACA- and ACA/Mic-treated mice in the order of 46.7%, 63.0% and 77.0%, respectively, when compared to the saline group (Fig. 9). Importantly, ACA/Mic-treated group showed the highest reductions in the parasite percentage in the evaluated organs, when compared to the other experimental groups.
Fig. 8.

Analysis of parasite load by limiting dilution technique. L. infantum-infected mice (n = 12 per group) were treated and, one and 15 days post-therapy, n = 6 per group were euthanized and their livers, spleens, bone marrows (BM) and draining lymph nodes (dLN) were collected for further parasite load quantification by a limiting dilution technique. Bars indicate the mean ± standard deviation of the groups. (*) indicate statistically significant difference in relation to the saline and B/Mic groups (P < 0.05). (**) indicate statistically significant difference in relation to the Miltefosine group (P < 0.05). (***) indicate statistically significant difference in relation to the ACA group (P < 0.05)
Fig. 9.
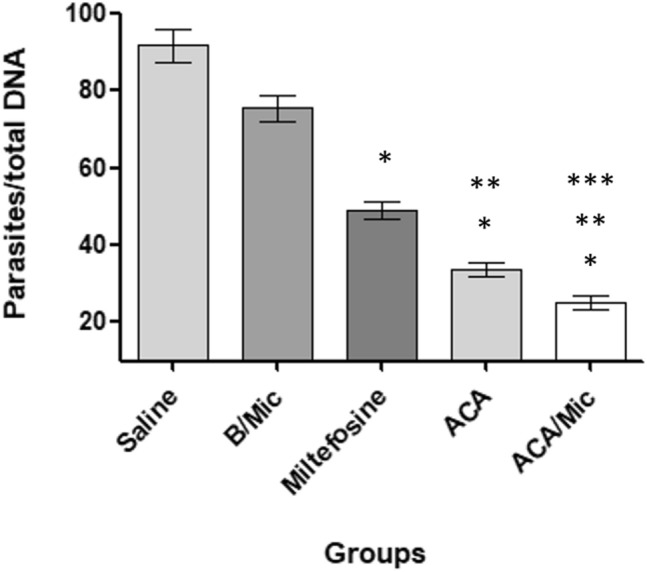
Evaluation of splenic parasite load by quantitative PCR (qPCR) technique. L. infantum-infected BALB/c mice (n = 12 per group) were treated and, 15 days post-treatment, animals (n = 6 per group) were euthanized, their spleen collected and the parasite load was estimated by qPCR technique. Results are expressed as the number of parasites per total DNA. Bars indicate the mean ± standard deviation of the groups. (*) indicate statistically significant difference in relation to the saline and B/Mic groups (P < 0.05). (**) indicate statistically significant difference in relation to the Miltefosine group (P < 0.05). (***) indicate statistically significant difference in relation to the ACA group (P < 0.05)
Evaluation of toxicity in vivo
The hepatic and cardiac toxicity was evaluated after treatment. Results showed higher levels of these markers in control group mice. By comparison of the distinct treatment schedules, Miltefosine-treated mice presented a slight increase in AST, ALT, direct bilirubin, total bilirubin and CK-MB levels, when compared to mice groups treated with ACA or ACA/Mic (Fig. 10). A comparison between these groups showed that micelles reduced the hepatic and cardiac toxicity attributed to ACA in the treated animals, showing values near to those found when sera samples of uninfected and untreated mice were used.
Fig. 10.

Evaluation of toxicity in vivo. Levels of creatine kinase muscle brain fraction (CK-MB; in a) alanine aminotransferase (ALT; in b), aspartate aminotransferase (AST; in c), direct bilirubin (in d) and total bilirubin (in e) were measured in sera samples collected from treated and infected mice, one and 15 days post-therapy. Sera samples of naive (uninfected and untreated) mice were used as control. Bars indicate the mean ± standard deviation of the groups. (*) indicate statistically significant difference in relation to the saline and B/Mic groups (P < 0.05). (**) indicate statistically significant difference in relation to the Miltefosine group (P < 0.05). (***) indicate statistically significant difference in relation to the ACA group (P < 0.05)
Discussion
Treatment against VL is toxic, expensive, lengthy and its efficacy is not warranted. In addition, parasite resistance to commonly used antileishmanial drugs has also been reported [9]. Thus, there is an urgent need to identify new antileishmanial targets. Discovery of new compounds is a long and expensive task, hence, drug repositioning or repurposing should be considered [1]. In the present study, we evaluated the potential antileishmanial activity of ACA, an approved drug for treating type-2-diabetes [22], against L. infantum, main species responsible for most VL cases in the Americas [37]. We analyzed both in vitro and in vivo the possibility of repurposing ACA as a potential therapeutic agent against VL.
Miltefosine was used as a positive control drug for the experiments in vivo. This drug has long been effectively utilized as an orally administered agent for VL treatment [32, 49]. However, disease relapses and parasite resistance have been reported, mainly due to prolonged therapy durations and the drug presenting with a long half-life [33, 35, 43, 48]. In the present study, therapeutic action of Miltefosine was found to be satisfactory. However, treatment of L. infantum-infected mice with ACA, administered either in its free format or incorporated into a micelle delivery system, resulted in significantly lower parasite loads in spleen, liver, BM and dLN than when treating with Miltefosine, tested both by limiting dilution technique and qPCR. The lowest parasitism values were found when infected animals received ACA incorporated into the polymeric micelle system; suggesting this composition should be considered in further studies for its clinical application to treat VL.
Higher efficacy has been observed with antileishmanial agents, such as well-characterized AmpB or newer candidates, such as quinolines, flavonoids, among others, when administered within a delivery system [13, 46, 57]. In this report, and in agreement with other studies, incorporation of ACA into a Poloxamer 407-based micelle system resulted in a significantly better immunological and parasitological response than when using the free molecule. Similarly, Espuelas et al. [16] tested AmpB-coated polymeric micelles against L. donovani species, reporting a synergic action between the drug and the micellar composition, with increases in anti-parasite activity of up to 100 times, when compared to the use of the drug in its free form. In a more recent study, Singh et al. [45] used an AmpB-containing chitosan-coated Poloxamer 407 micelle system, and found that drug delivery within the micelles presented better antileishmanial activity and lower toxicity, in comparison to the use of the free drug. The authors reported the development of a more polarized Th1-type immune response, as well as higher reductions in parasite burden in L. donovani-infected and treated hamsters when compared to treatment with the free drug.
In the present study, Miltefosine-, ACA- or ACA/Mic-treated mice developed a Th1-type immune response profile, characterized by significantly higher levels of IFN-γ, IL-12 and GM-CSF, as well as low IL-4 and IL-10 levels, than the corresponding controls. An in vitro assay using monoclonal antibodies to block T cell subtypes showed that both CD4+ and CD8+ T cell subtypes were responsible for IFN-γ production after treatment. This Th1-type response was related also to the induction of nitrite production by stimulated spleen cells of infected and treated animals, suggesting activation of host macrophages to kill the parasite. Others have likewise shown the development of similar immune profiles in response to treatment of Leishmania-infected mice with antileishmanial molecules, evidencing their relevance for the mammalian host to combat the infection [11, 19, 41].
With the aim to select novel, non-toxic antileishmanial agents, ACA toxicity was evaluated in vivo by means of biochemical dosage of hepatic and cardiac damage markers in sera samples collected from infected and treated animals. Results showed high levels of these enzymatic damage markers in control groups mice, while a slight increase was found in Miltefosine-treated mice when compared to data obtained in the ACA or ACA/Mic groups. In a similar fashion, others have showed also limitations to prolonged usage of Miltefosine related to organic side-effects [5, 18, 47]. Our results showed that treatment with ACA-containing micelles was not toxic to the host and thus, suggest ACA/Mic formulation as a promising replacement for a safer treatment against VL.
Leishmania mitochondria is a primary target for therapeutic candidates since it is intimately related to the parasite’s metabolism and signaling pathways, such as ATP production, regulation of antioxidant machinery, maintenance of ionic homeostasis and special sterol composition [17]. In our study, treatment with ACA resulted in impairment of L. infantum mitochondrial function, causing a disturbance in the mitochondrial membrane potential and a significant increase in ROS production. In addition, ACA caused also the formation and accumulation of lipid bodies in the parasites. Since lipid droplets are related to organelles that regulate the storage of neutral lipids, composed of triacylglycerols and sterol esters [30], this observation could imply cellular stress [26, 40, 44]. Concordantly, treatment with ACA significantly increased the levels of neutral lipids, suggesting the occurrence of cell disorders followed by parasite death. Despite the fact that preliminary results imply ACA targets the Leishmania mitochondria, additional studies are certainly necessary to confirm its mechanism of action.
Limitations of this study include the absence of an evaluation of therapeutic efficacy at different time points post-treatment, as well as the lack of comparison of different dose schedules. This point will be relevant, mainly due to reports of hepatoxicity caused by ACA in humans [8, 23]. In this context, lower doses of this drug, when incorporated in micelles as a delivery system, could help to reach maximum antileishmanial effect but minimum organ toxicity in treated hosts. As a consequence, additional studies are necessary to be performed aiming to solve this question. Nevertheless, the data here presented suggest both in vitro and in vivo antileishmanial activity of ACA against L. infantum, mainly when administered within Pluronic® F127-based polymeric micelles. Results showed specific production of Th1-type cytokines, with consequential significant reduction of parasitism in distinct organs. In conclusion, ACA/Mic is a promising candidate drug which merits further consideration in future studies for VL treatment advancement.
Acknowledgements
This work was supported by Grant MR/R005850/1 from the Medical Research Council (VAccine deveLopment for complex Intracellular neglecteD pAThogEns—VALIDATE), UK, and Grant APQ-408675/2018-7 from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil. The authors also thank the Brazilian agencies Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), CNPq and the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) for the student scholarships.
Declarations
Conflict of interest
The authors confirm that they have no conflicts of interest in relation to this work.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Andrade-Neto VV, Cunha-Junior EF, Faioes VS, Pereira TM, Silva RL, Leon LL, Torres-Santos EC. Leishmaniasis treatment: update of possibilities for drug repurposing. Front Biosci. 2018;23:967–996. doi: 10.2741/4629. [DOI] [PubMed] [Google Scholar]
- 2.Antinarelli LMR, Souza IO, Capriles PVZ, Gameiro J, Britta EA, Nakamura CV, Lima WP, Silva AD, Coimbra ES. Antileishmanial activity of a 4-hydrazinoquinoline derivative: induction of autophagy and apoptosis-related processes and effectiveness in experimental cutaneous leishmaniasis. Exp Parasitol. 2018;195:78–86. doi: 10.1016/j.exppara.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Antonia AL, Wang L, Ko DC. A real-time PCR assay for quantification of parasite burden in murine models of leishmaniasis. PeerJ. 2018;6:e5905. doi: 10.7717/peerj.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araújo VE, Morais MH, Reis IA, Rabello A, Carneiro M. Early clinical manifestations associated with death from visceral leishmaniasis. PLoS Negl Trop Dis. 2012;6:e1511. doi: 10.1371/journal.pntd.0001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharya SK, Sinha PK, Sundar S, Thakur CP, Jha TK, Pandey K, Das VR, Kumar N, Lal C, Verma N, Singh VP, Ranjan A, Verma RB, Anders G, Sindermann H, Ganguly NK. Phase-4 trial of miltefosine for the treatment of Indian visceral leishmaniasis. J Infect Dis. 2007;196:591–598. doi: 10.1086/519690. [DOI] [PubMed] [Google Scholar]
- 6.Brito RCF, Ruiz JC, Cardoso JMO, Ostolin T, Reis LES, Mathias FAS, Aguiar-Soares RDO, Roatt BM, Corrêa-Oliveira R, Resende DM, Reis AB. Chimeric vaccines designed by immunoinformatics-activated polyfunctional and memory T cells that trigger protection against experimental visceral leishmaniasis. Vaccines (Basel) 2020;8:e252. doi: 10.3390/vaccines8020252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet. 2018;392:951–970. doi: 10.1016/S0140-6736(18)31204-2. [DOI] [PubMed] [Google Scholar]
- 8.Carrascosa M, Pascual F, Aresti IS. Acarbose-induced acute severe hepatotoxicity. Lancet. 1997;349:698–699. doi: 10.1016/S0140-6736(05)60134-1. [DOI] [PubMed] [Google Scholar]
- 9.Chakravarty J, Sundar S. Current and emerging medications for the treatment of leishmaniasis. Exp Opin Pharm. 2019;20:1251–1265. doi: 10.1080/14656566.2019.1609940. [DOI] [PubMed] [Google Scholar]
- 10.Chávez-Fumagalli MA, Lage DP, Tavares GSV, Mendonça DVC, Dias DS, Ribeiro PAF, Ludolf F, Costa LE, Coelho VTS, Coelho EAF. In silico Leishmania proteome mining applied to identify drug target potential to be used to treat against visceral and tegumentary leishmaniasis. J Mol Graph Modell. 2019;87:89–97. doi: 10.1016/j.jmgm.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Chouhan G, Islamuddin M, Want MY, Ozbak HA, Hemeg HA, Sahal D, Afrin F. Leishmanicidal activity of Piper nigrum bioactive fractions is interceded via apoptosis in vitro and substantiated by Th1 immunostimulatory potential in vivo. Front Microb. 2015;6:1368. doi: 10.3389/fmicb.2015.01368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coelho EAF, Tavares CA, Carvalho FA, Chaves KF, Teixeira KN, Rodrigues RC, Charest H, Matlashewski G, Gazzinelli RT, Fernandes AP. Immune responses induced by the Leishmania (Leishmania) donovani A2 antigen, but not by the LACK antigen, are protective against experimental Leishmania (Leishmania) amazonensis infection. Infect Immun. 2003;71:3988–3994. doi: 10.1128/IAI.71.7.3988-3994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunha-Júnior EF, Martins TM, Canto-Cavalheiro MM, Marques PR, Portari EA, Coelho MG, Netto CD, Costa PR, Sabino KC, Torres-Santos EC. Preclinical studies evaluating subacute toxicity and therapeutic efficacy of LQB-118 in experimental visceral leishmaniasis. Antimicrob Agents Chemother. 2016;60:3794–3801. doi: 10.1128/AAC.01787-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dayakar A, Chandrasekaran S, Kuchipudi SV, Kalangi SK. Cytokines: key determinants of resistance or disease progression in visceral leishmaniasis: opportunities for novel diagnostics and immunotherapy. Front Immunol. 2019;10:670. doi: 10.3389/fimmu.2019.00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duarte MC, Lage LM, Lage DP, Martins VT, Carvalho AM, Roatt BM, Menezes-Souza D, Tavares CA, Alves RJ, Barichello JM, Coelho EA. Treatment of murine visceral leishmaniasis using an 8-hydroxyquinoline-containing polymeric micelle system. Parasitol Int. 2016;65:728–736. doi: 10.1016/j.parint.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Espuelas S, Legrand P, Loiseau PM, Bories C, Barratt G, Irache JM. In vitro reversion of amphotericin B resistance in Leishmaniadonovani by Poloxamer 188. Antimicrob Agents Chemother. 2000;44:2190–2192. doi: 10.1128/AAC.44.8.2190-2192.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fidalgo LM, Gille L. Mitochondria and trypanosomatids: targets and drugs. Pharm Res. 2011;28:2758–2770. doi: 10.1007/s11095-011-0586-3. [DOI] [PubMed] [Google Scholar]
- 18.Georgiadou SP, Makaritsis KP, Dalekos GN. Leishmaniasis revisited: current aspects on epidemiology, diagnosis and treatment. J Transl Int Med. 2015;3:43–50. doi: 10.1515/jtim-2015-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gogulamudi VR, Dubey ML, Kaul D, Hubert DJ, Kandimalla R, Sehgal R. Vitamins (A&D) and isoprenoid (Chenodeoxycholic acid) molecules are accompanied by Th1 immunostimulatory response and therapeutic cure in vivo: possible antileishmanial drugs. Sci Rep. 2019;9:8531. doi: 10.1038/s41598-019-44630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimaldi G, Jr, Tesh RB. Leishmaniases of the New World: current concepts and implications for future research. Clin Microb Rev. 1993;6:230–250. doi: 10.1128/CMR.6.3.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanefeld M, Schaper F. Acarbose: oral anti-diabetes drug with additional cardiovascular benefits. Exp Rev Card Ther. 2008;6:153–163. doi: 10.1586/14779072.6.2.153. [DOI] [PubMed] [Google Scholar]
- 22.He K, Shi JC, Mao XM. Safety and efficacy of acarbose in the treatment of diabetes in Chinese patients. Ther Clin Risk Man. 2014;10:505–511. doi: 10.2147/TCRM.S50362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsiao SH, Liao LH, Cheng PN, Wu TJ. Hepatotoxicity associated with acarbose therapy. Ann Pharmacother. 2006;40:151–154. doi: 10.1345/aph.1G336. [DOI] [PubMed] [Google Scholar]
- 24.Kedzierski L, Evans KJ. Immune responses during cutaneous and visceral leishmaniasis. Parasitology. 2014;30:1–19. doi: 10.1017/S003118201400095X. [DOI] [PubMed] [Google Scholar]
- 25.Lage LM, Barichello JM, Lage DP, Mendonça DV, Carvalho AM, Rodrigues MR, Menezes-Souza D, Roatt BM, Alves RJ, Tavares CA, Coelho EA, Duarte MC. An 8-hydroxyquinoline-containing polymeric micelle system is effective for the treatment of murine tegumentary leishmaniasis. Parasitol Res. 2016;115:4083–4095. doi: 10.1007/s00436-016-5181-4. [DOI] [PubMed] [Google Scholar]
- 26.Macedo-Silva ST, Visbal G, Urbina JA, Souza W, Rodrigues JC. Potent in vitro antiproliferative synergism of combinations of ergosterol biosynthesis inhibitors against Leishmania amazonensis. Antim Agents Chem. 2015;59:6402–6418. doi: 10.1128/AAC.01150-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mbui J, Olobo J, Omollo R, Solomos A, Kip AE, Kirigi G, Sagaki P, Kimutai R, Were L, Omollo T, Egondi TW, Wasunna M, Alvar J, Dorlo TPC, Alves F. Pharmacokinetics, safety, and efficacy of an allometric miltefosine regimen for the treatment of visceral leishmaniasis in Eastern African children: an open-label, phase II clinical trial. Clin Infect Dis. 2019;68:1530–1538. doi: 10.1093/cid/ciy747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendonça DVC, Martins VT, Lage DP, Dias DS, Ribeiro PAF, Carvalho AMRS, Dias ALT, Miyazaki CK, Menezes-Souza D, Roatt BM, Tavares CAP, Barichello JM, Duarte MC, Coelho EAF. Comparing the therapeutic efficacy of different amphotericin B-carrying delivery systems against visceral leishmaniasis. Exp Parasitol. 2018;186:24–35. doi: 10.1016/j.exppara.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Mendonça DVC, Tavares GSV, Lage DP, Soyer TG, Carvalho LM, Dias DS, Ribeiro PAF, Ottoni FM, Antinarelli LMR, Vale DL, Ludolf F, Duarte MC, Coimbra ES, Chávez-Fumagalli MA, Roatt BM, Menezes-Souza D, Barichello JM, Alves RJ, Coelho EAF. In vivo antileishmanial efficacy of a naphthoquinone derivate incorporated into a Pluronic® F127-based polymeric micelle system against Leishmania amazonensis infection. Biomed Pharmacother. 2019;109:779–787. doi: 10.1016/j.biopha.2018.10.143. [DOI] [PubMed] [Google Scholar]
- 30.Miranda N, Volpato H, Rodrigues JHS, Caetano W, Ueda-Nakamura T, Silva SO, Nakamura CV. The photodynamic action of pheophorbide a induces cell death through oxidative stress in Leishmania amazonensis. J Potochem Photobiol. 2017;174:342–354. doi: 10.1016/j.jphotobiol.2017.08.016. [DOI] [PubMed] [Google Scholar]
- 31.Mohamed-Ahmed AH, Brocchini S, Croft SL. Recent advances in development of amphotericin B formulations for the treatment of visceral leishmaniasis. Curr Opin Infect Dis. 2012;25:695–702. doi: 10.1097/QCO.0b013e328359eff2. [DOI] [PubMed] [Google Scholar]
- 32.Monge-Maillo B, López-Vélez R. Miltefosine for visceral and cutaneous leishmaniasis: drug characteristics and evidence-based treatment recommendations. Clin Infect Dis. 2015;60:1398–1404. doi: 10.1093/cid/civ004. [DOI] [PubMed] [Google Scholar]
- 33.Pandey K, Ravidas V, Siddiqui NA, Sinha SK, Verma RB, Singh TP, Dhariwal AC, Gupta RK, Das P. Pharmacovigilance of miltefosine in treatment of visceral leishmaniasis in endemic areas of Bihar, India. Am J Trop Med Hyg. 2016;95:1100–1105. doi: 10.4269/ajtmh.16-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellosi DS, Moret F, Fraix A, Marino N, Maiolino S, Gaio E, Hioka N, Reddi E, Sortino S, Quaglia F. Pluronic® P123/F127 mixed micelles delivering Sorafenib and its combination with Verteporfin in cancer cells. Int J Nanomed. 2016;11:4479–4494. doi: 10.2147/IJN.S103344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pérez-Victoria FJ, Sánchez-Cañete MP, Seifert K, Croft SL, Sundar S, Castanys S, Gamarro F. Mechanisms of experimental resistance of Leishmania to miltefosine: implications for clinical use. Drug Resist Update. 2006;9:26–39. doi: 10.1016/j.drup.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Pijpers J, den Boer ML, Essink DR, Ritmeijer K. The safety and efficacy of miltefosine in the long-term treatment of post-kala-azar dermal leishmaniasis in South Asia—a review and meta-analysis. PLoS Negl Trop Dis. 2019;13:e0007173. doi: 10.1371/journal.pntd.0007173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pratlong F, Lami PP, Ravel C, Balard Y, Dereure J, Serres G, Baidouri FE, Dedet JP. Geographical distribution and epidemiological features of Old World Leishmania infantum and Leishmaniadonovani foci, based on the isoenzyme analysis of 2277 strains. Parasitology. 2013;140:423–434. doi: 10.1017/S0031182012001825. [DOI] [PubMed] [Google Scholar]
- 38.Rahman R, Goyal V, Haque R, Jamil K, Faiz A, Samad R, Ellis S, Balasegaram M, Boer MD, Rijal S, Strub-Wourgaft N, Alves F, Alvar J, Sharma B. Safety and efficacy of short course combination regimens with Am Bisome®, miltefosine and paromomycin for the treatment of visceral leishmaniasis (VL) in Bangladesh. PLoS Negl Trop Dis. 2017;11:e0005635. doi: 10.1371/journal.pntd.0005635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raja MRC, Velappan AB, Chellappan D, Debnath J, Mahapatra SK. Eugenol derived immunomodulatory molecules against visceral leishmaniasis. Eur J Med Chem. 2017;139:503–518. doi: 10.1016/j.ejmech.2017.08.030. [DOI] [PubMed] [Google Scholar]
- 40.Rebello KM, Andrade-Neto VV, Zuma AA, Motta MCM, Gomes CRB, Souza MVN, Atella GC, Branquinha MH, Santos ALS, Torres-Santos EC, d'Avila-Levy CM. Lopinavir, an HIV-1 peptidase inhibitor, induces alteration on the lipid metabolism of Leishmaniaamazonensis promastigotes. Parasitology. 2018;145:1304–1310. doi: 10.1017/S0031182018000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reguera RM, Álvarez-Velilla R, Domínguez-Asenjo B, Gutiérrez-Corbo C, Balaña-Fouce R, Cushman M, Pérez-Pertejo Y. Antiparasitic effect of synthetic aromathecins on Leishmania infantum. BMC Vet Res. 2019;15:405. doi: 10.1186/s12917-019-2153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reis TAR, Oliveira-da-Silva JA, Tavares GSV, Mendonça DVC, Freitas CS, Costa RR, Lage DP, Martins VT, Machado AS, Ramos FF, Silva AM, Ludolf F, Antinarelli LMR, Brito RCF, Chávez-Fumagalli MA, Humbert MV, Roatt BM, Coimbra ES, Coelho EAF. Ivermectin presents effective and selective antileishmanial activity in vitro and in vivo against Leishmania infantum and is therapeutic against visceral leishmaniasis. Exp Parasitol. 2020;221:108059. doi: 10.1016/j.exppara.2020.108059. [DOI] [PubMed] [Google Scholar]
- 43.Rijal S, Ostyn B, Uranw S, Rai K, Bhattarai NR, Dorlo T, Beijnen JH, Vanaerschot M, Decuypere S, Dhakal SS, Das ML, Karki P, Singh R, Boelaert M, Dujardin JC. Increasing failure of miltefosine in the treatment of Kala-azar in Nepal and the potential role of parasite drug resistance, reinfection, or noncompliance. Clin Infect Dis. 2013;56:1530–1538. doi: 10.1093/cid/cit102. [DOI] [PubMed] [Google Scholar]
- 44.Sangenito LS, Menna-Barreto RFS, Oliveira AC, d'Avila-Levy CM, Branquinha MH, Santos ALS. Primary evidence of the mechanisms of action of HIV aspartyl peptidase inhibitors on Trypanosoma cruzi trypomastigote forms. Int J Antimicrob Agents. 2018;52:185–194. doi: 10.1016/j.ijantimicag.2018.03.021. [DOI] [PubMed] [Google Scholar]
- 45.Singh PK, Pawar VK, Jaiswal AK, Singh Y, Srikanth CH, Chaurasia M, Bora HK, Raval K, Meher JG, Gayen JR, Dube A, Chourasia MK. Chitosan coated Pluronic F127 micelles for effective delivery of amphotericin B in experimental visceral leishmaniasis. Int J Biol Macromol. 2017;105:1220–1231. doi: 10.1016/j.ijbiomac.2017.07.161. [DOI] [PubMed] [Google Scholar]
- 46.Sousa JKT, Antinarelli LMR, Mendonça DVC, Lage DP, Tavares GSV, Dias DS, Ribeiro PAF, Ludolf F, Coelho VTS, Oliveira-da-Silva JA, Perin L, Oliveira BA, Alvarenga DF, Chávez-Fumagalli MA, Brandão GC, Nobre V, Pereira GR, Coimbra ES, Coelho EAF. A chloroquinoline derivate presents effective in vitro and in vivo antileishmanial activity against Leishmania species that cause tegumentary and visceral leishmaniasis. Parasitol Int. 2019;73:101966. doi: 10.1016/j.parint.2019.101966. [DOI] [PubMed] [Google Scholar]
- 47.Sundar S, Olliaro PL. Miltefosine in the treatment of leishmaniasis. Clinical evidence for informed clinical risk management. Ther Clin Risk Manag. 2007;3:733–740. [PMC free article] [PubMed] [Google Scholar]
- 48.Sundar S, Singh A, Rai M, Prajapati VK, Singh AK, Ostyn B, Boelaert M, Dujardin JC, Chakravarty J. Efficacy of miltefosine in the treatment of visceral leishmaniasis in India after a decade of use. Clin Infect Dis. 2012;55:543–550. doi: 10.1093/cid/cis474. [DOI] [PubMed] [Google Scholar]
- 49.Sundar S, Singh A, Chakravarty J, Rai M. Efficacy and safety of miltefosine in treatment of post-kala-azar dermal leishmaniasis. Sci World J. 2015;2015:414378. doi: 10.1155/2015/414378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sundar S, Singh A. Chemotherapeutics of visceral leishmaniasis: present and future developments. Parasitology. 2018;145:481–489. doi: 10.1017/S0031182017002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sundar S, Singh A, Agrawal N, Chakravarty J. Effectiveness of single-dose liposomal amphotericin B in visceral leishmaniasis in Bihar. Am J Trop Med Hyg. 2019;101:795–798. doi: 10.4269/ajtmh.19-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tavares GSV, Mendonça DVC, Lage DP, Granato JDT, Ottoni FM, Ludolf F, Chávez-Fumagalli MA, Duarte MC, Tavares CAP, Alves RJ, Coimbra ES, Coelho EAF. Antileishmanial activity, cytotoxicity and mechanism of action of clioquinol against Leishmania infantum and Leishmaniaamazonensis species. Basic Clin Pharm Toxicol. 2018;123:236–246. doi: 10.1111/bcpt.12990. [DOI] [PubMed] [Google Scholar]
- 53.Tavares GSV, Mendonça DVC, Miyazaki CK, Lage DP, Soyer TG, Carvalho LM, Ottoni FM, Dias DS, Ribeiro PAF, Antinarelli LMR, Ludolf F, Duarte MC, Coimbra ES, Chávez-Fumagalli MA, Roatt BM, Menezes-Souza D, Barichello JM, Alves RJ, Coelho EAF. A Pluronic® F127-based polymeric micelle system containing an antileishmanial molecule is immunotherapeutic and effective in the treatment against Leishmania amazonensis infection. Parasitol Int. 2019;68:63–72. doi: 10.1016/j.parint.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 54.Tavares GSV, Mendonça DVC, Pereira IAG, Oliveira-da-Silva JA, Ramos FF, Lage DP, Machado AS, Carvalho LM, Reis TAR, Perin L, Carvalho AMRS, Ottoni FM, Ludolf F, Freitas CS, Bandeira RS, Silva AM, Chávez-Fumagalli MA, Duarte MC, Menezes-Souza D, Alves RJ, Roatt BM, Coelho EAF. A clioquinol-containing Pluronic® F127 polymeric micelle system is effective in the treatment of visceral leishmaniasis in a murine model. Parasite. 2020;27:29. doi: 10.1051/parasite/2020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang MZ, Zhu X, Srivastava A, Liu Q, Sweat JM, Pandharkar T, Stephens CE, Riccio E, Parman T, Munde M, Mandal S, Madhubala R, Tidwell RR, Wilson WD, Boykin DW, Hall JE, Kyle DE, Werbovetz KA. Novel arylimidamides for treatment of visceral leishmaniasis. Antimicrob Agents Chemother. 2010;54:2507–2516. doi: 10.1128/AAC.00250-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.World Health Organization (2018) Leishmaniasis, http://www.who.int/topics/leishmaniasis/en/. Accessed 2 Jun 2018
- 57.Yousuf M, Mukherjee D, Pal A, Dey S, Mandal S, Pal C, Adhikari S. Synthesis and biological evaluation of ferrocenylquinoline as a potential antileishmanial agent. ChemMedChem. 2015;10:546–554. doi: 10.1002/cmdc.201402537. [DOI] [PubMed] [Google Scholar]
- 58.Zhang N, Prasad S, Despointes CEH, Young J, Kima PE. Leishmania parasitophorous vacuole membranes display phosphoinositides that create conditions for continuous Akt activation and a target for miltefosine in Leishmania infections. Cell Microb. 2018;20:e12889. doi: 10.1111/cmi.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu X, Farahat AA, Mattamana M, Joice A, Pandharkar T, Holt E, Banerjee M, Gragg JL, Hu L, Kumar A, Yang S, Wang MZ, Boykin DW, Werbovetz KA. Synthesis and pharmacological evaluation of mono-arylimidamides as antileishmanial agents. Bioorg Med Chem Lett. 2016;26:2551–2556. doi: 10.1016/j.bmcl.2016.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]