

Summary
The chick embryo is a favored model for developmental studies owing to its accessibility and ease of manipulation. Ex ovo electroporation provides a highly efficient method for screening perturbation phenotypes using a variety of reagents, including CRISPR and morpholinos. Additionally, the chick system lends itself well to rapid medium-throughput enhancer screening. Constructs facilitating tissue-specific protein pull-down can also be transfected using this protocol. Furthermore, bilateral electroporation with control and experimental reagents provides a robust assay for accurately interpreting functional perturbations.
For complete details on the use and execution of this protocol, please refer to Williams et al. (2019).
Subject areas: High Throughput Screening, Model Organisms
Graphical abstract
Highlights
-
•
Highly efficient method for transfection of early chick embryos
-
•
Bilateral electroporation allows comparison of control and experimental conditions
-
•
Non-mosaic, highly reproducible results using various reagents
The chick embryo is a favored model for developmental studies owing to its accessibility and ease of manipulation. Ex ovo electroporation provides a highly efficient method for screening perturbation phenotypes using a variety of reagents, including CRISPR and morpholinos. Additionally, the chick system lends itself well to rapid medium-throughput enhancer screening. Constructs facilitating tissue-specific protein pull-down can also be transfected using this protocol. Furthermore, bilateral electroporation with control and experimental reagents provides a robust assay for accurately interpreting functional perturbations.
Before you begin
This protocol provides the user step-by-step instructions for electroporating early chick embryos ex ovo. This assay is highly efficient and enables transfection of plasmids carrying CRISPR reagents (Williams et al., 2018) or putative enhancer elements (Williams et al., 2019) without issues of high mosaicism and low reproducibility hampering other models. We detail the procedure to extract embryos from eggs and subsequent injection and electroporation of reagents. Bilateral electroporation of control and experimental reagents is also described, that provides an excellent experimental paradigm offering most accurate internal, staged matched control.
Incubate eggs
Timing: 18–24 h
-
1.
For electroporations into embryonic cell layers at gastrulation stage HH4 (Hamburger and Hamilton, 1951), incubate eggs vertically for 18–24∗ h at 37°C, in conditions of approximately 70% humidity (∗ timing can vary according to season and egg quality).
Prepare reagents to be electroporated
-
2.Plasmid preparations should be endotoxin free to ensure high electroporation efficiency and embryo survival. We recommend the Qiagen Endofree maxi prep kit (Cat. no. 12362), but we have also had success with the Omega BioTek EZNA Endofree mini II kit (Cat. No. D6950), when smaller DNA quantities for quick screening experiments are adequate.
-
a.Final concentration of reagents used will require optimization. We find 2.0 μg/μL to be an optimal concentration in fluorescent enhancer reporter assays, whereas 0.5-1.0 μg/μL yields adequate expression levels of CRISPR components, driven under control of ubiquitous promoters (Williams et al., 2018).
-
b.Vegetable dye mixed with DNA preparations to enable visualization during the injection step is diluted in Ringer’s and filter sterilized. If performing bilateral electroporation different colors of the dye should be used for experimental and control reagents to ensure and visually inspect that experimental and control reagents remain clearly separated during electroporation and hence avoid cross diffusion and reagent mixing.
-
a.
Prepare filter papers
-
3.
Cut Whatman filter paper into approximately 1.5–2.0 cm square pieces, make holes in the middle using regular office hole puncher and autoclave.
Prepare needles
-
4.
Pull needles from 0.8 – 1.1 mm diameter PYREX® capillary tubes.
Prepare Ringers solution
-
5.
Prepare stock solutions.
-
6.
Prepare aliquots of working solution.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Fertilized hen eggs | Henry Stewart & Co Ltd | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| “Vegetable dye” Blue 1 | Spectra Colors Corp. | Cat# 3844-45-9 |
| Critical commercial assays | ||
| Endofree Plasmid Maxi Kit | QIAGEN | Cat# 12362 |
| E.Z.N.A Endofree Plasmid Mini Kit II | VWR/Omega Bio-tek | Cat# D6950 |
| Experimental models: organisms/strains | ||
| Hen eggs come from Bovans brown chickens | Henry Stewart & Co Ltd | N/A |
| Other | ||
| OvoEasy 190 Advance Series II incubator | Brinsea Scientific | N/A |
| Dissecting microscope | N/A | N/A |
| 35 mm Petri dishes | Corning | Cat# 430165 |
| 92×16 mm Petri dishes (with cams) | Fisher Scientific | Cat# 82.1473.001 |
| PYREX® capillary tubes 0.8–1.1 × 100 mm (borosilicate glass) | Corning | Cat# 9530-2 |
| Microloader tips 0.5–20 μL | Eppendorf | Cat# F5242956003 |
| Whatman chromatography filter paper (3 mm) | VWR | Cat# 588-3113 |
Materials and equipment
CRITICAL: Use autoclaved glassware that has only been washed with hot water, not detergent, as this is not well-tolerated by chick embryos.
40× stock solution 1
| Amount (g) for 1 l | Reagent | Final concentration (M) |
|---|---|---|
| 144.0 | NaCl | 2.50 |
| 14.8 | KCl | 0.20 |
| 9.0 | CaCl2·2H2O | 0.06 |
| To a final volume of 1 l | ddH20 | n/a |
40× stock solution 2
| Amount (g) for 1 l | Reagent | Final concentration (M) |
|---|---|---|
| 144.0 | NaCl | 2.50 |
| 8.7 | NaHPO4·7H2O | 0.03 |
| 0.8 | KH2PO4 | 0.006 |
| To a final volume of 1 l | ddH20 | n/a |
| Adjust pH to 7.4 with HCl | ||
Stock solutions can be stored at 18°C–22°C for 6 months.
1× working Ringers solution
| Amount (mL) for 4 l | Reagent | Final concentration (X) |
|---|---|---|
| 100 | 40× solution 1 | 1 |
| 100 | 40× solution 2 | 1 |
| To a final volume of 4 l | ddH20 | N/A |
Final 1× working solution:
(123.2 mM NaCl, 1.53 m CaCl2, 4.96 mM KCl, 0.81 mM Na2HPO4, 0.15 mM KH2PO4)
Working solution can be stored at 18°C–22°C for 6 months but once opened should be used on the same day or discharged to avoid cross contamination between experiments.
Prepare dissection tools
Ensure all dissection tools are cleaned with hot water, ddH2O and ethanol. Tools used for live embryo work and those used for dissection of fixed tissue should be kept separate. Live embryo work tools should not be washed using detergent.
Incubators
Egg incubator(s) must enable maintaining 37°C temperature with ∼70% humidity. This can be achieved by placing trays of water in the incubator. It is also useful to acquire plug-in timers such that egg incubation time can be conveniently controlled.
Electroporation setup
Our electrodes were custom designed (by Tatjana Sauka-Spengler) and made at California Institute of Technology Bio-electronics shop. Here we describe the design such that it can be reproduced or appropriate alternatives can be sourced (Figure 1). The negative electrode chamber resembles a small (∼35 mm) petri-dish, and is made of a clear plastic resin (Figure 1A). The solid base is 4 mm in depth and features a central blunt-cone shaped cavity (∼9 mm diameter at the top, 4 mm at the base) (Figure 1B). The surrounding open surface of the chamber is sunken by ∼3 mm to allow the chamber to be filled with Ringers solution (Figure 1C). A platinum foil sheet (0.01 × 5 × 5 mm) is situated at the base of the central cavity and connected through the wall of the chamber to an exterior copper tab via a platinum wire, to a facilitate connection to the square-pulse electroporator (Figure 1C). The hand-held positive electrode is a pen-size/shape (Figure 1D) with a paddle shaped platinum (18ga) electrode (2 × 4 mm) attached through the ‘pen’ to 1.375 mm platinum wire (Figure 1E). Although these electrodes are not commercially available, they can be sourced by contacting Tim Heitzman (heitzman@caltech.edu).
Alternatives: These electrodes can be sourced from Sonidel™ Ltd, however we have not used these products and therefore optimization would be required for this application.
Figure 1.

Positive and negative electrodes
(A–C) The negative electrode is a thin piece of platinum foil, encased centrally within a resin chamber.
(D) The positive electrode is a platinum paddle housed in a pen-like structure.
(E) Close up of platinum paddle from (D).
Step-by-step method details
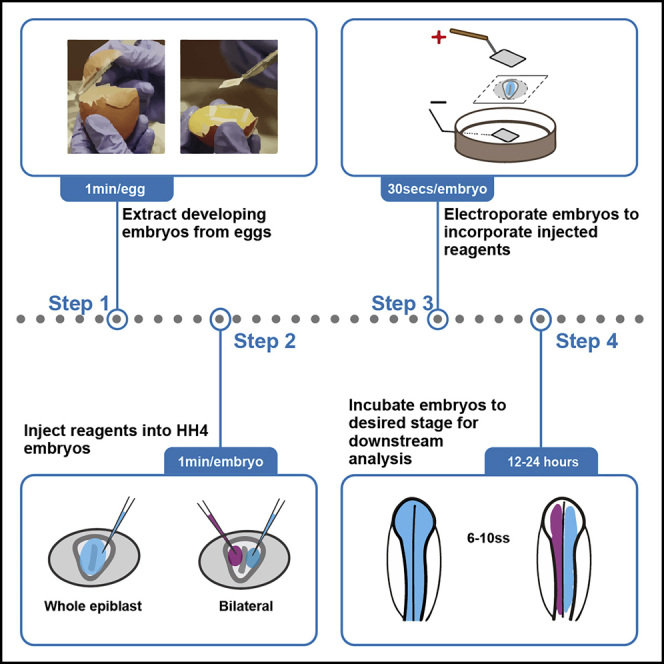
Extracting embryos from the egg
Here we describe the steps involved to remove developing embryos from the egg and prepare them for electroporation.
Note: Electroporation at HH4, targets ectodermal derivatives (neural, skin, placodes, and neural crest); if mesodermal derivatives are to be targeted electroporations should be conducted at HH3+, however this does reduce the over-night survival rate.
-
1.
Break a small hole in the shell in the top hemisphere of the egg.
-
2.
Remove the thick albumin by pouring/pulling with blunt serrated forceps. Collect thin albumin only into a 50 mL falcon tube to use as media for embryo incubation. Take care not to break the yolk during this process as this will disrupt the embryo.
-
3.
Break the eggshell away until none is visible over the yolk (Figures 2A and 2B).
-
4.
Locate the developing embryo; this may require gently turning the yolk using horizontally positioned blunt forceps.
-
5.
Place a piece of filter paper on the yolk such that the embryo is positioned in the middle of the hole (Figure 2C).
-
6.
Carefully cut each edge of the filter paper, and using dissecting forceps gently lift the filter square with the embryo attached to it off the yolk, sliding it in the horizontal direction (Figure 2D).
-
7.
Invert the filter paper, hence holding the ventral side of the embryo (with hypoblast on the top and dorsal side, covered with the vitelline membrane to the bottom) and place it into Ringer’s solution (Figure 2E). Move the filter paper and the attached embryo around the petri dish gently to remove any excess yolk still attached to the upper surface and transfer it to a clean dish of Ringer’s.
Note: Embryos remain positioned ventral side up and can be left in Ringer’s at room temperature while collecting others. This time should ideally not exceed ∼1 h, as the embryos can detach from the paper.
Figure 2.

Ex ovo preparation and electroporation of gastrula stage chick embryos
(A and B) Egg shell is removed until none protrudes above the surface of the yolk.
(C and D) Hole-punched filter paper is used to extract the embryo from the yolk.
(E) Embryos are collected in Ringer’s solution.
(F–H) (F and G) Injection and (H) electroporation of HH4 embryo.
(I) Embryos are cultured on a thin layer of albumin in 35 mm petri dish.
Electroporation
During this step embryos are injected with required reagents (plasmids, morpholinos, CRISPR components etc) and electroporated using square-wave electric pulses, such that cell membranes are transiently depolarized and destabilized, allowing the reagents to move directionally into the cells.
-
8.
Secure the electroporation chamber containing the negative electrode to the microscope base using double-sided tape, ensuring the center of the chamber is central to the field of view. Fill with ∼1 mL clean 1× Ringer’s solution. Attach to the electroporator equipment using a crocodile clip.
-
9.
Place the embryo on filter paper into the center of the electroporation chamber, overlaying the chamber cavity and the electrode platinum sheet. Ventral side facing up and submerged in Ringer’s solution.
-
10.
Back fill a needle with reagents to be injected using a micro-loader tip and carefully break off the end of the needle using fine forceps to allow expulsion of reagents.
-
11.
Inject reagents into the cavity between the blastoderm and underlying vitelline membrane. We use an airflow system, whereby oxygen-free nitrogen is allowed to continuously escape through an outlet valve and serves as injection OFF switch. When outlet valve is covered by the user’s finger/thumb (ON-switch), the airflow is directed to the needle and consequently reagents are pushed into the region of interest. This can also be performed using mouth pipetting, but we do not recommend using the microinjector due to the difficulty of controlling the flow and intensity of the injection. Gently break the surface of the hypoblast with the needle at the targeted region ensuring not to push too far through and pierce the vitelline membrane (Figures 2F and 2G). Multiple injection sites can be created if required. The injected reagents (in color due to the presence of vegetable dye) will distribute radially and the injected “circle” should stably stay in place if the reagents were delivered appropriately.
Note: If performing bilateral electroporation use two separate needles to sequentially inject left and right sides of the embryo. Ensure the control and experimental reagents do not cross over the primitive streak.
-
12.
Ensuring the embryo is positioned centrally above the negative electrode at the base of the chamber, position the paddle shaped positive electrode directly above the embryo. Take care not to touch the embryo with the electrode but keeping it submerged in Ringer’s (Figure 2H). Apply the electric current, for our electroporation setup, we use a set of 5 pulses (5 V, 100 ms ON, 50 ms OFF). However, the exact conditions may vary depending on the equipment used, and we provide some optimization guidelines in the troubleshooting section. It is of note that, in general, electroporators producing square electric pulses work significantly better than those generating exponential decay wave.
-
13.
Place the embryo into a 35 mm petri dish containing 1 mL of the thin albumin, (Figure 2I) coat the inside edge of the lid with thin albumin such that a seal is created. We have not found it necessary to supplement albumin with antibiotics, but use of Penicillin/Streptomycin can be implemented at a final concentration of 1×, if infections in the cultures occur.
-
14.
Incubate embryos at 37°C to the desired stage (approximately 12–16 h from HH4 to 7ss).
Expected outcomes
Depending on the quality of eggs and reagents injected we routinely achieve survival rates >70% following up to 20 h incubation. This drops to ∼50% over 48 h.
We have quantitatively assessed this method by cell-counting and determined ∼80% of pre-migratory neural crest cells of the dorsal neural tube at 6ss and ∼60% of other epiblast derivatives received the electroporated constructs (Williams et al., 2018).
Limitations
Developmental time after electroporation is limited to ∼48 h, as embryos are grown flat on albumin which hinders development after HH16. As such this assay is not applicable if later stages are to be studied. Here we recommend using in ovo techniques for later stages.
Troubleshooting
Problem 1
Embryos detaching from paper during embryo extraction (step 7)
Potential solution
Ensure all albumin is removed during embryo preparation (step 4)
Check Ringer’s solution is made correctly.
Avoid keeping embryos prepared for electroporations for extended periods of time (step 7).
Problem 2
Poor survival of embryos
Potential solution
Electroporation conditions may need to be adjusted. Specifically, consider reducing the voltage from 5 V to 4.8 V or lower.
Titer down concentration of reagents injected. We have found excessive Cas9 (>1.5 μg/μL) causes toxicity. Some morpholinos can also be toxic. Total plasmid concentration above 6 μg/μL also effects survival.
Problem 3
Lack of expression of fluorescent constructs
Potential solution
Check that the constructs sequences are correct, ORF of interest starts with the first methionine after the promoter and is placed in-frame after the Kozak sequence.
Electroporations conditions can be adjusted, we recommend incrementally increasing voltage by 0.2 to maximum 6 V and assessing the effect, however this can cause a decrease in embryo survival.
The upper paddle electrode (positive) needs to be held approximately 3–5 mm above the embryo. If the upper electrode is held too close to the embryo, the embryonic tissue may be burnt and embryos will die (ideally embryonic tissue should not be touched). If the distance between the lower electrode (platinum sheet at the bottom of the electroporation cuvette) and the upper paddle electrode is too large, the electroporation will be ineffective.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tatjana Sauka-Spengler tatjana.sauka-spengler@imm.ox.ac.uk
Materials availability
This study did not generate new unique reagents
Data and code availability
This study did not generate/analyze [datasets/code.
Acknowledgments
This technique has evolved from other methods (Chapman et al., 2001) and has been applied in many subsequent studies (Betancur et al., 2010; Sauka-Spengler and Barembaum, 2008; Simoes-Costa et al., 2012). This work was supported by MRC (G0902418), The Lister Institute for Preventive Medicine Research Prize, John Fell Fund (131/038), and Leverhulme Trust (grant RPG-2015-026) to T.S.-S.
Author contributions
Methodology: R.M.W. and T.S.S.; resources, T.S.S.; writing – original draft, R.M.W.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Ruth M. Williams, Email: ruth.williams@imm.ox.ac.uk.
Tatjana Sauka-Spengler, Email: tatjana.sauka-spengler@imm.ox.ac.uk.
References
- Betancur P., Bronner-Fraser M., Sauka-Spengler T. Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc. Natl. Acad. Sci. U S A. 2010;107:3570–3575. doi: 10.1073/pnas.0906596107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman S.C., Collignon J., Schoenwolf G.C., Lumsden A. Improved method for chick whole-embryo culture using a filter paper carrier. Dev. Dyn. 2001;220:284–289. doi: 10.1002/1097-0177(20010301)220:3<284::AID-DVDY1102>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Hamburger V., Hamilton H.L. A series of normal stages in the development of the chick embryo. J. Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Sauka-Spengler T., Barembaum M. Gain- and loss-of-function approaches in the chick embryo. Methods Cell Biol. 2008;87:237–256. doi: 10.1016/S0091-679X(08)00212-4. [DOI] [PubMed] [Google Scholar]
- Simoes-Costa M.S., McKeown S.J., Tan-Cabugao J., Sauka-Spengler T., Bronner M.E. Dynamic and differential regulation of stem cell factor FoxD3 in the neural crest is Encrypted in the genome. PLoS Genet. 2012;8:e1003142. doi: 10.1371/journal.pgen.1003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.M., Candido-Ferreira I., Repapi E., Gavriouchkina D., Senanayake U., Ling I.T.C., Telenius J., Taylor S., Hughes J., Sauka-Spengler T. Reconstruction of the global neural crest gene regulatory network in vivo. Dev. Cell. 2019;51:255–276 e257. doi: 10.1016/j.devcel.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.M., Senanayake U., Artibani M., Taylor G., Wells D., Ahmed A.A., Sauka-Spengler T. Genome and epigenome engineering CRISPR toolkit for in vivo modulation of cis-regulatory interactions and gene expression in the chicken embryo. Development. 2018;145:dev160333. doi: 10.1242/dev.160333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze [datasets/code.