

The mechanisms by which antigens specifically sensitize lymphocytes to induce immunity are well recognized. The past decade has witnessed an increased awareness of the importance of specific sensitization of monocytes, natural killer cells, and dendritic cells. This sensitization results from long-lived epigenetic changes that profoundly enhance or suppress immune responses. Stated simply, immunologic stimuli — infections or immunizations — can have long-term consequences for the immune responsiveness of the host. Our purpose is to describe the mechanisms by which these epigenetic effects come about and their effects on responses to subsequent infection and vaccination. Epigenetic therapies have improved clinical outcomes in patients with cancer and are on the verge of regular clinical applicability in the management of infections and vaccine administration.
SPECIFICITY OF HUMORAL AND CELL-MEDIATED IMMUNITY
In the 1890s, Klemperer and Klemperer showed that inoculating rabbits with killed pneumococci protected them against pneumococcal challenge. Infusing serum from immune rabbits to nonimmune ones transferred protection, thus expressing the concept of humoral immunity. They further discovered that this inoculation did not protect against challenge with a different pneumococcal strain, thereby showing that humoral immunity was an immunologically specific phenomenon.1
The concept of cell-mediated immunity developed more slowly. Koch’s attempts to immunize against tuberculosis with tuberculin, a set of proteins extracted from Mycobacterium tuberculosis, were not successful. Von Pirquet showed that, in patients who had tuberculosis, injection of tuberculin caused a local inflammatory response; this delayed-type hypersensitivity was characterized by accumulation of lymphocytes and macrophages and was antigen-specific. In 1957, Dubos and Schaedler reported that injecting bacille Calmette–Guérin (BCG), an attenuated strain of Mycobacterium bovis, into mice enhanced their resistance to Staphylococcus aureus and M. fortuitum.2 They attributed this non–antigen-specific immunity to activation of monocytes, a phenomenon that was shown in vitro.3,4 Seminal experiments by Mackaness5,6 showed that infection of mice with intracellular organisms such as brucella or BCG protected them against antigenically unrelated bacteria such as listeria. The proposed mechanism was an antigenically specific stimulation that resulted in an enhanced nonspecific bactericidal capacity of macrophages.
Mackaness called this phenomenon acquired cellular resistance. Musher et al. reported that infection with listeria protected mice against the quintessentially extracellular organism Streptococcus pneumoniae7 and attributed this effect to increased oxidative metabolism in listeria-infected macrophages.8 Nonspecific acquired cellular resistance protected experimental animals against challenge with intracellular bacteria (listeria),6 viruses (influenza virus),9 and protozoa (babesia and plasmodium,10 toxoplasma,11 and leishmania12). A crucial set of observations in Mackaness’s experiments went unexplained. Macrophages from mice that were previously infected with listeria, brucella, or BCG retained enhanced antimicrobial capacity after the original infecting organisms had been eradicated, without reintroduction of the sensitizing antigen and independent of lymphocytes. This phenomenon is now understood to be due to long-lasting epigenetic changes in macrophages.
EPIGENETICS AND ANTIGEN-NONSPECIFIC TRAINED IMMUNITY
The mechanisms by which innate cells — macrophages, dendritic cells, natural killer cells, and reticuloendothelial cells — can become activated by acquiring antigen-nonspecific immune memory have been elucidated in the past decade. This adaptive characteristic, one that is independent of lymphocytes, has been termed “trained immunity.”13 The two most commonly studied models for inducing trained immunity are immunization with BCG or with β-glucan (a cell-wall component of Candida). The lymphocyte-independent nature of this training and antigen nonspecificity have been shown in a broad array of experiments (Table 1). Seven days after purified macrophages were incubated with BCG or with β-glucan in vitro, exposure to lipopolysaccharide, candida, or staphylococcus caused them to generate greater amounts of tumor necrosis factor than control macrophages.14 When mice that had severe combined immunodeficiency (SCID) and, therefore, lacked B or T cells were infected with BCG, their macrophages had more robust antigen-nonspecific secondary responses and they were able to survive challenge with a lethal dose of candida.14 Mice that were vaccinated with β-glucan had increased survival when given a lethal dose of S. aureus.19 Three months after humans were vaccinated with BCG, their macrophages generated greater amounts of tumor necrosis factor and interleukin-6 after exposure to staphylococcus or candida.14 Other experiments in laboratory animals have shown that macrophages can acquire antigen-nonspecific and cross-protective trained immunity against an array of pathogens, including bacteria (Escherichia coli, S. pneumoniae, and S. aureus),14,19–21 viruses (yellow fever and influenza),9,15 parasites (plasmodia),22 and fungi (Candida albicans).14,20,22,23 Within 5 days after initial exposure to BCG or β-glucan, innate cells begin to exhibit more robust responses that persisted for at least 1 year, long after the initial stimulus had been eradicated.21 Clearly, the trained immunity described in this body of experimental data is distinct from B-cell–mediated and T-cell–mediated antigen-specific adaptive immunity.
Table 1.
Evidence for Persistent, Beneficial Nonspecific Immunity.*
| Type of Evidence | Epigenetic Modifications | Reference |
|---|---|---|
| In vitro: incubating monocytes with BCG or β-glucan enhances microbicidal activity against candida, Escherichia coli, and Staphylococcus aureus | BCG increases H3K4me3 at TNF and IL6 promoter14; BCG increases H3K27ac in immune pathways related to Fc epsilon signaling, chemokine signaling, and Fc gamma–mediated phagocytosis15; training occurs through long noncoding RNA, such as UMLILO, to rearrange topologically associated domains of chromosomes16 | Kleinnijenhuis et al.,14 Arts et al.,15 Fanucchi et al.16 |
| Animal models: infection by intracellular bacteria (brucella, listeria, salmonella, toxoplasma, influenza, or BCG) induces nonspecific, cross-protective immunity against other intracellular pathogens | Historic studies from 1960–1970 did not evaluate epigenetic modifications | Dubos and Schaedler,2 Ratzan et al.,8 Spencer et al.,9 Clark et al.,10 Remington et al.,11 Murray et al.12 |
| Animal models: memory-like cells capable of protective recall develop in mice infected with MCMV | After infection, memory NK cells and CD8+ T cells share a common opening in chromatin accessibility in genes related to immune memory (Tcf7 and Bach2) and immune-effector function (granzymes and perforin)17 | Lau et al.17 |
| Animal models: infection with intracellular organisms induces nonspecific, cross-protective immunity against extracellular pathogens (E. coli, S. aureus, candida, and Streptococcus pneumoniae) | MTBVAC (attenuated, live Mycobacterium tuberculosis vaccine) increases H3K4me3 of TNF and IL6 promoters18; candida β-glucan increases H3K27ac and H3K4me3 of genes related to TNF, mTOR, and glycolysis (hexokinase, pyruvate kinase, TNF, IKKB, TSC1, and ribosomal protein S6)19; BCG increases H3K4me3 of TNF and IL6 and decreases H3K9me3 of genes related to glycolysis (GLUD, GLS, HK2, and MTOR)20 | Dubos and Schaedler,2 Mackaness,5 Mackaness,6 Musher et al.,7 Kleinnijenhuis et al.,14 Tarancon et al.,18 Cheng et al.,19 Arts et al.,20 Kleinnijenhuis et al.,21 Walk et al.,22 Kaufmann et al.23 |
| Humans: injection of BCG or β-glucan increases antibody responses to vaccines, increases clearance of yellow fever viremia, and increases symptoms after experimentally induced malaria infection | Elderly persons vaccinated with BCG have increased H3K27ac of the TNF and IL6 promoters24; β-glucan increases epigenome-wide chromatin accessibility (DNase accessibility assay), H3K27ac, and H3K4me3 in genes related to cytokines and chemokines25 | Arts et al.,15 Walk et al.,22 Giamarellos-Bourboulis et al.,24 Saeed et al.25 |
| Epidemiologic studies: BCG decreases neonatal sepsis, pneumonia, and all-cause mortality in developing countries | Humans vaccinated with BCG have increased H3K4me3 at TNF and IL6 promoters14 | Kleinnijenhuis et al.,14 Benn et al.26 |
| Humans: β-glucan reverses immune tolerance | β-glucan is able to restore beneficial H3K27ac marks in promoters and enhancers after LPS-induced tolerance27 | Novakovic et al.27 |
BCG denotes bacille Calmette–Guérin, H3K4me3 trimethylation of histone 3 at lysine 4, H3K27ac acetylation of histone 3 at lysine 27, LPS lipopolysaccharide, MCMV murine cytomegalovirus, mTOR mammalian target of rapamycin, NK natural killer, and TNF tumor necrosis factor.
Trained immunity occurs through epigenetic changes, a reconfiguration of how the genome is organized. Each human cell contains approximately 2 m of double-stranded DNA that is packed and tightly wrapped into the nucleus in nucleosomes consisting of groups of eight histones. The packing of the DNA is not haphazard; rather, it is organized into a three-dimensional structure such that the ability of the gene-expression machinery to access a specific gene promoter depends on the its location within the chromatin network. The chromatin can be reorganized by acetylating or methylating histones or methylating the 5′ cytosine residues of DNA (Fig. 1). Other layers of epigenetic regulation of transcription are methylation of DNA itself, which controls the binding of transcription factors to gene promoters, and the release of noncoding RNAs, which also regulate transcriptional processes.
Figure 1. Epigenetic Mechanisms of Antigen-Nonspecific Immune Memory (Trained Immunity).
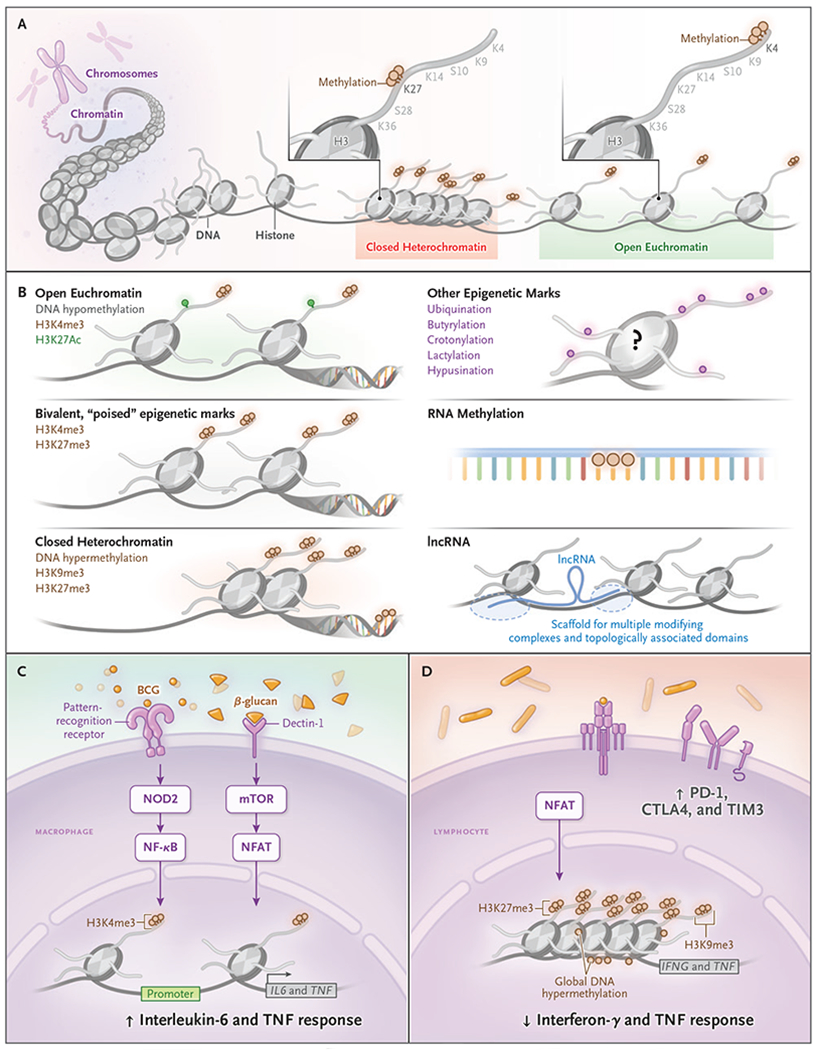
DNA is condensed into closed heterochromatin and open euchromatin by wrapping DNA around histone proteins (Panel A). Histone proteins have tails that can be modified, often with marks that make genes more accessible. Some common post-translational modifications include trimethylation of histone 3 at lysine 4 (H3K4me3), which promotes open chromatin. In contrast, trimethylation of histone 3 at lysine 27 (H3K27me3) promotes heterochromatin. The gene-expression implications of certain epigenetic marks are well established (Panel B). DNA methylation decreases transcription-factor binding and decreases gene expression. In contrast, H3K27 acetylation increases chromatin accessibility, which increases the capacity for gene expression. Genes can also be “poised” in a bivalent state simultaneously with both activating (i.e., H3K4me3) and repressive (i.e., H3K27me3) modifications. Epigenetic marks other than acetylation and methylation are not as well studied and are less understood. Long noncoding RNA (lncRNA) acts as a scaffolding to connect topologically associated domains of genes many kilobases apart. The role of histone ubiquination, crotonylation, butyrylation, or lactylation and of RNA acetylation and methylation in long-lasting immune function is still unknown. In macrophages (Panel C), bacille Calmette-Guerin (BCG) and β-glucan induce specific ligand-cell signaling pathways through nucleotide-binding oligomerization domain-containing protein 2 (NOD2) and mammalian target of rapamycin (mTOR), respectively, which results in increased H3K4me3 at the TNF and IL6 promoter. Thereby, when macrophages are exposed to an antigenically different, nonspecific stimulation, there is increased interleukin-6 and tumor necrosis factor (TNF) response. In both monocytes and lymphocytes (Panel D), sepsis, pneumonia, and chronic infections induce detrimental epigenetic scars, such as DNA hypermethylation and closed chromatin conformation that results in decreased effector function when the cells are challenged. CTLA-4 denotes cytotoxic T-lymphocyte–associated protein 4, H3K9me3 trimethylation of histone 3 at lysine 9, H3K27ac acetylation of histone 3 at lysine 27, NFAT nuclear factor of activated T cells, NF-κB nuclear factor κB, PD-1 programmed death 1, and TIM3 T-cell immunoglobulin mucin 3.
This reorganization, not of the DNA itself but “above” (epi) the DNA, is referred to as epigenetic modification. These modifications restructure accessibility to promoters and, therefore, accessibility to gene transcription (Fig. 1). The net result is a reorganization of the interactions within topologically associated domains that inhibits or stimulates expression of some genes; thus, the genes and domains become available for rapid coexpression on cellular activation.16 After stimulation, innate cells undergo epigenetic rewiring with increased chromatin accessibility that persists and allows the cell to respond more quickly and robustly to a second stimulus that is either similar or antigenically different. “Trained immunity” is the term given to these antigen-nonspecific, cross-protective, and long-lived innate immune responses.
When BCG or β-glucan stimulates innate cells, nucleotide-binding oligomerization domain-like receptors, C-type lectin receptors, and other pattern-recognition receptors induce a shift in cellular metabolism, which increases glycolysis, glutaminolysis, and cholesterol synthesis20 and triggers epigenetic changes.25,28 The best-described changes to date are increases in chromatin accessibility and histone 3 lysine trimethylation (H3K4me3) in interleukin-1β, interleukin-6, and tumor necrosis factor. The use of chemical inhibitors of histone methylation blocks BCG-induced epigenetic changes and trained immunity, which establishes a mechanistic relationship between epigenetic changes and trained immunity.14,15,19 In addition to BCG and β-glucan, exposure of mice to another live, attenuated vaccine derived from M. tuberculosis (called MTBVAC) induced similar epigenetic-mediated innate training and decreased mortality when vaccinated mice were challenged with S. pneumoniae.18 Epigenetic-mediated innate training was still detected 1 year after initial sensitization, even though terminally differentiated innate cells are thought to live only a few weeks. Preliminary evidence suggests that transcriptional, metabolic, and epigenetic changes in hematopoietic stem cells yield terminally differentiated monocytes with increased antimicrobial killing capacity,23,29–31 which establishes a potential mechanism for the long-lived nature of antigen-nonspecific training.
EPIDEMIOLOGY OF ANTIGEN-NONSPECIFIC IMMUNITY
These observations now explain epidemiologic studies that have shown a survival benefit from live, attenuated vaccines that is not specific to the targeted pathogen. In developing countries, children vaccinated with live vaccines — BCG, measles vaccine, or oral polio vaccine — have decreased all-cause mortality.26 (Some nonlive vaccines have been suggested to negate the benefit of live vaccines and increase the risk of unrelated, antigen-nonspecific infections, especially among girls.26) Results have been mixed, but with follow-up of 8 months to 5 years, measles or BCG vaccination decreased mortality from nontarget-related infections.26 A randomized, controlled trial showed that, in the year after BCG vaccination of South African adolescents, the risk of upper respiratory tract infections was reduced by 72%.32 A similarly designed trial involving elderly adults showed that BCG vaccination augmented antigen-nonspecific innate immunity that corresponded with beneficial epigenetic changes and that vaccination was associated with a 75% decrease in respiratory infections.24 The ability of BCG, the oral polio vaccine, and the measles vaccine to improve antigen-unrelated immunity and decrease nonspecific infections is being broadly evaluated as a means to decrease coronavirus disease 2019–related morbidity and mortality in multiple studies.33
DETRIMENTAL EPIGENETIC SOARS IN IMMUNE CELLS
Unfortunately, epigenetic changes can serve as a double-edged sword, because the same mechanisms that drive antigen-nonspecific, cross-protective beneficial immunity can also underlie detrimental, antigen-nonspecific immune suppression. In order to prevent exuberant, pathologic immunity, epigenetic machinery rearranges chromatin organization to limit immune-induced collateral damage to host tissue. This was illustrated in experiments in which blocking these epigenetic changes led to exuberant immune pathologic responses.34,35 Limiting exuberant immunity in the short term has advantages, but in the long term, the persistent immune suppression renders the host susceptible to a variety of microbial pathogens. Previous infections may increase the risk of future antigen-nonspecific infections. For example, in 1909 von Pirquet observed clinical flares of tuberculosis during recovery from measles and showed that this clinical finding correlated with suppressed responses to tuberculin on delayed-type hypersensitivity skin tests.36 Measles-induced immune suppression also increases the risk of secondary bacterial otitis media, tracheitis, and pneumonia.37 Similarly, hepatitis B and C have been associated with an increased risk of staphylococcal and pneumococcal infections.38–40
After recovery from the acute phase of sepsis, adaptive and innate immune function may be profoundly suppressed for a prolonged period, which can result in an increased risk of secondary bacterial infections.41,42 A propensity-matched cohort study showed that for at least 2 years after recovery from sepsis, the risk of death is increased 10 to 22%.43 Similarly, chronic helminth infections perturb host immunity and increase the risk of tuberculosis by a factor of 444 and the risk of human immunodeficiency virus (HIV) by a factor of 2 to 645,46 and cause a loss of vaccine immunogenicity.47 When Sandvall et al. reported that pneumococcal pneumonia is followed by a persistent increase in the risk of death that was proportional to the severity of the initial infection,48 their interpretation was that the initial infection was a marker of increased susceptibility41; long-lasting detrimental epigenetic changes that result from severe infection appear to offer a better explanation. Similarly, after successful treatment of tuberculosis, an increase in all-cause risk of infection-related death and non–infection-related death is noted.49,50
Mechanistic insight into these suppressed immune responses has been derived from a wide variety of experiments in which detrimental epigenetic scars in both innate and adaptive immune cells have been shown. The evidence for such changes is overwhelming (Table 2). In mice, infection due to chronic lymphocytic choriomeningitis virus (LCMV) blunts lymphocyte production of interleukin-2, tumor necrosis factor, and interferon-γ with a consequent decrease in T-cell proliferative capacity, delayed-type hypersensitivity, and antigen-nonspecific microbial killing capacity.51,54 Chronic LCMV infection induces DNA hypermethylation marks in the promoter region of IFNG that inhibits CD8+ T-cell immunity.52,55 Exposure of monocytes to lipopolysaccharide induces a closed chromatin conformation that epigenetically silences the gene encoding interleukin-6 (IL6), which results in decreased production of interleukin-6 (and other proinflammatory cytokines) on restimulation.25 In animal models of sepsis, 12 weeks after ligation of the colon, persistent epigenetic perturbations inhibit interleukin-12 production in dendritic cells, thereby diminishing type 1 helper T-cell immunity and skewing immunity toward a type 2 helper T-cell response (decreased interferon-γ and increased interleukin-4, -5, and -13).57 Experimental E. coli or influenza pneumonia in mice causes long-lasting epigenetic changes that tolerize macrophage function and decrease the capacity to phagocytose antigen-nonspecific, unrelated bacteria.41 One mechanism of the long-lived nature of epigenetic scarring is the development of detrimental epigenetic marks in progenitor, stem-cell memory populations.53,56,61 Specifically, chronic LCMV induces epigenetic-mediated exhausted memory T cells.53,56 Chronic M. avium infection in mice induces dysfunction in stem-cell differentiation,61 and macrophages derived after tolerization of hematopoietic stem cells have decreased production of tumor necrosis factor on secondary stimulation.62
Table 2.
Evidence for Persistent, Detrimental Effects on Nonspecific Immunity.*
| Type of Evidence | Epigenetic Modifications | Reference |
|---|---|---|
| In vitro: LPS causes decreased TNF and interleukin-6 production after exposure of monocytes to diverse challenges | LPS-tolerized monocytes decrease H3K4me3 and H3K27ac marks in proinflammatory genes and transcription factors (iRfs, STAT, and HIF-1α)27; LPS induces epigenome loss of H3K27ac marks25 | Saeed et al.,25 Novakovic et al.27 |
| Animal models: chronic LCMV infection inhibits TNF, interleukin-2, and interferon-γ production; decreases antigen-induced proliferation, and decreases nonspecific microbicidal activity | CD8+ T cells from chronic LCMV–infected mice have increased DNA methylation and decreased chromatin accessibility in Ifng, Myc, and genes related to T-cell receptor signaling and interleukin-7 and −2 signaling; increased chromatin accessibility in genes related to immune-checkpoint inhibition (PD-1, TIM3, and LAG3)51–53 | Seo et al.,51 Ghoneim et al.,52 Miller et al.,53 Ahmed et al.,54 Bengsch et al.,55 Yao et al.56 |
| Animal models: sepsis causes prolonged epigenetic inhibition of interleukin-12 and decreased microbial killing capacity | Six weeks after colon ligation–induced sepsis, interleukin-12 and both p35 and p40 promoters of dendritic cells have decreased H3K4me3 and increased H3K27me2; PRC remains bound and inhibits the interleukin-12 promoter57 | Roquilly et al.,41 Wen et al.57 |
| Animal models: infection with E. coli or influenza virus induces detrimental epigenetic marks and inhibits phagocytosis of unrelated organisms | Infection-cured mice have decreased H3K27ac of TLR5 and epigenomic overlap with human monocytes tolerized by LPS41 | Roquilly et al.41 |
| Humans: measles increases the risk of TB and other bacterial infections | No studies have yet evaluated whether epigenetic perturbations are part of the postmeasles ablation of adaptive immunity | Turk,36 Perry and Halsey37 |
| Humans: recovery from sepsis is followed by a persistent increased risk of death | No human studies have documented postsepsis induced immune tolerance and immune exhaustion, but no epigenetic studies involving humans post sepsis have yet been completed | van der Poll et al.,42 Prescott et al.43 |
| Humans: hepatitis B or C virus infection increases the risk of bacteremia or bacterial pneumonia | Patients with hepatitis have DNA hypermethylation and closed chromatin conformation that resembles chronic LCMV–induced immune exhaustion | Musher and McKenzie,38 Marrie et al.39 |
| Humans: helminths ablate vaccine immunogenicity | Six months after successful deworming, CD4+ T cells from children who had had schistosomiasis retained DNA hypermethylation of interleukin-12-interferon-y signaling and multiple transcription factors58 | Labeaud et al.,47 DiNardo et al.58 |
| Humans: chronic helminth infection increases the risk of TB or HIV infection | Memory CD4+ T cells in patients with HIV have DNA hypermethylation of interleukin-2; 2 years after successful aviremia, CD8+ T cells retain unmethylated DNA marks at PD-159 | Kroidl et al.,45 Downs et al.,46 Youngblood et al.59 |
| Humans: after successful therapy, patients with TB are at increased risk for recurrent TB and other infections and retain detrimental DNA hypermethylation marks | After TB therapy, NK cells, monocytes, and CD8+ and CD4+ T cells retain DNA hypermethylation of the interferon-γ, mTOR, TNF–NF-κB, and PI3K–AKT signaling pathways60 | Verver et al.,49 Romanowski et al.,50 DiNardo et al.60 |
HIF-1α denotes hypoxia-inducible factor 1α, IRF interferon regulatory factor, LAG3 lymphocyte-activation gene 3, LCMV lymphocytic choriomeningitis virus, NF-κB nuclear factor κB, PD-1 programmed death 1, PI3K phosphatidylinositol 3-kinase, PRC polycomb repressive complex, STAT signal transducer and activator of transcription, TB tuberculosis, TIM3 T-cell immunoglobulin mucin 3, and TLR5 toll-like receptor 5.
Just as with beneficial epigenetic marks, detrimental epigenetic scars are also persistently retained. After bacterial pneumonia or sepsis, epigenetic reprogramming inhibits macrophage phagocytic capacity for at least 6 months.41 Similarly, 6 months after children have been successfully treated for schistosomiasis, they retain detrimental DNA hypermethylation marks in CD4+ T cells that ablate immune responses to BCG.58 Despite successful antituberculosis therapy, detrimental epigenetic scars persist in both adaptive and innate immune cells for at least 6 months.60 After 2 years of successful antiretroviral therapy, HIV-infected persons retain detrimental epigenetic marks in the genes encoding interleukin-2 and programmed death 1 in CD8+ T cells.59
Detrimental epigenetic scars also occur as a consequence of physiologic aging. With aging, immune cells increase global DNA methylation and closed chromatin conformation. These age-related epigenetic changes occur preferentially in aspects of immunity that are required for the development of new immunologic memory63; DNA hypermethylation and chromatin closure at the interleukin-7 receptor and T-cell factor 1 (TCF1) appear to be responsible. Interleukin-7 is required for the survival of memory cells, and TCF1 is a transcription factor necessary for the development of effector memory immune formation. Therefore, detrimental epigenetic scars, whether from chronic infections or from aging, inhibit the development of new immune memory. These observations may explain why elderly persons lack strong immunologic responses, as shown by their increased susceptibility to infection and decreased responses to immunization. The implications of epigenetic scars for tumor immunology and susceptibility to autoimmune disease are apparent, but these problems are beyond the scope of the present article.
HARNESSING EPIGENETICS TO HEAL DETRIMENTAL SCARS
An understanding of epigenetic mechanisms shows why, in vaccine development, investigators cannot simply extrapolate results from experimental animals or healthy humans to results in countries where chronic infections are prevalent. An important question is whether it is possible to use knowledge of epigenetics to mend detrimental scars. Immune cells, and macrophages in particular, have plasticity in their fate and function,64 and initial evidence preliminarily suggests that detrimental epigenetic scars are mendable. In ex vivo studies, human monocytes that had decreased responsiveness to lipopolysaccharide were rescued by β-glucan, resulting in reversal of detrimental epigenetic chromatin marks and restoration of the production of tumor necrosis factor.27 In mice, decitabine, a hypomethylating agent, reversed chronic LCMV-induced detrimental DNA hypermethylating marks in CD8+ T cells and restored immune functionality.52 Additional evidence that epigenetic scars can be reversed comes from a trial involving elderly adults after hospital discharge, a population expected to have substantial epigenetic scars. This trial showed a benefit from BCG vaccination, as indicated by increased candida-induced cytokine production and decreased all-cause infections (especially respiratory infections), changes that were associated with epigenetic rewiring.24
IMPLICATIONS OF EPIGENETICS FOR VACCINE DEVELOPMENT
Ideally, vaccine development would simultaneously harness classic B-cell and T-cell adaptive immunity, as well as antigen-nonspecific innate immune training. With BCG bioengineered to lack urease production and include lysteriolysin O, a toxin that enables listeria to escape the phagosome, the VPM1002 strain of BCG increases T-cell immunogenicity while maintaining innate training.65 Similarly, MTBVAC enhances T-cell immunity while also inducing epigenetic-mediated trained immunity.18 Vaccination with M. tuberculosis–specific antigens (Rv0125 and Rv1196) together with the adjuvant AS01E induces both robust innate and adaptive immunity and provides substantial protection against progression of tuberculosis.66 Previous administration of a live vaccine augments humoral responses to influenza, hepatitis B, pneumococcal, and meningococcal vaccines.26 Clarifying and harnessing the mechanisms for these effects might help to enhance the efficacy of all existing vaccines. In contrast, the timing of vaccine administration needs to address the phenomenon by which administration of nonlive vaccines hinders immunity and increases the risk of nonrelated infections, particularly among young women.26
CONCLUSIONS AND IMPLICATIONS FOR THE FUTURE
Studies over the past decade have revealed a new epigenetic basis for acquired immunity in both innate and adaptive immune cells. These studies provide a mechanistic explanation for the antigen-nonspecific immune enhancement that has been shown in numerous studies during the past 60 years. They show that innate training works in concert with, and augments, specific T- and B-cell responses. The metaphor of a double-edged sword reflects the fact that those same mechanisms that underlie beneficial epigenetic innate training also underlie detrimental epigenetic scarring that occurs after severe infections or with aging. Identifying safe mechanisms to reverse detrimental epigenetic scars, for example by injection of β-glucan or BCG vaccination, could have broad implications for decreasing mortality after serious infections, protecting elderly persons against infection, improving treatment approaches for tuberculosis and other chronic infections, and implementing effective vaccines in developing countries.
Reversing detrimental epigenetic scarring and applying beneficial innate training are on the verge of clinical applicability but require continued basic and translational research. Cellular metabolic shifts trigger the epigenetic changes that underlie both beneficial innate training and detrimental epigenetic scarring. Identifying the metabolic processes that drive epigenetic changes will clarify methods to manipulate the system and identify potential means to overcome immune suppression.51,55,67 The differences in epigenetic effects according to sex suggest that we will need to clarify how hormones influence epigenetic changes before they can be broadly used.26,68 Detrimental scars that are induced by chronic LCMV in mice are well-characterized, and this understanding needs to be applied to chronic infections in humans. The documentation of detrimental and beneficial epigenetic marks is only beginning. Oncologists have established public database repositories (the Cancer Genome Atlas and Gene Expression Omnibus) that allow data to be merged and mined, thereby aiding in the identification of new therapeutic options. Because of similarities between chronic infection and cancer-induced immune suppression,51 tools identified to reverse cancer-induced detrimental epigenetic scars69 have the potential to reverse postinfectious epigenetic scars.
In 1966, Fazekas de St. Groth and Webster presciently stated: “Response to vaccine depends not only on the nature of the antigen itself but also on the immunological history of the recipient.”70 Up to one third of the world’s population has been affected by malaria, helminth infections, HIV infection, tuberculosis, or another chronic infection. The effect of the persistent epigenetic scars from these chronic infections increases susceptibility to other infections and decreases vaccine immunogenicity; how this can be reversed is unknown. To date, vaccine development has been focused nearly exclusively on the vaccine and the adjuvant, not on the host receiving the vaccine. Detrimental epigenetic scars that occur after chronic infection or with aging will probably inhibit robust vaccine immunogenicity. Finding a mechanism to reverse detrimental epigenetic immune scarring may be the key to better treatment of infections in elderly persons and for successful application of vaccines in countries where malaria, helminthic infection, tuberculosis, and other acute or chronic infections are so unfortunately widespread.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
REFERENCES
- 1.Gray G, Musher DM. The history of pneumococcal disease. Washington, DC: ASM Press, 2008. [Google Scholar]
- 2.Dubos RJ, Schaedler RW. Effects of cellular constituents of mycobacteria on the resistance of mice to heterologous infections I. Protective effects. J Exp Med 1957;106:703–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loebel RO, Shorr E, Richardson HB. The influence of adverse conditions upon the respiratory metabolism and growth of human tubercle bacilli. J Bacteriol 1933;26:167–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elberg SS, Schneider P, Fong J. Cross-immunity between Brucella melitensis and Mycobacterium tuberculosis; intracellular behavior of Brucella melitensis in monocytes from vaccinated animals. J Exp Med 1957;106:545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackaness GB. Cellular resistance to infection. J Exp Med 1962;116:381–406. [PubMed] [Google Scholar]
- 6.Mackaness GB. The immunological basis of acquired cellular resistance. J Exp Med 1964;120:105–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musher DM, Ratzan KR, Weinstein L. The effect of Listeria monocytogenes on resistance to pneumococcal infection. Proc Soc Exp Biol Med 1970;135:557–60. [PubMed] [Google Scholar]
- 8.Ratzan KR, Musher DM, Keusch GT, Weinstein L. Correlation of increased metabolic activity, resistance to infection, enhanced phagocytosis, and inhibition of bacterial growth by macrophages from Listeria- and BCG-infected mice. Infect Immun 1972;5:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spencer JC, Ganguly R, Waldman RH. Nonspecific protection of mice against influenza virus infection by local or systemic immunization with Bacille Calmette-Guerin. J Infect Dis 1977;136:171–5. [DOI] [PubMed] [Google Scholar]
- 10.Clark IA, Allison AC, Cox FE. Protection of mice against Babesia and Plasmodium with BCG. Nature 1976;259:309–11. [DOI] [PubMed] [Google Scholar]
- 11.Remington JS, Krahenbuhl JL, Mendenhall JW. A role for activated macrophages in resistance to infection with Toxoplasma. Infect Immun 1972;6:829–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray HW, Rubin BY, Rothermel CD. Killing of intracellular Leishmania donovani by lymphokine-stimulated human mononuclear phagocytes: evidence that interferon-gamma is the activating lymphokine. J Clin Invest 1983;72:1506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe 2011;9:355–61. [DOI] [PubMed] [Google Scholar]
- 14.Kleinnijenhuis J, Quintin J, Preijers F, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A 2012;109:17537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arts RJW, Moorlag SJCFM, Novakovic B, et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe 2018;23(1):89–100.e5. [DOI] [PubMed] [Google Scholar]
- 16.Fanucchi S, Fok ET, Dalla E, et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat Genet 2019;51:138–50. [DOI] [PubMed] [Google Scholar]
- 17.Lau CM, Adams NM, Geary CD, et al. Epigenetic control of innate and adaptive immune memory. Nat Immunol 2018;19: 963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarancón R, Domínguez-Andrés J, Uranga S, et al. New live attenuated tuberculosis vaccine MTBVAC induces trained immunity and confers protection against experimental lethal pneumonia. PLoS Pathog 2020;16(4):e1008404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014;345:1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arts RJW, Carvalho A, La Rocca C, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep 2016;17:2562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleinnijenhuis J, Quintin J, Preijers F, et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun 2014;6:152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walk J, de Bree LCJ, Graumans W, et al. Outcomes of controlled human malaria infection after BCG vaccination. Nat Commun 2019;10:874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaufmann E, Sanz J, Dunn JL, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 2018;172(1-2):176–190.e19. [DOI] [PubMed] [Google Scholar]
- 24.Giamarellos-Bourboulis EJ, Tsilika M, Moorlag S, et al. Activate: randomized clinical trial of BCG vaccination against infection in the elderly. Cell 2020;183(2):315–323.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saeed S, Quintin J, Kerstens HHD, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014;345:1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benn CS, Fisker AB, Rieckmann A, Sørup S, Aaby P. Vaccinology: time to change the paradigm? Lancet Infect Dis 2020;20(10):e274–e283. [DOI] [PubMed] [Google Scholar]
- 27.Novakovic B, Habibi E, Wang S-Y, et al. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 2016;167(5):1354–1368.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verma D, Parasa VR, Raffetseder J, et al. Anti-mycobacterial activity correlates with altered DNA methylation pattern in immune cells from BCG-vaccinated subjects. Sci Rep 2017;7:12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitroulis I, Ruppova K, Wang B, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 2018;172(1–2):147–161.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Laval B, Maurizio J, Kandalla PK, et al. C/EBPβ-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell 2020;26(5):657–674.e8. [DOI] [PubMed] [Google Scholar]
- 31.Cirovic B, de Bree LCJ, Groh L, et al. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe 2020; 28(2):322–334.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemes E, Geldenhuys H, Rozot V, et al. Prevention of M. tuberculosis infection with H4:IC31 vaccine or BCG revaccination. N Engl J Med 2018;379:138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mantovani A, Netea MG. Trained innate immunity, epigenetics, and Covid-19. N Engl J Med 2020;383:1078–80. [DOI] [PubMed] [Google Scholar]
- 34.Hu L, Yu Y, Huang H, et al. Epigenetic regulation of interleukin 6 by histone acetylation in macrophages and its role in paraquat-induced pulmonary fibrosis. Front Immunol 2017;7:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, Zhao K, Shen Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015;525:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turk JL. Von Pirquet, allergy and infectious diseases: a review. J R Soc Med 1987;80:31–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perry RT, Halsey NA. The clinical significance of measles: a review. J Infect Dis 2004;189:Suppl 1:S4–S16. [DOI] [PubMed] [Google Scholar]
- 38.Musher DM, McKenzie SO. Infections due to Staphylococcus aureus. Medicine (Baltimore) 1977;56:383–409. [DOI] [PubMed] [Google Scholar]
- 39.Marrie TJ, Tyrrell GJ, Majumdar SR, Eurich DT. Concurrent infection with hepatitis C virus and Streptococcus pneumoniae. Emerg Infect Dis 2017;23:1118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaka AS, Filice GA, Kuskowski M, Musher DM. Does active hepatitis C virus infection increase the risk for infection due to Staphylococcus aureus? Eur J Clin Microbiol Infect Dis 2017;36:1217–23. [DOI] [PubMed] [Google Scholar]
- 41.Roquilly A, Jacqueline C, Davieau M, et al. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol 2020;21:636–48. [DOI] [PubMed] [Google Scholar]
- 42.van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol 2017;17:407–20. [DOI] [PubMed] [Google Scholar]
- 43.Prescott HC, Osterholzer JJ, Langa KM, Angus DC, Iwashyna TJ. Late mortality after sepsis: propensity matched cohort study. BMJ 2016;353:i2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elias D, Mengistu G, Akuffo H, Britton S. Are intestinal helminths risk factors for developing active tuberculosis? Trop Med Int Health 2006;11:551–8. [DOI] [PubMed] [Google Scholar]
- 45.Kroidl I, Saathoff E, Maganga L, et al. Effect of Wuchereria bancrofti infection on HIV incidence in southwest Tanzania: a prospective cohort study. Lancet 2016; 388:1912–20. [DOI] [PubMed] [Google Scholar]
- 46.Downs JA, van Dam GJ, Changalucha JM, et al. Association of schistosomiasis and HIV infection in Tanzania. Am J Trop Med Hyg 2012;87:868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Labeaud AD, Malhotra I, King MJ, King CL, King CH. Do antenatal parasite infections devalue childhood vaccination? PLoS Negl Trop Dis 2009;3(5):e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sandvall B, Rueda AM, Musher DM. Long-term survival following pneumococcal pneumonia. Clin Infect Dis 2013; 56:1145–6. [DOI] [PubMed] [Google Scholar]
- 49.Verver S, Warren RM, Beyers N, et al. Rate of reinfection tuberculosis after successful treatment is higher than rate of new tuberculosis. Am J Respir Crit Care Med 2005;171:1430–5. [DOI] [PubMed] [Google Scholar]
- 50.Romanowski K, Baumann B, Basham CA, Ahmad Khan F, Fox GJ, Johnston JC. Long-term all-cause mortality in people treated for tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis 2019;19:1129–37. [DOI] [PubMed] [Google Scholar]
- 51.Seo H, Chen J, González-Avalos E, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc Natl Acad Sci U S A 2019;116:12410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghoneim HE, Fan Y, Moustaki A, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 2017;170(1):142–157.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller BC, Sen DR, Al Abosy R, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 2019;20:326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice: role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med 1984;160:521–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bengsch B, Johnson AL, Kurachi M, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity 2016;45:358–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao C, Sun H-W, Lacey NE, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat Immunol 2019;20:890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wen H, Dou Y, Hogaboam CM, Kunkel SL. Epigenetic regulation of dendritic cell-derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood 2008;111:1797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DiNardo AR, Nishiguchi T, Mace EM, et al. Schistosomiasis induces persistent DNA methylation and tuberculosis-specific immune changes. J Immunol 2018;201:124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Youngblood B, Noto A, Porichis F, et al. Cutting edge: prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol 2013;191:540–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DiNardo AR, Rajapakshe K, Nishiguchi T, et al. DNA hypermethylation during tuberculosis dampens host immune responsiveness. J Clin Invest 2020;130:3113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matatall KA, Jeong M, Chen S, et al. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep 2016;17:2584–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yáñez A, Hassanzadeh-Kiabi N, Ng MY, et al. Detection of a TLR2 agonist by hematopoietic stem and progenitor cells impacts the function of the macrophages they produce. Eur J Immunol 2013;43: 2114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Márquez EJ, Chung C-H, Marches R, et al. Sexual-dimorphism in human immune system aging. Nat Commun 2020;11:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marakalala MJ, Martinez FO, Plüddemann A, Gordon S. Macrophage heterogeneity in the immunopathogenesis of tuberculosis. Front Microbiol 2018;9:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaufmann SHE. Vaccination against tuberculosis: revamping BCG by molecular genetics guided by immunology. Front Immunol 2020;11:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tait DR, Hatherill M, Van Der Meeren O, et al. Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. N Engl J Med 2019;381:2429–39. [DOI] [PubMed] [Google Scholar]
- 67.Ryan DG, O’Neill LAJ. Krebs cycle reborn in macrophage immunometabolism. Annu Rev Immunol 2020;38:289–313. [DOI] [PubMed] [Google Scholar]
- 68.Koeken VA, de Bree LCJ, Mourits VP, et al. BCG vaccination in humans inhibits systemic inflammation in a sex-dependent manner. J Clin Invest 2020;130:5591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bates SE. Epigenetic therapies for cancer. N Engl J Med 2020;383:650–63. [DOI] [PubMed] [Google Scholar]
- 70.Fazekas de St Groth S, Webster RG. Disquisitions on original antigenic sin. II. Proof in lower creatures. J Exp Med 1966; 124:347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]