

Abstract
Background
Corylus heterophylla Fisch. is a species of the Betulaceae family native to China. As an economically and ecologically important nut tree, C. heterophylla can survive in extremely low temperatures (–30 to –40 °C). To deepen our knowledge of the Betulaceae species and facilitate the use of C. heterophylla for breeding and its genetic improvement, we have sequenced the whole genome of C. heterophylla.
Findings
Based on >64.99 Gb (∼175.30×) of Nanopore long reads, we assembled a 370.75-Mb C. heterophylla genome with contig N50 and scaffold N50 sizes of 2.07 and 31.33 Mb, respectively, accounting for 99.23% of the estimated genome size (373.61 Mb). Furthermore, 361.90 Mb contigs were anchored to 11 chromosomes using Hi-C link data, representing 97.61% of the assembled genome sequences. Transcriptomes representing 4 different tissues were sequenced to assist protein-coding gene prediction. A total of 27,591 protein-coding genes were identified, of which 92.02% (25,389) were functionally annotated. The phylogenetic analysis showed that C. heterophylla is close to Ostrya japonica, and they diverged from their common ancestor ∼52.79 million years ago.
Conclusions
We generated a high-quality chromosome-level genome of C. heterophylla. This genome resource will promote research on the molecular mechanisms of how the hazelnut responds to environmental stresses and serves as an important resource for genome-assisted improvement in cold and drought resistance of the Corylus genus.
Keywords: Corylus heterophylla Fisch, Genome sequences, Nanopore, Hi-C
Background
The Corylus genus, a member of the birch family Betulaceae that includes economically and ecologically important nut tree species, is widely distributed throughout temperate regions of the Northern Hemisphere [1]. As a valuable nut crop, hazelnut provides the predominant flavor in a variety of cakes, candies, chocolate spreads, and butters. There is a high content of unsaturated fatty acids and several essential vitamins in hazelnut oil.
The number of Corylus species recognized by taxonomists ranges from 7 to 25, depending on different morphological and molecular classifications [2, 3]. Among these, the European hazelnut, Corylus avellana L., is the most widely commercially cultivated species, with >400 cultivars having been described [4]. Commercial cultivation of C. avellana is limited to regions with climates moderated by large bodies of water that have cool summers and mild, humid winters, such as the slopes on the Black Sea of Turkey or the Willamette Valley of Oregon [5, 6]. Inadequate cold hardiness is a major factor limiting the expansion of commercial production into northern and inland areas. When C. avellana was introduced into China, twigs withered and died almost every winter owing to the cold, windy, and dry climate in northern China. In southern China, however, European hazelnut trees seemed to grow well but bore few nuts, and abortive kernels were observed frequently [7].
Eight species and 2 botanical varieties of Corylus are reported to be native to China [5]. The Asian hazel Corylus heterophylla (NCBI:txid80754) is one of the most important economic Corylus species. Among the 1.67 million ha of wild Corylus in China, C. heterophylla occupies 90% of the geographic area [8]. Wild C. heterophylla is mainly distributed in the mountains from northern to northeastern China. The geographical distribution range is 36.78–51.73 (°N) and 100.57–132.20 (°E), where the main climate type is temperate. Compared with C. avellana, C. heterophylla can be adapted to regions with low temperatures (–30 to –40 °C) and drought conditions. Therefore, the cold and drought resistance characteristics of C. heterophylla can be used as parent materials for cross-breeding with other hazel species.
In the present study, to better understand the molecular mechanism of how hazelnuts respond to environmental stress, we assembled a high-quality genome of C. heterophylla using a combination of the Oxford Nanopore high-throughput sequencing technology and the high-throughput chromosome conformation capture (Hi-C) technique. Long reads were de novo assembled into 1,328 polished contigs with a total size of 370.75 Mb and contig N50 and scaffold N50 values of 2.07 and 31.33 Mb, respectively, which is in line with genome sizes estimated using flow cytometry and k-mer analysis. A total of 361.90 Mb contigs were anchored into 11 chromosomes, representing 97.61% of the assembled genome. Our results provide a high-quality, chromosome-level genome assembly of C. heterophylla, which will support breeding programs leading to genetic improvement of hazelnuts. Furthermore, it will facilitate understanding of the special position of Corylus and Betulaceae in plant evolution.
Data Description
Sample collection
Fresh and healthy leaves were collected from a single wild C. heterophylla tree in Yanqing, Beijing, China (40.54 N; 116.06 E; Fig. 1). The fresh leaf tissue was flash-frozen in liquid nitrogen for 30 min and then stored at –80 °C. DNA was extracted from leaf tissues following a previously published protocol [9]. Different tissues, including root, stem, staminate inflorescence, and leaf, were sampled and flash-frozen in liquid nitrogen for total RNA sequencing. Total RNA was extracted using the modified CTAB method [10].
Figure 1:

Morphological characteristics of the Asian hazelnut variety, C. heterophylla. Mature plants in (A) and (B), female inflorescence (C), staminate inflorescence (D), fruit with husk (E), and nuts (F) are shown.
Library preparation and whole-genome sequencing
Genomic DNA for library construction was isolated from leaf tissues using the DNeasy Plant Mini Kit (Qiagen, Beijing, China) according to the manufacturer's instructions. DNA concentrations and quality were measured using a NanoDrop 2000 (Thermo Fisher, Waltham, MA, USA) and Qubit Fluorometer (Thermo Fisher, Waltham, MA, USA), respectively. The genomic DNA was sheared to ∼500-bp fragments using an S2 Focused-Ultrasonicator (Covaris Inc., Woburn, MA, USA). Paired-end (PE) libraries were prepared using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, CA, USA) according to the Illumina standard protocol. After quality control by an Agilent 2100 Bioanalyzer and qPCR, all PCR-free libraries were sequenced on an Illumina HiSeq X Ten system (Illumina, San Diego, CA, USA) (Illumina HiSeq X Ten, RRID:SCR_016385) with a 350-bp PE sequencing strategy according to the manufacturer's instructions. A total of 38.02 Gb (∼102.55-fold coverage) clean reads were generated for the genome survey and Nanopore genome polishing (Supplementary Table S1a).
Estimation of genome size and heterozygosity analysis
Before genome assembly, we estimated the C. heterophylla genome's size using Jellyfish (Jellyfish, RRID:SCR_005491) [11] with an optimal k-mer size. A total of 38.02 Gb short reads (∼102.55×) were processed by Jellyfish to assess their k-mer distribution (k-mer value = 19). Theoretically, the k-mer frequency follows a Poisson distribution. We selected k = 19 for the genome size estimation in this study. Genome sizes were calculated from the following equation: Genome size = 19-mer number/19-mer depth, where 19-mer number is the total counts of each unique 19-mer and 19-mer depth is the highest frequency that occurred (Supplementary Fig. S1). The estimated genome size of C. heterophylla is 373.61 Mb.
Nanopore, RNA, and Hi-C sequencing
Genomic DNA was extracted and sequenced following the instructions of the Ligation Sequencing Kit (Oxford Nanopore Technologies, Oxford, UK). DNA quality was assessed by agarose gel electrophoresis and NanoDrop 2000c spectrophotometry, followed by Thermo Fisher Scientific Qubit fluorometry. After quality control, genomic DNA was size-selected using a Blue Pippin BLF7510 cassette (Sage Science, Beverly, MA, USA). Libraries (fragments >20 kb) were prepared using the standard Ligation Sequencing kit (SQK-LSK109; Oxford Nanopore Technologies, Oxford, UK) and sequenced on the GridION X5 platform (Oxford Nanopore Technologies, Oxford, UK) with FLOMIN106 (R9.4) flow cells. Raw ONT reads (fastq) were extracted from base-called FAST5 files using poretools [12]. Then, the short reads (<5 kb) and reads having low-quality bases and adapter sequences were removed. A total of 64.99 Gb (∼175.30-fold coverage) Nanopore long reads with an N50 length of 27.17 kb were produced for genome assembly (Supplementary Fig. S2, Supplementary Tables S1b and S1c).
Different tissues, including leaf, stem, root, and staminate inflorescence, were harvested and flash-frozen in liquid nitrogen for total RNA sequencing. Each sample was subjected to poly(A) purification using oligo-dT beads (Thermo Fisher, Waltham, MA, USA) followed by ribosomal RNA (rRNA) removal using the Ribo-Zero rRNA Removal Kit (Illumina, San Diego, CA, USA). The RNA quality was measured by 2100 RNA Nano 6000 Assay Kit (Agilent Technologies, Santa Clara, CA, USA) and pooling together. The resulting RNA sample was used for complementary DNA library construction using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA). The quantified libraries were then prepared for sequencing on the Illumina HiSeq X Ten system, producing 9.66 Gb PE reads (Supplementary Table S1d).
Hi-C experiments were performed as described with some modifications [13, 14]. Briefly, 2 g of freshly harvested leaves were cut into 2- to 3-mm pieces and infiltrated in 2% formaldehyde before cross-linking was stopped by adding glycine. The tissue was ground to powder and suspended in nuclei isolation buffer to obtain a nuclei suspension. The procedure for the Hi-C experiment, including chromatin digestion, labeling of DNA ends, DNA ligation, purification, and fragmentation, was performed as described previously [15]. The cross-linked DNA was digested with HindIII as previously described and marked by incubating with Klenow enzyme and biotin-14-dCTP overnight at 37°C [15]. The 5′ overhang of the fragments was repaired and labeled using biotinylated nucleotides, followed by ligation with T4 DNA polymerase. After reversal of cross-linking, ligated DNA was purified and sheared to 300–700 bp fragments using an S2 Focused-Ultrasonicator (Covaris Inc., MA, USA). The linked DNA fragments were enriched with streptavidin beads and prepared for Illumina HiSeq X Ten sequencing, producing 231.31 Mb (totaling ∼69.11 Gb) Hi-C link data (Supplementary Table S1e).
De novo genome assembly and pseudo-chromosome construction
After the self-error correction using the error correction model in Canu (Canu, RRID:SCR_015880) v1.5 [16], the Nanopore long reads were assembled into contigs using WTDBG2 (WTDBG, RRID:SCR_017225) v1.0 [17]. Two rounds of consensus correction were performed using Racon (Racon, RRID:SCR_017642) v1.32 [18] with corrected Nanopore long reads, and the resulting assembly was further polished using Pilon (Pilon, RRID:SCR_014731) [19] with 38.02 Gb Illumina short reads (Supplementary Table S1a). The assembled length of 1,291 contigs of C. heterophylla is 370.71 Mb, accounting for 99.22% of the estimated genome size (373.61 Mb). The contigs N50 and N90 were 2.11 Mb and 138.6 kb, respectively.
The pseudo-chromosomes were constructed using Hi-C link data. The clean Hi-C reads were mapped to the consensus contigs using BWA [20] (BWA, RRID:SCR_010910) v0.7.17, and only uniquely mapped read pairs were considered as high-quality read pairs in Hi-C analysis. The reads were removed if the mapped positions in the reference genome were farther than 500 bp from the nearest restriction enzyme site. The quality assessment and normalization were performed using HiC-Pro (HiC-Pro, RRID:SCR_017643) [21]. There were 109,306,012 uniquely mapped PE reads, of which 58.33% (63,755,940) uniquely mapped reads were considered valid interaction pairs for chromosome construction (Supplementary Table S2). The contigs were then clustered, ordered, and oriented into 11 pseudo-chromosomes using LACHESIS (LACHESIS, RRID:SCR_017644) [21]. Finally, we obtained a high-quality chromosome-level reference genome with a total size of 370.75 Mb. The contig N50 and scaffold N50 values were 2.07 and 31.33 Mb, respectively (Table 1). A total of 361.90 Mb contigs were anchored into 11 chromosomes, representing 97.61% of the assembled genome (Table 2).
Table 1:
Statistics of assembly results of C. heterophylla genome
| Feature | C. heterophylla |
|---|---|
| Genome size (bp) | 370,750,808 |
| Contig | |
| No. | 1,328 |
| Maximum length (bp) | 9,680,353 |
| N50 (bp) | 2,068,510 |
| L50 | 48 |
| N90 (bp) | 125,301 |
| Scaffold | |
| No. | 951 |
| Maximum length (bp) | 46,514,939 |
| N50 (bp) | 31,328,411 |
| L50 | 5 |
| N90 (bp) | 21,561,575 |
| GC content (%) | 35.84 |
| Genes | |
| No. | 27,591 |
| Length (bp) | 123,431,253 |
| Mean length (bp) | 4,473.61 |
| Exons | |
| No. | 138,886 |
| Length (bp) | 33,679,425 |
| Introns | |
| No. | 138,885 |
| Length (bp) | 89,751,828 |
| Pseudogenes | |
| No. | 2,988 |
| Length (bp) | 7,166,319 |
Note: only sequences of length >1 kb are considered.
Table 2:
Summary of 11 pseudo-chromosomes for C. heterophylla
| Chromosome | Clustered sequences | Ordered sequences | ||
|---|---|---|---|---|
| No. | Length (bp) | No. | Length (bp) | |
| LG01 | 114 | 49,577,893 | 56 | 46,509,439 |
| LG02 | 113 | 48,019,691 | 49 | 44,425,769 |
| LG03 | 67 | 37,395,073 | 33 | 36,016,943 |
| LG04 | 95 | 38,562,170 | 53 | 36,392,613 |
| LG05 | 85 | 34,656,877 | 37 | 31,324,811 |
| LG06 | 76 | 31,263,564 | 31 | 28,814,739 |
| LG07 | 103 | 29,494,057 | 36 | 25,003,895 |
| LG08 | 45 | 23,716,498 | 23 | 22,749,571 |
| LG09 | 41 | 23,427,462 | 17 | 22,292,654 |
| LG10 | 41 | 23,093,417 | 25 | 22,249,747 |
| LG11 | 53 | 22,694,573 | 28 | 21,558,875 |
| Total (%) | 833 (62.73) | 361,901,275 (97.61) | 388 (46.58) | 337,339,056 (93.21) |
Genome quality assessment
Genome completeness was assessed using the plants dataset of the BUSCO (BUSCO, RRID:SCR_015008) database v1.22 [22], with e-value < 1e−5. The BUSCO database detected 93.47% and 1.18% of complete and partial gene models, respectively, in the C. heterophylla assembly results (Table 3). The core eukaryotic gene-mapping approach (CEGMA, RRID:SCR_015055) [23] provides a method to rapidly assess genome completeness because it comprises a set of highly conserved, single-copy genes, present in all eukaryotes, containing 458 core eukaryotic genes (CEGs). We identified CEGs using the CEGMA (CEGMA, RRID:SCR_015055) v2.3 pipeline [23] and found that 430 (93.89%) CEGs could be found in the assembly results (Supplementary Table S3a). The PE short libraries, including 103,392,992 paired reads, were remapped to the assembly genome with BWA-MEM (BWA, RRID:SCR_010910) [24] to assess the completeness of the assembly results. More than 98.47% of these reads could be accurately mapped into genome sequences (Supplementary Table S3b). Additionally, the heat map of the Hi-C interaction frequency was selected to visually assess the assembled accuracy of the C. heterophylla genome. The interaction heat map was displayed at 100-kb resolution. LG01–LG11 represent the 11 chromosomes of the C. heterophylla genome ordered by chromosome length. The horizontal and vertical coordinates represent the order of each “bin” on the corresponding chromosome. The signal intensities clearly divide the “bins” into 11 distinct groups (LG01–LG11), demonstrating the high quality of the chromosome assignment (Fig. 2). These observations suggest the high quality and completeness of this chromosome-level reference genome for C. heterophylla.
Table 3:
Genome completeness assessment by BUSCO
| BUSCO categories | No. (%) |
|---|---|
| Complete | 1,346 (93.47) |
| Complete and single-copy | 1,296 (90.00) |
| Complete and duplicated | 50 (3.47) |
| Fragmented | 17 (1.18) |
| Missing | 77 (5.35) |
| Total groups searched | 1,440 (100) |
Figure 2:
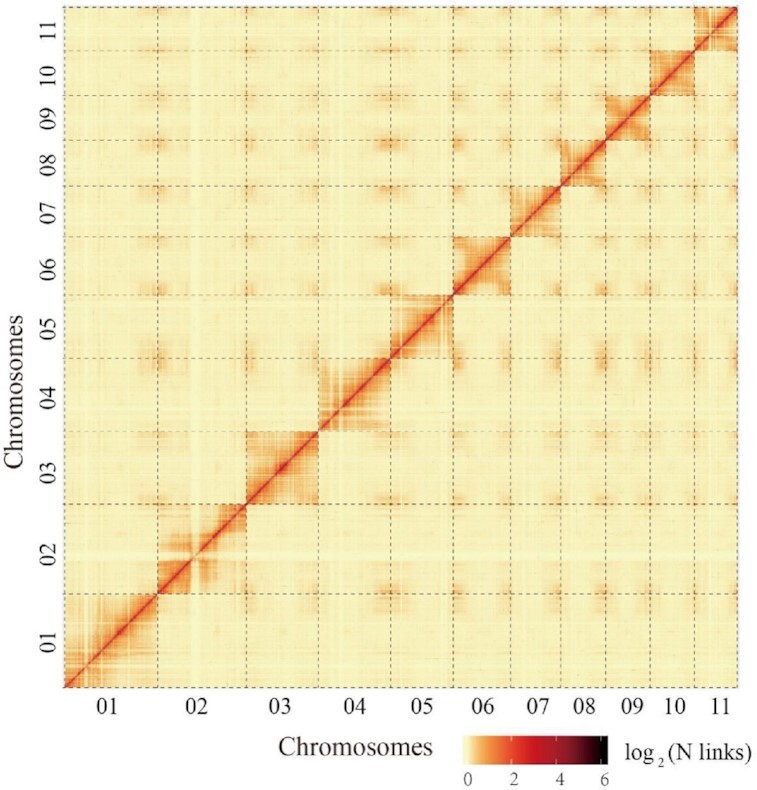
Interaction frequency distribution of Hi-C links among 11 chromosomes. Genome-wide Hi-C map of C. heterophylla. We scanned the genome by 500-kb nonoverlapping window as a bin and calculated valid interaction links of Hi-C data between any pair of bins. The log2 of link number was transformed. The color key of heat map ranging from light yellow to dark red represents the frequency of Hi-C interaction links from low to high (0–6).
Repetitive elements and protein-coding gene annotation
Repetitive elements in the C. heterophylla genome were identified using a combined strategy of de novo and homology-based approaches at the DNA and protein levels. Tandem repeats were annotated using TRF. A repeat library was constructed using MITE-Hunter (MITE-Hunter, RRID:SCR_020946) [25], LTR-FINDER (LTR_Finder, RRID:SCR_015247) v1.05 [26], RepeatScout (RepeatScout, RRID:SCR_014653) v1.0.5 [27], and PILER (PILER, RRID:SCR_017333) [28] for de novo repeat content annotation. The de novo repeat library was classified through PASTEClassifier (PASTEClassifier, RRID:SCR_017645) v1.0 package [29] with default parameters and then integrated with Repbase (RepeatMasker, RRID:SCR_012954) v19.06 [30] to build a new repeat library. Finally, RepeatMasker (RepeatMasker, RRID:SCR_012954) v4.0.6 [31] with parameters of “-nolow -no_is -norna -engine wublast” was selected to identify and classify the genomic repetitive elements of C. heterophylla. In total, 210.26 Mb of repetitive sequences were identified, accounting for 56.71% of C. heterophylla genome sequences (Table 4). The top 3 classes of repetitive elements were Class I/LARD, Class I/LTR/Gypsy, and Class I/LTR/Copia, occupying 20.51%, 11.14%, and 10.44% of assembled genome sequences, respectively (Table 4).
Table 4:
Repetitive elements in the C. heterophylla genome
| Class | No. | Length (bp) | Percent (%) |
|---|---|---|---|
| Class I | 584,311 | 169,738,018 | 45.78 |
| Class I/DIRS | 18,638 | 7,059,337 | 1.9 |
| Class I/LARD | 303,288 | 76,033,830 | 20.51 |
| Class I/LINE | 60,182 | 18,890,786 | 5.1 |
| Class I/LTR/Copia | 101,158 | 38,719,023 | 10.44 |
| Class I/LTR/Gypsy | 83,300 | 41,302,761 | 11.14 |
| Class I/LTR/Unknown | 1,953 | 1,080,718 | 0.29 |
| Class I/PLE | 5,600 | 4,125,513 | 1.11 |
| Class I/SINE | 5,344 | 1,058,985 | 0.29 |
| Class I/TRIM | 3,828 | 1,023,113 | 0.28 |
| Class I/Unknown | 1,020 | 244,561 | 0.07 |
| Class II | 77,407 | 24,382,510 | 6.58 |
| Class II/Crypton | 455 | 109,226 | 0.03 |
| Class II/Helitron | 27,254 | 8,348,317 | 2.25 |
| Class II/MITE | 1,112 | 194,088 | 0.05 |
| Class II/Maverick | 754 | 165,986 | 0.04 |
| Class II/TIR | 44,403 | 15,342,483 | 4.14 |
| Class II/Unknown | 3,429 | 459,116 | 0.12 |
| Potential host gene | 46,369 | 9,994,181 | 2.7 |
| SSR | 1,135 | 265,113 | 0.07 |
| Unknown | 116,728 | 26,584,597 | 7.17 |
| Total | 825,950 | 210,255,221 | 56.71 |
DIRS: dictyostelium intermediate repeat sequence; LARD: large retrotransposon derivative; LINE: long interspersed nuclear element; LTR: long terminal repeat; MITE: miniature inverted-repeat transposable element; PLE: Penelope-like element; SINE: short interspersed nuclear element; SSR: simple sequence repeat; TIR: terminal inverted repeat; TRIM: terminal-repeat retrotransposons in miniature.
Gene annotation was performed using a combination of ab initio prediction, homology-based gene prediction, and transcript evidence from RNA-seq data. The de novo approach was implemented using Augustus (Augustus, RRID:SCR_008417) v3.2.3 [32], GeneID (Entrez Gene, RRID:SCR_002473) v1.4.4 [33], GlimmerHMM (GlimmerHMM, RRID:SCR_002654) v3.52 [34], GenScan (GENSCAN, RRID:SCR_012902) [35], and SNAP (SNAP, RRID:SCR_007936) [36]. For homology-based prediction, TBLASTN (TBLASTN, RRID:SCR_011822) v2.2.31 [37] was used to align predicted protein sequences of Arabidopsis thaliana, Betula pendula, Juglans regia, and Ostrya chinensis to the C. heterophylla genome with an e-value threshold of 1e−5. Then, GeMoMa (GeMoMa, RRID:SCR_017646) v1.3.1 [38] was used for homology-based gene prediction. The transcriptome data from pooled tissues of leaf, stem, root, and staminate inflorescence from C. heterophylla were assembled into unigenes using HISAT (HISAT, RRID:SCR_015530) v2.0.4 [39] and StringTie (StringTie, RRID:SCR_016323) v1.2.3 [40]. Then unigenes were used to predict gene structures using TransDecoder (TransDecoder, RRID:SCR_017647) v2.0 [41], GeneMarkS-T (GeneMarkS-T, RRID:SCR_017648) v5.1 [42], and PASA (PASA, RRID:SCR_014656) v2.0.2 [43]. Finally, the gene models obtained from the above 3 approaches were integrated into a consensus gene set using EVidenceModeler (EVidenceModeler, RRID:SCR_014659) v1.1.0 [44] with default parameters. PASA (PASA, RRID:SCR_014656) v2.0.2 [43] was then used to annotate the gene structures, including untranslated regions and alternative-splice sites (Supplementary Fig. S3, Supplementary Table S4a). A total of 27,591 non-redundant protein-coding genes were predicted for the C. heterophylla genome (Table 1). Gene models were annotated by homologous searching against several databases using BLASTP (BLASTP, RRID:SCR_001010) from the BLAST+ package [37] (e-value = 1e−5), including NR [45], KOG [46], TrEMBL (Universal Protein Resource, RRID:SCR_002380) [47], and KEGG (KEGG, RRID:SCR_012773) [48] databases. InterProScan (InterProScan, RRID:SCR_005829) v4.3 [49] was used to annotate the protein motifs and domains. The Blast2GO (Blast2GO, RRID:SCR_005828) [50, 51] pipeline was used to obtain GO terms annotation from the NCBI NR database. In total, 25,389 protein-coding genes (92.02%) were successfully assigned into corresponding functions (Supplementary Table S4b).
Genome-wide pseudogene identification was carried out for C. heterophylla. Only candidate pseudogenes containing frameshifts and/or premature stop codons in their coding regions were considered as reliable pseudogenes. C. heterophylla proteins were aligned to the reference genome using GenBlastA (GenBlastA, RRID:SCR_020951) v1.0.4 [52] to detect candidate homolog regions. Then, the candidate pseudogenes were identified using GeneWise (GeneWise, RRID:SCR_015054) v2.4.1 [53]. Finally, 2,988 pseudogenes were identified in C. heterophylla genome sequences (Table 1).
Different types of non-coding RNA in the C. heterophylla genome were identified and classified as family and subfamily. The tRNAscan-SE (tRNAscan-SE, RRID:SCR_010835) v1.23 [54] was applied to detect transfer RNAs (tRNAs). MicroRNAs (miRNAs) were identified by homolog searching miRBase (microRNA database (miRBase), RRID:SCR_003152) v21 [55] against the C. heterophylla genome with 1 mismatch. Then, secondary structures of the putative sequences were predicted by miRDeep2 (miRDeep, RRID:SCR_010829) [56]. Finally, putative miRNAs with hairpin structures were considered as reliable ones. Other types of non-coding RNA were detected using Infernal (Infernal, RRID:SCR_011809) [57] (e-value ≤ 0.01) based on the Rfam database (Rfam, RRID:SCR_007891) v12.0 [58]. In total, 92 miRNAs, 617 tRNAs, and 622 rRNAs were annotated in C. heterophylla genome sequences (Supplementary Table S4c).
Gene family identification and phylogenetic tree construction
In the gene family and phylogenetic analysis, the protein-coding genes of O. sativa, A. thaliana, Populus trichocarpa, Quercus variabilis, J. regia, B. pendula, Ostrya japonica, and C. heterophylla were downloaded from Genbank or Ensembl databases. The longest transcripts were selected to represent the protein-coding genes. Protein sequence clustering was performed using OrthoMCL (OrthoMCL DB: Ortholog Groups of Protein Sequences, RRID:SCR_007839) v2.0 [59] with default parameters to identify the orthologous groups. The result showed that C. heterophylla has 16,811 orthologous groups, including 5,150 single-copy genes, 6,040 multiple-copy genes, and 582 specific genes. Notably, 222 species-specific families were identified for C. heterophylla, which might contribute to its unique features (Fig. 3A). To construct the phylogenetic analysis, 1,182 single-copy orthologs were identified from 1-copy families of selected species. The protein sequences of single-copy orthologs were aligned using MUSCLE (MUSCLE, RRID:SCR_011812) v3.8.31 [60], and low-quality alignment regions were removed using Gblocks (Gblocks, RRID:SCR_015945) v0.91b [61] with default parameters. A phylogenetic tree was constructed using the maximum-likelihood method with the JTT amino acid substitution model implemented in the PhyML (PhyML, RRID:SCR_014629) v3.3 package [62]. The divergence time was estimated using the MCMCtree program in the PAML (PAML, RRID:SCR_014932) v4.7b package [63]. An age of 51.2–66.7 million years ago (Mya) was used to calibrate the crown nodes of the family Betulaceae [64]. The monocot-dicot split time (152–160 Mya) obtained from the TimeTree database was also used to calibrate the time estimation [65]. The result showed that C. heterophylla is close to O. japonica, and they diverged from their common ancestor ∼52.79 Mya (Fig. 3B).
Figure 3:
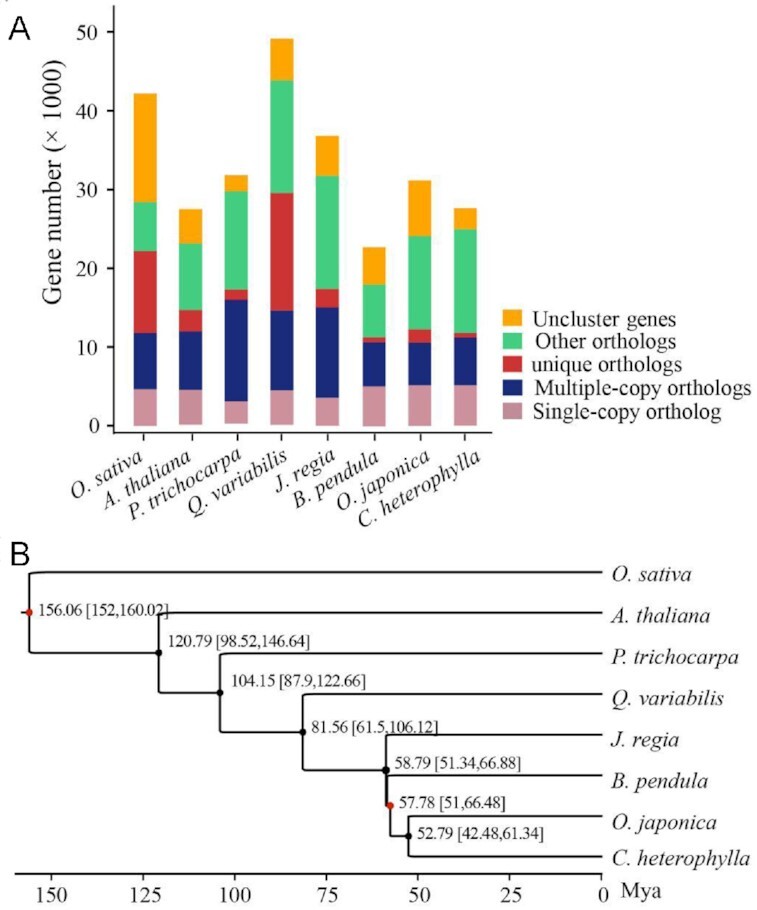
Genome evolution analysis of C. heterophylla. (A) Summary of gene family clustering of C. heterophylla and 7 related species. Single-copy orthologs: 1-copy genes in ortholog group. Multiple-copy orthologs: multiple genes in ortholog group. Unique orthologs: species-specific genes. Other orthologs: the rest of the clustered genes. Uncluster genes: number of genes out of cluster. (B) Phylogenetic relationship and divergence time estimation. The O. sativa was considered as outgroup in phylogenetic tree construction. The red dots indicate the fossil correction time of O. sativa vs P. trichocarpa (152–160 Mya) and crown nodes of family Betulaceae (51.2–66.7 Mya), respectively.
Conclusion
To our knowledge, this is the first report of a chromosome-level genome assembly of C. heterophylla using the third-generation sequencing technologies of Nanopore and Hi-C. C. heterophylla has 210.26 Mb of repetitive sequences, accounting for 56.71% of genomic sequences. A total of 25,389 high-quality protein-coding genes were annotated by integrating evidence from de novo prediction, homologous protein prediction, and transcriptome data. Phylogenetic analysis showed that Corylus is closely related to Ostrya, and they diverged from their common ancestor ∼52.79 Mya. This work provides valuable chromosome-level genomic data for studying hazelnut traits. The genomic data should promote research on the molecular mechanisms of hazelnut responses to environmental stress and provides a valuable resource for genome-assisted improvements in Corylus breeding.
Data Availability
The genome sequence data underlying this article are available in NCBI and can be accessesd with accession JADOBO000000000. Raw reads of Nanopore, whole-genome sequencing, Hi-C, and RNAseq, and genome assembly sequences of the C. heterophylla genome have been deposited at the Nucleotide Sequence Archive and GenBank in NCBI under BioProject PRJNA655406 and BioSample Accessions of SAMN15734705 and SAMN15734794. The SRA accessions are SRR12458330, SRR12458329, SRR12458328, and SRR12458327. Additional supporting data and materials, including annotations, RNA-seq data, and phylogenetic trees, are available in the GigaScience databse, GigaDB [66].
Additional Files
Supplementary Figure S1: Genome survey analysis of C. heterophylla based on k-mer = 19.
Supplementary Figure S2: Fragment size distribution of Hi-C read pairs.
Supplementary Figure S3: Venn plot of predicted genes generated from ab initio, RNAseq, and homology methods.
Supplementary Table S1a: Summary of Illumina data for genome survey and genome polishing.
Supplementary Table S1b: Statistics of Nanopore long reads.
Supplementary Table S1c: Distribution of Nanopore long-read lengths.
Supplementary Table S1d: Summary of pooled transcriptome data used for gene prediction.
Supplementary Table S1e: Summary of Hi-C data for error correction and chromosome construction.
Supplementary Table S2: Valid interaction pairs of Hi-C sequencing data.
Supplementary Table S3a: Completeness analysis based on the CEG database.
Supplementary Table S3b: Genome completeness assessment based on Illumina sequencing reads.
Supplementary Table S4a: Summary of gene predictions resulting from different evidence.
Supplementary Table S4b: Gene function annotated by different databases.
Supplementary Table S4c: Non-coding RNA identification.
Abbreviations
BLAST: Basic Local Alignment Search Tool; bp: base pairs; BUSCO: Benchmarking Universal Single-Copy Orthologs; BWA: Burrows-Wheeler Aligner; CEGMA: Core Eukaryotic Genes Mapping Approach; CTAB: hexadecyltrimethylammonium bromide; Gb: gigabase pairs; GeMoMa: Gene Model Mapper; GO: Gene Ontology; Hi-C: high-throughput chromosome conformation capture; HiSeq: high-throughput sequencing; HMM: hidden Markov model; kb: kilobase pairs; KEGG: Kyoto Encyclopedia of Genes and Genomes; KOG: EuKaryotic Orthologous Groups; LG: linkage group; LTR: long terminal repeat; Mb: megabase pairs; miRNA: microRNA; MITE: miniature inverted-repeat transposable element; MUSCLE: MUltiple Sequence Comparison by Log-Expectation; Mya: million years ago; NCBI: National Center for Biotechnology Information; NR: non-redundant; PAML: Phylogenetic Analysis of Maximum-Likelihood; PASA: Program to Assemble Spliced Alignments; PE: paired-end; PhyML: Phylogeny Maximum Likelihood; RNA-seq: RNA sequencing; rRNA: ribosomal RNA; SNAP: Semi-HMM-based Nucleic Acid Parser; TIR: terminal inverted repeat; TrEMBL: a database of translated proteins from European Bioinformatics Institute; TRF: Tandem Repeats Finder; tRNA: transfer RNA.
Competing Interests
The authors declare that they have no competing interests.
Funding
This work was financed by the Special Investigation on Basic Resources of Science and Technology (Grant No. 2019FY100801_03) and the Special Fund for Basic Scientific Research Business of Central Public Research Institutes of the Chinese Academy of Forestry, China (Grant No. RIF-12 and CAFYBB2017ZA004–9).
Authors’ Contributions
T.Z., Z.Y., W.M., Q.M., and L.W. designed and conceived the study; W.M., L.L., and G.W. helped to collect the samples; T.Z., Z.Y., L.L., Q.M., and L.W. performed the experiments; T.Z., W.M., Z.Y., Q.M., X.C., and L.W. wrote and revised the manuscript. All authors read and approved the manuscript.
Supplementary Material
Kelly Vining -- 12/9/2020 Reviewed
Jarkko Salojarvi, DSc (tech) -- 1/27/2021 Reviewed
ACKNOWLEDGEMENTS
We thank Changmian Ji from the Chinese Academy of Tropical Agricultural Sciences for assistance with bioinformatics analysis and discussion.
Contributor Information
Tiantian Zhao, Research Institute of Forestry, Chinese Academy of Forestry/Key Laboratory of Tree Breeding and Cultivation of the State Forestry and Grassland Administration, No.1 Dongxiaofu, Xiangshan Road, Haidian District, Beijing 100091, China; National Hazelnut Industry Innovation Alliance/Hazelnut Engineering and Technical Research Center of the State Forestry and Grassland Administration, Xiangshan Road, Haidian District, Beijing 100091, China.
Wenxu Ma, Research Institute of Forestry, Chinese Academy of Forestry/Key Laboratory of Tree Breeding and Cultivation of the State Forestry and Grassland Administration, No.1 Dongxiaofu, Xiangshan Road, Haidian District, Beijing 100091, China; National Hazelnut Industry Innovation Alliance/Hazelnut Engineering and Technical Research Center of the State Forestry and Grassland Administration, Xiangshan Road, Haidian District, Beijing 100091, China.
Zhen Yang, Research Institute of Forestry, Chinese Academy of Forestry/Key Laboratory of Tree Breeding and Cultivation of the State Forestry and Grassland Administration, No.1 Dongxiaofu, Xiangshan Road, Haidian District, Beijing 100091, China; National Hazelnut Industry Innovation Alliance/Hazelnut Engineering and Technical Research Center of the State Forestry and Grassland Administration, Xiangshan Road, Haidian District, Beijing 100091, China.
Lisong Liang, Research Institute of Forestry, Chinese Academy of Forestry/Key Laboratory of Tree Breeding and Cultivation of the State Forestry and Grassland Administration, No.1 Dongxiaofu, Xiangshan Road, Haidian District, Beijing 100091, China; National Hazelnut Industry Innovation Alliance/Hazelnut Engineering and Technical Research Center of the State Forestry and Grassland Administration, Xiangshan Road, Haidian District, Beijing 100091, China.
Xin Chen, National Hazelnut Industry Innovation Alliance/Hazelnut Engineering and Technical Research Center of the State Forestry and Grassland Administration, Xiangshan Road, Haidian District, Beijing 100091, China; Shandong Institute of Pomology, Shandong Academy of Agricultural Sciences, No. 66 Longtan Road, Taishan District, Taian 271000, China.
Guixi Wang, Research Institute of Forestry, Chinese Academy of Forestry/Key Laboratory of Tree Breeding and Cultivation of the State Forestry and Grassland Administration, No.1 Dongxiaofu, Xiangshan Road, Haidian District, Beijing 100091, China; National Hazelnut Industry Innovation Alliance/Hazelnut Engineering and Technical Research Center of the State Forestry and Grassland Administration, Xiangshan Road, Haidian District, Beijing 100091, China.
Qinghua Ma, Research Institute of Forestry, Chinese Academy of Forestry/Key Laboratory of Tree Breeding and Cultivation of the State Forestry and Grassland Administration, No.1 Dongxiaofu, Xiangshan Road, Haidian District, Beijing 100091, China; National Hazelnut Industry Innovation Alliance/Hazelnut Engineering and Technical Research Center of the State Forestry and Grassland Administration, Xiangshan Road, Haidian District, Beijing 100091, China.
Lujun Wang, National Hazelnut Industry Innovation Alliance/Hazelnut Engineering and Technical Research Center of the State Forestry and Grassland Administration, Xiangshan Road, Haidian District, Beijing 100091, China; Anhui Academy of Forestry, No. 820 Changjiangxi Road, Shushan District, Hefei 230031, China.
References
- 1. Zong JW, Zhao TT, Ma QH, et al. Assessment of genetic diversity and population genetic structure of Corylus mandshurica in China using SSR markers. PLoS One. 2015;10(9):e0137528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehlenbacher SA. Hazelnuts. In: Fulbright D, ed. A Guide to Nut Tree Culture in North America. Northern Nut Growers Association; 2003:183-215.; [Google Scholar]
- 3. Boccacci P, Beltramo C, Sandoval Prando MA, et al. In silico mining, characterization and cross-species transferability of EST-SSR markers for European hazelnut (Corylus avellana L.). Mol Breed. 2015;35(1):21. [Google Scholar]
- 4. Gürcan K, Mehlenbacher S, Botta R, et al. Development, characterization, segregation, and mapping of microsatellite markers for European hazelnut (Corylus avellana L.) from enriched genomic libraries and usefulness in genetic diversity studies. Tree Genet Genomes. 2010;6(4):513–31. [Google Scholar]
- 5. Zhang YH, Liu L, Liang WJ, et al. China Fruit's Monograph-Chestnut and Hazelnut. Beijing: China Forestry Publishing House; 2005:187-209. [Google Scholar]
- 6. Molnar TJ. Corylus. In: Kole C, ed. Wild Crop Relatives: Genomic and breeding resources. 1st ed. Berlin, Heidelberg: Springer; 2011:15–48. [Google Scholar]
- 7. Wang GX. Studies on the cultivation and utilization of Corylus resources in China (Ⅰ) - Corylus germplasm resources. For Sci Res. 2018;31:105–12. [Google Scholar]
- 8. Wang GX, Ma QH, Zhao TT, et al. Resources and production of hazelnut in China. Acta Hortic. 2018;1226(1226):59–64. [Google Scholar]
- 9. Mayjonade B, Gouzy J, Donnadieu C, et al. Extraction of high-molecular-weight genomic DNA for long-read sequencing of single molecules. Biotechniques. 2016;61(4):203–5. [DOI] [PubMed] [Google Scholar]
- 10. Doyle J, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- 11. Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27(6):764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Loman NJ, Quinlan AR. Poretools: A toolkit for analyzing nanopore sequence data. Bioinformatics. 2014;30(23):3399–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Belton JM, McCord RP, Gibcus JH, et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 2012;58(3):268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grob S, Schmid MW, Grossniklaus U. Hi-C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol Cell. 2014;55(5):678–93. [DOI] [PubMed] [Google Scholar]
- 15. Xie T, Zheng JF, Liu S, et al. De novo plant genome assembly based on chromatin interactions: A case study of Arabidopsis thaliana. Mol Plant. 2015;8(3):489–92. [DOI] [PubMed] [Google Scholar]
- 16. Koren S, Walenz BP, Berlin K, et al. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17(2):155–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaser R, Sović I, Nagarajan N, et al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27(5):737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walker BJ, Abeel T, Shea T, et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burton JN, Adey A, Patwardhan RP, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31(12):1119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seppey M, Manni M, Zdobnov EM. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol Biol. 2019;1962:227–45. [DOI] [PubMed] [Google Scholar]
- 23. Parra G, Bradnam K, Korf I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7. [DOI] [PubMed] [Google Scholar]
- 24. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han Y, Wessler SR. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010;38(22):e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu Z, Wang H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35(Web Server):W265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl 1):i351–8. [DOI] [PubMed] [Google Scholar]
- 28. Edgar RC, Myers EW. PILER: Identification and classification of genomic repeats. Bioinformatics. 2005;21(Suppl 1):i152–8. [DOI] [PubMed] [Google Scholar]
- 29. Hoede C, Arnoux S, Moisset M, et al. PASTEC: An automatic transposable element classification tool. PLoS One. 2014;9(5):e91929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bao W, Kojima KK, Kohany O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;Chap 4:Unit 4.10, doi: 10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 32. Stanke M, Morgenstern B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33(Web Server):W465–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alioto T, Blanco E, Parra G, et al. Using geneid to identify genes. Curr Protoc Bioinformatics. 2018;64(1):e56. [DOI] [PubMed] [Google Scholar]
- 34. Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–9. [DOI] [PubMed] [Google Scholar]
- 35. Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268(1):78–94. [DOI] [PubMed] [Google Scholar]
- 36. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Camacho C, Coulouris G, Avagyan V, et al. Blast+: Architecture and applications. BMC Bioinformatics. 2009;10(1):421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keilwagen J, Wenk M, Erickson JL, et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016;44(9):e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim D, Langmead B, Salzberg SL. Hisat: A fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pertea M, Pertea GM, Antonescu CM, et al. Stringtie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. TransDecoder (find coding regions within transcripts). Version 5.5.0. https://github.Com/transdecoder/transdecoder. Accessed 10 February 2020. [Google Scholar]
- 42. Tang S, Lomsadze A, Borodovsky M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015;43(12):e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Campbell MA, Haas BJ, Hamilton JP, et al. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics. 2006;7(1):327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haas BJ, Salzberg SL, Zhu W, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deng YY, Li JQ, Wu SF, et al. Integrated NR database in protein annotation system and its localization. Comput Eng. 2006;32(5):71–2. [Google Scholar]
- 46. Koonin EV, Fedorova ND, Jackson JD, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5(2):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boeckmann B, Bairoch A, Apweiler R, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31(1):365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–d61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zdobnov EM, Apweiler R. InterProScan–An integration platform for the signature-recognition methods in interPro. Bioinformatics. 2001;17(9):847–8. [DOI] [PubMed] [Google Scholar]
- 50. Conesa A, Götz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008, doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Götz S, García-Gómez JM, Terol J, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. She R, Chu JS, Wang K, et al. GenBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res. 2009;19(1):143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Birney E, Durbin R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000;10(4):547–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Friedländer MR, Mackowiak SD, Li N, et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nawrocki EP, Burge SW, Bateman A, et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015;43(D1):D130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li L, Stoeckert CJ Jr., Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–77. [DOI] [PubMed] [Google Scholar]
- 62. Guindon S, Dufayard JF, Lefort V, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst Biol. 2010;59(3):307–21. [DOI] [PubMed] [Google Scholar]
- 63. Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;24(8):1586–91. [DOI] [PubMed] [Google Scholar]
- 64. Takhtajan AL. Outline of the classification of flowering plants (Magnoliophyta). Bot Rev. 1980;46(3):225–359. [Google Scholar]
- 65. Timetree database. http://www.timetree.org/. Accessed 10 February 2020. [Google Scholar]
- 66. Zhao T, Ma W, Yang Z, et al. Supporting data for “A chromosome-level reference genome of the hazelnut, Corylus heterophylla Fisch.”. GigaScience Database. 2021. 10.5524/100877 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Zhao T, Ma W, Yang Z, et al. Supporting data for “A chromosome-level reference genome of the hazelnut, Corylus heterophylla Fisch.”. GigaScience Database. 2021. 10.5524/100877 [DOI] [PMC free article] [PubMed]
Supplementary Materials
Kelly Vining -- 12/9/2020 Reviewed
Jarkko Salojarvi, DSc (tech) -- 1/27/2021 Reviewed
Data Availability Statement
The genome sequence data underlying this article are available in NCBI and can be accessesd with accession JADOBO000000000. Raw reads of Nanopore, whole-genome sequencing, Hi-C, and RNAseq, and genome assembly sequences of the C. heterophylla genome have been deposited at the Nucleotide Sequence Archive and GenBank in NCBI under BioProject PRJNA655406 and BioSample Accessions of SAMN15734705 and SAMN15734794. The SRA accessions are SRR12458330, SRR12458329, SRR12458328, and SRR12458327. Additional supporting data and materials, including annotations, RNA-seq data, and phylogenetic trees, are available in the GigaScience databse, GigaDB [66].