

Abstract
The AMP-activated protein kinase (AMPK) is a molecular sensor to maintain energy homeostasis. The two isoforms of the AMPK catalytic subunit (AMPKα1 and α2) are both expressed in brains, but their roles in cognition are unknown. We generated conditional knockout mice in which brain AMPKα isoforms are selectively suppressed (AMPKα1/α2 cKO), and determined the isoform-specific effects in mice of either sex on cognition and synaptic plasticity. AMPKα2 cKO but not AMPKα1 cKO displayed impaired cognition and hippocampal late long-term potentiation (L-LTP). Further, AMPKα2 cKO mice exhibited decreased dendritic spine density and abnormal spine morphology in hippocampus. Electron microscope imaging demonstrated reduced postsynaptic density formation and fewer dendritic polyribosomes in hippocampi of AMPKα2 cKO mice. Biochemical studies revealed unexpected findings that repression of AMPKα2 resulted in increased phosphorylation of mRNA translational factor eIF2α and its kinase PERK. Importantly, L-LTP failure and cognitive impairments displayed in AMPKα2 cKO mice were alleviated by suppressing PERK activity pharmacologically or genetically. In summary, we demonstrate here that brain-specific suppression of AMPKα2 isoform impairs cognition and hippocampal LTP by PERK-mediated eIF2α phosphorylation, providing molecular mechanisms linking metabolism, protein synthesis, and cognition.
Introduction
Accurate control of energy metabolism to balance energy demand and supply is critically important for function and survival of all organisms. Mounting evidence has established that the AMP-activated protein kinase (AMPK) is a key sensor/regulator at the molecular level to maintain energy homeostasis [1–3]. Mammalian AMPK is a heterotrimeric complex composed of a catalytic α subunit, and regulatory β and γ subunits [2, 4]. AMPK is activated in response to low energy states, and can be activated via either allosteric activation mediated by the γ subunit or phosphorylation of the α subunit at Thr172. Phosphorylation is quantitatively much more important than the allosteric activation because it causes at least a 50- to 100-fold increase in AMPK activation [5]. The major kinases phosphorylating Thr172 include LKB1, a tumor suppressor that appears to be constitutively active; the Ca2+/calmodulin-dependent protein kinase CaMKKβ, stimulated by increased cytosolic Ca2+; and TAK1 (also known as MAP3K7 or MEKK7), a protein kinase activated by cytokines. Furthermore, AKT is a negative regulator of AMPK, leading to decreased phosphorylation of Thr172 [3, 6].
The brain has an extremely high metabolic rate and functions of neurons depend on sustaining energy levels [7]. Several studies indicated that AMPK is involved in hippocampal long-term potentiation (LTP), a major form of synaptic plasticity and cellular model for memory [8–10]. Emerging evidence also links AMPK dysregulation to diverse neurological diseases characterized by cognitive impairments, including Alzheimer’s disease (AD), albeit with conflicting results about whether activation/phosphorylation of AMPKα in brain is beneficial or detrimental for cognition and synaptic function [4, 11–13]. The kinase catalytic subunit of AMPK exists in two isoforms: α1 and α2, which are encoded by distinct genes located in different chromosomes [3]. Both the α1 and α2 isoforms are expressed in most tissues, including the brain, for regulation of energy homeostasis. Previous work, mainly in non-neuronal systems, indicated isoform-specific substrate specificity [14–17]. However, very little is known about subcellular localization of AMPK isoforms in brain. One study in mouse brain revealed that AMPKα1 is present mainly in the neuropil, while AMPKα2 is expressed more extensively in both soma (cytoplasm and nuclear) and neuropil, and presumably is the predominant isoform in brain [18]. However, the specific molecular targets and roles of AMPKα isoforms in the central nervous system remain unclear, and whether there is an isoform-specific role of the AMPK catalytic subunits in cognitive functions and synaptic plasticity is unknown.
Given its central role as a molecular energy sensor regulating fundamental cellular functions, it is not surprising that AMPK has “crosstalk” with multiple signaling cascades and has numerous downstream effectors participating in biosynthetic pathways [19]. Among the many downstream effects of AMPK, regulation of de novo protein synthesis (mRNA translation) is of particular interest. Substantial studies have demonstrated that de novo protein synthesis is indispensable for long-term synaptic plasticity and memory formation [20–22]. Previous studies indicated that AMPK activation may lead to suppression of mRNA translation by (1) inhibition of the mammalian target of rapamycin complex 1 (mTORC1) pathway, which controls cap-dependent translation initiation and synthesis of the translational machinery; and (2) phosphorylation and activation of eukaryotic elongation factor 2 kinase (eEF2K), leading to eEF2 phosphorylation and consequently disruption of the elongation step in mRNA translation [3]. Phosphorylation of eIF2α, particularly by PERK (one of its 4 kinases), results in suppression of general protein synthesis and has been implicated in neurodegenerative diseases such as Alzheimer’s disease (AD) and prion disease [23–26].
Here, we generated brain- and isoform-specific conditional AMPKα1 and α2 knockout mice (AMPKα1 cKO and AMPKα2 cKO), and performed behavioral and electrophysiological experiments to investigate the isoform-specific role of AMPKα in regulating memory formation and synaptic plasticity. Furthermore, we have carried out studies with multiple approaches including electron microscopy and proteomics analyses to elucidate detailed cellular/molecular mechanisms potentially associated with the isoform-specific behavioral and electrophysiology phenotypes in the mutant mice. Our findings revealed the importance of the brain AMPKα2 isoform in long-term synaptic plasticity and memory formation. Surprisingly, such roles of AMPKα2 are associated with PERK-mediated eIF2α phosphorylation.
Materials and methods
Key reagents and resources are summarized in Table 1.
Table 1.
Key resources.
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Monoclonal Anti-ß-Actin antibody produced in mouse ((1:10000)) | Sigma | Cat#A2228 |
| GAPDH (14C10) Rabbit mAb (1:5000) | Cell Signaling | Cat#2118 S |
| Anti-AMPK alpha 1 antibody (1:1000) | Abcam | Cat#ab3759 |
| Anti-AMPK alpha 2 antibody (1:1000) | Abcam | Cat#ab3760 |
| AMPKa1 Antibody (1:500, IHC) | Cell Signaling | Cat#2795 S |
| AMPKa2 Antibody (1:250, IHC) | Cell Signaling | Cat#2757 S |
| Human/Mouse/Rat AMPK alpha 1 Affinity Purified Pab (Goat) (7.5 μg/mL) | R&D Systems | Cat#AF3197 |
| PSD95 (D27E11) XP® Rabbit mAb (1:1000) | Cell Signaling | Cat#3450 P; RRID:AB_10828082 |
| mTOR (7C10) Rabbit mAb (1:1000) | Cell Signaling | Cat#2983 S; RRID:AB_2105622 |
| Phospho-mTOR (Ser2448) (D9C2) XP® Rabbit mAb (1:1000) | Cell Signaling | Cat#5536 S; RRID:AB_10691552 |
| Phospho-mTOR (Ser2481) Antibody (1:1000) | Cell Signaling | Cat#2974 S; RRID:AB_2231885 |
| p70 S6 Kinase (49D7) Rabbit mAb (1:1000) | Cell Signaling | Cat#2708 S; RRID:AB_390722 |
| Phospho-p70 S6 Kinase (Thr389) (108D2) Rabbit mAb (1:1000) | Cell Signaling | Cat#9234 S; RRID:AB_2269803 |
| Phospho-p70 S6 Kinase (Ser371) Antibody, Rabbit (1:1000) | Cell Signaling | Cat#9208 S; RRID:AB_330990 |
| Phospho-p70 S6 Kinase (Thr421/Ser424) Antibody (1:1000) | Cell Signaling | Cat#9204 S; RRID:AB_2265916 |
| 4E-BP1 (53H11) Rabbit mAb (1:1000) | Cell Signaling | Cat#9644 S; RRID:AB_2097841 |
| Phospho-4E-BP1 (Thr37/46) (236B4) Rabbit mAb (1:1000) | Cell Signaling | Cat#2855 S; RRID:AB_560835 |
| Akt (pan) (C67E7) Rabbit mAb (1:1000) | Cell Signaling | Cat#4691 S; RRID:AB_915783 |
| Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb (1:2000) | Cell Signaling | Cat#4060 S; RRID:AB_2315049 |
| eEF2 Antibody (1:1000) | Cell Signaling | Cat#2332 S; RRID:AB_10693546 |
| Phospho-eEF2 (Thr56) Antibody (1:1000) | Cell Signaling | Cat#2331 S; RRID:AB_10015204 |
| eIF2α Antibody (1:1000) | Cell Signaling | Cat#9722 S; RRID:AB_2230924 |
| Phospho-eIF2α (Ser51) (D9G8) XP® Rabbit mAb(1:1000) | Cell Signaling | Cat#3398 S; RRID:AB_2096481 |
| Phospho-PERK (Thr980) (16F8) Rabbit mAb (1:1000) | Cell Signaling | Cat#3179 S;RRID:AB_2095853 |
| PERK Antibody (H-300) (1:200) | Santa cruz | sc-13073;RRID:AB_2230863 |
| GSK-3β (D5C5Z) XP® Rabbit mAb (1:1000) | Cell Signaling | Cat#12456 S |
| Phospho-GSK-3β (Ser9) (D85E12) XP® Rabbit mAb (1:1000) | Cell Signaling | Cat#5558 S; RRID:AB_10013750 |
| CaMKII (pan) Antibody (1:1000) | Cell Signaling | Cat#3362 S;RRID:AB_10692639 |
| Phospho-CaMKII (Thr286) (D21E4) Rabbit mAb (1:1000) | Cell Signaling | Cat#12716; RRID:AB_2713889 |
| PKM ζ Antibody (H-1) (1:10000) | Santa Cruz | Cat#sc-17781; RRID:AB_628148 |
| Anti-Kv4.2 (1:1000) | Alomone Labs | Cat#APC-023; RRID:AB_2040176 |
| p-Kv4.2 Antibody (C-6) (1:300) | Santa Cruz | Cat#sc-377574 |
| AMPA Receptor (GluR 1) (D4N9V) Rabbit mAb (1:1000) | Cell Signaling | Cat#13185 S |
| ATF4 (D4B8) Antibody (1:1000) | Cell Signaling | Cat#11815 S |
| Oligophrenin-1 Antibody (1:1000) | Cell Signaling | Cat#11939 S |
| Anti-Puromycin Antibody, clone 12D10 (1:5000) | EMD Millipore | MABE343 |
| Goat Anti-Mouse IgG (H + L)-HRP Conjugate (1:5000) | Bio-Rad | Cat#170-6516; RRID:AB_11125547 |
| Goat Anti-Rabbit IgG (H + L)-HRP Conjugate (1:5000) | Bio-Rad | Cat#170-6515; RRID:AB_11125142 |
| Goat anti-Rabbit IgG (H + L) Secondary Antibody, Alexa Fluor® 488 conjugate (1:250) | Life Technology | Cat#A-11008; RRID: AB_143165 |
| Donkey anti-Goat IgG (H + L) Secondary Antibody, Alexa Fluor® 633 conjugate (1:250) | Life Technology | Cat#A-21082; RRID:AB_2535739 |
| Biotinylated Goat Anti-Rabbit IgG Antibody | Vector Laboratories | Cat#BA-1000; RRID:AB_2313606 |
| Biotinylated Horse Anti-Goat IgG Antibody | Vector Laboratories | Cat#BA-9500; RRID:AB_2336123 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories | Cat#H-1200 |
| Bacterial and Virus Strains | N/A | |
| Biological Samples | ||
| Human hippocampal, cerebellum brain blocks | University of Washington Brain Bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Pierce™ T-PER™ Tissue Protein Extraction Reagent | Thermo Fisher | Cat#78510 |
| Halt™ Protease Inhibitor Cocktails (with EDTA) | Pierce | Cat#87786 |
| Halt™ Phosphatase Inhibitor Cocktail | Pierce | Cat#78420 |
| Restore™ Western Blot Stripping Buffer | Pierce | Cat#PI21059 |
| SuperBlock (TBS) Blocking Buffer | Pierce | Cat#PI37581 |
| 2-Mercaptoethanol (Reagent), Fisher Chemical | Thermo Fisher | Cat#O3446I-100 |
| SYBR Safe DNA Gel Satin 10,000× | Invitrogen | Cat#S33102 |
| PERK Inhibitor I, GSK2606414 - CAS 1337531-89-1 - Calbiochem | Millipore | Cat#516535 |
| Critical Commercial Assays | ||
| FD Rapid GolgiStain™ Kit | FD NeuroTechnologies, Inc. | Cat#PK401 |
| Deposited data | ||
| Experimental Models: Cell Lines | N/A | |
| Experimental Models: Organisms/Strains | ||
| Mouse:B6.Cg-Tg(Camk2a-cre)T29-1Stl/J | The Jackson Laboratory | Stock No: 005359 | T29-1 |
| Mouse:STOCK Prkaa1tm1.1Sjm/J | The Jackson Laboratory | Stock No: 014141 | Prkaa1 fl |
| Mouse:STOCK Prkaa2tm1.1Sjm/J | The Jackson Laboratory | Stock No: 014142 | Prkaa2fl |
| Oligonucleotides | ||
| Primer: Camk2a-Cre-oIMR1084 Forward: GCG GTC TGG CAG TAA AAA CTA TC | The Jackson Laboratory | Stock No: 005359 | T29-1 |
| Primer: Camk2a-Cre-oIMR1085 Reverse: GTG AAA CAG CAT TGC TGT CAC TT | The Jackson Laboratory | Stock No: 005359 | T29-1 |
| Primer: Prkaa1-11528 CCC ACC ATC ACT CCA TCT CT | The Jackson Laboratory | Stock No: 014141 | Prkaa1 fl |
| Primer: Prkaa1-11529 AGC CTG CTT GGC ACA CTT AT | The Jackson Laboratory | Stock No: 014141 | Prkaa1 fl |
| Primer: Prkaa2-11530 GCA GGC GAA TTT CTG AGT TC | The Jackson Laboratory | Stock No: 014141 | Prkaa1 fl |
| Primer: Prkaa2-11531 TCC CCT TGA ACA AGC ATA CC | The Jackson Laboratory | Stock No: 014141 | Prkaa1 fl |
| Recombinant DNA | N/A | |
| Software and algorithms | ||
| ImageJ (Version1.48) | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism (Version 6.05) | GraphPad Software Inc | http://www.graphpad.com/scientific-software/prism/ |
| EthoVision XT (Version 7) | Noldus | http://www.noldus.com/EthoVision-XT/more-about-ethovision-xt |
Mice
All mice are housed in a barrier facility dedicated to transgenic mice at Wake Forest School of Medicine. Randomization was not used in animal studies. The facility operates in accordance with standards and policies of the US Department of Agriculture’s Animal Welfare Information Center (AWIC), and the NIH Guide for Care and Use of Laboratory Animals. The facility is kept on a 12 h light/dark cycle, with a regular feeding and cage-cleaning schedule. Mice of both sexes were used. The following mice were purchased from the Jackson Laboratory (Bar Harbor, ME): B6.Cg-Tg (Camk2a-cre)T29-1Stl/J (Camk2a-cre mice), stock No. 005359; Prkaa1tm1.1Sjm/J (loxP-flanked Prkaa1 mice), stock No. 014141; Prkaa2tm1.1Sjm/J (loxP-flanked Prkaa2 mice), stock No. 014142. Generation of PERK cKO mice was as described [27]. The genotypes were verified by polymerase chain reaction (PCR). Mice of 3–6-month old were used for all experiments. Investigators were not blind to the group allocation during the experiments.
Hippocampal slice preparation and electrophysiology
Acute 400 μm transverse hippocampal slices were prepared using a Leica VT1200S vibratome as described previously [28]. Slices were maintained before experimentation at room temperature for at least 2 h in artificial cerebrospinal fluid (ACSF) containing (in mM) 118 NaCl, 3.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.25 NaH2PO4, 5 NaHCO3, and 15 glucose, bubbled with 95% O2/5% CO2. For electrophysiology, monophasic, constant-current stimuli (100 μs) were delivered with a bipolar silver electrode placed in the stratum radiatum of area CA3. Field excitatory postsynaptic potentials (fEPSPs) were recorded using a glass microelectrode from the stratum radiatum of area CA1. Late-LTP (L-LTP) was induced using high-frequency stimulation (HFS) consisting of two 1s 100 Hz trains separated by 60 s, each delivered at 70–80% of the intensity that evoked spiked fEPSPs. Early LTP (E-LTP) was induced with one train HFS (100 Hz).
Drug treatment
The PERK inhibitor GSK2606414 (Calbiochem/Millipore) was prepared as a stock solution of 1 mM in DMSO and diluted to final concentration of 1 μM in ACSF before experiments. Incubation of hippocampal slices was performed in recording chambers.
Morris water maze (MWM) and visible maze (VM)
The MWM test was performed as previously described [26]. The training paradigm for the hidden platform version of the Morris water maze consists of 4 trials (60 s maximum; interval 15 min) each day for 5 consecutive days. The probe trial was carried 2 h after the completion of training on day 5. The visible platform task consists of 4 trials each day for 2 consecutive days with the escape platform marked by a visible cue and moves randomly between four locations. The trajectories were recorded with a video tracking system (Nodulus Ethovision XT).
Open field test (OF) and novel object recognition (NOR)
OF and NOR behavioral tests were performed as described previously [29]. For OF, mice were allowed to explore freely in a 40 × 40 cm2 arena for 15 min. The distance traveled and the duration spent in the center (20 × 20 cm2) and in the periphery were counted by the Noldus software (Ethovision XT). The NOR test is based on the natural tendency of mice to explore a novel object rather than a familiar object. The amount of time spent exploring the novel or familiar object was normalized by the total time spent exploring both objects. Object interaction was monitored and recorded by Noldus software (Ethovision XT).
Y-water maze reversal task
The reversal Y-water maze test was conducted as described previously [27]. On the day of training, the mouse was trained to pick up one side (arm) of the Y-shape maze, where a platform was hidden. The memory test phase began after a delay of 24 h, which included 5 trials. For mice chose the correct arm, the escape platform was switched to the opposite arm, and the mice were trained to learn the new location of the platform.
Immunofluorescence and confocal microscopy
Hippocampal slices were fixed overnight in ice-cold 4% paraformaldehyde in PBS. Free floating sections were subsectioned to 40 μm using a Leica VT1200S vibratome and permeabilized with 0.3% TritonX-100. Sections were incubated with signal enhancer for 30 m, blocked with 10% normal goat serum, 0.1% sodium azide, and 1% BSA in PBS for 5 h, and incubated overnight at 4 °C with primary antibody for AMPKα1 (7.5 μg/mL, AF3197, R&D Systems) and AMPKα2 (1:400, ab3760, Abcam). Alexa Fluor 488 secondary antibody (Thermo Fisher Catalog #: R37120, RRID:AB_2556548) was used. The sections were imaged using an Olympus FV1200 Confocal microscope at 63×. All parameters (pinhole, contrast, gain, and offset) were held constant for all sections across the same experiment.
Immunohistochemistry for mouse tissue
Mice were sacrificed and brains were hemisected and fixed overnight in ice-cold PFA and transferred to 70% ethanol. Paraffin embedding was performed by the Department of Pathology, Wake Forest School of Medicine. Paraffin embedded mouse sections were deparaffinized in xylene and rehydrated through graded ethanol series. Antigen retrieval utilized citrate buffer (pH 6.0) in a standard 10 min microwave procedure. Blocking was done for 2 h with 10% NGS in 1% BSA/TBS. Slides were incubated in a humidified chamber in primary antibody for AMPKα1 (1:250; Abcam) or AMPKα2 (1:250, Abcam) overnight at 4 °C. Following 15 min 3% hydrogen peroxide blocking, sections were then incubated in biotinylated anti-rabbit secondary antibody (1:200; Vector Laboratories) for 1 h at room temperature followed by Vectastain Elite ABC reagent (PK-6100, Vector Laboratories) for another 30 min. Primary and secondary antibodies as well as ABC reagent were diluted in 1% BSA/TBS. Diaminobenzidine (DAB, SK-4105, Vector Laboratories) was diluted in Tris buffer (pH 7.7) and 3% H2O2 for a working DAB solution. Sections were developed in DAB for 30 s to 3 min with monitoring. Slides were counterstained using Mayer’s hematoxylin for 60 s and blued with 0.2% lithium carbonate for 20 s. In between each step of immunohistochemistry, sections were rinsed using TBS + 2.5% TritonX-100 (pH 7.4). Negative controls were incubated in 1% BSA with no primary antibody. Sections were dehydrated in an alcohol series and cleared with xylene, coverslipped with mounting media, and dried overnight. Imaging was performed using BZ-X710 all-in-one fluorescent microscope (Keyence, Japan).
Western immunoblotting
Tissues were removed from appropriate structures and flash-frozen on dry ice. Tissues were then homogenized in an appropriate lysis buffer and quantified. Samples were loaded on 4–15% TGX™ Precast Gels (Bio-Rad), and transferred to nitrocellulose membranes. Membranes were blocked and then probed overnight at 4 °C using primary antibodies of interest. Blots were washed and HRP-labeled secondary antibodies were added. The blots were visualized using chemiluminescence (Clarity™ ECL; Bio-Rad) and the Bio-Rad ChemiDoc™ MP Imaging System. Densitometric analysis was performed using ImageJ software. Data were normalized to β–actin or GAPDH (for total protein analysis) or relevant total proteins (for phospho-protein analysis) unless otherwise specified.
Dendritic spine morphology assay with Golgi staining
Rapid Golgi Kit (FD NeuroTechnologies, MD, USA) was used for the staining procedure. Briefly, the whole brain was isolated, rinsed in cold PBS and then hemisected, quickly immersed into impregnation solution (A + B), stored at room temperature in the dark for two weeks. After three day in Solution C, 100 μm sections were cut with a vibratome, processed, and mounted following the protocol provided with the kit. Hippocampal sections were imaged on a BZ-X710 all-in-one fluorescent microscope (Keyence, Japan) with a 100×/NA 1.45 oil lens. Ten different view fields per mouse were taken, and the z-stack and all-focus functions were applied to generate the final images. Dendritic spine density was calculated by the number of spines of all types divided by the corresponding dendritic length. Spines were morphologically classified based on guidelines published previously [30, 31].
Transmission electron microscopy
The freshly dissected mouse hippocampus CA1 was fixed with 1% PFA + 2.5% glutaraldehyde in 0.1 M Millonig’s phosphate buffer (pH 7.3) for overnight at room temperature. Subsequently, the samples were washed 3× in buffer and post-fixed with 1% osmium tetroxide in phosphate buffer for 1 h. After 3× washes in buffer the samples were dehydrated through a graded series of ethanols (10 min each). For preparation of resin infiltration the samples were incubated in propylene oxide for two changes of 15 min each. Finally, the samples were gradually infiltrated with 1:1, 1:2 and pure solutions of Spurr’s resin after which they were allowed to cure in a 70 °C oven overnight. Ninty nanometers sections were obtained with a Reichert-Jung Ultracut E ultramicrotome, stained with lead citrate and uranyl acetate and viewed with a FEI Tecnai Spirit TEM operating at 80 kV. Images were obtained with an AMT 2Vu CCD camera. Three mice were used for each genotype, and for each mouse, twenty different images, covering the whole CA1 area, were taken at a magnification of 11000. ImageJ software (NIH, USA) was used to analyze the PSD size and active zone length. Clusters of polyribosomes consisted of at least three distinct opaque spots, which are usually in the range of 0.01–0.03 μm in diameter, was considered.
SUnSET protein synthesis assay
As described [32], hippocampal slices were incubated with puromycin (1 μg/ml) for 60 min. At the end of the experiment slices were harvested and flash-frozen on dry ice, and protein lysates were prepared for Western blotting. Puromycin-labeled proteins were identified using anti-puromycin antibody (1:5000; Millipore Cat# AB3258, RRID:AB_91423). Protein synthesis levels were assessed by taking total lane density in the molecular weight of 10–250 kDa.
Statistical analyses
Data are presented as mean ± SEM. For comparisons between two groups, a two-tailed unpaired Student’s t-test was used. For comparisons between multiple groups, one-way ANOVA was used followed by individual post hoc tests when applicable. Error probabilities of p < 0.05 were considered statistically significant. Sample size was chosen following previous publications. Variance was similar between the groups that were being statistically compared based on our observation. Data were analyzed using GraphPad Prism software. Outliers were determined via Grubbs test with a criteria of p < 0.05 for removal.
Results
Generation and characterization of Forebrain- and isoform-specific AMPKα knockout mice
We first performed immunohistochemistry/confocal microscopy experiments to study subcellular localization of AMPKα isoforms, demonstrating that both AMPKα1 and α2 isoforms were expressed and co-localized within soma and dendrites of hippocampus from wild-type mice (Fig. 1a). Further, mice harboring loxP-flanked Prkaa1 or Prkaa2 (genes that encode AMPK α1 or α2 subunits, respectively) were bred with mice expressing a brain-specific Cre recombinase (Camk2a-cre) [26] to generate AMPKα1 or α2 conditional knockout mice (AMPKα1 cKO or AMPK α2 cKO). In these mutant mice, AMPKα1 or α2 is conditionally removed in excitatory neurons in the forebrain and hippocampus late in development. The presence of the Prkaa1loxP, Prkaa2loxP, and Cre transgene was determined using PCR-specific primers (Fig. S1a). By performing Western blotting assays in different brain regions, we confirmed that protein levels of AMPKα1 and α2 in the corresponding conditional knockout mice were significantly reduced in hippocampus (HIP) and prefrontal cortex (PFC), but unaltered in cerebellum (CER) (Fig. 1b–d). Moreover, immunohistochemical staining confirmed neuronal reduction of AMPKα1 or α2 levels in PFC and hippocampus in corresponding mutant mice (Fig. 1e). We also determined that levels of both AMPKα isoforms were unaltered in organ/tissue outside of the brain, including heart, liver and skeletal muscle (Fig. S1b–d). In addition, data from Hematoxylin & Eosin (H&E) and Nissl-staining revealed no gross alterations of forebrain (hippocampus and prefrontal cortex) morphology in conditional knockout mice compared with control littermates (Cre+/−) (Fig. 1f, S1E, and data not shown). Therefore, we successfully generated brain- and isoform-specific AMPKα knockout mice, which will enable us to study the precise role of brain AMPKα isoforms in cognitive functions and synaptic plasticity.
Fig. 1. Characterization of Forebrain- and Isoform-specific AMPKα knockout mice.
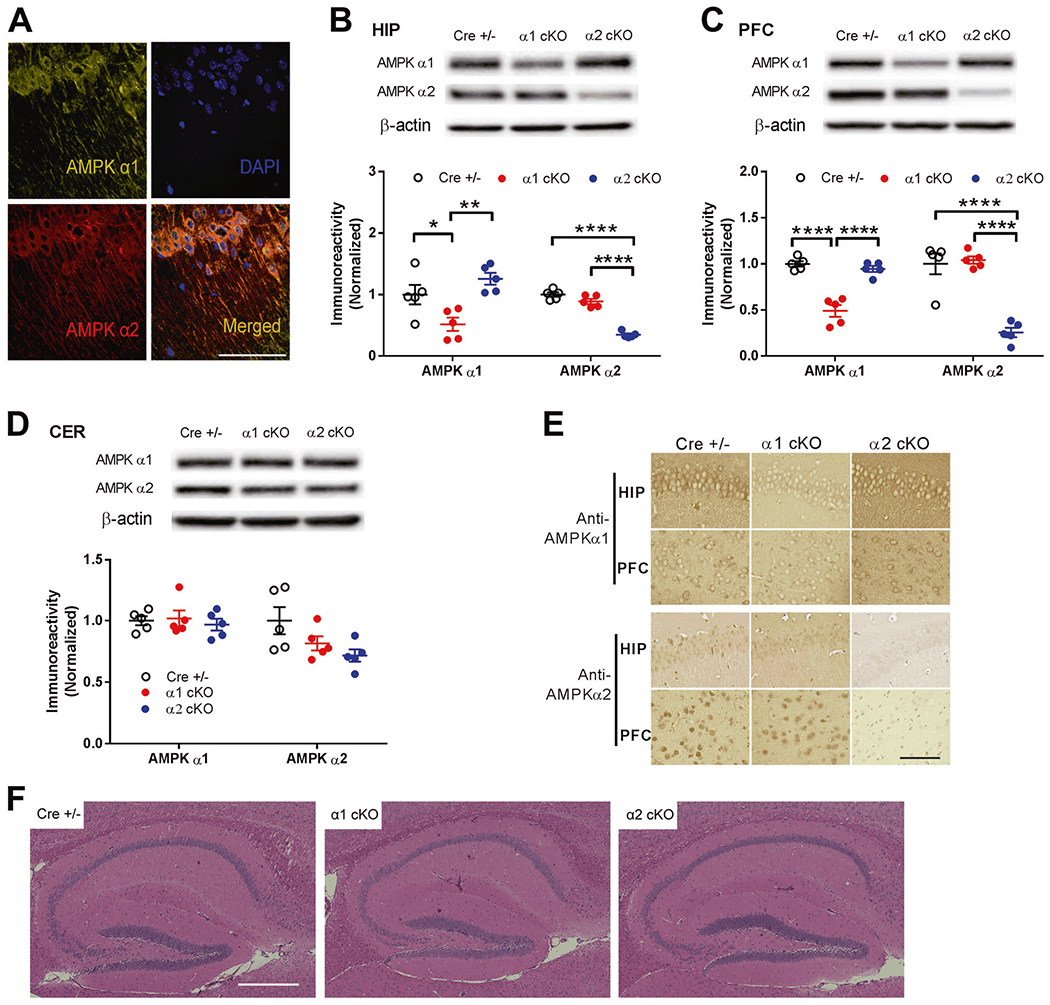
a Immunofluorescence/confocal microscopy data indicated expression and co-localization of AMPKα1 and α2 isoforms within soma and dendrites of hippocampus from CRE+/− mice. Scale bar, 100 μm. Representative images from three independent experiments are shown. b-d Conditional knockout (using CaM-KIIα-driven Cre-lox system) of Prkaa1 or Prkaa2 genes resulted into corresponding reduction of AMPKα1 or AMPKα2 protein levels in hippocampus (HIP) and prefrontal cortex (PFC), but not in cerebellum (CER). Representative Western blot gels and cumulative data for quantification presented in bar graphs are shown (n = 5, *p <0.05, **p <0.01, ****p <0.0001, one-way ANOVA, followed by Tukey’s post hoc test). e Immunohistochemical staining on hippocampus and prefrontal cortex demonstrated decreased neuronal expression of AMPKα1 and AMPKα2 in correlated conditional knockout mouse model compared with CRE+/− control groups. Representative images from three sets of independent experiments are shown. Scale bars: 100 μm. f Hematoxylin and eosin (h, e) staining revealed normal gross hippocampal morphology in AMPKα1 cKO or AMPKα2 cKO mice compared with CRE+/− littermates. Scale bars: 400 μm.
Learning and memory are impaired in AMPKα2 cKO mice but unaffected in AMPKα1 cKO mice
We performed several behavioral experiments to evaluate effects of the AMPKα isoform on cognitive function. To assess general locomotor activity and anxiety, we performed open field test (OF) and did not observe any difference between Cre+/−, AMPKα1 cKO or α2 cKO mice (Fig. 2a, b). To further investigate potential spatial learning and memory alterations, we tested the mice in the hidden platform version of the Morris water maze (MWM) behavioral task [26]. Compared with CRE+/− mice, AMPKα2 cKO mice displayed impaired spatial learning and memory, as indicated by longer day-to-day escape latency during the acquisition phase (Fig. 2c), lower “platform” crossing frequency, and target quadrant occupancy in probe trials (Figs. 2d and S2). In contrast, AMPKα1 cKO mice demonstrated normal “learning” and “memory” compared with CRE+/− mice, as indicated by similar learning curves and “platform” crossing frequency (Fig. 2c, d). To determine whether the above findings can be attributed to certain memory-independent effects associated with deletion of AMPKα isoforms such as vision, motivation and swimming ability, mice were tested on a visible platform task in which the escape platform is labelled with a visible cue and moved from trial to trial [33]. Performance of AMPKα1 cKO and α2 cKO mice did not differ from CRE+/− mice in latency to locate the platform (Fig. 2e). Taken together, suppression of AMPKα2 but not α1 isoform in brain leads to impairment of spatial learning and memory.
Fig. 2. Learning and memory are impaired in AMPKα2 cKO mice but unaffected in AMPKα1 cKO mice.

a, b Open field test (OF) showed normal travel distance and center duration in AMPKα1 cKO or AMPKα2 cKO mice, compared with CRE+/− control. n = 15 for Cre+/− and AMPKα2 cKO, n = 17 for AMPKα1 cKO. p > 0.05, one-way ANOVA. c Hidden platform Morris water maze (MWM) test showed longer escape latency (plotted against training days) in AMPKα2 cKO mice (filled circles), compared with Cre+/− (open squares) and AMPKα1 cKO mice (open circles). n = 24 for Cre+/−, n = 28 for AMPKα1 cKO, n = 23 for AMPKα2 cKO. On each individual day, one-way ANOVA, followed by Tukey’s post hoc test was applied. **p <0.01, ***p <0.001. Learning curves were compared with repetitive one-way ANOVA, ****p <0.001. d Probe trial of MWM test showed reduced “platform” crossing in AMPKα2 cKO mice compared with Cre+/− and AMPKα1 cKO mice. n = 24 for Cre+/−, n = 28 for AMPKα1 cKO, n = 23 for AMPKα2 cKO. **p < 0.01, ****p < 0.0001, one-way ANOVA, followed by Tukey’s post hoc test. e Visible platform test showed normal performance in AMPKα1 cKO and AMPKα2 cKO mice compared with CRE+/− controls. n = 23 for Cre+/−, n = 31 for AMPKα1 cKO, n = 16 for AMPKα2 cKO. f Novel object recognition test (NOR) showed that recognition of novel object over familiar object was normal in Cre+/− mice (left) and AMPKα1 cKO mice (middle), but impaired in AMPKα2 cKO mice (right). n = 10 for Cre+/−, n = 15 for AMPKα1 cKO, n = 12 for AMPKα2 cKO *p < 0.05, ****p < 0.0001, two-tailed independent Student t-test.
Next we tested the mice with the novel object recognition (NOR) task to assess long-term hippocampus-dependent recognition memory [29]. As expected, CRE+/− mice exhibited preference for the novel object over the familiar object in the test day (Day 2), as demonstrated by significant more exploration/interaction with the novel object (Fig. 2f, left). Similarly, AMPKα1 cKO mice also displayed a preference for the novel object in the test day (Fig. 2f, middle). In contrast, AMPKα2 cKO mice showed an inability to discriminate object novelty, as demonstrated by similar exploration/interaction with both novel and familiar objects (Fig. 2f, right). These findings are consistent with those from the MWM experiments (Fig. 2c, d), indicating impaired long-term recognition memory in AMPKα2 cKO mice.
We also assessed the impact of brain AMPKα isoform reduction on cognitive and memory flexibility by testing mice with a Y-water maze reversal task [27]. In brief, mice were trained to locate an escape platform in one arm of a water-based Y maze, and the escape platform was switched to the opposite arm after 24 h. AMPKα1 cKO and α2 cKO mice showed similar ability to learn the new escape location compared with CRE+/− littermates, indicating normal cognitive and memory flexibility (Fig. S2b–e).
Hippocampal L-LTP is impaired in AMPKα2 cKO mice but unaltered in AMPKα1 cKO mice
Next we investigated how reduction of the AMPKα isoform affects hippocampal LTP, one of the mostly intensively studied forms of synaptic plasticity and putative cellular mechanism for learning and memory [34, 35]. We conducted synaptic electrophysiology experiments to measure LTP at the CA3-CA1 synapses of acute hippocampal slices [36]. Consistent with the findings from behavioral tests, protein synthesis-dependent, late LTP (L-LTP) induced by a strong high-frequency stimulation (HFS) protocol was impaired in AMPKα2 cKO mice, but unaltered in AMPKα1 cKO mice, compared with L-LTP performance in CRE+/− mice (Fig. 3a–c). Interestingly, a previous study showed normal L-LTP in a global AMPKα2 KO mice [11]. A potential mechanism contributing to the different L-LTP performance is that global knockout of AMPKα2 subunit may induce compensated upregulation of the other subunit i.e. AMPKα1. Indeed, results from Western blot demonstrated significant increased levels of AMPKα1 in hippocampus of AMPKα2 KO mice (Fig. S3), in contrast to unaltered AMPKα1 expression in AMPKα2 conditional knockout mice (Fig. 1b). We also examined protein synthesis-independent, early-LTP (E-LTP) elicited by a weak HFS protocol [37]. Hippocampal E-LTP of AMPKα1 cKO or α2 cKO mice was indistinguishable from that of CRE+/− mice (Fig. 3d–f). Moreover, we measured paired-pulse facilitation (PPF), a form of calcium-dependent presynaptic plasticity evoked by two temporally linked stimuli at various intervals [38]. We observed no differences in PPF performance among AMPKα1 cKO, α2 cKO, or CRE+/− mice (Fig. 3g). We also investigated basal synaptic transmission function by eliciting synaptic responses with a range of stimulus intensities, and observed decreased synaptic input-output relationships in AMPKα2 cKO cKO mice, compared with CRE+/− mice (Fig. 3h). Taken together, hippocampal L-LTP is specifically impaired with genetic repression of the AMPKα2 isoform but not the a1 isoform; reduction of either isoform has no apparent effect on transient, protein synthesis-independent E-LTP.
Fig. 3. Hippocampal L-LTP is impaired in AMPKα2 cKO mice but unaltered in AMPKα1 cKO mice.

a L-LTP induced by two-train HFS was impaired in hippocampal slices from AMPKα2 cKO mice (filled circles, n = 8), compared with normal L-LTP expression in slices from either CRE+/− mice (open squares, n = 18) or AMPKα1 cKO mice (open circles, n = 14). b Representative traces of fEPSPs before and after HFS for L-LTP experiments shown in a. c Cumulative data showing measurement of mean fEPSP slopes 90 min after delivery of HFS based on L-LTP experiments in a. *p < 0.05, one-way ANOVA followed by Tukey’s post hoc test. d Hippocampal E-LTP induced by one-train HFS was unchanged in AMPKα1 cKO (open circles, n = 5) or AMPKα2 cKO mice (filled circles, n = 10) mice, compared with Cre+/− group (open squares, n = 10). e Representative traces of fEPSPs before and after HFS for E-LTP experiments shown in d. f Cumulative data showing measurement of mean fEPSP slopes 90 min after delivery of HFS based on E-LTP experiments in d. g Performance of paired-pulse facilitation (PPF) was unaltered in AMPKα1 cKO (open circles, n = 5) or AMPKα2 cKO (filled circles, n = 9) mice, compared with Cre+/− group (open squares, n = 16). h Basal synaptic transmission assessed by input-output relationships plotted as fiber volley amplitude vs. fEPSP slope in AMPKα1 cKO (open circles, n = 15) or AMPKα2 cKO (filled circles, n = 15) mice, compared with Cre+/− group (open squares, n = 16). *p < 0.05, **p < 0.01, ****p <0.0001, two-way ANOVA.
AMPKα2 cKO mice display abnormal hippocampal dendritic spine morphology and impaired postsynaptic density formation
Dendritic spines function as a storage site for synaptic strength. Regulation of spine morphology is an essential indicator of synaptic integrity and transmissions, and has been associated with memory formation and synaptic plasticity [31, 39]. Therefore we investigated the effects of brain AMPKα isoform deletion on dendritic spine morphology in hippocampus using the rapid Golgi staining protocol [30]. We assessed spine density in apical dendritic branches of hippocampus and spine morphology types based on published guidelines [30]. Compared with CRE+/− mice, overall dendritic spine density was unaltered in AMPKα1 cKO mice, but significantly decreased in AMPKα2 cKO mice (Fig. 4a, b). Further analysis of spine morphological classification revealed that density of “mature” spines (stubby, mushroom and branched) in AMPKα1 cKO mice was significantly higher than those in AMPKα2 cKO mice. Interestingly, there was also a trend of increase in mature spine density in AMPKα1 cKO mice (p = 0.08), compared with CRE+/− group (Fig. 4c, left). Moreover, density of immature spines (thin and filopodia) was significantly lower in AMPKα1 cKO and α2 cKO mice, compared with CRE+/− mice (Fig. 4c, right).
Fig. 4. AMPKα2 cKO mice display abnormal hippocampal dendritic spine morphology and impaired postsynaptic density formation.
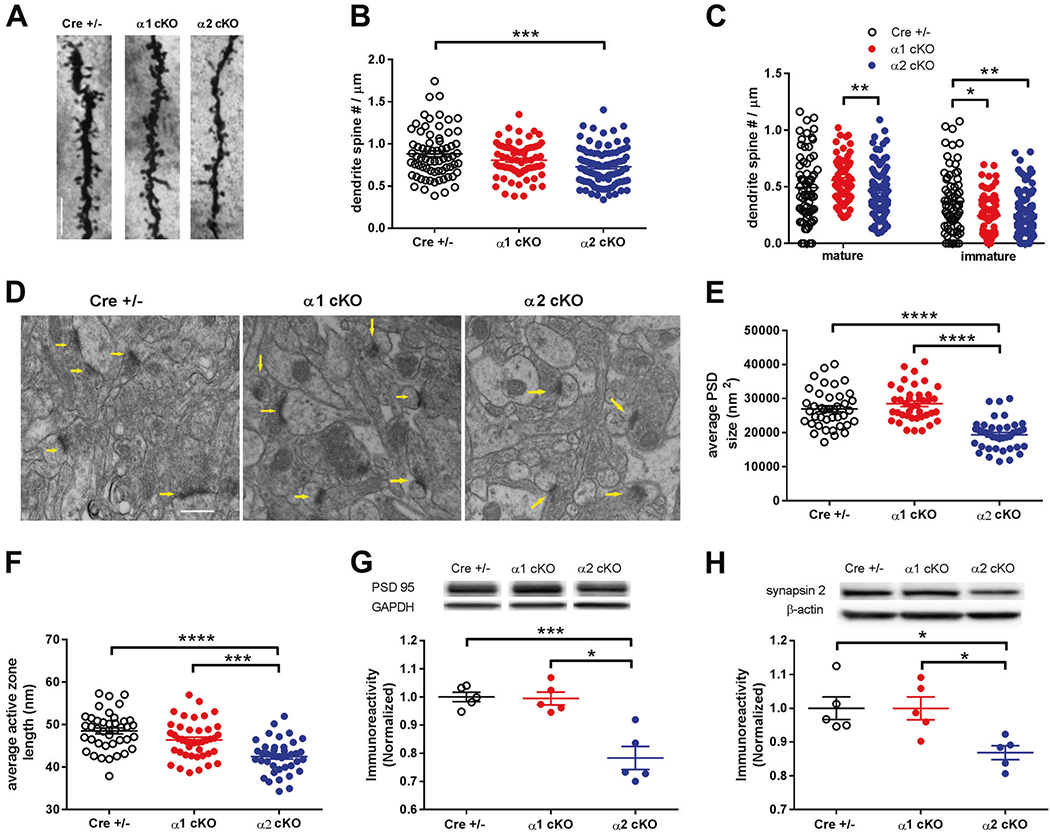
a Representative images of hippocampal CA1 apical dendrites and spines from AMPKα1/α2 cKO and Cre+/− mice visualized with Golgi-Cox staining. Scale bar: 10 μm. b Total spine counts on hippocampal dendrites (numbers per μm length) were decreased in AMPKα2 cKO mice but unaltered in AMPKα1 cKO mice, compared with control. For Cre+/−, n = 69 dendrites from 3 mice; for AMPKα1 cKO, n = 73 dendrites from 3 mice; and for AMPKα2 cKO, n = 99 dendrites from 4 mice; **p <0.01, one-way ANOVA followed by Tukey’s post hoc test. c Analysis of spine morphology showed that density of “mature” spines (stubby, mushroom, and branched) in AMPKα1 cKO mice was higher than those in AMPKα2 cKO mice (left). Density of immature spines (thin and filopodia) was significantly lower in either AMPKα1 cKO or α2 cKO mice, compared with CRE+/− group (right). For Cre+/−, n = 69 dendrites from 3 mice; for AMPKα1 cKO, n = 73 dendrites from 3 mice; and for AMPKα2 cKO n = 99 dendrites from 4 mice; *p <0.05, **p <0.01, one-way ANOVA followed by Tukey’s post hoc test. d Representative transmission electron microcopy (EM) images of hippocampal CA1 area of AMPKα1/α2 cKO and CRE+/− mice. Arrows indicate synapses. Scale bar: 100 nm. e Analysis and quantification of the EM data revealed decreased hippocampal postsynaptic density (PSD) size in AMPKα2 cKO mice, compared with CRE+/− or AMPKα1 cKO mice. n = 40 for Cre+/− and AMPKα2 cKO, n = 42 for AMPKα1 cKO; ****p < 0.0001, one-way ANOVA followed by Tukey’s post hoc test. f Analysis and quantification of the EM data showed that AMPKα2 cKO led to shorter length of synaptic active zone, compared with CRE+/− or AMPKα1 cKO mice. n = 40 for Cre+/− and AMPKα2 cKO, n = 42 for AMPKα1 cKO; ***p <0.001, ****p <0.0001, one-way ANOVA followed by Tukey’s post hoc test. g Western blot experiments showed that protein levels of PSD95 in hippocampus of AMPKα2 cKO mice were lower than those of CRE+/− or AMPKα1 cKO mice. Representative Western blot gels and cumulative data for quantification presented in bar graphs are shown (n = 5, *p <0.05, ***p < 0.001, one-way ANOVA followed by Tukey’s post hoc test). h Western blot experiments showing protein levels of synapsin 2 in hippocampus of AMPKα2 cKO mice were lower than those of CRE+/− or AMPKα1 cKO mice. Representative Western blot gels and accumulative data for quantification presented in bar graphs are shown (n = 5, *p <0.05, one-way ANOVA followed by Tukey’s post hoc test).
Using transmission electron microscopy (EM) techniques, we next investigated effects of AMPKα isoform reduction on postsynaptic density (PSD), which usually is located at the head of spine and critically involved in synaptic functions [31, 40]. Ultrastructural analysis revealed that reduction of the AMPKα2 isoform was associated with significantly reduced PSD density, compared with either CRE+/− or AMPKα1 cKO mice (Fig. 4d, e, S4a). In contrast, loss of the AMPKα1 isoform did not change PSD density, compared with CRE+/− mice (Fig. 4d, e, S4a). Further analysis of the EM data revealed that suppression of the AMPKα2 isoform led to decreased length of the active zone, which is the synaptic vesicle release site [41] (Fig. 4f). Western blotting experiments also demonstrated significantly decreased protein levels of PSD95 (a key component of PSD) and synapsin 2 (a presynaptic marker) in hippocampi of AMPKα2 cKO mice, compared with CRE+/− and AMPKα1 cKO mice (Fig. 4g, h). We also performed Western blot experiments to investigate regulation of proteins that have been implicated in synaptic plasticity and memory, including calcium/calmodulin-dependent protein kinase II (CAMKII), AMPA receptor subunit GluA1, protein kinase M zeta (PKMζ), and potassium channel Kv4.2 [42–44]. We observed no significant effects of AMPKα isoform reduction on levels of any of these proteins (Fig. S4b–e).
In summary, genetic suppression of the AMPKα2 isoform in brain is associated with reduced spine density, impaired spine morphology, decreased PSD area and active zone length in hippocampus.
Isoform-specific dysregulation of dendritic polyribosome assembly in AMPKα cKO mice
Polyribosomes (three or more ribosomes co-occurring) are commonly considered the principal sites of mRNA translation. The presence of polyribosomes in dendrites indicates new protein synthesis, and their regulation has been associated with LTP expression and memory formation [45, 46]. To understand the effects of suppressing the AMPKα isoform on de novo protein synthesis, we performed transmission EM experiments to detect and quantify polyribosomes in dendrites of area CA1 of hippocampus. Compared with CRE+/− mice, there were significantly fewer dendritic polyribosomes in AMPKα2 cKO mice. In contrast, repression of AMPKα1 isoform did not alter presence of polyribosomes (Fig. 5a, b). We further examined de novo protein synthesis by performing SUnSET experiments on acute hippocampal slices [11, 47]. Interestingly, no significant alterations were observed in AMPKα1/2 cKO mice compared with the control group (Fig. 5c)
Fig. 5. Isoform-specific regulation of protein synthesis in AMPKα cKO mice.
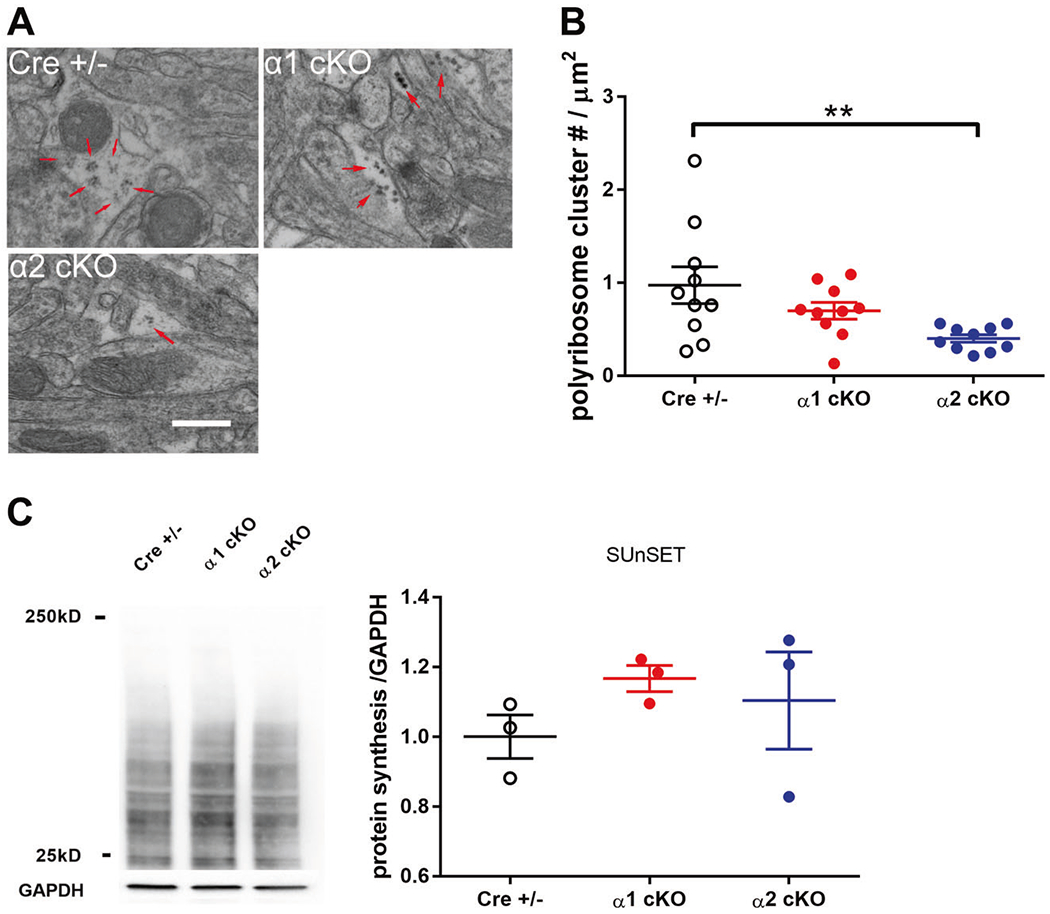
a Representative EM images of hippocampal CA1 area of AMPKα1/α2 cKO and CRE+/− mice. Polyribosomes were indicated with arrows. Scale bar: 500 nm. b Cumulative data showing that the overall polyribosome cluster number per μm2 dendritic area was lower in AMPKα2 cKO mice than CRE+/− mice. n = 3 mice; **p <0.01, one-way ANOVA followed by Tukey’s post hoc test. c Representative images and quantification from the SUnSET de novo protein synthesis assay. n = 3 mice. One-way ANOVA.
Increased phosphorylation of eIF2α and PERK in AMPKα2 cKO mice
To further elucidate the molecular mechanisms underlying AMPKα isoform-specific behavioral and LTP phenotypes, we investigated potential alterations of signaling pathways controlling de novo protein synthesis that could be associated with AMPKα1 or α2 repression. Previous studies indicate that inhibition of AMPK activity leads to activation of mTORC1 signaling, a central regulator of mRNA translation initiation that has an established role in memory formation and LTP expression [48, 49]. To our surprise, we did not detect any significant effects of AMPKα isoform suppression on mTORC1 signaling, as indicated by no change in the levels of mTOR phosphorylation (Fig. 6a) or mTORC1 activity measured by levels of phosphorylation of its two established downstream effectors: p70S6K and 4EBP1 (Fig. 6b, c). In agreement, Western blot experiments also showed no effects of AMPKα isoform reduction on activity (as assessed by phosphorylation levels) of AKT and GSK3β, two established upstream regulators of mTORC1 ((Fig. 6d, e). AMPK activation has been linked to inhibition (via phosphorylation) of eEF2, a translational factor critical for elongation of newly synthesized peptide and is involved in memory formation and synaptic plasticity [50]. However, results from Western blot showed no significant alterations of eEF2 phosphorylation in AMPKα1 cKO or α2 cKO mice (Fig. 6f).
Fig. 6. Increased phosphorylation of eIF2α and PERK in AMPKα2 cKO mice.
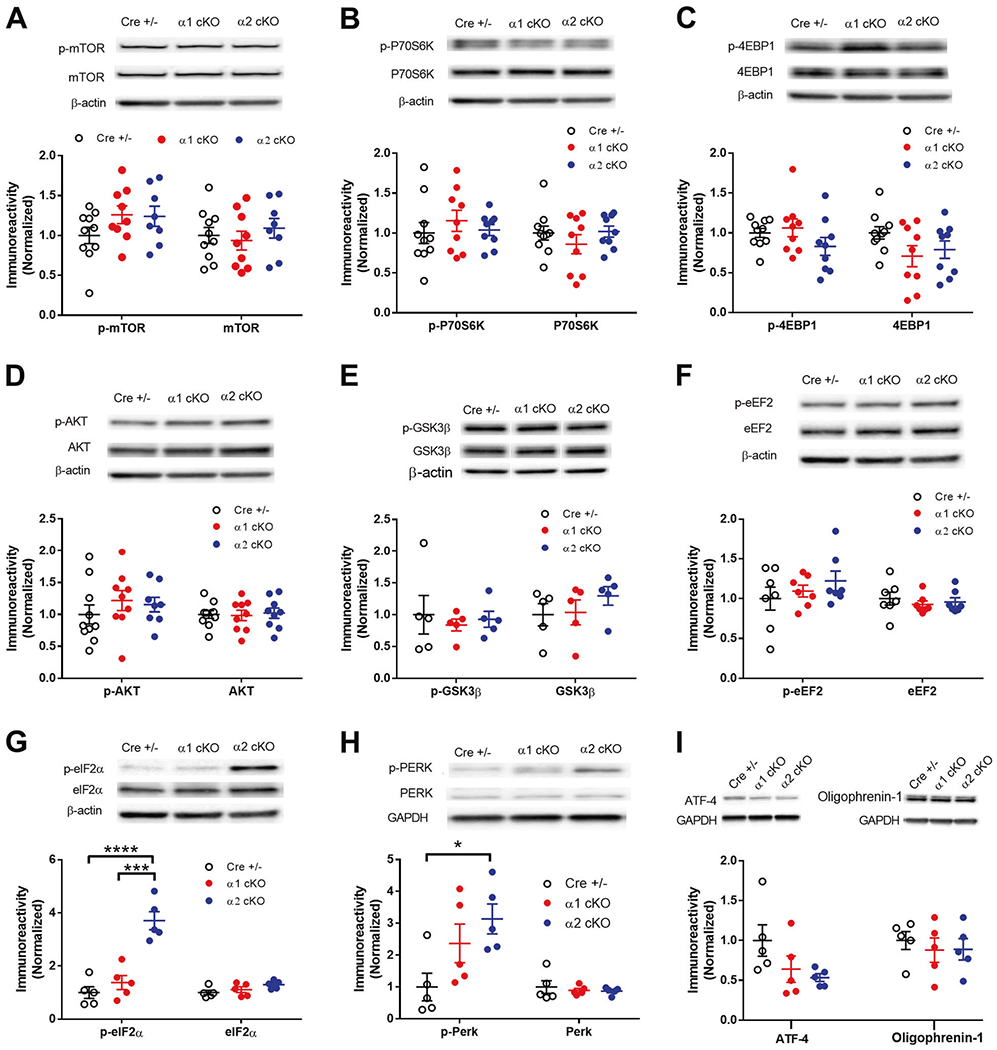
a–f Western blot experiments on hippocampus from AMPKα1 cKO or AMPKα2 cKO mice showed no change in phosphorylation levels of mTOR (Ser2448), p70S6K (Thr389), 4EBP1 (Thr37/46), AKT (Ser473), GSK3β (Ser9), and eEF2 (Thr56). Representative Western blot gels and cumulative data for quantification presented in bar graphs are shown (n = 5, p >0.05, one-way ANOVA). g Levels of phospho-eIF2α (Ser51) were increased in hippocampus of AMPKα2 cKO mice, compared with AMPKα1 cKO or CRE+/− mice. Representative Western blot gels and cumulative data for quantification presented in bar graphs are shown. (n = 5, ***p < 0.001, ****p < 0.0001, one-way ANOVA followed by Tukey’s post hoc test). h Levels of phospho-PERK (Thr980) were increased in hippocampus of AMPKα2 cKO mice, compared with CRE+/− mice. Representative Western blot gels and cumulative data for quantification presented in bar graphs are shown. (n = 5 mice per group, *p < 0.05, one-way ANOVA followed by Tukey’s post hoc test). i Levels of ATF-4 or Oligophrenin-1 were not significantly altered in AMPK α1/α2 cKO mice. (n = 5 per group, one-way ANOVA).
Another important translational mechanism for maintenance of memory and neuronal plasticity involves mRNA translational initiation factor 2 (eIF2) [51]. Phosphorylation of eIF2 α subunit (eIF2α) by one of its four known kinases, PKR, HRI, GCN2, and PERK, leads to general protein synthesis inhibition [23, 52]. Of note, recent studies link overactive PERK activity and consequently eIF2α hyperphosphorylation to memory loss and synaptic plasticity impairments associated with neurodegenerative diseases such as AD and frontotemporal dementia [24–26, 53]. To our surprise, the levels of eIF2α phosphorylation were markedly increased in hippocampus of AMPKα2 cKO mice, compared with CRE+/− controls (Fig. 6g). Meanwhile, hippocampal eIF2α phosphorylation was not altered in AMPKα1 cKO mice (Fig. 6g). In agreement, phosphorylation of PERK was significantly increased in AMPKα2 cKO mice (Fig. 6h). We also examined levels of ATF4 and oligophrenin, both proteins are potential downstream effectors of eIF2α phosphorylation [27, 54]. We did not find any significant changes of protein levels in hippocampus for either protein associated with AMPKα isoform suppression (Fig. 6i).
Suppression of PERK activity alleviates L-LTP failure and cognitive impairments in AMPKα2 cKO mice
We further investigated the relationship between PERK/eIF2α signaling dysregulation and impairments of synaptic plasticity and cognition in AMPKα2 cKO mice. First, we performed L-LTP experiments with GSK2606414 (1 μM), a selective PERK inhibitor that effectively reduces eIF2α phosphorylation [28]. Application of the PERK inhibitor significantly alleviated the hippocampal L-LTP impairments in AMPKα2 cKO mice (Fig. 7a, c). Moreover, the PERK inhibitor did not affect hippocampal L-LTP in CRE+/− mice (Fig. 7b, c). Next, we crossed AMPKα2 cKO mice to a transgenic mouse line in which PERK protein expression and eIF2α phosphorylation are selectively repressed in brain (PERK cKO) [27]. Four genotypes were studied: Cre+/−, PERK cKO, AMPKα2 cKO (a2 cKO), and PERK cKO/AMPKα2 cKO double mutant mice (PERK/α2). First we confirmed that increased phosphorylation of eIF2α in hippocampus of AMPKα2 cKO mice was blunted by brain-specific genetic reduction of PERK, demonstrated by decreased levels of p-eIF2α in hippocampus of the double mutant mice (PERK/α2) (Fig. 7d). No differences were observed in OF test; indicating normal general locomotor activity and anxiety level among the four groups (Fig. 7e). Mice were then subjected to the NOR assay to assess their cognitive performance. Consistent with findings described above, AMPKα2 cKO mice exhibited cognitive impairments, spending similar amount of time with novel and familiar objects (Fig. 7f). Notably, PERK/α2 mice showed normal cognition during NOR test, spending significantly more time with novel than with familiar objects (Fig. 7f). Worth mentioning is that PERK cKO mice displayed cognitive impairments in the NOR test (Fig. 7f), and the observation is consistent with a previous report [27]. In brief, these findings strongly suggest that eIF2α hyperphosphorylation caused by PERK could be a key molecular mechanism for synaptic failure and cognitive impairments associated with genetic reduction of the brain AMPKα2 isoform.
Fig. 7. Suppression of PERK activity alleviates L-LTP failure and cognitive impairments in AMPKα2 cKO mice.
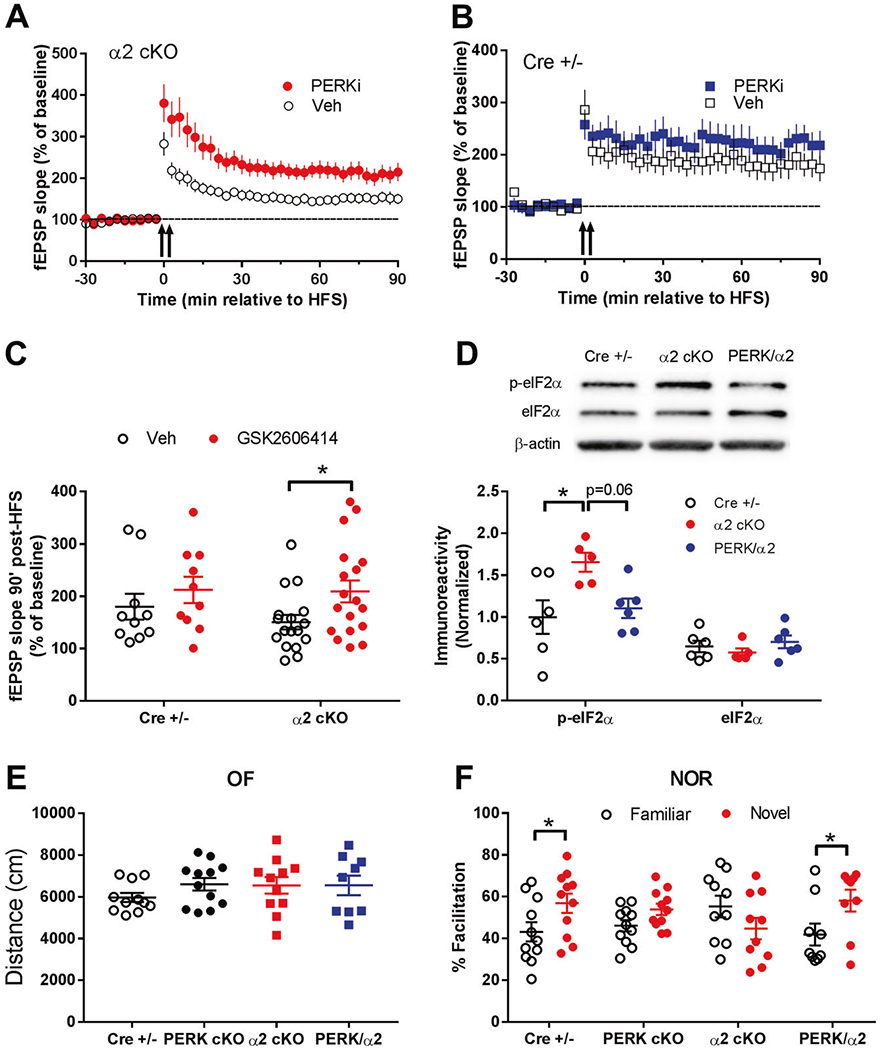
a Impairments of hippocampal L-LTP in AMPKα2 cKO mice (open circles, n = 17) were improved by treatment with the selective inhibitor of eIF2α kinase PERK (1 μM GSK2606414, filled circles, n = 18). b Hippocampal L-LTP in CRE+/− mice was not altered by the PERK inhibitor (filled squares, n = 10), compared with vehicle-treated controls (open squares, n = 10). c Cumulative data showing measurement of mean fEPSP slopes 90 min after delivery of HFS based on L-LTP experiments in h and i. *p <0.05, two-tailed unpaired Student’s t-test. d Increased hippocampal levels of phospho-eIF2α (Ser51) in AMPKα2 cKO (α2 cKO) mice were blunted in PERK cKO/AMPKα2 cKO double mutant mice (PERK/α2). Representative Western blot gels and cumulative data for quantification presented in bar graphs are shown. (n = 11 for Cre+/− and α2 cKO, n = 9 for PERK/α2, *p < 0.05, one-way ANOVA followed by Tukey’s post hoc test). e Open field test (OF) showed similar performance (travel distance) between Cre+/−, PERK cKO, AMPKα2 cKO (α2 cKO), and PERK cKO/AMPKα2 cKO double mutant mice (PERK/α2). p > 0.05, one-way ANOVA. f Novel object recognition test (NOR) showed that recognition of novel object over familiar object was normal in Cre+/− mice but impaired in PERK cKO and AMPKα2 cKO mice. PERK cKO/AMPKα2 cKO double mutant mice (PERK/α2) showed normal recognition memory. n = 11 for Cre+/−, n = 12 for PERK cKO, n = 11 for α2 cKO, n = 9 for PERK/α2 *p <0.05, two-tailed independent Student t-test.
Discussion
Advances in understanding of cellular/molecular mechanisms regulating neural plasticity and memory formation could yield insights into the pathophysiology of cognitive syndromes. AMPK is a key metabolic sensor and effector at the molecular level to maintain cellular energy homeostasis, disruption of which has been linked to neuronal diseases with cognitive impairments [4, 11, 55, 56]. In the current study, we took advantage of the Cre-lox system to achieve spatially and temporally restricted deletion of the genes encoding the two isoforms of the AMPK catalytic subunit (AMPKα1 and α2). Our study reveals a previously unrecognized isoform-specific role of AMPK in the central nervous system, demonstrating the importance of the AMPKα2 isoform in long-lasting forms of learning, memory, and LTP.
What molecular mechanisms could be responsible for these isoform-specific findings from the behavioral and LTP studies? In addition to isoform-specific effects on neuronal structures including spine morphology and PSD status as revealed by our data, it is likely that AMPKα isoforms are differentially recruited for neuronal signaling pathways and thus play distinct role in key biological processes underlying cognition and synaptic plasticity. AMPK functions as a “hub” to incorporate multiple signaling pathways including the mTORC1 and eEF2K/eEF2 signaling cascades. Through regulation of mRNA translation initiation and elongation, respectively, mTORC1 and eEF2K/eEF2 signaling controls de novo protein synthesis, which is indispensable for maintenance of long-lasting forms of memory and synaptic plasticity [20, 21]. Nevertheless, we did not observe any significant alterations in mTORC1 or eEF2K/eEF2 signaling pathways in either AMPKα1 cKO or α2 cKO mice, indicating alternative mechanism(s) involved. To our surprise, reduction of AMPKα2, but not AMPKα1, lead to hyper-phosphorylation of mRNA translational factor eIF2α and its kinase PERK. Increased phosphorylation of eIF2α by PERK is associated with inhibition of general protein synthesis [26], and integral protein synthesis is critical for long-term memory and synaptic plasticity. Importantly, L-LTP failure and cognitive impairments associated with AMPKα2 reduction were significantly improved by suppressing PERK activity pharmacologically (with a selective antagonist) or genetically (brain-specific PERK reduction in AMPKα2 cKO mice), providing evidence that eIF2α hyper-phosphorylation (by PERK) and consequently protein synthesis dysregulation might be the molecular underpinnings for cognition impairments and synaptic failure in AMPKα2 cKO mice. It is worth mentioning that a previous study showed that PERK cKO mice display preference/preservation for familiar object during the NOR test [27], a behavioral phenotype not observed in our current study (Fig. 7f). A couple differences between the two studies may explain such “inconsistent” results. First, in the study by Trinh et al., only male mice were used, while mice of both sex were used in our study. Second, the age of mice is 2–5 month in the Trinh paper, compared with 3–6 months in our study. It would be an interesting research topic in the future to investigate how these factors such as sex and age can affect behavioral phenotypes in the PERK cKO mice and other mutant mice. Moreover, while PERK is known as a kinase for eIF2α, it may affect cognition through other substrates such as CREB, calcineurin, and Gq protein-coupled intracellular Ca2+ alterations [57–59]. Previous studies show that PERK and eIF2α phosphorylation may mediate induction of metabotropic glutamate receptor (mGluR)-dependent long-term depression (LTD), another form of synaptic plasticity that is linked to cognitive function [37, 54]. In brief, the exact roles of PERK/eIF2α phosphorylation in synaptic and behavioral phenotypes associated with AMPKα isoform mutant mice could be complex and would require future studies to elucidate.
Interestingly, while reduction of AMPKα2 results in decreased levels of dendritic polyribosomes, overall de novo protein synthesis assessed by SUnSET is not altered (Fig. 5). A fraction of polyribosomes in neuronal dendrites are known to be “stalled polyribosomes” that are associated with repression of mRNA translation [60, 61], and this may contribute to the seemingly inconsistent findings between polyribosome counts and protein synthesis measurement. Furthermore, a most recent study indicates previously unrecognized roles of monosomes (as compared with polyribosomes) in mediating local protein synthesis [62, 63], and we did not measure monosomes in the current study.
How AMPK signaling is connected to eIF2α phosphorylation remains unclear. It was reported in non-neuronal systems that AMPK activation leads to inhibition of endoplasmic reticulum (ER) stress [64, 65]. ER stress often is associated with accumulation of misfolded proteins in the ER and accompanying unfolded protein response (UPR). PERK activation represents a crucial component of the UPR, resulting in eIF2α phosphorylation and global protein synthesis inhibition, a short-term protective strategy for cells to conserve energy and cope with cellular stress under physiological and pathophysiological conditions [23, 53, 66]. Therefore, it is possible that repression of AMPKα2 increases ER stress, which in turn activates PERK and phosphorylation of eIF2α. Notably, we did not observe (in hippocampus of AMPKα2 cKO mice) significant alteration of general protein synthesis measured by SUnSET (Fig. 5). Expression of UPR-related proteins such as ATF4 was not affected either with suppression of AMPKα2 (Fig. 6i). One potential explanation for such findings is that the AMPKα2 conditional knockout strategy in our study leads to partial but not full activation of the UPR-PERK-eIF2α axis. To the best of our knowledge, it is unknown whether there exists an isoform-specific role of AMPK in ER stress. Meanwhile, our data demonstrate that eIF2α phosphorylation is unchanged in AMPKα1 cKO mice, suggesting that the AMPKα1 isoform might not be involved in brain UPR induced by ER stress. Future studies are warranted to elucidate the detailed mechanisms involved in isoform-specific roles of AMPKα in ER stress/UPR in the central nervous system. Further, multiple recent studies indicate that over-activation of PERK and resulting eIF2α phosphorylation may represent key molecular mechanisms underlying cognitive impairments in neurodegenerative diseases, including prion disease and AD [25, 53, 67]. These findings are in line with our observations that the homeostasis of AMPKα isoform expression is disrupted in AD [68], and thus offer insights into etiology of neurological diseases with dementia syndromes.
In conclusion, findings from the current study not only help contribute to our understanding of the basic cellular and molecular mechanisms for physiological learning and memory, but also provide insights into pathophysiology of neurological diseases with cognitive syndromes, thus pointing toward potential new therapeutic avenues for treating human cognitive syndromes in neuronal diseases, in which homeostasis of brain AMPKα isoforms is disrupted.
Supplementary Material
Acknowledgements
We thank Dr Eric Klann of the New York University for providing the original breeders for the PERK cKO mice. We thank Mr Kenneth Grant for technical help on EM imaging. We thank Karen Klein, MA, in the Wake Forest Clinical and Translational Science Institute (UL1 TR001420) for editing the manuscript. This work was supported by National Institutes of Health grants K99/R00 AG044469, R01 AG055581, R01 AG056622 (TM), F31AG055264 (HRZ), the Alzheimer’s Association grant NIRG-15-362799 (TM), the BrightFocus Foundation grant A2017457S (TM), Wake Forest Alzheimer’s Disease Core Center (ADCC) pilot grant (TM), and a Wake Forest Clinical and Translational Science Institute (CTSI) pilot grant (TM). The mass spectrometry analysis was performed by the Proteomics and Metabolomics Shared Resource at Wake Forest School of Medicine, which is partly supported by the Comprehensive Cancer Support Grant (P30CA012197).
Footnotes
Supplementary information The online version of this article (https://doi.org/10.1038/s41380-020-0739-z) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardie DG. AMPK-sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–78. [DOI] [PubMed] [Google Scholar]
- 4.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, et al. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardie DG. The AMP-activated protein kinase pathway-new players upstream and downstream. J Cell Sci. 2004;117:5479–87. [DOI] [PubMed] [Google Scholar]
- 6.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–9. [DOI] [PubMed] [Google Scholar]
- 7.Magistretti P J, Allaman I A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883–901. [DOI] [PubMed] [Google Scholar]
- 8.Potter WB, O’Riordan KJ, Barnett D, Osting SMK, Wagoner M, Burger C, et al. Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS ONE. 2010;5:e8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma T, Tzavaras N, Tsokas P, Landau EM, Blitzer RD. Synaptic stimulation of mTOR is mediated by Wnt signaling and regulation of glycogen synthetase kinase-3. J Neurosci. 2011;31:17537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malenka RC. The long-term potential of LTP. Nat Rev Neurosci. 2003;4:923–6. [DOI] [PubMed] [Google Scholar]
- 11.Ma T, Chen Y, Vingtdeux V, Zhao H, Viollet B, Marambaud P, et al. Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid β. J Neurosci. 2014;34:12230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD. Neuro-protective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke. 2007;38:2992–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salminen A, Kaarniranta K, Haapasalo A, Soininen H, Hiltunen M. AMP-activated protein kinase: a potential player in Alzheimer’s disease. J Neurochem. 2011;118:460–74. [DOI] [PubMed] [Google Scholar]
- 14.Michell BJ, Stapleton D, Mitchelhill KI, House CM, Katsis F, Witters LA, et al. Isoform-specific purification and substrate specificity of the 5’-AMP-activated protein kinase. J Biol Chem. 1996;271:28445–50. [DOI] [PubMed] [Google Scholar]
- 15.Woods A, Salt I, Scott J, Hardie DG, Carling D. The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett. 1996;397:347–51. [DOI] [PubMed] [Google Scholar]
- 16.Viollet B, Andreelli F, Jørgensen SB, Perrin C, Geloen A, Flamez D, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jørgensen SB, Viollet B, Andreelli F, Frøsig C, Birk JB, Schjerling P, et al. Knockout of the alpha2 but not alpha1 5’-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–9. [DOI] [PubMed] [Google Scholar]
- 18.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–16. [DOI] [PubMed] [Google Scholar]
- 19.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004;5:931–42. [DOI] [PubMed] [Google Scholar]
- 21.Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alberini C. The role of protein synthesis during the labile phases of memory: revisiting the skepticism. Neurobiol Learn Mem. 2008;89:234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wek RC, Jiang H-Y, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34(Pt 1):7–11. [DOI] [PubMed] [Google Scholar]
- 24.Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. 2012;485:507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015;130:633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, et al. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci. 2013;16:1299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR, Klann E. Brain-specific disruption of the eIF2α kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep. 2012;1:676–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W, Zhou X, Cavener HZD, Klann E, Ma T. Repression of the eIF2α kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2016;41:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, et al. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Risher WC, Ustunkaya T, Alvarado JS, Eroglu C. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS ONE. 2014;9:e107591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–8. [DOI] [PubMed] [Google Scholar]
- 32.Beckelman BC, Yang W, Kasica NP, Zimmermann HR, Zhou X, Keene CD, et al. Genetic reduction of eEF2 kinase alleviates pathophysiology in Alzheimer’s disease model mice. J Clin Invest. 2019;129:820–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma T, Du X, Pick JE, Sui G, Brownlee M, Klann E. Glucagon-like peptide-1 cleavage product GLP-1 (9-36) amide rescues synaptic plasticity and memory deficits in alzheimer’s disease model mice. J Neurosci. 2012;32:13701–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. [DOI] [PubMed] [Google Scholar]
- 35.Malenka RC, Nicoll RA. Long-term potentiation–a decade of progress? Science. 1999;285:1870–4. [DOI] [PubMed] [Google Scholar]
- 36.Ma T, Hoeffer CA, Capetillo-Zarate E, Yu F, Wong H, Lin MT, et al. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS ONE. 2010;5:e12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trinh MA, Ma T, Kaphzan H, Bhattacharya A, Antion MD, Cavener DR, et al. The eIF2α kinase PERK limits the expression of hippocampal metabotropic glutamate receptor-dependent long-term depression. Learn Mem. 2014;21:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sala C, Segal M. Dendritic spines: the locus of structural and functional plasticity. Physiol Rev. 2014;94:141–88. [DOI] [PubMed] [Google Scholar]
- 40.Okabe S. Molecular anatomy of the postsynaptic density. Mol Cell Neurosci. 2007;34:503–18. [DOI] [PubMed] [Google Scholar]
- 41.Clarke GL, Chen J, Nishimune H. Presynaptic active zone density during development and synaptic plasticity. Front Mol Neurosci. 2012;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sacktor TC. How does PKMζ maintain long-term memory? Nat Rev Neurosci. 2011;12:9–15. [DOI] [PubMed] [Google Scholar]
- 43.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–90. [DOI] [PubMed] [Google Scholar]
- 44.Chen X, Yuan L-L, Zhao C, Birnbaum SG, Frick A, Jung WE, et al. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–45. [DOI] [PubMed] [Google Scholar]
- 46.Ostroff LE, Botsford B, Gindina S, Cowansage KK, LeDoux JE, Klann E, et al. Accumulation of polyribosomes in dendritic spine heads, but not bases and necks, during memory consolidation depends on cap-dependent translation initiation. J Neurosci. 2017;37:1862–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–7. [DOI] [PubMed] [Google Scholar]
- 48.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graber TE, McCamphill PK, Sossin WS. A recollection of mTOR signaling in learning and memory. Learn Mem. 2013;20:518–30. [DOI] [PubMed] [Google Scholar]
- 50.Taha E, Gildish I, Gal-Ben-Ari S, Rosenblum K. The role of eEF2 pathway in learning and synaptic plasticity. Neurobiol Learn Mem. 2013;105:100–6. [DOI] [PubMed] [Google Scholar]
- 51.Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, et al. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007;129:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trinh MA, Klann E. Translational control by eIF2α kinases in long-lasting synaptic plasticity and long-term memory. Neurobiol Learn Mem. 2013;105:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma T, Klann E. PERK: a novel therapeutic target for neurodegenerative diseases? Alzheimers Res Ther. 2014;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prisco GVD, Huang W, Buffington SA, Hsu C-C, Bonnen PE, Placzek AN, et al. Translational control of mGluR-dependent long-term depression and object-place learning by eIF2α. Nat Neurosci. 2014;17:1073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marinangeli C, Didier S, Vingtdeux V. AMPK in neurodegenerative diseases: implications and therapeutic perspectives. Curr Drug Targets. 2016;17:890–907. [DOI] [PubMed] [Google Scholar]
- 56.Vingtdeux Vr, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011;121:337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sen T, Gupta R, Kaiser H, Sen N. Activation of PERK elicits memory impairment through inactivation of CREB and down-regulation of PSD95 after traumatic brain injury. J Neurosci. 2017;37:5900–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu S, McGrath BC, Bai Y, Tang X, Cavener DR. PERK regulates Gq protein-coupled intracellular Ca2+ dynamics in primary cortical neurons. Mol Brain. 2016;9:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang R, McGrath BC, Kopp RF, Roe MW, Tang X, Chen G, et al. Insulin secretion and Ca2+ dynamics in β-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. J Biol Chem. 2013;288:33824–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Graber TE, Hébert-Seropian S, Khoutorsky A, David A, Yewdell JW, Lacaille J-C, et al. Reactivation of stalled polyribosomes in synaptic plasticity. Proc Natl Acad Sci USA. 2013;110:16205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sossin WS, DesGroseillers L. Intracellular trafficking of RNA in neurons. Traffic. 2006;7:1581–9. [DOI] [PubMed] [Google Scholar]
- 62.Rangaraju V, Dieck S, Schuman EM. Local translation in neuronal compartments: how local is local? EMBO Rep. 2017;18:693–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.E.Heyer E, J.Moore M. Redefining the translational status of 80S monosomes. Cell. 2016;164:757–69. [DOI] [PubMed] [Google Scholar]
- 64.Kim H, Moon SY, Kim J-S, Baek CH, Kim M, Min JY, et al. Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am J Physiol Ren Physiol. 2015;308:F226–236. [DOI] [PubMed] [Google Scholar]
- 65.Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paschen W, Proud CG, Mies G. Shut-down of translation, a global neuronal stress response: mechanisms and pathological relevance. Curr Pharm Des. 2007;13:1887–902. [DOI] [PubMed] [Google Scholar]
- 67.Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013;5:206ra138. [DOI] [PubMed] [Google Scholar]
- 68.Zimmermann HR, Yang W, Kasica NP, Zhou X, Wang X, Beckelman BC, et al. Brain-specific repression of AMPKα1 alleviates pathophysiology in Alzheimer’s model mice. J Clin Invest. 2020E. 10.1172/JCI133982 [pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.