

Abstract
N4-acetylcytidine (ac4C) is a highly conserved modified RNA nucleobase whose formation is catalyzed by the disease-associated N-acetyltransferase 10 (NAT10). Here we report a sensitive chemical method to localize ac4C in RNA. Specifically, we characterize the susceptibility of ac4C to borohydride-based reduction and show this reaction can cause introduction of noncognate base pairs during reverse transcription (RT). Combining borohydride-dependent misincorporation with ac4C’s known base-sensitivity provides a unique chemical signature for this modified nucleobase. We show this unique reactivity can be used to quantitatively analyze cellular RNA acetylation, study adapters responsible for ac4C targeting, and probe the timing of RNA acetylation during ribosome biogenesis. Overall, our studies provide a chemical foundation for defining an expanding landscape of cytidine acetyltransferase activity and its impact on biology and disease.
The N4-acetylcytidine (ac4C) is a highly conserved RNA nucleobase whose formation is catalyzed by N-acetyltransferase 10 (NAT10), an acetyltransferase implicated in cancer and premature aging syndromes.1,2 Studies in human cell lines and yeast indicate that NAT10 and its homologues regulate ribosome biogenesis as well as tRNA stability.3,4 However, studying the mechanistic role of RNA acetylation in these processes remains challenging due to limited methods for site-specific ac4C analysis. To date, the only method that has been used to localize ac4C to specific sites in cellular RNA is mung bean nuclease cleavage coupled to UV-HPLC analysis.4,5 While this method has advanced the field, it suffers from poor sensitivity due to a lack of signal amplification and requires the synthesis of tiling oligonucleotides, limiting throughput. This contrasts with other highly conserved RNA modifications such as 2′-O-methylation (Nm) and pseudouridine (Ψ), for whom chemistries have been developed that allow site-specific analysis by primer extension and/or sequencing.6–8 Similar methods to analyze ac4C have the potential to greatly expand our understanding of RNA acetylation by facilitating the study of ac4C dynamics,9,10 defining how adapter molecules such as snoRNAs guide NAT10 to its RNA targets,4 and allowing the localization of ac4C in organisms where its presence has been reported, but specific sites are not known.11
In developing a chemical method to detect ac4C in RNA, we were inspired by early studies which observed that the electron-deficient nature of the ac4C pyrimidine renders it susceptible to addition of sodium borohydride across its 5,6-double bond.12,13 Addition of two hydride equivalents forms the reduced nucleotide N4-acetyl-3,4,5,6-tetrahydrocytidine (“reduced ac4C”; Figure 1). On the basis of analogy with other methods,6–8,14–16 we hypothesized the altered structure of reduced ac4C may cause pausing or misincorporation during reverse transcription (RT) of ac4C-containing RNA, generating a unique PCR-amplifiable chemical signature that could be used to detect the modified nucleotide.
Figure 1.
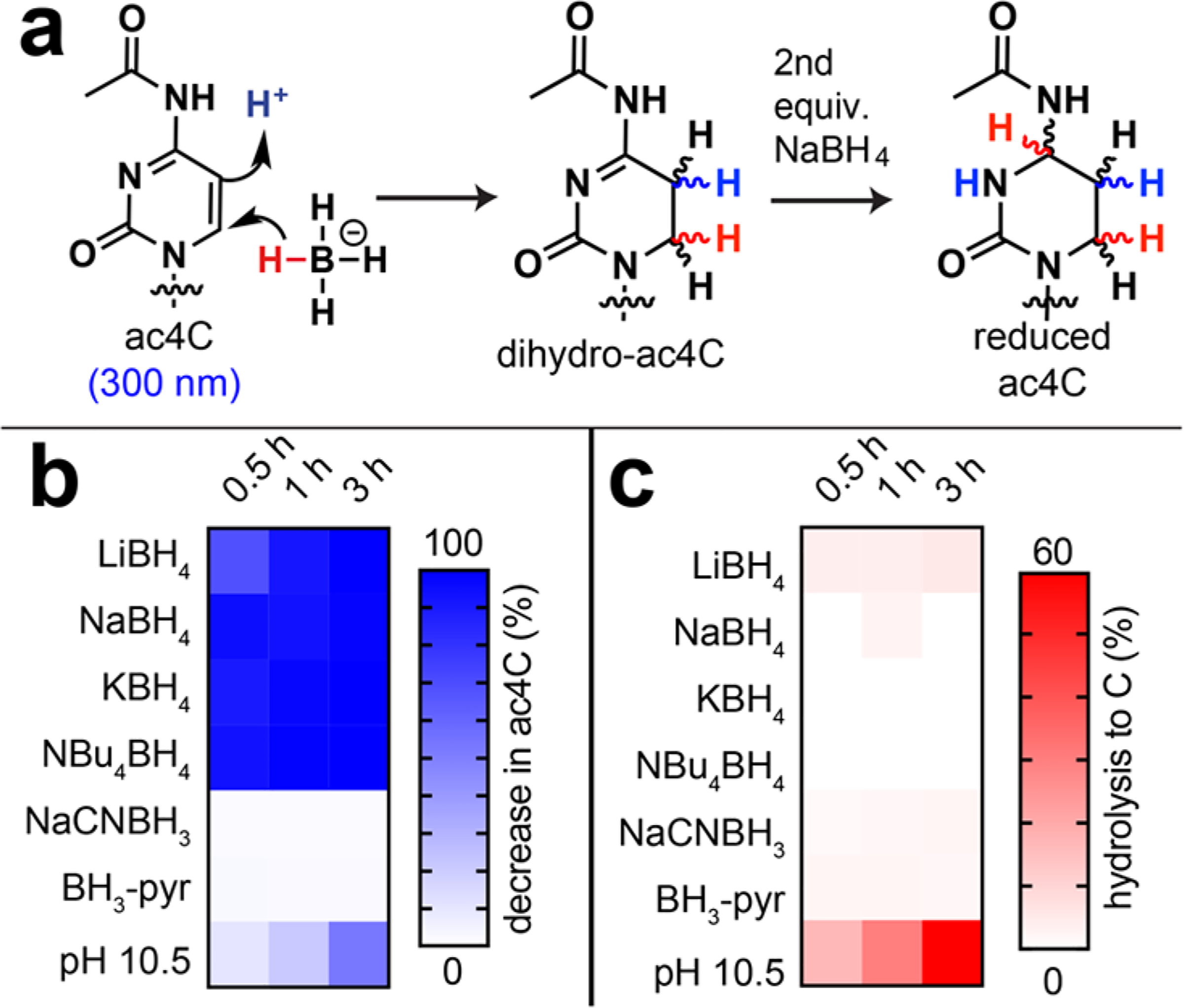
(a) Chemical reduction of N4-acetylcytidine to tetrahydro-N4-acetylcytidine. (b) Heat map of UV analysis testing different hydride donors for ablation of ac4C absorbance at 300 nm, indicative of reduction or deacetylation. (c) Heat map of UV analysis testing different hydride donors for production of C (280 nm) from ac4C, indicative of deacetylation. Reaction conditions: ac4C (0.1 mM), reductant (20 mM), H2O. Additional UV data is provided in Figure S1.
To explore this approach, we first compared the propensity of different hydride donors to stimulate reduction of ac4C versus chemical deacetylation, the latter which is known to occur in alkali solutions.17 Differentiating ac4C reduction from deacetylation is critical, as the latter erases the information content of the acetylated nucleobase and could limit its sensitive detection. To study these processes we used UV spectroscopy to analyze the ac4C nucleoside, monitoring reduction and deacetylation in parallel (Figure S1). Several hydride donors were identified that rapidly reduced ac4C (loss of 300 nm) but did not cause measurable cytidine formation (gain of 270 nm; Figure 1b,c). These studies indicate reduction occurs rapidly relative to deacetylation and establish an information-rich chemical transformation of ac4C.
Next, we assessed the reactivity of borohydride reagents with ac4C in RNA polynucleotides. For these studies synthetic RNA containing a single ac4C site was produced via in vitro transcription, and reduction of ac4C was analyzed using a recently reported anti-ac4C antibody (Figure 2a).17 Control experiments indicate the antibody does not interact with reduced ac4C, validating its ability to monitor this transformation (Figure S2). Consistent with studies of the free nucleoside, sodium borohydride caused time- and concentration-dependent reduction of ac4C RNAs (Figure 2b). LCMS analysis confirmed loss of ac4C from these RNAs (Figure S3). Importantly, ablation of ac4C was not accompanied by a concomitant increase in cytidine, consistent with preferential reduction versus deacetylation (Figure S3). Borohydride reactivity was observed in the presence of complementary oligonucleotides spanning the ac4C site, suggesting ac4C reduction can occur in duplex RNA (Figure S4a). Furthermore, borohydride-treated ac4C RNAs did not show increased cleavage in the presence of aniline (Figure S4b). This distinguishes ac4C from dihydrouridine, 3-methylcytidine, and 7-methylguanosine, which have each been shown to be susceptible to aniline-catalyzed strand scission following borohydride reduction.18
Figure 2.
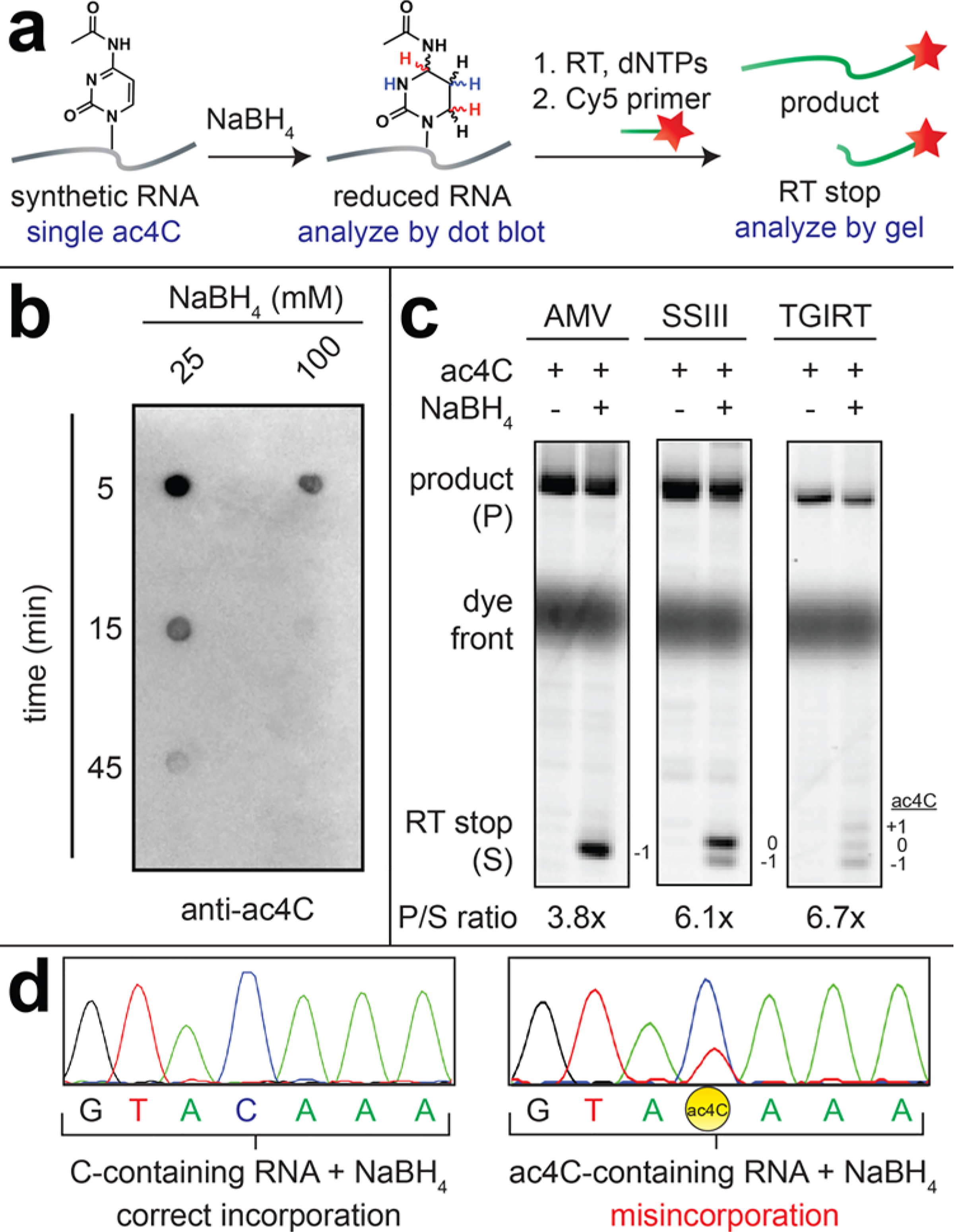
Chemical reduction of ac4C in polynucleotide RNA. (a) Schematic of synthetic ac4C/C-containing RNA. (b) Dot blot analysis of ac4C reduction in a synthetic RNA substrate (37 °C). (c) Primer extension analysis of ac4C-containing RNAs following NaBH4 treatment (100 mM, 37 °C, 1 h). (d) Sanger sequencing analysis of PCR-amplified cDNAs (sense strand) generated from ac4C RNAs following NaBH4 treatment (100 mM, 37 °C, 1 h) and TGIRT RT.
Having established the borohydride reactivity of ac4C in RNA, we next examined the ability of the reduced nucleobase to alter RT (Figure 2c). In theory, reduced ac4C could cause RT enzymes to stop, as occurs at sites of Ψ upon chemical derivatization, or increase incorporation of noncognate nucleotide triphosphates, causing mutations readily detectible during cDNA sequencing.6,19 To assess RT stop we tested the effect of borohydride on extension of a labeled (Cy5) primer during RT of our model ac4C RNA substrate. Since polymerases display different intrinsic sensitivities to modified nucleobases,15,20 we screened three RT enzymes for their ability to report on the presence of reduced ac4C: Avian Myeloblastosis Virus (AMV), Superscript III (SSIII; a derivative of MMLV RT), and thermostable group II intron (TGIRT). All three RT enzymes generated a mixture of full length and borohydride-dependent RT stop products when tested against the ac4C model RNA (Figure 2c). The relative abundance of full-length product (P) versus stop (S) followed the rough rank order TGIRT > Superscript ≫ AMV. RT stop primarily occurred −1, 0, and +1 relative to the position of the modified ac4C, with the specific distribution of stops highly dependent on the RT enzyme used. Of note, no RT stop was observed when the analogous cytidine-containing RNA was treated with borohydride (Figure S5a).
To further assess whether reduced ac4C caused misincorporation during RT, full length cDNA products from ac4C/borohydride reactions were PCR amplified, gel-extracted, and analyzed by Sanger sequencing. This revealed a borohydride-dependent mismatch in the ac4C-containing model RNA which was not observed in similarly treated cytidine-containing RNA (Figure 2d). The mutational signal was partial and manifested as incorporation of adenosine into the antisense cDNA during RT opposite the reduced ac4C. The mixture of G and A indicates a preference for incorporation of a purine nucleotides across from reduced ac4C in the template strand. The mutational bias of reduced ac4C was highest with TGIRT, consistent with previous reports of its application as a highly processive reverse transcriptase.21 It is important to note that our survey of RTs was not exhaustive, and screening of additional enzymes may further improve ac4C detection.20,22 Misincorporation was found to proceed to a slightly greater extent at lower dGTP concentrations (Figure S5c). To assess the influence of sequence context on this assay we compared four synthetic RNAs containing a single site of ac4C but distinguishable flanking sequences (Figure S5d). The presence of ac4C in these four RNAs caused comparable, but statistically distinct, mismatch signal (25–31%). This indicates flanking nucleobases may fine-tune the magnitude of mutational signature observed at ac4C sites, as has been observed for other modifications.23 These studies define a sensitive, RT-based method to detect ac4C in RNA.
Next, we sought to exploit this unique chemical reactivity for site-specific detection of endogenous cellular ac4C. Human 18S rRNA contains ac4C at two sites: helix 34 (C1337) and helix 45 (C1842) (Figure 3). Primers for RT of these sites were designed to overlap the two N6-dimethyladenosine (m26A) residues present in helix 45, which are known to disrupt RT.24 Borohydride-treatment of total RNA isolated from HeLa cells followed by TGIRT RT, PCR, and sequencing led to the reproducible observation of ~50% G → A mutations at each site, relative to <10% in each untreated control (Figure 3b,c, S6). As a further proof of ac4C at these sites, we preincubated HeLa RNA in alkali buffer (pH 10.5), which is known to stimulate chemical deacetylation.17 In contrast, misincorporation at ac4C flanking bases was not borohydride-dependent, and was characterized by a low signal to standard deviation ratio. Alkali pretreatment diminished the rate of borohydride-dependent mutations observed at C1337 and C1842, but not at adjacent sites (Figure 3b–e, S6). To define the quantitative aspect of this assay, we prepared different stoichiometries of acetylated and deacetylated cytidine (100:0, 80:20, 60:40, etc.) using RNA from wild-type cells (assumed 100% modified)5 and cells deficient in the U13 snoRNA responsible for C1842 acetylation (0% modified).4 Linear, stoichiometry-dependent misincorporation was observed at the known site of ac4C in human helix 45 (Figure 3d). An analogous experiment with the synthetic ac4C RNA demonstrated similar linearity and slope (Figure 3e). Together, these results suggest that borohydride-dependent misincorporation is well-suited for the quantitative analysis of individual ac4C sites, with the caveat that intrinsic differences in mismatch rate caused by sequence (Figure S5d) or structural effects may limit quantitative comparisons of distinct ac4C sites.22 Overall, these studies establish alkali-sensitive, borohydride-dependent misincorporation as a unique and quantitative chemical signature that can be used to study ac4C in RNA.
Figure 3.
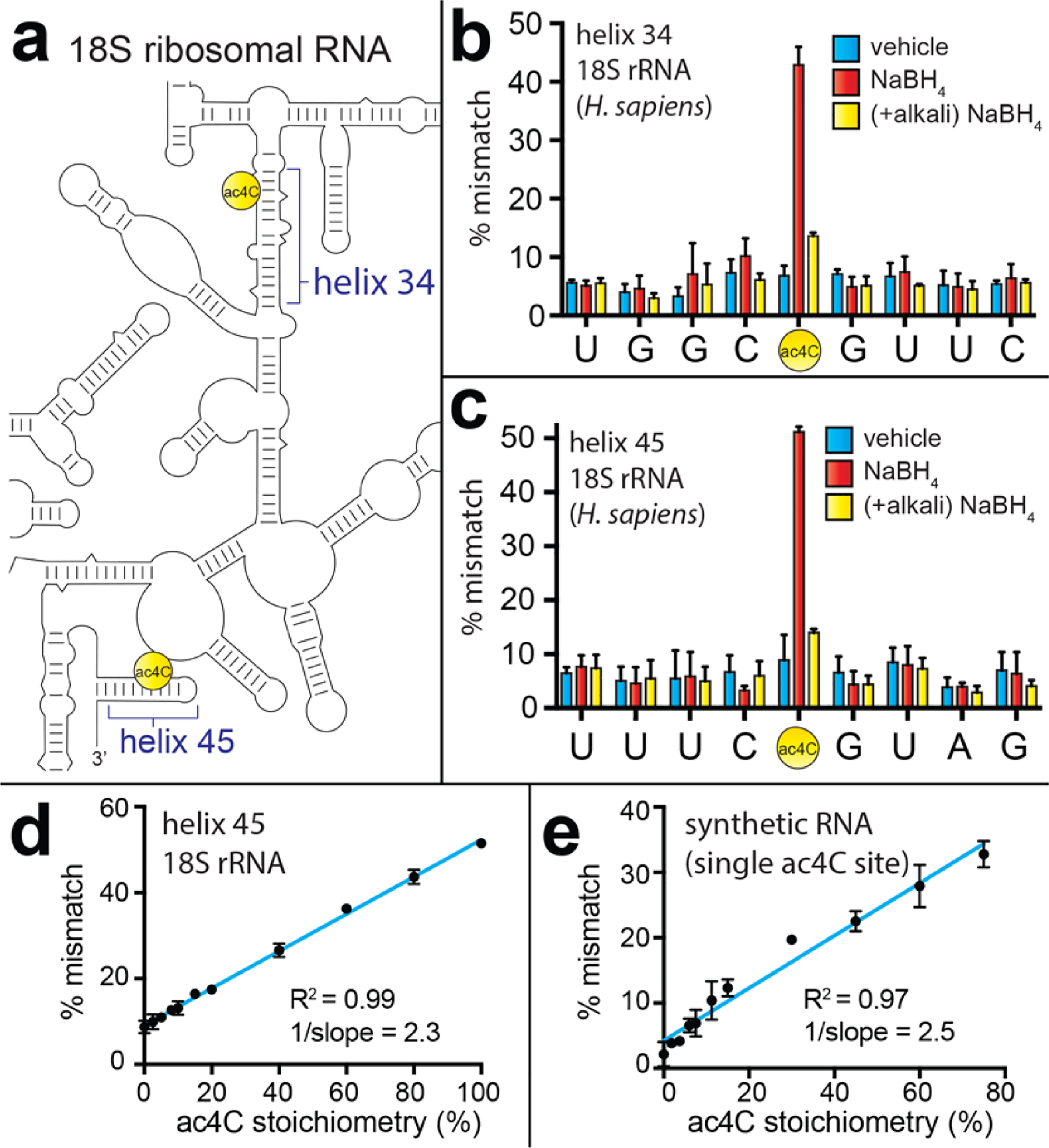
(a) Schematic of human rRNA ac4C sites. (b) Sequencing analysis of ac4C-dependent mismatches in cDNAs generated from human rRNA helix 34 and (c) helix 45 following borohydride treatment and RT. Sequence corresponds to the cDNA sense strand. Vehicle = water; NaBH4 = 100 mM sodium borohydride; (+alkali) NaBH4 = alkali pretreatment (100 mM NaCO3, pH 10, 60 °C, 1 h), precipitation, then borohydride. (d) Relationship between misincorporation signal and stoichiometry of ac4C in rRNA and (e) a synthetic RNA harboring ac4C in an “ACA” sequence context. For all data, error bars indicate the standard deviation (n = 3). Primary data in the form of full sequencing traces is provided in the Supporting Information.
Finally, we applied our method to understand the timing of cytidine acetylation in ribosome biogenesis. The apical site of ac4C in helix 45 of human rRNA occurs eight base pairs from two m26A sites (Figure 4a). Previous studies have shown extension of an RT primer specific for the 21S-C preribosomal (and its precursors) is halted by m26A, providing evidence that this methylation occurs in the nucleus.24 However, similar studies of ac4C have not been performed due to a lack of methods. To address this, we assayed for ac4C-dependent misincorporation using a 21S-C specific primer (Figure 4b). Our hypothesis was that if ac4C is installed in this species prior to m26A, it should allow read-through, and ac4C detection. Indeed, hydride reduction of total RNA followed by RT, PCR, and sequencing revealed clear misincorporation at C1842 using the 21S-C primer (Figure 4c). While the precise species upstream of 21S-C where ac4C is incorporated remains to be determined (Figure S6), this straightforward experiment indicates that installation of ac4C can (1) occur in the nucleus, and (2) occur prior to m26A dimethylation in at least a subset of pre-rRNAs. Also of note, this streamlined approach avoids fractionation, radioactivity, and requires several orders of magnitude less material (pg versus μg) than traditional HPLC methods for ac4C analysis.
Figure 4.
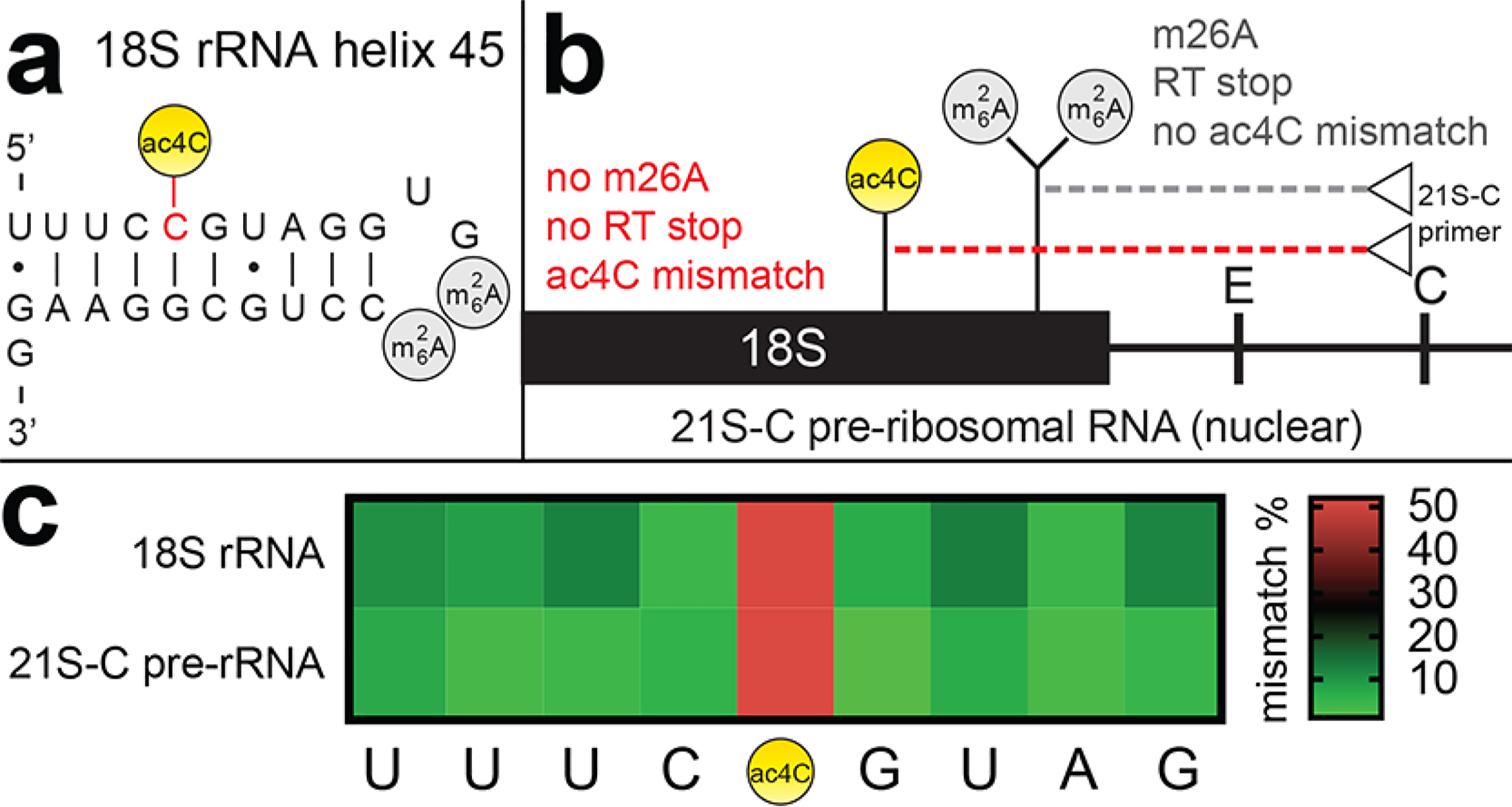
Applying borohydride-dependent misincorporation to study ac4C in rRNA biogenesis. (a) RNA modifications in human 18S helix 45. (b) RT of 21S-C pre-rRNA depends on relative timing of m26A and ac4C modifications. “C” and “E” represent sites at which the pre-rRNA is cleaved. Extension of the 21S-C primer, which forms cDNA from pre-rRNAs in which these cleavage sites are intact, may be impeded by m26A and reduced ac4C. (c) ac4C-dependent mismatch can be detected using an RT primer specific for the 21S-C pre-rRNA and its precursors.
In summary, here we have reported a chemical signature for cytidine acetylation in RNA. We find that ac4C can be efficiently reduced by borohydride-based reagents in model nucleosides, nucleotides, and endogenous rRNA substrates. While sodium borohydride is known to also react with other modified RNA nucleobases, ac4C is differentiated by its unique alkali sensitivity. Reduced ac4C interferes with RT and produces a unique mutational signature that was applied to quantitatively study snoRNA-dependent NAT10 rRNA acetylation. Applying this method to study pre-rRNAs provided evidence for the installation of ac4C in the nucleus, prior to adenine dimethylation of helix 45’s hairpin loop (Figure S6). Beyond the findings demonstrated here, in the future this method should enable multiple additional applications. First, the high sensitivity of this assay (enabled by PCR amplification) will be extremely useful for assessing ac4C dynamics in response to stimuli such as the metabolic state of the cell.9,10 Second, cytidine acetyltransferases are known to use adapter snoRNAs to modify their RNA substrates, some of whom remain uncharacterized.25 The ability to query ac4C sensitively and site-specifically will be an essential component in genetic screens aimed at identifying snoRNA adapters for cytidine acetyltransferases, and potentially also help define rules for enzymatic ac4C-targeting.26 Finally, this method may be useful for identifying putative sites of ac4C in coding RNAs, which have recently been proposed to exist in yeast.27 One limitation of our method as currently constituted is an inability to analyze ac4C in densely modified targets such as tRNAs, whose sequencing is challenged by the presence of nucleobases that disrupt Watson–Crick base-pairing and limit RT procession.28 In the future, we anticipate this challenge may be addressed by integrating borohydride-based reduction with recently reported ligation-based tRNA sequencing methods as well as by exploring more processive RT enzymes such as HIV and Marathon reverse transcriptase.22 Overall, our studies provide a chemical foundation for the site-specific analysis of ac4C dynamics in settings where pathogenic NAT10 activity has been identified, as well as novel explorations of how and where this RNA modification is targeted.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Cancer Institute, Center for Cancer Research (ZIA BC011488–04). This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06636.
Gels, full sequencing traces, supporting materials, and methods (PDF)
REFERENCES
- (1).Larrieu D; Britton S; Demir M; Rodriguez R; Jackson SP Science 2014, 344 (6183), 527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tschida BR.; Temiz NA.; Kuka TP.; Lee LA.; Riordan JD.; Tierrablanca CA.; Hullsiek R.; Wagner S.; Hudson WA.; Linden MA.; Amin K.; Beckmann PJ.; Heuer RA.; Sarver AL.; Yang JD.; Roberts LR.; Nadeau JH.; Dupuy AJ.; Keng VW.; Largaespada DA. Cancer Res. 2017, 77 (23), 6576–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Johansson MJ; Bystrom AS RNA 2004, 10 (4), 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sharma S; Langhendries JL; Watzinger P; Kotter P; Entian KD; Lafontaine DL Nucleic Acids Res. 2015, 43 (4), 2242–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sharma S; Marchand V; Motorin Y; Lafontaine DL J. Sci. Rep 2017, 7 (1), 11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Motorin Y; Muller S; Behm-Ansmant I; Branlant C Methods Enzymol. 2007, 425, 21–53. [DOI] [PubMed] [Google Scholar]
- (7).Lei Z; Yi C Angew. Chem., Int. Ed 2017, 56 (47), 14878–14882. [DOI] [PubMed] [Google Scholar]
- (8).Birkedal U; Christensen-Dalsgaard M; Krogh N; Sabarinathan R; Gorodkin J; Nielsen H Angew. Chem., Int. Ed 2014, 54 (2), 451–455. [DOI] [PubMed] [Google Scholar]
- (9).Montgomery DC; Garlick JM; Kulkarni RA; Kennedy S; Allali-Hassani A; Kuo YM; Andrews AJ; Wu H; Vedadi M; Meier JL J. Am. Chem. Soc 2016, 138 (20), 6388–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sun C; Jora M; Solivio B; Limbach PA; Addepalli B ACS Chem. Biol 2018, 13 (3), 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Noon KR; Bruenger E; McCloskey JA J. Bacteriol 1998, 180 (11), 2883–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cerutti P; Miller NJ Mol. Biol 1967, 26 (1), 55–66. [DOI] [PubMed] [Google Scholar]
- (13).Miller N; Cerutti PA J. Am. Chem. Soc 1967, 89 (11), 2767–2768. [Google Scholar]
- (14).Hong T; Yuan Y; Chen Z; Xi K; Wang T; Xie Y; He Z; Su H; Zhou Y; Tan ZJ; Weng X; Zhou XJ Am. Chem. Soc 2018, 140 (18), 5886–5889. [DOI] [PubMed] [Google Scholar]
- (15).Harcourt EM; Ehrenschwender T; Batista PJ; Chang HY; Kool ET J. Am. Chem. Soc 2013, 135 (51), 19079–19082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shu X; Dai Q; Wu T; Bothwell IR; Yue YA; Zhang ZZ; Cao J; Fei QL; Luo MK; He C; Liu JZ J. Am. Chem. Soc 2017, 139 (48), 17213–17216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sinclair WR; Arango D; Shrimp JH; Zengeya TT; Thomas JM; Montgomery DC; Fox SD; Andresson T; Oberdoerffer S; Meier JL ACS Chem. Biol 2017, 12 (12), 2922–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Caprara M Cold Spring Harb. Protoc 2013, 2013 (11), pdb.prot078485. [DOI] [PubMed] [Google Scholar]
- (19).Kietrys AM; Velema WA; Kool ET J. Am. Chem. Soc 2017, 139 (47), 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sexton AN; Wang PY; Rutenberg-Schoenberg M; Simon MD Biochemistry 2017, 56 (35), 4713–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Safra M; Sas-Chen A; Nir R; Winkler R; Nachshon A; Bar-Yaacov D; Erlacher M; Rossmanith W; Stern-Ginossar N; Schwartz S Nature 2017, 551 (7679), 251–255. [DOI] [PubMed] [Google Scholar]
- (22).Zhou KI; Clark WC; Pan DW; Eckwahl MJ; Dai Q; Pan T RNA Biol 2018, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hauenschild R; Tserovski L; Schmid K; Thuring K; Winz ML; Sharma S; Entian KD; Wacheul L; Lafontaine DL; Anderson J; Alfonzo J; Hildebrandt A; Jaschke A; Motorin Y; Helm M Nucleic Acids Res 2015, 43 (20), 9950–9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zorbas C; Nicolas E; Wacheul L; Huvelle E; Heurgue-Hamard V; Lafontaine DL Mol. Biol. Cell 2015, 26 (11), 2080–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sharma S; Yang J; van Nues R; Watzinger P; Kotter P; Lafontaine DLJ; Granneman S; Entian KD PLoS Genet. 2017, 13 (5), e1006804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cavaille J; Nicoloso M; Bachellerie JP Nature 1996, 383 (6602), 732–5. [DOI] [PubMed] [Google Scholar]
- (27).Tardu M; Lin Q; Koutmou K biorxiv.org 2018, 10.1101/327585. [DOI]
- (28).Zheng G; Qin Y; Clark WC; Dai Q; Yi C; He C; Lambowitz AM; Pan T Nat. Methods 2015, 12 (9), 835–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.