

Summary
Improving effector activity of antigen specific T cells is a major goal in cancer immunotherapy. Despite the identification of several effector T cell (TEFF)-driving transcription factors (TF), the transcriptional coordination of TEFF biology remains poorly understood. We developed an in vivo T cell CRISPR screening platform and identified a key mechanism restraining TEFF biology through the ETS family TF, Fli1. Genetic deletion of Fli1 enhanced TEFF responses without compromising memory or exhaustion precursors. Fli1 restrained TEFF lineage differentiation by binding to cis-regulatory elements of effector-associated genes. Loss of Fli1 increased chromatin accessibility at ETS:RUNX motifs allowing more efficient Runx3-driven TEFF biology. CD8+ T cells lacking Fli1 provided substantially better protection against multiple infections and tumors. These data indicate that Fli1 safeguards the developing CD8+ T cell transcriptional landscape from excessive ETS:RUNX-driven TEFF cell differentiation. Moreover, genetic deletion of Fli1 improves TEFF differentiation and protective immunity in infections and cancer.
Graphical Abstract
Introduction
Understanding the mechanisms that regulate effector CD8+ T cell (TEFF) differentiation is crucial to improve therapeutic approaches for cancer and other diseases. Activation of naïve CD8+ T cells (TN) during acutely resolved infections or following vaccination results in differentiation into TEFF cells accompanied by transcriptional and epigenetic remodeling. After antigen clearance, a terminally differentiated subset of TEFF cells dies over the ensuing days to weeks, while a small proportion of memory precursors (TMP) differentiates into long-term memory CD8+ T cells (TMEM) (Kaech and Cui, 2012). During chronic infections and cancers, however, CD8+ T cell differentiation is diverted down a path of exhaustion. Under these conditions, TEFF cells become over-stimulated and persist poorly (Angelosanto et al., 2012; Chen et al., 2019b; Khan et al., 2019), whereas a population of activated precursors differentiates into exhausted CD8+ T cells (TEX)(Chen et al., 2019b; McLane et al., 2019). TEX cells have high expression of multiple inhibitory receptors including PD-1, decreased effector functions, altered homeostatic regulation compared to TMEM cells, and a distinct transcriptional and epigenetic program (Wherry and Kurachi, 2015). Blocking inhibitory receptors such as PD-1 can reinvigorate TEX temporarily restoring proliferation and some effector-like properties (Barber et al., 2006; Huang et al., 2017; Pauken et al., 2016), with clinical benefit demonstrated in multiple cancer types (Topalian et al., 2015). Despite of the success of checkpoint blockade, however, most patients do not achieve durable clinical benefit (Sun et al., 2018) and there is a great need to augment T cell differentiation and effector-like activity following checkpoint blockade or during cellular therapies in cancer or other diseases.
There has been considerable interest in defining the populations of T cells responding to checkpoint blockade (Ribas and Wolchok, 2018; Wei et al., 2018) and interrogating the optimal differentiation states for cellular therapies (Brown and Mackall, 2019). TEX cells are prominent in human tumors and likely represent a major source of tumor reactive T cells (Duhen et al., 2018; Huang et al., 2017; Simoni et al., 2018). PD-1 pathway blockade mediates clinical benefit, at least in part, due to reinvigoration of TEX cells allowing these cells to re-access parts of the TEFF cell program (Huang et al., 2017; 2019; Pauken et al., 2016). However, limited therapeutic efficacy is associated with suboptimal reinvigoration of TEX cells (Huang et al., 2017; 2019; Koyama et al., 2016). Therapeutic failures for CAR T cells are also associated with exhaustion (Chen et al., 2019a; Fraietta et al., 2018) and approaches that antagonize exhaustion are actively being investigated (Long et al., 2015; Lynn et al., 2019; Wei et al., 2019). However, a key to both response to checkpoint blockade and cellular therapies to control cancer is the ability to effectively engage a robust effector program, including numerical expansion and elicitation of effector activity. Understanding the underlying molecular mechanisms that control this effector activity is needed to effectively design therapeutic interventions for chronic infections and cancer.
The role of transcription factors (TFs) in regulating differentiation of TEFF versus TMEM or TEX has received considerable attention (Kaech and Cui, 2012; Wherry and Kurachi, 2015). For example, the TFs Batf and Irf4 have an early role in T cell activation and also induce the second wave of transcriptional induction of effector genes (Kurachi et al., 2014; Man et al., 2013). Runx3 induces TEFF gene expression through T-bet and Eomes (Cruz-Guilloty et al., 2009) and is important for the tissue resident memory CD8+ T cells (TRM)(Milner et al., 2017). Runx3 also antagonizes a follicular-like CD8+ T cell fate by inhibiting TCF-1 expression (Shan et al., 2017). Runx1, in contrast, is antagonized by Runx3 during TEFF differentiation (Cruz-Guilloty et al., 2009). Most TEFF-associated genes and their cognate cis-regulatory regions are inaccessible in the TN state linking the role of effector-driving TFs to chromatin accessibility changes that occur during the TN to TEFF transition. Indeed, there is evidence that some of these early operating TFs, such as Batf, may contribute to TEFF gene accessibility through chromatin remodeling (Pham et al., 2019), but other mechanisms of control remain to be defined.
In addition to TF that foster TEFF formation, opposing mechanisms temper complete commitment to effector differentiation to preserve more durable T cell populations for future or ongoing responses. The two alternate cell fates, TMEM and TEX, cannot form from fully committed TEFF (Angelosanto et al., 2012; Chen et al., 2019b; Joshi et al., 2007), suggesting that parts of the TEFF program must be antagonized to allow TMEM and TEX to differentiate. The high mobility group (HMG) TF, TCF-1, for example, is essential for development and maintenance of both TMEM and TEX (Chen et al., 2019b; Im et al., 2016; Utzschneider et al., 2016; Wu et al., 2016; Zhou et al., 2010). TCF-1 represses TEFF-driving TF such as T-bet and Blimp-1(Tiemessen et al., 2014), and may foster epigenetic changes (Xing et al., 2016). Moreover, a second HMG TF, Tox, is essential for the development of the TEX cell fate and represses TEFF lineage differentiation (Alfei et al., 2019; Khan et al., 2019; Scott et al., 2019; Seo et al., 2019; Yao et al., 2019). Despite this work, mechanisms that safeguard against commitment to TEFF differentiation remain poorly understood. Such information could enable immunotherapies for cancer and chronic infections. However, whereas inactivating pathways like TCF-1 or Tox that would de-repress the entire program of TEFF differentiation are of interest, such approaches result in terminal TEFF and may have limited therapeutic benefit because such cells cannot sustain durable responses. Thus, the discovery of mechanisms that selectively de-repress key aspects of TEFF differentiation, particularly those involved in control of numerical expansion and/or protective immunity would be of considerable interest.
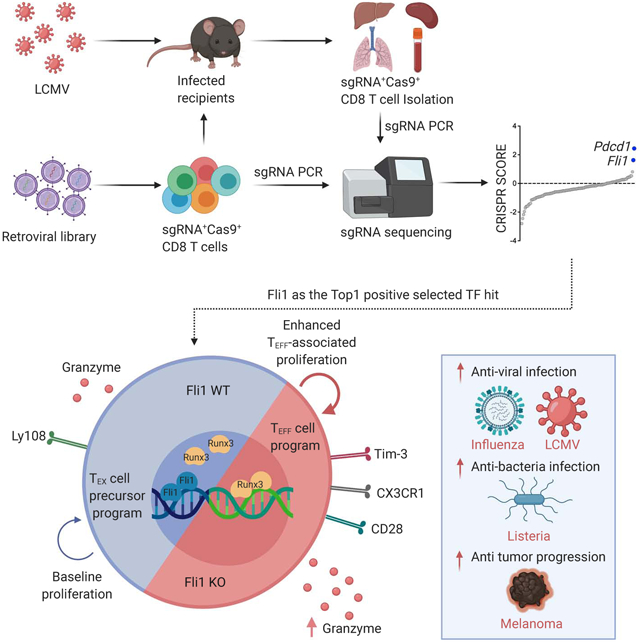
Here, we used in vivo CRISPR-Cas9 screening in antigen-specific CD8+ T cells responding to acute or chronic viral infection to identify key regulators of TEFF and TEX. In particular, we aimed to identify genes that resulted in gain-of-function, improving TEFF differentiation (i.e. an “Up” screen (Kaelin, 2017)). The CRISPR-Cas9 system has been used to interrogate anti-cancer immune responses through screening in cancer cells (Gerlach et al., 2016; Ishizuka et al., 2019; Manguso et al., 2017; Pan et al., 2018; Wang et al., 2019), in vitro in human T cells (Shifrut et al., 2018) or in vivo in mouse T cells (Dong et al., 2019; LaFleur et al., 2019; Wei et al., 2019; Ye et al., 2019). Many of the targets identified appear to function by modulating the activity state of the cell through altered signaling or RNA biology. However, the ability to discover fundamental regulators of cellular differentiation state and/or cellular programming via in vivo CRISPR/Cas9 screening in CD8+ T cells relevant for immunotherapy remains a key goal. Previous in vivo CRISPR screening approaches have used Cas9-edited bone marrow progenitors (LaFleur et al., 2019) or genome-wide screening approaches employing large numbers of CD8+ T cells in vivo (Dong et al., 2019; Ye et al., 2019). Although insights have been gained using these strategies, here, we developed a platform that circumvented impacts on immune system development and was capable of using physiological numbers of CD8+ T cells in vivo to avoid altered T cell differentiation and/or viral pathogenesis that occurs when high numbers of antigen-specific T cells are used in vivo (Badovinac et al., 2007; 2004; Blackburn et al., 2008; Blattman et al., 2009; Marzo et al., 2005; Wherry et al., 2005). Our CD8+ T cell CRISPR screening platform used Cas9+ antigen specific CD8+ T cells combined with an optimized retroviral (RV) based-sgRNA expression strategy (named OptimizedT cell In vivo CRISPR Screening, OpTICS). We focused on TFs to identify genes with central regulatory roles in fate decisions in TEFF versus TEX differentiation. This approach identified known key TFs essential for TEFF and TEX differentiation including Batf, Irf4 and Myc as well as known repressors of TEFF differentiation including Tcf7, Smad2 and Tox. However, this screen also revealed central regulators of TEFF differentiation including many that limit optimal differentiation, such as Irf2, Erg and Fli1, where CRISPR perturbation led to gain-of-function and improved TEFF responses. We further interrogated the role of Fli1, an ETS family TF with roles in hematopoiesis and other developmental pathways in non-immune and immune cell types (Kruse et al., 2009; Pimanda et al., 2007; Tijssen et al., 2011). Here, we discovered a central role for Fli1 in TEFF responses where this TF specifically antagonized the genome-wide function of ETS:RUNX activity and prevented Runx3-driven TEFF biology. Indeed, genetic loss of Fli1 resulted in robustly improved TEFF responses whereas enforced Fli1 expression restrained differentiation. Fli1 prevented chromatin accessibility specifically at ETS:RUNX motifs and loss of Fli1 enabled transcriptional induction of the TEFF program in a Runx3-driven manner. Moreover, loss of Fli1 improved TEFF biology and protective immunity not only during LCMV infection, but also following infection with influenza virus or Listeria monocytogenes. In addition, deletion of Fli1 potently improved anti-tumor immunity. Thus, Fli1 safeguards the developing activated CD8+ T cell epigenome from excessive ETS:RUNX-driven TEFF differentiation and disruption of Fli1 activity improved TEFF activity and protective immunity to infections and cancer.
Results
Optimized CRISPR-Cas9 for gene editing in mouse primary T in vivo.
To enable gene editing in antigen specific primary CD8+ T cells, we crossed LSL-Cas9+ mice (Platt et al., 2014) to CD4CRE+P14+ mice bearing CD8+ T cells specific for the LCMV DbGP33-41 epitope (termed Cas9+P14, or C9P14). We expressed the backbone-optimized Cas9 single guide RNA (sgRNA, Grevet et al., 2018) with a fluorescence marker in a retroviral (RV) vector (Figure S1A). To evaluate gene editing efficiency in vivo, we transduced C9P14 cells ex vivo with either negative control sgRNA (sgCtrl, Table S1) or sgRNA targeting Pdcd1 (Encoding PD-1, sgPdcd1, Table S1) using an optimized RV transduction protocol (Kurachi et al., 2017) (Figure S1B). The double-positive populations of sgRNA (mCherry+) and Cas9 (GFP+) CD8+ T cells (Figure S1C) were adoptively transferred into congenic recipient mice infected with the chronic strain of LCMV (clone13; Cl13) (Figure S1B). On day 9 post infection (p.i.), sgRNA+C9P14 cells were isolated and evaluated. As expected, sgPdcd1 induced antigen specific CD8+ T cell expansion 5 fold greater than the control sgRNA (Figure S1D), consistent with the genetic knockout of Pdcd1 (Odorizzi et al., 2015). The sgPdcd1 also resulted in a robust decrease of PD-1 protein expression (Figure S1E) and indel mutations in the Pdcd1 locus (Figure S1F). Furthermore, we confirmed the high gene editing efficiency of our system by designing sgRNAs targeting Klrg1 and Cxcr3 (Figure S1G, Table S1). Collectively, this in vitro sgRNA RV transduction in C9P14 followed by in vivo adoptive transfer system provides a robust platform to investigate genetic regulatory network of mouse CD8+ T cells in vivo.
OpTICS enables pooled genetic screening in CD8+ T cells in vivo.
To enable in vivo pooled genetic screening in the LCMV infection system, we further optimized the C9P14 and RV sgRNA platform (Figure 1A). First, we determined a physiological number of adoptively transferred CD8+ T cells for screening, as the number of T cells transferred can influence progression of cancer in tumor models or the outcome of infection in vivo (Blattman et al., 2009). We, therefore, limited the number of adoptively transferred RV-transduced CD8+ T cells to 1x105 per mouse (~1x104 after take), a number previously optimized in chronic infection (Chen et al., 2019b; Kao et al., 2011). Next, to evaluate the performance of our system targeting a focused set of 29 TFs in CD8+ T cells in vivo using the LCMV model. Previously, we found that sgRNAs targeting functionally important protein-coding domains can substantially improve genetic screening efficiency, as both in frame and frameshift mutations contribute to generating loss-of-function alleles (Shi et al., 2015). We designed and cloned a sgRNA library targeting the DNA-binding domains of 29 TFs and other control genes (e.g. non-selected control sgRNAs and Pdcd1) with 4-5 sgRNAs per target. An average input coverage of ~400 cells per sgRNA after CD8+ T cell engraftment improved the signal-to-noise ratio, and successfully identify hits compared to 100 cells per sgRNA (Figure S1H). Third, we assessed the performance of our in vivo screen using P14 cells expressing heterozygous versus homozygous alleles of the LSL-Cas9 transgene. Cas9 heterozygous P14 cells outperformed Cas9 homozygous P14 cells in terms of signal-to-noise and consistency between independent screens (Figure S1H-S1I) perhaps due to reduced off-target DNA damage in the heterozygous setting as we have noted for CRE (Kurachi et al., 2019). From these preliminary optimization screens, we identified Batf, Irf4, and Myc as essential for early T cell activation because genetic targeting of these genes potently inhibited T cell activation in vivo (Figure S1H), consistent with known roles for Batf and Irf4 (Grusdat et al., 2014; Kurachi et al., 2014; Man et al., 2013; 2017; Quigley et al., 2010), and Myc (Lindsten et al., 1988; Wang et al., 2011) in TEFF biology. This system was highly efficient with up to ~100-fold enrichment for genes essential for CD8+ T cell responses (Figure S1H) and nearly 20-fold enrichment for genes that repress T cell activation and differentiation (Figure S1J).
Figure 1. Dissecting transcriptional programs of CD8+ T cells using the OpTICS system.

A. Experimental design for Optimized T cell In vivo CRISPR Screening (OpTICS). On Day 0(D0), CD8+ T cells were isolated from CD45.2+ C9P14 mice and activated in vitro; CD45.1+ WT recipient mice were infected with LCMV. On D1 p.i., activated C9P14 cells were transduced with the RV-sgRNA library for 6 hours. On D2 p.i., Cas9+sgRNA+ P14 cells were purified, 5-10% of the sorted cells frozen for D2 baseline (T0 time point), and the rest were adoptively transferred into LCMV-infected recipient mice. Cas9+sgRNA+ P14 cells were isolated from recipient mice on the indicated days by MACS and FACS (T1 time point). Targeted PCR with sequencing adaptors for the sgRNA cassettes was performed and PCR products were sequenced. The CRISPR Score (CS) was calculated as shown.
B. The CS comparing T1 time point to T0 time point (D2 baseline) for target genes from Cas9+sgRNA+ cells from spleen on D8 or D15 p.i. of Arm, D9 or D14 p.i. of Cl13. X-axis shows targeted genes; y-axis shows the CS of each targeted gene (using 4-5 sgRNAs).
C. Heatmap of CS for targeted genes. Heatmap ranks the geometric means of CS for each gene. See Star Methods.
D. Distribution of Ctrl, Pdcd1 and Fli1 sgRNAs. Axis represents log2 fold change (FC). Histogram shows distribution of all sgRNAs. Red bars represent targeted sgRNAs, grey bars represent all other sgRNAs.
E. Western blot for Fli1 protein from sorted Cas9+sgFli1+ P14 cells and paired Cas9+sgFli1− P14 cells from spleen. Two Fli1-sgRNAs (sgFli1_290 and sgFli1_360) were used. Pooled mice were used (3-5 mice for Arm, 10-15 mice for Cl13). Bar graph represents the normalized band intensity of Fli1. Fli1 was first normalized to GAPDH, then a ratio between Cas9+sgFli1+ versus Cas9+sgFli1− was calculated and is shown.
F. Normalized Cas9+sgRNA (VEX)+ cell numbers from spleen for Ctrl-sgRNA(sgCtrl) and 2 Fli1-sgRNA (sgFli1_290 and sgFli1_360) groups on D8 p.i. of Arm, D15 p.i. of Arm, D9 p.i. of Cl13 and D14 p.i. of Cl13. Cell numbers normalized to the sgCtrl group based on D2 in vitro transduction efficiency (see Figure S3B and S3D).
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (One-Way Anova analysis). Data are representative of 4 independent experiments (mean±s.e.m.) with at least 4 mice/group for F.
OpTICS identifies TF involved in TEFF and TEX cell differentiation.
To identify TFs governing TEFF and TEX cell differentiation, we constructed another domain-focused sgRNA library against 120 TFs (Table S2, see Star Methods). This library has 675 sgRNAs total, including 4-5 sgRNAs per DNA-binding domain, positive selection controls (sgPdcd1), and non-selection controls (e.g. sgAno9, sgRosa26, etc.) (Figure 1A). With this 120-TF targeting sgRNA library, we next interrogated in vivo selection and sgRNA enrichment at 1 or 2 weeks p.i. with acutely resolving LCMV Arm (Arm) or chronic Cl13 (Figure 1A). We examined C9P14 cells from different organs (PBMC, spleen, liver and lung) to identify TFs broadly important for CD8+ T cell responses to infection. In general, groups clustered by time point and infection, rather than anatomical location (Figure S1K). At 2 weeks p.i. the data for Arm and Cl13 diverged (Figure S1K), consistent with distinct trajectories of T cell differentiation during acutely resolved versus chronic infections (Wherry et al., 2007). Focusing on the spleen, Batf, Irf4 and Myc emerged as some of the strongest negatively selected hits at both time points in both infections (Figure 1B-1C). We also confirmed several other known effector-driving TFs including Tbx21 (encoding T-bet), Id2, Stat5a, Stat5b, and components of the NF-kB complex (Figure 1B-1C). In addition, several TFs with potential roles in T cell activation and differentiation were revealed including Smad4, Smad7 and Mybl2 (Figure 1B-1C).
We also used the OpTICS system as an “UP” screen (Kaelin, 2017) to identify genes that repressed optimal T cell activation and TEFF cell differentiation. Such genes, like Pdcd1, represent potential immunotherapy targets for improving T cell responses in cancer or infections. PD-1 served as a prototypical positive control where, as expected, Pdcd1-sgRNAs were strongly positively selected across infections, time points and in all tissues (Figure 1B-1D). This screen also identified TFs that antagonized robust CD8+ T cell responses (Figure 1B-1C). Among these, Smad2 has been shown to limit TEFF cell responses during both acute and chronic infection (Tinoco et al., 2009). We also identified Nfatc2 and Nr4a2 (Figure 1C), both of which have been implicated in fostering T cell exhaustion and thus limiting TEFF responses (Chen et al., 2019a; Martinez et al., 2015). In addition, Gata3 identified here has been implicated in driving T cell dysfunction and inhibiting TEFF cell response (Singer et al., 2016). Thus, this screen identified key TFs known to restrain TEFF differentiation and, in some cases, promote exhaustion.
This OpTICS screen also identified additional TFs that restrained optimal TEFF differentiation. This set of genes included Atf6, Irf2, Erg and Fli1, with Fli1 among the strongest hits in repressing TEFF differentiation. The identification of Fli1 as a repressor of TEFF differentiation occurred similarly in Arm and Cl13 infections (Figure 1B-1D) indicating a common role for this TF in restraining TEFF biology. We next selected 2 Fli1-sgRNAs (sgFli1_290 and sgFli1_360) and confirmed that these sgRNAs effectively edited the Fli1 gene (70%-80% editing; Figure S2A) leading to reduced protein expression (Figure 1E). Targeting Fli1 using these individual Fli1-sgRNAs in C9P14 cells in vivo resulted in 5-20 fold greater expansion at 1 and 2 weeks p.i. with either Arm or Cl13 (Figure 1F and S2B-S2E). These data indicate repression of robust CD8+ T cell expansion by Fli1 in acutely resolving or developing chronic infection.
Genetic deletion of Fli1 promotes robust TEFF-differentiation during acutely resolved infection.
We next interrogated the differentiation state of Fli1-sgRNA (sgFli1) or Ctrl-sgRNA (sgCtrl) transduced C9P14 cells during acutely resolved infection. On Day 8 p.i., Fli1 deletion reduced the proportion of the CD127Hi memory precursors (TMP), whereas the frequency KLRG1Hi terminal effector (TEFF) population remained unchanged and the CD127LoKLRG1Lo population slightly increased (Figure 2A and S3A). These effects were more dramatic at Day 15 p.i. when the frequency of the CD127Hi TMP population was reduced and the KLRG1Hi TEFF cell population increased (Figure 2A). However, at both time points the absolute number of both TMP and TEFF was increased ~2-10-fold due to the proliferative expansion of Fli1-deficient CD8+ T cells (Figure 2A). This skewing of T cell differentiation towards TEFF-like populations was also observed using CX3CR1 and CXCR3 (Gerlach et al., 2016), with sgFli1 significantly enriching for the CX3CR1+CXCR3− TEFF population compared to the CX3CR1−CXCR3+ early TMEM subset (Figure 2B). C9P14 cells with increased cytotoxic potential (GzmB+TCF-1−) were also increased in the sgFli1+ group (Figure S3B), though the proportion of cells expressing T-bet or Eomes was similar between the groups (Figure S3C). The frequency of C9P14 cells producing IFN-γ after ex vivo peptide stimulation was slightly lower in the sgFli1+ versus sgCtrl+ group, but the absolute number of IFN-γ producing cells, or cells co-producing multiple effector molecules simultaneously was increased (Figure S3D).
Figure 2. Fli1 restrains TEFF cell proliferation and differentiation during acute infection.

A. Flow cytometry plots and statistical analysis of KLRG1HiCD127Lo Terminal Effectors (TEs) and KLRG1LoCD127Hi Memory Precursors (MPs). Frequencies (left) and numbers (right) from spleen for sgCtrl and 2 sgFli1 groups on D8 and D15 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA(VEX)+ P14 cells.
B. Flow cytometry plots and statistical analysis of CX3CR1+CXCR3− TEFF cells and CX3CR1−CXCR3+ early TMEM cell frequencies (left) and numbers (right) from spleen for sgCtrl and 2 sgFli1 groups on D8 and D15 p.i. of Arm. Gated on Cas9+sgRNA+ P14 cells.
(C-E) Experimental design. On D0, CD45.1+ P14 cells were activated and recipient mice were infected with Arm; On D1 p.i., activated P14 cells were transduced with Empty-RV or Fli1-over expressing (OE)-RV. On D2 p.i., VEX+ P14 cells were purified for each group, and 5 x 104 cells were adoptively transferred into infected recipient mice.
C. Flow cytometry plots of CD45.2+VEX+ cell frequency and statistical analysis of CD45.2+VEX+ cell number for Empty-RV and Fli1-OE-RV conditions.
D. Flow cytometry plots and statistical analysis of KLRG1HiCD127Lo TE and KLRG1LoCD127Hi MP frequencies from spleen for Empty-RV and Fli1-OE-RV groups on D8 and D15 p.i. of Arm. Gated on VEX+ P14 cells.
E. Flow cytometry plots and statistical analysis of CX3CR1+CXCR3− TEFF cell and CX3CR1−CXCR3+ early TMEM cell frequencies from spleen for Empty-RV and Fli1-OE-RV groups on D8 and D15 p.i. of Arm. Gated on VEX+ P14 cells.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (two-tailed Student’s t-test and One-Way Anova analysis). Data are representative of 2-4 independent experiments (mean±s.e.m.) with at least 3 mice/group.
One concern with fostering an increase in TEFF is the preventing the formation of TMEM. Thus, we next examined formation of Fli1-deficient TMEM 1 month p.i. Indeed, the number of KLRG1Hi effector memory C9P14 cells remained higher in the sgFli1+ C9P14 group compared to the sgCtrl+ group on Day 29 p.i. The number of CD127HiTMEM at this time point was similar between the groups (Figure S3E-S3F), indicating that the robust increase in the KRLG1HI TEFF populations generated in the absence of Fli1 did not compromise the formation of TMEM. To further interrogate the effects of Fli1 deficiency on TMEM we examined expression of pro-and anti-apoptotic molecules, Bcl-2, Bcl-XL and Bim. On Day 15 p.i. sgFli1+ C9P14 cells had lower expression of both Bcl-2 and Bim but no change in Bcl-XL expression compared to the sgCtrl+ group (Figure S3G-S3H), resulting in a trend to a higher BclXL/Bim ratio in the sgFli1+ C9P14 group (Figure S3H). There were no differences in expression of Bcl-2, Bcl-XL or Bim in TEFF or TMP subsets (Figure S3I-S3J) suggesting that the differences in total C9P14 populations reflect, in part, different proportions of terminal TEFF and TMP. These observations may be consistent with the observation that the Bcl-XL/Bim ratio may be more important in terminal TEFF survival than the Bcl-2/Bim ratio (Chen et al., 2017).
We next enforced Fli1 expression in WT LCMV-specific P14 cells using an RV based overexpression (OE) system (Kurachi et al., 2017). A ~5-fold reduction in responding Fli1-OE-RV transduced P14 cells was observed Day 8 and Day 16 p.i. compared to the empty vector control (Figure 2C). Furthermore, enforced Fli1 expression diverted the responding P14 cells towards TMP differentiation (Figure 2D-E). Together, these data reveal a role for Fli1 in restraining TEFF differentiation and promoting TMP development during acute infection.
Fli1 antagonizes TEFF-like differentiation during chronic infection.
During chronic viral infection, there is an early fate bifurcation for CD8+ T cell responses where antiviral CD8+ T cells develop into either terminal TEFF-like cells or form TEX precursors that ultimately seed the mature TEX population (Chen et al., 2019b; Khan et al., 2019). We therefore investigated the role of Fli1 in this cell fate decision early during chronic infection. As in acutely resolving infection, genetic perturbation of Fli1 skewed the virus-specific CD8+ T cell response towards the TEFF pathway, defined as TCF-1−GrzmB+ or Ly108−CD39+ cells (Figure 3A and (Chen et al., 2019b)). Due to the 5-10-fold increase in total Fli1-deficient cells, the cell numbers of both the TEFF-like and TEX precursor populations were increased (Figure S4A-S4B). We also investigated the TF circuitry known to be involved in early TEX formation. TCF-1 at this early time point drives expression of Eomes, another TF central to TEX formation (Chen et al., 2019b) and, indeed, Eomes was reduced in the absence of Fli1 (Figure S4C). Expression of T-bet, another T-box TF, was similar between sgCtrl+ and sgFli1+ groups (Figure S4C). Loss of Fli1 was also associated with lower expression of the TEX master regulator Tox on Day 15 p.i. with Cl13, suggesting that Fli1-deficiency antagonized the development of TEX and instead fostered TEFF-like differentiation (Figure S4C). This skewing towards the TEFF fate was associated with increased expression of cytotoxic molecules (Figure 3A) and higher cell numbers that translated to increased numbers of cytokine producing cells (Figure S4D). Moreover, genetic perturbation of Fli1 resulted in an increased proportion of CX3CR1+ and Tim3+ TEFF-like cells in chronic infection (Figure 3B and (Chen et al., 2019b; Zander et al., 2019)). In contrast, enforced Fli1 expression had the opposite effect and not only resulted in lower cell numbers (Figure S4E), but also fostered formation of Ly108+CD39− or TCF-1+GrzmB− TEX precursors and fewer CX3CR1+ cells (Figure S4F-S4H). Notably, PD-1 expression was unchanged and, unlike acutely resolved infection, KLRG1 expression was unaffected (Figure 3B).
Figure 3. Fli1 antagonizes TEFF-like cell differentiation during chronic infection.

A. Flow cytometry plots and statistical analysis of Ly108−CD39+ or TCF-1−Gzmb+ TEFF-like cells and Ly108+CD39− or TCF-1+Gzmb− TEX precursor frequencies from spleen for sgCtrl and 2 sgFli1 groups on D8 and D15 p.i. of Cl13. Gated on the Cas9(GFP)+sgRNA(VEX)+ P14 cells.
B. Statistical analysis of CX3CR1+ and Tim-3+ frequencies, and KLRG1 and PD-1 MFI from spleen for sgCtrl and 2 sgFli1 groups on D8 and D15 p.i. of Cl13. Gated on the Cas9+sgRNA+ P14 cells.
C. Heatmap of differentially expressed genes between sgCtrl and 2 sgFli1 groups.
D. Gene Ontology (GO) enrichment analysis for the sgFli1 groups.
E. GO enrichment analysis for the sgCtrl groups.
F. Gene Set Enrichment Analysis (GSEA) of TEX precursor signature (adapted from Chen et al. 2019) between sgCtrl and sgFli1 groups.
G. GSEA of TEFF-like signature (adapted from Bengsch et al. 2018) between sgCtrl and sgFli1 groups.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (two-tailed Student’s t-test and One-Way Anova analysis). Data are representative of 4 independent experiments (mean±s.e.m.) with at least 4 mice/group for A and B.
To dissect the underlying mechanisms, we performed RNA-seq on sorted sgCtrl+ or sgFli1+ C9P14 cells on Day 9 of Cl13 infection. Both sgRNAs targeting Fli1 resulted in a similar transcriptional effect (Figure 3C, S5A). The sgFli1+ C9P14 cells were transcriptionally distinct from the sgCtrl+ C9P14 cells with over 1400 genes differentially expressed between the two conditions (Figure 3C, S5A). Effector-associated genes such as Prf1, Gzmb, Cd28, Ccl3 and Prdm1 were robustly increased in the sgFli1+ C9P14 cells, whereas the sgCtrl+ C9P14s were enriched in TEX precursor genes such as Tcf7, Cxcr5, Slamf6 and Id3 (Figure 3C). Gene Ontology enrichment analysis also identified cell division-associated and T cell activation-associated pathways in sgFli1+ C9P14 cells (Figure 3D), whereas sgCtrl+ C9P14s enriched in metabolic pathways, in particular nucleotide, nucleoside and purine biosynthesis (Figure 3E). We next used gene set enrichment analysis (GSEA) to examine the early phase of chronic infection, when a divergence of differentiation into either TEFF-like or TEX precursor cell fates occurs. Indeed, the TEX precursor signature (Chen et al., 2019b) was strongly enriched in sgCtrl+ C9P14 cells compared to the sgFli1+ C9P14 population whereas a TEFF gene signature (Bengsch et al., 2018) was strongly enriched in the sgFli1+ C9P14 population (Figure 3F-3G). Thus, Fli1 repressed optimal TEFF differentiation in both acutely resolving and chronic infection and loss of Fli1 antagonized the development of TEX cells. However, although genetic perturbation of Fli1 drove an increase in expression of effector-associated genes at Day 9 of chronic infection, Tox, Tox2 and Cd28 were also increased. This effect may suggest that although loss of Fli1 might enhance TEFF-like biology this effect might not be at the expense of genes necessary to sustain responses in chronic infection or cancer.
Fli1 remodels the epigenetic landscape of CD8+ T cells and antagonizes TEFF-associated gene expression.
In acute myeloid leukemia, FLI1 co-localizes with the chromatin remodeler BRD4 (Roe et al., 2015). The EWS-Fli1 fusion oncoprotein driving Ewing’s sarcoma can trigger de novo enhancer formation via chromatin remodeling and can inactivate existing enhancers by displacing ETS family members (Riggi et al., 2014). It is unclear, however, how Fli1 affects epigenetic landscape changes in developing TEFF, TMEM or TEX cells.
To examine the role of Fli1 in supporting the epigenetic landscape of CD8+ T cells, we performed ATAC-seq on sgFli1+ and sgCtrl+ C9P14 cells on Day 9 p.i. with Cl13. Compared to sgCtrl+ C9P14 cells, the sgFli1+ group had considerable changes in chromatin accessibility (Figure 4A). Over 5000 open chromatin regions (OCRs) differed between the control and Fli1-perturbed groups with approximately equal numbers of peaks gained or lost (Figure 4B-4D). Most of these changes were located in intronic or intergenic regions consistent with cis-regulatory or enhancer elements (Figure 4C).
Figure 4. Fli1 reshapes the epigenetic profile of CD8+ T cells and inhibits TEFF-associated gene expression.

A. PCA plot of ATAC-seq data for sgCtrl, sgFli1_290 and sgFli1_360 groups on D9 p.i. of Cl13.
B. Overall open chromatin region (OCR) peak changes for sgFli1 groups compared to sgCtrl group.
C. Categories of Cis-element OCR peaks that changed between sgCtrl and sgFli1 groups. Left plot represents all changes; right plot represents changes for increased or decreased accessibility.
D. Heatmap shows differentially accessible peaks between sgCtrl group and 2 sgFli1 groups (adjusted p-value<0.05, Log10 Fold Change>0.6). Selected genes assigned to the peaks are indicated.
E. Overlapping Venn plot of the genes with differentially accessible (DA) peaks and differentially expressed genes from Figure 3C.
F. Pearson correlation of the peak accessibility of the nearest genes versus the differential expression of the genes.
G. Transcription factor (TF) motif gain or loss associated with loss of Fli1. X-axis represents the logP-value of the motif enrichment. Y-axis represents the fold change of the motif enrichment. Targeted motifs in the changed OCR between the sgCtrl and sgFli1 groups were compared to the whole genome background to calculate p-value and fold change.
H. IgG or Fli1 binding signals from CUT&RUN on P14 cells on D8 p.i. of Cl13 and OCR signals detected by ATAC-seq for sgCtrl-sgRNA, sgFli1_290 and sgFli1_360 groups at the Cd28, Cx3cr1, and Havcr2 loci.
I. Histogram of CD28 staining and statistical analysis for sgCtrl, sgFli1_290 and sgFli1_360 groups on D8 Cl13 p.i. Grey shows CD28 staining of CD44− naïve T cell.
J. Heatmap shown differentially accessible (DA) peaks overlapped with the Fli1 CUT&RUN binding peaks between sgCtrl group and 2 sgFli1 groups. Selected genes assigned to the peaks are indicated.
K. Top 4 enriched TF motifs in the Fli1 CUT&RUN peaks are shown.
*P<0.05, **P<0.01 versus control (One-Way Anova analysis). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 5 mice/group for I.
We next assigned each OCR to the nearest gene to estimate genes that could be regulated by these cis-regulatory elements. TEFF-associated genes, such as Ccl3, Ccl5, Cd28, Cx3cr1 and Prdm1, gained chromatin accessibility in the sgFli1+ group (Figure 4D), consistent with the RNA-seq data. In contrast, there was decreased chromatin accessibility near genes involved in T cell progenitor biology such as Tcf7, Slamf6, Id3 and Cxcr5 (Figure 4D) in the sgFli1+ group. Moreover, the sgFli1+C9P14 cells had altered accessibility in the Tox (and Tox2) locus, but these changes included both increased and decreased accessibility for different peaks (Figure 4D). These changes in chromatin accessibility corresponded to changes in gene expression with ~1/3 of the genes that changed transcriptionally associated with a differentially accessible chromatin region (402 out of 1467) (Figure 4E). In general, increased accessibility correlated with increased transcription though there was a clear subset of regions where decreased accessibility corresponded to increased transcription (Figure 4F).
We next defined the TF motifs present in the OCRs that were dependent on Fli1 for altered accessibility. Among the OCRs that decreased in accessibility in the absence of Fli1, the most enriched TF motifs were for IRF1 and IRF2 (Figure 4G), potentially linking Fli1 to IRF1 and IRF2 downstream of IFN signaling or to the regulation of cell cycle by these TFs (Choo et al., 2006). In the group of OCRs that increased in accessibility in the absence of Fli1, ETS and RUNX motifs were highly enriched (Figure 4G). The composite ETS:RUNX motif was by far the most changed with 18-fold enrichment (Figure 4G). These observations suggested that Fli1 may limit the activity of other ETS family members (e.g. ETS1, ETV1 or ELK1) or alter accessibility at ETS:RUNX binding sites (Figure 4G). Runx3 is a central driver of TEFF differentiation and functions by directly regulating effector gene expression, coordinating and enabling effector gene regulation via T-bet and Eomes and antagonizing TCF-1 expression (Cruz-Guilloty et al., 2009; Shan et al., 2017; Wang et al., 2018). Thus, a potential role for Fli1 in Runx3 biology would provide a mechanistic link between loss of Fli1 and improved TEFF differentiation.
We tested how Fli1 genomic binding was related to changes in chromatin accessibility and TEFF biology, using Fli1 CUT&RUN (Skene et al. 2017). At Day 9 p.i. with Cl13, >90% of the identified Fli1 binding sites were contained in OCRs detected by ATAC-seq (Figure S5B-S5D). Specifically, Fli1 bound to OCRs of TEFF-like genes such as Cx3cr1, Cd28 and Havcr2. Chromatin accessibility increased at these locations upon Fli1 deletion (Figure 4H), resulting in increased transcription (Figure 3C) and protein expression (Figure 3B and 4I). In contrast, for genes involved in progenitor biology that were decreased in expression in the absence of Fli1 such as Tcf7 and Id3, direct binding of Fli1 was not observed (Figure S5D), likely indicating that the major role of Fli1 is to safeguard against an overly robust TEFF program rather directly enabling memory/progenitor biology. Furthermore, 78% of the sites where Fli1 was defined to bind by CUT&RUN increased in chromatin accessibility in the absence of Fli1; in contrast, 22% decreased in accessibility (Figure 4J), suggesting that Fli1 mainly functions to repress chromatin accessibility. Analyzing the DNA binding motifs in the Fli1 CUT&RUN data revealed the expected FLI1 motif. However, SP2, NFY1 and RUNX1 motifs were also significantly enriched where Fli1 bound (Figure 4K). Together with the increase in ETS:RUNX motifs in the Fli1-deficient ATAC-seq observations above, these data support a model where Fli1 coordinates with RUNX family members to control TEFF differentiation.
Enforced Runx3 expression synergizes with Fli1-deletion to enhance TEFF responses
Unlike TEFF biology, the roles of Runx1 and Runx3 in TEX development are less clear. Because TEFF and TEX are opposing fates in chronic infection (Chen et al., 2019b; Khan et al., 2019; Yao et al., 2019; Zander et al., 2019) and ETS:RUNX motifs become more accessible in the absence of Fli1, we hypothesized that a RUNX-Fli1 axis might influence TEFF versus TEX differentiation. We, therefore, tested whether Runx1 or Runx3 expression in Fli1-deficient CD8+ T cells would impact TEFF differentiation in early chronic infection.
Enforced expression of Runx1 in WT P14 cells reduced cell numbers at Day 7 p.i. with Cl13 infection (Figure S5E). Moreover, Runx1-OE fostered formation of Ly108+CD39− TEX precursors at the expense of the more TEFF-like Ly108−CD39+ population at this time point (Figure S5E). To interrogate the impact of enforced Runx1 expression in the absence of Fli1, we used a dual RV transduction approach and combined control or Fli1 sgRNA RV transduction with either empty or Runx1 expressing RVs. Singly versus dually transduced cells were distinguished using VEX (for sgRNA) and mCherry. C9P14 cells were efficiently singly or dually transduced (Figure S5F), adoptively transferred into Cl13-infected mice and the double transduced (i.e. GFP+VEX+mCherry+) C9P14 cells were analyzed on Day 8 p.i. (Figure 5A). In the sgFli1+Runx1-OE group the number of GFP+VEX+mCherry+ C9P14 cells was reduced and there were fewer Ly108−CD39+ cells. In contrast the sgFli1+Empty-RV group had an increase in the GFP+VEX+mCherry+ C9P14 cell population and these cells were skewed towards the Ly108−CD39+ TEFF-like fate (Figure 5B-5D, S5G) as above. However, in the Fli1-deficient setting where there was enhanced CD8+ T cell expansion, Runx1 overexpression reduced the magnitude of the response and partially reversed the skewing towards the Ly108−CD39+ TEFF-like fate caused by loss of Fli1 (Figure 5B-5D).
Figure 5. Overexpression of Runx1 or Runx3 in the context of Fli1-deficiency in CD8+ T cells.

A. Experimental design. On D0, CD8+ T cells were isolated from CD45.2+ C9P14 donor mice and activated; CD45.1+ WT recipient mice were infected with Cl13. On D1 p.i., activated C9P14 cells were transduced with sgRNA-RV or OE-RV and 1 x 105 transduced cells adoptively transferred into infected recipient mice.
B-D. Flow cytometry plots (B) and statistical analysis (C-D) of VEX+mCherry+ C9P14 cells and Ly108−CD39+/Ly108+CD39− C9P14 cells from spleen for sgCtrl-VEX + Empty-mCherry, sgCtrl-VEX + Runx1-mCherry, sgFli1_290-VEX + Empty-mCherry and sgFli1_290-VEX + Runx1-mCherry on D8 p.i. of Cl13. Gated on Cas9(GFP)+CD45.2+ P14 cells.
E-G. Flow cytometry plots (E) and statistical analysis (F-G) of VEX+mCherry+ C9P14 cells and Ly108−CD39+/Ly108+CD39− C9P14 cells from spleen for sgCtrl-mCherry + Empty-VEX, sgCtrl-mCherry + Runx3-VEX, sgFli1_290-mCherry + Empty-VEX and sgFli1_290-mCherry + Runx3-VEX at D8 p.i. of Cl13. Gated on Cas9+CD45.2+ P14 cells.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (One-Way Anova analysis). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 5 mice/group.
In contrast to the effect of Runx1, enforced expression of Runx3 alone (in the sgCtrl+ group), modestly increased the magnitude of the CD8+ T cell response but robustly skewed the GFP+VEX+mCherry+ C9P14 population towards a CD39+Ly108− TEFF-like population (Figure 5A, 5E-5G). These effects were more dramatic in the absence of Fli1 with greater numerical amplification and even further skewing towards CD39+Ly108− TEFF-like cells in the sgFli1+Runx3-OE enforced expression group (Figure 5E-5G). Whereas the sgFli1+Runx3-OE group had lower frequency of the TEX precursor population, the absolute number of this population remained unchanged compared to control (Figure 5E and 5G; S5I). Taken together, these data support a model where loss of Fli1 reveals ETS:RUNX motifs that can be used by Runx1 and/or Runx3. However, whereas Runx3 drives a more TEFF-like population, an effect amplified in the absence of Fli1, Runx1 appears to antagonize TEFF generation, in agreement with the opposing functions of Runx1 and Runx3 (Cruz-Guilloty et al., 2009). Thus, Fli1 restrains the TEFF promoting activity of Runx3 function by restricting genome access and protecting ETS:RUNX binding sites. These data reveal Fli1, Runx3 and perhaps Runx1 as key regulators of the fate choice between TEFF and TEX early after initial activation.
Augmented TEFF cell responses in the absence of Fli1 improve protective immunity against pathogens.
The data above provoke the question of whether loss of Fli1 would improve control of infections due to the augmented TEFF differentiation. To test this idea, we used LCMV Cl13 to investigate protective immunity during chronic infection and two models of acute infection with influenza virus (PR8) or Listeria monocytogenes (LM) each expressing the LCMV GP33-41 epitope (PR8GP33 and LMGP33) recognized by P14 cells (Figure 6A).
Figure 6. Fli1-deficiency in CD8+ T cells enhances protective immunity against infections.

A. Experimental design. On D0, CD8+ T cells were isolated from CD45.2+ C9P14 donor mice and activated; CD45.1+ WT recipient mice were infected with LCMV Cl13, influenza virus PR8-GP33 or Listeria monocytogenes-GP33 (LM-GP33). On D1 p.i., activated C9P14 cells were transduced with sgCtrl or sgFli1 RV. On D2 p.i., Cas9+sgRNA(VEX)+ P14 cells were purified by flow cytometry for sgCtrl or sgFli1 groups, and adoptively transferred into the infected recipient mice. For Cl13, 1.5 x 105 VEX+ C9P14 cells were transferred per mouse; for PR8-GP33 and LM-GP33, 1.0 x 105 VEX+ C9P14 cells were transferred per mouse.
B. LCMV viral load was measured by plaque assay on D15 p.i. with Cl13 in liver, kidney and serum of the indicated mice. Data pooled from 2 independent experiments.
C. Weight curve for PR8-GP33 infected mice from NT, sgCtrl+ cell-transferred, or sgFli1+ cell-transferred groups. Dashed line represents the time of C9P14 adoptive transfer.
D. PR8-GP33 viral RNA load in the lung of NT, sgCtrl+ or sgFli1+ C9P14 recipient mice. Dashed line indicates the limit of detection. Lung samples from naïve mice and spleen samples from PR8-GP33 infected mice were used as negative controls.
E. Adjusted survival curve of LM-GP33 infected mice for NT, sgCtrl+ or sgFli1+ C9P14 recipient mice. Dashed line represents the time of C9P14 adoptive transfer.
F. LM-GP33 bacteria load in spleen and liver of the surviving NT, sgCtrl+ or sgFli1+ C9P14 recipient mice on D7 p.i.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (One-Way Anova analysis for 6B-6D, 6F and Mantel-Cox test for 6E). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 3 mice/group.
During Cl13 infection, adoptive transfer of sgCtrl+ C9P14 cells conferred a moderate degree of viral control compared to the no transfer condition (NT, Figure 6B) as expected (Blattman et al., 2009). However, sgFli1+ C9P14 provided substantially improved control of viral replication compared to sgCtrl+ C9P14 cells at ~2 weeks p.i. (Figure 6B), demonstrating a benefit due to loss of Fli1 even in chronic viral infection where induction of exhaustion is a major barrier to effective protective immunity. Notably, in some experiments sgFli1+ C9P14, but not sgCtrl+ C9P14 recipient mice experienced severe disease and had to be euthanized between D7-D13 p.i., (Figure S6A), suggesting that Fli1 may safeguard from excessive TEFF-like differentiation and limit T cell mediated immunopathology.
We next evaluated the impact of loss of Fli1 during acutely resolving infections. During influenza-PR8GP33 infection, mice receiving sgFli1+ C9P14 cells lost less weight than control non-transferred mice or mice receiving sgCtrl+ C9P14 cells (Figure 6C). Decreased weight loss was associated with better control of viral replication in the lungs of mice receiving sgFli1+ C9P14 cells (Figure 6D). In this setting, there was variation in the magnitude of the sgFli1+ C9P14 expansion in the lungs following PR8GP33 infection (Figure S6B-S6D). This heterogeneity in the T cell responses was associated with differences in viral control, with some mice nearly eliminating viral RNA by this time point (Figure 6D) and recovering from weight loss. Indeed, the overall magnitude of the C9P14 response was lower in mice that had recovered, consistent with prolonged viral replication and higher antigen load driving increased T cell expansion in mice that had not yet controlled the infection (Figure S6B-S6C). Notably, 6 out of 12 of the mice receiving sgFli1+ C9P14 cells had controlled disease by this time point, compared to only 1 out of 11 mice receiving sgCtrl+ C9P14 (Figure 6D, S6C). In the group of mice still harboring viral RNA in the lungs, sgFli1+ C9P14 cells had expanded to substantially higher numbers than sgCtrl+ C9P14 cells (Figure S6C). A similar difference in TEFF cell expansion was also observed in the spleen (Figure S6D).
Loss of Fli1 conferred a similar advantage following LMGP33 infection. Although both sgCtrl+ and sgFli1+ C9P14 cells improved survival following a high dose LMGP33 challenge (Figure 6E), sgFli1+ C9P14 cells resulted in substantially better control of bacterial replication compared to the sgCtrl+ C9P14 cells at Day 7 p.i. (Figure 6F). Consistent with the influenza virus model, this improved protective immunity was associated with greater numerical expansion of sgFli1+ C9P14 cells compared to the sgCtrl+ group (Figure S6E). Thus, deficiency in Fli1 confers a substantial benefit on TEFF cell expansion and protective immunity during chronic Cl13 infection, respiratory influenza virus infection, and systemic infection with an intracellular bacterium.
Loss of Fli1 in CD8+ T cells enhances immunity to tumors.
We next asked whether Fli1 deficiency provided enhanced control of tumors. Thus, we employed a subcutaneous B16GP33 tumor model. Tumor-bearing mice received equal numbers of sgCtrl+ or sgFli1+ C9P14 cells on day 5 post tumor inoculation (p.t.) (Figure 7A). We used Rag2−/− recipient mice to isolate the effects of sgCtrl+ versus sgFli1+ C9P14 cells (Figure 7A). In this setting, sgFli1+ C9P14 cells robustly controlled tumor progression compared to non-transferred mice or the sgCtrl+ C9P14 group (Figure 7B). Furthermore, tumor weight was significantly lower in the sgFli1+ C9P14 group compared to either control group at endpoint (Figure 7C). Although there was not a clear difference in the number of C9P14 cells/gram of tumor, this tumor control was associated with a significant increase in Ly108−CD39+ donor C9P14 in the sgFli1+ group, consistent with a shift towards the more TEFF-like population (Figure 7D-7E). In the spleen, however, there was a significant increase in sgFli1+ C9P14 cell numbers, as well as the proportion of Ly108−CD39+ cells compared to the sgCtrl+ group (Figure 7F-7G). We extended these findings to immune competent mice. We used Cas9+ C57BL/6 recipient mice to prevent rejection of the C9P14 donor cells and allow responses to be analyzed for an extended period of time (Figure S7A). In this setting, sgFli1+ C9P14 cells again conferred substantial benefit on tumor control (Figure S7B-S7C) compared to sgCtrl+ C9P14 cells. Furthermore, this improved tumor control was associated with an increase in the Ly108−CD39+ TEFF-like population in the tumor, draining lymph node (dLN) and spleen, as well as increased C9P14 cell numbers in the dLN and spleen (Figure S7D-S7G). Thus, genetic deletion of Fli1 conferred a substantial benefit on tumor control indicating a central role for Fli1 in coordinating and restraining protective TEFF responses during tumor progression. Together, these data show that loss of Fli1 results in improved protective immunity, in the setting of systemic and local, acutely resolving and chronic infections, as well as tumor progression.
Figure 7. Loss of Fli1 in CD8+ T cells improved anti-tumor immunity.

A. Experimental design. On D0, CD45.2+Rag2−/− mice were inoculated with 1.0 x 105 B16-Dbgp33 cells. On D3 post tumor inoculation (p.t.), CD8+ T cells were isolated from CD45.1+ C9P14 mice and activated. On D4 p.t., activated C9P14 cells were transduced with sgCtrl or sgFli1 RVs. On D5 p.t., sgRNA (VEX)+Cas9 (GFP)+P14 cells were sorted from sgCtrl or sgFli1 groups, and 1 x 106 purified VEX+ C9P14 cells were adoptively transferred into tumor-bearing mice.
B. Tumor volume curve for mice receiving NT, sgCtrl+ or sgFli1+ C9P14 cells.
C. Tumor weight on D23 p.t. for mice receiving NT, sgCtrl+ or sgFli1+ C9P14 cells.
D-E. Flow cytometry plots (D) and statistical analysis (E) of CD45.1+ sgRNA(VEX)+Cas9+P14 cells and Ly108−CD39+/Ly108+CD39− C9P14 cells from tumor for sgCtrl or sgFli1 groups on D23 p.t.
F-G. Flow cytometry plots (F) and statistical analysis (G) of CD45.1+ sgRNA+Cas9+ P14 cells and Ly108−CD39+/Ly108+CD39− C9P14 cells from spleen for sgCtrl or sgFli1 groups at D23 p.t.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (two-tailed Student’s t-test and One-Way Anova analysis). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 5 mice/group.
Discussion
Considerable current efforts focus on improving immunotherapy for cancer and chronic infections through a better understanding of the biology of TEX and TEFF differentiation. In this study, we used a focused in vivo CRISPR screening approach to specifically interrogate the mechanisms governing TEX versus TEFF differentiation. We identified Fli1 as a key TF that safeguards the transcriptional and epigenetic commitment to full TEFF differentiation. Mechanistically, Fli1 limited epigenetic accessibility to ETS:RUNX sites, preventing Runx3 from fully enabling an effector program. As a result, deleting Fli1 robustly improved protective immunity in multiple models of acute infection, chronic infection and cancer, thus identifying Fli1 as a key regulator of the TEFF versus TEX differentiation programs and a target for future immunotherapy strategies.
Recent advances in CRISPR-based screening approaches have allowed the dissection of in vitro T cell activation (Shang et al., 2018; Shifrut et al., 2018) and in vivo responses to infections and tumors (Dong et al., 2019; LaFleur et al., 2019; Wei et al., 2019; Ye et al., 2019). To better understand the fate commitment of TEFF and TEX, we developed OpTICS, an in vivo CRISPR system that allows screening in mature CD8+ T cells using physiological T cell numbers that preserve disease pathogenesis and normal CD8+ T cell differentiation biology. In addition to identifying many known regulators of CD8+ T cell responses, this approach identified several previously unexplored negative regulators of TEFF differentiation, including Smad2, Erg and Fli1. Indeed, genetic loss of Fli1 improved protective immunity in multiple settings of acute or chronic infection and cancer. Moreover, unlike the effect seen with loss of the TEX-driving TF Tox, where CD8+ T cell responses cannot be sustained during chronic infection or cancer due to loss of TEX progenitor cells (Khan et al., 2019, Seo et al., 2019), deficiency in Fli1 did not diminish the TEX progenitor population.
Fli1 has a role in hematopoietic stem cell differentiation and co-localizes with other TFs such as Gata1/2 and Runx1 (Tijssen et al., 2011). Here, we find that genetic perturbation of Fli1 significantly increased chromatin accessibility at ETS:RUNX motifs in antigen-specific CD8+ T cells responding to viral infection. Moreover, the effect of enforced Runx3 expression is enhanced in the absence of Fli1. These observations suggest that Fli1 prevents accessibility to RUNX binding sites, restricting the activity of the effector-promoting TF Runx3. Moreover, Runx3 can coordinate epigenetic changes at loci encoding other effector-promoting TFs (Wang et al., 2018). Runx1 likely antagonizes Runx3 and vice versa, though Runx3 appears to dominate in settings of T cell activation (Cheng et al., 2008; Cruz-Guilloty et al., 2009). Our data also suggest that Fli1 can cooperate with Runx1 to restrain TEFF differentiation, perhaps with Fli1 and Runx1 co-binding at ETS-RUNX motifs. Together, these data suggest a model where Fli1, in combination with Runx1, prevents efficient genome accessibility or activity of Runx3 and thus restrain the full effector gene program that involves the positive feed-forward effector promoting activity of Runx3. Thus, genetic deletion of Fli1 de-represses TEFF differentiation, at least partially by creating opportunities for more efficient Runx3 activity.
Recent work has begun to define the transcriptional circuitry that directs fate decisions between terminal TEFF, TMEM and TEX. Many of these transcriptional mechanisms that promote one cell fate directly repress the opposing fate. For example, Tox promotes TEX while repressing TEFF (Alfei et al., 2019; Khan et al., 2019), TCF-1 promotes TMEM or TEX at the expense of TEFF (Chen et al., 2019b; Zhou et al. 2010) and Blimp-1, T-bet, Id2, and others drive TEFF and repress TMEM (Kaech and Cui, 2012). The identification of Fli1 as a type of genomic “safeguard” against over-commitment to effector differentiation reveals several concepts. First, during chronic infection, loss of TCF-1 or Tox results in an ability to sustain responses later in infection due to commitment to terminally differentiated TEFF (Alfei et al., 2019; Chen et al., 2019b; Im et al., 2016; Khan et al., 2019; Utzschneider et al., 2016). These observations provoke the question of whether fostering an increase in the TEFF cell fate necessarily comes at the expense of losing the TMEM or TEX lineage. Fli1 represents a distinct type of damper on an otherwise robust feed-forward effector transcriptional circuit. By restraining the Runx3 node in the effector wiring, Fli1 tempers a central step that not only directly controls expression of key effector genes but also positively reinforces other cooperating effector TFs. However, unlike TCF-1 and Tox, Fli1 is not required for progenitor biology and the number of both TEFF cells and TMP (in acutely resolved infections) or TEX progenitor cells (in chronic infection) were increased in the absence of Fli1. Thus, by interrupting this “damper” in the circuit, rather than deleting the master switch of TMP or TEX differentiation it may be possible to augment beneficial aspects of short-term protective immunity without compromising long-term immunity. Second, our data reveal a mechanism of competition for epigenetic access between Fli1 and other factors that bind at ETS:RUNX motifs. These effects may manifest because Fli1 occupies genomic locations that can be bound by ETS:RUNX family TFs that would catalyze chromatin accessibility changes. Alternatively, these effects may be due to chromatin changes coordinated by Fli1 itself. For example, the EWS-FLI1 fusion recruits the BAF complex to initiate chromatin changes in cancer cells (Boulay et al., 2017). Thus, the role of Fli1 in CD8+ T cells likely involves a chromatin accessibility-based mechanism to restrain ETS:RUNX driven effector biology, though other effects through IRF1/IRF2 may also exist.
The current studies demonstrate a major beneficial effect of loss of Fli1 on protective immunity in multiple settings of infection and cancer. The absence of Fli1 consistently improved protective immunity across models. Of particular relevance for immunotherapy, deleting Fli1 improved control of both tumor progression and chronic LCMV infection where the induction of exhaustion typically limits immunity. It will be interesting to determine in future studies whether this beneficial effect on tumor immunity is due to increased T cell numbers, the de-repression of TEFF differentiation by loss of Fli1, enhanced transcription of specific effector gene programs or a combination of these effects. It is also possible that enhanced Runx3 activity fosters more efficient tissue residency, a change that would be consistent with improved immunity in the influenza virus model. Finally, given the ability to apply CRISPR-mediated genetic manipulations in cellular therapy settings (Stadtmauer et al., 2020; Xu et al., 2019) it might be possible to envision clinical benefits with CAR T cells by targeting Fli1 or related pathways.
Thus, the OpTICS platform provides a highly robust in vivo platform to screen genes involved in regulating CD8+ T cell differentiation as it relates to tumor immunotherapy. This highly focused and optimized platform allows for a 20-100-fold enrichment of sgRNA detection and considerable resolution for gain-of-function screening. In addition to the role of Fli1 revealed here, many other potential targets for exploration exist from this screen. Moreover, using OpTICS to extend from this TF focused biology to other areas of cellular biology should provide a robust platform for future discovery.
STAR★Methods
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, E. John Wherry (wherry@pennmedicine.upenn.edu).
MATERIALS AVAILABILITY
All plasmids are deposited to Addgene for public requests (Addgene numbers in the key resource table). Transcriptional factor libraries will be available upon requests to the corresponding authors.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Mouse strains | ||
| C57BL/6 | Charles River | N/A |
| CD45.1+ C57BL/6 | Charles River | N/A |
| C57BL/6 | The Jackson Lab | N/A |
| CD4CRE | The Jackson Lab | Stock No. 022071 |
| TCRα−; P14 TCRVα2Vβ8 | The Jackson Lab | Stock No. 37394-JAX |
| Rag2−/− C57BL/6 | The Jackson Lab | Stock No: 008449 |
| LSL-Cas9-GFP | The Jackson Lab | Stock No: 026175 |
| Constitutive-Cas9-GFP | The Jackson Lab | Stock No: 026179 |
| Flow cytometry reagents | ||
| Live/Dead Aqua Dye | Thermofisher | Cat# L34957 |
| Live/Dead Zombie NIR Dye | BioLegend | Cat# 423106 |
| Anti-Mouse KLRG1(2F1) | BD Biosciences | Cat# 561619, RRID:AB_10898017 |
| Anti-Mouse CD127(A7R34) | BioLegend | Cat# 135016, RRID:AB_1937261 |
| Anti-Mouse CD8(53-6.7) | BioLegend | Cat# 100742, RRID:AB_2563056 |
| Anti-Mouse CD44(IM7) | BioLegend | Cat# 103059, RRID:AB_2571953 |
| Anti-Mouse CD45.1(A20) | BioLegend | Cat# 110724, RRID:AB_493733; Cat# 110716, RRID:AB_313505 |
| Anti-Mouse CD45.2(104) | BioLegend | Cat# 109828, RRID:AB_893350; Cat# 109823, RRID:AB_830788 |
| Anti-Mouse Ly108(330-AJ) | BioLegend | Cat# 134608, RRID:AB_2188093; Cat# 134605, RRID:AB_1659258 |
| Anti-Mouse Tim-3(RMT3-23) | BioLegend | Cat# 119721, RRID:AB_2616907 |
| Anti-Mouse CD39(24DMS1) | Thermofisher | Cat# 46-0391-80, RRID:AB_10717513 |
| Anti-Mouse PD-1(RMP1-30) | BioLegend | Cat# 109109, RRID:AB_572016 |
| Anti-Mouse CD28 | BioLegend | Cat# 644808, RRID:AB_1595479 |
| Anti-Mouse CX3CR1 | Thermofisher | Cat# 50-4875-80, RRID:AB_2574226 |
| Anti-Mouse CXCR3 | BioLegend | Cat# 652420, RRID:AB_2564285 |
| Anti-Mouse TCF-1(S33-966) | BD Biosciences | Cat# 564217, RRID:AB_2687845 |
| Anti-Mouse Gzmb (GB11) | Thermofisher | Cat# GRB17, RRID:AB_2536540 |
| Anti-Mouse T-bet(4B10) | BioLegend | Cat# 644808, RRID:AB_1595479 |
| Anti-Mouse Eomes(Dan11mag) | Thermofisher | Cat# 50-4875-80, RRID:AB_2574226 |
| Anti-Human(Mouse) Tox(REA473) | Miltenyl. Biotec. | Cat# 130-118-335, RRID:AB_2751485 |
| Anti-Mouse Bcl-2(A19-3) | BD Biosciences | Cat# 556537, RRID:AB_396457 |
| Anti-Mouse Bim(C34C5) | Cell Signaling Technology | Cat# 2933, RRID:AB_1030947 |
| Anti-Mouse Bcl-xL(H-5) | Santa Cruz Biotech. | Cat# sc-8392, RRID:AB_626739 |
| Anti-Mouse CD107a(1D4B) | BioLegend | Cat# 121606, RRID:AB_572007 |
| Anti-Mouse TNFα(MP6-XT22) | BioLegend | Cat# 506328, RRID:AB_2562902 |
| Anti-Mouse IFNγ(XMG1.2) | BD Biosciences | Cat# 560661, RRID:AB_1727534 |
| Anti-Mouse MIP-1α(39624) | R&D Systems | Cat# IC450P, RRID:AB_2244085 |
| BD GolgiStop | Thermofisher | Cat# 554724 |
| BD GolgiPlug | Thermofisher | Cat# 555029 |
| LCMV DbGP33 tetramer | NIH | Conjugated in house |
| Foxp3 Transcription Factor Staining Buffer Kit | Thermofisher | Cat# A25866A |
| Experimental Models | ||
| LCMV Clone13 (Cl13) | Rafi Ahmed | Grew up in house |
| LCMV Armstrong (Arm) | Rafi Ahmed | Grew up in house |
| Listeria Monocytogenes-DbGP33 | Hao Shen | Grew up in house |
| Influenza-PR8-DbGP33 | Richard J. Webby | Grew up at St. Jude Children’s Hospital |
| Influenza PA protein sense primer | (Laidlaw et al., 2013) | CGGTCCAAATTCCTGCTGAT |
| Influenza PA protein anti-sense primer | (Laidlaw et al., 2013) | CATTGGGTTCCTTCCATACA |
| Influenza PA protein probe | (Laidlaw et al., 2013) | 6FAMCCAAGTCATGAAGGAGAGGGAATACCGCTTAMRA |
| B16-DbGP33 | (Prévost-Blondel et al., 1998) | Grew up in house |
| In vitro culture and retroviral transduction reagents | ||
| Recombinant human IL-2 | NIH | N/A |
| Anti-Mouse CD3(145-2C11) | BioLegend | Cat# 100302, RRID:AB_312667 |
| Anti-Mouse CD28(37.51) | Thermofisher | Cat# 16-0281-82, RRID:AB_468921 |
| EasySep™ Mouse CD8+ T Cell Isolation Kit | STEMCELL Technologies | Cat# 19853 |
| RPMI-1640 medium | Corning/Mediatech | Cat# 10-040-CV |
| DMEM medium | Corning/Mediatech | Cat# 10-017-CV |
| HI Fetal Bovine Serum | Thermofisher | Cat# 26170-043 |
| HEPES | Thermofisher | Cat# 15630080 |
| Non-Essential Amino Acids | Thermofisher | Cat# 11140050 |
| Penicillin-Streptomycin | Thermofisher | Cat# 15140122 |
| β-mercaptoethanol | Sigma-Aldrich | Cat# M6250-500ML |
| Opti-MEM | Thermofisher | Cat# 31985088 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003-G |
| Lipofectamine™ 3000 Transfection Reagent | Thermofisher | Cat# L3000001 |
| Molecular constructs | ||
| Runx1 overexpression vector | Nancy A. Speck; Addgene | N/A; Addgene Cat#80157 |
| Runx3 overexpression vector | In this paper | N/A |
| Fli1 overexpression vector | In this paper | N/A |
| Empty-VEX retroviral vector | (Kurachi et al. 2017) | N/A |
| Empty-mCherry retroviral vector | (Kurachi et al. 2017) | N/A |
| pSL21-VEX | In this paper | Addgene Cat#158230 |
| pSL21-mCherry | In this paper | Addgene Cat#164410 |
| LRG2.1 | Addgene | Cat#108098 |
| MSCV Retroviral Expression System | Takara Bio. | Cat#634401 |
| BbsI | NEB | Cat#R0539L |
| Esp3I(BsmBI) | Thermofisher | Cat#ER0451 |
| Phusion Flash High Fidelity PCR Master Mix with HF buffer | Thermofisher | Cat#F531L |
| Gibson Assembly Master Mix | NEB | Cat#E2611L |
| T4 DNA Polymerase | NEB | Cat#M0203L |
| DNA Polymerase I, Large (Klenow) Fragment | NEB | Cat#M0210L |
| T4 polynucleotide kinase | NEB | Cat#M0201L |
| Klenow Fragment | NEB | Cat#M0212L |
| PrimeSTAR HS DNA Polymerase | Takara Bio. | Cat#R040A |
| Quick T4 DNA Ligase | NEB | Cat#M2200 |
| Antibodies for biochemical experiments | ||
| Anti-Mouse Fli1 | Abcam | Cat# ab15289 |
| Guinea Pig anti-Rabbit IgG (Heavy & Light Chain) Antibody | Antibodies-online | Cat# ABIN101961 |
| Anti-Mouse GAPDH | Cell Signaling Technology | Cat# 14C10 |
| IRDye 800 CW Goat anti-Rabbit Antibody | Licor | #92632211 |
| RNA-Sequencing Processing Reagents | ||
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing (24 rxn) | Takara Bio | Cat# 634889 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat# FC-131-1024 |
| Agencourt AMPure XP | Beckman | Cat# A63880 |
| HSD5000 ScreenTape | Agilent | Cat# 5067-5592 |
| HSD1000 ScreenTape | Agilent | Cat# 5067-5584 |
| ATAC-Sequencing Processing Reagents | ||
| IGEPAL-CA-630 | Sigma-Aldrich | Cat# I8896 |
| Tn5 Transposes | Illumina | Cat# FC-121-1030 |
| MinElute PCR Purification Kit | Qiagen | Cat# 28004 |
| NEBNext High-Fidelity 2x PCR Master Mix | New England Labs | Cat# M0541 |
| CUT&RUN Processing Reagents | ||
| Digitonin | Millipore | Cat# 300410-1GM |
| BioMag Plus Concanavalin A (10mL) | Bangs laboratories | Cat# BP531 |
| Complete, EDTA-free Protease Inhibitor Cocktail | Sigma | Cat# 4693132001 |
| Glycogen | Thermo | Cat# R0561 |
| Proteinase K | Denville Scientific | Cat# CB3210-5 |
| RNase A | Thermo | Cat# EN0531 |
| Spermidine | Sigma | Cat# 85558-1G |
| NEB Next Ultra II DNA library prep kit | NEB | Cat# E7645L |
| Illumina Sequencing Reagents | ||
| NEBNext Library Quant Kit for Illumina | NEB | Cat# E7630L |
| NextSeq 500/550 High Output Kit (75 cycles) v2.5 kit | Illumina | Cat# 20024906 |
| NextSeq 500/550 High Output Kit (150 cycles) v2.5 kit | Illumina | Cat# 20024907 |
| Software and Algorithms | ||
| FlowJo v10.4.2 | TreeStar | https://www.flowjo.com/solutions/flowjo/downloads |
| Prism Version 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| IGV v2.4.16 | The Broad Institute | https://software.broadinstitute.org/software/igv/download |
| Deseq2 | (Love et al., 2014) | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Great (v3.0) | (McLean et al., 2010) | http://great.stanford.edu/public/html/ |
| GSEA (v3.0) | The Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| ClusterProfiler | (Yu et al., 2012) | https://guangchuangyu.github.io/2016/01/go-analysis-using-clusterprofiler/ |
| Homer (v4.6) | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/download.html |
| Bowtie2 v2.3.4.1 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Picard v1.96 | The Broad Institute | https://broadinstitute.github.io/picard/index.html |
| Samtools v1.1 | (Li et al., 2009) | http://samtools.sourceforge.net |
| Bedtools v2.28.0 | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/# |
| bedGraphToBigWig (UCSC) | UCSC Genome | http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/ |
| MACS v2.1 | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| HOMER v4 | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| ChIPpeakAnno | (Zhu et al., 2010) | https://bioconductor.org/packages/release/bioc/html/ChIPpeakAnno.html |
| ComplexHeatmap | (Gu et al., 2016) | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Datasets | ||
| Blacklist | ENCODE | https://sites.google.com/site/anshulkundaje/projects/blacklists |
| TEFF gene signature | (Bengsch et al., 2018) | N/A |
| TEX precursor gene signature | (Chen et al., 2019b) | GSE131535 |
| sgCtrl vs sgFli1 RNA sequencing at D8 Cl13 p.i. |
In this paper | GSE149838 |
| sgCtrl vs sgFli1 ATAC sequencing at D9 Cl13 p.i. |
In this paper | GSE149836 |
| IgG vs Fli1-ab(ab15289) CUT&RUN Sequencing at D8 Cl13 p.i. |
In this paper | GSE149837 |
DATA AND CODE AVAILABILITY
All the genomic data, including RNA-Seq, ATAC-Seq and CUN&RUN data are available on GEO indicated in the key resource table. The full transcription factor CRISPR screening data will be available upon requests to the corresponding authors.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
CD4CRE, LSL-Cas9-GFP and Constitutive-Cas9-GFP mice were purchased from Jackson Laboratory. LSL-Cas9-GFP mice were bred to CD4CRE mice and TCR transgenic P14 C57BL/6 mice (TCR specific for LCMV DbGP33–41) and back crossed for more than 6 generations before use. Constitutive-Cas9-GFP mice were bred to TCR transgenic P14 C57BL/6 mice. Constitutive-Cas9-GFP mice for recipient use were bred in house. 6-8 week-old C57BL/6 Ly5.2CR (CD45.1) or C57BL/6 (CD45.2) mice were purchased from NCI. 6 week-old C57BL/6 (CD45.2) mice were purchased from Jackson Laboratory. 5-7 week-old Rag2−/− C57BL/6 mice were purchased from Jackson Laboratory. Recipient mice for LCMV challenge were from NCI unless otherwise noted in the figure legends. Both male and female mice were used unless otherwise noted. All mice were used in accordance with Institutional Animal Care and Use Committee guidelines for the University of Pennsylvania.
Experimental models
LCMV Infection:
Mice were infected intraperitoneally (i.p.) with 2 × 105 plaque-forming units (PFU) Arm or intravenously (i.v.) with 4 × 106 PFU Cl13. Plaque assay for LCMV Cl13 to detect viral load was processed as previously described(Pauken et al., 2016). Basically, tissues were homogenized, and either 1:10, 1:102, 1:103 dilution of serum or 1:10, 1:102, 1:103, 1:104, 1:105 and 1:106 dilution of homogenized tissue-contained media were incubated on adherent Vero cells for 1 hr. Cells were overlaid with a 1:1 mixture of medium and 1% agarose and cultured for 4 days. Plaques (PFUs) were counted after overlaying with a 9.5:9.5:1 mixture of medium:1% agarose:neutral red for 16 hr.
Listeria Monocytogenes (LM) infection:
LM expressing DbGP33(LMGP33) concentration was measured by optical density (OD) after overnight culture in brain heart infusion (BHI) media (1 OD refers to 8 × 108 LMGP33). Each recipient mouse was infected intravenously (i.v.) with 1 × 105 CFU LM-gp33. Male mice were used for these LMGP33 experimentts. Adjusted survival was based on mice remaining above the mandatory Institutional Animal Care and Use Committee (IACUC) euthanasia cut off of 30% weight loss. LMGP33 colony formation per unit calculation for bacteria load was calculated using 2% agarose plate with complete BHI media. Infected organs are smashed in 1ml BHI media and 20ul(2%) of the organs are taken out. 1:10, 1:102, 1:103, 1:104, 1:105 and 1:106 dilution of the infected organ BHI media are made and plated on the BHI agarose plates. Colonies are counted after 16 hours incubation of the plates in 37°C incubator.
Influenza PR8 infection:
Mice were infected intranasally (i.n.) with PR8 strain expressing DbGP33 (PR8GP33) at a dose of 3.0 LD50. Male mice were used for these PR8GP33 experiments. Mice were anesthetized before i.n. infection. PR8 viral qPCR detection for viral RNA amount was calculated as previously described(Laidlaw et al., 2013). Total RNA (including host and viral RNA) was purified from lungs of PR8GP33infected mice as well as the paired spleen, followed by reverse transcriptions with random primers. Real-time quantitative PCR was performed on cDNA targeting the influenza PA protein with technical triplicate. Influenza viral RNA amount was standardized using influenza PA protein cDNA standards.
Tumor transfer:
B16F10 melanoma cells expressing the LCMV GP33-41 epitope (B16F10-gp33,(Prévost-Blondel et al., 1998)) were maintained at 37 °C in DMEM medium supplemented with 10% FBS, penicillin, streptomycin and L-glutamine. Tumor cells were injected subcutaneously into the flanks of Rag2−/− mice at 1 x 105 cells/recipient and of Cas9+ B6 mice at 2 x 105 cells/recipient. Activated sgRNA+ C9P14 cells are sorted and transferred into recipient mice at a dose of 1 x 106 cells/recipient (for Rag2−/−) or 3 x 106 cells/recipient (for Cas9+). Tumor size was measured using digital calipers every 2-3 days after inoculation.
METHOD DETAILS
Vector construction and sgRNA cloning
In this study, SpCas9 sgRNA was expressed using pSL21-VEX or pSL21-mCherry (U6-sgRNA-EFS-VEX or U6-sgRNA-EFS-mCherry, will be available through Addgene). To generate the pSL21-VEX or pSL21-mCherry, U6-sgRNA expression cassette was PCR cloned from LRG2.1 into a retroviral vector MSCV-Neo, followed by swapping the Neo selection marker with a VEX or mCherry fluorescence reporter. sgRNAs were cloned by annealing two DNA oligos and T4 DNA ligation into a Bbs1- digested pSL21-VEX or pSL21-mCherry vector. To improve U6 promoter transcription efficiency, an additional 5’ G nucleotide was added to all sgRNA oligo designs that did not already start with a 5’ G. Runx1 and Runx3 constructs are built on the MIGR or MSCV-mCherry constructs, empty MIGR or MSCV-mCherry are used as the controls for these vectors.
Cell culture and in vitro stimulation
CD8+ T cells were purified from spleens by negative selection using EasySep Mouse CD8+ T Cell Isolation Kit (STEMCELL Technologies) according to manufacturer’s instructions. Cells were stimulated with 100 U/mL recombinant human IL-2, 1 μg/mL anti-mouse CD3ε, and 5 μg/mL anti-mouse CD28 in RPMI-1640 medium with 10% fetal bovine serum (FBS), 10 mM HEPES , 100 μM non-essential amino acids (NEAA), 50 U/mL penicillin, 50 μg/mL streptomycin, and 50 μM β-mercaptoethanol.
Retroviral vector (RV) experiments
RVs were produced in 293T cells with MSCV and pCL-Eco plasmids using Lipofectamine 3000. RV transduction was performed as described (Kurachi et al., 2017). Briefly, CD8+ T cells were purified from spleens of P14 mice using EasySep™ Mouse CD8+ T Cell Isolation Kit. After 18-24 hrs of in vitro stimulation, P14 cells were transduced with RV in the presence of polybrene (0.5 μg/ml) during spin infection (2,000 g for 60 min at 32°C) following incubation at 37°C for 6 hrs for single RV and sgRNA library, or 12 hrs for double RV. RV-transduced P14 cells were adoptively transferred into recipient mice that were infected 24-48 hrs prior to transfer.
Flow cytometry and sorting
For mouse experiments, tissues were processed, single cell suspensions obtained, and cells were stained as described (Wherry et al., 2003). Mouse cells were stained with LIVE/DEAD cell stain (Invitrogen) and antibodies targeting surface or intracellular proteins. Intracellular cytokine staining was performed after 5 hrs ex vivo stimulation with GP33-41 peptide in the presence of GolgiPlug, GolgiStop and anti-CD107a. After stimulation, cells were stained with surface antibodies, followed by fixation with Fixation/Permeabilization Buffer and then stained with intracellular antibodies for TNF, IFN-γ and MIP1α using Permeabilization Wash Buffer according to manufacturer’s instructions. Flow cytometry was performed with an LSRII. Cell sorting experiments were performed with a BD-Aria sorter, with 70-micron nozzle and a 4°C circulating cool-down system for sequencing, western and TIDE assays.
For sorting RV+ cells optimized sorting in the transfer experiments, the BD Aria Sorter was set at 37°C and 100-micron nozzle, with a flow rate lower than 3.0. 3 X 106 Cells are concentrated in 300ul 10% complete RPMI with 100 U/mL recombinant human IL-2 during sorting. 37°C pre-warmed collection tubes with 10% complete RPMI (100 U/ml IL-2) are used. Sorted cells are washed by 37°C warm pure RPMI before transferring into recipients.
TIDE Assay
At least 1 x 104 Cas9+sgRNA+ T cell pellets were frozen down. Genomic DNA was isolated from these samples using QIAmp DNA Mini Kit. A TIDE PCR, using 2x Phusion Flash High-Fidelity PCR Master Mix and primers designed around the genome region of the sgRNA target part was run for each sample to extract the guide region from the genome DNA; the resulting products were then gel verified, PCR purified, and sent for Sanger sequencing.
Western Blot
2 x 105 T cells were sorted using FACS machine and the pellets were frozen down. Protein from these samples was extracted and denatured by boiling at 95°C in 2X working loading sample buffer (1M Tris-HCl, 10% SDS, Glycerol, 10% Bromophenol blue). Lysate was run on a 10% SDS-PAGE gel and then transferred to a nitrocellulose membrane. Primary Fli1 (1:200) and GAPDH (1:1000) antibodies were staining over night, followed 1:5000 secondary antibody staining on the next day.
OpTICS screening
• sgRNA candidate selection
271 TFs that met the following criteria were selected 1) Among the top 50 differentially expressed across (Doering et al., 2012) and (Philip et al., 2017), 2) Among the top 10 differentially open TF motifs across Naïve, D8 Arm and D8 Cl13 in the previous described(Sen et al., 2016), 3) Involved in the top immune-regulatory families, such as IRF and STAT proteins. 120 TFs were manually chosen to be included in the TF library with the following principles: 1) TFs with known functions in CD8+ T cells; 2) TF family members of those with related to immune functions, e.g. IRFs, STATs and Smads; 3) TF with the most significant differential RNA expression across published CD8+ T cell datasets; 4) TF with the highest motif enrichment in ATAC-seq data from previous CD8+ T cell data sets.
• Library construction
4-5 sgRNA were designed against individual DNA binding domains or other functional domains of each TF based on the domain sequence information retrieved from NCBI Conserved Domains Database. All of the sgRNA oligos, including positive and negative control sgRNAs, were synthesized by Integrated DNA Technologies (IDT) and pooled in equal molarity. The pooled sgRNA oligos were then amplified by PCR and cloned into BsmBI-digested SL21 vector using Gibson Assembly Kit. To verify the identity and relative representation of sgRNAs in the pooled plasmids, a deep-sequencing analysis was performed on a MiSeq instrument. We confirmed that 100% of the designed sgRNAs were cloned in the SL21 vector and the abundance of >95% of individual sgRNA constructs was within 5-fold of the mean (data not shown).
• Mouse Experimental Workflow
On day 0, C9P14 cells were isolated from the spleens and lymph nodes of CD45.2+ C9P14 mice and processed to standard T cell activation protocol using anti-CD3/CD28 and IL-2; on the same day, naïve CD45.1+ recipient mice were infected by LCMV. On D1 p.i., activated C9P14 cells were transduced by RV-sgRNA library and incubated for 6 hours before washing out the RV supernatant. 18-24 hours later, the transduced sgRNA+Cas9+ cells were sorted. Then, 10% of the sgRNA+Cas9+ T cells were frozen down as a D2 baseline (T0 time point) control prior to any selection, while 90% of the cells are transfer to the infected recipients (maximum 1X105 cells/recipient). On the T1 time point (D8 in the graph), sgRNA+Cas9+ CD45.2+ T cells were sorted out from multiple organs of the recipients. For the test screening, both male and female donor/recipient mice were used; for the 120-TF library screening, male mice were used.
• Isolated library construction and MiSeq processing
To quantify the sgRNA abundance of reference and end time points, the sgRNA cassette was PCR amplified from genomic DNA using high-fidelity polymerase. The PCR product was end-repaired by T4 DNA polymerase, DNA Polymerase I, Large (Klenow) Fragment, and T4 polynucleotide kinase. Next, a 3’ A-overhang was then added to the ends of blunted DNA fragments with Klenow Fragment (3’−5’ exo-). The DNA fragments were ligated to diversity-increased custom barcodes with Quick ligation kit. Illumina paired-end sequencing adaptors were attached to the barcoded ligated products through PCR reaction with high-fidelity polymerase. The final product was quantified by Bioanalyzer Agilent DNA 1000 and pooled together in equal molar ratio and pair-end sequenced by using MiSeq (Illumina) with MiSeq Reagent Kit V3 150-cycle (Illumina).
• Data processing
The sequencing data was de-multiplexed and trimmed to contain only the sgRNA sequence cassettes. The read count of each individual sgRNA was calculated with no mismatches and compared to the sequence of reference sgRNA as described previously(Shi et al., 2015). Data of each sample were normalized to the same read account. Waterfall plots (Figure 1B): For each gene, mean of the log2 fold changes from multiple sgRNA was computed. Heatmap (Figure 1C): For each gene, mean of the log10 fold changes from multiple sgRNA was computed. In the matrix of genes by conditions, quantile normalization was performed across conditions such that each condition has the same distribution of values. Genes were ordered by the mean value of each row. Histogram (Figure 1D): For each condition, the background (grey bars and the histogram) was plotted for sgRNAs of all the genes. The 5% and 95% intervals were extracted by using the 5th percentile and 95th percentile of background values. The red bars show the log folds change for sgRNAs for one gene (or the control)
RNA-Sequencing
• Experiment workflow
At D8 p.i. with Cl13, CD8+ T cells were isolated from spleens of infected recipients. Male mice were used for these experiments. VEX+GFP+ cells are sorted using FACS with >95% purity. RNA was isolated using the QIAGEN RNeasy Micro Kit with 2 x 104 cell per sample. cDNA libraries were generated using SMARTSeq V4 Ultra Low kit. Libraries were quantified by qPCR using a KAPA Library Quant Kit (KAPA Biosystems). Normalized libraries were pooled, diluted to 1.8pg/ml loaded onto a TG NextSeq 500/550 Mid Output Kit v2 (150 cycles, 130M reads, Illumina) and paired-end sequencing was performed on a NextSeq 550 (Illumina). The estimated read depth per sample is 15M reads.
• Data processing
Raw FASTQ files from RNAseq paired-end sequencing were aligned to the GRCm38/mm10 reference genome using Kallisto (https://pachterlab.github.io/kallisto/). Sequencing reads were read in for 19357 genes and 8 samples. Genes with zero read count in more than three conditions were filtered out. 13628 genes remained after this step. Then, differential expression analysis was run using DESeq 2 package. The expression of 1440 genes were found to significantly differ between the two conditions at a BH corrected P-value < 0.05. GO enrichment analysis was performed using ClusterProfiler. The top 20 most enriched pathways are shown in the plot. GSEA was performed using the Broad Institute software (https://www.broadinstitute.org/gsea/index.jsp). Enrichment scores were calculated by comparing sgCtrl to sgFli1 groups. TEX precursor gene signature was from(Chen et al., 2019b). TEFF gene signature was from(Bengsch et al., 2018).
ATAC-Sequencing
• Experimental Workflow
ATACseq sample preparation was performed as described with minor modifications (Buenrostro et al., 2013). Male mice were used for these experiments. VEX+GFP+ cells were sorted using FACS with >95% purity. Sorted cells (2.5 x 104) were washed twice in cold PBS and resuspended in 50μl of cold lysis buffer (10nM Tris-HCl, pH 7.4, 10mM NaCl, 3mM MgCl2, 0.1% Tween). Lysates were centrifuge (750xg, 10min, 4°C) and nuclei were resuspended in 50μl of transposition reaction mix (TD buffer [25μl], Tn5 Transposase [2.5μl], nuclease-free water [22.5μl]; (Illumina)) and incubated for 30min at 37°C. Transposed DNA fragments were purified using a Qiagen Reaction MiniElute Kit, barcoded with NEXTERA dual indexes (Illumina) and amplified by PCR for 11 cycles using NEBNext High Fidelity 2x PCR Master Mix (New England Biolabs). PCR products were purified using a PCR Purification Kit (Qiagen) and amplified fragment sizes were verified on a 2200 TapeStation (Agilent Technologies) using High Sensitivity D1000 ScreenTapes (Agilent Technologies). Libraries were quantified by qPCR using a KAPA Library Quant Kit (KAPA Biosystems). Normalized libraries were pooled, diluted to 1.8pg/ml loaded onto a TG NextSeq 500/550 Mid Output Kit v2 (150 cycles, 130M reads, Illumina) and paired-end sequencing was performed on a NextSeq 550 (Illumina). Estimation read depth per sample is 10M reads.
• Data processing
Raw ATACseq FASTQ files from paired-end sequencing were processed using the script available at the following repository (https://github.com/wherrylab/jogiles_ATAC). Samples were aligned to the GRCm38/mm10 reference genome using Bowtie2. We used samtools to remove unmapped, unpaired, mitochondrial reads. ENCODE blacklist regions were also removed (https://sites.google.com/site/anshulkundaje/projects/blacklists). PCR duplicates were removed using Picard. Peak calling was performed using MACS v2 (FDR q-value 0.01). For each experiment, we combined peaks of all samples to create a union peak list and merged overlapping peaks with BedTools merge. The number of reads in each peak was determined using BedTools coverage. Differentially accessible regions were identified following DESeq2 normalization using an FDR cut-off < 0.05 unless otherwise indicated. Motif enrichment was calculated using HOMER (default parameters) on peaks differentially accessible across sgCtrl group and sgFli1 group. Transcription binding site prediction analysis was performed using known motif discovery strategy.
CUT&RUN
• Experimental Workflow
CUT&RUN experiments were performed as previously described(Skene et al., 2018) with modifications. Briefly, 2x105 sorted cells were washed twice with 1 ml of cold wash buffer (20 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, and protease inhibitor cocktails from Sigma) in 1.5ml tubes. Cells were then resuspended in 1 ml of cold wash buffer and incubated with 10 μl of BioMagPlus Concanavalin A (Bangs laboratories) by rotating at 4°C for 25 min to allow the cells to bind. Tubes were placed on a magnetic stand and liquid was removed after the solution turned clear. Primary antibody in 250 μl of cold antibody buffer (20 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 2 mM EDTA, 0.1% digitonin, and protease inhibitor cocktails from Sigma) was added to the tubes and rotated at 4°C overnight. The next day, after washing cells once with 1 ml of cold wash buffer, protein A-MNase (pA-MN) in 250 μl of cold digitonin buffer (20 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 0.1% digitonin, and protease inhibitor cocktails from Sigma) was added to the tube and rotate at 4°C for 1 h. To wash away unbound pA-MN, cells were washed twice with 1 ml of cold digitonin buffer, and then resuspended in 150 μl of cold digitonin buffer. The tubes were placed on a pre-cooled metal block. To initiate pA-MN digestion, 3 μl of 0.1 M CaCl2 was mixed with cells in 150 μl cold digitonin buffer by gently flicking the tubes 10 times. Tubes were immediately placed back in the metal block. After 30 min incubation, the digestion was stopped by adding 150 μl of 2x stop buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.02% Digitonin, 50 μg/ml RNase A, 50 μg/ml Glycogen, and 4 pg/ml yeast heterologous spike-in DNA). Target chromatin was released by incubating the tubes on a heat block at 37°C for 10 min. Supernatant was spun at 16,000 g for 5 min at 4°C and transferred to a new tube. Chromatin was incubated with 3 μl of 10% SDS and 2.5 μl of 20 μg/ml proteinase K at 70°C for 10 min, followed by phenol:chloroform:isoamyl alcohol extraction. Upper phase containing DNA was mixed with 20 mg of glycogen and incubated with 750 μl of cold 100% ethanol at −20°C overnight. DNA was precipitated by centrifugation at 20,000 g for 30 min at 4°C. DNA pellets was washed once by cold 100% ethanol, air-dried, and stored at −20°C for library preparation. Protein A-MNase (batch 6, use at 1:200) and yeast heterologous spike-in DNA were kindly provided by Dr. Steve Henikoff. The antibodies used were: Fli1, ab15289, used at 1:50 (abcam) and guinea pig anti-rabbit IgG, used at 1:100, ABIN101961 (antibodies-online).
CUT&RUN DNA library was prepared as previously descried(Liu et al., 2018) with slight modifications. Briefly, all DNA precipitated from pA-MN digestion was used for library preparation using NEBNext Ultra II DNA Library Prep Kit (NEB). The adaptor was diluted to 1:25 for adaptor ligation. DNA was barcoded and amplified for 14 PCR cycle, and DNA library was cleaned up by AMPure XP beads(Liu et al., 2018). The library quality was checked with Qubit and bioanalyzer, and the quantity of the library was determined by qPCR using NEBNext Library Quant Kit for Illumina (NEB) according to manufacture’s instruction. Eighteen barcoded libraries were pooled at equal molarity and sequenced in the NextSeq 550 platform with NextSeq 500/550 High Output Kit (75 cycles) v2.5 kit. Paired-end sequencing was carried out (42:6:0:42).
• Data processing
Paired-end reads were aligned to mm10 reference genome using Bowtie2 v2.3.4.1 with options suggested by Henikoff (Skene et al., 2018). Picard tools v1.96 was used to remove presumed PCR duplicates using the MarkDuplicates command. Bam files containing uniquely mapped reads were created using Samtools v1.1. For downstream analysis, biological replicates (3 per condition) were merged at this step. Bedtools v2.28.0 was used to generate fragment BED files with size 40bp-500bp. Blacklist regions, random chromosomes, and mitochondria were removed. Filtered BED files were used for downstream analysis. Read per million (RPM) normalized bigwig files were created using bedGraphToBigWig (UCSC) and were used to visualize binding signals. Peaks were called using MACS v2.1 using the broadPeak setting with p-value cutoff of 1e-8, -f BEDPE and IgG as controls. Genes proximal to peaks were annotated against the mm10 genome using annotatePeaks.pl from HOMER v4. Fli1 binding motifs were identified using findMotifsGenome.pl from HOMER v4. Venn diagram of comparison with ATAC-Seq peaks was plotted using Bioconductor package ChIPpeakAnno. Heatmap was generated using Bioconductor package ComplexHeatmap.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was calculated with unpaired two-tailed student’s t-test or one-way ANOVA with Tukey’s multiple comparisons test by Prism 7 (GraphPad Software). The number of mice used per experiment, number of experimental repeats and P values are indicated in the figure legends.
Supplementary Material
Figure S1 High-efficiency in vivo gene editing and screening using retroviral-transduced sgRNA in Cas9+ antigen specific CD8+ T cells. (Related to Figure 1)
A. Optimizing sgRNA backbone compared to the original sgRNA.
B. Experimental design for in vivo gene editing test. At Day 0 (D0), CD8+ T cells were isolated from CD45.1+ LSL-Cas9+CD4CRE+P14 (C9P14) donor mice and activated with anti-CD3, anti-CD28 and IL-2; CD45.2+ WT recipient mice were infected with Cl13. On D1 p.i., activated C9P14 cells were transduced with either Ctrl-sgRNA(sgCtrl) or Pdcd1-sgRNA (sgPdcd1). Six hours after transduction, 5 X 104 activated donor cells were adoptively transferred into infected recipient mice. C9P14 cells were then isolated, at the indicated times, from different organs of recipient mice for analysis.
C. D2 in vitro transduction efficiency of activated C9P14 cells with sgRNA vector (mCherry). Gates are set based on the non-transduced control.
D. Flow cytometry plots of the Cas9(GFP)+sgRNA(mCherry)+ population in the spleen at D9 p.i. for the sgCtrl group and sgPdcd1 groups.
E. Histogram of PD-1 expression and statistical analysis of the PD-1+ population from the Cas9+sgRNA+ P14 cells in the blood (D7 p.i.), spleen (D9 p.i.) or liver (D9 p.i.).
F. Sanger sequencing results for the Pdcd1 locus from FACS purified Cas9+sgRNA+ P14 cells from the sgCtrl group (pooled 5 mice) or the sgPdcd1 group (pooled 2 mice).
G. Histogram of KLRG1 or CXCR3 expression and statistical analysis of KLRG1+ or CXCR3+ populations from Cas9+sgRNA+ P14 cells from the spleen (D8 p.i. of Arm) between targeted-sgRNA and sgCtrl groups.
H. Log2 fold change (L2FC) of D8 p.i.(T1) to D2 baseline(T0) across different sgRNAs from spleen or liver under 3 conditions during Arm infection. The x-axis represents different sgRNAs, y-axis represents L2FC of D8 p.i.(T1) to D2 baseline(T0). Condition 1: No optimized sorting, average input coverage ~100 cells/sgRNA, Cas9+/+ P14 donor. Condition 2: Optimized sorting (in Star Methods), average input coverage ~400 cells/sgRNA, Cas9+/+ P14 donor. Condition 3: Optimized sorting, average input coverage ~400 cells/sgRNA, Cas9+/− P14 donor. Example target genes are highlighted with the indicated color.
I. Rank correlation of targeted genes between 2 independent screenings of Cas9+/+ or Cas9+/− donor P14 groups. The mean values of sgRNA L2FC from each targeted gene were calculated and ranked from the independent screenings. Pearson correlation of the rankings was calculated.
J. Fold change enrichment of sgPdcd1 at D14 p.i. of Cl13 in the spleen. Data from the screening performed in Figure 1A-1C.
K. Pearson correlation of different samples from the screening performed in Figure 1A-1C. Sample collection time (days p.i.), LCMV infection and organ of sorted Cas9+sgRNA+ cells are presented.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (two-tailed Student’s t-test). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 3 mice/group.
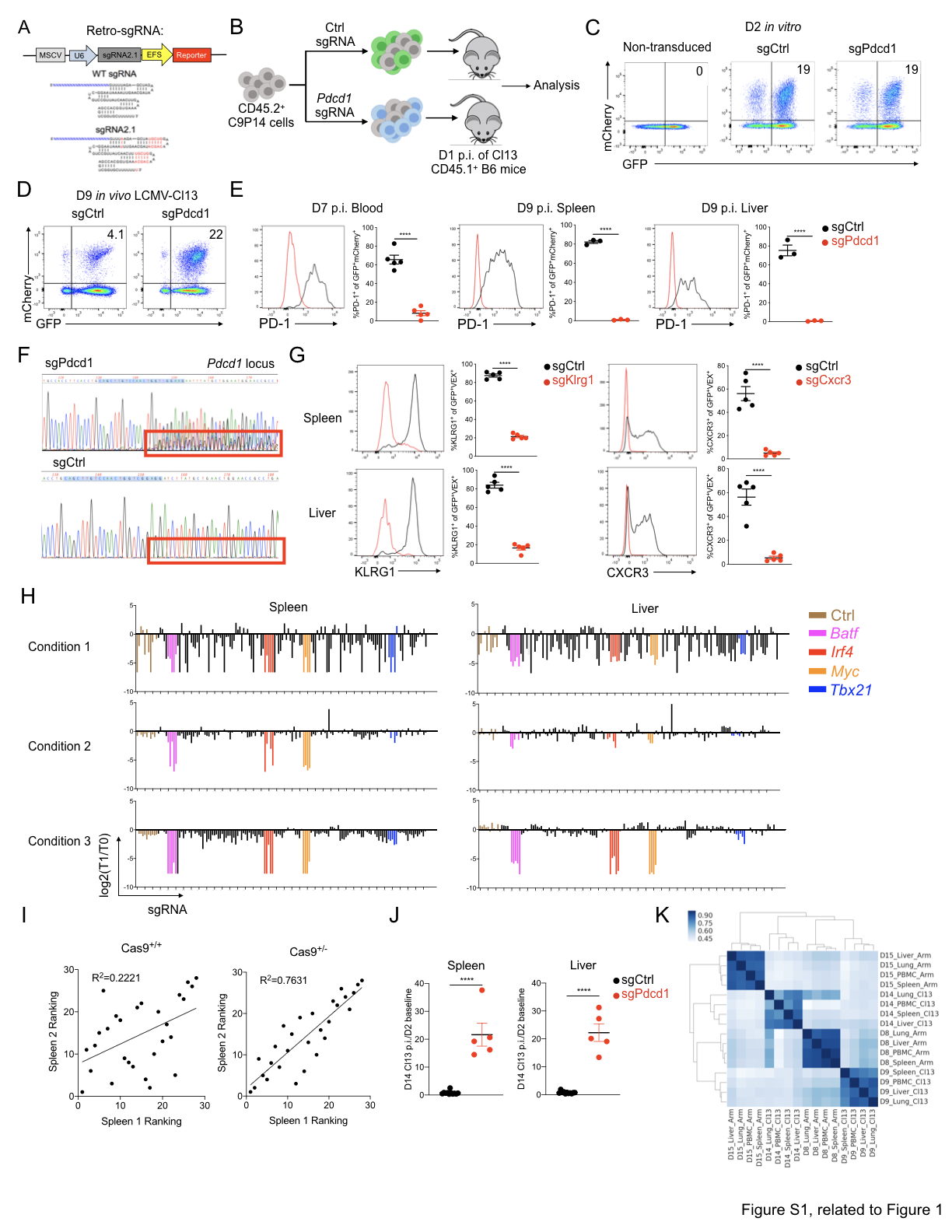{kind=link}
Figure S2 Genetic deletion of Fli1 leads to a greater T cell expansion. (Related to Figure 1)
A. TIDE assay results showing genome disruption efficiency of the Fli1 locus for the Cas9+Fli1-sgRNA (sgFli1)+ cells. Genome disruption was detected by sanger sequencing.
B. Flow cytometry plots (gated on donor P14 cells) of the Cas9 (GFP)+sgRNA(VEX)+ population on D2 after in vitro transduction; D8 and D15 p.i. of Arm from splenocytes from sgCtrl or 2 Fli1-sgRNA(sgFli1_290 and sgFli1_360) groups. 5 X 104 activated donor cells were adoptively transferred into congenic infected recipient mice on D1 p.i.
C. Normalized Cas9+sgRNA+ cell numbers from blood, liver and lung from sgCtrl and the two sgFli1 groups at D8 and D15 p.i. of Arm. Cell numbers normalized to the sgCtrl group based on D2 in vitro transduction efficiency.
D. Flow cytometry plots (gated on donor P14 cells) of Cas9+sgRNA+ P14 cells on D2 post in vitro transduction, D9 and D15 p.i. with Cl13 (speen) for sgCtrl and the two sgFli1 groups. 5 X 104 activated donor P14 cells were adoptively transferred into the infected recipient mice on D1 p.i.
E. Normalized Cas9+sgRNA+ cell numbers from blood, liver and lung from sgCtrl and the two sgFli1 groups at D9 and D15 p.i. of Cl13. Cell number normalized to the sgCtrl group based on D2 in vitro transduction efficiency.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (One-Way Anova analysis). Data are representative of 2-4 independent experiments (mean±s.e.m.) with at least 3 mice/group.
{kind=link}
Figure S3 Fli1 inhibits terminal TEFF differentiation without impacting TMEM cell formation. (Related to Figure 1)
A. Flow cytometry plots and statistical analysis of KLRG1LoCD127Lo cells. Frequencies (left) and numbers (right) from spleen for sgCtrl and 2 sgFli1 groups on D8 and D15 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
B. Flow cytometry plots and statistical analysis of GzmB+ and TCF-1+ C9P14 cells. Frequencies from spleen for sgCtrl and sgFli1 (2 sgRNA combined) groups on D8 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
C. Histogram and statistical analysis of T-bet and Eomes expression in C9P14 cells from spleen for sgCtrl and sgFli1 (2 sgRNA combined) groups on D8 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells. Naïve T cell (CD44−) staining shown in grey.
D. Flow cytometry plots and statistical analysis of cytokine producing C9P14 cells after 5 hours of stimulation (see Star Methods). IFNγ+, IFNγ+TNF+ and MIP1α+CD107a+ frequencies (left) and numbers (right) from spleen for non-stim P14 cells, sgCtrl and sgFli1 (2 sgRNA combined) groups on D8 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
E. Statistical analysis of total, terminal effector (TE) or memory precursor (MP) C9P14 cell numbers in the blood of sgCtrl and sgFli1 (2 sgRNA combined) groups on D8, D20 and D29 p.i. with Arm. Data normalized to sgCtrl+ D2 p.i. transduction efficiency. Normalized sgRNA+ C9P14 cell number is around 75 cells/1X106 PBMCs on the transfer day (D1 p.i.)
F. Statistical analysis of TE or MP C9P14 frequency and total, TE or MP C9P14 cell number in the spleen of sgCtrl and sgFli1 (2 sgRNA combined) groups on D29 p.i. of Arm. Data normalized to sgCtrl+ D2 p.i. transduction efficiency.
G. Histograms of Bcl-2, Bcl-XL and Bim expression of total C9P14 cells from spleen of sgCtrl and sgFli1 (2 sgRNA combined) groups on D15 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells. Naïve T cell (CD44−) staining shown in grey.
H-J. Statistical analysis of Bcl-2, Bcl-XL, Bim expression and Bcl-2/Bim and Bcl-XL/Bim ratios for total (H), TE (I) and MP(J) C9P14 cells from spleen for sgCtrl and sgFli1 (2 sgRNA combined) groups on D15 p.i. with Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (One-Way Anova analysis). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 4 mice/group.
{kind=link}
Figure S4 Fli1 restrains TEFF-like differentiation during chronic infection. (Related to Figure 3 and Figure 4)
A-B. Statistical analysis of Ly108−CD39+ or TCF-1−GzmB+ TEFF-like cells and Ly108+CD39− or TCF-1+GzmB− TEX precursor cell numbers from spleen for sgCtrl and the two sgFli1 groups at D8 (A) and D15 (B) p.i. with Cl13. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
C. Histogram and statistical analysis of Eomes, T-bet and Tox expression in C9P14 cells from spleen for sgCtrl and sgFli1 groups on D8 and D15 p.i. with Cl13. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells. Naïve T cell (CD44−) staining shown in grey.
D. Flow cytometry plots and statistical analysis of cytokine producing C9P14 cells after 5 hours of stimulation (see Star Methods). IFNγ+, IFNγ+TNF+ and MIP1α+CD107a+ frequencies (left) and numbers (right) from spleen for non-stim P14 cells, sgCtrl and sgFli1 (2 sgRNA combined) groups on D8 p.i. with Cl13. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
E. Flow cytometry plots and statistical analysis of cell number of CD45.2+VEX+ P14 for Empty-RV and Fli1-OE-RV groups at D8 and D16 p.i. with Cl13. At D0, CD45.2+ P14 cells were activated and CD45.1+ recipient mice were infected with Cl13. On D1 p.i., activated P14 were transduced with either Empty-RV or Fli1-OE-RV for 6 hours. On D2 p.i., VEX+ P14 cells were sorted from each RV transduced group and 1X105 cells were adoptively transferred into the infected recipients.
F-G. Flow cytometry plots and statistical analysis of Ly108−CD39+ or TCF-1−Gzmb+ TEFF-like cells and Ly108+CD39− or TCF-1+Gzmb−TEX precursor frequencies for Empty-RV and Fli1-OE-RV groups on D8 and D16 p.i. of Cl13. Gated on VEX+ P14 cells.
H. Statistical analysis of CX3CR1+ and Tim-3+ frequencies for Empty-RV and Fli1-OE-RV groups on D8 and D16 p.i. of Cl13. Gated on VEX+ P14 cells.
*P<0.05, **P<0.01, ***P<0.001 versus control (One-Way Anova analysis). Data are representative of 3 independent experiments (mean±s.e.m.) with at least 4 mice/group for A-D.
{kind=link}
Figure S5. Transcriptional and epigenetic profiling addresses Fli1 inhibiting TEFF-like differentiation by coordinating with Runx1 and antagonizing Runx3 function. (Related to Figure 5)
A. PCA plot for RNA-seq data ffor sgCtrl, sgFli1_290 and sgFli1_360 groups on D8 p.i. with Cl13.
B. Overlap of all CUT&RUN Fli1 binding peaks with ATAC-seq detected peaks in sgCtrl and sgFli1 groups.
C. Histogram of all CUT&RUN peaks co-localized with the ATAC-seq peaks. Peaks co-localized with the ATAC-seq peaks are red; peaks not co-localized are blue.
D. CUT&RUN IgG or Fli1 binding signals for P14 cells on D9 p.i. and open chromatin region signals detected by ATAC-seq for sgCtrl, sgFli1_290 and sgFli1_360 groups in the Tcf7 and Id3 loci.
E. Flow cytometry plots and statistical analysis of CD45.1+mCherry+ and Ly108−CD39+/ Ly108+CD39− P14 cell numbers for Empty-RV or Runx1-OE-RV on D2 in vitro and D7 p.i. On D0, CD45.1+ P14 cells were activated and CD45.2+ recipient mice were infected with Cl13; On D1 p.i., activated P14 cells were transduced with Empty-RV or Runx1-OE-RV for 6 hours, and 1X105 transduced P14 cells were adoptively transferred into infected recipient mice. Flow cytometry plots gated on the CD45.1+ P14 cells.
F. D2 in vitro transduction efficiency of sgCtrl-VEX+Empty-mCherry, sgCtrl-VEX+Runx1-mCherry, sgFli1_290-VEX+Empty-mCherry, and sgFli1_290-VEX+Runx1-mCherry C9P14 cells.
G. Statistical analysis of VEX+mCherry+ C9P14 cells and Ly108−CD39+/Ly108+CD39− cell numbers from spleen for sgCtrl-VEX+Empty-mCherry, sgCtrl-VEX+Runx1-mCherry, sgFli1_290-VEX+Empty-mCherry, and sgFli1_290-VEX+Runx1-mCherry groups on D7 p.i. with Cl13. Gated on Cas9(GFP)+CD45.2+ P14 cells.
H. D2 in vitro transduction efficiency of sgCtrl-mCherry+Empty-VEX, sgCtrl-mCherry+Runx3-VEX, sgFli1_290-mCherry+Empty-VEX, and sgFli1_290-mCherry+Runx3-VEX C9P14 cells.
I. Statistical analysis of VEX+mCherry+ C9P14 cells and Ly108−CD39+/Ly108+CD39− cell numbers from spleen for sgCtrl-mCherry+Empty-VEX, sgCtrl-mCherry+Runx3-VEX, sgFli1_290-mCherry+Empty-VEX and sgFli1_290-mCherry+Runx3-VEX at D8 p.i. with Cl13. Gated on Cas9+CD45.2+ P14 cells.
*P<0.05, **P<0.01 versus control (two-tailed Student’s t-test). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 5 mice/group.
{kind=link}
Figure S6. Fli1-deficiency results in CD8+ T cell expansion during influenza virus or Listeria monocytogenes infection. (Related to Figure 6)
A. Adjusted survival curve of LCMV Cl13 infected mice for sgCtrl+, sgFli1_290+ and sgFli1_360+ C9P14 recipient mice (1.5 x 105 cell/mouse, N=5 per group) Note, JAX recipient mice were used here instead of NCI recipients resulting in the difference in pathogenesis. Dashed line represents the time of C9P14 adoptive transfer.
B. Flow cytometry plots showing sgRNA(VEX)+ C9P14 cells in the lungs of influenza virus (PR8-GP33) infected mice, comparing non-transfer(NT), sgCtrl and sgFli1 groups at D8 p.i. “Recovered” was defined by complete weight recovery on D8 p.i.
C. Correlation of sgRNA+ C9P14 cell numbers and weight ratio (D8 p.i./D2 p.i.) during the PR8-GP33 infection. In the “weight not recovered” group (weight ratio between 0.7 and 1.0), sgRNA+ C9P14 cell numbers were further compared.
D. Flow cytometry plots and statistical analysis of sgRNA+ C9P14 cells in the spleens of PR8-GP33-infected recipient mice for non-transfer (NT), sgCtrl and sgFli1 groups on D8 p.i.
E. Flow cytometry plots and statistical analysis of sgRNA+ C9P14 cells in the spleens of Listeria monocytogenes-infected recipients for non-transfer (NT), sgCtrl and sgFli1 groups on D7 p.i.
*P<0.05, **P<0.01 versus control (two-tailed Student’s t-test and One-Way Anova analysis). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 6 mice/group.
{kind=link}
Figure S7. Deleting Fli1 in CD8+ T cells leads to better tumor protection in an immune competent mice. (Related to Figure 7)
A. Experimental design. On D0, CD45.2+Cas9+P14− mice were inoculated with 2 x 105 B16-Dbgp33 cells. On D3 post tumor inoculation (p.t.), CD8+ T cells were isolated from CD45.2+ C9P14 donor mice and activated. The next day, activated C9P14 cells were transduced with sgCtrl or sgFli1 RV for 6 hours. On D5 p.t., sgRNA (VEX)+ P14 cells were sorted from sgCtrl or sgFli1 groups, and 3 x 106 purified VEX+ C9P14 cells were adoptively transferred into tumor-bearing mice.
B. Tumor volume curve of tumor-bearing mice from NT, sgCtrl+ and sgFli1+ C9P14 cell transferred groups.
C. Tumor weight from NT, sgCtrl+ and sgFli1+ C9P14 transferred mice on D24 p.t.
D-E. Flow cytometry plots (D) and statistical analysis (E) of sgRNA (VEX)+ C9P14 cells and Ly108−CD39+/Ly108+CD39− populations from tumor for sgCtrl and sgFli1 groups on D24 p.t.
F-G. Statistical analysis of sgRNA+ C9P14 cells and Ly108−CD39+ or Ly108+CD39− populations from draining lymph node (dLN, F) and spleen (G) for sgCtrl and sgFli1 groups on D24 p.t.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (two-tailed Student’s t-test and One-Way Anova analysis). Data are representative of 3 independent experiments (mean±s.e.m.) with at least 6 mice/group.
{kind=link}
Table S1 sgRNAs used for single gene editing, related to Figure S1.
Table S2 sgRNA library targeting transcription factors for in vivo screening, related to Figure 1.
Highlights:
OpTICS In vivo CD8+ T cell CRISPR screening platform developed for focused gene discovery
OpTICS identifies genes involved in effector versus exhausted CD8+ T cells
The Fli1 transcription factor represses optimal effector CD8+ T cells during exhaustion
Fli1 antagonizes epigenetic accessibility at ETS-RUNX sites limiting Runx3 activity
An in vivo T cell CRISPR screening platform identifies Fli1 as a factor modulating CD8+ T cell effector differentiation without effects on memory or exhaustion.
Acknowledgement
We thank the Wherry and Shi Labs for discussions. This work was supported by grants from NIH (AI105343, AI117950, AI082630, AI112521, AI115712, AI108545, CA210944), Stand Up 2 Cancer and the Mark Foundation to EJW; NIH grant CA234842 to Z Chen; NIH grant CA009140 to JRG; NIH grant MH109905, HG010480 and Searle Scholar’s Program to AB. EJW is supported by the Parker Institute for Cancer Immunotherapy which supports the cancer immunology program at UPenn; SFN is supported by an Australia NH&MRC CJ Martin Fellowship (1111469) and the Mark Foundation Momentum Fellowship; JRG is supported by Cancer Research Institute-Mark Foundation Fellowship.
Footnotes
Declaration of Interests
E.J.W. has consulting agreements with and/or is on the scientific advisory board for Merck, Elstar, Janssen, Related Sciences, Synthekine and Surface Oncology. E.J.W. is a founder of Surface Oncology and Arsenal Biosciences. E.J.W. has a patent licensing agreement on the PD-1 pathway with Roche/Genentech. O.K. is an employee and shareholder of Arsenal Biosciences.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, Hösslin, von M, Cullen JG, Fan Y, et al. (2019). TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2017 545:7652 1. [DOI] [PubMed] [Google Scholar]
- Angelosanto JM, Blackburn SD, Crawford A, and Wherry EJ (2012). Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol 86, 8161–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badovinac VP, Haring JS, and Harty JT (2007). Initial T Cell Receptor Transgenic Cell Precursor Frequency Dictates Critical Aspects of the CD8+ T Cell Response to Infection. Immunity 26, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badovinac VP, Porter BB, and Harty JT (2004). CD8+ T cell contraction is controlled by early inflammation. Nature Immunology 2006 7:12 5, 809–817. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, and Ahmed R (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2017 545:7652 439, 682–687. [DOI] [PubMed] [Google Scholar]
- Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O’Brien S, Gherardini PF, Herati RS, Huang AC, Chang K-M, et al. (2018). Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 48, 1029–1045.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Freeman GJ, and Wherry EJ (2008). Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. Pnas 105, 15016–15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattman JN, Wherry EJ, Ha S-J, van der Most RG, and Ahmed R (2009). Impact of Epitope Escape on PD-1 Expression and CD8 T-Cell Exhaustion during Chronic Infection. J. Virol 83, 4386–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, Awad ME, Rengarajan S, Volorio A, McBride MJ, et al. (2017). Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, and Mackall CL (2019). CAR T cell therapy: inroads to response and resistance. Nature Reviews Immunology 2012 12:11 19, 73–74. [DOI] [PubMed] [Google Scholar]
- Chen J, López-Moyado IF, Seo H, Lio C-WJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, and Rao A (2019a). NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2017 545:7652 567, 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, et al. (2019b). TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Stelekati E, Kurachi M, Yu S, Cai Z, Manne S, Khan O, Yang X, and Wherry EJ (2017). miR-150 Regulates Memory CD8 T Cell Differentiation via c-Myb. Cell Reports 20, 2584–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CK, Li L, Cheng SH, Lau KM, Chan NPH, Wong RSM, Shing MMK, Li CK, and Ng MHL (2008). Transcriptional repression of the RUNX3/AML2 gene by the t(8;21) and inv(16) fusion proteins in acute myeloid leukemia. Blood 112, 3391–3402. [DOI] [PubMed] [Google Scholar]
- Choo A, Palladinetti P, Passioura T, Shen S, Lock R, Symonds G, and Dolnikov A (2006). The Role of IRF1 and IRF2 Transcription Factors in Leukaemogenesis. Cgt 6, 543–550. [DOI] [PubMed] [Google Scholar]
- Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, Groner Y, and Rao A (2009). Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. Journal of Experimental Medicine 206, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, and Wherry EJ (2012). Network Analysis Reveals Centrally Connected Genes and Pathways Involved in CD8+ T Cell Exhaustion versus Memory. Immunity 37, 1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, Park JJ, Kim HR, Errami Y, Guzman CD, et al. (2019). Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 178, 1189–1204.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, et al. (2018). Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nature Communications 2017 8 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang W-T, et al. (2018). Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nature Medicine 2018 24, 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, la Torre, de JC, and Andrian, von UH (2016). The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 45, 1270–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevet JD, Lan X, Hamagami N, Edwards CR, Sankaranarayanan L, Ji X, Bhardwaj SK, Face CJ, Posocco DF, Abdulmalik O, et al. (2018). Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grusdat M, McIlwain DR, Xu HC, Pozdeev VI, Knievel J, Crome SQ, Robert-Tissot C, Dress RJ, Pandyra AA, Speiser DE, et al. (2014). IRF4 and BATF are critical for CD8+ T-cell function following infection with LCMV. Cell Death and Differentiation 2009 16:11 21, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, Manne S, Kraya AA, Wubbenhorst B, Dorfman L, et al. (2019). A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nature Medicine 2018 25, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017 545:7652 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2017 545:7652 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2017 545:7652 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity 27, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, and Cui W (2012). Transcriptional control of effector and memory CD8+ T cell differentiation. Nature Reviews Immunology 2012 12:11 12, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG (2017). Common pitfalls in preclinical cancer target validation. Nature Reviews Cancer 2012 13:1 17, 441–450. [DOI] [PubMed] [Google Scholar]
- Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali M-AA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, et al. (2011). Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nature Immunology 2006 7:12 12, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2017 545:76521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. (2016). Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nature Communications 2017 8 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse EA, Loughran SJ, Baldwin TM, Josefsson EC, Ellis S, Watson DK, Nurden P, Metcalf D, Hilton DJ, Alexander WS, et al. (2009). Dual requirement for the ETS transcription factors Fli-1 and Erg in hematopoietic stem cells and the megakaryocyte lineage. Pnas 106, 13814–13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, et al. (2014). The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nature Immunology 2006 7:12 15, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M, Kurachi J, Chen Z, Johnson J, Khan O, Bengsch B, Stelekati E, Attanasio J, McLane LM, Tomura M, et al. (2017). Optimized retroviral transduction of mouse T cells for in vivo assessment of gene function. Nature Protocols 2017 12:9 12, 1980–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M, Ngiow SF, Kurachi J, Chen Z, and Wherry EJ (2019). Hidden Caveat of Inducible Cre Recombinase. Immunity 51, 591–592. [DOI] [PubMed] [Google Scholar]
- LaFleur MW, Nguyen TH, Coxe MA, Yates KB, Trombley JD, Weiss SA, Brown FD, Gillis JE, Coxe DJ, Doench JG, et al. (2019). A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nature Communications 2017 8 10, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw BJ, Decman V, Ali M-AA, Abt MC, Wolf AI, Monticelli LA, Mozdzanowska K, Angelosanto JM, Artis D, Erikson J, et al. (2013). Cooperativity between CD8+ T cells, non-neutralizing antibodies, and alveolar macrophages is important for heterosubtypic influenza virus immunity. PLOS Pathogens 9, e1003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 2017 14:3 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, June CH, and Thompson CB (1988). Multiple mechanisms regulate c - myc gene expression during normal T cell activation. The EMBO Journal 7, 2787–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. (2018). Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. (2015). 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature Medicine 2018 21, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al. (2019). c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2017 545:7652 576, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, et al. (2017). Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47, 1129–1141.e5. [DOI] [PubMed] [Google Scholar]
- Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, Pellegrini M, Belz GT, Smyth GK, Febbraio MA, et al. (2013). The transcription factor IRF4 is essential for TCR affinity–mediated metabolic programming and clonal expansion of T cells. Nature Immunology 2006 7:12 14, 1155–1165. [DOI] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017 545:7652 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, et al. (2015). The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 42, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo AL, Klonowski KD, Le Bon A, Borrow P, Tough DF, and Lefrançois L (2005). Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nature Immunology 2006 7:12 6, 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLane LM, Abdel-Hakeem MS, and Wherry EJ (2019). CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol 37, 457–495. [DOI] [PubMed] [Google Scholar]
- Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. (2017). Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 2017 545:7652 552, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi PM, Pauken KE, Paley MA, Sharpe A, and Wherry EJ (2015). Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. Journal of Experimental Medicine 212, 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, de Andrade LF, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham D, Moseley CE, Gao M, Savic D, Winstead CJ, Sun M, Kee BL, Myers RM, Weaver CT, and Hatton RD (2019). Batf Pioneers the Reorganization of Chromatin in Developing Effector T Cells via Ets1-Dependent Recruitment of Ctcf. Cell Reports 29, 1203–1220.e1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017 545:7652 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimanda JE, Ottersbach K, Knezevic K, Kinston S, Chan WYI, Wilson NK, Landry J-R, Wood AD, Kolb-Kokocinski A, Green AR, et al. (2007). Gata2, Fli1, and Scl form a recursively wired gene-regulatory circuit during early hematopoietic development. Pnas 104, 17692–17697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prévost-Blondel A, Zimmermann C, Stemmer C, Kulmburg P, Rosenthal FM, and Pircher H (1998). Tumor-infiltrating lymphocytes exhibiting high ex vivo cytolytic activity fail to prevent murine melanoma tumor growth in vivo. The Journal of Immunology 161, 2187–2194. [PubMed] [Google Scholar]
- Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, Julg B, Jesneck JL, Brosnahan K, Imam S, et al. (2010). Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nature Medicine 2018 16, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suvà ML, Rossetti NE, Boonseng WE, Oksuz O, Cook EB, et al. (2014). EWS-FLI1 Utilizes Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell 26, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe J-S, Mercan F, Rivera K, Pappin DJ, and Vakoc CR (2015). BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Molecular Cell 58, 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara SJ, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2017 545:7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao H-W, Godec J, LaFleur MW, Brown FD, et al. (2016). The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, et al. (2019). TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. U.S.a 116, 12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, Kurup SP, Van Braeckel-Budimir N, Su Y, Martin MD, et al. (2017). The transcription factor Runx3 guards cytotoxic CD8+ effector T cells against deviation towards follicular helper T cell lineage. Nature Immunology 2006 7:12 18, 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang W, Jiang Y, Boettcher M, Ding K, Mollenauer M, Liu Z, Wen X, Liu C, Hao P, Zhao S, et al. (2018). Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Pnas 115, E4051–E4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, and Vakoc CR (2015). Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nature Biotechnology 2015 33:6 33, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. (2018). Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2017 545:7652 557, 575–579. [DOI] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2016). A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 166, 1500–1511.e1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Elife SH, 2017. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Cdn.Elifesciences.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nature Protocols 2017 12:9 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science eaba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Mezzadra R, and Schumacher TN (2018). Regulation and Function of the PD-L1 Checkpoint. Immunity 48, 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiemessen MM, Baert MRM, Kok L, van Eggermond MCJA, van den Elsen PJ, Arens R, and Staal FJT (2014). T Cell Factor 1 Represses CD8+ Effector T Cell Formation and Function. The Journal of Immunology 193, 5480–5487. [DOI] [PubMed] [Google Scholar]
- Tijssen MR, Cvejic A, Joshi A, Hannah RL, Ferreira R, Forrai A, Bellissimo DC, Oram SH, Smethurst PA, Wilson NK, et al. (2011). Genome-wide Analysis of Simultaneous GATA1/2, RUNX1, FLI1, and SCL Binding in Megakaryocytes Identifies Hematopoietic Regulators. Developmental Cell 20, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco R, Alcalde V, Yang Y, Sauer K, and Zuniga EI (2009). Cell-Intrinsic Transforming Growth Factor-β Signaling Mediates Virus-Specific CD8+ T Cell Deletion and Viral Persistence In Vivo. Immunity 31, 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, and Pardoll DM (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Wang D, Diao H, Getzler AJ, Rogal W, Frederick MA, Milner J, Yu B, Crotty S, Goldrath AW, and Pipkin ME (2018). The Transcription Factor Runx3 Establishes Chromatin Accessibility of cis-Regulatory Landscapes that Drive Memory Cytotoxic T Lymphocyte Formation. Immunity 48, 659–674.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Chow RD, Bai Z, Zhu L, Errami Y, Dai X, Dong MB, Ye L, Zhang X, Renauer PA, et al. (2019). Multiplexed activation of endogenous genes by CRISPRa elicits potent antitumor immunity. Nature Immunology 2006 7:12 168, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. (2011). The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, Wang Y, Wang Y-D, Qian C, Xu B, et al. (2019). Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 2017 545:7652 576, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Duffy CR, and Allison JP (2018). Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M (2015). Molecular and cellular insights into T cell exhaustion. Nature Reviews Immunology 2012 12:11 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, and Ahmed R (2005). Low CD8 T-cell proliferative potential and high viral load limit the effectiveness of therapeutic vaccination. J. Virol 79, 8960–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 27, 670–684. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, Andrian, von UH, and Ahmed R (2003). Lineage relationship and protective immunity of memory CD8 T cell subsets. Nature Immunology 2006 7:12 4, 225–234. [DOI] [PubMed] [Google Scholar]
- Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, et al. (2016). The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Science Immunology 1, eaai8593–eaai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, Li Y, Phillips FC, Maina PK, Qi HH, et al. (2016). Tcf1 and Lef1 transcription factors establish CD8+ T cell identity through intrinsic HDAC activity. Nature Immunology 2006 7:12 17, 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Wang J, Liu Y, Xie L, Su B, Mou D, Wang L, Liu T, Wang X, Zhang B, et al. (2019). CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med 381, 1240–1247. [DOI] [PubMed] [Google Scholar]
- Yao C, Sun H-W, Lacey NE, Ji Y, Moseman EA, Shih H-Y, Heuston EF, Kirby M, Anderson S, Cheng J, et al. (2019). Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nature Immunology 2006 7:12 20, 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, Du Y, Guo J, Dai X, Wang G, et al. (2019). In vivo CRISPR screening in CD8 T cells with AAV− Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nature Biotechnology 2015 33:6 39, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, and Cui W (2019). CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset that Protects against Chronic Infection and Cancer. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Yu S, Zhao D-M, Harty JT, Badovinac VP, and Xue H-H (2010). Differentiation and Persistence of Memory CD8+ T Cells Depend on T Cell Factor 1. Immunity 33, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, and Green MR (2010). ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 High-efficiency in vivo gene editing and screening using retroviral-transduced sgRNA in Cas9+ antigen specific CD8+ T cells. (Related to Figure 1)
A. Optimizing sgRNA backbone compared to the original sgRNA.
B. Experimental design for in vivo gene editing test. At Day 0 (D0), CD8+ T cells were isolated from CD45.1+ LSL-Cas9+CD4CRE+P14 (C9P14) donor mice and activated with anti-CD3, anti-CD28 and IL-2; CD45.2+ WT recipient mice were infected with Cl13. On D1 p.i., activated C9P14 cells were transduced with either Ctrl-sgRNA(sgCtrl) or Pdcd1-sgRNA (sgPdcd1). Six hours after transduction, 5 X 104 activated donor cells were adoptively transferred into infected recipient mice. C9P14 cells were then isolated, at the indicated times, from different organs of recipient mice for analysis.
C. D2 in vitro transduction efficiency of activated C9P14 cells with sgRNA vector (mCherry). Gates are set based on the non-transduced control.
D. Flow cytometry plots of the Cas9(GFP)+sgRNA(mCherry)+ population in the spleen at D9 p.i. for the sgCtrl group and sgPdcd1 groups.
E. Histogram of PD-1 expression and statistical analysis of the PD-1+ population from the Cas9+sgRNA+ P14 cells in the blood (D7 p.i.), spleen (D9 p.i.) or liver (D9 p.i.).
F. Sanger sequencing results for the Pdcd1 locus from FACS purified Cas9+sgRNA+ P14 cells from the sgCtrl group (pooled 5 mice) or the sgPdcd1 group (pooled 2 mice).
G. Histogram of KLRG1 or CXCR3 expression and statistical analysis of KLRG1+ or CXCR3+ populations from Cas9+sgRNA+ P14 cells from the spleen (D8 p.i. of Arm) between targeted-sgRNA and sgCtrl groups.
H. Log2 fold change (L2FC) of D8 p.i.(T1) to D2 baseline(T0) across different sgRNAs from spleen or liver under 3 conditions during Arm infection. The x-axis represents different sgRNAs, y-axis represents L2FC of D8 p.i.(T1) to D2 baseline(T0). Condition 1: No optimized sorting, average input coverage ~100 cells/sgRNA, Cas9+/+ P14 donor. Condition 2: Optimized sorting (in Star Methods), average input coverage ~400 cells/sgRNA, Cas9+/+ P14 donor. Condition 3: Optimized sorting, average input coverage ~400 cells/sgRNA, Cas9+/− P14 donor. Example target genes are highlighted with the indicated color.
I. Rank correlation of targeted genes between 2 independent screenings of Cas9+/+ or Cas9+/− donor P14 groups. The mean values of sgRNA L2FC from each targeted gene were calculated and ranked from the independent screenings. Pearson correlation of the rankings was calculated.
J. Fold change enrichment of sgPdcd1 at D14 p.i. of Cl13 in the spleen. Data from the screening performed in Figure 1A-1C.
K. Pearson correlation of different samples from the screening performed in Figure 1A-1C. Sample collection time (days p.i.), LCMV infection and organ of sorted Cas9+sgRNA+ cells are presented.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (two-tailed Student’s t-test). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 3 mice/group.
Figure S2 Genetic deletion of Fli1 leads to a greater T cell expansion. (Related to Figure 1)
A. TIDE assay results showing genome disruption efficiency of the Fli1 locus for the Cas9+Fli1-sgRNA (sgFli1)+ cells. Genome disruption was detected by sanger sequencing.
B. Flow cytometry plots (gated on donor P14 cells) of the Cas9 (GFP)+sgRNA(VEX)+ population on D2 after in vitro transduction; D8 and D15 p.i. of Arm from splenocytes from sgCtrl or 2 Fli1-sgRNA(sgFli1_290 and sgFli1_360) groups. 5 X 104 activated donor cells were adoptively transferred into congenic infected recipient mice on D1 p.i.
C. Normalized Cas9+sgRNA+ cell numbers from blood, liver and lung from sgCtrl and the two sgFli1 groups at D8 and D15 p.i. of Arm. Cell numbers normalized to the sgCtrl group based on D2 in vitro transduction efficiency.
D. Flow cytometry plots (gated on donor P14 cells) of Cas9+sgRNA+ P14 cells on D2 post in vitro transduction, D9 and D15 p.i. with Cl13 (speen) for sgCtrl and the two sgFli1 groups. 5 X 104 activated donor P14 cells were adoptively transferred into the infected recipient mice on D1 p.i.
E. Normalized Cas9+sgRNA+ cell numbers from blood, liver and lung from sgCtrl and the two sgFli1 groups at D9 and D15 p.i. of Cl13. Cell number normalized to the sgCtrl group based on D2 in vitro transduction efficiency.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (One-Way Anova analysis). Data are representative of 2-4 independent experiments (mean±s.e.m.) with at least 3 mice/group.
Figure S3 Fli1 inhibits terminal TEFF differentiation without impacting TMEM cell formation. (Related to Figure 1)
A. Flow cytometry plots and statistical analysis of KLRG1LoCD127Lo cells. Frequencies (left) and numbers (right) from spleen for sgCtrl and 2 sgFli1 groups on D8 and D15 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
B. Flow cytometry plots and statistical analysis of GzmB+ and TCF-1+ C9P14 cells. Frequencies from spleen for sgCtrl and sgFli1 (2 sgRNA combined) groups on D8 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
C. Histogram and statistical analysis of T-bet and Eomes expression in C9P14 cells from spleen for sgCtrl and sgFli1 (2 sgRNA combined) groups on D8 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells. Naïve T cell (CD44−) staining shown in grey.
D. Flow cytometry plots and statistical analysis of cytokine producing C9P14 cells after 5 hours of stimulation (see Star Methods). IFNγ+, IFNγ+TNF+ and MIP1α+CD107a+ frequencies (left) and numbers (right) from spleen for non-stim P14 cells, sgCtrl and sgFli1 (2 sgRNA combined) groups on D8 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
E. Statistical analysis of total, terminal effector (TE) or memory precursor (MP) C9P14 cell numbers in the blood of sgCtrl and sgFli1 (2 sgRNA combined) groups on D8, D20 and D29 p.i. with Arm. Data normalized to sgCtrl+ D2 p.i. transduction efficiency. Normalized sgRNA+ C9P14 cell number is around 75 cells/1X106 PBMCs on the transfer day (D1 p.i.)
F. Statistical analysis of TE or MP C9P14 frequency and total, TE or MP C9P14 cell number in the spleen of sgCtrl and sgFli1 (2 sgRNA combined) groups on D29 p.i. of Arm. Data normalized to sgCtrl+ D2 p.i. transduction efficiency.
G. Histograms of Bcl-2, Bcl-XL and Bim expression of total C9P14 cells from spleen of sgCtrl and sgFli1 (2 sgRNA combined) groups on D15 p.i. of Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells. Naïve T cell (CD44−) staining shown in grey.
H-J. Statistical analysis of Bcl-2, Bcl-XL, Bim expression and Bcl-2/Bim and Bcl-XL/Bim ratios for total (H), TE (I) and MP(J) C9P14 cells from spleen for sgCtrl and sgFli1 (2 sgRNA combined) groups on D15 p.i. with Arm. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (One-Way Anova analysis). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 4 mice/group.
Figure S4 Fli1 restrains TEFF-like differentiation during chronic infection. (Related to Figure 3 and Figure 4)
A-B. Statistical analysis of Ly108−CD39+ or TCF-1−GzmB+ TEFF-like cells and Ly108+CD39− or TCF-1+GzmB− TEX precursor cell numbers from spleen for sgCtrl and the two sgFli1 groups at D8 (A) and D15 (B) p.i. with Cl13. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
C. Histogram and statistical analysis of Eomes, T-bet and Tox expression in C9P14 cells from spleen for sgCtrl and sgFli1 groups on D8 and D15 p.i. with Cl13. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells. Naïve T cell (CD44−) staining shown in grey.
D. Flow cytometry plots and statistical analysis of cytokine producing C9P14 cells after 5 hours of stimulation (see Star Methods). IFNγ+, IFNγ+TNF+ and MIP1α+CD107a+ frequencies (left) and numbers (right) from spleen for non-stim P14 cells, sgCtrl and sgFli1 (2 sgRNA combined) groups on D8 p.i. with Cl13. Gated on Cas9(GFP)+ sgRNA (VEX)+ P14 cells.
E. Flow cytometry plots and statistical analysis of cell number of CD45.2+VEX+ P14 for Empty-RV and Fli1-OE-RV groups at D8 and D16 p.i. with Cl13. At D0, CD45.2+ P14 cells were activated and CD45.1+ recipient mice were infected with Cl13. On D1 p.i., activated P14 were transduced with either Empty-RV or Fli1-OE-RV for 6 hours. On D2 p.i., VEX+ P14 cells were sorted from each RV transduced group and 1X105 cells were adoptively transferred into the infected recipients.
F-G. Flow cytometry plots and statistical analysis of Ly108−CD39+ or TCF-1−Gzmb+ TEFF-like cells and Ly108+CD39− or TCF-1+Gzmb−TEX precursor frequencies for Empty-RV and Fli1-OE-RV groups on D8 and D16 p.i. of Cl13. Gated on VEX+ P14 cells.
H. Statistical analysis of CX3CR1+ and Tim-3+ frequencies for Empty-RV and Fli1-OE-RV groups on D8 and D16 p.i. of Cl13. Gated on VEX+ P14 cells.
*P<0.05, **P<0.01, ***P<0.001 versus control (One-Way Anova analysis). Data are representative of 3 independent experiments (mean±s.e.m.) with at least 4 mice/group for A-D.
Figure S5. Transcriptional and epigenetic profiling addresses Fli1 inhibiting TEFF-like differentiation by coordinating with Runx1 and antagonizing Runx3 function. (Related to Figure 5)
A. PCA plot for RNA-seq data ffor sgCtrl, sgFli1_290 and sgFli1_360 groups on D8 p.i. with Cl13.
B. Overlap of all CUT&RUN Fli1 binding peaks with ATAC-seq detected peaks in sgCtrl and sgFli1 groups.
C. Histogram of all CUT&RUN peaks co-localized with the ATAC-seq peaks. Peaks co-localized with the ATAC-seq peaks are red; peaks not co-localized are blue.
D. CUT&RUN IgG or Fli1 binding signals for P14 cells on D9 p.i. and open chromatin region signals detected by ATAC-seq for sgCtrl, sgFli1_290 and sgFli1_360 groups in the Tcf7 and Id3 loci.
E. Flow cytometry plots and statistical analysis of CD45.1+mCherry+ and Ly108−CD39+/ Ly108+CD39− P14 cell numbers for Empty-RV or Runx1-OE-RV on D2 in vitro and D7 p.i. On D0, CD45.1+ P14 cells were activated and CD45.2+ recipient mice were infected with Cl13; On D1 p.i., activated P14 cells were transduced with Empty-RV or Runx1-OE-RV for 6 hours, and 1X105 transduced P14 cells were adoptively transferred into infected recipient mice. Flow cytometry plots gated on the CD45.1+ P14 cells.
F. D2 in vitro transduction efficiency of sgCtrl-VEX+Empty-mCherry, sgCtrl-VEX+Runx1-mCherry, sgFli1_290-VEX+Empty-mCherry, and sgFli1_290-VEX+Runx1-mCherry C9P14 cells.
G. Statistical analysis of VEX+mCherry+ C9P14 cells and Ly108−CD39+/Ly108+CD39− cell numbers from spleen for sgCtrl-VEX+Empty-mCherry, sgCtrl-VEX+Runx1-mCherry, sgFli1_290-VEX+Empty-mCherry, and sgFli1_290-VEX+Runx1-mCherry groups on D7 p.i. with Cl13. Gated on Cas9(GFP)+CD45.2+ P14 cells.
H. D2 in vitro transduction efficiency of sgCtrl-mCherry+Empty-VEX, sgCtrl-mCherry+Runx3-VEX, sgFli1_290-mCherry+Empty-VEX, and sgFli1_290-mCherry+Runx3-VEX C9P14 cells.
I. Statistical analysis of VEX+mCherry+ C9P14 cells and Ly108−CD39+/Ly108+CD39− cell numbers from spleen for sgCtrl-mCherry+Empty-VEX, sgCtrl-mCherry+Runx3-VEX, sgFli1_290-mCherry+Empty-VEX and sgFli1_290-mCherry+Runx3-VEX at D8 p.i. with Cl13. Gated on Cas9+CD45.2+ P14 cells.
*P<0.05, **P<0.01 versus control (two-tailed Student’s t-test). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 5 mice/group.
Figure S6. Fli1-deficiency results in CD8+ T cell expansion during influenza virus or Listeria monocytogenes infection. (Related to Figure 6)
A. Adjusted survival curve of LCMV Cl13 infected mice for sgCtrl+, sgFli1_290+ and sgFli1_360+ C9P14 recipient mice (1.5 x 105 cell/mouse, N=5 per group) Note, JAX recipient mice were used here instead of NCI recipients resulting in the difference in pathogenesis. Dashed line represents the time of C9P14 adoptive transfer.
B. Flow cytometry plots showing sgRNA(VEX)+ C9P14 cells in the lungs of influenza virus (PR8-GP33) infected mice, comparing non-transfer(NT), sgCtrl and sgFli1 groups at D8 p.i. “Recovered” was defined by complete weight recovery on D8 p.i.
C. Correlation of sgRNA+ C9P14 cell numbers and weight ratio (D8 p.i./D2 p.i.) during the PR8-GP33 infection. In the “weight not recovered” group (weight ratio between 0.7 and 1.0), sgRNA+ C9P14 cell numbers were further compared.
D. Flow cytometry plots and statistical analysis of sgRNA+ C9P14 cells in the spleens of PR8-GP33-infected recipient mice for non-transfer (NT), sgCtrl and sgFli1 groups on D8 p.i.
E. Flow cytometry plots and statistical analysis of sgRNA+ C9P14 cells in the spleens of Listeria monocytogenes-infected recipients for non-transfer (NT), sgCtrl and sgFli1 groups on D7 p.i.
*P<0.05, **P<0.01 versus control (two-tailed Student’s t-test and One-Way Anova analysis). Data are representative of 2 independent experiments (mean±s.e.m.) with at least 6 mice/group.
Figure S7. Deleting Fli1 in CD8+ T cells leads to better tumor protection in an immune competent mice. (Related to Figure 7)
A. Experimental design. On D0, CD45.2+Cas9+P14− mice were inoculated with 2 x 105 B16-Dbgp33 cells. On D3 post tumor inoculation (p.t.), CD8+ T cells were isolated from CD45.2+ C9P14 donor mice and activated. The next day, activated C9P14 cells were transduced with sgCtrl or sgFli1 RV for 6 hours. On D5 p.t., sgRNA (VEX)+ P14 cells were sorted from sgCtrl or sgFli1 groups, and 3 x 106 purified VEX+ C9P14 cells were adoptively transferred into tumor-bearing mice.
B. Tumor volume curve of tumor-bearing mice from NT, sgCtrl+ and sgFli1+ C9P14 cell transferred groups.
C. Tumor weight from NT, sgCtrl+ and sgFli1+ C9P14 transferred mice on D24 p.t.
D-E. Flow cytometry plots (D) and statistical analysis (E) of sgRNA (VEX)+ C9P14 cells and Ly108−CD39+/Ly108+CD39− populations from tumor for sgCtrl and sgFli1 groups on D24 p.t.
F-G. Statistical analysis of sgRNA+ C9P14 cells and Ly108−CD39+ or Ly108+CD39− populations from draining lymph node (dLN, F) and spleen (G) for sgCtrl and sgFli1 groups on D24 p.t.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.001 versus control (two-tailed Student’s t-test and One-Way Anova analysis). Data are representative of 3 independent experiments (mean±s.e.m.) with at least 6 mice/group.
Table S1 sgRNAs used for single gene editing, related to Figure S1.
Table S2 sgRNA library targeting transcription factors for in vivo screening, related to Figure 1.
Data Availability Statement
All the genomic data, including RNA-Seq, ATAC-Seq and CUN&RUN data are available on GEO indicated in the key resource table. The full transcription factor CRISPR screening data will be available upon requests to the corresponding authors.