

Abstract
Progression of intracranial hemorrhage (PICH) is a significant cause of secondary brain injury in patients with traumatic brain injury (TBI). Previous studies have implicated a variety of mediators that contribute to PICH. We hypothesized that patients with PICH would display either a hypocoagulable state, hyperfibrinolysis, or both. We conducted a prospective study of adult trauma patients with isolated TBI. Blood was obtained for routine coagulation assays, platelet count, fibrinogen, thrombelastography, markers of thrombin generation, and markers of fibrinolysis at admission and 6, 12, 24, and 48 h. Univariate analyses were performed to compare baseline characteristics between groups. Linear regression models were created, adjusting for baseline differences, to determine the relationship between individual assays and PICH. One hundred forty-one patients met entry criteria, of whom 71 had hemorrhage progression. Patients with PICH had a higher Injury Severity Score and Abbreviated Injury Scale score (head), a lower Glasgow Coma Scale score, and lower plasma sodium on admission. Patients with PICH had higher D-dimers on admission. After adjusting for baseline differences, elevated D-dimers remained significantly associated with PICH compared to patients without PICH at admission. Hypocoagulation was not significantly associated with PICH in these patients. The association between PICH and elevated D-dimers early after injury suggests that fibrinolytic activation may contribute to PICH in patients with TBI.
Keywords: coagulopathy, fibrinolysis, thrombelastography, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is uniquely associated with a coagulopathic state, though the mechanism is incompletely understood.1–3 In patients with TBI, the extent of brain damage is determined by the severity of the primary mechanical injury as well as the degree of secondary brain injury. Coagulopathy has been recognized as a major factor contributing to progression of intracranial hemorrhage, a major cause of secondary brain injury, and worse outcome after TBI.4 Overall, up to 50% of patients with TBI develop progression of intracranial hemorrhage (ICH), as evidenced by a new or increasing size lesion on repeat head computed tomography (CT) scans.5
The brain is rich in tissue factor, and the release of tissue factor in TBI leads to activation of the coagulation system and may eventually contribute to disseminated intravascular coagulation, consumptive coagulopathy, microvascular thrombosis, and the acute coagulopathy of trauma.1–3,6–8 Eighty-five percent of patients with at least one abnormal routine coagulation test will develop progression of ICH, yet even in patients with normal coagulation assays, one third of patients will still develop progression.4 This highlights the controversial nature of correlating routine coagulation tests and progression of ICH, as shown in the literature. Stein and colleagues demonstrated that an abnormal international normalized ratio (INR), activated partial thromboplastin time (aPTT), and platelet count are independently correlated with progression of intracranial hemorrhage.9 Oertel and colleagues showed that only the aPTT correlated with injury progression,10 whereas Schnuriger and colleagues showed that decreased platelet count is associated with increased ICH progression.5
Although the relative impact of individual clotting abnormalities on progression of ICH continues to be the subject of debate, it is clear that patients with demonstrated coagulopathy after TBI have a significantly increased rate of ICH progression compared to those without coagulopathy.11 Given that progression of ICH is independently associated with increased need for surgical intervention, poorer long-term neurological outcome, and a 5-fold increase in mortality, a better understanding of the mechanism involved has the potential to lead to the development of an intervention that could limit ICH progression and have a major impact on patient outcomes.12
Thrombelastography (TEG) has been used to study trauma patients.13 In a prospective study, hyperfibrinolysis occurred in 6% of the study population.14 In severely injured trauma patients receiving massive transfusions, the incidence of fibrinolysis may be as high as 34%.15 Further, hyperfibrinolysis (lysis at 30 min [LY30] >3%) in trauma patients was associated with a 76% mortality rate.16 We sought to characterize the coagulation profile in patients with ICH to determine the relationship between specific coagulation pathways and PICH. We hypothesized that patients with PICH would display either a hypocoagulable state, hyperfibrinolysis, or both.
Methods
Patients and laboratory assays
We conducted a single-center, prospective, observational study of major trauma patients presenting to an urban Level I trauma center from October 2011 to December 2014 in accordance with the Declaration Helsinki code of ethics. The primary aim of this study was to examine the association between progression of intracranial hemorrhage and TEG values. The association between intracranial hemorrhage and coagulation parameters was a secondary analysis. Adult trauma patients with isolated blunt TBI (head Abbreviated Injury Scale [AIS] ≥3 and ≤2 in other regions) were enrolled. Exclusion criteria included age <15 years, pregnancy, warfarin or clopidogrel use within 30 days of the injury, red cell, platelet, fresh frozen plasma, or cryoprecipitate transfusion in the first 6 h after admission, use of recombinant factor VIIa, or the presence of a known coagulation disorder. Consent was obtained under an institutional review board–approved waiver of informed consent. Attempts were made to consent the patient, or a legally authorized representative if the patient was not able to provide consent, as soon as possible. In the situation where no legally authorized representative was available or the patient did not become consentable, the patient was excluded from the study.
Patient characteristics, including demographics, admission physiological and laboratory values, AIS scores, and Injury Severity Score (ISS), were recorded. Blood was obtained at admission, 6, 12, 24, and 48 h for coagulation assays, prothrombin time (PT), INR, aPTT, fibrinogen, and D-dimer (STAGO compact; Diagnostica Stago S.A.S., Asnières-sur-Seine, France), TEG (TEG 5000; Haemonetics Corporation, Boston, MA), thrombin-antithrombin complexes (MyBioSource, San Diego, CA), prothrombin fragments 1 + 2, (MyBioSource), plasminogen activator inhibitor 1 (PAI-1), and tissue plasminogen activator (tPA; Procarta Biosystems Ltd., Norwich, UK). TEG was performed at the same time points. Fresh whole-blood specimens in kaolin-activated cups were used. Standard TEG measurements, including R value, K value, maximum amplitude, α angle, and LY30, were obtained.
Imaging
Computerized axial tomography (CAT) of the head was performed at admission and 6 h after admission in all patients, per institutional protocol. Volumes of subdural, epidural, and intraparenchymal hemorrhages were quantified using the previously validated ABC/2 method.17 Briefly, the CAT slice with the largest area of hemorrhage was identified. The largest diameter (A) of the hemorrhage on this slice was measured. The largest diameter 90 degrees to A on the same slice was measured next (B). Finally, the approximate number of 10-mm slices on which the ICH was seen was calculated (C).17,18 Progression of hemorrhage was defined as ≥30% increase in the total hemorrhage volume, or by attending radiologist interpretation in the case of subarachnoid hemorrhage. Previous literature has established that hemorrhage growth of ∼30% on serial head CAT is a conservative definition for progression of intracranial hemorrhage.19
Statistical analyses
Baseline patient characteristics were compared between patients with and without progression of intracranial hemorrhage. Median values of each lab value were plotted over time by progression status. Labs at admission and 6 h were compared between patients with and without progression of intracranial hemorrhage. To determine whether there was a significant difference in lab values between patients with and without ICH progression, linear regression was conducted with the lab value as the outcome and progression status as the primary predictor. The analysis was conducted separately for admission and 6 h. Patient covariates were included in this regression analysis if p < 0.25 when comparing those who progressed and those who did not. The covariates included in the linear regression model were sex, aspirin use within week preceding injury, age, ISS, Glasgow Coma Scale (GCS), and AIS head.
Labs at admission and 6 h were compared between patients who progressed and did not progress. To determine whether there was a significant difference in lab values between patients who did and did not have ICH progression, linear regression was conducted with the lab value as the outcome and progression status as the primary predictor. Lab values were log transformed when heavily right skewed. This analysis was conducted separately for admission and 6 h. Patient covariates were included in this regression analysis if they were thought to be clinically meaningful and p < 0.25 when comparing between progressers and non-progressers; therefore, the covariates included were sex, aspirin in week preceding age, ISS, GCS, and AIS head. In order to correct for multiple comparisons, a Bonferroni correction was used and a p value of <0.002 was considered significant (13 labs, two times, 26 comparisons, 0.05/26 = 0.0019, ∼0.002).
Descriptive statistics and plots over time were performed for all time points (admission, 6 h, 12 h, 24 h, and 48 h). A receiver operating characteristic (ROC) curve was generated to illustrate the performance of log-transformed D-dimer at admission and 6 h in classifying patients with and without progression. No other patient characteristics were controlled for in the generation of these ROC curves.
Patient data were deidentified and maintained in a Microsoft Excel database (Microsoft Corporation, Redmond, WA). Data are presented as mean ± standard deviation or median (interquartile rage; IQR) or percentage, as appropriate. Univariate comparisons were made using Student's t-test for normally distributed data, Mann-Whitney U tests for non-normally distributed data, and chi-square or Fischer's exact test for proportions. Analyses were performed using SAS software (SAS Institute Inc., Cary, NC). This study was approved by the Institutional Review Board at Oregon Health & Science University and followed the ethical principles of the Declaration of Helsinki.
Results
Patients
One hundred forty-one patients met entry criteria, of which 71 demonstrated PICH on repeat head CT at 6 h (50%). Patients with PICH had a higher ISS (25.77 [9.78] vs. 18.99 [9.12]) and AIS head (4 [4–5] vs. 4 [3–4]), a lower GCS (14 [4–14] vs. 14 [14–15]), slightly lower blood sodium levels (139.28 [3.18] vs. 140.36 [2.41]), and larger 6-h hemorrhage volumes (1.64 [0.60–4.03] vs. 0.36 [0.14–0.77]). No other differences in patient characteristics at admission were observed (Table 1).
Table 1.
Patient Characteristics by Progression
| Patient characteristics at admission | Progression status |
p value | |
|---|---|---|---|
| No progression (n = 70) |
Progression (n = 71) |
||
| Sex, n (%) | |||
| Male | 47 (67.1) | 54 (76.1) | 0.24 |
| Female | 23 (32.9) | 17 (23.9) | |
| ASA in week before, n (%) | |||
| Yes | 8 (11.4) | 12 (16.9) | 0.65 |
| No | 61 (87.1) | 58 (81.7) | |
| Unknown | 1 (1.4) | 1 (1.4) | |
| Age, mean (SD) | 47.1 (20.75) | 53.07 (21.58) | 0.10 |
| SBP, mean (SD) | 151.53 (32.47) | 150.93 (26.33) | 0.90 |
| ISS, mean (SD) | 18.99 (9.12) | 25.77 (9.78) | <0.001 |
| GCS | 14 (14–15) | 14 (4–14) | <0.001 |
| AIS head | 4 (3–4) | 4 (4–5) | <0.001 |
| Sodium, mean (SD) | 140.36 (2.41) | 139.28 (3.18) | 0.04 |
| 6-h hemorrhage volume (mL) | 0.36 (0.14–0.77) (n = 39) | 1.64 (0.60–4.03) (n = 61) | <0.001 |
| Total hemorrhage volume (mL) | 0.35 (0.16–0.76) (n = 37) | 0.59 (0.33–0.91) (n = 65) | 0.06 |
| TBI mortality, n | 0 | 2 | 0.1 |
Tests used: t-test for two samples: age, SBP, ISS, sodium; Wilcoxon's two-sample test: AIS head, GCS; Wilcoxon's rank-sum test: hemorrhage volumes; chi-square: sex; Fisher's exact test: ASA. Bold italicized values are used to indicate significant findings (p < 0.05). Interquartile range (25th–75th percentile).
ASA, American Society of Anesthesiologists; SD, standard deviation; SBP, systolic blood pressure; ISS, Injury Severity Score; GCS, Glasgow Coma Scale; AIS, Abbreviated Injury Scale; TBI, traumatic brain injury.
Coagulation parameters
There were no significant differences in coagulation assays, including INR, fibrinogen, platelet count, or aPTT at admission, 6, 12, 24, or 48 h between patients with PICH and those without (Table 3). INR values showed a trend toward prolongation in both groups over time (Supplementary Fig. S1). Similar to the INR, the aPTT values showed a trend toward prolongation in both groups (Supplementary Fig. S2). Interestingly, fibrinogen levels tended to increase in both groups over time, which may reflect the fact that fibrinogen is an acute phase reactant that increases during inflammation (Supplementary Fig. S3). In contrast, platelet levels tended to decrease over time in both groups, suggestive of consumption (Supplementary Fig. S4).
Table 3.
Linear Regression Models
| |
Admission |
6 h |
||
|---|---|---|---|---|
| Coefficient (95% CI) | p value | Coefficient (95% CI) | p value | |
| INRa | 1.02 (0.98–1.05) | 0.32 | 1 (0.97–1.03) | 0.99 |
| FIBb | 0.69 (−19.79 to 21.17) | 0.95 | 2.23 (−20.79 to 25.24) | 0.85 |
| D-DIa | 1.27 (1.01–1.60) | 0.04 | 1.18 (0.98–1.42) | 0.08 |
| Pltb | –3.05 (−16.13 to 10.03) | 0.65 | 0.97 (−18.36 to 20.30) | 0.92 |
| aPTTa | 0.99 (0.96–1.03) | 0.63 | 1.01 (0.98–1.04) | 0.60 |
| F1 + 2a | 0.91 (0.75–1.09) | 0.28 | 0.86 (0.70–1.06) | 0.15 |
| TATa | 0.93 (0.78–1.12) | 0.45 | 0.95 (0.79–1.14) | 0.60 |
| PAI-1a | 1.1 (0.92–1.32) | 0.30 | 1.11 (0.95–1.30) | 0.20 |
| tPAa | 0.93 (0.77–1.12) | 0.43 | 0.99 (0.84–1.18) | 0.94 |
| LY30a | 1 (0.76–1.30) | 0.98 | 1.22 (0.90–1.64) | 0.19 |
Bolded italicized values are used to indicate significant findings (p < 0.05).
Log-transformed value used in linear regression. Coefficients are in terms of the ratio of the geometrical mean.
Raw value used in linear regression. Coefficients are on the original scale of the lab.
All models controlled for sex, ASA in week before, age, ISS, GCS, and AIS head.
INR, international normalized ratio; FIB, fibrinogen; D-DI, D-dimer; Plt, platelets; aPTT, activated partial thromboplastin time; F1 + 2, prothrombin fragments 1 + 2; TAT, thrombin-antithrombin; PAI-1, plasminogen activator inhibitor 1; tPA, tissue plasminogen activator; LY30, lysis at 30 min; CI, confidence interval; ASA, American Society of Anesthesiologists; ISS, Injury Severity Score; GCS, Glasgow Coma Scale; AIS, Abbreviated Injury Scale.
No trends were observed in the temporal profile of thrombin-antithrombin complexes (TAT) or prothrombin fragments 1 + 2 [F1 + 2] between patients with PICH and those without (Supplementary Figs. S5 and S6; Table 3). These results suggest that hemorrhage progression was not attributable to alternations in thrombin generation. We therefore investigated markers of fibrinolysis to see whether there was an association between fibrinolytic markers and PICH.
Fibrinolysis parameters
The p value for the association between the initial volume and D-dimers was 0.35, with an R2 value of 0.01, suggesting that there was no significant association between the initial hematoma volume and levels of D-dimer. In addition, the p value for the association between the change in volume and change in D-dimer concentrations from admission to 6 h was 0.31, with an R2 value of 0.01, suggesting that there was no significant association between the change in volume and change in D-dimer concentrations from admission to 6 h.
In contrast to coagulation markers, there was a significant association between D-dimers and hemorrhage progression. Patients with PICH had higher D-dimers on admission (p = 0.04). D-dimers peaked at 6 h and progressively decreased over 48 h in patients with PICH while remaining low at all time points in non-PICH patients (Fig. 1). These results indicate that patients with PICH initially had increased fibrinolysis at admission.
FIG. 1.
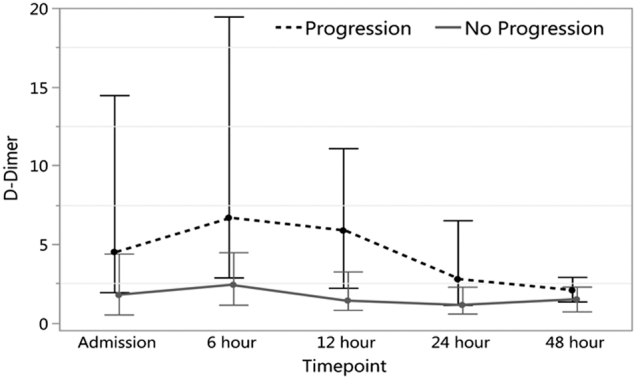
D-dimer values admission to 48 h (median with IQR). IQR, interquartile range.
In order to ascertain whether decreased PAI-I levels or increased tPA levels contributed to increased fibrinolysis, we measured both of these fibrinolytic components at all time points. Both PAI-1 and tPA were highest at admission and tended to decrease in both groups over 48 h (Supplementary Figs. S8 and S9). However, there was no significant difference in PAI-I or tPA between patients with PICH and those without. There was no difference in the TEG LY30 at any time point between patients with PICH and those without (Table 2; Supplementary Fig. S9). This may be attributable to the relative insensitivity of the LY30 to changes in fibrinolysis compared to D-dimers.
Table 2.
Median (IQR) of Labs by Progression
| |
Admission |
6 h |
||||||
|---|---|---|---|---|---|---|---|---|
| |
No progression |
Progression |
No progression |
Progression |
||||
| n | Median (IQR) | n | Median (IQR) | n | Median (IQR) | n | Median (IQR) | |
| INR | 67 | 0.94 (0.89–1.03) | 62 | 0.99 (0.94–1.12) | 59 | 1 (0.93–1.07) | 54 | 1.08 (0.98–1.16) |
| PT (sec) |
67 | 12.8 (12.3–13.7) | 62 | 13.25 (12.78–14.55) | 59 | 13.4 (12.7–14.1) | 54 | 14.2 (13.18–15.03) |
| aPTT (sec) |
67 | 25.4 (23.1–28.3) | 62 | 25.8 (23.93–28.53) | 60 | 26.4 (23.40–28.25) | 54 | 27.1 (24.50–29.53) |
| FIB (mg/dL) |
67 | 307 (256–367) | 63 | 284 (236–377) | 60 | 281 (224–364) | 55 | 290 (230–352) |
| Plt ( × 103/μL) |
68 | 242 (193–270) | 69 | 217 (160–267) | 29 | 217 (171–243) | 33 | 181 (139–252) |
| F1 + 2 (pg/mL) |
40 | 273 (210–349) | 46 | 264 (156–373) | 39 | 271 (193–353) | 43 | 233 (158–394) |
| TAT (μg/mL) |
47 | 409 (186–524) | 47 | 320 (188–510) | 43 | 338 (180–514) | 42 | 340 (166–490) |
| D-DI (μg/mL) |
64 | 0.57 (−0.64 to 1.48) | 67 | 1.64 (0.77–2.68) | 61 | 0.88 (0.14–1.50) | 60 | 1.9 (1.07–2.91) |
| PAI-1 (pg/mL) |
67 | 9355 (7201–21,800) | 68 | 9346 (5934–25,904) | 61 | 7033 (5557–16,455) | 60 | 10,408 (7303–19,843) |
| tPA (pg/mL) |
67 | 1306 (578–2183) | 68 | 1464 (740–2685) | 61 | 794 (396–1319) | 61 | 922 (636–1401) |
| LY30 (%) |
69 | 0.6 (0–2) | 69 | 0.3 (0.00–1.35) | 58 | 0.35 (0.00–1.43) | 59 | 0.4 (0.0–1.8) |
INR, international normalized ratio; PT, prothrombin time; aPTT, activated partial thromboplastin time; FIB, fibrinogen; Plt, platelets; F1 + 2, prothrombin fragments 1 + 2; TAT, thrombin-antithrombin; D-DI, D-dimer; PAI-1, plasminogen activator inhibitor 1; tPA, tissue plasminogen activator; LY30, lysis at 30 min; IQR, interquartile range.
The temporal relationship between PICH and D-dimers is also important because the second CT scan was performed ∼6 h after admission. Therefore, the timing of the D-dimer differences correlates with the timing of the CT scan. The area under the ROC curve (AUROC) indicates that log-transformed D-dimer discriminates between patients with and without PICH (AUROC = 0.69 at admission; 0.72 at 6 h; Table 3; Figs. 2 and 3).
FIG. 2.
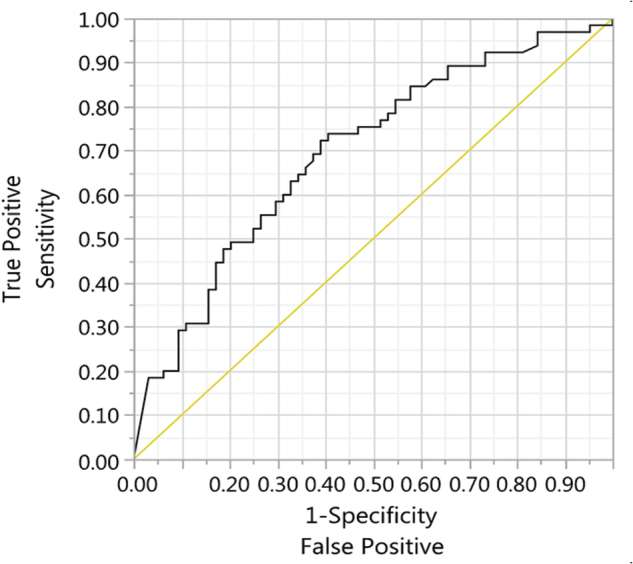
ROC curve for the classification of progression using log-transformed admission D-dimers. ROC, receiver operating characteristic.
FIG. 3.
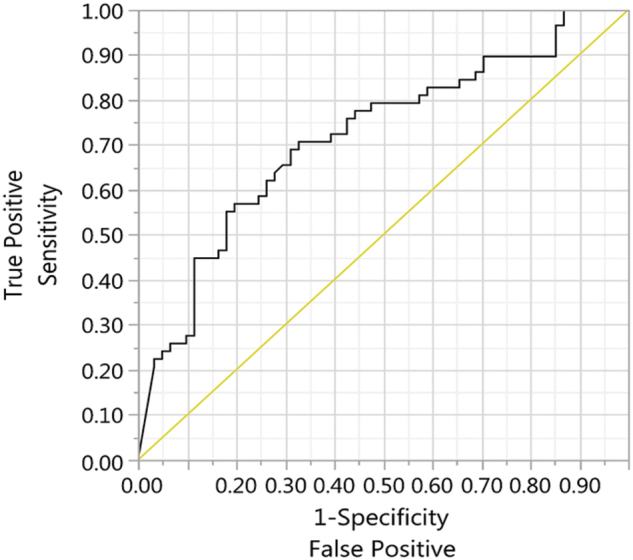
ROC curve for the classification of progression using log-transformed 6-h D-dimers. ROC, receiver operating characteristic.
Discussion
TBI has been associated with a coagulopathic state since Penick and McLendon described a case of a coagulopathic newborn who suffered TBI during delivery.20 Patients with TBI are at risk for developing abnormalities of both coagulation and fibrinolysis, and there is evidence that the extent of injured brain tissue correlates with the degree of coagulopathy.21,22 Recent findings from the PROPPR trial suggest that coagulopathy induced by TBI is substantial because patients with combined TBI and hemorrhagic shock had worse coagulopathy compared to those who endured either condition alone.23
Although the mechanism of coagulopathy after TBI has been investigated extensively, the numerous hypotheses that have been proposed indicate that the mechanisms are complex and remain unclear. In 1974, Goodnight and colleagues hypothesized that tissue thromboplastin released from damaged brain (now known as tissue factor) enters the circulation, activates the extrinsic coagulation pathway, and produces fibrin clot.3,12 Cerebral endothelial cells are rich in tissue factor,24 which is considered the primary initiator of the extrinsic pathway, and its release can activate the coagulation system excessively in patients with TBI.3,25 Additional proposed mechanisms involve depletion of platelets and clotting factors after blood loss or consumption from disseminated intravascular coagulation.26,27 A more recent hypothesis suggests that TBI alone does not cause early coagulopathy, but must be coupled with hypoperfusion to lead to coagulation derangements associated with the activated protein C pathway.28
Depletion of the fibrinolytic system may also contribute to the coagulopathy after TBI. Based on this hypothesis, several studies have investigated whether indices of fibrinolytic degradation, particularly D-dimer, may be useful markers to assess coagulopathy in this patient population. A cumulative assessment of the current literature indicates that D-dimer levels may be a useful prognostic tool for predicting PICH, but not all studies are in full agreement.29 Whereas several studies have shown that the D-dimer value is a strong predictor of PICH,30–33 others did not observe a significant association.34
In agreement with the majority of the literature, we demonstrated that PICH on serial head CT was associated with elevated D-dimers in patients with isolated blunt TBI. When plotted during the first 48 h after injury, D-dimers peaked at 6 h and progressively decreased in patients with PICH while remaining low at all time points in patients without PICH. D-dimer is a fibrin degradation product that is present in the blood when the coagulation system is activated (as in thrombosis and disseminated intravascular coagulation). Given that fibrinogen is upregulated relatively slowly with a half-life of ∼3–4 days,35 these finding suggest that this is a result of clot breakdown. To explain the observed differences in D-dimers, we examined markers of coagulation as well as markers of the fibrinolytic pathway. To determine whether the observed elevation in D-dimer in progressors was a result of prothrombotic fibrin generation, we examined prothrombin fragments (F1 + 2) as well as thrombin-antithrombin complexes.
No differences were found between progressors and non-progressors at any time point in either marker, suggesting that the elevated D-dimers were not a result of excess thrombin generation. This provides confirmation of previous studies, such as those of Folkerson and colleagues36 and Karri and colleagues.37 Of note, a slight decrease in both groups occurred during the first 48 h after injury, suggesting that thrombin generation decreased in both groups, and D-dimer formation was not attributable to differences in initiation of clot formation.
We then examined mediators involved in the fibrinolytic pathway, which is regulated by several activators and inhibitors (Fig. 4). The primary enzyme involved in the fibrinolytic cascade is plasmin. Plasmin is formed when tPA and plasminogen bind fibrin, resulting in the proteolytic cleavage of plasminogen by tPA to its active form, plasmin. Plasmin then cleaves fibrin, leading to the production of various products, including D-dimers. We first looked at tPA, which is primarily responsible for the conversion of plasminogen to plasmin. tPA was found to progressively decrease over time in both groups, indicating that fibrinolysis was occurring; however, given that no differences were observed between progressors and non-progressors, the differences observed in D-dimer formation are not likely the result of differences in tPA. PAI-1 is another primary regulator of the fibrinolytic system and its synthesis is highly regulated.38 PAI-1 inhibits tPA; thus, higher levels of active PAI-1 would be expected to be associated with decreased fibrinolysis. As with tPA, however, no differences were observed between progressors and non-progressors in PAI-1 at any time point, suggesting that similar to tPA, differences in PAI-1 are not likely to be responsible for the observed differences in D-dimers.
FIG. 4.

Fibrinolysis pathway (courtesy of Thomas Deloughery, MD). PAI-1, plasminogen activator inhibitor 1; TAFI, thrombin activatable fibrinolysis inhibitor; tPA, tissue plasminogen activator.
Although we propose that elevated D-dimer in patients with PICH in this study is attributable to increased fibrinolysis, no differences were observed in the lysis of clot based on TEG results at 30 min (LY30). However, LY30 is a relatively insensitive marker and continues to undergo examination for appropriate cut-off points (hyperfibrinolysis vs. fibrinolysis shutdown, etc.). Others have demonstrated similar findings. In one study of fibrinolytic activation in trauma patients, fibrinolysis (as demonstrated by increased plasmin-antiplasmin complexes, elevated tPA, and elevated D-dimers) was associated with more severe injury and worse outcome; however, 90% of the patients in the study did not meet criteria for hyperfibrinolysis based on viscoelastic testing. The researchers concluded that thrombelastography was an insensitive measure of endogenous fibrinolytic activity, which they attributed to rapid hydrolysis of free plasmin.39
Given that our findings suggest that hyperfibrinolysis, rather than impaired coagulation, is responsible for PICH, this further supports the use of tranexamic acid (TXA) as a treatment to prevent PICH in TBI patients. TXA prevents fibrinolysis by inhibiting the conversion of plasminogen to plasmin. The CRASH-2 trial showed that the use of TXA attenuates PICH in patients with moderate-to-severe TBI.40 Further studies, including a large multi-center study involving our institution, are currently ongoing to elucidate the precise mechanisms by which TXA prevents PICH. These future studies will additionally have sufficient power to allow for analyses across all parameters concurrently to determine whether relationships exist between coagulation parameters.
There are several limitations of this study. First, there are multiple regulators of fibrinolysis that we did not measure, including α2-plasmin inhibitor and factor XIII. Additionally, although we were able to measure progression of intracranial hemorrhage using serial head CT and determine its association with a marker of fibrinolysis, we were unable to make any association between mortality in our population and coagulopathy because our overall mortality rate in this study was low. Finally, although we sought to select patients with isolated TBI, study patients with progression of intracranial hemorrhage did have significantly higher ISS. Thus, the elevated D-dimers could be partially attributable to increased injury severity and were therefore included in the linear regression modeling.
In conclusion, the association between PICH and elevated D-dimers at admission suggests that fibrinolytic activation may, in part, be responsible for PICH in patients with TBI. D-dimer levels may therefore be useful as a predictor of PICH in clinical settings, which could be used to justify more aggressive treatment to prevent PICH, such as TXA.
Supplementary Material
Acknowledgments
We thank the staff of the Trauma Research Laboratory at Oregon Health & Science University. We also like to thank Dr. Thomas Deloughery for the use of his illustration in Figure 4.
An abstract entitled “Fibrinolytic Activation in Patients with Progressive Intracranial Hemorrhage Early after TBI” was presented at the 2016 Annual Meeting of the American Association for the Surgery of Trauma as an oral presentation.
Funding Information
The project described was supported by Award Number 5K12HLK108974-03 from the National Heart, Lung, and Blood Institute. This publication was supported by the Oregon Clinical and Translational Research Institute (OCTRI; grant number UL1TR000128) from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH).
Author Disclosure Statement
OHSU and Dr. David Farrell have a significant interest in Gamma Therapeutics, a company that may have a commercial interest in the results of this research and technology. This potential individual and institutional conflict of interest has been reviewed and managed by OHSU.
Supplementary Material
References
- 1. Stein, S.C., and Smith, D.H. (2004). Coagulopathy in traumatic brain injury. Neurocrit. Care 1, 479–488 [DOI] [PubMed] [Google Scholar]
- 2. McCully, S.P., and Schreiber, M.A. (2013). Traumatic brain injury and its effect on coagulopathy. Semin. Thromb. Hemost. 39, 896–901 [DOI] [PubMed] [Google Scholar]
- 3. Goodnight, S.H., Kenoyer, G., Rapaport, S.I., Patch, M.J., Lee, J.A., and Kurze, T. (1974). Defibrination after brain-tissue destruction: a serious complication of head injury. N. Engl. J. Med. 290, 1043–1047 [DOI] [PubMed] [Google Scholar]
- 4. Carrick, M.M., Tyroch, A.H., Youens, C.A., and Handley, T. (2005). Subsequent development of thrombocytopenia and coagulopathy in moderate and severe head injury: support for serial laboratory examination. J. Trauma 58, 725–730 [DOI] [PubMed] [Google Scholar]
- 5. Schnüriger, B., Inaba, K., Abdelsayed, G.A., Lustenberger, T., Eberle, B.M., Barmparas, G., Talving P, and Demetriades D. (2010). The impact of platelets on the progression of traumatic intracranial hemorrhage. J. Trauma 68, 881–885 [DOI] [PubMed] [Google Scholar]
- 6. Laroche, M., Kutcher, M.E., Huang, M.C., Cohen, M.J., and Manley, G.T. (2012). Coagulopathy after traumatic brain injury. Neurosurgery 70, 1334–1345 [DOI] [PubMed] [Google Scholar]
- 7. Nekludov, M., Antovic, J., Bredbacka, S., and Blombäck, M. (2007). Coagulation abnormalities associated with severe isolated traumatic brain injury: cerebral arterio-venous differences in coagulation and inflammatory markers. J. Neurotrauma 24, 174–180 [DOI] [PubMed] [Google Scholar]
- 8. Howard, B.M., Miyazawa, B.Y., Dong, W., Cedron, W.J., Vilardi, R.F., Ruf, W., and Cohen, M.J. (2015). The tissue factor pathway mediates both activation of coagulation and coagulopathy after injury. J. Trauma Acute Care Surg. 79, 1009–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stein, S.C., Young, G.S., Talucci, R.C., Greenbaum, B.H., and Ross, S.E. (1992). Delayed brain injury after head trauma: significance of coagulopathy. Neurosurgery 30, 160–165 [DOI] [PubMed] [Google Scholar]
- 10. Oertel, M., Kelly, D.F., McArthur, D., Boscardin, W.J., Glenn, T.C., Lee, J.H., Gravori, T., Obukhov, D., McBride, D.Q., and Martin, N.A. (2002). Progressive hemorrhage after head trauma: predictors and consequences of the evolving injury. J. Neurosurg. 96, 109–116 [DOI] [PubMed] [Google Scholar]
- 11. Brohi, K., Singh, J., Heron, M., and Coats, T., (2003). Acute traumatic coagulopathy. J. Trauma 54, 1127–1130 [DOI] [PubMed] [Google Scholar]
- 12. Olson, J.D., Kaufman, H.H., Moake, J., O'Gorman, T.W., Hoots, K., Wagner, K., Brown, C.K., and Gildenberg, P.L. (1989). The incidence and significance of hemostatic abnormalities in patients with head injuries. Neurosurgery 24, 825–832 [DOI] [PubMed] [Google Scholar]
- 13. Holcomb, J.B., Minei, K.M., Scerbo, M.L., Radwan, Z.A., Wade, C.E., Kozar, R.A., Gill, B.S., Albarado, R., McNutt, M.K., Khan, S., Adams, P.R., McCarthy, J.J., and Cotton, B.A. (2012). Admission rapid thrombelastography can replace conventional coagulation tests in the emergency department: experience with 1974 consecutive trauma patients. Ann. Surg. 256, 476–486 [DOI] [PubMed] [Google Scholar]
- 14. Levrat, A., Gros, A., Rugeri, L., Inaba, K., Floccard, B., Negrier, C., and David, J.S. (2008). Evaluation of rotation thrombelastography for the diagnosis of hyperfibrinolysis in trauma patients. Br. J. Anaesth. 100, 792–797 [DOI] [PubMed] [Google Scholar]
- 15. Kashuk, J.L., Moore, E.E., Sawyer, M., Wohlauer, M., Pezold, M., Barnett, C., Biffl, W.L., Burlew, C.C., Johnson, J.L., and Sauaia, A. (2010) Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Ann Surg. 252, 434–444 [DOI] [PubMed] [Google Scholar]
- 16. Cotton, B.A., Harvin, J.A., Kostousouv, V., Minei, K.M., Radwan, Z.A., Schöchl, H., Wade, C.E., Holcomb, J.B., and Matijevic, N. (2012). Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J. Trauma Acute Care Surg. 73, 365–370 [DOI] [PubMed] [Google Scholar]
- 17. Kothari, R.U., Brott, T., Broderick, J.P., Barsan, W.G., Sauerbeck, L.R., Zuccarello, M., Khoury, J. (1996). The ABCs of measuring intracerebral hemorrhage volumes. Stroke 27, 1304–1305 [DOI] [PubMed] [Google Scholar]
- 18. Khan, M., Baird, G.L., Elias, R., Rodriguez-Srednicki, J., Yaghi, S., Yan, S., Collins, S., Thompson, B.B., Wendell, L.C., Potter, N.S., Fehnel, C., Saad, A., and Silver, B. (2016). Comparison of intracerebral hemorrhage volume calculation methods and their impact on scoring tools. J. Neuroimaging 27, 144–148 [DOI] [PubMed] [Google Scholar]
- 19. Brott, T., Broderick, J., Kothari, R., Barsan, W., Tomsick, T., Sauerbeck, L., Spilker J., Duldner, J., and Khoury, J. (1997). Early hemorrhage growth in patients with intracerebral hemorrhage. Stroke 28, 1–5 [DOI] [PubMed] [Google Scholar]
- 20. Penick, G.D., and McLendon, W.W. (1960). Disorders of the hemostatic mechanism. Int. Rec. Med. 173, 491–496 [PubMed] [Google Scholar]
- 21. Hulka, F., Mullins, R.J., and Frank, E.H. (1996). Blunt brain injury activates the coagulation process. Arch. Surg. 131, 923–928 [DOI] [PubMed] [Google Scholar]
- 22. Scherer, R.U., and Spangenberg, P. (1998). Procoagulant activity in patients with isolated severe head trauma. Crit. Care Med. 26, 149–156 [DOI] [PubMed] [Google Scholar]
- 23. Galvagno, S.M.Jr., Fox, E.E., Appana, S.N., Baraniuk, S., Bosarge, P.L., Bulger, E.M., Callcut, R.A., Cotton, B.A., Goodman, M., Inaba. K., O'Keeffe, T., Schreiber, M.A., Wade, C.E., Scalea, T.M., Holcomb, J.B., Stein, D.M.; and PROPPR Study Group. (2017). Outcomes after concomitant traumatic brain injury and hemorrhagic shock: a secondary analysis from the Pragmatic, Randomized Optimal Platelets and Plasma Ratios trial. J. Trauma Acute Care Surg. 83, 668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Østerud, B., and Bjørklid, E. (2006). Sources of tissue factor. Semin. Thromb. Hemost. 32, 11–23 [DOI] [PubMed] [Google Scholar]
- 25. Giesen, P.L., and Nemerson, Y. (2000). Tissue factor on the loose. Semin. Thromb. Hemost. 26, 379–384 [DOI] [PubMed] [Google Scholar]
- 26. Harhangi, B.S., Kompanje, E.J., Leebeek, F.W., and Maas, A.I. (2008). Coagulation disorders after traumatic brain injury. Acta Neurochir. (Wien) 150, 165–175 [DOI] [PubMed] [Google Scholar]
- 27. Kutcher, M.E., Ferguson, A.R., and Cohen, M.J. (2013). A principal component analysis of coagulation after trauma. J. Trauma Acute Care Surg. 74, 1223–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen, M.J., Brohi, K., Ganter, M.T., Manley, G.T., Mackersie, R.C., and Pittet, J.F. (2007). Early coagulopathy after traumatic brain injury: the role of hypoperfusion and the protein C pathway. J. Trauma 63, 1254–1262 [DOI] [PubMed] [Google Scholar]
- 29. Zhang, J., He, M., Song, Y., and Xu, J. (2018). Prognostic role of D-dimer level upon admission in patients with traumatic brain injury. Medicine. 97, e11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tian, H.L., Chen, H., Wu, B.S., Cao, H.L., Xu, T., Hu, J., Wang G, Gao, W.W., Lin, Z.K., and Chen, S.W. (2010). D-dimer as a predictor of progressive hemorrhagic injury in patients with traumatic brain injury: analysis of 194 cases. Neurosurg. Rev. 33, 359–365; discussion, 365–366 [DOI] [PubMed] [Google Scholar]
- 31. Yuan, F., Ding, J., Chen, H., Guo, Y., Wang, G., Gao, W.W., Chen, S.W., and Tian, H.L. (2012). Predicting progressive hemorrhagic injury after traumatic brain injury: derivation and validation of a risk score based on admission characteristics. J. Neurotrauma 29,2137–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Knisel, W., Müller, M., Besenthal, I., di Nicuolo, A., Rebstock, M., Risler, T., and Eggstein, M. (1991). Application of a new LDL apheresis system using two dextran sulfate cellulose columns in combination with an automatic column-regenerating unit and a blood cell separator. J. Clin. Apher. 6, 11–15 [DOI] [PubMed] [Google Scholar]
- 33. Kuo, J.R., Lin, K.C., Lu, C.L., Lin, H.J., Wang, C.C., and Chang, C.H. (2007). Correlation of a high D-dimer level with poor outcome in traumatic intracranial hemorrhage. Eur. J. Neurol. 14, 1073–1078 [DOI] [PubMed] [Google Scholar]
- 34. Juratli, T.A., Zang, B., Litz, R.J., Sitoci, K.H., Aschenbrenner, U., Gottschlich, B., Daubner, D., Schackert, G., and Sobottka, S.B. (2014). Early hemorrhagic progression of traumatic brain contusions: frequency, correlation with coagulation disorders, and patient outcome: a prospective study. J. Neurotrauma 31, 1521–1527 [DOI] [PubMed] [Google Scholar]
- 35. Stein, T.P., Leskiw, M.J., and Wallace, H.W. (1978), Measurement of half-life of human plasma fibrinogen. Am. J. Physiol. 234, D504–D510 [DOI] [PubMed] [Google Scholar]
- 36. Folkerson, L.E., Sloan, D., Cotton, B.A., Holcomb, J.B., Tomasek, J.S., and Wade, C.E. (2015). Predicting progressive hemorrhagic injury from isolated traumatic brain injury and coagulation. Surgery 158, 655–661 [DOI] [PubMed] [Google Scholar]
- 37. Karri, J., Cardenas, J.C., Matijevic, N., Wang, Y.W., Choi, S., Zhu, L., Cotton, B.A., Kitagawa, R., Holcomb, J.B., and Wade, C.E. (2017). Early fibrinolysis associated with hemorrhagic progression following traumatic brain injury. Shock 48, 644–650 [DOI] [PubMed] [Google Scholar]
- 38. Dellas, C., and Loskutoff, D.J. (2005). Historical analysis of PAI-1 from its discovery to its potential role in cell motility and disease. Thromb. Haemost. 93, 631–640 [DOI] [PubMed] [Google Scholar]
- 39. Raza, I., Davenport, R., Rourke, C., Platton, S., Manson, J., Spoors, C., Khan, S., De'Ath, H.D., Allard, S., Hart, D.P., Pasi, K.J., Hunt, B.J., Stanworth, S., MacCallum, P.K., and Brohi, K. (2013). The incidence and magnitude of fibrinolytic activation in trauma patients. J. Thromb. Haemost. 11, 307–314 [DOI] [PubMed] [Google Scholar]
- 40. CRASH-2 Collaborators, Intracranial Bleeding Study. (2011). Effect of tranexamic acid in traumatic brain injury: a nested randomised, placebo controlled trial (CRASH-2 Intracranial Bleeding Study). BMJ 343, d3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.