

Abstract
Endometrial cancer (EC) is the most common gynecologic malignancy and still remains clinically challenging. We aimed to explore the potential biomarkers of EC and provide a theoretical basis for early screening and targeted therapy. The available transcriptome data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) were analyzed to identify differentially expressed genes. Immunohistochemistry was performed to detect gene expression. We analyzed the associations of MYBL2 with clinicopathological features and survival time and the biological effect of MYBL2 on the proliferation of EC cells. The effect of MYBL2 silencing on the transcriptome of EC cell model was analyzed by RNA-Seq. MYBL2 was significantly upregulated with obvious copy number alteration in EC. Copy number amplification significantly increased MYBL2 mRNA expression, which led to a poor prognosis and severe pathological types of EC. Additionally, MYBL2 silencing significantly inhibited proliferation and induced apoptosis and G1-phase cell cycle arrest in EC cell lines. Our results indicate that MYBL2 is closely related to the cell cycle and apoptosis pathways in EC. The findings in this study provide evidence that MYBL2 can serve as a new candidate prognostic marker and a target for future therapeutic intervention in EC.
Key words: Endometrial carcinoma, MYBL2, copy number, proliferation, prognosis
Introduction
Endometrial carcinoma (EC) is one of the most lethal gynecological cancers and is the most common malignant tumor of the female genital tract in the world. There are 319,500 new cases each year, with a high mortality rate that exceeds 23%.1 Surgery is the main treatment for EC patients, while radiotherapy, hormone therapy, chemotherapy and other auxiliary methods are also used.2,3 However, the poor prognosis, high local recurrence rate, high metastasis rate, and chemoresistance of EC remain to be addressed.3 Hence, it is urgent to explore the potential molecular mechanisms of EC and to identify novel biomarkers and therapeutic targets. Genetic factors play an important role in the etiology of EC. Cancer evolution is a complex developmental process that is mainly related to the activation of proto-oncogenes and the loss of suppressor genes.4,5 The identification of proto-oncogenes or tumor suppressor genes can partly elucidate the pathogenesis of tumors, including chromosomal alterations, copy number alterations (CNAs), and epigenetic changes.6,7
Copy number amplification is a form of structural variation defined as the CNA of DNA fragments from 1 Kb to several Mb in the human genome.6 DNA deletion, insertion, replication and complex multisite mutations are involved in copy number alteration (CNA).7 CNA is a hallmark of cancer, leading to oncogene activation or inactivation of tumor suppressor genes in various malignant tumors.8 Previous research has found that high-risk germline mutations are likely to occur during the mismatch repair of the MSH2, MSH6 and PMS2 genes, and some rare germline copy number deletions will also occur in the early phase of EC.9 The study found that GSTT1 copy number amplification increased EC risk with substrate specificity. The intermediates produced by GSTT1 may be genotoxic to cells.10
In recent years, through analysis of data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) for comprehensive genomic and transcriptome analysis of EC, new molecular types of EC have been discovered. The classification system greatly contributes to the development of targeted therapy for specific genetic abnormalities.11 In this study, based on the EC mRNA expression profiles in the TCGA and GEO databases, we conducted a comprehensive analysis to identify potential biomarkers of EC and provided a potential target for EC therapy.
Materials and Methods
Clinical samples
According to the minimum sample rule of mathematical statistics, thirty-pairs of EC tissues and the adjacent tissues were acquired from the Second Affiliated Hospital of Nanchang University. The tissues were frozen in liquid nitrogen and then immediately stored at -80°C. The Ethics Committee of the Second Affiliated Hospital of Nanchang University approved the protocol with approval number: Review [2018] No. (014), and all patients provided consent for the utilization of their tissue samples in this study. The patients were registered in the Ethics Committee of our hospital; thus, we got the approval of the ethic approval document. The study was conformed to the provisions of the Declaration of Helsinki (as revised in Edinburgh 2000).
Data collection and comprehensive analysis
We downloaded EC RNA-seq data and phenotype data from the TCGA database (https://xenabrowser.net/datapages/), which includes 174 EC specimens and 13 normal endometrial specimens. According to Diskin’s research, the clinical information, CNA spectrum, and gene sequencing details of EC patients were obtained.12 Moreover, two gene expression profile matrix files were downloaded from the GEO database. GSE63678 includes 7 EC tissues and 5 normal endometrial tissues.13 GSE17025 includes 91 EC tissues and 12 normal endometrial tissues.14 The platforms for these two data sets are GPL571 and GPL570. According to the annotation information on the platform, the probe is converted into the corresponding gene symbol, and log2 conversion is performed on all gene expression data.
We used the method of linear model empirical Bayes statistics in the limma package in R software to perform nonspecific filtering on the EC data set to screen out the differentially expressed genes (DEGs) between tumor and adjacent tissues.15 We used GEO2R to screen DEGs in GSE63678 and GSE17025, where genes with p values <0.05 and log (FC) >1 were considered DEGs.
Furthermore, the mRNA expression and clinical data were extracted from the TCGA with cBioPortal (www.cbioportal.org). These data were preprocessed and analyzed. To prove the relationship between MYBL2 copy number status or mRNA expression level and survival rate, the mean expression value of each gene was compared between CNA groups, and the significance analysis was assessed with t-test. A Kaplan-Meier diagram was constructed to prove the relationship between genetic disorders and patient survival. Additionally, various clinical and pathological features were also detected, including the age of initial diagnosis, menstrual conditions, pathological type, FIGO stage and pathological grade. The data were subjected to the chi-square test after quantification and analysis.
Immunohistochemistry
We stained the tumor tissue samples using rabbit monoclonal antibodies against MYBL2 (1:200, Abcam, Cambridge, UK), CDK1, and CCNB1 (1:200, Proteintech, Rosemont, IL, USA) based on the standard Immunohistochemistry (IHC) protocol. Briefly, 5-μm-thick paraffin-embedded tissue sections were deparaffinized with xylene and graded alcohol, and endogenous peroxidase activity was quenched with 3% H2O2 in methanol. We washed the tissues with Tris-borate saline Tween-20 (TBST). After washing, slides were incubated with DAB (3,3'-diaminobenzidine tetrahydrochloride) (Sigma-Aldrich, St. Louis, MO, USA) and immediately washed under tap water after color development. Slides were then counterstained with hematoxylin. Slides were mounted with dibutyl phthalate xylene (DPX) and were then observed under a light microscope (Carl Zeiss, Oberkochen, Germany). Two independent pathologists evaluated the slices. Positive staining of MYBL2 was observed in the nucleus and cytoplasm. Five fields of view were randomly selected for each assessment and viewed at 400 times magnification (each field had at least 200 cells). MYBL2, CDK1 and CCNB1 staining were scored based on the percentage of positive cells and fluorescence intensity (0, negative; 1, weak; 2, medium; 3, strong). The product of the intensity score and the distribution score was used as the staining score. A specimen with a final staining score of 0-1 was considered negative, and a specimen with a score of 2 was considered positive.
Cell culture
We used HEC-1-B and AN3CA cell lines for in vitro experiments, and they were purchased from Shanghai Enzyme Biotechnology (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fresh fetal bovine serum, 100 μg/ml penicillin and 100 μg/ml streptomycin. Cells in media were placed at 37°C with 5.0% CO2 in a humidified incubator and sub-cultured when the cells reached 70% confluence.
RNA extraction, transfection and qRT-PCR
HEC-1-B and AN3CA cells were plated from cell culture flasks into 6-well plates. The shRNA plasmid was transfected into HEC-1-B and AN3CA cells with TurboFect transfection reagent (Life Technologies Co., Carlsbad, CA, USA) when the cells grew to 50%-60%. The cells were then stained with EdU reagent and Hoechst reagent after 48 h. The shRNA sequences targeting MYBL2 were shMYBL2-1, GUUAUGGACCAAAGAGGAATT and shMYBL2-2, CCGUCCCUCCUACCAUAAATT. The positive control shRNA (shPC) sequence was UUCUCCGAACGUGUCACGUTT.
The total RNA of each group of cells was extracted according to the TRIzol reagent instructions (Invitrogen), and a reverse transcription kit (TaKaRa, Dalian, Liaoning, China) was used to reverse transcribe the RNA. qRT-PCR was performed with SYBR green (Applied Biosystems, Foster City, CA, USA) by means of a ViiA7 real-time PCR system (Applied Biosystems). The reaction system of qRT-PCR was as follows: 10 μL SYBR, 0.4 μL ROX enzyme, 1.6 μL upstream and downstream primers, 6 μL DEPC water, and 2 μL cDNA template. We calculated the relative mRNA expression by the comparative Ct method using glyceraldehyde-3- phosphate dehydrogenase (GAPDH) as an internal control. The following primers were used for GAPDH: forward, 5’- TGACTTCAACAGCGACACCCA-3’ and reverse, 5’-CACCCTGTTGCTGTAGCCAAA- 3’. The following primers were used for MYBL2: forward, 5’-CCGGAGCAGAGGGATAGCA-3’ and reverse, 5’-CAGTGCGGTTAGGGAAGTGG-3’.
Western blotting
We extracted proteins from each group of transfected cells by using radioimmunoprecipitation lysis buffer (Roche, Nutley, NJ, USA). Protein lysates were quantified with a BioRad kit (Bio-Rad, Hercules, CA, USA). Microgram protein samples were separated via 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then electrophoretically transferred to a polyvinylidene difluoride membrane (Millipore, Burlington, MA, USA). Then, the NC membrane was blocked with 5% skim milk at 37°C for 1 h. The membranes were immunoblotted with primary antibodies overnight at 4°C followed by incubation with the respective secondary antibodies for 4 h. The ECL kit was used to detect the bands. The antibodies used were as follows: MYBL2 (1:1,000; Absin Bioscience, Shanghai, PRC); GAPDH (1:5,000; Epitomics, Burlingame, CA, USA); and anti-rabbit IgG antibody (1:4,000; Epitomics).
Cell proliferation and clone formation
EC cells were cultured in DMEM with 10% (v/v) FBS (Gibco). Cells in culture flasks were placed in an incubator (37°C, 5% CO2) and subcultured every 3 days. To perform the cell proliferation assay, we divided EC cells into 96-well plates at a density of 3×103 cells/well and cultured them with 0.5 mL DMEM (with 10% FBS) for 48 h. Then, the cells were transfected with constructed shRNA expression vectors. After transfection, the EdU assay was performed according to the manufacturer’s protocol for the EdU colorimetric cell proliferation assay kit (RiboBio, Guangzhou, China). Cell activity was detected by CCK8 colorimetry. Three thousands (3000) cells per well were inoculated in a 96-well plate, cultured for 48 h after transfection, 10 μL CCK8 reagent was added to each well, and incubated for two hours. The absorbance was then measured with a microplate reader at 562 nm. The growth curve generated according to the OD values. For the clonogenic assay, EC cells (3×103 per well) were plated in 6-well plates and cultured for 2 weeks. All cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet (w/v) at room temperature, and the colony numbers were determined. Cell cycle distribution was analyzed by propidium iodide (PI) staining and flow cytometry.
Figure 1.
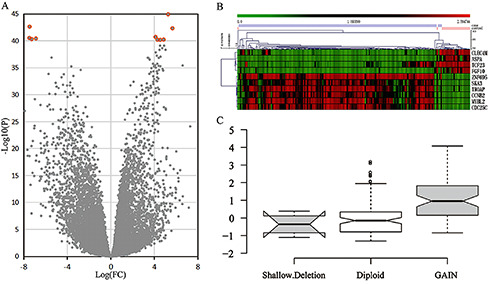
Summary of EC genome-wide integration analysis based on TCGA and GEO data sets. A) In the screening of DEGs in EC, red circles represent ten significant genes. B) In the TCGA data set, the top ten genes expressed most prominently in EC patients were identified; each column represents a sample, while each row represents the expression level of the gene; the color scale represents the original Z score, ranging from green (low expression) to red (high expression). C) Among the top 10 DEGs, MYBL2 had the most significant CNA in EC, and copy number amplification led to upregulated expression levels of MYBL2.
Cell cycle
We collected EC cells (2×106) with or without sh-MYBL2 treatment. Half of these cells were fixed with 500 μl 70% cold ethanol for 2 h overnight according to the manual for the cell cycle detection kit (Keygen Biotech Co., Ltd., Nanjing, Jiangsu, China) and stored at 4°C. The cells were centrifuged by vortexing at 1500 rpm for 5 min, the supernatant was removed, and the hydrated cells were resuspended for 10 min in 0.5 mL PBS (precooled at 4°C). RNase was also added at 37°C for 10 min. We added 400 μL of PI (final concentration: 50 μg/mL) and incubated it for 15 min at room temperature in the dark.
The cell cycle was detected on a flow cytometer (BD Accuri C6, Ann Arbor, MI, USA) after cell digestion by assessing red fluorescence at a wavelength of 480 nm and analyzed by FlowJo software. We performed at least 3 independent experiments to determine the percentage of cells in G0, G1, and G2/M.
RNA-seq analysis
We extracted and purified total RNA from cells under various culture conditions, removing rRNA to build libraries. We collected 10 Gb of sequencing data for each condition after ABI SOLiD sequencing for data processing. We identified all the transcription units (TUs) in the genome and calculated the expression abundance. The expression levels of the genes were calculated from the reads per kilobase of transcript per million mapped reads (RPKM) value, and the TU shear mode was determined by comparing the sequences. DEGseq software was used to compare the expression levels of each group of genes to determine the DEGs and to analyze the biological pathways.
Statistics
Data analyses were performed using SPSS 22.0 software (SPSS Inc., Chicago, IL, USA). We used Student’s t-test or analysis of variance for the statistical analyses; p-values <0.05 were considered significant.
Results
MYBL2 was overexpressed in EC according to wholegenome screening and clinical experiments
We downloaded the EC data set from the TCGA, which contains information on 20,530 genes. After screening, we selected 12,047 significant genes (p<0.05). We compared the gene expression levels between EC tissue and nontumor tissue, and the DEGs were screened out. Among the top 10 most significant DEGs, the downregulated genes in EC were CLEC4M, ASPS, TCF23, and FGF10. The remaining DEGs were highly expressed, including ZNF695, SKA3, MYBL2, TROAP, CCNB2, and CDC25C (Figure 1A and 1B). In addition, the DEGs between EC tissues and nontumor tissues were assessed in the GEO data set after normalization and analysis. The results showed that ASPA, MYBL2, TROAP, CCNB2, and CDC25C were the most significantly altered genes (p<0.05). We verified that MYBL2 was highly expressed in EC by assessing these two different data sets. Moreover, copy number variation data of these top 10 significant DEGs from the TCGA were analyzed by GISTIC2.0 online software (Table S1). Among them, MYBL2 had obvious CNA in EC (p<0.001), which caused the expression of MYBL2 mRNA to significantly increase (p<0.05; Figure 1C). Therefore, MYBL2 may be involved in EC occurrence through CNA.
Figure 2.
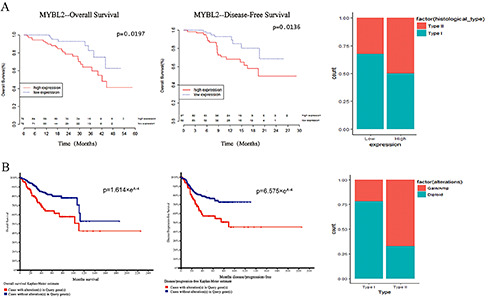
The association between MYBL2 overexpression and amplification and clinical data analysis. A) The overexpression of MYBL2 affects the OS and DFS, pathological type and pathological stage of patients with EC. B) CNA of the MYBL2 leads to a significant deterioration in the prognosis of EC patients, and CNA is more significant in type II EC than in type 1 EC.
We also used clinical experiments to identify MYBL2 expression in EC tissue via IHC. Thirty EC tissue samples and 30 nontumor tissue samples were collected. The IHC results revealed that MYBL2 was overexpressed in tumor tissues (Figure S1).
MYBL2 overexpression and amplification were related to the clinicopathological characteristics of EC
To explore the relationship between MYBL2 and the clinicopathological features and prognosis of EC, we analyzed the clinical data, expression profiles, and CNA data in the TCGA. Cox regression analysis showed that the high expression of MYBL2 was positively correlated with the pathological type (p=0.034) and pathological stage (p<0.001) of EC (Table 1). We also performed overall survival (OS) and disease-free survival (DFS) analyses of MYBL2, and they were significantly different in EC patients with MYBL2 overexpression (p<0.05, Table S2). The EC patients with high expression of MYBL2, especially those with high-grade and type II EC, had a worse prognosis (p<0.05) than those with low expression (Figure 2A). Interestingly, the CNA of MYBL2 also led to a significant deterioration in the prognosis of EC patients, and this effect was more significant in type II EC than in other types (Figure 2B). Thus, overexpression of MYBL2 and CNA may be prognostic factors for EC.
MYBL2 knockdown inhibited EC cell proliferation
To further investigate the biological effect of MYBL2 on the proliferation of EC cells, we chose two EC cell lines (HEC-1-B and AN3CA) and designed two shRNAs to silence MYBL2 expression. Quantitative RT-PCR and Western blotting (WB) were used for assessment. qRT-PCR and WB confirmed that sh-MYBL2 can significantly down-regulate the expression of MYBL2 in EC cell lines (Figure 3 A,B). Moreover, EdU and Cell Counting Kit-8 (CCK-8) data analysis also showed that knockdown of MYBL2 inhibited the proliferation of the two EC cell lines, especially HEC-1-B cells with sh-MYBL2-2, which had a 32% reduction in the expression level of MYBL2 compared to that in control cells (Figure 3 C,D). Similarly, compared with negative control cells (sh-NC), colony formation was decreased after knockdown (p=0.0012; Figure 3E). Together, these results indicate that MYBL2 may play an important role in regulating the proliferation of EC cells.
Downregulation of MYBL2 inhibits the expression of genes involved in cell proliferation
To explore the molecular mechanism of MYBL2 expression in EC, we used RNA-seq to analyze the effect of MYBL2 silencing on the transcriptome of EC cell models. The expression of a total of 16,238 genes was detected in EC cells treated with sh-NC and sh- MYBL2 (RPKM> 0 in any group). Comparison and screening of sh-NC and sh-MYBL2 groups revealed 1,367 DEGs (p<0.05), accounting for 8.42% of the total expressed genes; these DEGs included 644 upregulated genes (logFC >0) and 723 downregulated genes (logFC <0; Figure 4A). To deeply understand the biological significance of MYBL2 silencing on the gene expression regulation of EC cells, the research team conducted pathway enrichment analysis based on 1,367 DEGs. WebGestalt analysis showed that these DEGs were enriched in 20 functional pathways (FDR <0.05). We selected the top 10 enriched pathways for further study. Among them, MYBL2 was closely related to the two pathways of the cell cycle and apoptosis (Figure 4B). Further analysis of cell cycle-related genes revealed that 22 genes were related to the cell cycle (Figure 4C). Consistent with these results, downregulation of MYBL2 could induce EC cell apoptosis, inhibit cell cycle progression, and block most cells in the G1 phase (Figure 4 D,E). These results indicated that MYBL2 silencing can inhibit the proliferation and growth of EC cells by blocking cell cycle progression.
Table 1.
Association of MYBL2 expression levels with clinicopathologic characteristics in EC.
| Factors | MYBL2 protein expression | p | |||
|---|---|---|---|---|---|
| Low (n=68) | High (n=72) | ||||
| Count | % | Count | % | ||
| Age (years) | |||||
| >60 | 17 | 12.10 | 18 | 12.90 | 1 |
| ≤60 | 51 | 36.40 | 54 | 38.60 | |
| Menopause status* | |||||
| Peri/Pre | 5 | 3.60 | 6 | 4.30 | 0.829 |
| Fost | 63 | 45.00 | 66 | 47.10 | |
| Histological type | |||||
| Type I | 46 | 32.90 | 26 | 25.70 | 0.034 |
| Type II | 22 | 15.70 | 36 | 25.70 | |
| Clinical stage | |||||
| Type I | 42 | 30.00 | 38 | 27.10 | 0.283 |
| Type II-IV | 26 | 18.60 | 34 | 24.30 | |
| Histologic Grade | |||||
| G1-G2 | 23 | 16.40 | 5 | 3.60 | <0.001 |
| G3-G4 | 45 | 32.10 | 67 | 47.90 | |
*Peri, 6-12 months since last menstrual period (LMP); Pre,<6 months since LMP and no prior bilateral ovariectomy and not on estrogen replacement; Post, prior bilateral ovariectomy or >12 months since LMP with no prior hysterectomy.
Figure 3.
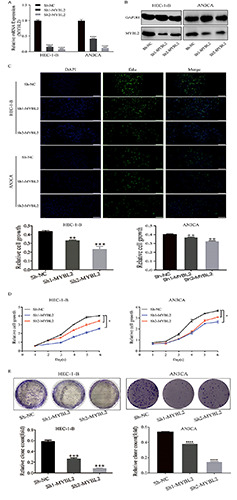
Cell proliferation assay in MYBL2 knockdown EC cell lines. A) The capability of different shRNAs to downregulate MYBL2 expression. B) The ability of different shRNAs to inhibit the expression of MYBL2 protein. C) The cell proliferation rate in HEC-1-B and AN3CA cells at 48 h after MYBL2 knockdown; after transfecting the two cell lines with MYBL2 shRNA, MYBL2 expression was downregulated, and the cell proliferation of HEC-1-B and AN3CA cells was inhibited dramatically compared to that of the sh-NC cells. D) CCK-8 cell growth curve. E) Knockdown of MYBL2 significantly reduced the colony forming ability of HEC-1-B and AN3CA cell lines. **p<0.01; ***p<0.001.
Figure 4.
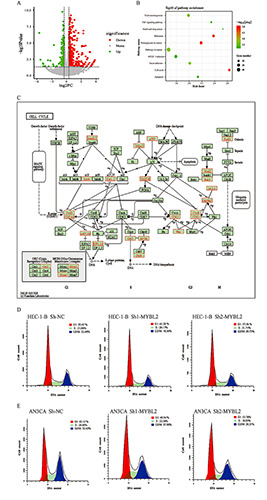
Pathway enrichment analysis of DEGs in MYBL2 knockdown EC cells. A) Analysis of DEGs in EC cell untreated and treated groups. Volcano plots were used to summarize the DEGs. B) Pathway enrichment analysis of the top 10 DEGs; the negative log10 Q value from pathway enrichment analysis is plotted, and the pathways are sorted based on the p-value of DEGs and enrichment based on the core DEG data set. C) Heat map of genes related to the cell cycle. The heat map showed that after knocking out MYBL2, 22 genes in EC cells were significantly differentially expressed. D-E) Cell cycle analysis of EC cells 48 h after transfection with the indicated plasmids.
Discussion
In our study, based on the transcriptome and survival data of EC in the TCGA and GEO databases. After screening, it was determined that the DEGs were ASPA, MYBL2, TROAP, CCNB2 and CDC25C. MYBL2 is highly expressed in EC (Figure 1B). IHC results also verified the high expression of MYBL2 in EC tissues (Figure S1). We used the Phenotype database to analyze the survival of the above five candidate genes. The results showed that only the high expression of MYBL2 was positively correlated with the poor prognosis of EC patients (Figure 2A; Table S2). MYBL2 has obvious CNAs in EC (p<0.001), and CNAs significantly increased the expression of MYBL2 gene mRNA level (p<0.05) (Figure 1C; Table S1). On the other hand, the clinical data of TCGA shows that the overexpression of MYBL2 in EC is related to clinicopathological characteristics (such as poor survival, high pathological type and pathological stage) (Table1). In addition, through the data in cBio, we found that the high expression of MYBL2 is closely related to its CNAs, and CNAs is closely related to clinical indicators (Figure 2B). In summary, MYBL2 does have an impact on EC, so the target gene MYBL2 was finally screened out. But the exploration of its mechanism will require further experimental verification.
The proto-oncogene MYBL2, also known as B-MYB, is a member of the Myb family of transcription factors, which also includes A-MYB and C-MYB. C-MYB was first identified as highly expressed in hematopoietic stem cells16 and in the brain17 and colon.18 A-MYB is mainly expressed in the testis, with extremely low expression in the ovary, spleen and brain.19 MYBL2 is usually expressed in rapidly dividing cells, especially in adult hematopoietic and embryonic stem cells and is involved in the proliferation, differentiation and apoptosis of various cells.20 Studies have confirmed that MYBL2 is highly expressed in malignant tumors such as colorectal cancer, pancreatic ductal adenocarcinoma and lung cancer.21 Previous research also indicates that MYBL2 is associated with the prognosis of many tumors.22 Possible mechanisms of MYBL2 dysregulation in cancer were assumed. Moreover, the amplification of chr20q13 can cause changes in MYBL2 expression, and the amplification of this chromosomal region has been reported in breast and ovarian cancers.23,24 Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer and can lead to an increase in MYBL2. These findings suggest that MYBL2 plays a vital role in the occurrence and development of various tumors. However, the expression of MYBL2 and its biological functions in EC have not been studied. Previous reports prompted us to explore the role of MYBL2 in the progression of EC.
MYBL2 is widely expressed in all proliferating cells, and its expression level is related to the cell proliferation capacity.25 Liang and colleagues found that after knocking down MYBL2, the proportion of cells in the G2/M phase increased, and cell cycle arrest tended to occur. Overexpression of MYBL2 increased the proportion of S-phase cells. The same results were observed in liver cancer and breast cancer cell lines, and the mechanism may be related to the downregulation of CCNB1, CCNA2, CDC2, and Plk1 protein after inhibition of MYBL2 protein.26 The downregulation of MYBL2 leads to the inhibition of cell cycle progression, and MYBL2, together with MuvB and FoxM1, regulates the expression of several key proteins required for the G2/M transition.27 Therefore, we speculated that the transcription factor MYBL2 can promote cell cycle progression to S phase and M phase by affecting the transcription of important genes related to the cell cycle, which could promote the proliferation of cancer cells. Studies have shown that MYBL2 knockout in mice indicates that the colony formation of blastocysts is blocked. These results indicated that MYBL2 is an essential gene that regulates the cell cycle and cell survival.28 Previous studies have shown that the mechanism of oncogenesis is related to the cell cycle.7 These data demonstrate the important role of MYBL2 in promoting cell proliferation. Nevertheless, the mechanism of MYBL2 in EC is still unknown.
Therefore, we established a cell model of MYBL2 down-regulation to explore the role of MYBL2 in EC. We selected two EC cell lines (HEC-1-B and AN3CA) and constructed two shRNAs to down-regulate MYBL2 expression. MYBL2 mRNA and protein expression levels were significantly down-regulated in sh-MYBL2 groups (Figure 3 A,B). Moreover, EdU and CCK-8 data analysis also showed that sh-MYBL2 inhibited the proliferation of the two EC cell lines (Figure 3C and 3D). Similarly, colony formation was significantly reduced (p=0.0012, Figure 3E). Consistent with these results, pathway enrichment analysis based on RNAseq data shows that MYBL2 is closely related to the two pathways of cell cycle and apoptosis, and can activate gene expression related to cell proliferation (Figure 4 B,C). These results indicate that MYBL2 may play an important role in regulating the proliferation of EC cells.
Based on our research, we innovatively found that the MYBL2 gene had CNA in EC, which affects MYBL2 expression. Analysis of clinical data showed that patients with MYBL2 overexpression have a poor prognosis. In addition, the results of the cell proliferation assay also demonstrated that MYBL2 plays a vital role in the proliferation of EC cells. We provided evidence that MYBL2 may contribute to the progression of EC by regulating the cell cycle and promoting cell proliferation. Therefore, MYBL2 may be used as a new biomarker for the prognostication and treatment of EC patients.
Funding Statement
Funding: This work was supported by the National Natural Science Foundation of China (82060474) and the Major Project of Jiangxi Natural Science Foundation (20161ACB20022).
References
- 1.Felix AS, Yang HP, Bell DW, Sherman ME. Epidemiology of endometrial carcinoma: Etiologic importance of hormonal and metabolic influences. Adv Exp Med Biol 2017;943:3-26. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton CA, Cheung MK, Osann K, Chen L, Teng NN, Longacre TA, et al. Uterine papillary serous and clear cell carcinomas predict for poorer survival compared to grade 3 endometrioid corpus cancers. Br J Cancer 2016;94:642-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bestvina CM, Fleming GF. Chemotherapy for endometrial cancer in adjuvant and advanced disease settings. Oncologist 2016;21:1250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viet CT, Schmidt BL. Understanding oral cancer in the genome era. Head Neck 2010;32:1246-68. [DOI] [PubMed] [Google Scholar]
- 5.Sischo L, Broder HL. Oral health-related quality of life: what, why, how, and future implications. J Dent Res 2011;90:1264-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 1998;20:207-11. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144:646-74. [DOI] [PubMed] [Google Scholar]
- 8.Kumaran M, Cass CE, Graham K, Mackey JR, Hubaux R, Lam W, et al. Germline copy number variations are associated with breast cancer risk and prognosis. Sci Rep 2017;7:14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moir-Meyer GL, Pearson JF, Lose F. Australian National Endometrial Cancer Study Group Scott RJ, McEvoy M, et al. Rare germline copy number deletions of likely functional importance are implicated in endometrial cancer predisposition. Hum Genet 2015; 134:269-78. [DOI] [PubMed] [Google Scholar]
- 10.Karageorgi S, Prescott J, Wong JYY, Lee IM, Buring JE, De Vivo I. GSTM1 and GSTT1 copy number variation in population- based studies of endometrial cancer risk. Cancer Epidemiol Biomarkers Prev 2011;20:1447-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McAlpine J, Leon-Castillo A, Bosse T. The rise of a novel classification system for endometrial carcinoma; integration of molecular subclasses. J Pathol 2018;244:538-49. [DOI] [PubMed] [Google Scholar]
- 12.Diskin SJ, Eck T, Greshock J, Mosse YP, Naylor T, Stoeckert CJ, et al. STAC: A method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. Genome Res 2006;16:1149-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pappa KI, Polyzos A, Jacob-Hirsch J, Amariglio N, Vlachos GD, Loutradis D, et al. Profiling of discrete gynecological cancers reveals novel transcriptional modules and common features shared by other cancer types and embryonic stem cells. PLoS One 2015;10:e0142229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Day RS, McDade KK, Chandran UR, Lisovich A, Conrads TP, Hood BL, et al. Identifier mapping performance for integrating transcriptomics and proteomics experimental results. BMC Bioinformatics 2011;12:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNAsequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandberg ML, Sutton SE, Pletcher MT, Wiltshire T, Tarantino LM, Hogenesch JB, et al. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev Cell 2015;8:53-66. [DOI] [PubMed] [Google Scholar]
- 17.Malaterre J, Mantamadiotis T, Dworkin S, Lightowler S, Yang Q, Ransome MI, et al. c-Myb is required for neural progenitor cell proliferation and maintenance of the neural stem cell niche in adult brain. Stem Cells 2008;26:173-81. [DOI] [PubMed] [Google Scholar]
- 18.Malaterre J, Carpinelli M, Ernst M, Alexander W, Cooke M, Sutton S, et al. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proc Natl Acad Sci USA 2007;104:3829-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mettus RV, Litvin J, Wali A, Toscani A, Latham K, Hatton K, et al. Murine A-myb: Evidence for differential splicing and tissue- specific expression. Oncogene 1994;9:3077-86. [PubMed] [Google Scholar]
- 20.Tarasov K V., Tarasova YS, Tam WL, Riordon DR, Elliott ST, Kania G, et al. B-MYB is essential for normal cell cycle progression and chromosomal stability of embryonic stem cell. PLoS One 2008;3:e2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren F, Wang L, Shen X, Xiao X, Liu Z, Wei P, et al. MYBL2 is an independent prognostic marker that has tumor-promoting functions in colorectal cancer. Am J Cancer Res 2015;5:1542-52. [PMC free article] [PubMed] [Google Scholar]
- 22.Guan Z, Cheng W, Huang D, Wei A. High MYBL2 expression and transcription regulatory activity is associated with poor overall survival in patients with hepatocellular carcinoma. Curr Res Transl Med 2018;66:27-32. [DOI] [PubMed] [Google Scholar]
- 23.Tanner MM, Grenman S, Koul A, Johannsson O, Meltzer P, Pejovic T, et al. Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res 2000;6:1833-9. [PubMed] [Google Scholar]
- 24.Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006;10:529-41. [DOI] [PubMed] [Google Scholar]
- 25.Liang H-B, Cao Y, Ma Q, Shu YJ, Wang Z, Zhang F, et al. MYBL2 is a potential prognostic marker that promotes cell proliferation in gallbladder cancer. Cell Physiol Biochem 2017;41:2117-31. [DOI] [PubMed] [Google Scholar]
- 26.Thomas C, Robinson C, Dessauvagie B, Wood B, Sterrett G, Harvey J, et al. Expression of proliferation genes in formalinfixed paraffin-embedded (FFPE) tissue from breast carcinomas. Feasibility and relevance for a routine histopathology laboratory. J Clin Pathol 2017;70:25-32. [DOI] [PubMed] [Google Scholar]
- 27.Musa J, Aynaud MM, Mirabeau O, Delattre O, Grünewald TG. MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis 2017;8:e2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka Y, Patestos NP, Maekawa T, Ishii S. B-myb is required for inner cell mass formation at an early stage of development. J Biol Chem 1999;274:28067-70. [DOI] [PubMed] [Google Scholar]