

Abstract
Rhabdomyosarcoma (RMS) is the most common pediatric soft tissue sarcoma and outcomes have stagnated, highlighting a need for novel therapies. Genomic analysis of RMS has revealed that alterations in the receptor tyrosine kinase (RTK)/RAS/PI3K axis are common and that FGFR4 is frequently mutated or overexpressed. Although FGFR4 is a potentially druggable receptor tyrosine kinase, its functions in RMS are undefined. This study tested FGFR4-activating mutations and overexpression for the ability to generate RMS in mice. Murine tumor models were subsequently used to discover potential therapeutic targets and to test a dual PI3K/mTOR inhibitor in a preclinical setting. Specifically, we provide the first mechanistic evidence of differential potency in the most common human RMS mutations, V550E or N535K, compared to FGFR4wt overexpression as murine myoblasts expressing FGFR4V550E undergo higher rates of cellular transformation, engraftment into mice, and rapidly form sarcomas that highly resemble human RMS. Murine tumor cells overexpressing FGFR4V550E were tested in an in vitro dose–response drug screen along with human RMS cell lines. Compounds were grouped by target class, and potency was determined using average percentage of area under the dose–response curve (AUC). RMS cells were highly sensitive to PI3K/mTOR inhibitors, in particular, GSK2126458 (omipalisib) was a potent inhibitor of FGFR4V550E tumor-derived cell and human RMS cell viability. FGFR4V550E-overexpressing myoblasts and tumor cells had low nanomolar GSK2126458 EC50 values. Mass cytometry using mouse and human RMS cell lines validated GSK2126458 specificity at single-cell resolution, decreasing the abundance of phosphorylated Akt as well as decreasing phosphorylation of the downstream mTOR effectors 4ebp1, Eif4e, and S6. Moreover, PI3K/mTOR inhibition also robustly decreased the growth of RMS tumors in vivo. Thus, by developing a preclinical platform for testing novel therapies, we identified PI3K/mTOR inhibition as a promising new therapy for this devastating pediatric cancer.
Introduction
Rhabdomyosarcoma (RMS) is predominantly a pediatric sarcoma that is characterized by the expression of myogenic transcription factors (e.g., MYOD1, MYOG/MYF4). RMS is thought to arise due to dysregulation of skeletal muscle development [1], and myoblasts, which are the skeletal muscle progenitor cells, are considered a putative RMS cell-of-origin [2]. The two most common RMS subtypes are embryonal (ERMS) or alveolar (ARMS), the latter of which is defined by PAX3-FOXO1 or PAX7-FOXO1 gene fusions [3–6]. ERMS, in contrast, is genetically heterogeneous. Despite an absence of pathognomonic fusion genes, the majority of ERMS contain mutations that dysregulate receptor tyrosine kinase (RTK), phosphatidyl-inositol 3 kinase (PI3K), and RAS signaling pathways [7]. Mutations within the p53 pathway are also common as at least 60% of ERMS tumors contain a “p53 OFF” genotype [8]. Compared to adult cancers, pediatric malignancies like RMS contain relatively few mutations [7, 9, 10], and yet there is limited understanding of how specific mutations affect tumorigenesis. Furthermore, survival rates—30% in high-risk RMS patients—remain largely unchanged in the past decade, despite implementation of more aggressive therapy [11, 12]. Thus it is imperative that the molecular events that drive rhabdomyosarcomagenesis are defined so that more effective, less toxic treatments can be developed.
Alterations in FGFR4 signaling commonly occur in ARMS patients by overexpression, as FGFR4 is a transcriptional target of the PAX3-FOXO1 fusion gene and in ERMS patients due to FGFR4-activating mutations or amplification. Whether these three distinct mechanisms regulating FGFR4 activity directly impact RMS biology and/or patient outcomes is currently undefined. FGFR4 expression is significantly higher in ARMS tumors [13], and recent epigenetic studies have shown that the PAX3-FOXO1 oncoprotein regulates FGFR4 expression through super enhancers [14]. Activating FGFR4 mutations occur in approximately 10% of ERMS patients [7, 15, 16], most commonly in the tyrosine kinase domain at residues V550E or V550L or N535K. Furthermore, expression of FGFR4-activating V550E and N535K mutations in NIH 3T3 fibroblasts caused development of more aggressive tumors than expression of wild-type FGFR4 [16]. However, no difference in tumor biology was noted between the V550E vs. N535K mutations. Although the RTK inhibitor ponatinib reduced xenograft growth in this model, toxicity of this multi-targeted tyrosine kinase inhibitor has limited further development [17]. Thus it is critical that key pathways and specific targets that are required for RMS growth and progression are defined in order to identify promising drug candidates for this disease.
We have recently established a novel approach to model high-grade sarcomas by genetically modifying skeletal muscle precursor cells followed by orthotopic injection into syngeneic, immunocompetent hosts. Specifically, we demonstrated that constitutive Ras activation (KrasG12D) in p53-deficient (p53−/−) myoblasts generated high-grade sarcomas, but in contrast to human ERMS and ARMS, they lacked the expression of muscle-specific genes [18]. In this study, we examined whether expression of human ERMS-associated FGFR4-activating mutations would induce RMS from myoblasts injected into muscle. Specifically, we discovered that expression of the FGFR4-activating mutation FGFR4V550E rapidly induced primary sarcomas with myogenic differentiation. Myoblasts expressing FGFR4V550E exhibited mammalian target of rapamycin (mTOR) pathway activation, providing a preclinical platform for drug discovery and validation. In a comprehensive drug screen, we identified the dual PI3K/mTOR inhibitor GSK2126458 (Omipalisib [19]) as a potent inhibitor of murine and human RMS cell growth. Furthermore, mass cytometry confirmed inhibition of PI3K/mTOR signaling by GSK2126458 at the single-cell level in RMS cell lines. Finally, inhibition of mTOR signaling impaired growth of FGFR4V550E-expressing tumors to a greater extent than standard chemotherapy. Collectively, our findings demonstrate that FGFR4 activation drives rhabdomyosarcomagenesis and that inhibition of mTOR activity holds therapeutic promise in this devastating pediatric cancer.
Results
FGFR4-activating mutations transform p53−/− myoblasts
Myoblast lines M25.FGFR4wt, M25.FGFR4N535K, M25. FGFR4V550E, and M25.EV were generated after transduction of the p53-deficient murine myoblast Myo25 cell line [18] with lentivirus particles encoding wild-type FGFR4 (FGFR4wt), activating mutations of FGFR4 (FGFR4N535K, FGFR4V550E) [16], or an empty vector (EV) control, followed by antibiotic selection (Fig. 1a). Proviral integration was confirmed using transgene-specific PCR primers and DNA sequencing (Fig. 1b, c). Human FGFR4 proteins were overexpressed in M25.FGFR4wt, M25.FGFR4N535K, and M25.FGFR4V550E myoblasts but were not detected in the parental Myo25 and M25.EV myoblasts (Fig. 1d).
Fig. 1.
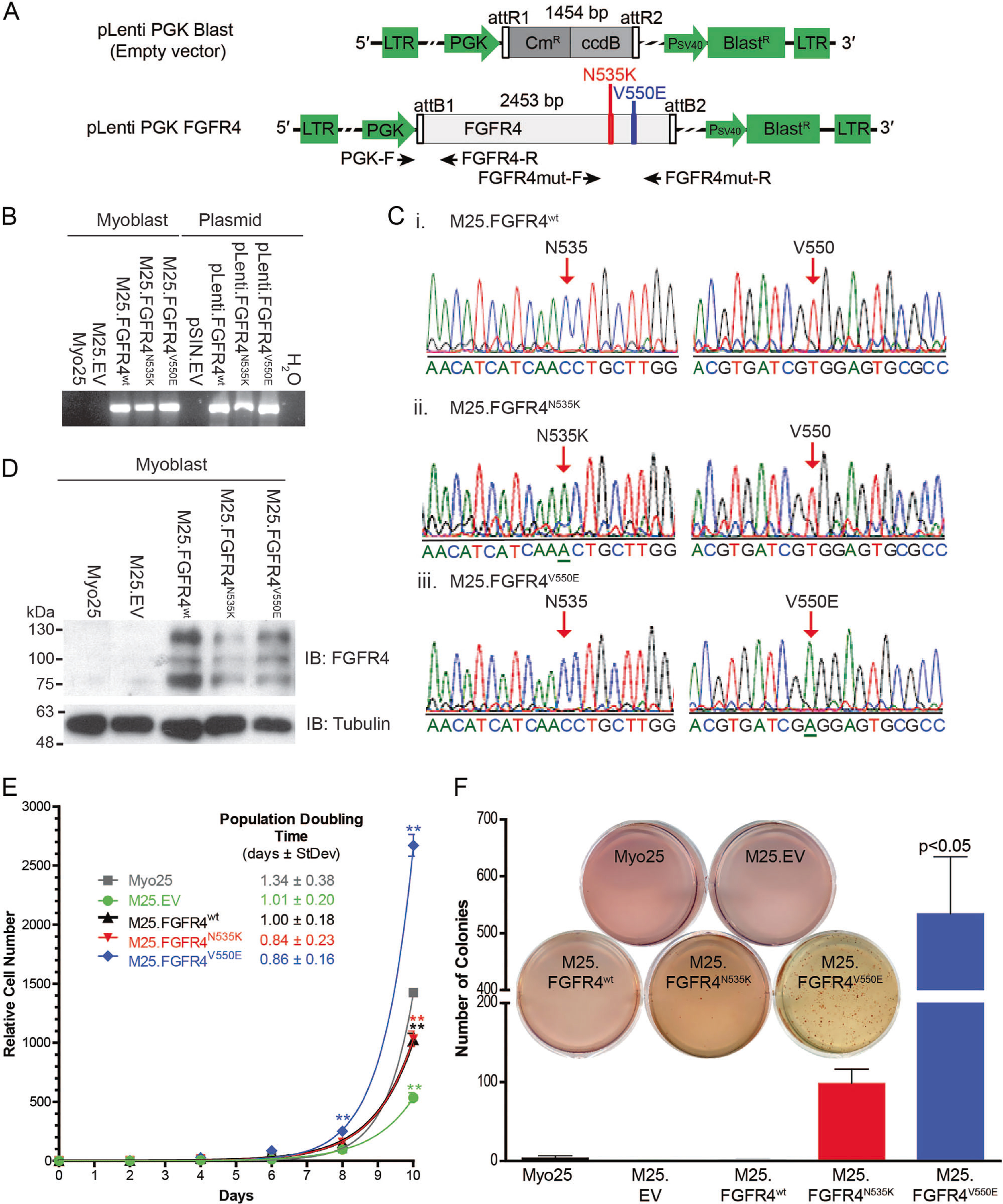
Engineering human FGFR4 overexpressing p53−/− murine myoblasts. a Lentiviral particles were generated using constructs encoding human FGFR4wt, FGFR4N535K, and FGFR4V550E and empty vector (EV). Primer-binding sites for proviral integration as indicated (black arrows). b Control myoblasts (Myo25, M25.EV) did not contain provirus encoding FGFR4. Transgene-specific PCR confirmed proviral integration in populations transduced with the constructs: FGFR4wt, FGFR4N535K, and FGFR4V550E. c FGFR4 sequencing confirmed that M25.FGFR4wt, M25.FGFR4N535K, and M25.FGFR4V550E expressed the correct mutations. d Western blot analysis demonstrated human FGFR4 protein expression in M25.FGFR4wt M25.FGFR4N535K, and M25.FGFR4V550E in contrast to Myo25 and M25. EV myoblasts. e Proliferation analysis demonstrated increased M25.FGFR4V550E cell number at days 8 and 10 (**p < 0.001) and reduced M25.FGFR4wt, M25.FGFR4N535K, and M25.EV cell number at day 10, when compared to Myo25 in a representative assay. Population doubling times calculated from combined assays (n = 3) did not reveal statistically significant differences. f Colony quantification and compilation of biological replicates (n = 3) illustrated the ability of M25.FGFR4N535K and M25.FGFR4V550E to grow in an anchorage-independent manner. A representative assay is shown (inset). Overexpressing FGFR4wt did not transform cells in this assay. Parental Myo25 and empty vector myoblasts (M25.EV) did not form many colonies over the 3-week time course. Bars represent mean ± SD
To examine whether FGFR4 overexpression resulted in p53−/− myoblast transformation, we measured cell proliferation and anchorage-independent growth. M25. FGFR4V550E demonstrated significantly increased proliferation at days 8 and 10 (p < 0.001); however, M25. FGFR4wt and M25.FGFR4N535K had significantly reduced cell number compared to control (p < 0.001) (Fig. 1e). Finally, M25.FGFR4N535K and M25.FGFR4V550E were capable of anchorage-independent growth (Fig. 1f) but only FGFR4V550E-overexpressing myoblasts formed significantly more colonies than parental myoblasts (5.5-fold, p < 0.05). Collectively, these results demonstrate that FGFR4 mutational status affects myoblast proliferation and growth and that different FGFR4 mutations influence cellular transformation.
FGFR4 activated p53−/− myoblasts form tumors in mice
To determine whether FGFR4-activating mutations differentially impact tumorigenesis compared to wild-type overexpression, myoblast cell lines expressing FGFR4N535K (n = 11) or FGFR4V550E (n = 12) were orthotopically engrafted into skeletal muscle of the right hind limb of neonatal p53+/− syngeneic mice and M25.FGFR4wt-overexpressing myoblasts were injected into left limbs (Fig. 2, Table 1, Supplemental Fig. 1). Another cohort (n = 7) was generated by injecting M25.FGFR4wt into right hind limbs vs. control M25.EV myoblasts injected into contralateral limbs. We observed rapid tumor formation in mice injected with M25.FGFR4V550E-expressing myoblasts compared to mice engrafted with either M25.FGFR4N535K (p < 0.001) or M25.FGFR4wt (p < 0.01). Specifically, M25.FGFR4V550E-injected mice developed tumors with a median latency of 11 weeks (range 9–26 weeks), which was highly penetrant as 11/12 mice (92%) reached end point (Fig. 2c, d, Table 1). The majority of mice injected with M25.FGFR4wt formed tumors (5/7, 71%) but with a longer median latency of 37 weeks (range 19–51 weeks) than the M25.FGFR4V550E cohort (p ≤ 0.01). One of the 11 (9%) mice injected with M25.FGFR4N535K developed a tumor, whereas control mice M25.EV (n = 10) remained disease free. Another tumor from this cohort formed in a left hind limb that had been injected with M25.FGFR4wt and tumor DNA sequencing confirmed that it encoded wild-type FGFR4 (Supplemental Fig. 2). No metastatic disease was observed during necropsy. FGFR4 constructs were confirmed by sequencing, and FGFR4wt and FGFR4V550E tumors also expressed human FGFR4 proteins (Fig. 2d, e). Collectively, these data suggest that FGFR4V550E is significantly more tumorigenic than FGFR4N535K and FGFR4wt, which has not been previously demonstrated.
Fig. 2.
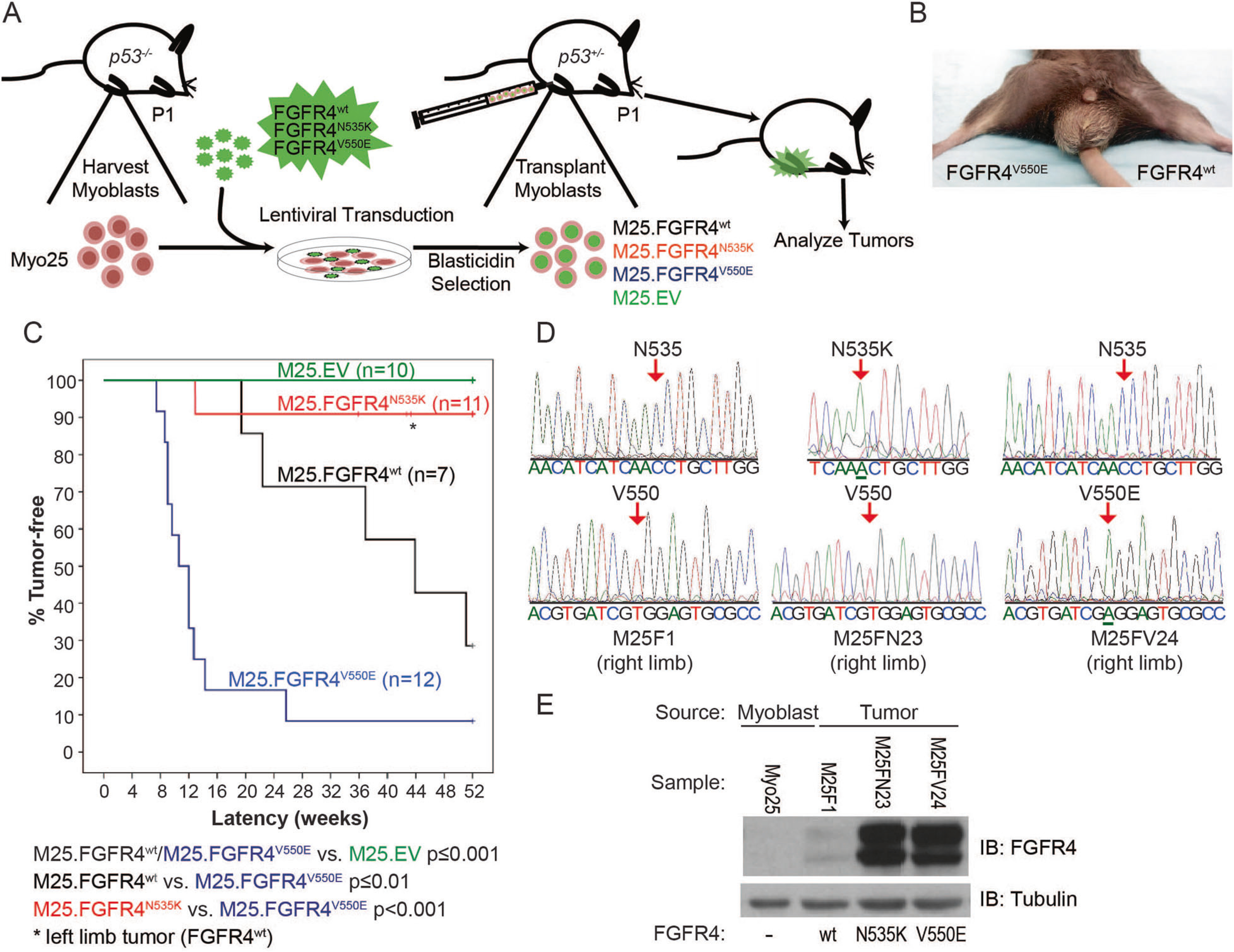
FGFR4V550E p53−/− myoblasts engrafted into murine skeletal muscle generate a high incidence of tumors. a Murine model schema depicts p53−/− myoblast harvest, transduction, injection into p53+/− host hind limbs, and tumor analysis. b A murine tumor following M25. FGFR4V550E injection into the right hind limb. c Kaplan–Meier curves depicting tumor-free survival illustrate statistically significant differences among tumor models expressing different FGFR4 constructs. M25.EV did not generate tumors. M25.FGFR4V550E- and M25. FGFR4wt-injected mice formed tumors with statistically different penetrance and latencies compared to each other and to the M25.EV control cohort. Two mice injected with M25.FGFR4N535K developed tumors, one encoded FGFR4N535K while the second tumor was shown to encode FGFR4wt (asterisk). d Amplicons from tumor tissue were sequenced and shown to encode the correct FGFR4 transgenes. M25F1 represents tumor tissue from the M25.FGFR4wt cohort, M25FN23 from M25.FGFR4N535K, and M25FV24 from M25. FGFR4V550E. e Protein expression was confirmed in tumor lysates using immunoblot and human FGFR4-specific antibodies. Parental myoblasts, Myo25, did not express human FGFR4 proteins
Table 1.
Penetrance and latencies of experimental cohorts
| Injected myoblasts |
Host mice | Tumors |
|||||
|---|---|---|---|---|---|---|---|
| Transgene |
Penetrance (n) | Mean latency (weeks) | Median latency (weeks) | ||||
| Parental cell line | Genotype | Right limb | Left limb | Genotype | (Range) | ||
| Myo25 | p53−/− | FGFR4wt | Empty vector | p53+/−, p53+/+ | 71% | 34.7 | 36.9 |
| (5/7) | (19–51 weeks) | ||||||
| Myo25 | p53−/− | FGFR4N535K | FGFR4wt | p53+/− | 9% | 12.9 | 12.9 |
| (1/11) | — | ||||||
| Myo25 | p53−/− | FGFR4V550E | FGFR4wt | p53+/− | 92% | 11.9 | 10.6 |
| (11/12) | (9–26 weeks) | ||||||
| Myo25 | p53−/− | — | Empty vector | p53+/− | — | — | — |
| (0/10) | — | — | |||||
FGFR4V550E-overexpressing myoblasts generate sarcomas with muscle differentiation
To establish whether RMS was generated in our model system, we performed pathologic assessment of tumors using the WHO Classification for Soft Tissue Sarcomas [20]. Hematoxylin and eosin assessment revealed well-defined spindle–epithelioid masses with necrosis and brisk mitotic activity, which were compatible with high-grade sarcomas (Fig. 3). Additionally, some tumors contained large cells with rhabdoid morphology.
Fig. 3.
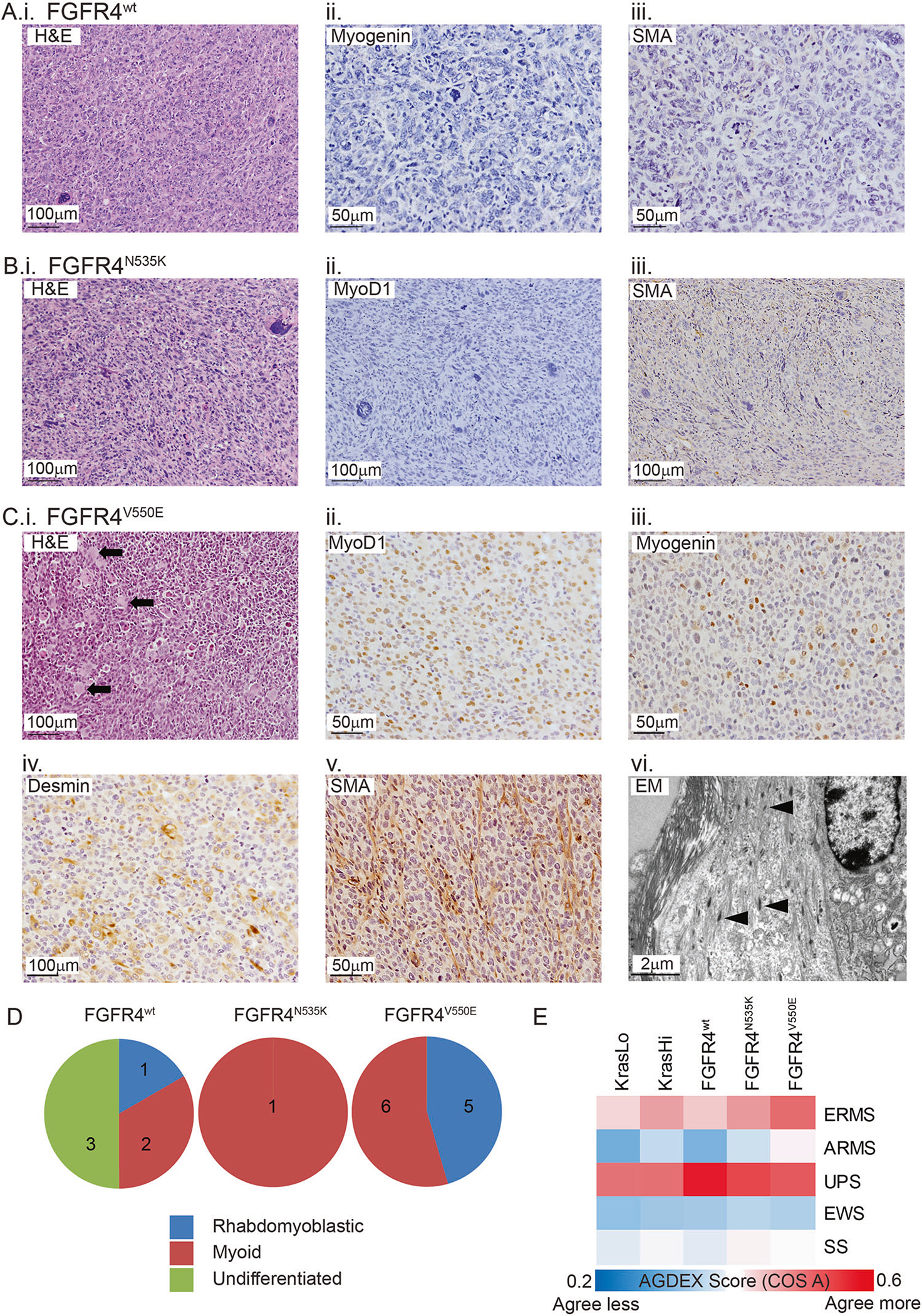
Histopathology of FGFR4-expressing sarcomas is consistent with rhabdomyoblastic differentiation. a Tumors were subjected to morphological analysis using H&E-stained sections and classified following immunohistochemical labeling with a panel of diagnostic markers. Half of the tumors generated following M25. FGFR4wt injection were undifferentiated (i) and did not commonly express the muscle-specific proteins Myogenin (ii) and SMA (iii). b The only M25. FGFR4N535K tumor was classified as a high-grade sarcoma (i) with myoid differentiation based on the absence of Myod1 protein (ii) and presence of SMA (iii). c All tumors generated from M25. FGFR4V550E-injected host mice were high-grade sarcomas (i) with muscle differentiation (myoid/rhabdomyoblastic) based on the presence of rhabdomyoblasts (black arrows) and expression of Myod1 (ii), Myogenin (iii), Desmin (iv), or SMA (v). Transmission electron microscopy showed tumor cells containing immature muscle fibers (vi, black arrowheads). d Pie charts illustrate classification of the high-grade sarcomas among the three injected cohorts. e Heat map reporting the transcriptomic agreement (AGDEX score) in comparisons between KRAS- or FGFR4-driven mouse sarcomas and five subtypes of human sarcoma. EWS represents Ewing sarcoma and SS represents synovial sarcoma
Immunohistochemistry was used to define tumor cell differentiation, with heterogeneity identified among cohorts injected with myoblasts expressing different FGFR4 constructs (Fig. 3, Supplemental Fig. 3). Specifically, tumors (3/6) generated following M25.FGFR4wt injection were undifferentiated (i.e., undifferentiated pleomorphic sarcoma (UPS)), based on a null immunophenotype (Fig. 3a, ii, iii). However, the remaining FGFR4wt tumors had myogenic differentiation, which included Myod1-positive tumor cells indicating rhabdomyoblastic differentiation (n = 1) or smooth muscle actin (SMA) staining, a marker of smooth muscle or myoid differentiation (n = 2). Also, the single M25.FGFR4N535K tumor highly expressed SMA (Fig. 3b, iii). All M25.FGFR4V550E-expressing tumors (11/11) expressed at least one myoid marker (e.g., SMA, Desmin, Myod1, and/or Myogenin). Many FGFR4V550E-expressing tumors (5/11) displayed rhabdomyoblastic differentiation (Myogenin and Desmin) including eosinophilic rhabdomyoblasts (Fig. 3c, i, iii, iv), while Myod1-positive cells were observed in four of these five FGFR4V550E tumors (Fig. 3c, ii). The remaining M25.FGFR4V550E tumors (6/11) expressed SMA (Fig. 3c, v). For ultrastructural evidence of skeletal muscle differentiation, transmission electron microscopy confirmed immature muscle fibers in two FGFR4V550E tumors (Fig. 3c, vi). Importantly, this demonstrates that the degree of myogenic differentiation observed varied with different FGFR4 mutations (Fig. 3d) while FGFR4V550E tumors had evidence of muscle differentiation many of which were consistent with a diagnosis of RMS.
FGFR4V550E tumor gene expression patterns highly correlate with human RMS
To determine whether FGFR4V550E tumors faithfully model human RMS, we used the differential expression (AGDEX) algorithm to correlate ortholog expression profiles of five oncogene-driven syngeneic mouse sarcoma models, including tumors from previously published models (KRASLo, KRASHi [18]) as well as FGFR4wt-, FGFR4N535K- and FGFR4V550E-expressing tumors, with human sarcomas (ERMS, ARMS, UPS, Ewing sarcoma (EWS) and synovial sarcoma (SS)). AGDEX confirmed correlative gene expression signatures between each of the mouse models studied and both ERMS and UPS (Fig. 3e). Overall, the FGFR4V550E model had the highest correlation with ERMS (AGDEX score 0.5, p = 0.004). In contrast, the FGFR4wt tumors were most highly correlated with UPS (AGDEX score 0.58, p < 0.001). These data confirm that FGFR4V550E tumors resemble human ERMS at the gene expression level as well as the histological level.
FGFR4 mutant myoblasts maintain terminal differentiation capacity
As some degree of muscle-specific differentiation was identified in most murine sarcomas, we next sought to characterize the differentiation capacity of myoblasts expressing wild-type or mutant FGFR4 in vitro (Fig. 4a). Assay quantification demonstrated M25.FGFR4wt and M25. FGFR4V550E differentiated at levels similar to Myo25 cells (Fig. 4b). However M25.FGFR4N535K differentiation was approximately twofold lower when compared to Myo25 (p < 0.001), and M25.FGFR4N535K-overexpressing myoblasts had barely detectable levels of MyHC by immunoblotting (Fig. 4c), illustrating reduced terminal muscle differentiation.
Fig. 4.
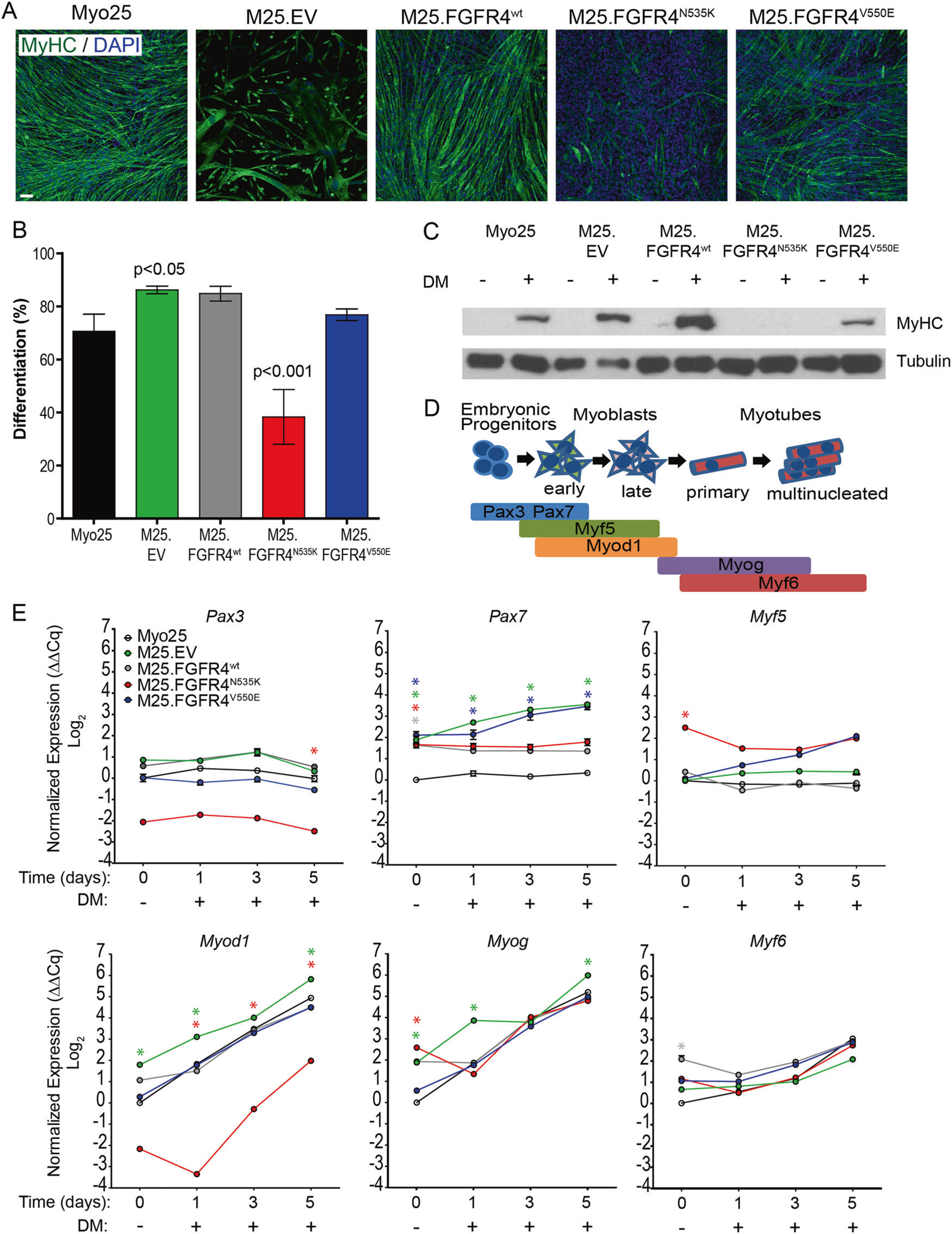
FGFR4-transduced myoblasts retain the ability to terminally differentiate. a Myoblasts immunolabeled with myosin heavy chain (MyHC), counterstained with DAPI, and visualized with fluorescence microscopy illustrated varying patterns and levels of terminal differentiation. Scale bar represents 100 μm. b Differentiation was similar among most myoblast populations, with the exception of M25. FGFR4N535K, which had significantly reduced percentage of differentiation compared to parental Myo25. Bars represent mean ± SD. c Myoblast lysates were collected and immunoblotted to demonstrate the presence of MyHC. After 5 days of differentiation, MyHC immunopositivity can be seen in all myoblast lysates except M25.FGFR4N535K. d Schema illustrating the sequential activation of myogenic regulatory factors as myogenic precursor cells differentiate into mature muscle fibers. e cDNA was generated from myoblasts collected after 0 (MGM) or following 1, 3, or 5 day differentiation and used to measure the gene expression of Pax3, Pax7, Myf5, Myod1, Myogenin, and Myf6. Graphs show representative results from one of three biological replicates. Each experiment consisted of technical triplicates. Statistical significance was analyzed by one-way ANOVA with Tukey’s multiple comparisons test (*p < 0.001) compared to Myo25 at the same time point. Asterisk color represents significance for the corresponding myoblasts
Skeletal muscle differentiation genes were also quantified during differentiation assays (Fig. 4d) to determine how FGFR4 activity may influence myogenic regulation. Overall, no significant differences in the expression of Pax3 and Myf5, early markers of skeletal muscle differentiation, in Myo25, M25.EV, M25.FGFR4wt, or M25.FGFR4V550E myoblasts (Fig. 4e) were observed. Pax7 gene expression was higher in all transduced myoblasts at day 0 (p < 0.001), which continued to rise in myoblasts expressing mutated FGFR4 compared to Myo25 (p < 0.001). Strikingly, FGFR4N535K-overexpressing myoblasts had low Myod1 levels initially and throughout the assay (p < 0.001). Together, these results demonstrate that, although there are different expression patterns of some myogenic regulatory factors, all of the transduced myoblasts maintained some ability to differentiate in vitro. Importantly, the ability of FGFR4V550E-overexpressing myoblasts to differentiate was confirmed in vitro and in vivo, as most tumors generated from these myoblasts expressed myoid and rhabdomyoblastic markers.
FGFR4V550E-overexpressing cells derived from primary tumors generate secondary tumors
Since a hallmark of tumorigenesis is the ability of transformed cells to self-renew, we generated cell lines from two different FGFR4V550E tumors and determined that secondary tumors developed following engraftment of M25.FV24c or M25.FV28c cells into host mice (Supplemental Fig. 4). Strikingly, tumorigenesis was accelerated in both secondary tumor models compared to primary tumors (median latency 4 vs. 10.6 weeks) with 100% penetrance. Thus we demonstrate that cell lines derived from FGFR4V550E tumors have the capacity to generate secondary tumors.
Inhibition of PI3K and mTOR reduces viability in murine and human RMS cell lines
In order to discover novel compounds that might be effective in inhibiting RMS growth, we next used our myoblast cell lines and secondary tumor models for preclinical drug development. Specifically, M25.FV24c were functionally tested along with human RMS cell lines in a single-agent dose–response drug screen using the MIPE-v4 library, which contains 1912 drugs composed of oncology-focused, mechanistically annotated agents that are prioritized for clinical relevance [21]. Viability of M25.FV24c cells was measured by Cell Titer Glo 48 h after addition of compounds. We then grouped compounds by target class where the potency of each class was defined as the average percentage of area under the dose–response curve (AUC). Next, we grouped target classes by potency (Fig. 5a) and discovered that PI3K inhibitors were among the most potent inhibitors of M25.FV24c viability. The dual PI3K/mTOR inhibitor, GSK2126458 (GSK212) was particularly potent in M25.FV24c cells, with an AUC of 25% (Supplemental Table 2). GSK212 also displayed similar potency in human ERMS RMS559 (FGFR4V550L) and ARMS RH30 (PAX3-FOXO1-positive) cell lines, with EC50 values <10 nM (Fig. 5b). Furthermore, the non-transformed fibroblast cell line 7250 had an EC50 >200 nM when treated with GSK212, which is an encouraging therapeutic index for further drug development. Finally, we verified that M25.FGFR4V550E myoblasts had a statistically significant EC50 reduction compared to control (mean EC50 = 10 nM vs. mean EC50 = 36 nM, respectively; p < 0.05) (Fig. 5c). Collectively, these data show that, by functionally screening and grouping class potency of compounds, we identified that targeting the PI3K/mTOR pathway as a potential therapeutic approach for FGFR4-activated RMS.
Fig. 5.

FGFR4V550E tumor-derived cells are sensitive to PI3K/mTOR inhibition in vitro. a Bubble plot displaying ranked potency of the target classes of compounds found in the MIPEv4 screen of M25. FV24c. Bubble size is proportional to the number of compounds found in each target class. b EC50 values for cell lines treated with increasing concentrations of GSK2126458. M25.FV24c, human RMS559 (FGFR4V550L), and RH30 (PAX3/FOXO1) cell lines were sensitive to GSK2126458 with EC50 ≤ 10 nM. Normal fibroblast line, 7250, demonstrated an EC50 of 220 nM, illustrating RMS-specific sensitivity to PI3K/mTOR inhibition. c Cellularity was measured to assess sensitivity of transduced myoblasts to GSK2126458. M25.FGFR4V550E were most sensitive to the dual PI3K/mTOR inhibitor compared to M25.EV (*p < 0.05). d, e Cell lines treated with 40 nM GSK212 or vehicle were stained with metal-tagged antibodies specific for pAKT (S473), p4EBP1(Thr37/46), and pS6(Ser235/236). Histogram overlays depict phospho-protein abundance after treatment of M25.FV24c vs. human K562 leukemia cells (d) or RMS559 vs. RH30 cells (e). Histogram coloration reflects Log2 FC ratio (Veh/GSK) of the median metal intensity, which are also shown on each histogram, demonstrating similar inhibition of p4EBP1 and pS6. pAKT did not demonstrate decreased phosphorylation, suggesting that GSK2126458 primarily inhibits signaling downstream of mTOR but not Ras/Mapk
GSK212 selectively inhibits mTOR signaling in murine and human RMS cells
To more fully characterize the impact of GSK on PI3K/mTOR as well as other signaling pathways in RMS cells, we performed single-cell analyses using mass cytometry time of flight (CyTOF), a platform that allows more highly multi-plexed single-cell analysis than conventional flow cytometry [22, 23]. Murine and human RMS cells were incubated with either vehicle alone (dimethyl sulfoxide (DMSO)) or 40 nM GSK212 for 30 min and then probed with a panel of metal-tagged antibodies that recognize phosphorylated signaling proteins active in several pathways. Phosphorylation of extracellular signal–regulated kinase (ERK) (pERK), p38 mitogen-activated protein kinase (MAPK), pSRC, phosphorylation of signal transducer and activator of transcription factor 1 (pSTAT1), pSTAT3, and pSTAT5 was very low to moderate in murine and human RMS cells and was not decreased by GSK212 treatment (Supplemental Fig. 5a), suggesting that PI3K/mTOR signaling does not activate RAS/MAPK, SRC, or Janus-activated kinase signaling in these cells. Nonetheless, pervanadate treatment robustly increased phosphorylation of these signaling proteins, demonstrating that these pathways could be activated in RMS cells (Supplemental Fig. 5B).
Phosphorylation of AKT at S473, an mTORC2 substrate [24, 25], was detected but was minimally affected by GSK212 in mouse and human RMS cells (Fig. 5d, e). The amount of phosphorylated S6, an mTORC1 substrate, was low in murine RMS cells and was also minimally affected by GSK212 treatment, whereas pS6 was essentially undetectable in both human RMS cells lines (Fig. 5d, e). In striking contrast, both murine and human RMS cells expressed high amounts of p4EBP1, another mTORC1 substrate (Fig. 5d, e). Moreover, p4EBP1 was potently and acutely reduced by pretreatment with 40 nM GSK212 (Log2 [Veh/GSK] fold-change of −2.13 to −4.83). Nonetheless, both p4EBP1 and pS6 levels were sensitive to GSK212 in K562 leukemia cells (Fig. 5d), demonstrating that this drug can inhibit both effector arms of mTOR signaling. Collectively, these data demonstrate that RMS cells with two different activating FGFR4 mutations (V550E or V550L) as well as PAX3-FOXO1 gene fusions exhibit active mTOR signaling and suggest that GSK212 selectively targets the 4EBP1/EIF4E cap-dependent translation arm of mTORC1 signaling in these cells.
GSK212 reduces growth of established FGFR4V550E RMS tumors in vivo
Finally, we employed our secondary tumor model in a preclinical drug study to evaluate whether GSK212 would inhibit RMS in vivo (Fig. 6a). Ten days after injection of M25.FV24c tumor-derived cells into the hind limbs of male and female syngeneic hosts, experimental animals were randomized into four treatment arms: Arm 1, control group—drug vehicles (e.g., DMSO, ethanol); Arm 2, vincristine, the backbone of RMS chemotherapy (0.38 mg/kg, i.p., 1 × weekly); Arm 3, GSK212 (3 mg/kg, p.o., weekly, 5 days on, 2 days off); and Arm 4, combination of vincristine and GSK212 at the described doses. Treatments were administered for 3 weeks (day 21). End point was defined as 34 days, at which point all mice in the control cohort (Arm 1) were sacrificed due to tumor size.
Fig. 6.
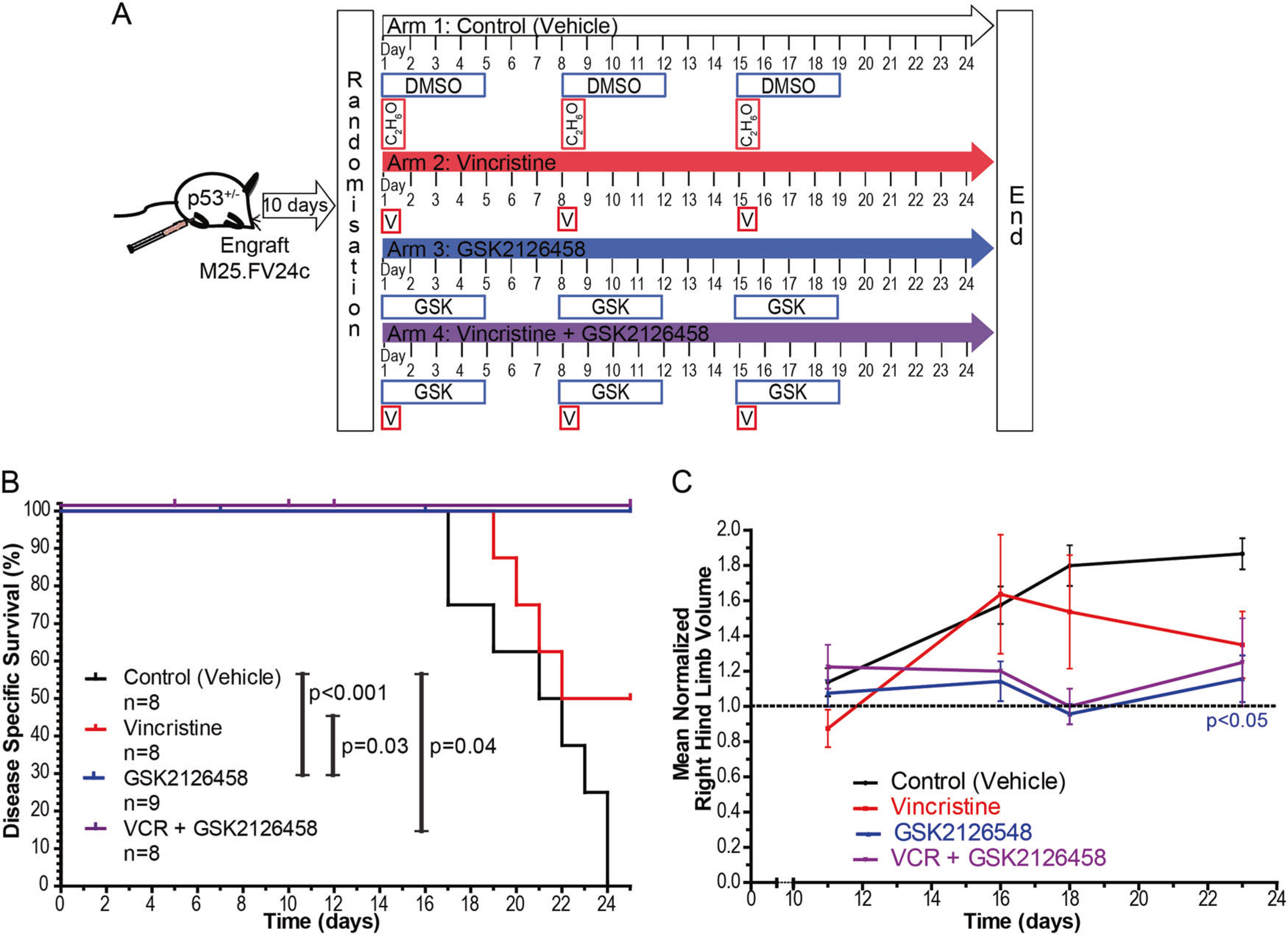
Dual inhibition of PI3K and mTOR reduces tumor growth in a preclinical RMS mouse model. a Schema depicting preclinical study design to determine whether GSK2126458 is effective in inhibiting M25.FGFR4V550E tumor growth. b Disease-specific survival (DSS) illustrates statistically significant increased survival in mice treated with GSK2126458 when compared to vehicle-treated (control) (p < 0.001) or vincristine-treated cohorts (p = 0.03). Murine cohorts treated with the combination of vincristine and GSK2126458 also significantly increased DSS (p = 0.04). c Normalized hind limb volume calculations demonstrate variability among individual mice in the four cohorts. Treatment with GSK2126458 significantly reduced tumor size compared to vehicle-treated controls (p < 0.05). Statistical significance was analyzed with two-tailed t-test. Points represent mean ± SEM
Disease-specific survival (DSS) was significantly prolonged by the use of GSK212 (Arm 3—Fig. 6b) compared to control mice (Arm 1, p < 0.001) and vincristine-treated mice (Arm 2, p = 0.03). Fifty percent of mice in Arm 2 (vincristine) survived until day 34, although this increase in DSS compared to control animals did not reach statistical significance (Fig. 6b). Furthermore, there was a statistically significant decrease in tumor size (p < 0.05) in mice treated with GSK212 compared to control mice (Fig. 6c). Six out of the eight mice (75%) in Arm 4 (vincristine plus GSK212) lost >10% of their body weight during the experiment, suggesting that they died from toxicity (Supplemental Fig. 6A). Tumor growth was reduced in the surviving two mice compared to controls, although this did not reach statistical significance likely due to limited numbers (Fig. 6c). Overall, treatment of FGFR4V550E-expressing tumors with the dual PI3K/mTOR inhibitor GSK212 decreased tumor growth and increased survival when compared vehicle-treated controls and mice treated with vincristine alone, supporting its efficacy in vivo.
Discussion
FGFR4 is a genetic driver of RMS
In this study, we report that FGFR4 dysregulation combined with p53 deficiency results in sarcoma with variable myogenic differentiation. Furthermore, we discovered that different FGFR4-activating mutations (V550E vs. N535K) affected the expression of key myogenic differentiation factors, which likely accounted for the spectrum of myogenic tumors observed. Importantly, drug screening and single-cell validation by mass cytometry demonstrated that GSK212 selectively inhibited mTOR signaling in murine and human RMS cell lines and reduced growth of established FGFR4-driven RMS in mice.
To date, comprehensive genome-wide analysis of RMS samples revealed frequent mutations within RTK/RAS/PI3K pathways and FGFR4 represents the most commonly mutated RTK in ERMS [7, 9, 15]. In silico analysis suggests that FGFR4 mutation occurs early in the clonal evolution of RMS [26] and high FGFR4 expression correlates with decreased survival of RMS patients [16]. It has also been reported that human ERMS cells have increased FGFR4 mRNA expression compared to normal human myoblasts, and FGFR4 pathway blockade decreases proliferation [27]. In fusion gene-positive ARMS, the same FGFR4-regulatory elements bound by PAX3 are direct targets of the PAX3-FOXO1 fusion product providing insight into mechanisms responsible for increased FGFR4 expression in this RMS subtype [28]. Furthermore, copy number alterations are common in ERMS and are associated with FGFR4 amplification [29]. These findings collectively suggest that FGFR4 dysregulation occurs in both ARMS and ERMS, albeit by different mechanisms.
In our murine model system, transgenic overexpression of human wild-type (FGFR4wt) or mutant FGFR4 (i.e., FGFR4N535K, FGFR4V550E) had distinct effects on myoblasts in vitro and in vivo. Specifically, FGFR4-activating mutations transformed p53−/− mouse myoblasts in vitro and FGFR4wt-, FGFR4N535K- and FGFR4V550E-overexpressing p53−/− myoblasts generated tumors following engraftment into neonatal p53+/− host skeletal muscle. While FGFR4N535K mutant myoblasts generated a single high-grade myoid sarcoma (1/11, Fig. 3d), the most penetrant mutation was FGFR4V550E, generating 11 myogenic sarcomas including 5 tumors with classic rhabdomyoblastic differentiation (Fig. 3). Surprisingly, wild-type FGFR4 overexpression did not support anchorage-independent growth in vitro, yet in vivo, murine sarcomas driven by FGFR4wt expression developed with high penetrance (71%). Thus aberrant FGFR4 signaling, either by activating mutation and/or receptor overexpression, is a driver of sarcoma formation.
Dysregulation of FGFR4 signaling in skeletal muscle development
The FGF axis has a known regulatory role in skeletal muscle development, which may provide insight into how FGFR4 dysregulation drives the growth of skeletal muscle tumors. Specifically, Fgfr4 is required for chick embryo myogenic cell differentiation [30] and ablation of Fgfr4 impairs murine adult skeletal muscle regeneration [31]. Fgfr4 expression precedes Myod1 expression but is not expressed in differentiated skeletal muscle fibers, demonstrating the importance of this growth factor receptor in muscle progenitors [32].
Previously, we demonstrated that transgenic overexpression of activated KrasG12D in p53−/− myoblasts (Myo25) impaired in vitro differentiation and resulted in high-grade undifferentiated sarcomas [18]. In contrast, FGFR4wt- and FGFR4V550E-overexpressing myoblasts maintained their capacity to differentiate while myoblasts expressing FGFR4N535K had some impaired differentiation. Interestingly, FGFR4N535K expression decreased Myod1 mRNA levels during differentiation and the sole FGFR4N535K-expressing tumor did not show Myod1 immunopositivity. Increasing this cohort size is necessary to determine whether FGFR4N535K truly drives formation of undifferentiated sarcomas (i.e., UPS) perhaps due to impaired Myod1 activity. Finally, FGFR4V550E-expressing myoblasts generated tumors containing rhabdomyoblasts and expressing RMS diagnostic markers Myod1 and Myog and had the strongest correlation with human RMS using AGDEX scoring (Fig. 3e). Taken together, the ability of FGFR4 overexpressing or V550E myoblasts to differentiate in vitro correlated with tumor differentiation. Further studies are required to determine why FGFR4N535K myoblasts appear to have impaired skeletal muscle development.
FGFR4 activation and development of novel therapies in RMS
FGFR4 inhibition has emerged as an attractive therapeutic target; however, how to effectively target its activity has not yet been realized [33]. Although the RTK inhibitor ponatinib blocked FGFR4 activation and was shown to reduce xenograft tumor growth [17], its utility has been limited by serious toxicity likely due to off-target effects. Since FGF receptors are known to activate multiple effectors, including RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways, we performed a functional drug screen using a well-characterized oncology-based drug library on FGFR4V550E-overexpressing tumor cells as well as human ARMS and ERMS cell lines. PI3K/mTOR inhibitors caused the most potent viability reduction, and GSK2126458, a dual PI3K/mTOR inhibitor, robustly decreased cell numbers in RMS lines in vitro and in vivo. Furthermore, in single-cell assays, GSK2126458 exhibited a highly selective reduction in signaling downstream of mTOR activity but not other major pathways. Interestingly, all RMS cell lines tested exhibited high basal levels of p4EBP1 but pS6 levels were more variable, suggesting differential activation of mTORC1 effector pathways. Furthermore, GSK2126458 decreased p4EBP1 more potently than pS6 in RMS cells. Collectively, these data suggest that proliferation of mouse and human RMS cells requires mTORC1/4EBP1-dependent translation and identify mTOR inhibition as a promising therapy for RMS.
RMS mouse models inform clinical trials of PI3K/mTOR inhibitors
Our mouse models employ primary murine myoblasts as the tumor cell-of-origin, allowing for genetically modified myoblasts to be interrogated in vitro, as well as in vivo. In a previous study, the FGFR4-activating mutations, N535K and V550E, also drove tumor formation in xenograft models employing NIH 3T3 fibroblasts transgenically expressing FGFR4 and engrafted into immunodeficient host mice [16]. Mosaic mouse models represent a novel method to probe for the effects of FGFR4 pathway activation in vivo and may better recapitulate human tumor development as tumors develop within immunocompetent and syngeneic host skeletal muscle. Thus our models have further substantiated the role of FGFR4 mutation and p53 pathway inactivation as drivers of RMS.
Effective preclinical models require in vivo models that recapitulate the human disease [34]. A current Children’s Oncology Group Phase III clinical trial is examining the effectiveness of the mTOR inhibitor temsirolimus in combination with standard chemotherapy (VAC/VI) in RMS patients (NCT02567435). In our study, the dual PI3K/mTOR inhibitor GSK2126458 impaired tumor growth and improved survival in FGFR4V550E-overexpressing murine RMS, providing preclinical evidence that inhibition of these key pathways may prove therapeutically useful for RMS with FGFR4 mutations. Despite the apparent toxicity associated with the combination of vincristine and GSK2126458, additional animal studies and Phase I clinical trials will determine effective dosing [19]. Future studies should also be undertaken to test additional promising inhibitors of PI3K and mTOR signaling pathways, in the hopes of defining more effective treatment regimes for RMS patients.
Materials and methods
Myoblast transformation, engraftment, and pathology
Primary myoblast cell line Myo25 was generated as previously published [18]. Wild-type and mutant human FGFR4 constructs in pDONR vectors were cloned into pLenti PGK Blasticidin (Addgene: 19,065 [35]) using Gateway Technology (Invitrogen). Myoblasts were transduced with lentiviral particles generated using previously published protocols [18], using a multiplicity of infection of 10. Myo25 was incubated with MGM containing lentiviral particles for 16 h and then expanded in 10 μg/ml Blasticidin (Invitrogen). Proliferation and colony-forming ability in semi-soft agar of myoblasts was assessed using standard protocols [18].
Experiments were performed using male and female C57Bl/6p53tm1/Tyj mutant mice [18]. Animal protocols were performed in accordance with the Animal Care Committee at The Centre for Phenogenomics. Myoblasts were injected intramuscular into syngeneic hosts as previously described [36] and were sacrificed when tumors measured 1.5 cm3 or other experimental end points were reached. Gross necropsy was performed on all animals. Tumor and contralateral limb skeletal muscle were harvested and underwent standard fixation or snap frozen in liquid nitrogen. Antibodies are described in Supplemental Table 1. Pathology was reviewed by a dedicated sarcoma pathologist (BCD).
Gene expression microarray analysis
To evaluate global gene expression, RNA was harvested from FGFR4wt (n = 3), FGFR4N535K (n = 1), and FGFR4V550E (n = 2) tumors using the RNAeasy MINI Kit (Qiagen), in vitro transcribed, hybridized, and applied to Affymetrix Mouse 430A arrays. FGFR4 and Kras transcriptomes [18] were compared to human tumors using the AGDEX statistic as previously described [37, 38]. Published microarray gene expression data sets included mouse normal tissue (GSE10246), human normal tissue (GSE3526), and human tumors (GSE8840 for ARMS and ERMS, GSE12102 for EWS, GSE20196 for SS, and GSE21050 for UPS).
Assessment and quantification of myoblast in vitro differentiation
Myoblast differentiation was examined according to previously published protocols [18], and samples were imaged on INCell Analyzer 6000 (GE Healthcare), equipped with 10×/0.45 NA Plan Apo objective and 2048 × 2048 sCMOS camera. Twenty-five fields with five z-planes were acquired. Individual focal plane image for each z-series was automatically selected based on sharpness. Image analysis algorithm and PCR is described in Supplementary Methods.
Mass cytometry
For single-cell analysis of phosphoprotein abundance, described cells lines were stained with a panel of phosphor-specific antibodies and performed CyTOF analysis using the published methods [22]. Cells (2 × 106 cells/condition) were precultured in serum phenol-red-free Dulbecco’s modified Eagle’s medium (Gibco) for 2 h and lifted with Versene (Life Technologies) prior to addition of GSK2126548 (40 nM) or vehicle (0.001% DMSO). After incubation for 27 min at 37 °C, 195Cisplatin (BioVision Inc., USA) was added (3 μM, 3 min). Cells were immediately fixed (10 min, room temperature (RT)) with equal volume of 3.2% formaldehyde. Cells were pelleted (500 × g, 5′), washed (1 ml phosphate-buffered saline (PBS)+1% bovine serum albumin (BSA)) and permeabilized (1 ml Perm III buffer (Becton Dickinson, Canada)) for 10 min on ice. Cells were stored at −20 °C, prior to washing, rehydration in PBS/BSA, and staining with the metal-tagged phospho-specific antibodies (Supplemental Table 1). Antibodies (BD Biosciences, Canada) were metal tagged using the MaxPar Conjugation Kits (Fluidigm, Markham, ON, Canada). Antibody dilutions were determined by titration on cells treated with appropriate signaling inducers or inhibitors as previously described [23]. For some experiments, cells in each treatment group were individually barcoded using the 20-Plex Pd Barcoding Kit (Fluidigm) following the manufacturer’s protocol, then combined for multi-plexed antibody labeling. Briefly, cell pellets were re-suspended in antibody cocktail with metal-tagged antibodies, incubated (30 min, RT), washed in staining buffer (PBS+1% BSA), and re-suspended in PBS containing 0.3% saponin, 1.6% formaldehyde, and 0.05 μM 191/193Ir (Fluidigm) for 1–48h at 4 °C. Cells were then washed in PBS, followed by ddH20, and suspended at ~2–5 × 105/ml prior to adding EQ normalization beads (Fluidigm) and running on a Helios CyTOF (Fluidigm). Normalization and de-barcoding was performed using the Helios software. Displays are shown pregated on 191/193Ir+ 195Cisplatinlo cleaved caspase3− events.
Drug studies
For the high-throughput drug screen, 1000 M25.FV24c cells per well in 5 μl of RPMI media were dispensed into 1536-well plates preplated with the MIPE-v4 compound library as previously described [39]. Cells were treated for 48 h prior to addition of Cell-Titer Glo. Luminescence was measured on a ViewLux instrument. AUC from the resultant dose–response curves was calculated using a standard trapezoidal method.
Lyophilized GSK2126458 (Toronto Research Chemicals) was reconstituted in DMSO and vincristine sulfate (Toronto Research Chemicals) reconstituted in 40% ethanol. Drugs were added to cell lines in 96-well plates, incubated for 48 (myoblasts) or 72 h (RMS cell lines), then assayed using ATPLite (Perkin-Elmer), and bioluminescence measured with the PerkinElmer Enspire 2300. For in vivo studies, p53+/− mice were injected at weaning age with 1 × 104 M25.FV24c. Sample size was estimated from previous studies in our laboratory [18]. Animals were randomly assigned into four experimental arms and, after 10 days, were subsequently treated for 3 weeks. GSK2126458 was diluted in 30% polyethylene glycol (PEG)/1% Tween 20 and vincristine in PBS. Mice were monitored daily in a blinded manner and sacrificed at end points (e.g., >20% loss in body weight, lack of mobility, ≥1.5 cm tumor size) or at experiment completion.
Statistics
Statistical analyses were performed with SPSS (IBM, version 19) or GraphPad Prism Software (v5.02). Proliferation assays were performed in technical triplicates, doubling time calculations were made from at least three independent assays, and anchorage-independent growth assays were performed in biologic triplicates. The above assays and muscle differentiation assays were analyzed with one-way analysis of variance and Tukey’s multiple comparison tests. All analyzed assays met statistical assumptions. Survival curves were calculated using the Kaplan–Meier method (SPSS).
Supplementary Material
Acknowledgements
We acknowledge Dr. M. Bhaskhurov (LTRI) for high content image analysis, S. Burtenshaw for statistical analysis and manuscript preparation, Dr. D. Holmyard (Mount Sinai Hospital) for electron microscopy, Dr. E. Stewart (St. Jude’s) for preclinical study design and Dr. J. Fletcher (Brigham and Women’s Hospital) for use of RMS559.
Funding CJG was supported by funding from the Canadian Cancer Society Research Institute and the Cancer Stem Cells Program of the Ontario Institute for Cancer Research; RAG was supported by a Clinical Investigator Award from Ontario Institute for Cancer Research.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41388–017-0122-y) contains supplementary material, which is available to authorized users.
References
- 1.Tenente IM, Hayes MN, Ignatius MS, McCarthy K, Yohe M, Sindiri S, et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. Elife. 2017;6:e19214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kikuchi K, Rubin BP, Keller C. Developmental origins of fusion-negative rhabdomyosarcomas. Curr Top Dev Biol 2011;96:33–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barr FG. The role of chimeric paired box transcription factors in the pathogenesis of pediatric rhabdomysarcoma. Cancer Res 1999;59:1711s–15. [PubMed] [Google Scholar]
- 4.Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet 1993;5:230–5. [DOI] [PubMed] [Google Scholar]
- 5.Linardic CM. PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett 2008;270:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williamson D, Missiaglia E, de Reynies A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 2010;28:2151–8. [DOI] [PubMed] [Google Scholar]
- 7.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 2014;4:216–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abraham J, Nunez-Alvarez Y, Hettmer S, Carrio E, Chen HI, Nishijo K, et al. Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev 2014;28:1578–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chmielecki J, Bailey M, He J, Elvin J, Vergilio JA, Ramkissoon S, et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res 2017;77:509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer. 2009;115:4218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weigel BJ, Lyden E, Anderson JR, Meyer WH, Parham DM, Rodeberg DA, et al. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol 2016;34:117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 2001;7:673–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov 2017;7:884–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun. 2015;6:7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor JGT, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest 2009;119:3395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li SQ, Cheuk AT, Shern JF, Song YK, Hurd L, Liao H, et al. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS ONE. 2013;8:e76551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKinnon T, Venier R, Dickson BC, Kabaroff L, Alkema M, Chen L, et al. Kras activation in p53-deficient myoblasts results in high-grade sarcoma formation with impaired myogenic differentiation. Oncotarget 2015;6:14220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett 2010;1:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F. WHO classification of tumours, vol. 5. International Agency for Research on Cancer (IARC): Lyon, France, 2013. [Google Scholar]
- 21.Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci USA. 2014;111:2349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–96.21551058 [Google Scholar]
- 23.Perova T, Grandal I, Nutter LM, Papp E, Matei IR, Beyene J, et al. Therapeutic potential of spleen tyrosine kinase inhibition for treating high-risk precursor B cell acute lymphoblastic leukemia. Sci Transl Med 2014;6:236ra262. [DOI] [PubMed] [Google Scholar]
- 24.Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014;13:140–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L, Shern JF, Wei JS, Yohe ME, Song YK, Hurd L, et al. Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet 2015;11:e1005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crose LE, Etheridge KT, Chen C, Belyea B, Talbot LJ, Bentley RC, et al. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin Cancer Res 2012;18:3780–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, et al. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res 2010;70:6497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulson V, Chandler G, Rakheja D, Galindo RL, Wilson K, Amatruda JF, et al. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosomes Cancer. 2011;50:397–408. [DOI] [PubMed] [Google Scholar]
- 30.Marics I, Padilla F, Guillemot JF, Scaal M, Marcelle C. FGFR4 signaling is a necessary step in limb muscle differentiation. Development. 2002;129:4559–69. [DOI] [PubMed] [Google Scholar]
- 31.Zhao P, Caretti G, Mitchell S, McKeehan WL, Boskey AL, Pachman LM, et al. Fgfr4 is required for effective muscle regeneration in vivo. Delineation of a MyoD-Tead2-Fgfr4 transcriptional pathway. J Biol Chem 2006;281:429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao P, Hoffman EP. Embryonic myogenesis pathways in muscle regeneration. Dev Dyn 2004;229:380–92. [DOI] [PubMed] [Google Scholar]
- 33.Lewin J, Siu LL. Development of fibroblast growth factor receptor inhibitors: kissing frogs to find a prince? J Clin Oncol 2015;33:3372–4. [DOI] [PubMed] [Google Scholar]
- 34.Kashi VP, Hatley ME, Galindo RL. Probing for a deeper understanding of rhabdomyosarcoma: insights from complementary model systems. Nat Rev Cancer. 2015;15:426–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE. 2009;4:e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol 1994;125:1275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010;466:632–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poschl J, Stark S, Neumann P, Grobner S, Kawauchi D, Jones DT, et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol 2014;128:123–36. [DOI] [PubMed] [Google Scholar]
- 39.Ceribelli M, Kelly PN, Shaffer AL, Wright GW, Xiao W, Yang Y, et al. Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci USA. 2014;111:11365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.