

Summary
Human induced pluripotent stem cells (hiPSCs) have emerged as a promising platform for pharmacogenomics and drug development. In cardiology, they make it possible to produce unlimited numbers of patient-specific human cells that reproduce hallmark features of heart disease in the culture dish. Their potential applications include the discovery of mechanism-specific therapeutics, the evaluation of safety and efficacy in a human context before a drug candidate reaches patients, and the stratification of patients for clinical trials. Although this new technology has the potential to revolutionize drug discovery, translational hurdles have hindered its widespread adoption for pharmaceutical development. Here we discuss recent progress in overcoming these hurdles that should facilitate the use of hiPSCs to develop new medicines and individualize therapies for heart disease.
Keywords: hiPSC, cardiomyocyte, drug attrition, screen, drug discovery, HTS, heart disease, disease modeling
eTOC Blurb
The use of human induced pluripotent stem cell (hiPSC) technology could revolutionize cardiovascular drug discovery. Hnatiuk et al. review the first few large scale studies and discuss the lessons learned that are important for adopting this technology.
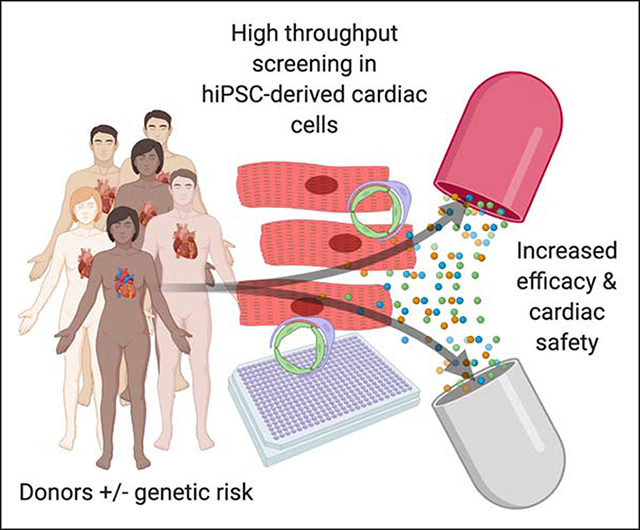
Graphical Abstract
Introduction
Among the most notable achievements of modern biology has been the development of a highly reproducible and simple procedure to reprogram adult human somatic cells, typically blood or skin, to a pluripotent state resembling that of the early embryo shortly after implantation in the uterus. Once reprogrammed, human induced pluripotent stem cells (hiPSCs) can be directed to differentiate into any cell type of the human body. Importantly, hiPSCs retain the genetic makeup of their human donors, so they have the potential to recapitulate essential aspects of genetic diseases or mimic drug responses in vitro. Thus, it is now possible to create essentially limitless numbers of human cells, harboring a diversity of patient genetics, that previously were unobtainable other than as primary cultures derived from tissue biopsies and post-mortem donors. From a drug development perspective, hiPSC-based technology is an alternative to reductionist or finicky cell culture models. hiPSCs make it possible to assess drug responses across a target population that is typically only evaluated by human trials. The technology has the potential to impact multiple points in the development pipeline – including therapeutic target and early stage drug discovery, safety pharmacology, and enrichment of clinical trial participants for likely responders (Figure 1) – and therefore could revolutionize how new medicines are developed.
Figure 1. Value added benefit of hiPSC-CMs for drug development.

The potential benefit of hiPSC-CMs for drug development includes disease models that bring the human context to discovery and preclinical stages. hIPSC-CM based studies could also be used to discriminate drug-responsive patients with a specific disease that would allow a more precise Phase II clinical trial to be performed. Together, these technologies might improve the efficiency of drug development. In addition, hiPSC-based phenotyping, combined with multi-omics analyses of patient samples, would aid clinicians in prescribing medicines on the basis of mechanism and drug-responsiveness.
There have been a number of excellent reviews of hiPSC-based modelling of cardiovascular (CV) and metabolic disorders (Moretti et al., 2013; Paik et al., 2020). These reviews summarized impressive studies showing that hiPSC-derived cardiomyocytes (hiPSC-CMs) can reproduce disease-relevant effects of gene variants, metabolic dysfunction and drug responses. Building on these advances, there is considerable support by governmental regulatory and funding agencies, pharmaceutical companies and academics for developing realistic hiPSC-based models for drug discovery.
Given the successes in modeling heart disease, hiPSC technology seems poised to transform the discovery of new medicines (Paik et al., 2020; Savoji et al., 2019). The impact could be enormous. Heart disease remains the major cause of human mortality worldwide, yet fewer than 300 of the nearly 5,000 ongoing clinical trials, and only 1 of the 48 drug approvals by the FDA, in 2019 were for heart drugs, reflecting a prioritization of other areas of pharmaceutical development such as oncology (Mullard, 2020). Contrary to expectations, however, hiPSC-based technology has not yet had the expected transformative effect on drug discovery. Thus, the objective of this review is to delineate the technical and conceptual challenges facing its adoption for the development of new heart disease medicines. We will relate the lessons learned from the few large-scale drug discovery and medicinal chemistry campaigns that have used hiPSCs for therapeutic target discovery and drug development. In our view, understanding these advances and addressing the associated challenges will hasten the adoption of realistic hiPSC disease models for discovery of novel therapeutics for heart disease, ultimately yielding medicines that target specific pathologic and etiologic mechanisms to increase the precision, effectiveness and safety of drug treatments.
Using hiPSCs to model congenital heart disease
There have been many instances of using hiPSC-CMs to model heart disease, as reviewed (Garg et al., 2018; Moretti et al., 2013). A partial list includes Long QT syndrome (LQTS) (Itzhaki et al., 2011; Liang et al., 2016; McKeithan et al., 2017; Moretti et al., 2010), arrhythmogenic right ventricular dysplasia (ARVD) (Kim et al., 2013; Ma et al., 2012), Timothy syndrome (Yazawa et al., 2011), catecholaminergic polymorphic ventricular tachycardia (CPVT) (Penttinen et al., 2015), dilated cardiomyopathy (DCM) (Briganti et al., 2020; Sun et al., 2012) and hypertrophic cardiomyopathy (HCM) (Lan et al., 2013; Seeger et al., 2019; Smith et al., 2018). In these and many other cases, the hiPSC-CMs reproduced many cellular electrophysiologic and contractile phenotypes as expected from the patients’ clinical presentation. Many studies have recapitulated the expected physiological effects of drugs, for instance, the action of antiarrhythmic drugs on action potential morphology in hiPSC-CMs carrying causal mutations for LQTS (Itzhaki et al., 2011; Liang et al., 2016; McKeithan et al., 2017; Moretti et al., 2010) and CPVT (Itzhaki et al., 2012; Penttinen et al., 2015). Most impressively, hiPSC-CM models have aided the development of potential therapeutic strategies, such as exon skipping for muscular dystrophy (Min et al., 2020; Ortiz-Vitali and Darabi, 2019; Piga et al., 2019).
Accumulating experience indicated the necessity to control for inter-line variability and reliably determine the influence of genetics on disease processes (Onder and Daley, 2012). hiPSCs, like donors, exhibit considerable individual genomic variability and phenotypic heterogeneity in their physiological responses (Nityanandam and Baldwin, 2015), meaning that large numbers of patient-derived hiPSC lines might be needed to infer the effect of a rare variant on a clinical phenotype or drug response (Falk et al., 2016). The accepted solution to this problem is to create control hiPSCs with identical genomic sequence except for the variant of interest thanks to new genomic editing tools of which clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9) is the most common. The technique uses a guide RNA to directs the Cas9 endonuclease to the genomic locus of interest. Cas9 then cuts the DNA and homologous recombination with a template DNA edits the nucleotide sequence. Phenotypic differences between the patient and the genome corrected isogenic control (or normal individual cell versus variant-introduced) simplifies attribution of causality to the gene variant itself and is now regarded as the gold standard of hiPSC-based disease modeling.
Addressing Physiological Immaturity.
An attractive feature of hiPSC-CMs as a drug discovery tool is that they express nearly all of the components of cardiac contractility of adult cardiomyocytes, including ion currents for transmembrane potential depolarization and repolarization, which together produce action potential (AP) waveforms (Karakikes et al., 2015), the central components of cardiac excitation-contraction (E-C) coupling (Uzun et al., 2016; Zhang et al., 2013), as well as structures and proteins for sarcoplasmic reticulum (SR) calcium release and uptake (Germanguz et al., 2011; Zhang et al., 2013). Thus, the fundamental mechanisms of cardiomyocyte contraction and its regulation can be probed in a realistic human context.
Nonetheless, hiPSC-CMs as commonly produced resemble fetal cardiomyocytes rather than mature adult cardiomyocytes. Although their relatively short time in culture undoubtedly plays a part, the major problem is most likely that differentiation in vitro lacks the structural and hemodynamic inputs of normal cardiac development. Consequently, hiPSC-CMs are smaller and lack the elongated, rectangular shape of adult CMs. They also exhibit characteristic spontaneous beating unlike their adult counterparts. Although hiPSC-CMs’ action potential waveforms resemble those of adult ventricular CMs, they have depolarized diastolic transmembrane potential, slower upstroke velocity and decreased repolarization reserve. Immature hiPSC-CMs have poorly organized myofilaments and develop considerably less contractile force than do adult cardiomyocytes (Shinozawa et al., 2012). Physiological immaturity could mask disease mechanisms. Conversely, exploration of drug actions and therapeutic targets carries the risk of uncovering biology relevant to a fetal rather than adult disease manifestation.
Physiological immaturity has been mitigated by culture systems that mimic mechanical and electrical inputs normally present in the developing heart (Table 1). Culture as three-dimensional, strip-format engineered heart tissues (EHT) improves ultrastructural organization of sarcomeres and mitochondria in the hiPSC-CMs as well as evidence of rudimentary T-tubules that can be further enhanced by mechanical and electrical stimulation, including by recent microelectromechanical systems (MEMS) devices (Table 1). Furthermore, engineered microtissues and heart-on-chip microdevices, especially MEMS-based microfluidics chips amenable to moderate throughput applications, are intriguing biomimetic models for assessing drug effects (Abulaiti et al., 2020; Mathur et al., 2015; Oleaga et al., 2018; Wang et al., 2014).
Table 1.
Common approaches to improve hiPSC-CM maturation.
| Maturation stimuli | Reported Improvements | Representative References |
|---|---|---|
| Micropatterned, grooved or rigid 2D surfaces | Structural, mechanical and electrophysiological. | (Kim et al., 2010; Ribeiro et al., 2015; Wang et al., 2011) |
| Shift from glycolytic to oxidative substrates | Metabolic, mechanical, structural and electrophysiological. | (Feyen et al., 2020; Horikoshi et al., 2019; Lin et al., 2017; Yang et al., 2019) |
| 3D engineered tissues | Structural, mechanical and electrophysiological (generally enhanced over 2D) | (Lemoine et al., 2017; Mannhardt et al., 2016; Turnbull et al., 2014; Ulmer et al., 2018) |
| 3D plus mechanical | Structural, mechanical and electrophysiological. | (Mihic et al., 2014; Tulloch et al., 2011; Zimmermann et al., 2004) |
| 3D plus electromechanical | Structural, mechanical and electrophysiological (reported enhanced over 3D alone) | (Eng et al., 2016; Lieu et al., 2013; Nunes et al., 2013; Ronaldson-Bouchard et al., 2018; Ruan et al., 2016) |
| MEMS | Structural and electrophysiological. | (Abulaiti et al., 2020; Dostanic et al., 2020) |
Three dimensional systems, however, are complex and not readily amenable to high throughput applications. Consequently, methods to improve the maturational status of two dimensional cultures in high density format are important to advance the utility of hiPSC-CMs. A hallmark of mature cardiomyocytes is their ability to efficiently metabolize both glycolytic and oxidative substrates, especially fatty acids, whereas immature cardiomyocytes typically utilize glycolysis. Several recent studies have shown that enhancing the natural developmental switch from glycolysis to mitochondrial oxidative metabolism not only promotes metabolic maturation but also electrophysiological, structural and mechanical properties (Feyen et al., 2020; Horikoshi et al., 2019; Lin et al., 2017; Yang et al., 2019). We recently described a metabolic maturation media that enabled reliable modelling of LQTS type 3 and RBM20 mutant DCM (Feyen et al., 2020). The pertinent disease phenotypes depend on substantial Na+ current and Ca2+ cycling, both of which are not readily apparent in hiPSC-CMs cultured under conventional conditions (Feyen et al., 2020). Metabolic maturation aided in the development of models of ischemia reperfusion injury (Hidalgo et al., 2018) and diabetic cardiomyopathy (Drawnel et al., 2014).
State of the art hiPSC-based disease modeling.
Modern preparations of hiPSC-cardiomyocytes are nearly pure populations of ventricular cardiomyocytes (Cyganek et al., 2018), and so the influence of other cells, such as vascular cells and fibroblasts, is lacking in most studies. On the other hand, single cell RNA sequencing has revealed heterogeneity among the ventricular cardiomyocytes produced (Friedman et al., 2018; Schmid et al., 2020), it is not yet clear how these relate to particular subpopulations in the human heart. This should be resolved soon once single cell transcriptomic and proteomic datasets are compiled into detailed cell atlases of the intact heart (Litvinukova et al., 2020). These will be used to correlate in vivo cell populations with individual hiPSC-CMs in culture. More challenging, however, is the question of how to obtain pure subpopulations of cells and engineer in vitro models of the ventricular wall that preserve the correct spatial organization of distinct cell types. Such models would be invaluable for modeling disease involving complex intercellular communication. For instance, reproducing the normal relationship of endocardial, mid and epicardial LV myocytes would allow the studies of drug effects and genetics on dispersion of repolarization that is a substrate for arrhythmia (Han and Moe, 1964; Surawicz, 1997).
A pragmatic solution to the current limitations has been to focus on disease manifestations that have a cardiomyocyte-autonomous etiology that is not predicated on structurally complicated three-dimensional architecture or interactions between different cell types. Accordingly, most congenital heart disease models to date reflect mutations in ion channels or contractile proteins, thus the salient clinical phenotypes can be recapitulated in isolated or monolayers of pure cardiomyocytes. This has led to faithful models (as listed above) for numerous congenital electrophysiological disorders for which there is clear cell autonomy. However, not all apparently myocyte-autonomously acting genetic mutations present clear phenotypes. An interesting example is hypertrophic cardiomyopathy, HCM, which is characterized by thickening of the left ventricle, myofibrillar disarray and left ventricular outflow tract obstruction. HCM mutants have had variable hiPSC-CM phenotypic presentations, in particular with respect to contractility (Eschenhagen and Carrier, 2019). Although early studies often lacked isogenic controls, and hence could not rule out confounding effects caused by the donors’ genetics, evaluation of isogenically controlled studies suggests that hiPSC-CM contractility phenotype is not uniform across mutations. For instance, in recent studies, MYH7 p.R453C was hypocontractile (Bhagwan et al., 2020) while p. R403Q was hypercontractile (Cohn et al., 2019). These mutants have distinct effects on the myosin motor biophysics (Sarkar et al., 2020; Sommese et al., 2013), and it will be interesting to determine if the manifestation reflects fundamental properties of sarcomere biophysics and/or culture and maturational conditions.
Accumulating evidence shows that acquired disease can also be evoked in vitro using hiPSC-CMs. Diabetic cardiomyopathy is an excellent example of using hiPSC-CMs to assess contractile dysfunction in response to acquired disease. Diabetic cardiomyopathy is a common long-term effect of diabetes that afflicts just under half a billion people globally (Saeedi et al., 2019). Elevated levels of circulating fatty acids in diabetes cause inefficient metabolism and, ultimately, cardiomyopathy with systolic and diastolic dysfunction. Insulin resistance and decreased mitochondrial respiratory characteristic of diabetic cardiomyopathy were mimicked in healthy donor hiPSC-CMs by modifying the culture conditions to mimic a diabetic environment (Drawnel et al., 2014; Graneli et al., 2019; Liu et al., 2017). Hallmark features of diabetic cardiomyopathy were apparent in the in vitro models, including elevated BNP, troponin release and disordered sarcomeres.
hiPSC-CMs in drug discovery
A major benefit of iPSC-based screening is that it can be implemented at early stages in drug discovery when therapeutic targets and hit compounds are being identified or when hit and lead compounds are being chemically optimized (Figure 1). These stages precede the ‘valley of death’ that spans preclinical to early clinical activities when the greatest attrition of investigational molecules occurs and contributes substantively to the high cost of developing new drugs (Paul et al., 2010; Waring et al., 2015). While many factors contribute to drug attrition during development, a quantitative decision theory analysis of pharmaceutical attrition concluded that there are currently too few preclinical models with high predictive value to sustain productivity (Scannell and Bosley, 2016). Concerns with conventional models include overly reductionist assays and poor generalizability of preclinical data, including animal data (Gintant et al., 2016; Gromo et al., 2014). Thus, we believe that implementing realistic hiPSC-based assays when compounds are being optimized and selected for later stage development could help mitigate the productivity crisis.
The technologies used for hiPSC-CM screening compatible with implementation early in the drug discovery pipeline have been reviewed recently (Del Alamo et al., 2016). These include fixed endpoint assays to evaluate static properties of cardiac cells, such as cell death, troponin release, mitochondrial transmembrane potential, and are read out using a wide range of plate reader or high content imaging modalities. Kinetic readouts are used to measure complex physiologically phenomena of importance for cardiomyocyte function, including action potential and Ca2+ transient morphology and contractility (Cerignoli et al., 2012; McKeithan et al., 2017; Sala et al., 2018). In our view, optical modalities have the greatest opportunity for higher throughput physiological recording thanks to recent advances in small molecule and genetically encoded voltage and Ca2+ sensors for recording intracellular and subcellularly compartmentalized Ca2+ concentration, transmembrane voltage changes as well as cardiomyocyte contractility from two dimensional hiPSC-CM cultures. It is also possible to read out force generation, albeit with lower throughput, by monitoring the displacement of fluorescent beads embedded in deformable substrata and calculating the generated force based on the known material properties of the substrata (Ferrari, 2019; Yoshie et al., 2018). Popular alternatives to optical recording include impedance or field potentials [e.g., multi-electrode array, MEA, devices (Kussauer et al., 2019)] that have the advantage of plug and play commercial instrumentation but also throughput limiting disadvantages of expensive and low density plate formats.
Below, we discuss four applications of hiPSC-based screening: therapeutic target discovery, early phase drug discovery, clinical trials in a dish (CTiD), and cardiotoxicity.
Therapeutic Target Discovery.
Comprehensive interrogation of the genome by functional genomics (i.e., siRNA knockdown or CRISPR knockout technologies) has the potential to identify novel cardiomyocyte-relevant targets for further drug development. Large-scale hiPSC-CM functional genomics screens have identified regulators of cardiomyocyte proliferation (Diez-Cunado et al., 2018) and differentiation (Ihry et al., 2019), so it is reasonable to expect success in identifying candidate targets in disease models (Figure 2A). Several recent low throughput studies support this idea. For instance, pharmacological upregulation of the splicing factor RBM20 restored contractility in DCM hiPSC-CMs carrying a disease-causing RBM20 variant (Briganti et al., 2020) and PDGF agonists decreased arrhythmia DCM hiPSC-CMs carrying a mutation in the gene LMNA that encodes the nuclear laminA/C (Lee et al., 2019). In both cases, the therapeutic mechanisms were unanticipated, illustrating the potential of phenotypic screening for therapeutic strategies.
Figure 2. Comparison Between Arrayed and Pooled Functional Genomics Screens.

A) hiPSC-CMs expressing Cas9 can be plated into microtiter plates and wells transfected with individual CRISPR guide RNAs, siRNAs, or miRNAs. Each well can then be assayed for cardiomyocyte function, such as calcium handling or contractility, or marker expression to call “hits”.
B) In pooled CRISPR-based screening, hiPSC-CMs are transduced with a lentiviral library of CRISPR guides. A selection is applied to enrich or deplete a phenotype of interest. The populations are then pooled, DNA is extracted, and guides are counted using Next Generation Sequencing. Enriched guides are called as “hits”.
C) The two approaches can be combined in series. This allows for the identification of functionally relevant “hits”.
Probing of therapeutic target space is often done by arrayed screening, in which siRNAs, CRISPR guide RNAs or other reagents are tested in individual wells in multiwell format (Figure 2A). Pooled screening of CRISPR guide RNAs has advantages over arrayed library screening that it is more readily scaled to interrogate the entire genome for potential therapeutic targets. Pooled CRISPR screening has been used extensively to perform unbiased, genome-wide screens to identify novel regulators of cancer and other and disease processes (Fellmann et al., 2017; Shalem et al., 2015). In this approach (Figure 2B), cells expressing a CRISPR effector are transfected with a large mixed library of tens of thousands of guide RNAs targeting the entire proteome so that, on average, each cell receives one guide RNA and, therefore, only one gene will be affected. Depending on the effector used, this library will either cause gene knockouts (CRISPR nuclease), gene inactivation (CRISPRi), or gene activation (CRISPRa) (Adli, 2018). After further culture and separation of cells that received a therapeutic benefit, followed by next generation sequencing to identify the guide templates, hits are determined by the relative enrichment or depletion of guides in the populations of interest (Doench, 2018) (Figure 2B). One of the limitations of implementing a pooled screen is that, unlike arrayed well-based screens, it is difficult to use a functional test such as contractility to physically enrich the cells containing the “hit” guides. However, a serial approach that combines pooled screening with a surrogate marker followed by arrayed secondary screening for physiological function should be tenable (Figure 2C).
Early phase drug discovery.
The past few years have seen the first large hiPSC-based campaigns that guided the medicinal chemistry optimization of new or existing drugs (Table 2). In the first example, Fiedler et al. (Fiedler et al., 2019) recreated aspects of myocardial ischemia, which is the most prevalent etiology of heart failure (Benjamin et al., 2019). The simulated ischemia model was used to establish the cardioprotective activity of an initial screen ‘hit’ and, subsequently, evaluate optimized analogues. In the induced phenotypic surrogate model of diabetic cardiomyopathy described above, Drawnel et al. (Drawnel et al., 2014) performed a chemical biology exploration of possible disease mechanisms. Several hit compounds pointed to dysregulated calcium cycling and signaling for causing the phenotypic effects.
Table 2.
Examples of large-scale hiPSC-CM chemical screens.
| Description | Compounds and Results | hiPSC-CMs | References |
|---|---|---|---|
| Cardioprotection in models of diabetic cardiomyopathy | 480 bioactive compounds. Identification of candidate pathways to ameliorate structural disarray. | 1 commercial healthy donor and 2 diabetic donors hiPSC-CMs. | (Drawnel et al., 2014) |
| Cardioprotection in a simulated ischemia model | Confirmation of initial screen ‘hi’ and, subsequently, evaluation of optimized analogues that inhibited MAP4K4 and protect against cell death. | 2 commercial healthy donors | (Fiedler et al., 2019) |
| Action potential shortening in LQT3 and arrhythmia in healthy donor hiPSC-CMs using high content imaging of transmembrane voltage potentials | ~170 mexiletine analogues. Refined analogues had increased on target potency and selectivity for desirable on target effects with decreased proarrhythmic liability. | 2 LQTS3 hiPSC-CMs plus drug-induced LQTS3 using 3 healthy donor hiPSC-CMs. | (McKeithan et al., 2020) |
| Proarrhythmic drug screening of field potentials by MEA recording | Kitaguchi et al., and Blinova et al. reference compounds of high, intermediate and low/no arrhythmic risk. Predictivity was in range of 80–90%; greatest for discrimination of high + intermediate from low/no risk compounds. | 2 commercial healthy donor hiPSC-CMs. | (Blinova et al., 2018; Kitaguchi et al., 2016) |
| Cardiotoxicity using kinetic high content imaging of contractility | 21 chemotherapeutic kinase inhibitors Good correlation was observed between the in vitro cardiotoxicity (cell viability and loss of contractility) with clinical incidence of cardiotoxicity. | 11 healthy donor hiPSC-CMs. | (Sharma et al., 2017) |
| Proarrhythmic drug screening using kinetic high content imaging of Ca2+ transients | 108 reference compounds of high, intermediate and low/no arrhythmic risk. Analysis of individual cells within the fields of view increased sensitivity of detection of proarrhythmic phenotypes such as EADs. | 1 commercial healthy donor hiPSC-CM. | (Pfeiffer et al., 2016) |
While these studies simulated disease in genetically healthy hiPSC-CMs, the presence of disease-causing gene variants has the potential to guide the development of mechanism specific therapeutics. To test this idea, McKeithan et al. (McKeithan et al., 2020) used hiPSC-CMs from patients with LQTS type 3, which is due to defects in the cardiac sodium channel, to direct the medicinal chemistry optimization of a drug used to treat LQTS3 patients. The drug, mexiletine, blocks the pathological effect of the mutated channel, but also has undesired properties. The approach of using patient hiPSC-CMs to guide iterative cycles of synthesis and testing succeeded in increasing potency and selectivity of mexiletine while decreasing its undesired proarrhythmic activity. The results illustrated the use of hiPSC-CMs to facilitate the rapid medicinal chemical refinement of a drug to improve therapeutic potential and reduce adverse liability.
Clinical Trials in a Dish (CTiD).
Drug testing using hiPSC-CMs from a cohort of patients are termed clinical trials in a dish (CTiD). By predicting the clinical effect of a drug candidate before it reaches patients, CTiDs could address safety and efficacy issues that contribute to the high attrition rate of drugs in clinical development (Figure 3). Because hiPSCs represent individual donor genetics, not only can they serve as a proxy for a clinical trial, but there is no severe limit on the number of different “people” that could be tested. Therefore, in principle it is possible to adequately power CTiDs to estimate the incidence of a drug’s effect in a target patient population (Figure 3A) that might not be achievable using animal models or Phase I human safety trials that use few subjects. This is conceptually important because certain medications cause life-threatening adverse reactions with a low incidence, and whereas early stage clinical testing is powered to determine the average response to drugs, it can be underpowered to predict rare, and potentially lethal, events. Fermini et al. (Fermini et al., 2018) recently discussed sample size considerations for CTiDs, and predicted that 22 individual lines, roughly the number of individuals in a Phase I clinical trial, would achieve a 90% probability of predicting events that occur in a 10% of the population. With 250 lines, the assay could predict events in 1% of the population with 90% probability (Fermini et al., 2018). Performing hiPSC-CM physiological assays with 250 or more hiPSC lines is certainly feasible. It is also worth considering strategies to incorporate lines carrying rare genetic predisposition to adverse effects (e.g., genetic variants that increase risk for arrhythmia) (Figure 3B). Although not predictive of the general population, the panel would increase sensitivity for anticipated events and could be useful to probe mechanisms for sensitivity to adverse effects and test whether particular genetic variants should be contraindicated for certain drugs. As will be discussed further below, hiPSC-derived cells carrying disease-causing gene variants are already in use to evaluate the efficacy of drugs (Figure 3C).
Figure 3. In Vitro hiPSC-CM Testing for Predicting Drug Safety and Efficacy in Humans.

A) hiPSC-CMs from healthy donors can be used in a clinical trial in a dish (CTiD) to assess susceptibility to drug-induced toxicity. Cardiac toxicity is the most frequent adverse drug reaction.
B) hiPSC-CMs carrying predisposing genetic risk factors can be used to predict the effect in human carriers. The incidence of such effects might be undetected in clinical trials due to the rarity of the predisposing genetic variants.
C) hiPSC-CM models of the target patient population can be used to predict the therapeutic effects of an investigational molecule. In the case of a genetic model, genome-corrected isogenic hiPSC-CMs are important controls for genetic background and hiPSC interline variation.
Similar studies could be used to enhance patient selection for clinical trials, for instance by distinguishing “non-responders” from “responders”. A large cohort of patient iPSCs would be evaluated for in vitro phenotype, and the people corresponding to the “responder” iPSCs would be stratified for inclusion in the human trial. Such enrichment could increase the effect size in the human trial allowing it to be run with fewer subjects.
In practice, validating hiPSC-CM based CTiDs might be challenging, as suggested by a recent study of 16 healthy subjects that failed to find a significant correlation between the in vitro iPSC-CM and corresponding subject-specific responses to human Ether-a-go-go Related Gene (hERG) blockers dofetilide and moxifloxin (Blinova et al., 2019). Good correlation had been observed in earlier reports – one on moxiflocin comprising 10 donors (Shinozawa et al., 2017), and another comparing high versus non-responders (20 total) to the hERG blocker Sotolol (Stillitano et al., 2017). Top among the likely issues that impede recapitulation of subject-specific drug effects in healthy individuals include variation in the iPSC cultures (e.g., differentiation batch and hiPSC line variation, including epigenetics).
Large-scale drug cardiotoxicity studies.
Over 400 medicinal products were withdrawn from sale between 1950–2015 mostly because of CV, hepatic, and nervous system toxicities (Onakpoya et al., 2016a, b; Valentin and Delaunois, 2018). Recent analysis of post-market withdrawals has revealed that primary cardiac toxicity accounted for 74% of withdrawals, including arrhythmia (35%), cardiac injury (cardiomyopathy) (38%) and CV (22%) events (Onakpoya et al., 2016a; Valentin and Delaunois, 2018). Similarly, during drug development (preclinical to marketing/post-approval), CV problems account for 22% of failures, a rate that is comparable to hepatoxicity (21%) (Laverty et al., 2011). Arrhythmic liabilities caused by QT prolongation have decreased thanks to mandated testing for inhibition of the hERG and the thorough QT/QTc study (TQTS) conducted in humans for all systematically bioavailable drugs prior to market authorization (Gintant, 2011). However, the association of hERG inhibition with lethal arrhythmia is not robust; thus, many safe drugs have consequently have been prevented from reaching the market (Gintant, 2011). For this reason, major near-term application of hiPSC-CM drug screening is assessing the risk of drug candidates. In vitro proarrhythmia screening with electrophysiological methods on adult cardiomyocytes or heterologous systems have been practiced for decades and development efforts continue as exemplified by the comprehensive in vitro proarrhythmia assay CiPA initiative that involves a collaboration between pharmaceutical industry, academics and regulatory agencies. In depth functional cardiovascular testing of drug candidates are typically assessed by animal studies only late in preclinical development (Berridge et al., 2016; Gintant et al., 2016; Yang and Papoian, 2018). hiPSC-CMs fit into this landscape by offering a human cardiomyocyte model that can be readily implemented in high throughput early in the development process when many compounds are being compared and their structures are being optimized (e.g., hit to lead or lead optimization) (Figure 1).
There are already several examples of hiPSC-CM testing of drug-induced toxicity that show their potential for guiding drug development. A good example is the anthracycline doxorubicin, which is a commonly used chemotherapeutic agent that causes cardio-toxicity, both acute and long-term. The long-term toxicity is the greater problem clinically because of its unpredictability. hiPSC-CM studies have been used to recapitulate individual patient susceptibility to doxorubicin treatment, reflected in disorganized myofilament structure and cell death compared to unaffected patient lines (Burridge et al., 2016; Sakamoto et al., 2019). More recent molecularly targeted therapeutics such as small molecule kinase inhibitors are also associated with cardiotoxic effects that include left ventricular dysfunction, myocardial infarction and arrhythmia (Force et al., 2007). Sharma et al. (Sharma et al., 2017) screened a panel of 21 chemotherapeutic kinase inhibitor (KI) drugs using 11 healthy donor lines. The study revealed a good correlation between the in vitro cardiotoxicity (cell viability and loss of contractility) and clinical incidence of cardiotoxicity (Sharma et al., 2017). Interestingly, this study revealed cardiotoxicity of compounds such as ponatinib that are considered to act primarily on the cardiac vasculature, suggesting that there might be underappreciated activity on cardiomyocytes. Nonetheless, the importance of the vasculature in drug-induced cardiotoxicity must not be underestimated; thus, it would be valuable to incorporate assays that quantify drug-induced vascular dysfunction (Prigozhina et al., 2011; Vazao et al., 2017) and validate them using reference compounds. Vascular dysfunction is not only a function of endothelial cells but might involve other cells such as pericytes (Chintalgattu et al., 2013), hence specialized multicellular models that involve fluid flow through patent vascular networks are under development and have the potential to enhance drug testing (Savoji et al., 2019).
The prediction of proarrhythmic liability of drugs remains a major objective of cardiotoxicity testing even after its major historic culprit, inhibition of the hERG channel encoded by KCNH2, is now routinely evaluated during drug development. Moderate scale testing of benchmark compounds has shown that proarrhythmia testing using hiPSC-CM is highly reproducible across sites and show promise over current methods of determining cardiac safety liabilities of drug candidates (Table 2). For example, consortia working with the CiPA (Blinova et al., 2018) and the Japanese Consortium for Safety Assessment using Human iPS cells initiative (CSAHi) (Kitaguchi et al., 2016) initiatives used MEA recording and commercially produced healthy donor hiPSC-CMs as an in vitro proarrhythmia model to test 20–30 reference compounds of low, intermediate, and high risk of causing the lethal ventricular tachyarrhythmia known as Torsades de Pointes (TdP) for its characteristic twisting of the waveforms on the surface ECG. The results showed good agreement across multiple sites in discerning intermediate and high risk from safe and low risk drugs. Similar findings were detected by Pfeiffer et al. (Pfeiffer et al., 2016) who evaluated a larger set of (108) reference compounds using a kinetic optical recording modality for proarrhythmic effects on Ca2+ transients in healthy donor hiPSC-CMs. This study analyzed individual cells within the fields of view, which increased sensitivity of proarrhythmia detection because it detected rare and heterogeneous events such as early after depolarizations (EADs) that are obscured by techniques that examine the ensemble average of a field of view or whole well such as MEA.
To date, all published large-scale drug screens used hiPSCs (typically commercially sourced) from only a few healthy donors. However, individual genetic variation, such as in ion channels or sarcomeric proteins, predisposes certain people to drug-induced arrhythmia. Developing large-scale testing that incorporates susceptibility loci could address the challenge of predicting adverse effects of particular drugs to particular people. Given their rarity in the population, predisposing variants might not be reflected in a cohort used for clinical testing, so incorporating them in hiPSC-based assays would complement clinical trials.
Perspectives
Despite substantial technological innovation, there is general consensus that the biopharmaceutical industry has been experiencing a productivity crisis over the past half century, and that this is especially true for cardiovascular therapeutics (Paul et al., 2010; Waring et al., 2015). The decline in efficiency is likely to reflect the exhaustion of highly predictive disease models rather than the depletion of ‘low hanging fruit’ (Scannell and Bosley, 2016), thus the rate of predictive models is insufficient to sustain productivity. If, as Scannell and Bosley concluded (Scannell and Bosley, 2016), small increases in model predictivity can have an even greater impact than recent advances in technological efficiency, it would seem that investment in hiPSC-based models should have a substantial impact on sustaining drug discovery.
hiPSC models are only just now gaining traction in the pharmaceutical industry, most notably for safety pharmacology (Blinova et al., 2018; Kitaguchi et al., 2016; Pfeiffer et al., 2016; Sharma et al., 2017). In addition, there have been several examples of using hiPSC-CMs for guiding discovery or medicinal chemistry refinement of drugs (Drawnel et al., 2014; Fiedler et al., 2019; McKeithan et al., 2020). These studies have demonstrated statistical robustness suitable for directing medicinal chemistry campaigns. To facilitate adoption in biopharmaceutical settings, we believe that more effort must be devoted to validating the predictivity of hiPSC-based models for clinical efficacy. Several limitations to predictivity are being addressed currently. Key among them is the physiological immaturity of hiPSC-CMs (or other derivatives) and the architectural simplicity of the culture systems relative to their adult counterparts. Both problems will be mitigated, at least partly, by three dimensional tissue engineering to more faithfully model normal physiology and hemodynamics combined with micro-instrumentation to read out clinically relevant phenotypes. However, it remains daunting to implement complex engineering solutions in high throughput, thus for now hiPSC-based models appear best suited for reading out disease manifestations with cell-autonomous mechanisms, such as diseases with cellular electrophysiological, metabolic and contractile etiologies. Such models are important since numerous congenital diseases have few or no effective treatments and the approaches developed for rare diseases might be relevant to acquired diseases as well. Lastly, there is a critical need for many more hiPSC lines from healthy donors and patients with congenital heart disease to generalize discovery efforts and enhance the predictive power of hiPSC-based studies. In summary, the past decade has seen enormous strides in the development of hiPSC-based models, and continued development and validation efforts should be highly impactful for the discovery of new therapeutics for heart disease.
Highlights.
Human iPSC disease modeling represents a paradigm shift for drug discovery
Initial large projects demonstrate potential as well as challenges
Recent studies provide lessons for pre-clinical and clinical drug development
Acknowledgements
MM gratefully acknowledges the Joan and Sanford I. Weill Scholar Endowment and grant funding from the National Institutes of Health (P01HL141084, R01HL138539, R01HL130840). DWS is a recipient of a fellowship funded by T32 5T32HL094274. Portions of the figures use art from BioRender.com.
Footnotes
Declaration of Interests
MM has equity in Vala Sciences, Inc. and is on its scientific advisory board. The authors declare no other competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abulaiti M, Yalikun Y, Murata K, Sato A, Sami MM, Sasaki Y, Fujiwara Y, Minatoya K, Shiba Y, Tanaka Y, et al. (2020). Establishment of a heart-on-a-chip microdevice based on human iPS cells for the evaluation of human heart tissue function. Sci Rep 10, 19201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M (2018). The CRISPR tool kit for genome editing and beyond. Nat Commun 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, et al. (2019). Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 139, e56–e528. [DOI] [PubMed] [Google Scholar]
- Berridge BR, Schultze AE, Heyen JR, Searfoss GH, and Sarazan RD (2016). Technological Advances in Cardiovascular Safety Assessment Decrease Preclinical Animal Use and Improve Clinical Relevance. ILAR J 57, 120–132. [DOI] [PubMed] [Google Scholar]
- Bhagwan JR, Mosqueira D, Chairez-Cantu K, Mannhardt I, Bodbin SE, Bakar M, Smith JGW, and Denning C (2020). Isogenic models of hypertrophic cardiomyopathy unveil differential phenotypes and mechanism-driven therapeutics. J Mol Cell Cardiol 145, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinova K, Dang Q, Millard D, Smith G, Pierson J, Guo L, Brock M, Lu HR, Kraushaar U, Zeng H, et al. (2018). International Multisite Study of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Drug Proarrhythmic Potential Assessment. Cell Rep 24, 3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinova K, Schocken D, Patel D, Daluwatte C, Vicente J, Wu JC, and Strauss DG (2019). Clinical Trial in a Dish: Personalized Stem Cell-Derived Cardiomyocyte Assay Compared With Clinical Trial Results for Two QT-Prolonging Drugs. Clin Transl Sci 12, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briganti F, Sun H, Wei W, Wu J, Zhu C, Liss M, Karakikes I, Rego S, Cipriano A, Snyder M, et al. (2020). iPSC Modeling of RBM20-Deficient DCM Identifies Upregulation of RBM20 as a Therapeutic Strategy. Cell Rep 32, 108117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmstrom A, Chang AC, Coronado MJ, Ebert AD, et al. (2016). Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med 22, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerignoli F, Charlot D, Whittaker R, Ingermanson R, Gehalot P, Savchenko A, Gallacher DJ, Towart R, Price JH, McDonough PM, et al. (2012). High throughput measurement of Ca(2)(+) dynamics for drug risk assessment in human stem cell-derived cardiomyocytes by kinetic image cytometry. Journal of pharmacological and toxicological methods 66, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J, Dunner K Jr., Pati S, Bankson JA, Pasqualini R, et al. (2013). Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med 5, 187ra169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn R, Thakar K, Lowe A, Ladha FA, Pettinato AM, Romano R, Meredith E, Chen YS, Atamanuk K, Huey BD, et al. (2019). A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations. Stem Cell Reports 12, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyganek L, Tiburcy M, Sekeres K, Gerstenberg K, Bohnenberger H, Lenz C, Henze S, Stauske M, Salinas G, Zimmermann WH, et al. (2018). Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Alamo JC, Lemons D, Serrano R, Savchenko A, Cerignoli F, Bodmer R, and Mercola M (2016). High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim Biophys Acta 1863, 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Cunado M, Wei K, Bushway PJ, Maurya MR, Perera R, Subramaniam S, Ruiz-Lozano P, and Mercola M (2018). miRNAs that Induce Human Cardiomyocyte Proliferation Converge on the Hippo Pathway. Cell Rep 23, 2168–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG (2018). Am I ready for CRISPR? A user’s guide to genetic screens. Nat Rev Genet 19, 67–80. [DOI] [PubMed] [Google Scholar]
- Dostanic M, Windt LM, Stein JM, Meer B.J.v., Bellin M, Orlova V, Mastrangeli M, Mummery CL, and Sarro PM (2020). A miniaturized EHT platform for accurate measurements of tissue contractile properties. Journal of Microelectromechanical Systems 29, 881–887. [Google Scholar]
- Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, Gerard R, Badi L, Kam-Thong T, Bu L, et al. (2014). Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep 9, 810–821. [DOI] [PubMed] [Google Scholar]
- Eng G, Lee BW, Protas L, Gagliardi M, Brown K, Kass RS, Keller G, Robinson RB, and Vunjak-Novakovic G (2016). Autonomous beating rate adaptation in human stem cell-derived cardiomyocytes. Nat Commun 7, 10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenhagen T, and Carrier L (2019). Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes-a systematic review. Pflugers Arch 471, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk A, Heine VM, Harwood AJ, Sullivan PF, Peitz M, Brustle O, Shen S, Sun YM, Glover JC, Posthuma D, et al. (2016). Modeling psychiatric disorders: from genomic findings to cellular phenotypes. Mol Psychiatry 21, 1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Gowen BG, Lin PC, Doudna JA, and Corn JE (2017). Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov 16, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermini B, Coyne KP, and Coyne ST (2018). Challenges in designing and executing clinical trials in a dish studies. J Pharmacol Toxicol Methods 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Ferrari A (2019). Recent technological advancements in traction force microscopy. Biophys Rev 11, 679–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyen DAM, McKeithan WL, Bruyneel AAN, Spiering S, Hormann L, Ulmer B, Zhang H, Briganti F, Schweizer M, Hegyi B, et al. (2020). Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep 32, 107925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler LR, Chapman K, Xie M, Maifoshie E, Jenkins M, Golforoush PA, Bellahcene M, Noseda M, Faust D, Jarvis A, et al. (2019). MAP4K4 Inhibition Promotes Survival of Human Stem Cell-Derived Cardiomyocytes and Reduces Infarct Size In Vivo. Cell Stem Cell 24, 579–591 e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force T, Krause DS, and Van Etten RA (2007). Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer 7, 332–344. [DOI] [PubMed] [Google Scholar]
- Friedman CE, Nguyen Q, Lukowski SW, Helfer A, Chiu HS, Miklas J, Levy S, Suo S, Han JJ, Osteil P, et al. (2018). Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 23, 586–598 e588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg P, Oikonomopoulos A, Chen H, Li Y, Lam CK, Sallam K, Perez M, Lux RL, Sanguinetti MC, and Wu JC (2018). Genome Editing of Induced Pluripotent Stem Cells to Decipher Cardiac Channelopathy Variant. J Am Coll Cardiol 72, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germanguz I, Sedan O, Zeevi-Levin N, Shtrichman R, Barak E, Ziskind A, Eliyahu S, Meiry G, Amit M, Itskovitz-Eldor J, et al. (2011). Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. J Cell Mol Med 15, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gintant G (2011). An evaluation of hERG current assay performance: Translating preclinical safety studies to clinical QT prolongation. Pharmacol Ther 129, 109–119. [DOI] [PubMed] [Google Scholar]
- Gintant G, Sager PT, and Stockbridge N (2016). Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov 15, 457–471. [DOI] [PubMed] [Google Scholar]
- Graneli C, Hicks R, Brolen G, Synnergren J, and Sartipy P (2019). Diabetic Cardiomyopathy Modelling Using Induced Pluripotent Stem Cell Derived Cardiomyocytes: Recent Advances and Emerging Models. Stem Cell Rev Rep 15, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromo G, Mann J, and Fitzgerald JD (2014). Cardiovascular drug discovery: a perspective from a research-based pharmaceutical company. Cold Spring Harb Perspect Med 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, and Moe GK (1964). Nonuniform Recovery of Excitability in Ventricular Muscle. Circ Res 14, 44–60. [DOI] [PubMed] [Google Scholar]
- Hidalgo A, Glass N, Ovchinnikov D, Yang SK, Zhang X, Mazzone S, Chen C, Wolvetang E, and Cooper-White J (2018). Modelling ischemia-reperfusion injury (IRI) in vitro using metabolically matured induced pluripotent stem cell-derived cardiomyocytes. APL Bioeng 2, 026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi Y, Yan Y, Terashvili M, Wells C, Horikoshi H, Fujita S, Bosnjak ZJ, and Bai X (2019). Fatty Acid-Treated Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes Exhibit Adult Cardiomyocyte-Like Energy Metabolism Phenotypes. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihry RJ, Salick MR, Ho DJ, Sondey M, Kommineni S, Paula S, Raymond J, Henry B, Frias E, Wang Q, et al. (2019). Genome-Scale CRISPR Screens Identify Human Pluripotency-Specific Genes. Cell Rep 27, 616–630 e616. [DOI] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Gepstein A, Arbel G, Caspi O, Miller L, Belhassen B, Nof E, Glikson M, et al. (2012). Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. Journal of the American College of Cardiology 60, 990–1000. [DOI] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, et al. (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229. [DOI] [PubMed] [Google Scholar]
- Karakikes I, Ameen M, Termglinchan V, and Wu JC (2015). Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res 117, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, et al. (2013). Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Lipke EA, Kim P, Cheong R, Thompson S, Delannoy M, Suh KY, Tung L, and Levchenko A (2010). Nanoscale cues regulate the structure and function of macroscopic cardiac tissue constructs. Proc Natl Acad Sci U S A 107, 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaguchi T, Moriyama Y, Taniguchi T, Ojima A, Ando H, Uda T, Otabe K, Oguchi M, Shimizu S, Saito H, et al. (2016). CSAHi study: Evaluation of multi-electrode array in combination with human iPS cell-derived cardiomyocytes to predict drug-induced QT prolongation and arrhythmia--effects of 7 reference compounds at 10 facilities. J Pharmacol Toxicol Methods 78, 93–102. [DOI] [PubMed] [Google Scholar]
- Kussauer S, David R, and Lemcke H (2019). hiPSCs Derived Cardiac Cells for Drug and Toxicity Screening and Disease Modeling: What Micro- Electrode-Array Analyses Can Tell Us. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, et al. (2013). Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 12, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverty H, Benson C, Cartwright E, Cross M, Garland C, Hammond T, Holloway C, McMahon N, Milligan J, Park B, et al. (2011). How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br J Pharmacol 163, 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Termglinchan V, Diecke S, Itzhaki I, Lam CK, Garg P, Lau E, Greenhaw M, Seeger T, Wu H, et al. (2019). Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine MD, Mannhardt I, Breckwoldt K, Prondzynski M, Flenner F, Ulmer B, Hirt MN, Neuber C, Horvath A, Kloth B, et al. (2017). Human iPSC-derived cardiomyocytes cultured in 3D engineered heart tissue show physiological upstroke velocity and sodium current density. Sci Rep 7, 5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang P, Sallam K, Wu H, Li Y, Itzhaki I, Garg P, Zhang Y, Vermglinchan V, Lan F, Gu M, et al. (2016). Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J Am Coll Cardiol 68, 2086–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu DK, Fu JD, Chiamvimonvat N, Tung KC, McNerney GP, Huser T, Keller G, Kong CW, and Li RA (2013). Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol 6, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Lin X, Stachel M, Wang E, Luo Y, Lader J, Sun X, Delmar M, and Bu L (2017). Culture in Glucose-Depleted Medium Supplemented with Fatty Acid and 3,3’,5-Triiodo-l-Thyronine Facilitates Purification and Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Front Endocrinol (Lausanne) 8, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, et al. (2020). Cells of the adult human heart. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Steinbusch LKM, Nabben M, Kapsokalyvas D, van Zandvoort M, Schonleitner P, Antoons G, Simons PJ, Coumans WA, Geomini A, et al. (2017). Palmitate-Induced Vacuolar-Type H(+)-ATPase Inhibition Feeds Forward Into Insulin Resistance and Contractile Dysfunction. Diabetes 66, 1521–1534. [DOI] [PubMed] [Google Scholar]
- Ma D, Wei H, Lu J, Ho S, Zhang G, Sun X, Oh Y, Tan SH, Ng ML, Shim W, et al. (2012). Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. European heart journal. [DOI] [PubMed] [Google Scholar]
- Mannhardt I, Breckwoldt K, Letuffe-Breniere D, Schaaf S, Schulz H, Neuber C, Benzin A, Werner T, Eder A, Schulze T, et al. (2016). Human Engineered Heart Tissue: Analysis of Contractile Force. Stem Cell Reports 7, 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur A, Loskill P, Shao K, Huebsch N, Hong S, Marcus SG, Marks N, Mandegar M, Conklin BR, Lee LP, et al. (2015). Human iPSC-based cardiac microphysiological system for drug screening applications. Sci Rep 5, 8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeithan WL, Feyen DAM, Bruyneel AAN, Okolotowicz KJ, Ryan DA, Sampson KJ, Potet F, Savchenko A, Gomez-Galeno J, Vu M, et al. (2020). Reengineering an Antiarrhythmic Drug Using Patient hiPSC Cardiomyocytes to Improve Therapeutic Potential and Reduce Toxicity. Cell Stem Cell 27, 813–821 e816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeithan WL, Savchenko A, Yu MS, Cerignoli F, Bruyneel AAN, Price JH, Colas AR, Miller EW, Cashman JR, and Mercola M (2017). An Automated Platform for Assessment of Congenital and Drug-Induced Arrhythmia with hiPSC-Derived Cardiomyocytes. Front Physiol 8, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic A, Li J, Miyagi Y, Gagliardi M, Li SH, Zu J, Weisel RD, Keller G, and Li RK (2014). The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials 35, 2798–2808. [DOI] [PubMed] [Google Scholar]
- Min YL, Chemello F, Li H, Rodriguez-Caycedo C, Sanchez-Ortiz E, Mireault AA, McAnally JR, Shelton JM, Zhang Y, Bassel-Duby R, et al. (2020). Correction of Three Prominent Mutations in Mouse and Human Models of Duchenne Muscular Dystrophy by Single-Cut Genome Editing. Mol Ther 28, 2044–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, et al. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 363, 1397–1409. [DOI] [PubMed] [Google Scholar]
- Moretti A, Laugwitz KL, Dorn T, Sinnecker D, and Mummery C (2013). Pluripotent stem cell models of human heart disease. Cold Spring Harb Perspect Med 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A (2020). 2019 FDA drug approvals. Nat Rev Drug Discov 19, 79–84. [DOI] [PubMed] [Google Scholar]
- Nityanandam A, and Baldwin KK (2015). Advances in reprogramming-based study of neurologic disorders. Stem Cells Dev 24, 1265–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, Jiang J, Masse S, Gagliardi M, Hsieh A, et al. (2013). Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods 10, 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleaga C, Riu A, Rothemund S, Lavado A, McAleer CW, Long CJ, Persaud K, Narasimhan NS, Tran M, Roles J, et al. (2018). Investigation of the effect of hepatic metabolism on off-target cardiotoxicity in a multi-organ human-on-a-chip system. Biomaterials 182, 176–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onakpoya IJ, Heneghan CJ, and Aronson JK (2016a). Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med 14, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onakpoya IJ, Heneghan CJ, and Aronson JK (2016b). Worldwide withdrawal of medicinal products because of adverse drug reactions: a systematic review and analysis. Crit Rev Toxicol 46, 477–489. [DOI] [PubMed] [Google Scholar]
- Onder TT, and Daley GQ (2012). New lessons learned from disease modeling with induced pluripotent stem cells. Curr Opin Genet Dev 22, 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Vitali JL, and Darabi R (2019). iPSCs as a Platform for Disease Modeling, Drug Screening, and Personalized Therapy in Muscular Dystrophies. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik DT, Chandy M, and Wu JC (2020). Patient and Disease-Specific Induced Pluripotent Stem Cells for Discovery of Personalized Cardiovascular Drugs and Therapeutics. Pharmacol Rev 72, 320–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, and Schacht AL (2010). How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nature reviews Drug discovery 9, 203–214. [DOI] [PubMed] [Google Scholar]
- Penttinen K, Swan H, Vanninen S, Paavola J, Lahtinen AM, Kontula K, and Aalto-Setala K (2015). Antiarrhythmic Effects of Dantrolene in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia and Replication of the Responses Using iPSC Models. PLoS One 10, e0125366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer ER, Vega R, McDonough PM, Price JH, and Whittaker R (2016). Specific prediction of clinical QT prolongation by kinetic image cytometry in human stem cell derived cardiomyocytes. J Pharmacol Toxicol Methods 81, 263–273. [DOI] [PubMed] [Google Scholar]
- Piga D, Salani S, Magri F, Brusa R, Mauri E, Comi GP, Bresolin N, and Corti S (2019). Human induced pluripotent stem cell models for the study and treatment of Duchenne and Becker muscular dystrophies. Ther Adv Neurol Disord 12, 1756286419833478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigozhina NL, Heisel A, Wei K, Noberini R, Hunter EA, Calzolari D, Seldeen JR, Pasquale EB, Ruiz-Lozano P, Mercola M, et al. (2011). Characterization of a novel angiogenic model based on stable, fluorescently labelled endothelial cell lines amenable to scale-up for high content screening. Biol Cell 103, 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro AJ, Ang YS, Fu JD, Rivas RN, Mohamed TM, Higgs GC, Srivastava D, and Pruitt BL (2015). Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc Natl Acad Sci U S A 112, 12705–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, and Vunjak-Novakovic G (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan JL, Tulloch NL, Razumova MV, Saiget M, Muskheli V, Pabon L, Reinecke H, Regnier M, and Murry CE (2016). Mechanical Stress Conditioning and Electrical Stimulation Promote Contractility and Force Maturation of Induced Pluripotent Stem Cell-Derived Human Cardiac Tissue. Circulation 134, 1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, et al. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract 157, 107843. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Sakatoku K, Sugimoto S, Iwasaki N, Sano Y, Yamaguchi M, and Kurokawa J (2019). Continued exposure of anti-cancer drugs to human iPS cell-derived cardiomyocytes can unmask their cardiotoxic effects. J Pharmacol Sci 140, 345–349. [DOI] [PubMed] [Google Scholar]
- Sala L, van Meer BJ, Tertoolen LGJ, Bakkers J, Bellin M, Davis RP, Denning C, Dieben MAE, Eschenhagen T, Giacomelli E, et al. (2018). MUSCLEMOTION: A Versatile Open Software Tool to Quantify Cardiomyocyte and Cardiac Muscle Contraction In Vitro and In Vivo. Circ Res 122, e5–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SS, Trivedi DV, Morck MM, Adhikari AS, Pasha SN, Ruppel KM, and Spudich JA (2020). The hypertrophic cardiomyopathy mutations R403Q and R663H increase the number of myosin heads available to interact with actin. Sci Adv 6, eaax0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoji H, Mohammadi MH, Rafatian N, Toroghi MK, Wang EY, Zhao Y, Korolj A, Ahadian S, and Radisic M (2019). Cardiovascular disease models: A game changing paradigm in drug discovery and screening. Biomaterials 198, 3–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell JW, and Bosley J (2016). When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis. PLoS One 11, e0147215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid C, Wohnhaas CT, Hildebrandt T, Baum P, and Rast G (2020). Characterization of iCell cardiomyocytes using single-cell RNA-sequencing methods. J Pharmacol Toxicol Methods 106, 106915. [DOI] [PubMed] [Google Scholar]
- Seeger T, Shrestha R, Lam CK, Chen C, McKeithan WL, Lau E, Wnorowski A, McMullen G, Greenhaw M, Lee J, et al. (2019). A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation 139, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, and Zhang F (2015). High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 16, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, Churko JM, Kitani T, Wu H, Holmstrom A, et al. (2017). High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozawa T, Imahashi K, Sawada H, Furukawa H, and Takami K (2012). Determination of appropriate stage of human-induced pluripotent stem cell-derived cardiomyocytes for drug screening and pharmacological evaluation in vitro. J Biomol Screen 17, 1192–1203. [DOI] [PubMed] [Google Scholar]
- Shinozawa T, Nakamura K, Shoji M, Morita M, Kimura M, Furukawa H, Ueda H, Shiramoto M, Matsuguma K, Kaji Y, et al. (2017). Recapitulation of Clinical Individual Susceptibility to Drug-Induced QT Prolongation in Healthy Subjects Using iPSC-Derived Cardiomyocytes. Stem Cell Reports 8, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JGW, Owen T, Bhagwan JR, Mosqueira D, Scott E, Mannhardt I, Patel A, Barriales-Villa R, Monserrat L, Hansen A, et al. (2018). Isogenic Pairs of hiPSC-CMs with Hypertrophic Cardiomyopathy/LVNC-Associated ACTC1 E99K Mutation Unveil Differential Functional Deficits. Stem Cell Reports 11, 1226–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommese RF, Sung J, Nag S, Sutton S, Deacon JC, Choe E, Leinwand LA, Ruppel K, and Spudich JA (2013). Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human beta-cardiac myosin motor function. Proc Natl Acad Sci U S A 110, 12607–12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillitano F, Hansen J, Kong CW, Karakikes I, Funck-Brentano C, Geng L, Scott S, Reynier S, Wu M, Valogne Y, et al. (2017). Modeling susceptibility to drug-induced long QT with a panel of subject-specific induced pluripotent stem cells. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, et al. (2012). Patient-Specific Induced Pluripotent Stem Cells as a Model for Familial Dilated Cardiomyopathy. Science Translational Medicine 4, 130ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surawicz B (1997). Ventricular fibrillation and dispersion of repolarization. J Cardiovasc Electrophysiol 8, 1009–1012. [DOI] [PubMed] [Google Scholar]
- Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD, Pabon L, Reinecke H, and Murry CE (2011). Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ Res 109, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull IC, Karakikes I, Serrao GW, Backeris P, Lee JJ, Xie C, Senyei G, Gordon RE, Li RA, Akar FG, et al. (2014). Advancing functional engineered cardiac tissues toward a preclinical model of human myocardium. FASEB J 28, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer BM, Stoehr A, Schulze ML, Patel S, Gucek M, Mannhardt I, Funcke S, Murphy E, Eschenhagen T, and Hansen A (2018). Contractile Work Contributes to Maturation of Energy Metabolism in hiPSC-Derived Cardiomyocytes. Stem Cell Reports 10, 834–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzun AU, Mannhardt I, Breckwoldt K, Horvath A, Johannsen SS, Hansen A, Eschenhagen T, and Christ T (2016). Ca(2+)-Currents in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Effects of Two Different Culture Conditions. Front Pharmacol 7, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin J-P, and Delaunois A (2018). Developing solutions to detect and avoid cardiovascular toxicity in the clinic (S24–02). In 54th EUROTOX Annual Congress (Brussels, Belgium: Belgian Society of Toxicology and Ecotoxicology (Beltox)). [Google Scholar]
- Vazao H, Rosa S, Barata T, Costa R, Pitrez PR, Honorio I, de Vries MR, Papatsenko D, Benedito R, Saris D, et al. (2017). High-throughput identification of small molecules that affect human embryonic vascular development. Proc Natl Acad Sci U S A 114, E3022–E3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, et al. (2014). Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med 20, 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PY, Yu J, Lin JH, and Tsai WB (2011). Modulation of alignment, elongation and contraction of cardiomyocytes through a combination of nanotopography and rigidity of substrates. Acta Biomater 7, 3285–3293. [DOI] [PubMed] [Google Scholar]
- Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD, Wang J, et al. (2015). An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov 14, 475–486. [DOI] [PubMed] [Google Scholar]
- Yang X, and Papoian T (2018). Moving beyond the comprehensive in vitro proarrhythmia assay: Use of human-induced pluripotent stem cell-derived cardiomyocytes to assess contractile effects associated with drug-induced structural cardiotoxicity. J Appl Toxicol 38, 1166–1176. [DOI] [PubMed] [Google Scholar]
- Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, Ritterhoff J, Zhao L, Kolwicz SC Jr., Pabon L, et al. (2019). Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Reports 13, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, and Dolmetsch RE (2011). Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471, 230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshie H, Koushki N, Kaviani R, Tabatabaei M, Rajendran K, Dang Q, Husain A, Yao S, Li C, Sullivan JK, et al. (2018). Traction Force Screening Enabled by Compliant PDMS Elastomers. Biophys J 114, 2194–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Haviland S, Wei H, Saric T, Fatima A, Hescheler J, Cleemann L, and Morad M (2013). Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects. Cell Calcium 54, 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann WH, Melnychenko I, and Eschenhagen T (2004). Engineered heart tissue for regeneration of diseased hearts. Biomaterials 25, 1639–1647. [DOI] [PubMed] [Google Scholar]