

Significance Statement
The lack of understanding of inverted formin-2 (INF2)-related human diseases has hindered the development of effective therapy. To address the pathological features of protein mistrafficking in both INF2-related neuropathy and FSGS, experiments using in vitro and in vivo models of INF2-related podocytopathy confirmed the hypothesis that INF2 regulates dynein-mediated trafficking of nephrin, and that pathogenic INF2 mutations can disrupt this regulation, resulting in impaired slit diaphragm integrity, proteinuric podocytopathy, and FSGS. Dysregulated dynein-mediated trafficking may be a viable therapeutic target for INF2-related (and perhaps other forms) of FSGS.
Keywords: genetic renal disease, podocyte, cytoskeleton
Visual Abstract

Abstract
Background
FSGS caused by mutations in INF2 is characterized by a podocytopathy with mistrafficked nephrin, an essential component of the slit diaphragm. Because INF2 is a formin-type actin nucleator, research has focused on its actin-regulating function, providing an important but incomplete insight into how these mutations lead to podocytopathy. A yeast two-hybridization screen identified the interaction between INF2 and the dynein transport complex, suggesting a newly recognized role of INF2 in regulating dynein-mediated vesicular trafficking in podocytes.
Methods
Live cell and quantitative imaging, fluorescent and surface biotinylation-based trafficking assays in cultured podocytes, and a new puromycin aminoglycoside nephropathy model of INF2 transgenic mice were used to demonstrate altered dynein-mediated trafficking of nephrin in INF2 associated podocytopathy.
Results
Pathogenic INF2 mutations disrupt an interaction of INF2 with dynein light chain 1, a key dynein component. The best-studied mutation, R218Q, diverts dynein-mediated postendocytic sorting of nephrin from recycling endosomes to lysosomes for degradation. Antagonizing dynein-mediated transport can rescue this effect. Augmented dynein-mediated trafficking and degradation of nephrin underlies puromycin aminoglycoside-induced podocytopathy and FSGS in vivo.
Conclusions
INF2 mutations enhance dynein-mediated trafficking of nephrin to proteolytic pathways, diminishing its recycling required for maintaining slit diaphragm integrity. The recognition that dysregulated dynein-mediated transport of nephrin in R218Q knockin podocytes opens an avenue for developing targeted therapy for INF2-mediated FSGS.
The filtering function of the kidneys depends on the integrity of the slit diaphragm (SD), the specialized cell-cell junction between the interdigitated foot processes of podocytes that encapsulate the glomerular capillaries. On a molecular scale, the extracellular domains of the transmembrane protein nephrin form a molecular sieve of the SD.1–3 Therefore, the surface trafficking of nephrin is crucial to the integrity of the SD.4 The homeostasis of nephrin at the SD relies on the dynamic balance of endocytosis, recycling, and exocytosis, the disruption of which has been observed in proteinuric podocytopathies.5–10 These findings have stimulated research aimed at understanding vesicle trafficking in podocytes.
Mutations in the inverted formin-2 (INF2) gene are a major cause of autosomal dominant podocyte dysfunction and FSGS.11–15 A subset of affected individuals also develop Charcot-Marie-Tooth (CMT) neuropathy.16–18 INF2 is an atypical formin family member that is highly expressed in podocytes. INF2 is composed of an N-terminal diaphanous inhibitory domain (DID), formin homology domains, and a C terminal diaphanous activation domain (DAD).19 In vitro experiments have shown the actin polymerization function of INF2 can be “autoinhibited” by the binding of DID to the DAD.19,20 INF2-DID can also bind the DAD of diaphanous formins (mDia) and antagonize Rho/mDia–mediated actin polymerization by “heteroinhibition.”21 Despite the recognition of a cause-effect relationship between mutation and disease, the molecular pathogenesis of INF2-related glomerulopathy remains incompletely understood.
Although current studies of INF2 have focused largely on its actin-regulatory function, our in vivo studies have provided support for a role for INF2 in modulating the trafficking of nephrin: (1) immunostaining of kidney biopsy specimens of patients with INF2-related FSGS showed mistrafficking of nephrin from along the glomerular basement membrane (GBM)22; (2) zebrafish embryos lacking INF2 show mistrafficking of nephrin with impaired glomerular filtration barrier23; and (3) transgenic mice harboring a pathogenic R218Q mutation demonstrate impaired nephrin recycling and SD restoration after protamine perfusion,24 suggesting dysregulated trafficking mechanisms contribute to INF2-mediated disease.
The fact that all of the disease-causing mutations reside within the DID suggests a unique function of this domain.14,15,17,25 While screening for binding partners to explore the biologic function of INF2-DID, we identified dynein light chain 1 (Dynll1), a component of the dynein transport complex that mediates microtubule dependent transport,26–29 suggesting a regulatory role of INF2 in this trafficking mechanism. Dynein components have been found to be enriched in the SD, which is further enhanced in nephrosis models.30,31 Dysfunction of dynein-mediated trafficking causes neurodegenerative diseases including CMT.32–37 However, its significance in podocyte disease remains unclear. Cytoplasmic dynein is a multiunit complex containing1 dynein heavy chains, which contain the ATPase activity and are responsible for generating movement along the microtubule,2 light intermediate chains that bind to other molecules to regulate the activity of the dynein complex,3; and light chains that are required to maintain the integrity and function of the whole dynein complex.36,38 In addition, dynein binds to HDAC6, which subsequently bridges the cargo protein of dynein to the ubiquitin proteolytic system (UPS) for degradation,39–42 making dynein an important switch that determines the postendocytic sorting and ultimate fate of the cargo protein. We found that FSGS-causing mutations can disrupt the INF2/Dynll1 interaction, leading to the hypothesis that a dysregulated dynein-mediated trafficking underlies INF2-related glomerulopathy.
Here we demonstrate INF2 mutations that disrupt its interaction with Dynll1 lead to enhanced dynein-mediated trafficking and degradation of nephrin, resulting in the inadequate surface recycling of nephrin needed to maintain SD integrity.
Methods
Antibodies (Table 1), siRNAs (Table 2), plasmids (Table 3), and chemical compounds (Table 4), used in the study are listed below. Mouse Dynll1 for mouse Dynll1 is a pool of three siRNA duplexes (Santa Cruz, sc-36229 A, B&C). Non-targeting siRNA duplex (Santa Cruz sc-37007) serves as a control.
Table 1.
Antibodies
| Antibody | Company | Catalog No. |
|---|---|---|
| Rabbit anti-Rab5a | Cell Signaling | 9765 |
| Rabbit anti-Rab7a | Novus | NBP2–24591 |
| Goat anti-Rab11 | Santa Cruz | sc-6565 |
| Rabbit anti-HDAC6 | Novus | NBP1–78981SS |
| Rabbit anti-Ubiquitin | Abcam | AB7780 |
| Sheep anti-Dynactin 1 | Novus | AF6657 |
| Rabbit anti-nephrin | Invitrogen | PA5–91907 |
| Mouse anti-nephrin (G-8) | Santa Cruz | sc-376522 |
| Mouse anti-Dynll1 | Santa Cruz | sc-136287 |
| Rabbit anti-Dynll1 | Invitrogen | PA5–97920 |
| Rabbit anti-INF2 | Bethyl Laboratories | A303–427A |
| Rabbit anti-KAc alpha-Tubulin | Cell Signaling | 5335T |
| Alexa Fluor labeled second antibodies | Thermo Fisher |
Table 2.
siRNA sequences
| SiRNA target | Catalog number | Sense (5’-3’) | Antisense (5’-3’) |
|---|---|---|---|
| Dynll1 | sc-36229A | GGCCAUUCUUCUGUUCAAAtt | UUUGAACAGAAGAAUGGCCtt |
| sc-36229B | CACCUCGUUUGAAUCUGUUtt | AACAGAUUCAAACGAGGUGtt | |
| sc-36229C | GGCUUCAUUCUCUGUACAAtt | UUGUACAGAGAAUGAAGCCtt | |
| None | Control sc-37007 | UUCUCCGAACGUGUCACGUtt | ACGUGACACGUUCGGAGAAtt |
Table 3.
Plasmids
Table 4.
Chemicals
| Chemical | Chemical Abstracts Service Registry No. | Company |
|---|---|---|
| Ciliobrevin D | 250401 | Sigma-Calbiochem |
| Puromycin aminoglycoside | 58–60–6 | MedChemExpress |
| Ricolinostat | 1316214–52–4 | MedChemExpress |
Yeast Two-Hybrid Screening and Yeast Mating
Yeast two-hybridization screening using INF2-DID as a bait to screen human kidney cDNA library and yeast mating to compare the interaction of Dynll1 and IFN2-DID (wild-type [WT] and mutants) were performed as described in our previous publication.21
Immunoblotting and Co-Immunoprescipitation
Cultured podocytes or homogenized kidney tissues were lysed at 4°C for 1 hour in Nonidet P40 lysis buffer with 0.5% deoxycholate, protease inhibitor mix (Roche), and phosphatase inhibitor (PhosSTOP; Sigma). For western blotting, proteins in the lysates were separated by SDS-PAGE and transferred to polyvinylidene fluoride or polyvinylidene difluoride (PVDF) membranes. The membrane was blocked with 5% nonfat milk, incubated with the primary antibody (1:1000) at 4°C overnight, and an appropriate peroxidase-labeled -secondary antibody (1:5000; Santa Cruz) after washes. Signals were detected using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher).
For co-immunoprescipitation (co-IP), the lysates were immunoprecipitated with the appropriate primary antibody and protein A/G beads. The precipitated proteins were separated by SDS-PAGE, transferred to a PVDF membrane, and detected by immunoblotting.
Transgenic Mice
The INF2 R218Q transgenic mice were created by homologous recombination to generate a R218Q knockin allele in C57Bl/6 embryonic stem cells per the VelociGene method at Charles River Laboratories (mouse strain C57B6, project ID VG5159/5161).24
Cell Lines
Podocytes were isolated from wt/wt or INF2 R218Q knockin (ki/ki) mice, and conditionally immortalized podocyte cell lines were generated by expression of a temperature-sensitive mutant of the SV40 Large-T antigen per Saleem’s protocol.43 Cells were maintained in RPMI 1640 with 10% FBS, insulin–transferrin–selenium (Gibco, Grand Island, NY), and 50 IU/ml penicillin/streptavidin.
In Vitro Nephrin Trafficking Model and Time-Lapse Imaging
Cells transfected with pcDNA3-nephrin (Table 3) were incubated with a mouse antibody against the extracellular domain of nephrin (mouse anti-nephrin [G8], Table 1), followed by a fluorophore-labeled secondary antibody to trigger crosslink and endocytosis of nephrin.44–46 Live imaging was performed immediately after cells completed the endocytosis. The cells were scanned every 5 seconds on an inverted Zeiss LSM 880 confocal microscope for 20 minutes. Single molecule tracking of nephrin was performed five times × five cells × three independent experiments. Using the Fiji plugin KymographClear, time-lapse tracking of endocytosed nephrin was stacked under Z-project and converted into a kymograph, with the x axis reflecting distance (μm) from time 0, and the y axis reflecting time (total 20 minutes). Using KymographDirect, the fraction and the average velocity of the endocytic (red), recycling (green), and static (blue) vesicle-trafficking events were calculated in each kymograph. As shown in the representative kymograph of WT cells treated with DMSO (Figure 4A), the velocity of a representative recycling vesicle was calculated as distance/time (μm per second).
Figure 4.
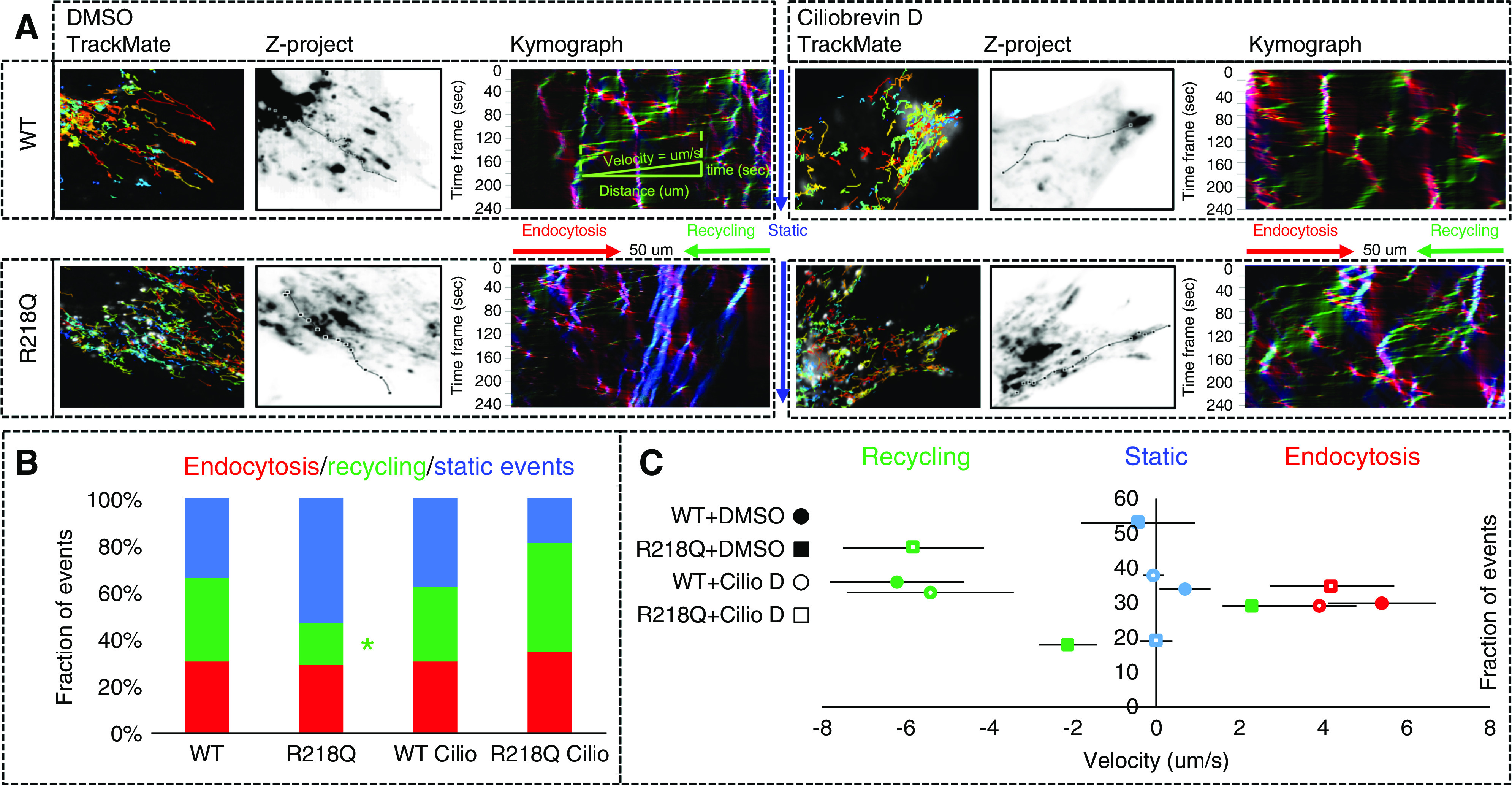
Time-lapse imaging and single-molecule tracking of nephrin using KymographClear, KymographDirect, and TrackMate. (A) The endocytic, recycling, and static events are labeled in red, green, and blue, respectively. Single-molecule tracking of nephrin was performed five times × five cells × three independent experiments. (B) The fractions and (C) average velocities of trafficking events in cells treated with Ciliobrevin (50 μM) and control cells treated DMSO (0.3%) were analyzed by using one-way ANOVA and the difference between groups analyzed by Student–Newman–Keuls (SNK) q test. *P<0.05 versus WT control (DMSO).
Fluorescent-Based Recycling Experiment
Fluorescent-based recycling experiment was performed as described in the literature.48–50Cells transfected with pcDNA3-nephrin were incubated with a mouse antibody against nephrin, followed by an Alexa Fluor 405–labeled secondary antibody to trigger crosslink and endocytosis of nephrin. After the endocytosis of nephrin, the cells were washed briefly in a low pH buffer (0.2 M acetic acid, 0.5 M NaCl, pH 2.5) to strip the uninternalized anti-nephrin antibody from the surface. To demonstrate successful surface stripping, a set of cells were washed, fixed, and stained with Alexa Fluor 594–conjugated secondary antibody. We saw no staining of residual surface anti-nephrin antibody. For the recycling assay, cells were washed, then incubated with an Alexa Fluor 594–conjugated secondary antibody, in the presence of different agents (Ciliobrevin D, Ricolinostat, DMSO used as a vehicle control) (Table 4). The recycled nephrin-anti-nephrin complex was chased by the Alexa Fluor 594–conjugated secondary antibody, and the fluorescent images were captured under confocal microscopy after 2 hours of recycling.
Surface Biotinylation–Based Recycling Experiment
The surface biotinylation–based recycling experiment was performed as described in the literature.51Podocytes transfected with pcDNA3-nephrin plasmid were incubated on ice in EZ link sulfo-NHS-SS-biotin (Pierce; Thermo Fisher Scientific) for 45 minutes. Cells were then returned to 37°C, the underwent endocytosis, followed by surface stripping by incubating with 100 mM mesna (Santa Cruz) at 4°C to remove biotin from surface nephrin that was not endocytosed. Cells were returned to 37°C to allow for recycling of the endocytosed nephrin for 2 hours, followed by a second surface stripping with mesna. Cells without the second mesna strip served as protein degradation control. The total nephrin in cell lysates was analyzed by immunoblotting. The biotinylated protein in cell lysates was pulled down by incubation with Streptavidin beads (Pierce) at 4°C overnight with rotating, and biotinylated nephrin in the Streptavidin pulldown was measured by immunoblotting using anti-nephrin.
Puromycin Aminoglycoside Nephropathy in INF2-Transgenic Mice
Puromycin aminoglycoside (PA, Table 4) diluted in normal saline (10 mg/ml) was injected interperitoneally into five littermates of 3 month–old, male, INF2-R218Q knockin (ki) mice (wt/wt, wt/ki, ki/ki) at the dose of 450 mg/kg body weight. Another five age- and sex-matched littermates without PA injection served as controls. Urine specimens were collected on day 0 (D0) (before injection), then D3, D7, D11, D14, and D21 after PA injection. Urine albumin was quantified in duplicate using an Albumin ELISA Quantitation kit according to the manufacturer’s protocol (Bethyl Laboratories Inc.). Plasma and urine creatinine were measured in duplicate using a colorimetric quantification kit (Bioassay Systems). The urine albumin-to-creatinine ratio was calculated for comparison of albuminuria. The urine albumin was also examined by a 10% PAGE gel and Coomassie stain.
Kidney Specimen Handling, Histology, and Quantification
Mice were anesthetized by isoflurane inhalation, followed by perfusion with EZ link sulfo-NHS-SS-biotin per an in vivo surface biotinylation protocol52 before sacrifice.
For light microscopy studies, kidney tissue was fixed in 10% formalin. The paraffinated slides were processed for periodic acid–Schiff and Masson’s trichrome staining. To evaluate glomerulosclerosis and interstitial fibrosis, kidney sections were analyzed by quantifying the ratio of sclerotic glomeruli/total glomeruli, and the ratio of tubulointerstitial area to total section area.53 For transmission electron microscopy evaluation, mouse kidney tissues were immersion fixed in 2.5% glutaraldehyde, 2% paraformaldehyde, in 0.1 M Cacodylate buffer pH 7.4 (modified Karnovsky’s fixative), poststained in 1% osmium tetroxide, dehydrated, and embedded in Epon for thin sectioning. Grids were imaged on a JEOL 1400 transmission electron microscope. We quantified the number of SDs per micrometer, and the fraction of capillary loops covered by healthy appearing podocytes with intact foot processes and SDs.24
Immunofluorescence Staining
Podocytes grown on coverslips were fixed with 4% paraformaldehyde for 15 minutes, followed by a standard immunofluorescent stain. Paraffin-embedded kidney sections were deparaffinized and rehydrated. Antigen was retrieved by incubating the slides in IHC-Tek Epitope Retrieving buffer in a steamer for 45 minutes, followed by standard immunofluorescent stain. The sections were imaged by confocal microscopy.
Bioinformatic Analysis
STRING54 (https://string-db.org/) and GeneMANIA55,56 (https://genemania.org/) were used independently to analyze the protein-protein interaction (PPI) networks on the basis of their functions related to physical interactions, coexpression, colocalization, pathway, genetic interaction, and shared protein domains. Using the molecular function–based weighting method, the GeneMANIA software generated a network on the basis of the 13 CMT-causing genes (including INF2) with Dynll1. Using the STRING algorithm, we generated PPI networks and then categorized them in two subnetworks using K-means clustering.
Statistical Analyses
Data analyses were performed using SPSS version 10.0. Urine albumin-to-creatinine ratio, serum creatinine, histology indices, and the densitometric analysis of immunoblotting using National Institutes of Health (NIH) Image J are expressed as mean±SEM. An independent sample t test was used to compare the difference between two groups. One-way ANOVA was used for comparisons among multiple groups, and a q test was used to compare the difference between groups. In a two-tailed test, P<0.05 was considered significant. The number of replicates for each experiment is shown in the figure legends.
Study Approval
The Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee (IACUC) approved all animal experiments. All work was carried out in accordance with the principles and procedures outlined in the NIH guidelines for the care and use of experimental animals, under the Animal Welfare Assurance #D16–00093 (A3153–01).
Results
FSGS-Causing Mutations Disrupt the Interaction of INF2-DID with Dynll1
A yeast two-hybrid screen using a human cDNA library to find DID-binding partners previously identified Dynll1,21 a key component of the dynein transport complex. By yeast mating, we confirmed the interaction of INF2-DID with Dynll1 and the disruption of this interaction by the FSGS-causing mutations E184K, S186P, and R218Q within the INF2-DID (Figure 1A). In podocytes coexpressing Myc-Dynll1 and Flag-INF2, we used co-IP to confirm the interaction of Dynll1 with INF2 is disrupted by these mutations (Figure 1C). In podocytes coexpressing Myc-Dynll1 and GFP-tagged truncations of INF2-DID (amino acid 1–547), delta-DID (amino acid 548–1249), or full-length INF2 (amino acid 1–1249),25 we confirmed the interaction of INF2 with Dynll1 is via the DID (Figure 1D). The interaction of endogenous INF2 and Dynll1 was confirmed in mouse kidney lysates by co-IP (Figure 1B).
Figure 1.
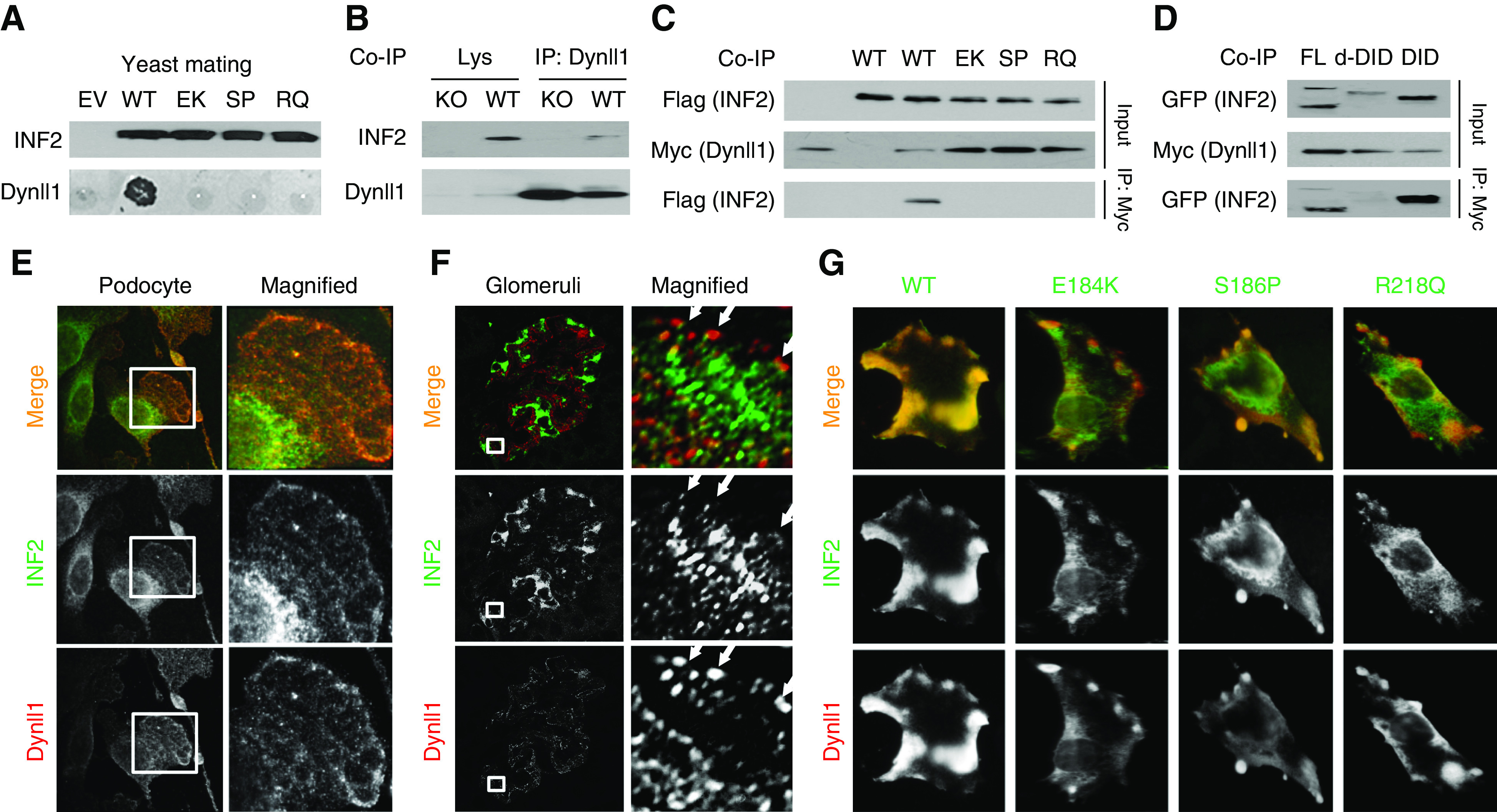
INF2 interacts with Dynll1. (A) The interaction of INF2-DID and Dynll1 is disrupted by FSGS-causing mutations (E184K, S186P, R218Q), as demonstrated by yeast mating. (B) An endogenous interaction of INF2 and Dynll1 in mouse kidney lysates (WT) was confirmed by co-IP, using kidney lysate of INF2 knockout (KO) mice as a negative control. (C) Mouse podocytes were cotransfected with flag full–length INF2 and Myc-Dynll1. The cell lysates immunoprecipitated with anti-Myc (Dynll1) were subjected to immunoblotting with anti-Flag (INF2). (D) Mouse podocytes were cotransfected with Myc-Dynll1 and GFP FL, d-DID, or DID of INF2. The cell lysates were immunoprecipitated with Myc (Dynll1) and subjected to immunoblotting with anti-GFP (INF2–d-DID or INF2-DID truncations). Coimmunofluorescent stain of INF2 and Dynll1 in WT mouse podocytes (E) and mouse kidney (F) showed a limited focal colocalization at the edge of podocytes (arrows). (G) Coimmunostaining of overexpressed Dynll1 and INF2 showed a decreased colocalization in cells expressing mutant INF2. EV, empty vector; EK, E184K; SP, S186P; RQ, R218Q; KO, knockout; FL, full length; d-DID, delta-DID.
Immunostaining of INF2 in cultured podocytes and mouse glomeruli showed an endoplasmic reticulum pattern in podocytes, with a small portion colocalizing with Dynll1 at the periphery of podocytes (Figure 1, E and F). In podocytes coexpressing Dynll1 and INF2, we observed a lamellipodial colocalization of Dynll1 with WT INF2, whereas there was only limited colocalization of Dynll1 with mutant INF2 at focal aggregates beneath the peripheral membrane (Figure 1G).
Bioinformatic Analysis of CMT-Enriched Genes Suggests a Role for INF2 in Dynein-Mediated Transport
Mutations in INF2 have also been recently recognized as a cause of CMT,16,17 a neurodegenerative disease with known genetic defects in dynein and proteolysis.37 To further elucidate mechanisms of INF2-mediated diseases, we used bioinformatic tools to explore the association among genes implicated in autosomal-dominant forms of CMT.
Using the molecular function–based weighting method, GeneMANIA55,56 generated a network showing the 13 CMT-causing genes including INF2 (ARHGEF10, DCTN1, DCTN2, DNM2, DYNC1H1, FGD4, INF2, KIF1B, KIF5A, NEFH, NEFL, PLEKHG5, RAB7A, http://ncbi.nlm.gov/books/NBK1358) and Dynll1 form PPI networks with functional associations related to cytoplasmic dynein, cytoskeletal-based transport, organelle fission, and surface protein presenting. This network supported physical interactions or functional connections of INF2 with dynein components (DCTN1/2, DYNC1H1, DYNC1LI2) and endocytosis machinery (DNM2) (Figure 2B). Using the STRING algorithm,54 we generated PPI networks with a significant enrichment (P<0.001).54 As shown in Figure 2C, we identified two subnetworks using K-means clustering, characterized by dynein-/microtubule-based lysosome transport, and small GTPase-/actin-/dynamin-related endocytosis, suggesting INF2 may participate in dynein-mediated, microtubule-dependent transport related to protein degradation.
Figure 2.
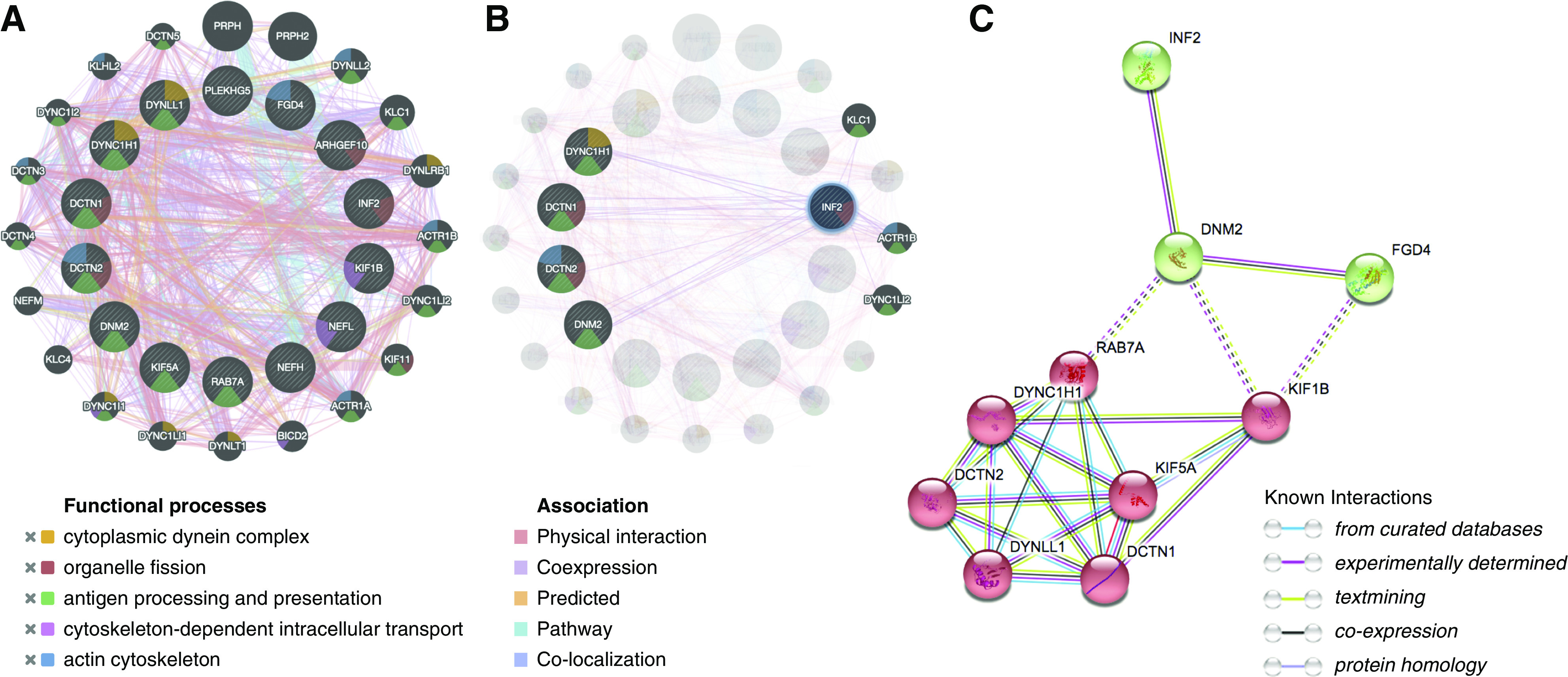
Bioinformatic analysis suggests INF2 participates in the dynein trafficking pathway. (A and B) PPI generated in GeneMANIA (13 CMT-causing genes ARHGEF10, DCTN1, DCTN2, DNM2, DYNC1H1, FGD4, INF2, KIF1B, KIF5A, NEFH, NEFL, PLEKHG5, RAB7A, and Dynll1 are drawn in the inner cycle, whereas 20 predicted genes by the molecular function–based weighting method are located in the outer circle. Distinct colors of the network edge indicate the bioinformatics methods applied: physical interactions, coexpression, predicted, colocalization, and pathway. The different colors used for the nodes indicate the biologic functions of the sets of enrichment genes: cytoplasmic dynein (orange), cytoskeletal-based intracellular transport (purple), organelle fission (red), and surface protein processing and presenting (green). (C) STRING generated a PPI with significant enrichment (PPI enrichment P<0.001). The network was further categorized into two subnetworks (in red and in green) by K-means clustering. The intercluster edges are represented by dashed lines.
INF2 R218Q Enhances Dynein-Mediated Postendocytic Sorting of Nephrin
We used a model of antibody-mediated nephrin crosslink and endocytosis to study the trafficking of nephrin in podocytes with different INF2 genotypes2,44–46 (Figure 3A).
Figure 3.
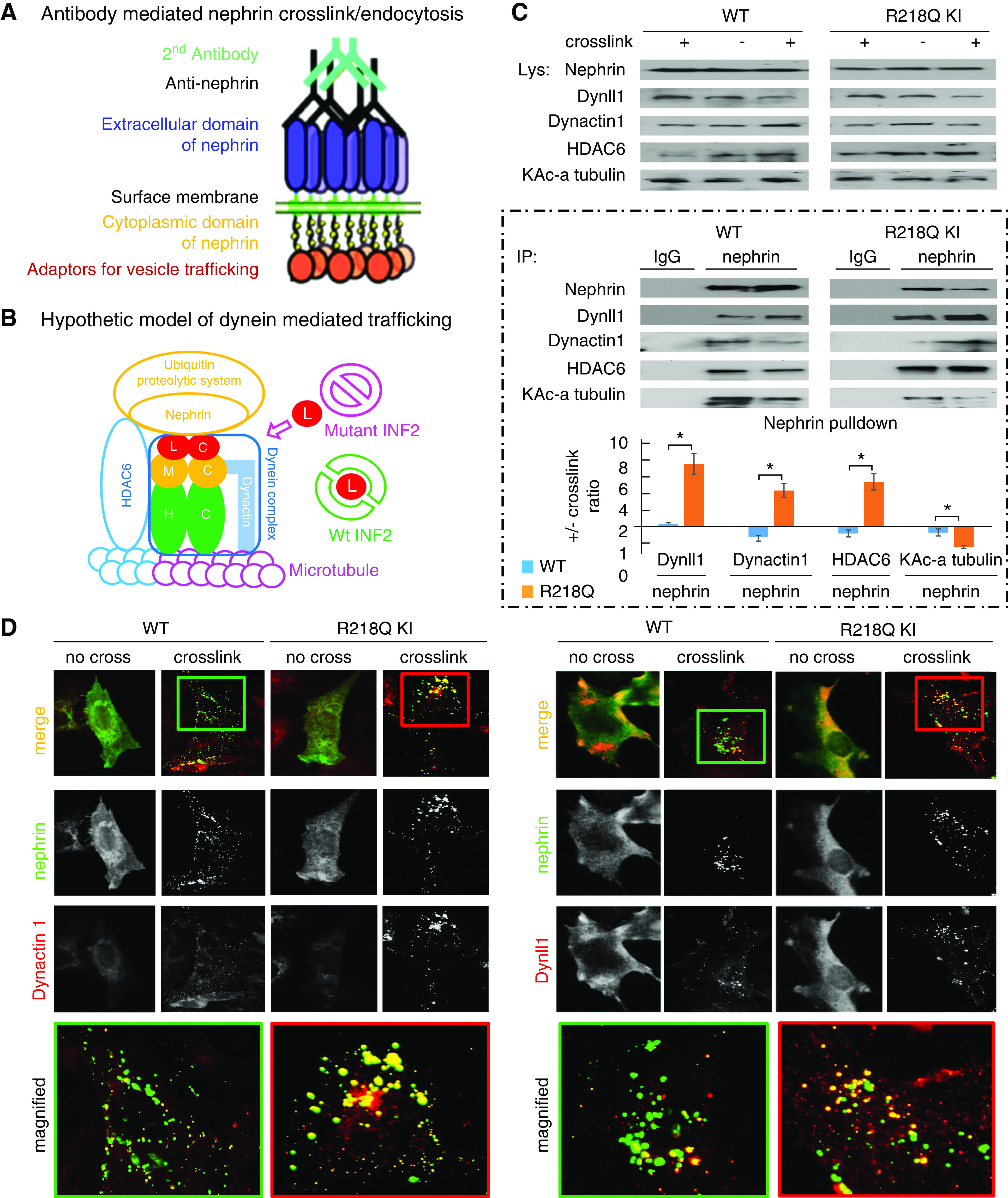
Enhanced dynein involvement in postendocytic sorting of nephrin. (A) Schematic of antibody-mediated nephrin crosslink and endocytosis. Podocytes overexpressing nephrin are incubated in media containing anti-nephrin antibody followed by Alexa Fluor 488–labeled secondary antibody to induce crosslinking and endocytosis of nephrin. (B) Hypothetic schematic model of dynein-mediated nephrin trafficking along the microtubule, coupled to UPS by the bridging of HDAC6. (L, Dynll1; M, dynein intermediate chains; H, dynein heavy chains; blue tubulin, deacetylated alpha tubulin; pink tubulin, acetylated alpha tubulin). (C) WT podocytes or R218Q KI podocytes with or without antibody-mediated nephrin crosslink were lysed and subjected to IP with anti-nephrin (using IgG as a negative control). The Dynll1, Dynactin 1, HDAC6, and the Lysine acetylated alpha tubulin in nephrin pulldown were analyzed by immunoblotting, quantified in Image J, and normalized to nephrin. The ratios of these normalized levels in cells with crosslink to those without crosslink were calculated. For statistical analysis, ratios of normalized Dynactin 1, HDAC6, and the Lysine acetylated alpha tubulin in cells +/- crosslink from three independent experiments were compared using independent sample t test. *P<0.05 R218Q versus WT. (D) Immunofluorescent-based colocalization illustrated the recruitment of Dynactin 1 and Dynll1 (red) to endocytosed nephrin (green) after antibody-mediated crosslinking.
In podocytes with (+) or without (-) anti-nephrin–mediated crosslinking, the recruitment of dynein components (Dynll1 and Dynactin 1) to nephrin was demonstrated by immunofluorescent labeling and localization assays (Figure 3D) and quantified by co-IP (Figure 3C). Compared with the WT podocytes (wt/wt), we demonstrated significant recruitment of Dynll1 and Dynactin 1 to crosslinked nephrin (Figure 3C) and enhanced colocalization of Dynll1 and Dynactin 1 to the endocytosed nephrin (Figure 3D) in podocytes with INF2-R218Q knockin (KI, ki/ki), suggesting the R218Q mutation of INF2 enhances the dynein-mediated postendocytic processing of nephrin. Additionally, we found this is associated with increased recruitment of HDAC6 and decreased KAc-alpha-tubulin to nephrin in R218Q KI cells. HDAC6 is a known partner of the dynein complex32,39,40,57 and a major deacetylase of the KAc-alpha tubulin.58 Therefore, these findings indicate that R218Q augmented the recruitment of HDAC6 with the dynein complex to the endocytosed nephrin, which subsequently deacetylates the alpha-tubulin and influences the efficiency of dynein-mediated, microtubule-based trafficking.
INF2 R218Q Mutant Causes Enhanced Dynein-Mediated Trafficking that Impairs Nephrin Recycling
We performed time-lapse imaging and single-particle tracking to assess the dynamic mediated trafficking of nephrin. Using TrackMate59 plugins of Fiji software (Figure 4A) to display the real-time velocity of the nephrin tracks in different colors (warm color = high velocity, cold color = low velocity), we demonstrated a much more static recycling traffic in R218Q KI cells compared with the WT cells, which was improved by incubation with Ciliobrevin D, a selective dynein inhibitor.60 Using KymographClear and KymographDirect,61 we found far fewer recycling events (Figure 4B) and much slower particle velocity (Figure 4C) in R218Q KI cells compared with the WT cells. This difference was rescued by incubation with Ciliobrevin D. These findings suggest the impaired recycling of nephrin caused by R218Q mutation is mediated through enhanced dynein-mediated retrograde trafficking that opposes the recycling force.
INF2 R218Q Mutant Diverts Endocytosed Nephrin from Recycling to the Degradation Pathway
To further investigate how the R218Q mutant alters the postendocytic sorting of nephrin, we compared the colocalization of nephrin with Rab 5 (an early endosome marker), Rab7 (a late endosome-lysosome marker), or Rab11 (a marker of recycling endosomes) in WT and R218Q KI cells. As shown in Figure 5, we found prominent Rab11 colocalization with endocytosed nephrin in WT cells, indicating a high baseline recycling of endocytosed nephrin. However, in R218Q KI cells, endocytosed nephrin showed greater colocalization with Rab7, suggesting that the R218Q mutation switches the postendocytic sorting of nephrin from recycling to degradation pathway. This is reflected by increased ubiquitination of the endocytosed nephrin. As a bridge between the dynein transport complex and the UPS, we noticed an enrichment of HDAC6 with endocytosed nephrin in R218Q KI cells, consistent with the hypothesis that a dynein mediated transport disinhibited by the R218Q mutation can divert the endocytosed nephrin to the UPS, leaving less nephrin to be recycled.
Figure 5.
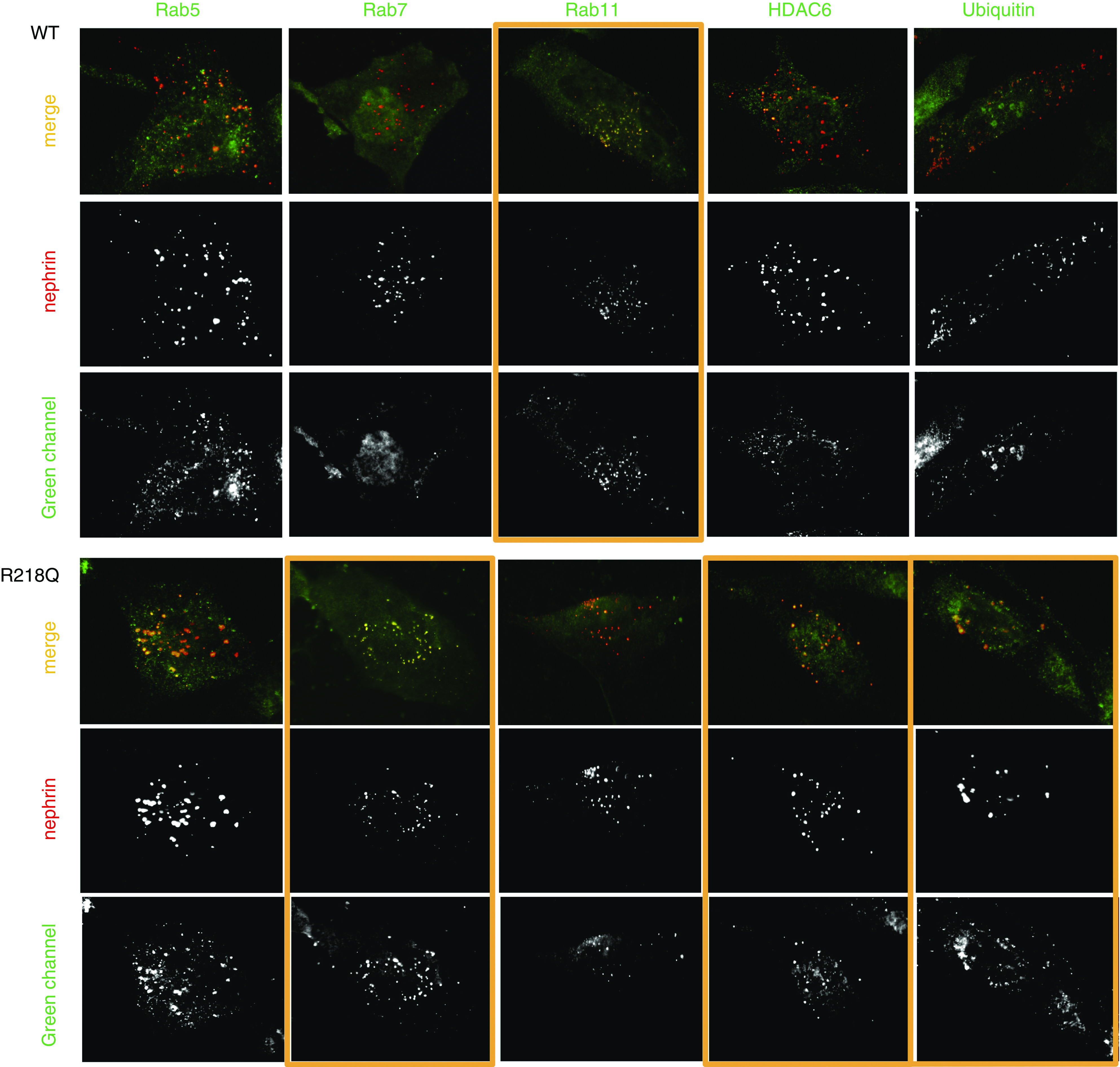
Immunostaining. Coimmunostaining of the nephrin signalosome (red) with Rab 5, Rab7, Rab 11, HDAC6, and ubiquitin (green) in WT podocytes and R218Q KI cells.
Targeting the Dynein-Mediated Trafficking Pathway Rescues the Altered Nephrin Recycling in R218Q KI Cells
We used a fluorescent-based recycling assay (Figure 6A)48 to observe nephrin recycling in cells with different interventions, which we then quantified using a surface biotinylation–based recycling assay (Figure 6C).51 We revealed an impaired recycling of nephrin in R218Q KI cells, which can be rescued by antagonizing the dynein-mediated trafficking. Specifically, the defective nephrin recycling in R218Q KI cells was improved by either coexpression of dominant negative dynactin 1 (DN DCTN147 or incubation with Ciliobrevin D), supporting the model that an uncontrolled dynein-mediated transport is responsible for the impaired nephrin recycling in R218Q KI cells. Altered nephrin recycling was rescued by knocking down Dynll1 (siRNA), demonstrating the availability of Dynll1 limits the activity of dynein-mediated nephrin transport; it was also rescued by coexpression of WT INF2-DID, consistent with the hypothesis that the interaction between INF2-DID and Dynll1 acts to sequester Dynll1, and thereby limits dynein-mediated processes. The HDAC6 inhibitor Ricolinostat62,63 rescues nephrin recycling, suggesting that HDAC6-dependent coupling of the dynein transport complex with the UPS plays an important role in INF2 R218Q–related podocytopathy with impaired surface recycling of nephrin.
Figure 6.
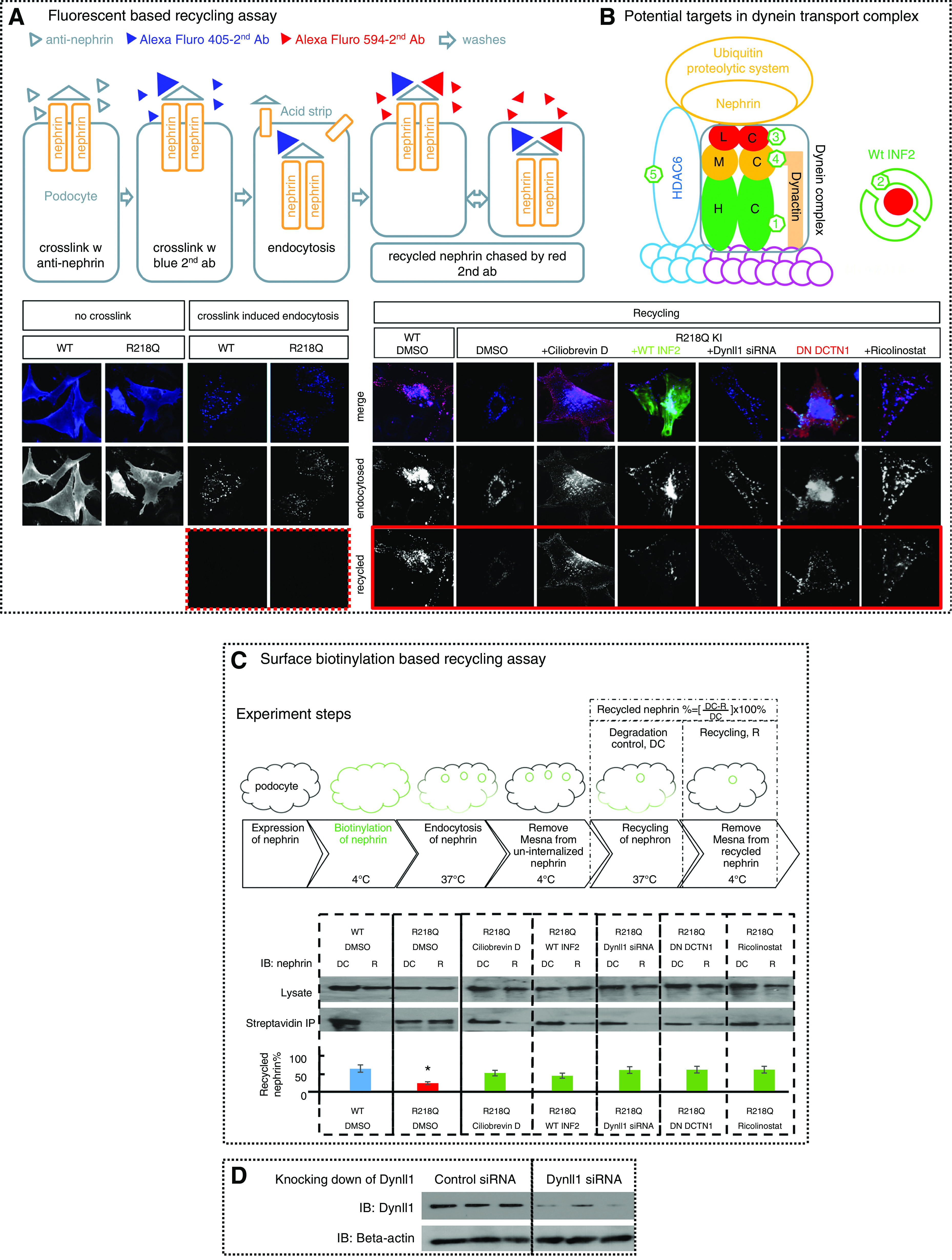
Antagonizing dynein trafficking pathway rescued impaired-nephrin recycling in R218Q KI cells. Nephrin recycling in WT and R218Q KI cells was visualized using (A) a fluorescent-based nephrin recycling assay and (C) quantified by surface biotinylation–based recycling assay. (A) As shown schematically, nephrin expressed on podocytes was crosslinked by anti-nephrin antibody, followed by Alexa Fluor 405–labeled second antibody. After endocytosis and acid strip, cells stained with Alexa Fluor 594–labeled second antibody showed successful removal of uninternalized anti-nephrin (dotted frame). Then the recycled nephrin was chased by Alexa Fluor 594–labeled second antibody (Alexa Fluor 488–labeled second antibody in cells transfected with DesRed DN-DCTN1) (solid frame). (B) A schematic of potential targets in the dynein transport complex. (1) Ciliobrevin D that inhibits the ATPase of the heavy chain (HC); (2) overexpression of GFP-WT INF2-DID that binds to and trap Dynll1; (3) Dynll1 siRNA; (4) dominant negative dynactin 1 (DesRed-DN-DCTN1); and (5) HDAC6 inhibitor (Ricolinostat). (C) Schematics of surface biotinylation–based recycling assay: cells went through a workflow from surface biotinylation of the surface nephrin, endocytosis, and recycling of the biotinylated nephrin, incubation with mesna to strip biotin off the recycled nephrin. Cells collected before (degradation control, DC) and after the mesna strip (recycling, R) were lyzed. The biotinylated nephrin in DC and in R were analyzed by streptavidin IP and normalized to total nephrin. The recycled nephrin is quantitated as biotinylated nephrin ([DC-R]/DC) ×100%. Data were collected from three independent experiments and analyzed using one-way ANOVA, and the differences between groups were analyzed by SNK q test. *P<0.05 versus WT control (DMSO). The impaired nephrin recycling in R218Q KI cells was rescued by coexpression of dominant negative dynactin 1(DN DCTN1), Dynll1 siRNA, WT INF2-DID, Ciliobrevin D (50 μM), or Ricolinostat (5 μM). (D) Knocking down of Dynll1 in R218Q KI cells using Dynll1 siRNA was demonstrated by western blotting, compared with cells treated with control siRNA with no targetable mRNA (sequences of these siRNA duplexes are listed in Methods).
PA Nephropathy in R218Q KI Mice
Having identified enhanced dynein-mediated postendocytic trafficking and degradation of nephrin as a cause of INF2 R218Q–mediated podocyte injury in vitro, we sought to identify pathologic insults that challenge this intrinsic alteration and trigger a proteinuric phenotype in INF2 R218Q KI mice generated on a C57BL/6 background, which have no obvious nephrosis phenotype when examined grossly, histologically, and ultrastructurally.24
We challenged INF2 R218Q KI mice with intraperitoneal injection of PA and successfully induced PA nephropathy (PAN).64,65 Compared with the WT littermates, both heterozygous and homozygous KI mice developed significant proteinuria (Figure 7, A and B) and elevated serum creatinine (Figure 7C) after PA injection.
Figure 7.
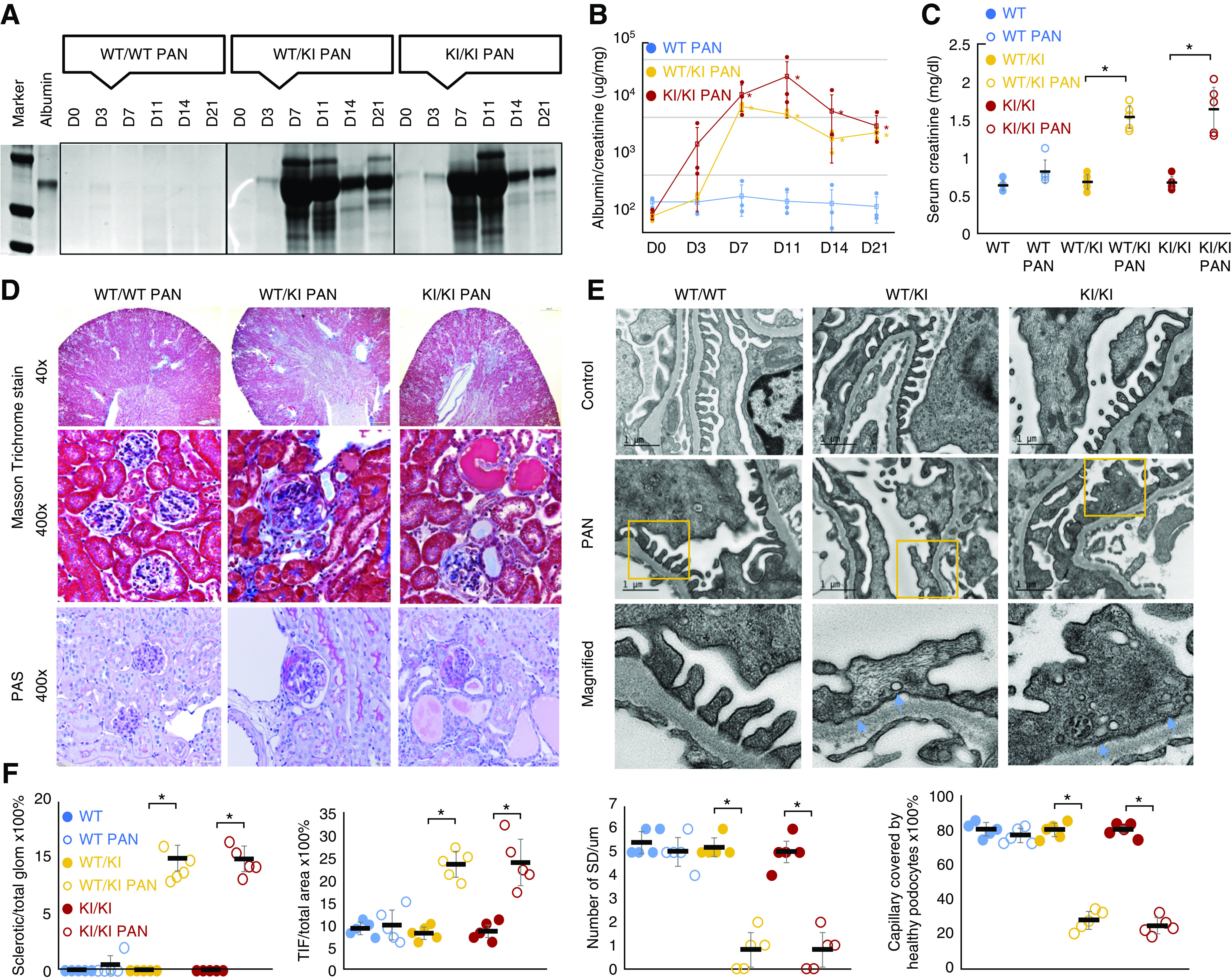
PAN of R218Q KI mice. PA-induced proteinuria in R218Q KI mice (wt/ki and ki/ki, but not in wt/wt mice) as demonstrated by Coomassie blue stain of a urine gel (A). (B) The trend of urine albumin to creatinine ratio in PAN of INF2-transgenic mice. Data were analyzed using repeated-measures ANOVA and the differences among wt/wt, wt/ki, and ki/k mice were analyzed by SNK q test. P<0.05 versus wt/wt mice at the same time point (n=3 for each genotype). (C) Kidney function was assessed by measured serum creatinine levels in blood samples collected at the time of animal sacrifice (D21). Data were analyzed using independent sample t test (n=5). P<0.05, PAN versus control mice of the same genotype without PA injection. (D) Masson trichrome and periodic acid–Schiff stain of mouse kidney sections showed PA induced segmental and global glomerulosclerosis in R218Q KI mice, and increased tubulointerstitial fibrosis. (E) Electron microscopy showed podocytopathy of PAN in R218Q KI mice characterized by foot process effacement with clusters of active vesiculation (orange arrows). (F) The glomerulosclerosis was quantified as the ratio of sclerotic glomeruli/total glomeruli, and tubulointerstitial fibrosis (TIF) was quantified as the ratio of TIF area to the whole section area. The podocytopathy was quantified by measuring the number of SDs per μm and capillary length covered by healthy appearing podocytes with intact foot processes and SDs (G). Data were analyzed using independent sample t test (n=5). P<0.05 PAN versus control mice of the same genotype without PA injection.
Histologically, PA induced FSGS in R218Q KI mice but not the WT mice (Figure 7D). By quantifying the percentages of sclerotic glomeruli and tubulointerstitial area,24,53 we demonstrated significant glomerulosclerosis and tubulointerstitial fibrosis in R218Q KI mice treated with PA, compared with control mice of the same genetic background (Figure 7F). Ultrastructurally, PA induced podocytopathy in R218Q KI mice, characterized by foot process effacement, microvillus formation, and unusual clusters of vesicles at the site of effacing foot processes (Figure 7, E and F), which may reflect active endocytosis.
Altered Dynein-Mediated Trafficking and Degradation of Nephrin in PAN of R218Q KI Mice
As shown in Figure 8A, we demonstrated that PA injection induced an increase in nephrin-bound Dynll1 and ubiquitinated nephrin examined by co-IP, and decreased surface nephrin measured by in vivo surface biotinylation assay in R218Q KI mice (both hetero- and homozygous). By immunofluorescent staining, we found that PA treatment enhanced the distribution of Dynll1 along the GBM, with increased colocalization with nephrin, which was more prominent in R218Q KI mice than in the WT littermates (Figure 8B). This pattern is consistent with our in vitro finding of enhanced recruitment of the dynein-transport complex to the endocytosed nephrin. In sclerosing glomeruli of the R218Q KI mice treated with PA, we observed increased Dynll1 and decreased nephrin along the GBM (Figure 8C).
Figure 8.
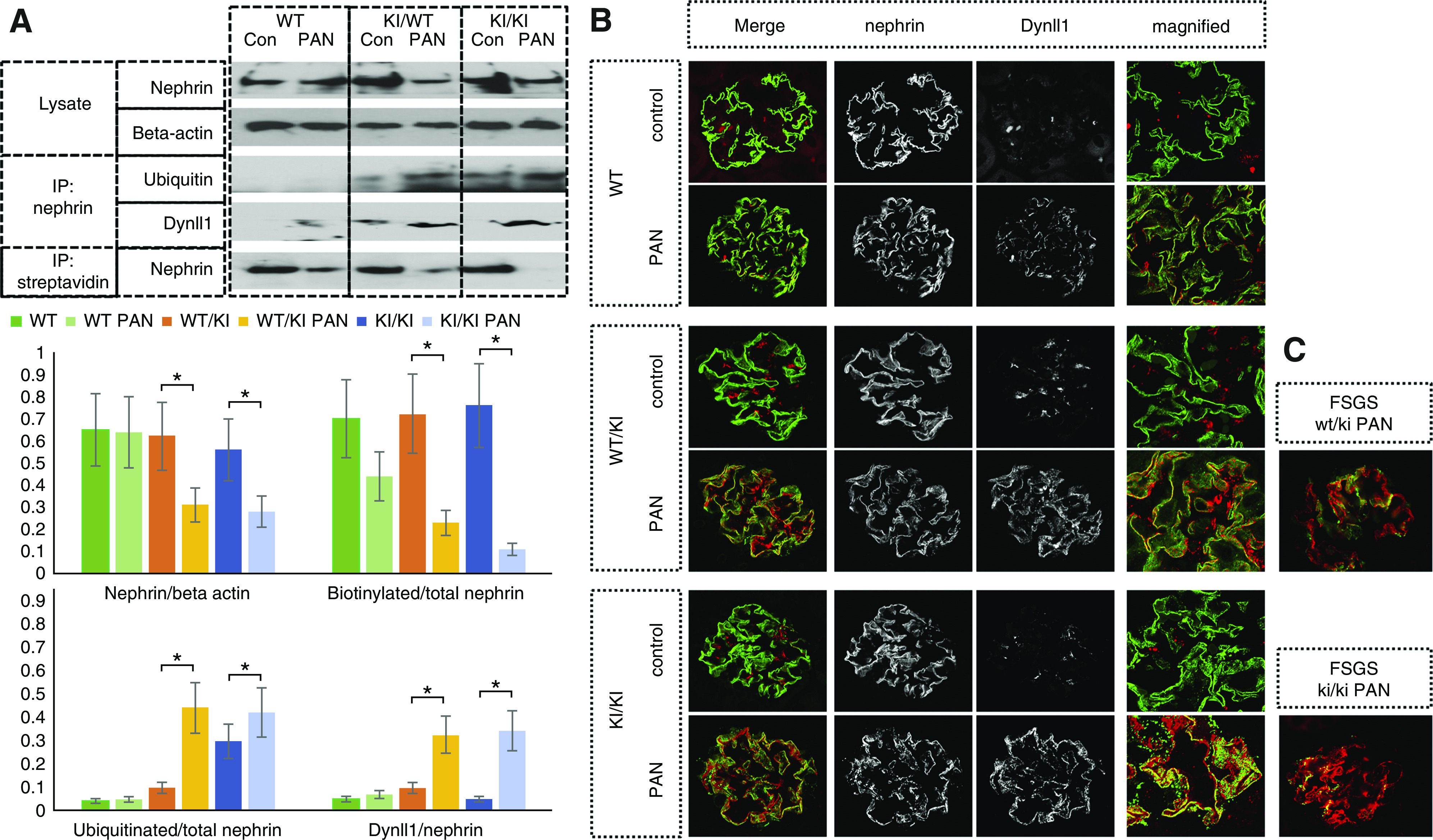
Enhanced dynein-mediated trafficking and degradation of nephrin in PAN model in R218Q KI mice. (A) Dynll1 and ubiquitinated nephrin were detected in the nephrin pulldown of kidney lysates by immunoblotting, and biotinylated nephrin was detected in streptavidin pulldown. Semiquantitative analysis was performed to compare Dynll1/total nephrin pulldown, biotinylated nephrin/total nephrin, ubiquitinated nephrin/total nephrin in mice (wt/wt, wt/ki and ki/ki), with PA or without PA injection using an independent sample t test (n=5). P<0.05 PAN versus control mice of the same genotype without PA injection. (B) Immunofluorescent stain showed increased colocalization of Dynll1 with nephrin in PAN of R218 KI mice (wt/ki and ki/ki) compared with the wt/wt mice. (C) Increased Dynll1 and decreased nephrin expression at the GBM of sclerosing glomeruli of the wt/ki and ki/ki mice with PAN.
Discussion
Our finding that INF2-related FSGS is characterized by a podocytopathy with mistrafficked SD proteins led to the hypothesis that INF2 regulates vesicle trafficking in podocytes. This is supported by our finding that INF2 is involved in closely coordinated Rho-mediated, actin-dependent, and dynein-mediated, microtubule-dependent vesicle trafficking. The regulatory function of INF2 is disrupted by pathogenic mutations, which leads to mistrafficking of SD proteins and disintegrity of the glomerular-filtration barrier.
Our previous work demonstrated that INF2 maintains the cortical actin mesh beneath the peripheral membrane by antagonizing Rho/mDia signaling. Pathogenic mutations disrupt this regulatory function of INF2, leading to the loss of cortical actin supporting the peripheral membrane where the SD proteins reside.22 This partially explains how INF2 might regulate the actin-dependent trafficking of the SD protein. However, the disease phenotype observed in podocytes harboring pathologic INF2 mutations was not fully rescued by inhibiting the Rho/mDia signaling,22,23 redirecting our attention to the dynein-mediated, microtubule-dependent trafficking pathway. In contrast to the actin-based endocytosis near plasma membranes,66,67 long-distance postendocytic sorting of cargo proteins dependent on microtubules40,68 remains to be elucidated. As in neuron and dendritic cells, microtubules form the backbone of podocyte primary processes that transfer cargo between the terminal foot processes and the soma of the cells. Evidence supports the notion that it is mediated by the antiparallel transport motors, kinesin and dynein. Previous work has shown that kinesin family proteins mediate anterograde (from soma to periphery) flow, supplying elements required for the elongation and growth of foot processes and membrane proteins such as nephrin for the integrity of the SD.69 The recruitment of dynein components to the SD during proteinuric injuries indicates a prevailing retrograde trafficking activity accompanying the contraction of the injured leading edge of cells.30 These pieces of evidence support our model that the dynein components normally residing at the cortex of cells are segregated from microtubules by the cortical actin mesh that is maintained by WT INF2; on the contraction of the cortical actin mesh induced by signals such as activation of Rho,22 dynein can access and facilitate microtubule-dependent membrane invaginations and initiate the retrograde trafficking from the periphery to the soma of cells.68 This model is also supported by studies in drosophila that demonstrated Exc6, the Drosophila orthologous homolog of INF2, coordinates actin and microtubules at the leading edge of epithelial cells. Moreover, this study found that disinhibited pathogenic INF2 mutations facilitate retrograde trafficking by counteracting kinesin-mediated anterograde trafficking.70
We demonstrated that INF2-DID interacts with Dynll1, an essential component for the integrity of the whole dynein complex; sequestration of Dynll1 can limit the activity of the dynein transport pathway.71,72 Thus, we hypothesize that INF2 limits the dynein-mediated trafficking of SD protein by binding to and sequestering Dynll1. The finding that pathogenic mutations within the DID of INF2 can disrupt the interaction with Dynll1 suggests these mutations alter INF2’s effect on dynein and cause enhanced dynein-mediated trafficking.
To test this hypothesis, in an in vitro nephrin-trafficking model, we demonstrated increased dynein recruitment to endocytosed nephrin in podocytes harboring the INF2 mutation R218Q, a well-described pathogenic mutation that disrupts the INF2/Dynll1 interaction. Then, using time-lapse imaging, we found a significant decrease in recycling events in R218Q KI cells, which was rescued by antagonizing dynein activity using Ciliobrevin D. Furthermore, knocking down Dynll1, or overexpressing WT INF2-DID that binds to and sequesters Dynll1 seemed to reduce the dynein-mediated trafficking and rescue nephrin recycling in R218Q KI cells. This finding supports the hypothesis that the restriction of Dynll1 by INF2 is a key to its regulation on dynein-mediated trafficking.
Further, we found a high baseline recycling of nephrin in WT cells, as reflected by significant nephrin localization in Rab11-positive recycling endosomes. The increased Rab7 labeling and less Rab11 labeling of nephrin in R218Q KI cells indicated that the R218Q mutation switched nephrin trafficking toward the lysosomal degradation pathway. HDAC6 binds to both ubiquitin and dynein, and bridges dynein-mediated cargo transport to UPS.32,39,41,73 The increased colabeling of endocytosed nephrin in R218Q KI cells with dynein, Rab7, ubiquitin, and HDAC6 may be the result of enhanced recruitment of dynein/HDAC6 complex to endocytosed nephrin, which then shuttles nephrin to the UPS of the lysosome. Taken together, the disruption of the INF2-Dynll1 interaction by R218Q impairs the ability of INF2 to modulate the dynein-transport pathway, which subsequently diverts endocytosed nephrin from the recycling pool to the degradation pathway, and ultimately resulting in inadequate recovery of surface nephrin. This model is supported by rescue of nephrin surface recycling in R218Q KI cells by interfering with different components of dynein transport complex, or by inhibiting HDAC6.
Do INF2 mutations affect dynein-mediated transport and recycling of nephrin in vivo? To answer this question, we used PA to challenge the intrinsic dysregulation of dynein-mediated trafficking of nephrin caused by INF2 R218Q mutation, and for the first time induced proteinuric podocytopathy and FSGS in R218Q KI mice.
PA is a podocyte toxin that causes nephropathy in rats. It remains puzzling why compared with rats, mice are more resistant to PAN. Although there has not been comparative genetic research to delineate this species-specific predisposition/resistance to glomerular disease, congenic studies in mice revealed that compared with BALB/c and FVB strains, C57BL/6 mice possess a gene expression regulatory network that increases the transcription of podocin, a molecule facilitating the docking of nephrin in the lift raft rich microdomain of SD. Therefore, C57BL/6 mice potentially have “stronger podocytes” with a higher reserve in the face of insults, such as in PAN.74–76 This different susceptibility to podocytopathy in hosts with different genetic backgrounds may help explain the clinical heterogeneity of families with kidney disease even caused by identical INF2 mutations: variable age of onset, clinical features, and course of kidney disease. Despite this resistance, PA has been used to facilitate the development of podocytopathy in certain susceptible transgenic mice models64,65 by augmenting nephrin endocytosis and upregulating the UPS in podocytes.77,78 On the basis of our in vitro findings, compared with the WT INF2, the R218Q mutant is more able to shunt the endocytosed and ubiquitinated nephrin induced by PA toward the ultimate path to degradation. Therefore, we speculate that the development of PAN in R218Q KI mice compared the WT mice is a consequence of PA pushing the dysregulated vesicle trafficking in the R218Q KI podocytes past some threshold needed to develop proteinuric podocytopathy in this normally resistant C57BL/6 strain of mice.
We validated in PAN R218Q mouse kidneys that there was an increased recruitment of Dynll1 to nephrin, reflecting enhanced dynein-mediated trafficking of nephrin, associated with increased nephrin ubiquitination and decreased surface expression of nephrin. This model demonstrated that the INF2 R218Q mutation increases the susceptibility of podocytes to PA toxicity by increasing dynein-mediated postendocytic sorting to a degradation pathway, thereby decreasing the functional recycling of SD proteins. Our findings support the notion that the PAN- and R218Q-related podocytopathies arise through this shared pathogenesis. The dynein/HDAC6/UPS is an attractive and targetable pathway in neurodegenerative diseases. We have identified a role for INF2 in regulating the dynein-mediated trafficking of nephrin, and demonstrate that INF2 mutations cause dysregulation of this pathway, which expands our understanding of the mechanism of not only INF2-related kidney disease but perhaps also of CMT. The PAN applied to R218Q KI mice is a genetically faithful model of human FSGS caused by the INF2 R218Q mutant, and therefore a platform to further test whether targeting the dynein/UPS pathway could be an effective therapeutic strategy for INF2-related FSGS.
Disclosures
Because Martin Pollak is an editor of the Journal of the American Society of Nephrology, he was not involved in the peer review process for this manuscript. A guest editor oversaw the peer review and decision-making process for this manuscript. All remaining authors have nothing to disclose.
Funding
This work was supported by National Institutes of Health grants R01DK088826-03 and T32 DK007726 NRSA.
Acknowledgments
We thank Mr. Aniket Gad, Mr. Kyle Smith, Dr. Lay-Hong Ang, Dr. Susan Hagen, and Ms. Suzanne White at Beth Israel Deaconess Medical Center core facilities for their help in the experiments.
Ms. Chandra Perez-Gill was responsible for the animal experiments and Dr. Balajikarthick Subramanian was responsible for the generation of mouse podocyte cell lines. Dr. Hua Sun was responsible for the design and performance of experiments, statistical analysis, and drafting the manuscript. Dr. Johannes Schlöndorff and Dr. Martin R. Pollak were responsible for design of experiments and writing of manuscript. All authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Garg P: A review of podocyte biology. Am J Nephrol 47[Suppl 1]: 3–13, 2018. [DOI] [PubMed] [Google Scholar]
- 2.Martin CE, Jones N: Nephrin signaling in the podocyte: An updated view of signal regulation at the slit diaphragm and beyond. Front Endocrinol (Lausanne) 9: 302, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holzman LB, St John PL, Kovari IA, Verma R, Holthofer H, Abrahamson DR: Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int 56: 1481–1491, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Liu L, Doné SC, Khoshnoodi J, Bertorello A, Wartiovaara J, Berggren PO, et al.: Defective nephrin trafficking caused by missense mutations in the NPHS1 gene: Insight into the mechanisms of congenital nephrotic syndrome. Hum Mol Genet 10: 2637–2644, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Teng B, Schroder P, Müller-Deile J, Schenk H, Staggs L, Tossidou I, et al.: CIN85 deficiency prevents nephrin endocytosis and proteinuria in diabetes. Diabetes 65: 3667–3679, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumont V, Tolvanen TA, Kuusela S, Wang H, Nyman TA, Lindfors S, et al.: PACSIN2 accelerates nephrin trafficking and is up-regulated in diabetic kidney disease. FASEB J 31: 3978–3990, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin CE, Petersen KA, Aoudjit L, Tilak M, Eremina V, Hardy WR, et al.: ShcA adaptor protein promotes nephrin endocytosis and is upregulated in proteinuric nephropathies. J Am Soc Nephrol 29: 92–103, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kampf LL, Schneider R, Gerstner L, Thünauer R, Chen M, Helmstädter M, et al.: TBC1D8B mutations implicate RAB11-dependent vesicular trafficking in the pathogenesis of nephrotic syndrome. J Am Soc Nephrol 30: 2338–2353, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorval G, Kuzmuk V, Gribouval O, Welsh GI, Bierzynska A, Schmitt A, et al.: TBC1D8B loss-of-function mutations lead to X-linked nephrotic syndrome via defective trafficking pathways. Am J Hum Genet 104: 348–355, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nihalani D, Solanki AK, Arif E, Srivastava P, Rahman B, Zuo X, et al.: Disruption of the exocyst induces podocyte loss and dysfunction. J Biol Chem 294: 10104–10119, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lipska BS, Iatropoulos P, Maranta R, Caridi G, Ozaltin F, Anarat A, et al.; PodoNet Consortium: Genetic screening in adolescents with steroid-resistant nephrotic syndrome. Kidney Int 84: 206–213, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Gbadegesin RA, Lavin PJ, Hall G, Bartkowiak B, Homstad A, Jiang R, et al.: Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int 81: 94–99, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Büscher AK, Celebi N, Hoyer PF, Klein HG, Weber S, Hoefele J: Mutations in INF2 may be associated with renal histology other than focal segmental glomerulosclerosis. Pediatr Nephrol 33: 433–437, 2018. [DOI] [PubMed] [Google Scholar]
- 14.Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, et al.: Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis [published correction appears in Nat Genet 42: 361, 2010]. Nat Genet 42: 72–76, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR: Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int 83: 316–322, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin S, Wang W, Wang R, Lv H, Zhang W, Wang Z, et al.: INF2 mutations associated with dominant inherited intermediate Charcot-Marie-Tooth neuropathy with focal segmental glomerulosclerosis in two Chinese patients. Clin Neuropathol 34: 275–281, 2015. [DOI] [PubMed] [Google Scholar]
- 17.Boyer O, Nevo F, Plaisier E, Funalot B, Gribouval O, Benoit G, et al.: INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N Engl J Med 365: 2377–2388, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Roos A, Weis J, Korinthenberg R, Fehrenbach H, Häusler M, Züchner S, et al.: Inverted formin 2-related Charcot-Marie-Tooth disease: Extension of the mutational spectrum and pathological findings in Schwann cells and axons. J Peripher Nerv Syst 20: 52–59, 2015. [DOI] [PubMed] [Google Scholar]
- 19.Gurel PS, Ge P, Grintsevich EE, Shu R, Blanchoin L, Zhou ZH, et al.: INF2-mediated severing through actin filament encirclement and disruption. Curr Biol 24: 156–164, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mu A, Fung TS, Kettenbach AN, Chakrabarti R, Higgs HN: A complex containing lysine-acetylated actin inhibits the formin INF2. Nat Cell Biol 21: 592–602, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun H, Schlondorff JS, Brown EJ, Higgs HN, Pollak MR: Rho activation of mDia formins is modulated by an interaction with inverted formin 2 (INF2). Proc Natl Acad Sci U S A 108: 2933–2938, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun H, Schlondorff J, Higgs HN, Pollak MR: Inverted formin 2 regulates actin dynamics by antagonizing Rho/diaphanous-related formin signaling. J Am Soc Nephrol 24: 917–929, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun H, Al-Romaih KI, MacRae CA, Pollak MR: Human kidney disease-causing INF2 mutations perturb Rho/Dia signaling in the glomerulus. EBioMedicine 1: 107–115, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian B, Sun H, Yan P, Charoonratana VT, Higgs HN, Wang F, et al.: Mice with mutant Inf2 show impaired podocyte and slit diaphragm integrity in response to protamine-induced kidney injury. Kidney Int 90: 363–372, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian B, Chun J, Perez-Gill C, Yan P, Stillman IE, Higgs HN, et al.: FSGS-causing INF2 mutation impairs cleaved INF2 N-fragment functions in podocytes. J Am Soc Nephrol 31: 374–391, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rapali P, Szenes Á, Radnai L, Bakos A, Pál G, Nyitray L: DYNLL/LC8: A light chain subunit of the dynein motor complex and beyond. FEBS J 278: 2980–2996, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Asthana J, Kuchibhatla A, Jana SC, Ray K, Panda D: Dynein light chain 1 (LC8) association enhances microtubule stability and promotes microtubule bundling. J Biol Chem 287: 40793–40805, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theerawatanasirikul S, Phecharat N, Prawettongsopon C, Chaicumpa W, Lekcharoensuk P: Dynein light chain DYNLL1 subunit facilitates porcine circovirus type 2 intracellular transports along microtubules. Arch Virol 162: 677–686, 2017. [DOI] [PubMed] [Google Scholar]
- 29.Wilson MJ, Salata MW, Susalka SJ, Pfister KK: Light chains of mammalian cytoplasmic dynein: Identification and characterization of a family of LC8 light chains. Cell Motil Cytoskeleton 49: 229–240, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Pierchala BA, Muñoz MR, Tsui CC: Proteomic analysis of the slit diaphragm complex: CLIC5 is a protein critical for podocyte morphology and function. Kidney Int 78: 868–882, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rayala SK, den Hollander P, Manavathi B, Talukder AH, Song C, Peng S, et al.: Essential role of KIBRA in co-activator function of dynein light chain 1 in mammalian cells. J Biol Chem 281: 19092–19099, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Wang N, Ma Q, Peng P, Yu Y, Xu S, Wang G, et al.: Autophagy and ubiquitin-proteasome system coordinate to regulate the protein quality control of neurodegenerative disease-associated DCTN1. Neurotox Res 37: 48–57, 2020. [DOI] [PubMed] [Google Scholar]
- 33.Jaarsma D, Hoogenraad CC: Cytoplasmic dynein and its regulatory proteins in Golgi pathology in nervous system disorders. Front Neurosci 9: 397, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eschbach J, Dupuis L: Cytoplasmic dynein in neurodegeneration. Pharmacol Ther 130: 348–363, 2011. [DOI] [PubMed] [Google Scholar]
- 35.Chen XJ, Xu H, Cooper HM, Liu Y: Cytoplasmic dynein: A key player in neurodegenerative and neurodevelopmental diseases. Sci China Life Sci 57: 372–377, 2014. [DOI] [PubMed] [Google Scholar]
- 36.Cianfrocco MA, DeSantis ME, Leschziner AE, Reck-Peterson SL: Mechanism and regulation of cytoplasmic dynein. Annu Rev Cell Dev Biol 31: 83–108, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tey S, Ahmad-Annuar A, Drew AP, Shahrizaila N, Nicholson GA, Kennerson ML: Mutation analysis of genes within the dynactin complex in a cohort of hereditary peripheral neuropathies. Clin Genet 90: 127–133, 2016. [DOI] [PubMed] [Google Scholar]
- 38.Reck-Peterson SL, Redwine WB, Vale RD, Carter AP: Publisher Correction: The cytoplasmic dynein transport machinery and its many cargoes. Nat Rev Mol Cell Biol 19: 479, 2018. [DOI] [PubMed] [Google Scholar]
- 39.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP: The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115: 727–738, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Oláh J, Szunyogh S, Szénási T, Szaniszló T, Szabó A, Lehotzky A, et al. Interactions between two regulatory proteins of microtubule dynamics, HDAC6, TPPP/p25, and the hub protein, DYNLL/LC8. Biochim Biophys Acta Mol Cell Res 1866: 118556, 2019 31505170 [DOI] [PubMed]
- 41.Yan J: Interplay between HDAC6 and its interacting partners: Essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol 33: 567–580, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Pinazza M, Ghisi M, Minuzzo S, Agnusdei V, Fossati G, Ciminale V, et al.: Histone deacetylase 6 controls Notch3 trafficking and degradation in T-cell acute lymphoblastic leukemia cells. Oncogene 37: 3839–3851, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keir LS, Firth R, May C, Ni L, Welsh GI, Saleem MA: Generating conditionally immortalised podocyte cell lines from wild-type mice. Nephron 129: 128–136, 2015. [DOI] [PubMed] [Google Scholar]
- 44.Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB: Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest 116: 1346–1359, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blasutig IM, New LA, Thanabalasuriar A, Dayarathna TK, Goudreault M, Quaggin SE, et al.: Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol 28: 2035–2046, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin XS, Tsukaguchi H, Shono A, Yamamoto A, Kurihara H, Doi T: Phosphorylation of nephrin triggers its internalization by raft-mediated endocytosis. J Am Soc Nephrol 20: 2534–2545, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quintyne NJ, Schroer TA: Distinct cell cycle-dependent roles for dynactin and dynein at centrosomes. J Cell Biol 159: 245–254, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blagojević Zagorac G, Mahmutefendić H, Maćešić S, Karleuša L, Lučin P: Quantitative analysis of endocytic recycling of membrane proteins by monoclonal antibody-based recycling assays. J Cell Physiol 232: 463–476, 2017. [DOI] [PubMed] [Google Scholar]
- 49.Perez Bay AE, Schreiner R, Benedicto I, Paz Marzolo M, Banfelder J, Weinstein AM, et al.: The fast-recycling receptor Megalin defines the apical recycling pathway of epithelial cells. Nat Commun 7: 11550, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winterstein C, Trotter J, Krämer-Albers EM: Distinct endocytic recycling of myelin proteins promotes oligodendroglial membrane remodeling. J Cell Sci 121: 834–842, 2008. [DOI] [PubMed] [Google Scholar]
- 51.Crupi MJ, Richardson DS, Mulligan LM: Cell surface biotinylation of receptor tyrosine kinases to investigate intracellular trafficking. Methods Mol Biol 1233: 91–102, 2015. [DOI] [PubMed] [Google Scholar]
- 52.Haase R, Potthoff SA, Meyer-Schwesinger C, Frosch C, Wiech T, Panzer U, et al.: A novel in vivo method to quantify slit diaphragm protein abundance in murine proteinuric kidney disease. PLoS One 12: e0179217, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inoue K, Gan G, Ciarleglio M, Zhang Y, Tian X, Pedigo CE, et al.: Podocyte histone deacetylase activity regulates murine and human glomerular diseases. J Clin Invest 129: 1295–1313, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al.: STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montojo J, Zuberi K, Rodriguez H, Bader GD, Morris Q: GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000 Res 3: 153, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Franz M, Rodriguez H, Lopes C, Zuberi K, Montojo J, Bader GD, et al.: GeneMANIA update 2018. Nucleic Acids Res 46[W1]: W60–W64, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, et al.: Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci 27: 3571–3583, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernández-Barrera J, Alonso MA: Coordination of microtubule acetylation and the actin cytoskeleton by formins. Cell Mol Life Sci 75: 3181–3191, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, et al.: TrackMate: An open and extensible platform for single-particle tracking. Methods 115: 80–90, 2017. [DOI] [PubMed] [Google Scholar]
- 60.Sainath R, Gallo G: The dynein inhibitor Ciliobrevin D inhibits the bidirectional transport of organelles along sensory axons and impairs NGF-mediated regulation of growth cones and axon branches. Dev Neurobiol 75: 757–777, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mangeol P, Prevo B, Peterman EJ: KymographClear and KymographDirect: Two tools for the automated quantitative analysis of molecular and cellular dynamics using kymographs. Mol Biol Cell 27: 1948–1957, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krukowski K, Ma J, Golonzhka O, Laumet GO, Gutti T, van Duzer JH, et al.: HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 158: 1126–1137, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang D, Wang B, Liu Y, Dong X, Su Y, Li S: Protective effects of ACY-1215 against chemotherapy-related cognitive impairment and brain damage in mice. Neurochem Res 44: 2460–2469, 2019. [DOI] [PubMed] [Google Scholar]
- 64.Rabe M, Schaefer F: Non-transgenic mouse models of kidney disease. Nephron 133: 53–61, 2016. [DOI] [PubMed] [Google Scholar]
- 65.Shimo T, Adachi Y, Yamanouchi S, Tsuji S, Kimata T, Umezawa K, et al.: A novel nuclear factor κB inhibitor, dehydroxymethylepoxyquinomicin, ameliorates puromycin aminonucleoside-induced nephrosis in mice. Am J Nephrol 37: 302–309, 2013. [DOI] [PubMed] [Google Scholar]
- 66.Fernandez-Borja M, Janssen L, Verwoerd D, Hordijk P, Neefjes J: RhoB regulates endosome transport by promoting actin assembly on endosomal membranes through Dia1. J Cell Sci 118: 2661–2670, 2005. [DOI] [PubMed] [Google Scholar]
- 67.Chi X, Wang S, Huang Y, Stamnes M, Chen JL: Roles of rho GTPases in intracellular transport and cellular transformation. Int J Mol Sci 14: 7089–7108, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laan L, Pavin N, Husson J, Romet-Lemonne G, van Duijn M, López MP, et al.: Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell 148: 502–514, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kobayashi N, Reiser J, Kriz W, Kuriyama R, Mundel P: Nonuniform microtubular polarity established by CHO1/MKLP1 motor protein is necessary for process formation of podocytes. J Cell Biol 143: 1961–1970, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shaye DD, Greenwald I: The disease-associated formin INF2/EXC-6 organizes lumen and cell outgrowth during tubulogenesis by regulating F-actin and microtubule cytoskeletons. Dev Cell 32: 743–755, 2015. [DOI] [PubMed] [Google Scholar]
- 71.Varma D, Dawn A, Ghosh-Roy A, Weil SJ, Ori-McKenney KM, Zhao Y, et al.: Development and application of in vivo molecular traps reveals that dynein light chain occupancy differentially affects dynein-mediated processes. Proc Natl Acad Sci U S A 107: 3493–3498, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Batlevi Y, Martin DN, Pandey UB, Simon CR, Powers CM, Taylor JP, et al.: Dynein light chain 1 is required for autophagy, protein clearance, and cell death in Drosophila. Proc Natl Acad Sci U S A 107: 742–747, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cristofani R, Crippa V, Rusmini P, Cicardi ME, Meroni M, Licata NV, et al.: Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 13: 1280–1303, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnstone DB, Ikizler O, Zhang J, Holzman LB: Background strain and the differential susceptibility of podocyte-specific deletion of Myh9 on murine models of experimental glomerulosclerosis and HIV nephropathy. PLoS One 8: e67839, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Papeta N, Zheng Z, Schon EA, Brosel S, Altintas MM, Nasr SH, et al.: Prkdc participates in mitochondrial genome maintenance and prevents Adriamycin-induced nephropathy in mice. J Clin Invest 120: 4055–4064, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Papeta N, Chan KT, Prakash S, Martino J, Kiryluk K, Ballard D, et al.: Susceptibility loci for murine HIV-associated nephropathy encode trans-regulators of podocyte gene expression. J Clin Invest 119: 1178–1188, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tojo A, Hatakeyama S, Kinugasa S, Fukuda S, Sakai T: Enhanced podocyte vesicle transport in the nephrotic rat. Med Mol Morphol 50: 86–93, 2017. [DOI] [PubMed] [Google Scholar]
- 78.Beeken M, Lindenmeyer MT, Blattner SM, Radón V, Oh J, Meyer TN, et al.: Alterations in the ubiquitin proteasome system in persistent but not reversible proteinuric diseases. J Am Soc Nephrol 25: 2511–2525, 2014. 24722446 [Google Scholar]