

Abstract
Hypertension and its comorbidities pose a major public health problem associated with disease-associated factors related to a modern lifestyle, such high salt intake or obesity. Accumulating evidence has demonstrated that aldosterone and its receptor, the mineralocorticoid receptor (MR), have crucial roles in the development of salt-sensitive hypertension and coexisting cardiovascular and renal injuries. Accordingly, clinical trials have repetitively shown the promising effects of MR blockers in these diseases. We and other researchers have identified novel mechanisms of MR activation involved in salt-sensitive hypertension and renal injury, including the obesity-derived overproduction of aldosterone and ligand-independent signaling. Moreover, recent advances in the analysis of cell-specific and context-dependent mechanisms of MR activation in various tissues—including a classic target of aldosterone, aldosterone-sensitive distal nephrons—are now providing new insights. In this review, we summarize recent updates to our understanding of aldosterone-MR signaling, focusing on its role in salt-sensitive hypertension and renal injury.
Keywords: mineralocorticoid receptor, aldosterone, pendrin, obesity, salt, hypertension
Aldosterone, secreted from adrenal glands by stimuli including angiotensin II (AngII) or hyperkalemia, binds to the mineralocorticoid receptor (MR) expressed on epithelial cells in the aldosterone-sensitive distal nephron (ASDN) to regulate sodium and potassium flux.1–3 According to the theory that renal handling of sodium is a key determinant of fluid volume and BP control,4,5 MR signaling in the connecting tubule and collecting duct is essential for the regulation of BP, especially in the development of salt-sensitive hypertension.6 Mutations in aldosterone synthase in the MR or in a major target of MR signaling, the epithelial sodium channel (ENaC), cause genetic disorders with hypertension or hypotension.7 Higher aldosterone levels within the physiologic range were associated with hypertension in a community-based study.8 Moreover, aldosterone administration in canines shifted the pressure-natriuresis curve, indicating salt-sensitive hypertension.9
In addition to the ASDN, MR is expressed in other tissues, including in the renal glomerulus,10 the myocardium,11,12 the vasculature,11,13,14 immune cells,15,16 and the brain.12 Accumulating evidence has shown that the aberrant activation of the MR is involved in the development of cardiovascular and renal injuries. Reports of clinical trials findings indicated that treatment with an MR blocker improved the prognosis of heart failure17–20 and reduced proteinuria in patients with CKD.21,22 Moreover, the pathogenicity of the aldosterone-MR system in hypertension and its comorbidities is associated with disease-associated conditions related to the modern lifestyle, such as obesity23,24 and aging.25
Given the benefit of MR blockers, investigation of the mechanism of MR activation may lead to the development of novel therapeutic strategies and targets. Previous studies reported that inappropriate aldosterone secretion10,26–29 and ligand-independent mechanisms of MR activation30–32 increased salt sensitivity of hypertension and end organ injuries. Moreover, recent studies demonstrated the tissue- or cell type–specific roles of MR activation in the myocardium,33,34 in vasculature,25,35–37 in immune cells,15,16,38 and in a classic target, the ASDN.39–43 These findings have updated our knowledge of the aldosterone-MR system and provided a new outlook for the use of MR blockers. Here, we summarize the recent research advances in the mechanism of MR activation in the development of salt-sensitive hypertension and renal injury.
Role of the Aldosterone-MR System in Metabolic Syndrome
Metabolic syndrome, a cluster of comorbidities that includes visceral obesity, hypertension, glucose intolerance, and dyslipidemia, is a major risk factor of cardiovascular and renal injury. Indeed, clinical studies have reported a higher incidence of CKD or proteinuria in individuals with metabolic syndrome than in those without it. Of note, hypertension and renal injury in obese patients are closely associated with high salt intake. Individuals with obesity reportedly showed greater salt-sensitivity of BP with shifted curve of pressure natriuresis, compared with lean individuals. Moreover, the shifted curve was normalized after weight-loss program, and resulted in BP reduction.44 The relationship between salt intake and urinary albumin excretion was steeper in obese study participants than in lean ones, suggesting increased salt sensitivity of renal injury in obesity.45
Multiple factors commonly observed in obese subjects, including compression of kidneys, hyperinsulinemia, hyperleptinemia, sympathetic overactivation, and activation of the renin-angiotensin-aldosterone system (RAAS), interdependently contribute to the impaired renal sodium handling and the resultant salt-sensitive hypertension in metabolic syndrome.46 Clinical studies have shown relatively high plasma aldosterone with low plasma renin activity in individuals of African descent with metabolic syndrome,47,48 indicating the role of aldosterone even in the presence of a blunted renin-angiotensin system (RAS). Moreover, primary aldosteronism49 and a variant in the promoter region of aldosterone synthase (CYP11B2) that leads to hyperaldosteronemia50 were associated with incident metabolic syndrome, suggesting an interaction between obesity and hyperaldosteronism.
Because excess aldosterone promotes salt-sensitive hypertension and end organ damage, we and others have explored its role in metabolic syndrome. We found in a study of obese SHR (SHR/NDmcr-cp) rats, a model of metabolic syndrome, that these animals had significantly greater proteinuria with podocyte damage compared with control lean SHR rats, despite both strains showing a similar BP.10 Obese SHR rats had a higher plasma aldosterone concentration compared with lean SHR rats, and aldosterone level was positively correlated with the amount of proteinuria. Accordingly, the expression of an MR target gene, serum and glucocorticoid-regulated kinase 1 (Sgk1), was increased in the glomerular fraction and a whole-kidney sample from obese SHR rats. Moreover, a selective MR blocker, eplerenone, reversed the proteinuria and podocyte injury in obese SHR rats, demonstrating the role of MR activation induced by aldosterone excess in renal injury in metabolic syndrome.
Of note, proteinuria and podocyte injury in obese SHR rats were markedly aggravated after salt loading.51 Because salt loading physiologically suppresses renin release in the kidney, plasma renin activity and aldosterone were suppressed in salt-loaded normotensive rats and lean SHR rats. However, plasma aldosterone levels in salt-loaded obese SHR rats were still higher than that in salt-loaded lean SHR rats, suggesting that aldosterone overproduction by escape from physiologic control was sustained in obese SHR rats. Furthermore, inappropriately high levels of aldosterone during salt loading are a key determinant of the pathogenicity of aldosterone. Thus, we hypothesized that the combination of imbalanced excess of aldosterone and salt loading in obese SHR induces the full activation of MR to exacerbate renal injury. Indeed, MR signaling, including the nuclear accumulation of MR protein and expression of Sgk1, was moderately increased in the kidneys of salt-loaded obese SHR rats. Moreover, eplerenone reversed the upregulation of Sgk1, resulting in the attenuation of salt-induced BP elevation and renal injury. Thus, MR activation by inadequate suppression of aldosterone production in the context of a high-salt diet plays a key role in the increased salt sensitivity of BP and renal injury in obese SHR rats.
Previous studies have suggested that obese adipose tissue is involved in the mechanism of aldosterone overproduction in metabolic syndrome. Goodfriend et al.26 demonstrated a positive correlation between plasma aldosterone level and the volume of visceral fat and suggested a role for the epoxy-keto derivates of linoleic acid.27 Ehrhart-Bornstein et al.28 showed that a secretory product from adipocytes isolated from obese subjects stimulated aldosterone production in cultured adrenal cells. We also observed that conditioned medium from obese SHR rat adipocytes, but not from lean SHR adipocytes, stimulated aldosterone production in cultured adrenal cells.51 Of note, these effects were not affected by the inhibition of the RAS with angiotensin-converting enzyme inhibitor/angiotensin receptor blocker treatment. This implies that these effects are different from physiologic stimuli, including AngII. However, the “aldosterone-releasing factors” from obese adipocytes have not been identified. Some researchers have suggested that complement-C1q TNF-related protein52 and leptin29 are candidate aldosterone-releasing factors. Taken together, aldosterone overproduction induced by aldosterone-releasing factors from obese adipocytes causes salt-sensitive hypertension and renal injury in metabolic syndrome (Figure 1).
Figure 1.

Mechanism of MR-mediated hypertension and renal injuries in metabolic syndrome. In metabolic syndrome, aldosterone (Aldo)-releasing factors from obese adipocytes, which are not identified, stimulate Aldo secretion in the adrenal glands, independent of RAS. Subsequently, activation of the MR in the kidney causes salt-sensitive hypertension and renal injury.
Alternative Mechanism of MR Activation by RAC1
In addition to aldosterone excess as a mechanism in activation of the MR, alternative pathways of MR activation have been suggested. The antihypertensive effect of eplerenone in a clinical trial did not depend on plasma renin-aldosterone profiles.53 In contrast to obese SHR rats, despite adequately suppressed plasma aldosterone, Dahl salt-sensitive (Dahl-S) rats, a classic model of salt-sensitive hypertension, develop hypertension and proteinuric renal injury after salt loading, along with increased MR signaling in kidneys that includes the upregulation of Sgk1 or ENaC.31,54,55
These observations implicate ligand-independent mechanisms of MR activation. The activity of nuclear receptors is affected by signaling molecules that modulate nuclear translocation, epigenetics, and the recruitment of coactivators or corepressors. A Rho family GTPase, Rac1, has multiple roles in intracellular signaling, including organization of the actin cytoskeleton, generation of reactive oxygen species (ROS), cell migration and adhesion, apoptosis, and modulation of the function of nuclear receptors.56–58 Rac1 functions as a molecular switch by cycling between an inactive GDP-bound and an active GTP-bound state. The transition is controlled positively by guanine nucleotide exchange factors and negatively by GTPase-activating proteins and GDP-dissociation inhibitors (GDIs)59,60; Rac1 activates downstream effectors, including p21-activated kinase, LIM kinase, and mitogen-activated protein kinases.56–58 Recently, we found that Rac1 mediates ligand-independent MR activation.
In our study using a stable line of human embryonic kidney cells (HEK293) and podocytes, the transfection of constitutively active Rac1 promoted the nuclear translocation of green fluorescent protein–tagged MR and enhanced MR-dependent transcriptional activity.30 A p21-activated kinase inhibitor reduced the Rac1-mediated activation of MR, indicating that Rac1 acts via activation of p21-activated kinase.30 We then further investigated the role of the Rac1-MR pathway in an in vivo model of Rac1 activation, RhoGDIα knockout mice. RhoGDIα is a negative regulator of Rac1 and expressed in kidney with relatively high abundance in podocytes61,62 and collecting ducts.63 Accordingly, RhoGDIα knockout mice showed renal activation of Rac1 along with the nuclear accumulation of MR and upregulation of Sgk1 in the kidney, despite normal aldosterone levels. Moreover, these mice developed profound proteinuria with podocyte damage and FSGS. Treatment with a selective Rac1 inhibitor (NSC23766) and eplerenone abolished the MR signaling in RhoGDIα knockout mice, attenuating glomerular injury, which suggests that the Rac1-mediated activation of MR induces proteinuric renal injury. The role of Rac1 activation in proteinuric renal disease was also reported in familial nephrotic syndrome with mutations in genes encoding Arhgap2464 and RhoGDIα,61,62 which induce Rac1 activation in podocytes. Moreover, an Rac1 inhibitor and eplerenone ameliorated the nephrotic phenotype recapitulated in RhoGDIα knockout zebrafish.61 These results strongly support the important role of the Rac1-MR pathway in glomerular injury, whereas some other Rac1 signaling, including cytoskeleton remodeling61,62,64 and crosstalk with transient receptor potential channel 5,65–67 would also be involved in the Rac1-dependent podocyte injuries. We also demonstrated the involvement of the Rac1-MR pathway in the development of proteinuric renal injury in animal models of RAAS activation68 and diabetic nephropathy.69
Of note, BP and glomerular injury in RhoGDIα knockout mice were salt sensitive, suggesting Rac1 activation has a pivotal role in the development of salt sensitivity.31 We assessed the Rac1-MR pathway in Dahl-S rats and their counterparts, Dahl salt-resistant (Dahl-R) rats. After salt loading, both strains showed similarly suppressed levels of plasma aldosterone. However, only the salt-loaded Dahl-S rats showed Rac1 activation. In contrast, the salt-loaded Dahl-R rats showed suppressed Rac1 activity and MR signaling, in accordance with decreased plasma aldosterone, leading to the absence of hypertension and glomerular injury.31 The phenotypes in salt-loaded Dahl-S rats were reversed by not only eplerenone but also NSC23766, associated with the significant reduction of Sgk1, a downstream molecule of MR signaling. These results indicated that the Rac1-MR pathway is a key determinant of salt sensitivity of BP and glomerular injury (Figure 2). Given that the knockdown of RhoGDIα in cultured principal cells of cortical collecting ducts caused Rac1 activation and induced ENaC activation,63 the activation MR-ENaC signaling in ASDN would be involved in the development of salt sensitivity of BP, whereas MR activation in non-ASDN might be also involved in the entire phenotype. The downstream effector of Rac1-MR activation in non-ASDN tissues remains unclear, but ROS are known to mediate MR signaling in the cardiovascular system25,33,70–72 and glomeruli.73–75
Figure 2.

Rac1 is a determinant of BP salt sensitivity through the on/off switching of MR activation. In both salt-resistant and salt-sensitive models, high salt intake suppressed plasma aldosterone (Aldo) concentration. In salt-resistant models, the salt loading suppresses renal Rac1 activity and subsequently, suppresses MR activity to maintain normal BP. In contrast, despite decreased plasma Aldo, high salt activates Rac1 in salt-sensitive models, which causes paradoxical activation of MR, leading to salt-induced BP elevation. The contrasting regulation of MR by Rac1 plays a key role in the development of salt-resistant and salt-sensitive phenotypes in each model.
The switching mechanism of Rac1 activity by salt loading in salt-sensitive and salt-resistant phenotypes is still unclear. One possible mechanism is genetic alteration in molecules that are involved in Rac1 activation. The quantitative trait loci involved in salt sensitivity of Dahl-S rats76–78 include some genes encoding GTPase-activating proteins. Another possibility is the change in the stimuli that activate Rac1. Rac1 is activated by various stimuli, including mechanical stretch,79,80 shear stress,81 inflammatory cytokines,82 growth factors,83 integrins,84 ROS,85 high glucose,86,87 sodium chloride88 or osmotic stress,89 AngII,90–92 and aldosterone.74,93 Tiam1, a guanine nucleotide exchange factor that is activated by some stimuli, including AngII,94 was upregulated by salt loading in Dahl-S rats but unchanged in Dahl-R rats.95 Given that intrarenal angiotensinogen was elevated by salt loading in Dahl-S rats,96 the local increase in AngII could induce Rac1 activation. Based upon the previous findings indicating that MR activation promotes ROS production and that ROS could activate Rac1 in turn, ROS also might play an important role in creating the vicious cycle of Rac1 and MR activation leading to salt-sensitive hypertension and renal injury. In AKI caused by ischemia-reperfusion, it is noteworthy that vascular ROS overproduction—which is attributed to Rac1 activation via the MR in vascular smooth muscle cells—causes endothelial dysfunction, resulting in tubular injury through reduced renal perfusion.97 Thus, intricate crosstalk between Rac1 and the MR among various cell types or tissues would also be involved in the MR-dependent renal injuries.
The Eplerenone Combination versus Conventional Agents to Lower Blood Pressure on Urinary Antialbuminuric Treatment Effect study reported that treatment with eplerenone significantly reduced BP and urinary albumin excretion in patients with CKD already receiving angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or both, independent of the plasma aldosterone level. Moreover, the antihypertensive and antialbuminuric effects of eplerenone were greater in patients with higher dietary salt intake as estimated by urinary sodium excretion.21,98 These results led us to the plausible hypothesis that patients with CKD have aldosterone-independent MR activation driven by high salt, possibly through Rac1 activation. Thus, the Rac1-MR pathway is a potential therapeutic target, especially for salt-sensitive hypertension and renal injury.
Cell Type–Specific and Context-Dependent Role of Aldosterone and the MR in the ASDN
Recent findings have also provided new insights into our understanding of the ASDN, which consists of distal convoluted tubules (DCTs), connecting tubules, and collecting ducts. In steady state, most of the sodium delivered to the distal convolution is reabsorbed in the DCT and connecting tubule; the late DCT and connecting tubule play a major role in potassium secretion.99,100 Classically, aldosterone binds to the MR in the principal cells of the connecting tubule and collecting duct to activate the ENaC via signaling pathways, including Sgk1 and neuronal precursor cell expressed developmentally down-regulated protein 4-2 (Nedd4–2).3 The resultant sodium reabsorption by the ENaC generates a lumen-negative potential, which promotes chloride reabsorption and the secretion of potassium.
Of note, both aldosterone and glucocorticoids, which circulate in plasma at concentrations 100–1000 times higher than that of aldosterone, bind to the MR with equivalent high affinity.1 In principal cells, however, 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which converts glucocorticoids to their inactive metabolites, guarantees the sensitivity of principal cells to aldosterone.101,102 In deficiency of 11β-HSD2, unconverted glucocorticoids should bind to the MR in principal cells and activate ENaC as a result. This mechanism is thought to be one of the causes of salt-sensitive hypertension observed in the syndrome of apparent mineralocorticoid excess, which involves a loss-of-function mutation in the gene encoding 11β-HSD2.103 Supporting this, mice with kidney-specific ablation of the gene encoding 11β-HSD2 develop salt-sensitive hypertension in association with ENaC activation in the kidney, and an ENaC inhibitor amiloride attenuates the hypertension.104
DCT cells express sodium chloride cotransporter (NCC), a target of thiazide diuretics. It has been reported that the activity of NCC is modulated differently in some conditions with aldosterone excess, such as volume depletion, hyperkalemia, and primary aldosteronism. During volume depletion that activates RAAS, AngII directly activates NCC via with-no-lysine kinase (WNK)–dependent pathways, enhancing sodium chloride reabsorption in the DCTs.105,106 Conversely, during high potassium intake, hyperkalemia directly suppresses NCC activity and increases urinary sodium excretion in the DCTs, despite the presence of hyperaldosteronism.107
It has been suggested that the NCC activation in volume depletion would reduce distal sodium delivery and flow rate, which may limit the ENaC-dependent generation of lumen-negative potential for potassium secretion and flow-dependent potassium excretion, and also that the NCC suppression on a high-potassium diet may exert the opposite effect to enhance potassium excretion.108,109 Supporting this hypothesis, acute hyperkalemia induced by intravenous infusion of potassium in mice was associated with both natriuresis and kaliuresis, along with significant suppression of NCC and redistribution of ENaC and the renal outer medullary potassium channel to the luminal membrane of principal cells.107 However, some recent studies have indicated that the influence of NCC activity upon potassium secretion is not solely explained by the alteration in distal sodium delivery and flow but would depend on remodeling of ASDN in some conditions.109,110 A recent study showed that the inhibition of NCC by acute administration of hydrochlorothiazide in mice induced natriuresis but did not caused kaliuresis.111 In another study, mice with DCT-specific expression of a constitutively active mutant of STE20/SPS1-related proline-alanine–rich protein kinase, which induced NCC activation, exhibited hyperkalemia in association with remodeling of ASDN; this remodeling involved a reduction of connecting tubule mass and a decrease in expression and apical localization of ENaC and renal outer medullary potassium.112 Hydrochlorothiazide treatment in these mutant mice immediately induced natriuresis but gradually caused kaliuresis to attenuate the hyperkalemia in association with reversal of the ASDN remodeling.112
Regarding the mechanism of NCC regulation by potassium, a recent study showed that changes in plasma potassium levels altered the polarization of DCT cells, and the resultant change in intracellular chloride level modulated WNK kinases to regulate NCC.113 In contrast to hyperkalemia, hypokalemia induces NCC activation. In primary aldosteronism, hypokalemia-induced NCC activation at the DCT, in cooperation with MR-ENaC activation at the connecting tubule and collecting duct, contributes to the aberrant sodium reabsorption and the resultant salt-sensitive hypertension. Supporting this, aldosterone infusion in rodents caused hypokalemia and NCC activation, but the correction of hypokalemia by an ENaC inhibitor or potassium supplementation reversed the NCC activation.114 Moreover, NCC inhibitors ameliorated salt-sensitive hypertension in mineralocorticoid-infused rats115 and in mice with the kidney-specific deletion of 11β-HSD2; in both cases, NCC is activated via hypokalemia induced by activation of MR-ENaC signals in principal cells.104 Of note, some studies offer evidence indicating a direct role of aldosterone in NCC regulation, although the modulation of NCC by aldosterone largely depends on change in plasma potassium. Late DCT, the marginal zone expressing both NCC and ENaC, expresses 11β-HSD2, although the expression level and area vary among species.116–118 Ackermann et al.2 have shown that suppression of aldosterone by dietary salt repletion in rats eliminated the nuclear localization of the MR in late DCT, demonstrating that the MR in late DCT is sensitive to the change in aldosterone. Some researchers have suggested that the aldosterone-MR cascade in DCT activates NCC by modulating interaction among Sgk1, Nedd4–2, and WNK1.119 In addition, a recent study showed that rapid aldosterone-mediated signaling activates NCC through an MR-independent, nongenomic mechanism via GPR30, a membrane-associated receptor.120
Emerging evidence has also shown that a chloride/bicarbonate exchanger, pendrin, expressed on β-intercalated cells (β-ICs) in the connecting tubule and cortical collecting duct, mediates chloride flux and is involved in sodium chloride reabsorption in coordination with a sodium-dependent chloride/bicarbonate exchanger.121–123 Pendrin knockout mice showed slightly reduced arterial pressure detectable by telemetric analysis under basal conditions,124,125 and they exhibited more severe hypotension with apparent fluid loss after salt depletion.121 In addition, mice with IC-specific pendrin overexpression developed salt-sensitive hypertension,126 demonstrating the important role of pendrin in fluid homeostasis and salt sensitivity of BP. Of note, it has been suggested that pendrin complements NCC. Some studies have indicated that NCC knockout mice showed compensatory activation of pendrin.127,128 Moreover, pendin/NCC double-knockout mice exhibited visibly enhanced fluid loss and hypotension with normal salt intake compared with mice that have a single knockout of pendrin or NCC.128 Similar to NCC, pendrin is upregulated by AngII during volume depletion129,130 and hypokalemic status during primary aldosterone excess.131 Interestingly, these upregulations of pendrin were suppressed by an MR blocker, suggesting the involvement of the MR.130,131
It is still unclear whether aldosterone is a sole ligand of the MR in ICs because in immunohistologic studies2,102 and single-cell transcriptome analysis,132 11β-HSD2 expression was absent or minimal in ICs, which would permit glucocorticoids to occupy the MR. Notably, recent studies suggest that ICs have a specific mechanism to regulate the ligand-binding affinity of MRs, which are associated with pendrin regulation. Shibata et al.95 reported that the MR in ICs was phosphorylated at S843 in the ligand-binding domain and that its phosphorylation prevented ligand binding in the steady state. AngII and hypokalemia dephosphorylate this site and increase the MR’s ligand-binding affinity, leading to the nuclear accumulation of MR in ICs and the upregulation of pendrin only when the ligand was present.95,130,131 These data indicate that the switching mechanism of MR affinity by phosphorylation in ICs is involved in the regulation of pendrin. Indeed, we and others demonstrated that mice with an IC-specific MR deletion (MRIC-KO) displayed reduced expression of pendrin after AngII infusion or endogenous AngII elevation by dietary salt depletion.42,43 Salt-depleted MRIC-KO mice showed normal BP related to the compensatory activation of NCC to maintain BP; they also exhibited fluid loss and hypotension after an additional treatment with an NCC inhibitor.42 Importantly, NCC inhibitor treatment of salt-depleted MRIC-KO mice resulted in hypokalemia, which is consistent with a recent finding indicating that pendrin has a potassium-sparing effect.133 Thus, the AngII-NCC and AngII-MR-pendrin cascades might coordinate to maintain fluid levels and BP and simultaneously maintain potassium balance during salt depletion.
We also observed that aldosterone administration to salt-loaded mice induced pendrin upregulation and NCC activation; this occurred in association with hypokalemia and metabolic alkalosis induced by activation of the MR-ENaC cascade in principal cells.42 In addition, immunohistologic analysis showed that aldosterone significantly increased the relative abundance of pendrin label in the cells’ apical membrane region.42,43,134 Correction of hypokalemic alkalosis by an ENaC inhibitor or potassium supplementation reversed the changes in pendrin and NCC. However, the aldosterone-induced upregulation of pendrin was only partially suppressed in MRIC-KO mice.42,43 A report described significant attenuation of apical membrane accumulation of pendrin label in MRIC-KO mice in immunohistochemical analysis43; however, another report found that aldosterone-induced change in subcellular distribution of pendrin is still observed in MRIC-KO mice.42 Despite this inconsistency, the correction of hypokalemic alkalosis by potassium supplementation in aldosterone-treated MRIC-KO mice significantly reduced pendrin expression.42 Thus, a substantial portion of aldosterone-induced regulation of pendrin is dependent on hypokalemic alkalosis but is independent of the MR in ICs.
We also examined whether hypokalemia or metabolic alkalosis was responsible for pendrin activation related to primary aldosterone excess. Because pendrin was originally reported as a mediator of bicarbonate excretion responding to alkali loading,135 we hypothesized that alkalosis drives the aldosterone-induced pendrin activation. Accordingly, we found that treating aldosterone-infused mice with a carbonic anhydrase inhibitor, acetazolamide, corrected alkalosis without a change in hypokalemia and reversed the upregulation of pendrin with no change in NCC activation.42 Thus, during aldosterone excess, hypokalemia and alkalosis—consequences of MR-ENaC pathway activation—induced the parallel activation of NCC and pendrin, respectively, which was independent of the MR.
On the basis of a previous finding that pendrin knockout attenuated hypertension induced by mineralocorticoid excess,134 pendrin upregulation induced by hypokalemic alkalosis contributed to aldosterone-induced hypertension. Furthermore, we observed that the correction of hypokalemic alkalosis by potassium supplementation suppressed pendrin expression and salt-sensitive hypertension in NCC knockout mice,42 indicating the role of pendrin as a complement pathway in NCC inhibition. As is well known, the clinical use of thiazide diuretics occasionally activates RAAS and hypokalemic alkalosis, resulting in thiazide resistance. Moreover, MR blockers effectively reduced the BP in a patient with resistant hypertension, defined as BP that remains above the target level despite treatment with at least three antihypertensive drugs, including a diuretic.136 Thus, the two pathways of pendrin upregulation—mediated through either the MR in ICs activated by RAS stimulation during volume depletion or hypokalemic alkalosis driven by the MR-ENaC cascade in principal cells during aldosterone excess (Figure 3)—may be therapeutic targets for thiazide-resistant hypertension.
Figure 3.
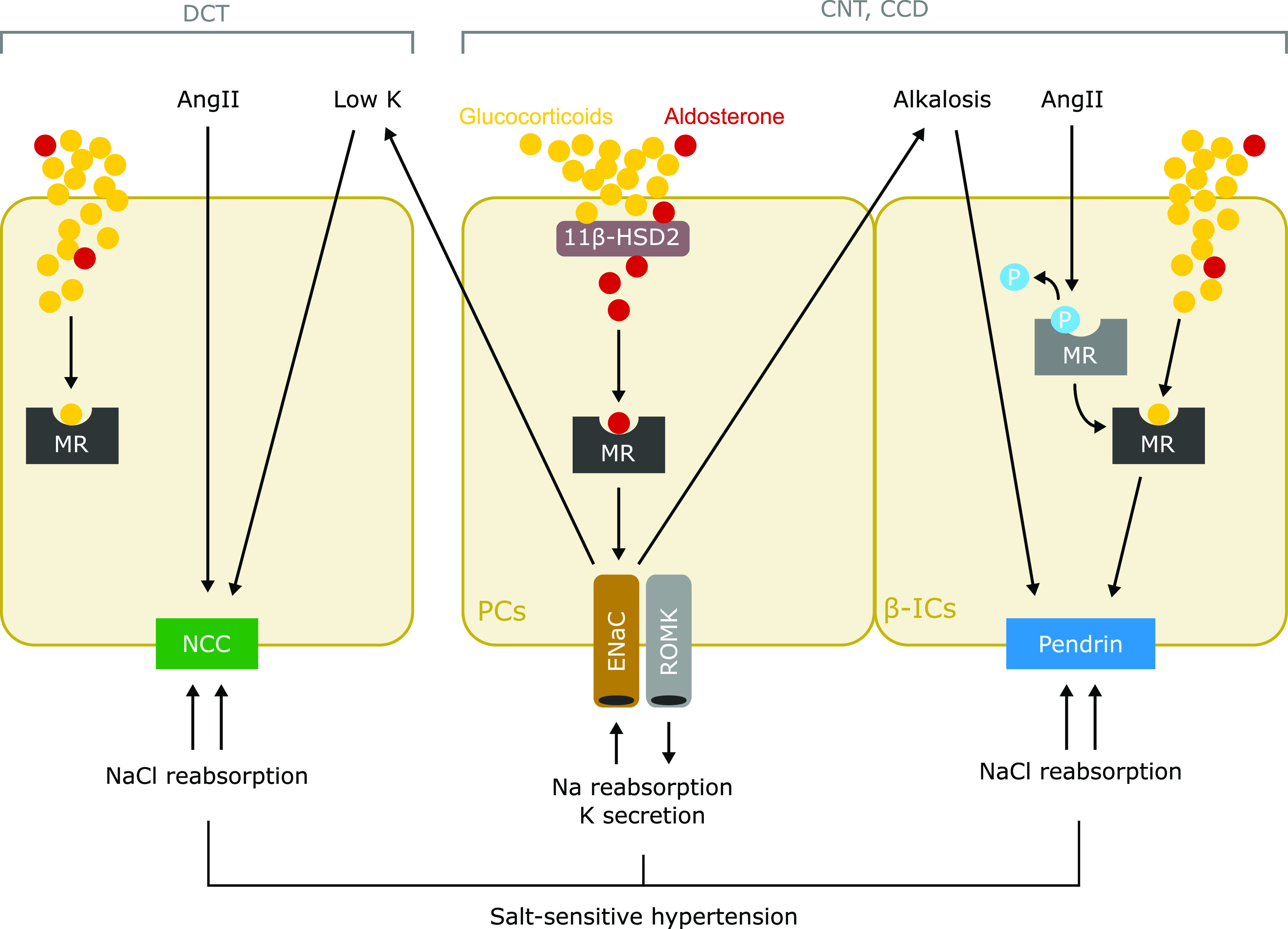
Putative mechanism of salt-sensitive hypertension in primary aldosteronism. Cell type–specific mechanisms of sodium reabsorption are mediated by MR in the ASDN. Classically, aldosterone activates the MR-ENaC pathway in principal cells (PCs) of the connecting tubules (CNTs) and cortical collecting ducts (CCDs) to reabsorb sodium and secrete potassium. The interaction between aldosterone and MR in PCs is guaranteed by 11β-HSD2 that converts glucocorticoids to inactive metabolites. NCC expressed in DCT is directly activated by AngII and hypokalemia, which is induced by the aldosterone-induced activation of MR-ENaC in PCs. In β-ICs in the CNTs and CCDs, AngII induces dephosphorylation of MR, which increases affinity of ligands, and resultantly causes MR activation, leading to upregulation of pendrin. In addition, metabolic alkalosis, which is induced by activation of MR-ENaC in PCs, directly induces pendrin upregulation in β-ICs. The contrasting activation of NCC and pendrin induced by aldosterone excess, in a coordinated manner, mediates sodium chloride reabsorption, leading to the development of salt-sensitive hypertension. P, phosphorylation of MR; ROMK, renal outer medullary potassium.
Conclusions
Updated information about the renal activation of the aldosterone-MR system in the development of salt-sensitive hypertension and renal injury will provide new insights into the use of MR antagonists for the treatment of resistant hypertension and renal injury. Recent studies have also revealed that the aldosterone-MR system in various tissues participates in the pathogenesis of cardiorenal diseases by both BP-dependent and BP-independent mechanims.15,16,33,34,137–140 Of note, clinical trials have demonstrated the efficacy and improved safety profiles of novel nonsteroidal MR antagonists, such as finerenone20,22,141,142 and esaxerenone,143 in cardiovascular and renal injuries in diabetic individuals. Given the promising effect of MR antagonists, further investigation of the aldosterone-MR system may lead to the discovery of therapeutic strategies and targets for hypertension and end organ injury.
Disclosures
T. Fujita reports research funding from Bayer Japan and Omron; honoraria from Daiichi-Sankyo; and scientific advisor or membership with Hypertension, Hypertension Research, the Japanese Society of Hypertension, and the Japanese Society of Nephrology. The remaining author has nothing to disclose.
Funding
This work was supported by Japan Society for the Promotion of Science grants KAKENHI JP20K08585 and KAKENHI JP15H05788.
Acknowledgments
We thank Dr. J. Ludovic Croxford from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.
This paper is on the basis of the 2019 American Society of Nephrology Homer W. Smith award lecture (T. Fujita).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al.: Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 237: 268–275, 1987 [DOI] [PubMed] [Google Scholar]
- 2.Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, et al.: In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: Differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol 299: F1473–F1485, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE: Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 10: 135–146, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall JE, Mizelle HL, Hildebrandt DA, Brands MW: Abnormal pressure natriuresis. A cause or a consequence of hypertension? Hypertension 15: 547–559, 1990 [DOI] [PubMed] [Google Scholar]
- 5.Guyton AC: Blood pressure control--special role of the kidneys and body fluids. Science 252: 1813–1816, 1991 [DOI] [PubMed] [Google Scholar]
- 6.Fujita T, Henry WL, Bartter FC, Lake CR, Delea CS: Factors influencing blood pressure in salt-sensitive patients with hypertension. Am J Med 69: 334–344, 1980 [DOI] [PubMed] [Google Scholar]
- 7.Lifton RP, Gharavi AG, Geller DS: Molecular mechanisms of human hypertension. Cell 104: 545–556, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N, et al.: Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med 351: 33–41, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Pan YJ, Young DB: Experimental aldosterone hypertension in the dog. Hypertension 4: 279–287, 1982 [DOI] [PubMed] [Google Scholar]
- 10.Nagase M, Yoshida S, Shibata S, Nagase T, Gotoda T, Ando K, et al.: Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: Possible contribution of fat-derived factors. J Am Soc Nephrol 17: 3438–3446, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Lombès M, Oblin ME, Gasc JM, Baulieu EE, Farman N, Bonvalet JP: Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ Res 71: 503–510, 1992 [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Sanchez CE, de Rodriguez AF, Romero DG, Estess J, Warden MP, Gomez-Sanchez MT, et al.: Development of a panel of monoclonal antibodies against the mineralocorticoid receptor. Endocrinology 147: 1343–1348, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Jaffe IZ, Mendelsohn ME: Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res 96: 643–650, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, et al.: Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res 102: 1359–1367, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ: Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt-induced cardiac fibrosis and increased blood pressure. Hypertension 54: 537–543, 2009 [DOI] [PubMed] [Google Scholar]
- 16.Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, et al.: Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest 120: 3350–3364, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al.: The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators N Engl J Med 341: 709–717, 1999 [DOI] [PubMed] [Google Scholar]
- 18.Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al.; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators: Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction [published correction appears in N Engl J Med 348: 2271, 2003]. N Engl J Med 348: 1309–1321, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, et al.; EMPHASIS-HF Study Group: Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 364: 11–21, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Filippatos G, Anker SD, Böhm M, Gheorghiade M, Køber L, Krum H, et al.: A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur Heart J 37: 2105–2114, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ando K, Ohtsu H, Uchida S, Kaname S, Arakawa Y, Fujita T; EVALUATE Study Group: Anti-albuminuric effect of the aldosterone blocker eplerenone in non-diabetic hypertensive patients with albuminuria: A double-blind, randomised, placebo-controlled trial [published correction appears in Lancet Diabetes Endocrinol 3: e3, 2015]. Lancet Diabetes Endocrinol 2: 944–953, 2014 [DOI] [PubMed] [Google Scholar]
- 22.Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, et al.; Mineralocorticoid Receptor Antagonist Tolerability Study–Diabetic Nephropathy (ARTS-DN) Study Group: Effect of finerenone on albuminuria in patients with diabetic nephropathy: A randomized clinical trial. JAMA 314: 884–894, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Sowers JR, Whaley-Connell A, Epstein M: Narrative review: The emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med 150: 776–783, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujita T: Mineralocorticoid receptors, salt-sensitive hypertension, and metabolic syndrome. Hypertension 55: 813–818, 2010 [DOI] [PubMed] [Google Scholar]
- 25.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, et al.: Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med 18: 1429–1433, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ: Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res 7: 355–362, 1999 [DOI] [PubMed] [Google Scholar]
- 27.Goodfriend TL, Ball DL, Egan BM, Campbell WB, Nithipatikom K: Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension 43: 358–363, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, Langenbach J, Willenberg HS, Barthel A, et al.: Human adipocytes secrete mineralocorticoid-releasing factors. Proc Natl Acad Sci U S A 100: 14211–14216, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huby AC, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, et al.: Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation 132: 2134–2145, 2015 [DOI] [PubMed] [Google Scholar]
- 30.Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, et al.: Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat Med 14: 1370–1376, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Shibata S, Mu S, Kawarazaki H, Muraoka K, Ishizawa K, Yoshida S, et al.: Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest 121: 3233–3243, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Treesaranuwattana T, Wong KYH, Brooks DL, Tay CS, Williams GH, Williams JS, et al.: Lysine-specific demethylase-1 deficiency increases agonist signaling via the mineralocorticoid receptor. Hypertension 75: 1045–1053, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraccarollo D, Berger S, Galuppo P, Kneitz S, Hein L, Schütz G, et al.: Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation 123: 400–408, 2011 [DOI] [PubMed] [Google Scholar]
- 34.Lother A, Berger S, Gilsbach R, Rösner S, Ecke A, Barreto F, et al.: Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension 57: 746–754, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard-Pascaud A, Berger S, et al.: Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension 63: 520–526, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rickard AJ, Morgan J, Chrissobolis S, Miller AA, Sobey CG, Young MJ: Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 63: 1033–1040, 2014 [DOI] [PubMed] [Google Scholar]
- 37.Mueller KB, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, et al.: Endothelial mineralocorticoid receptors differentially contribute to coronary and mesenteric vascular function without modulating blood pressure. Hypertension 66: 988–997, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun XN, Li C, Liu Y, Du LJ, Zeng MR, Zheng XJ, et al.: T-cell mineralocorticoid receptor controls blood pressure by regulating interferon-gamma. Circ Res 120: 1584–1597, 2017 [DOI] [PubMed] [Google Scholar]
- 39.Ronzaud C, Loffing J, Bleich M, Gretz N, Gröne HJ, Schütz G, et al.: Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 18: 1679–1687, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Canonica J, Sergi C, Maillard M, Klusonova P, Odermatt A, Koesters R, et al.: Adult nephron-specific MR-deficient mice develop a severe renal PHA-1 phenotype. Pflugers Arch 468: 895–908, 2016 [DOI] [PubMed] [Google Scholar]
- 41.Czogalla J, Vohra T, Penton D, Kirschmann M, Craigie E, Loffing J: The mineralocorticoid receptor (MR) regulates ENaC but not NCC in mice with random MR deletion. Pflugers Arch 468: 849–858, 2016 [DOI] [PubMed] [Google Scholar]
- 42.Ayuzawa N, Nishimoto M, Ueda K, Hirohama D, Kawarazaki W, Shimosawa T, et al.: Two mineralocorticoid receptor-mediated mechanisms of pendrin activation in distal nephrons. J Am Soc Nephrol 31: 748–764, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pham TD, Verlander JW, Wang Y, Romero CA, Yue Q, Chen C, et al.: Aldosterone regulates pendrin and epithelial sodium channel activity through intercalated cell mineralocorticoid receptor-dependent and -independent mechanisms over a wide range in serum potassium. J Am Soc Nephrol 31: 483–499, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rocchini AP, Key J, Bondie D, Chico R, Moorehead C, Katch V, et al.: The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N Engl J Med 321: 580–585, 1989 [DOI] [PubMed] [Google Scholar]
- 45.Verhave JC, Hillege HL, Burgerhof JG, Janssen WM, Gansevoort RT, Navis GJ, et al.; PREVEND Study Group: Sodium intake affects urinary albumin excretion especially in overweight subjects. J Intern Med 256: 324–330, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME: Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ Res 116: 991–1006, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bochud M, Nussberger J, Bovet P, Maillard MR, Elston RC, Paccaud F, et al.: Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension 48: 239–245, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Kidambi S, Kotchen JM, Grim CE, Raff H, Mao J, Singh RJ, et al.: Association of adrenal steroids with hypertension and the metabolic syndrome in blacks. Hypertension 49: 704–711, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Fallo F, Veglio F, Bertello C, Sonino N, Della Mea P, Ermani M, et al.: Prevalence and characteristics of the metabolic syndrome in primary aldosteronism. J Clin Endocrinol Metab 91: 454–459, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Russo P, Lauria F, Loguercio M, Barba G, Arnout J, Cappuccio FP, et al.; European Collaborative Group of the IMMIDIET Project: -344C/T Variant in the promoter of the aldosterone synthase gene (CYP11B2) is associated with metabolic syndrome in men. Am J Hypertens 20: 218–222, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T: Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: Role of oxidative stress. Hypertension 50: 877–883, 2007 [DOI] [PubMed] [Google Scholar]
- 52.Jeon JH, Kim KY, Kim JH, Baek A, Cho H, Lee YH, et al.: A novel adipokine CTRP1 stimulates aldosterone production. FASEB J 22: 1502–1511, 2008 [DOI] [PubMed] [Google Scholar]
- 53.Williams GH, Burgess E, Kolloch RE, Ruilope LM, Niegowska J, Kipnes MS, et al.: Efficacy of eplerenone versus enalapril as monotherapy in systemic hypertension. Am J Cardiol 93: 990–996, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Farjah M, Roxas BP, Geenen DL, Danziger RS: Dietary salt regulates renal SGK1 abundance: Relevance to salt sensitivity in the Dahl rat. Hypertension 41: 874–878, 2003 [DOI] [PubMed] [Google Scholar]
- 55.Aoi W, Niisato N, Sawabe Y, Miyazaki H, Tokuda S, Nishio K, et al.: Abnormal expression of ENaC and SGK1 mRNA induced by dietary sodium in Dahl salt-sensitively hypertensive rats. Cell Biol Int 31: 1288–1291, 2007 [DOI] [PubMed] [Google Scholar]
- 56.Takai Y, Sasaki T, Matozaki T: Small GTP-binding proteins. Physiol Rev 81: 153–208, 2001 [DOI] [PubMed] [Google Scholar]
- 57.Heasman SJ, Ridley AJ: Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701, 2008 [DOI] [PubMed] [Google Scholar]
- 58.Nagase M, Fujita T: Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat Rev Nephrol 9: 86–98, 2013 [DOI] [PubMed] [Google Scholar]
- 59.Rossman KL, Der CJ, Sondek J: GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6: 167–180, 2005 [DOI] [PubMed] [Google Scholar]
- 60.Bos JL, Rehmann H, Wittinghofer A: GEFs and GAPs: Critical elements in the control of small G proteins. Cell 129: 865–877, 2007 [DOI] [PubMed] [Google Scholar]
- 61.Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, et al.: ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 123: 3243–3253, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gupta IR, Baldwin C, Auguste D, Ha KC, El Andalousi J, Fahiminiya S, et al.: ARHGDIA: A novel gene implicated in nephrotic syndrome. J Med Genet 50: 330–338, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pavlov TS, Levchenko V, Staruschenko A: Role of Rho GDP dissociation inhibitor α in control of epithelial sodium channel (ENaC)-mediated sodium reabsorption. J Biol Chem 289: 28651–28659, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, et al.: Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest 121: 4127–4137, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, et al.: Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels [published correction appears in Sci Signal 3: er11, 2010]. Sci Signal 3: ra77, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Y, Castonguay P, Sidhom EH, Clark AR, Dvela-Levitt M, Kim S, et al.: A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 358: 1332–1336, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greka A, Mundel P: Balancing calcium signals through TRPC5 and TRPC6 in podocytes. J Am Soc Nephrol 22: 1969–1980, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kawarazaki W, Nagase M, Yoshida S, Takeuchi M, Ishizawa K, Ayuzawa N, et al.: Angiotensin II- and salt-induced kidney injury through Rac1-mediated mineralocorticoid receptor activation. J Am Soc Nephrol 23: 997–1007, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshida S, Ishizawa K, Ayuzawa N, Ueda K, Takeuchi M, Kawarazaki W, et al.: Local mineralocorticoid receptor activation and the role of Rac1 in obesity-related diabetic kidney disease. Nephron, Exp Nephrol 126: 16–24, 2014 [DOI] [PubMed] [Google Scholar]
- 70.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, et al.: Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med 13: 189–197, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hayashi H, Kobara M, Abe M, Tanaka N, Gouda E, Toba H, et al.: Aldosterone nongenomically produces NADPH oxidase-dependent reactive oxygen species and induces myocyte apoptosis. Hypertens Res 31: 363–375, 2008 [DOI] [PubMed] [Google Scholar]
- 72.Maron BA, Zhang YY, White K, Chan SY, Handy DE, Mahoney CE, et al.: Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation 126: 963–974, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miyata K, Rahman M, Shokoji T, Nagai Y, Zhang GX, Sun GP, et al.: Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol 16: 2906–2912, 2005 [DOI] [PubMed] [Google Scholar]
- 74.Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T: Podocyte as the target for aldosterone: Roles of oxidative stress and Sgk1. Hypertension 49: 355–364, 2007 [DOI] [PubMed] [Google Scholar]
- 75.Zhu C, Huang S, Yuan Y, Ding G, Chen R, Liu B, et al.: Mitochondrial dysfunction mediates aldosterone-induced podocyte damage: A therapeutic target of PPARγ. Am J Pathol 178: 2020–2031, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deng AY, Dutil J, Sivo Z: Utilization of marker-assisted congenics to map two blood pressure quantitative trait loci in Dahl rats. Mamm Genome 12: 612–616, 2001 [DOI] [PubMed] [Google Scholar]
- 77.Rapp JP, Garrett MR, Deng AY: Construction of a double congenic strain to prove an epistatic interaction on blood pressure between rat chromosomes 2 and 10. J Clin Invest 101: 1591–1595, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Palijan A, Dutil J, Deng AY: Quantitative trait loci with opposing blood pressure effects demonstrating epistasis on Dahl rat chromosome 3. Physiol Genomics 15: 1–8, 2003 [DOI] [PubMed] [Google Scholar]
- 79.Aikawa R, Nagai T, Tanaka M, Zou Y, Ishihara T, Takano H, et al.: Reactive oxygen species in mechanical stress-induced cardiac hypertrophy. Biochem Biophys Res Commun 289: 901–907, 2001 [DOI] [PubMed] [Google Scholar]
- 80.Ayuzawa N, Nagase M, Ueda K, Nishimoto M, Kawarazaki W, Marumo T, et al.: Rac1-mediated activation of mineralocorticoid receptor in pressure overload-induced cardiac injury. Hypertension 67: 99–106, 2016 [DOI] [PubMed] [Google Scholar]
- 81.Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, Li S, Chien S, et al.: Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J 21: 6791–6800, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Papaharalambus C, Sajjad W, Syed A, Zhang C, Bergo MO, Alexander RW, et al.: Tumor necrosis factor alpha stimulation of Rac1 activity. Role of isoprenylcysteine carboxylmethyltransferase. J Biol Chem 280: 18790–18796, 2005 [DOI] [PubMed] [Google Scholar]
- 83.Kurokawa K, Itoh RE, Yoshizaki H, Nakamura YO, Matsuda M: Coactivation of Rac1 and Cdc42 at lamellipodia and membrane ruffles induced by epidermal growth factor. Mol Biol Cell 15: 1003–1010, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Price LS, Leng J, Schwartz MA, Bokoch GM: Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell 9: 1863–1871, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nagase M, Ayuzawa N, Kawarazaki W, Ishizawa K, Ueda K, Yoshida S, et al.: Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: Role of small GTPase Rac1. Hypertension 59: 500–506, 2012 [DOI] [PubMed] [Google Scholar]
- 86.Lin CL, Wang JY, Ko JY, Surendran K, Huang YT, Kuo YH, et al.: Superoxide destabilization of beta-catenin augments apoptosis of high-glucose-stressed mesangial cells. Endocrinology 149: 2934–2942, 2008 [DOI] [PubMed] [Google Scholar]
- 87.Shen E, Li Y, Li Y, Shan L, Zhu H, Feng Q, et al.: Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes 58: 2386–2395, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Silva GB, Garvin JL: Rac1 mediates NaCl-induced superoxide generation in the thick ascending limb. Am J Physiol Renal Physiol 298: F421–F425, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE, et al.: Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol 5: 1104–1110, 2003 [DOI] [PubMed] [Google Scholar]
- 90.Schmitz U, Thömmes K, Beier I, Wagner W, Sachinidis A, Düsing R, et al.: Angiotensin II-induced stimulation of p21-activated kinase and c-Jun NH2-terminal kinase is mediated by Rac1 and Nck. J Biol Chem 276: 22003–22010, 2001 [DOI] [PubMed] [Google Scholar]
- 91.Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, et al.: Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest 108: 1429–1437, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nishida M, Tanabe S, Maruyama Y, Mangmool S, Urayama K, Nagamatsu Y, et al.: G alpha 12/13- and reactive oxygen species-dependent activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase by angiotensin receptor stimulation in rat neonatal cardiomyocytes. J Biol Chem 280: 18434–18441, 2005 [DOI] [PubMed] [Google Scholar]
- 93.Iwashima F, Yoshimoto T, Minami I, Sakurada M, Hirono Y, Hirata Y: Aldosterone induces superoxide generation via Rac1 activation in endothelial cells. Endocrinology 149: 1009–1014, 2008 [DOI] [PubMed] [Google Scholar]
- 94.Gong FH, Chen XL, Zhang Q, Xiao XQ, Yang YS, Song BJ, et al.: MicroRNA-183 as a novel regulator protects against cardiomyocytes hypertrophy via targeting TIAM1 [published online ahead of print September 1, 2020]. Am J Hypertens 10.1093/ajh/hpaa144 [DOI] [PubMed] [Google Scholar]
- 95.Shibata S, Rinehart J, Zhang J, Moeckel G, Castañeda-Bueno M, Stiegler AL, et al.: Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 18: 660–671, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kobori H, Nishiyama A, Abe Y, Navar LG: Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension 41: 592–597, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barrera-Chimal J, André-Grégoire G, Nguyen Dinh Cat A, Lechner SM, Cau J, Prince S, et al.: Benefit of mineralocorticoid receptor antagonism in AKI: Role of vascular smooth muscle Rac1. J Am Soc Nephrol 28: 1216–1226, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nishimoto M, Ohtsu H, Marumo T, Kawarazaki W, Ayuzawa N, Ueda K, et al.: Mineralocorticoid receptor blockade suppresses dietary salt-induced ACEI/ARB-resistant albuminuria in non-diabetic hypertension: A sub-analysis of evaluate study. Hypertens Res 42: 514–521, 2019 [DOI] [PubMed] [Google Scholar]
- 99.Meneton P, Loffing J, Warnock DG: Sodium and potassium handling by the aldosterone-sensitive distal nephron: The pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004 [DOI] [PubMed] [Google Scholar]
- 100.Loffing J, Korbmacher C: Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflugers Arch 458: 111–135, 2009 [DOI] [PubMed] [Google Scholar]
- 101.Funder JW, Pearce PT, Smith R, Smith AI: Mineralocorticoid action: Target tissue specificity is enzyme, not receptor, mediated. Science 242: 583–585, 1988 [DOI] [PubMed] [Google Scholar]
- 102.Kyossev Z, Walker PD, Reeves WB: Immunolocalization of NAD-dependent 11 beta-hydroxysteroid dehydrogenase in human kidney and colon. Kidney Int 49: 271–281, 1996 [DOI] [PubMed] [Google Scholar]
- 103.Bailey MA, Paterson JM, Hadoke PW, Wrobel N, Bellamy CO, Brownstein DG, et al.: A switch in the mechanism of hypertension in the syndrome of apparent mineralocorticoid excess. J Am Soc Nephrol 19: 47–58, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ueda K, Nishimoto M, Hirohama D, Ayuzawa N, Kawarazaki W, Watanabe A, et al.: Renal dysfunction induced by kidney-specific gene deletion of Hsd11b2 as a primary cause of salt-dependent hypertension. Hypertension 70: 111–118, 2017 [DOI] [PubMed] [Google Scholar]
- 105.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, et al.: Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci U S A 109: 7929–7934, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, Flagella M, et al.: Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl- cotransporter of the distal convoluted tubule. J Biol Chem 273: 29150–29155, 1998 [DOI] [PubMed] [Google Scholar]
- 107.Rengarajan S, Lee DH, Oh YT, Delpire E, Youn JH, McDonough AA: Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol 306: F1059–F1068, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hoover RS: Angiotensin II: A candidate for an aldosterone-independent mediator of potassium preservation during volume depletion. Kidney Int 79: 377–379, 2011 [DOI] [PubMed] [Google Scholar]
- 109.Kamel KS, Schreiber M, Halperin ML: Renal potassium physiology: Integration of the renal response to dietary potassium depletion. Kidney Int 93: 41–53, 2018 [DOI] [PubMed] [Google Scholar]
- 110.McCormick JA, Ellison DH: Nephron remodeling underlies hyperkalemia in familial hyperkalemic hypertension. J Am Soc Nephrol 28: 2555–2557, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hunter RW, Craigie E, Homer NZ, Mullins JJ, Bailey MA: Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol Renal Physiol 306: F457–F467, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grimm PR, Coleman R, Delpire E, Welling PA: Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2597–2606, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, et al.: Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, et al.: Direct and indirect mineralocorticoid effects determine distal salt transport. J Am Soc Nephrol 27: 2436–2445, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Takanohashi A, Tojo A, Kobayashi N, Yagi S, Matsuoka H: Effect of trichlormethiazide and captopril on nitric oxide synthase activity in the kidney of deoxycorticosterone acetate-salt hypertensive rats. Jpn Heart J 37: 251–259, 1996 [DOI] [PubMed] [Google Scholar]
- 116.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, et al.: 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules [published correction appears in J Am Soc Nephrol 9: 2179, 1998]. J Am Soc Nephrol 9: 1347–1358, 1998 [DOI] [PubMed] [Google Scholar]
- 117.Velázquez H, Náray-Fejes-Tóth A, Silva T, Andújar E, Reilly RF, Desir GV, et al.: Rabbit distal convoluted tubule coexpresses NaCl cotransporter and 11 beta-hydroxysteroid dehydrogenase II mRNA. Kidney Int 54: 464–472, 1998 [DOI] [PubMed] [Google Scholar]
- 118.Câmpean V, Kricke J, Ellison D, Luft FC, Bachmann S: Localization of thiazide-sensitive Na(+)-Cl(-) cotransport and associated gene products in mouse DCT. Am J Physiol Renal Physiol 281: F1028–F1035, 2001 [DOI] [PubMed] [Google Scholar]
- 119.Roy A, Al-Qusairi L, Donnelly BF, Ronzaud C, Marciszyn AL, Gong F, et al.: Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J Clin Invest 125: 3433–3448, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cheng L, Poulsen SB, Wu Q, Esteva-Font C, Olesen ETB, Peng L, et al.: Rapid aldosterone-mediated signaling in the DCT increases activity of the thiazide-sensitive NaCl cotransporter. J Am Soc Nephrol 30: 1454–1470, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, et al.: NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: Role in Cl- conservation. Hypertension 44: 982–987, 2004 [DOI] [PubMed] [Google Scholar]
- 122.Pech V, Wall SM, Nanami M, Bao HF, Kim YH, Lazo-Fernandez Y, et al.: Pendrin gene ablation alters ENaC subcellular distribution and open probability. Am J Physiol Renal Physiol 309: F154–F163, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, et al.: The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice [published correction appears in J Clin Invest 121: 1668, 2011]. J Clin Invest 120: 1627–1635, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lazo-Fernandez Y, Aguilera G, Pham TD, Park AY, Beierwaltes WH, Sutliff RL, et al.: Pendrin localizes to the adrenal medulla and modulates catecholamine release. Am J Physiol Endocrinol Metab 309: E534–E545, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, Duke BJ, et al.: Pendrin modulates ENaC function by changing luminal HCO3-. J Am Soc Nephrol 21: 1928–1941, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jacques T, Picard N, Miller RL, Riemondy KA, Houillier P, Sohet F, et al.: Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J Am Soc Nephrol 24: 1104–1113, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschênes G, et al.: Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol 17: 2153–2163, 2006 [DOI] [PubMed] [Google Scholar]
- 128.Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K, et al.: Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci U S A 109: 13368–13373, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Verlander JW, Hong S, Pech V, Bailey JL, Agazatian D, Matthews SW, et al.: Angiotensin II acts through the angiotensin 1a receptor to upregulate pendrin. Am J Physiol Renal Physiol 301: F1314–F1325, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hirohama D, Ayuzawa N, Ueda K, Nishimoto M, Kawarazaki W, Watanabe A, et al.: Aldosterone is essential for angiotensin II-induced upregulation of pendrin. J Am Soc Nephrol 29: 57–68, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xu N, Hirohama D, Ishizawa K, Chang WX, Shimosawa T, Fujita T, et al.: Hypokalemia and pendrin induction by aldosterone. Hypertension 69: 855–862, 2017 [DOI] [PubMed] [Google Scholar]
- 132.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, et al.: Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci U S A 114: E9989–E9998, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.López-Cayuqueo KI, Chavez-Canales M, Pillot A, Houillier P, Jayat M, Baraka-Vidot J, et al.: A mouse model of pseudohypoaldosteronism type II reveals a novel mechanism of renal tubular acidosis. Kidney Int 94: 514–523, 2018 [DOI] [PubMed] [Google Scholar]
- 134.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, et al.: Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: Role of pendrin in mineralocorticoid-induced hypertension. Hypertension 42: 356–362, 2003 [DOI] [PubMed] [Google Scholar]
- 135.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, et al.: Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci U S A 98: 4221–4226, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Václavík J, Sedlák R, Plachy M, Navrátil K, Plásek J, Jarkovsky J, et al.: Addition of spironolactone in patients with resistant arterial hypertension (ASPIRANT): A randomized, double-blind, placebo-controlled trial. Hypertension 57: 1069–1075, 2011 [DOI] [PubMed] [Google Scholar]
- 137.Jia G, Habibi J, DeMarco VG, Martinez-Lemus LA, Ma L, Whaley-Connell AT, et al.: Endothelial mineralocorticoid receptor deletion prevents diet-induced cardiac diastolic dysfunction in females. Hypertension 66: 1159–1167, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lother A, Fürst D, Bergemann S, Gilsbach R, Grahammer F, Huber TB, et al.: Deoxycorticosterone acetate/salt-induced cardiac but not renal injury is mediated by endothelial mineralocorticoid receptors independently from blood pressure. Hypertension 67: 130–138, 2016 [DOI] [PubMed] [Google Scholar]
- 139.Gueret A, Harouki N, Favre J, Galmiche G, Nicol L, Henry JP, et al.: Vascular smooth muscle mineralocorticoid receptor contributes to coronary and left ventricular dysfunction after myocardial infarction. Hypertension 67: 717–723, 2016 [DOI] [PubMed] [Google Scholar]
- 140.Amador CA, Bertocchio JP, Andre-Gregoire G, Placier S, Duong Van Huyen JP, El Moghrabi S, et al.: Deletion of mineralocorticoid receptors in smooth muscle cells blunts renal vascular resistance following acute cyclosporine administration. Kidney Int 89: 354–362, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ruilope LM, Agarwal R, Anker SD, Bakris GL, Filippatos G, Nowack C, et al.; FIGARO-DKD study investigators: Design and baseline characteristics of the finerenone in reducing cardiovascular mortality and morbidity in diabetic kidney disease trial. Am J Nephrol 50: 345–356, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, et al. ; FIDELIO-DKD Study Investigators : Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med 383: 2219–2229, 2020. [DOI] [PubMed] [Google Scholar]
- 143.Ito S, Shikata K, Nangaku M, Okuda Y, Sawanobori T: Efficacy and safety of esaxerenone (CS-3150) for the treatment of type 2 diabetes with microalbuminuria: A randomized, double-blind, placebo-controlled, phase II trial. Clin J Am Soc Nephrol 14: 1161–1172, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]