

Significance Statement
Renal ischemia-reperfusion injury is a major cause of AKI. The contribution of noncoding RNAs to this process is currently being explored. This study indicates that the long noncoding RNA H19 is silenced in adult mice and reactivated in ischemic AKI. Mechanistically, H19 functions as a competing endogenous RNA, ultimately activating important proangiogenic signaling pathways. Pharmacologic overexpression of H19 attenuates ischemic AKI by preserving capillary integrity. It may be used as a powerful therapeutic tool in the future care of patients with ischemic AKI.
Keywords: ischemic acute kidney injury, lncRNA, H19
Visual Abstract

Abstract
Background
Renal ischemia-reperfusion (I/R) injury is a major cause of AKI. Noncoding RNAs are intricately involved in the pathophysiology of this form of AKI. Transcription of hypoxia-induced, long noncoding RNA H19, which shows high embryonic expression and is silenced in adults, is upregulated in renal I/R injury.
Methods
Lentivirus-mediated overexpression, as well as antisense oligonucleotide-based silencing, modulated H19 in vitro. In vivo analyses used constitutive H19 knockout mice. In addition, renal vein injection of adeno-associated virus 2 (AAV2) carrying H19 caused overexpression in the kidney. Expression of H19 in kidney transplant patients with I/R injury was investigated.
Results
H19 is upregulated in kidney biopsies of patients with AKI, in murine ischemic kidney tissue, and in cultured and ex vivo sorted hypoxic endothelial cells (ECs) and tubular epithelial cells (TECs). Transcription factors hypoxia-inducible factor 1-α, LHX8, and SPI1 activate H19 in ECs and TECs. H19 overexpression promotes angiogenesis in vitro and in vivo. In vivo, transient AAV2-mediated H19 overexpression significantly improved kidney function, reduced apoptosis, and reduced inflammation, as well as preserving capillary density and tubular epithelial integrity. Sponging of miR-30a-5p mediated the effects, which, in turn, led to target regulation of Dll4, ATG5, and Snai1.
Conclusions
H19 overexpression confers protection against renal injury by stimulating proangiogenic signaling. H19 overexpression may be a promising future therapeutic option in the treatment of patients with ischemic AKI.
Ischemia-reperfusion (I/R) injury of the kidney is a major cause of AKI. Because of its high morbidity and mortality, it represents a major socioeconomic health problem.1 A variety of injurious insults in native kidneys may promote its development (e.g., during cardiac surgery). In addition, it is an unavoidable phenomenon during the kidney transplantation procedure.2 Noncoding RNAs, which comprise >98% of the human genome, may contribute to the induction or resolution of this process.3–7 They are divided into long noncoding RNAs (lncRNAs; ≥200 nucleotides) and small noncoding RNAs (≤200 nucleotides).
The adult kidney is characterized by an ability to respond to acute injury partly through activation of embryonic signaling pathways, among which include the Wnt, Bone Morphogenetic Protein, and retinoic acid signaling pathway, as well as the paired-box gene family among others.8 We identified a novel mechanism of renal regeneration involving the lncRNA H19, which we found to be highly induced in ischemic kidneys, as a proregenerative, anti-inflammatory, and antiapoptotic factor. Under physiologic conditions, H19 shows a high expression in embryonic kidneys, and is undetectable in adult life.9 We previously identified H19 to be upregulated in plasma samples of patients with AKI.10 However, the downstream mechanisms and therapeutic potential of H19 in kidney injury have never been investigated. Therapies aimed at restoring kidney function, and preventing inflammation and cell death, are an area of intense research initiatives to ameliorate I/R injury. Here, we provide evidence for the role and molecular mechanisms of H19 during renal regeneration in I/R injury of native kidneys. We show that H19 is highly expressed in embryonic kidneys and reactivated during ischemic AKI. We decipher the upstream regulators and downstream target genes of H19. Most importantly, overexpression of H19 in vivo protected from ischemic AKI.
Methods
Kidney Biopsies of Patients
In kidney transplant recipients, who presented with prolonged (707±73 minutes, n=5) and short (208±56 minutes, n=5) cold ischemia time (CIT), renal biopsy specimens and clinical and demographic data were collected. Patient characteristics are shown in Table 1.
Table 1.
Demographics of transplant patients with long and short CIT
| Demographics/underlying kidney disease | Short CIT | Long CIT |
|---|---|---|
| Recipient sex, male/female | 1/4 | 2/3 |
| Recipient age, yr | 46.2±3.9 | 47.0±4.7 |
| Cause of ESKD, n | ||
| GN | 3 | 2 |
| Hypertensive/diabetic nephropathy | 1 | 2 |
| Unknown | 1 | 1 |
| CIT, min | 208±56 | 707±73 |
Study Approval
Written informed consent was obtained from all patients. The local ethics committee approved the study.
Animals
Male C57BL/6 mice were housed under standard conditions. Mice that were 10–12 weeks old, weighing between 20 and 30 g, were used for all experiments. Constitutive H19 knockout mice (H19Δ3) were obtained from Luisa Dandolo (University Paris Descartes). The H19Δ3 strain carries a 3-kb deletion of the H19 transcription unit. For details regarding generation of these mice, please to refer to the initial description by Ripoche et al.11 Genotyping was done by PCR on tail DNA (see Supplemental Figure 17).
I/R Injury Protocol
After administration of isoflurane anesthesia, male C57BL/6 mice were subjected to median laparotomy; thereafter, the left renal pedicle was dissected and a vascular clamp was applied for 30 minutes (unilateral renal I/R-injury). H19 was overexpressed in vivo by injecting adeno-associated virus serotype 2 (AAV2) vectors (H19-AAV2) directly into the renal vein at the time of clamping. In this setting, the contralateral kidney serves as an internal control to the injured kidney (I/R kidney). These animals were euthanized on days 1 and 7 after renal I/R injury, and kidneys were harvested for further examination. In each experiment five mice were included per group. For measurement of renal function parameters, mice were subjected to bilateral renal I/R injury (ischemia time of 15 minutes). Sham-operated animals (n=5 per time point) were also included. Blood samples for analysis of kidney function parameters were drawn on days 0, 1, 3, 7, and 14 and either analyzed using a Thermo Fisher Scientific (EIABUN) colorimetric detection kit as per the manufacturer’s instructions (for BUN concentration), or on a Beckman Analyzer (Beckman Instruments GmbH, Munich, Germany) (for creatinine). In vivo studies conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (publication number 85–23, revised 1996). Animal experiments were approved by local authorities (Kantonales Veterinäramt). Constitutive H19 knockout mice and littermate wild-type controls were subjected to unilateral (ischemia time of 30 minutes) and bilateral renal I/R injury (ischemia time of 15 minutes), with five animals per group.
Histology and Immunohistochemical Staining
The kidney from each euthanized mouse was removed and either placed in optimal cutting temperature (OCT) compound and snapped frozen in liquid nitrogen, or placed in 10% formalin. The tissues were processed at the Histology Facility at the University of Zurich, and stained with periodic acid–Schiff (PAS). For immunofluorescence stainings, 5-µm-thick frozen sections were stained for F4/80 and CD3 analyses. Vascular cells were visualized by tomato lectin or endomucin staining, and proximal tubular cells by lotus tetragonolobus lectin staining. All tissues were incubated with the primary antibodies overnight and detected with fluorescence-labeled anti-species antibodies. The controls consisted of either no primary antibody or a nonspecific IgG. Fluorescence images were obtained using a Leica SP8 upright confocal microscope and LAS AF software, at the Center of Microscopy and Image Analysis, University of Zurich.
The severity of morphologic renal damage was assessed in a blinded manner, using an arbitrary score on the basis of PAS-stained kidney sections, following a modification of a protocol developed by Broekema et al.12 Briefly, the extent of four typical I/R injury–associated damage markers (i.e., dilatation, denudation, intraluminal casts, loss of brush border membrane, and cell flattening) was expressed in arbitrary units in a range of 0–4, according to the percentage of damaged tubules: 0, no damage; 1, <25% damage; 2, 25%–50% damage; 3, 50%–75% damage; and 4, >75% damage.
Construction and Production of AAV2 Vectors
Full-length H19 was amplified by PCR, using mouse genomic DNA; subsequently, two miR-122 binding sites (for detargeting in the liver) were added also by PCR, and the whole piece was cloned into the pTREK1 plasmid (kindly provided by Roger Hajjar, Icahn School of Medicine at Mount Sinai, New York). For virus production, HEK293-T cells were grown in triple flasks (DMEM, 10% FBS) for 24 hours before transfection. The transgene plasmid, together with the helper plasmid (pDP2rs, also kindly provided by Roger Hajjar), was transfected into HEK293-T cells using polyethylenimine (Sigma-Aldrich). After 72 hours, cell crude lysates were produced, treated with benzonase, and the virus particles were purified over an iodixanol density gradient (Optiprep; Sigma-Aldrich). Viral titers were determined by quantitative real-time PCR (qPCR) on vector genomes, using the FastStart Universal SYBR Green Master Mix (Roche). Mice were injected directly into the renal vein at the time of clamping.
Spatial Resolution of Renal lncRNA H19
Mus musculus H19 was in situ hybridized using RNAscope technology according to the manufacturer’s instructions (RNAscope; Advanced Cell Diagnostics, Hayward, CA). Briefly, the H19 target probe was designed to contain 20 distinct double Z probe pairs targeting the H19 RNA region between the nucleotides 762 and 1703 (catalog number 501671), and to be labeled with horseradish peroxidase. Mouse kidney tissues were fixed and paraffin embedded. Kidney paraffin blocks were cut into 4-μm sections and pretreated according to the manufacturer’s instructions. The H19 target probe was hybridized for 2 hours at 40°C, using the HybEZ oven by Advanced Cell Diagnostics. The H19 signals were amplified in a series of six distinct amplifications steps. The horseradish peroxidase–labeled H19 probe was finally detected by 3,3′-diaminobenzidine (2.5 HD reagent kit BROWN; Advanced Cell Diagnostics). To resolve H19 expression in context with the renal morphology, kidney sections were counterstained with 50% Gill’s hematoxylin and subsequently dehydrated by a 15-minute baking step at 60°C. Kidney sections were mounted using the xylene-based mounting medium Eukitt (Sigma-Aldrich).
Ex Vivo Cell Purification
The cellular origin of H19 after induction of I/R injury was investigated by FACS analysis, using specific antibodies and following a protocol by Chau et al.,13 with modifications. After clamping of the left renal pedicle and a reperfusion period of 24 hours, the kidney was extracted, decapsulated, homogenized, and then incubated at 37°C for 30 minutes, with collagenase II (81 U/ml) and DNase (100 U/ml; Roche) in serum-free DMEM. After centrifugation, cells were resuspended in 5 ml of PBS/1% BSA, and filtered (40 μm). Cells were separated using the following specific antibodies or lectins: rat anti–mouse-CD31-PE (1:400; BD Pharmingen), rat anti–mouse-CD45-PerCP-Cy5.5 (1:100; BD Pharmingen), lotus tetragonolobus lectin (1:200; DAKO), or aquaporin (Aqp1)-biotin for proximal epithelium; and anti–mouse Tim1-biotin (1:200; eBioscience) followed by streptavidin-APC (1:2000; BD Pharmingen) for injured proximal epithelium. Subsequently, RNA was isolated by the TRIzol method.
In Vitro Analyses
In vitro experiments were performed using human umbilical vein endothelial cells (HUVECs) and renal proximal tubular epithelial cells (RPTECs). Cells were exposed to normoxia and hypoxia (0.1% oxygen level).
Lentivirus-Mediated H19 Overexpression
The complete sequence of human H19 was amplified by PCR and cloned into VVPW lentiviral expression vector (kind gift from Luca Gusella, Mount Sinai Hospital, New York). HEK293-T cells grown to 70%–80% confluence were transfected with VVPW, the packaging plasmid psPAX2, and the envelope plasmid pCMV-VSVG (from Addgene), at a ratio of 3:2:1, respectively, using FuGENE HD transfection reagent, according to the manufacturer`s instructions.
qPCR
Real-time PCR was performed using a 7500 fast thermocycler (Applied Biosystems). Total RNA was isolated using a miRNeasy mini kit, and 1 μg of total RNA was reversed transcribed into cDNA using qScript cDNA supermix. Primers were designed using IDT primers designing tool software, purchased from IDT. Primers used in this study are listed in Supplemental Table 2 (human) and Supplemental Table 3 (mouse). Gene expression was normalized to Valosin-containing Protein (VCP) and Asparagine-Linked Glycosylation Protein 9 Homolog (ALG9). The normalized values (ΔCT) were then compared with untreated cells or control kidneys (ΔΔCT). The results are expressed as log10 (2–∆∆Ct).
microRNA Analyses
miR mimics, inhibitors, and primers were purchased at Thermo Fisher Scientific. For qPCR expression analysis of miR-30a-5p expression, miR-16 was used as a reference microRNA.
SDS-PAGE and Western Blotting
SDS-PAGE was done using 25 µg of cell lysate (cell lysis buffer: 50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 50 mM sodium fluoride, 0.5% Triton X-100, 1mM PMSF, 10 µg/ml leupeptin, 10 µg/ml aprotinin, and a protease inhibitor cocktail), and resolved using 12% acrylamide/Bis gels under denaturing conditions at 120 V for 90 minutes. Proteins were transferred onto nitrocellulose membrane (Bio-Rad) at 100 V for 60 minutes, and the membranes probed using the primary antibodies at the manufacturer’s recommended concentrations. The membranes were washed five times with TTBS, and probed with the appropriate HRP-labeled anti-species secondary antibodies for 1 hour. The membranes were washed three times with PBST, and developed using an ECL prime kit (GE Healthcare).
EdU Proliferation Assay
Cellular proliferation was evaluated using Click-iT EdU flow cytometry assay kit (C-10420; Invitrogen), as per the manufacturer’s instructions. Briefly, HUVECs or RPTECs treated with H19-expressing lentivirus or empty virus were incubated with 10 µM EdU, at 37°C and 5% CO2, for 24 hours. The cells were washed with wash buffer (PBS plus 1% BSA), and fixed with Click-iT fixative for 15 minutes, at room temperature, in the dark. The cells were later washed and permeabilized with Click-iT Triton X-100–based permeabilization buffer for 30 minutes, at room temperature, in the dark. The EdU was detected by the Alexa-Fluor 488 antibody provided by the kit, and the proliferating cells were analyzed by flow cytometer.
Chemotaxis/Boyden Chamber Assay
The cell migration assay was performed using 24-well Transwell plates with 8-µm pores (3422; Corning). HUVECs or RPTECs overexpressing H19 were loaded with the cell tracker CFMDA (Invitrogen), and seeded at a density of 105 cells in the insert in the presence of incomplete medium. Complete medium (with all growth factors and 10% FBS) or with 25 ng/ml recombinant human VEGF were placed in the bottom chamber. To eliminate the concentration gradient of the chemotactic factors, in some wells, complete medium was placed in the top insert and in the bottom well. The plate was incubated at 37°C and 5% CO2, for 24 hours. The next day, the filters were cut out of the insert, placed on a microscope slide, and visualized using a fluorescence microscope. Five fields of view per filter were counted.
Scratch Migration
RPTECs transduced with an empty vector or with lentivirus targeting H19 were seeded in a 96-well tissue culture plate, at a density of 103 cells/well, in triplicate, and cultured overnight at 37°C and 5% CO2. Thereafter, a scratch was placed using a 200-µl pipette tip in the middle of the cell monolayer. The cells were washed two times with PBS, and 100 µl of medium was added to the cells. Cells were visualized using an inverted microscope, and images of each well were captured. The area covered was calculated using ImageJ software.
Apoptosis Assay
HUVECs and/or RPTECs treated with H19 antisense oligonucleotides (ASOs) or lentivirus-mediated overexpression of H19 were subjected to an apoptotic stimulus including cycloheximide (25 ng/ml) and TNFα (20 ng/ml), for 3 hours, at 37°C and 5% CO2. The cells were washed two times with PBS, and apoptosis was determined using a Caspase-Glo 3/7 fluorescence assay (Promega), as per the manufacturer’s instructions.
MTT Cell Viability Assay
HUVECs overexpressing H19, lentivirus-GFP, empty vector, or untreated cells were seeded at 10,000 cells/well, in a 96-well tissue culture plate. The cells were treated with cycloheximide (25 ng/ml) and TNFα (20 ng/ml) overnight (approximately 16 hours). The cells were washed three times with PBS, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added before incubation for 4 hours, at 37°C and 5% CO2. The plate was centrifuged for 2 minutes at 1200 rpm, and the supernatants were discarded. Formazan crystals were solubilized with 100 µl dimethyl sulfoxide, and the absorbance was measured at 570 nm.
Small Interfering RNA Transfection
All small interfering RNA (siRNA) sequences were purchased from QIAGEN. The ASOs were purchased from IDT. The gene knockdown procedure was on the basis of the QIAGEN HiPerFect (301705) Transfection protocol. Cells were transfected with 50–100 nM siRNA, or with 1 nM ASOs at 50%–60% confluency. Knockdown efficiency was evaluated by Western blotting and/or real-time PCR, after 72 hours.
Luciferase Assay
H19 promoter was cloned into pXPG plasmid (a gift from Peter Cockerill, plasmid 71248; Addgene), and H19, Dll4, ATG5, and SNAI1 3′UTR were cloned into pIS2 plasmid (a gift from David Bartel, plasmid 12177; Addgene). HUVECs were transfected with 1 µg of either plasmid, using ViaFect (Promega) transfection reagent. In addition, cells were transfected with 10 nM of miR control, miR-30–5p mimic, or miR-30–5p inhibitor, using HiPerFect (Qiagen) transfection reagent. Luciferase activity was determined after 72 hours, using firefly (pXPG plasmid) or Renilla (pIS2) luciferase assay system (Promega), as per the manufacturer’s instructions. The background autoluminescence of the luciferase substrate was subtracted from all of the experimental samples, and the luciferase assay was normalized to protein concentrations determined by BCA protein assay. The calculation was performed according to reporter activity/total protein.
Three-Dimensional Angiogenesis Assay
The study of sprouting angiogenesis of HUVECs overexpressing H19 in vitro was determined using the protocol described by Nakatsu et al.,14 with some modifications. HUVEC were mixed with collagen-coated cytodex 3 microcarrier beads (C3275; Sigma-Aldrich), and incubated at 37°C and 5% CO2 for 4 hours, with shaking every 20 minutes. The coated beads were then transferred to T25 tissue culture flasks, and incubated for further 2 hours. The coated beads were washed two times with PBS, and mixed with 2 mg/ml fibrinogen (F4753; Sigma-Aldrich) and 0.15 U/ml aprotinin (A1153; Sigma-Aldrich). Next, 500 µl of the HUVEC-coated beads and fibrinogen solution was added to 0.625 U/ml thrombin (T4648; Sigma-Aldrich) in a 24-well tissue culture plate, and incubated at 37°C and 5% CO2 for approximately 15 minutes, until the fibrin gel hardened. Then, 1 ml of EBM2 was added to each well, and sprout formation was analyzed after 24 hours.
Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation (ChIP) of the transcription factors SPI1 and LHX8 was performed using MAGnify Chromatin Immunoprecipitation System (Thermo Fisher Scientific), as per the manufacturer’s instructions. For ChIP analysis, HUVECs were used. The mock IgG is a nonspecific rabbit IgG provided in the kit. The input DNA was normalized as described in the manual (Magnify immunoprecipitation; Invitrogen). Briefly, the replicate measurements for each immunoprecipitation were averaged. For each primer pair, the adjusted Ct for the input controls were calculated as follows: an input control that was 10% of the immunoprecipitation reaction was used as dilution factor of 10, and 3.32 cycles (log2 of 10) were subtracted from the Ct value of the input control. Then, the input control replicates were averaged. The normalized input was calculated with the equation 100×AE(average Ct input–average Ct immunoprecipitation), where AE is the amplification efficiency. To determine the amplification efficiency of the qPCR reaction, the input control DNA was prepared using ten-fold serial dilutions (1:1–1:1000). The efficiency range of the qPCR standard curve slopes was −3.3, and the AE was calculated using the equation 10(−1/slope), which, in this experiment, corresponded to 2.0009.
ATP Depletion
HUVECs were cultured in the presence of complete EBM2 (cEBM2) (single quots plus 10% FBS), until they reached 90% confluency. Cells were washed with PBS two times, and then cEBM2, incomplete EBM2, or PBS was added. Cells in cEBM2 and incomplete EBM2 were treated with 10 μM antimycin A plus 10 mM 2-deoxyglucose, overnight (approximately 16 hours). The cells were washed with PBS two times, and RNA was extracted.
Identification of microRNAs with Potential Binding to H19 and microRNA Target Prediction
To identify microRNA binding sites in the H19 sequence, we subjected the sequence of H19 to the PITA Catalogs of Predicted microRNA Targets Prediction Tool (Segal Laboratory of Computational Biology, Weizmann Institute, Rehovot, Israel; http://genie.weizmann.ac.il/pubs/mir07/index.html), which enables identification of potential microRNA binding sites because lncRNAs have been shown to sequester microRNAs, thereby functioning as so-called microRNA sponges. The microRNA databases and target prediction tools miRBase (http://microrna.sanger.ac.uk/), PicTar (http://pictar.mdc-berlin.de/), and TargetScan (http://www.targetscan.org/index.html) were used to identify potential microRNA targets.
Surface Plasma Resonance Measurements
Surface plasmon resonance (SPR) has previously been shown to serve as a highly sensitive tool to detect specific RNA-RNA interactions.15 SPR measurements were conducted at 20°C in HBS buffer (10 mM HEPES, 150 mM NaCl, 0.05% Tween-20, 3 mM EDTA; Teknova, Hollister, CA), using a Biacore T200 instrument (GE Healthcare). As a first step, neutravidin (Sigma) was immobilized on a CMD200M chip surface, according to a protocol recommended by the chip manufacturer (Xantec, Düsseldorf, Germany). The surface was activated using 50 mM EDC (Pierce)/100 mM NHS (Sigma) in water, for 550 seconds. Neutravidin was injected at a concentration of 0.1 mg/ml in 5 mM acetate buffer (pH 5.5), for 750 seconds. Unreacted NHS ester groups were blocked by injecting 1 M ethanolamine (Xantec) at pH 8.5, for 750 seconds. All coating steps and binding experiments were performed at a flow rate of 5 µl/min and 30 µl/min, respectively. Biotinylated oligonucleotides BIOmH19 (mouse) and BIOhH19 (human) were immobilized at 200 nM in HBS, resulting in a surface density of 715 RU and 680 RU, respectively. In a double-referenced binding experiment, a one-plus-one dilution series of eight concentrations of CY3-MIR30a-5p were injected in duplicate for 60 seconds, at a flow rate of 30 μl/min, in HBS buffer. Dissociation was measured for 300 seconds. After each measurement, the surface was regenerated with 0.1 M sodium hydroxide for 30 seconds. A neutravidin-coated flow cell of the sensor chip was used as a reference. Data were evaluated using Biacore software version 3.1. Sensorgrams were fitted using a one-plus-one kinetic model. Affinity was determined by fitting the concentration dependence of equilibrium values. Binding affinity is measured and reported by the equilibrium Kd, which is used to evaluate and rank order strengths of bimolecular interactions. The smaller the Kd value, the greater the binding affinity of the ligand for its target.
Statistical Analyses
Average data are presented as mean and SEM, unless otherwise stated. All statistical analyses were performed with GraphPad Prism software (GraphPad Prism Software Inc., San Diego, California). Two-sided P values <0.05 were considered statistically significant for all statistical procedures used. For statistical comparison of two groups, we used an unpaired two-tailed t test; for the comparison of three or more groups, we used one-way ANOVA followed by Tukey post hoc tests. In the figures, probability values are indicated by one (P<0.05), two (P<0.01), or three (P<0.001) asterisks.
Results
H19 Is Dysregulated in Hypoxic/Ischemic Kidney Injury in Humans, Mice, and Cells
We first assessed the expression level of H19 in kidney biopsies of transplant patients with long compared with short CIT, and found it to be highly increased, suggesting a possible induction by ischemia (Figure 1A). Patient characteristics are shown in Table 1. In murine kidney tissue H19 was found to be highly expressed in embryonic kidneys (E13.5), almost entirely repressed in adult kidneys, and reactivated in ischemic kidneys of mice (24 hours of reperfusion; Figure 1B). On day 7 after reperfusion, levels of H19 were still increased, but much lower compared with 24 hours of reperfusion (Figure 1B). To identify the cellular origin of H19 in ischemic AKI, we isolated CD31+/CD45− endothelial cells (Figure 1C), injured LTA+/KIM1+ proximal tubular epithelial cells (Figure 1D), and healthy LTA+/KIM1− cells (Figure 1E). Intriguingly, H19 was enriched in ischemic CD31+/CD45− endothelial cells and injured LTA+/KIM1+ proximal tubular epithelial cells, whereas its expression was unchanged in healthy LTA+/KIM1− proximal tubular epithelial cells. The tissue distribution of H19 was further investigated by RNAscope. Here, H19 was increased in peritubular capillaries and proximal tubular cells at 24 hours of reperfusion (Figure 1F). On day 7 after I/R injury, H19 was still increased in peritubular capillaries and proximal tubular cells (Figure 1G), albeit much lower compared with 24 hours of reperfusion. In line with this finding, H19 was induced in cultured endothelial cells (HUVECs, at 72 hours of hypoxia) and proximal tubular epithelial cells (RPTEC, at 24 hours of hypoxia) by hypoxia/reoxygenation (Figure 1, H and I). Moreover, H19 expression was upregulated in response to overnight (16 hours) serum starvation, ATP depletion, and, most prominently, in response to serum starvation in combination with ATP depletion (Figure 1J).
Figure 1.

The expression of H19 is increased in cells, humans, and mice under ischemic/hypoxic conditions. H19 expression was determined in kidney transplant patients with short compared with long CIT (A) (n=5 patients per group), in mouse embryonic (E13.5), adult, and I/R kidneys (B) (n=8 mice per group). FACS-based sorting of mouse kidney homogenates after I/R injury exposed to rat anti–mouse-CD31-PE/rat anti–mouse-CD45-PerCP-Cy5.5 antibody for endothelial cells (C), and injured or healthy proximal tubular cells positive for lotus tetragonolobus lectin for proximal epithelium and anti-mouse Tim1-biotin antibody followed by streptavidin-APC (D and E). RNAscope analysis of H19 expression in control mice and mice after I/R injury and 24 hours of reperfusion (F), and 7 days of reperfusion (G) (n=5 mice each). Arrows indicate endothelial expression, asterisk indicates tubular epithelial induction. Expression of H19 in cultured endothelial cells (HUVECs) (H) and proximal tubular cells (RPTECs) (I), under hypoxic condition over a 72-hour time course. H19 expression after 16 hours of serum starvation or ATP depletion (n=4) (J). Each bar represents the mean±SEM of duplicate cultures, and the data are from a representative experiment of six independent experiments (unless otherwise stated) that were performed. *P<0.05; **P<0.01; ***P<0.001. A/DG, actinomycin A/2-deoxyglucose; NT, no treatment; TPL, kidney transplantation.
Transcriptional Regulation of H19
The transcriptional regulation of H19 was determined under hypoxic conditions. The hypoxia-responsive transcription factors HIF1α and Endothelial PAS domain protein 1 (EPAS or HIF2α) were detectable at up to 24 hours of hypoxia in HUVECs (Supplemental Figure 1, A and B). siRNA silencing of HIF1α and EPAS resulted in a reduction of H19 expression after 72 hours of hypoxia (Figure 2A) in HUVECs, suggesting an indirect regulation of its expression, because HIF1α/EPAS were no longer detectable by hypoxia day 3 (Supplemental Figure 1, C and D).
Figure 2.
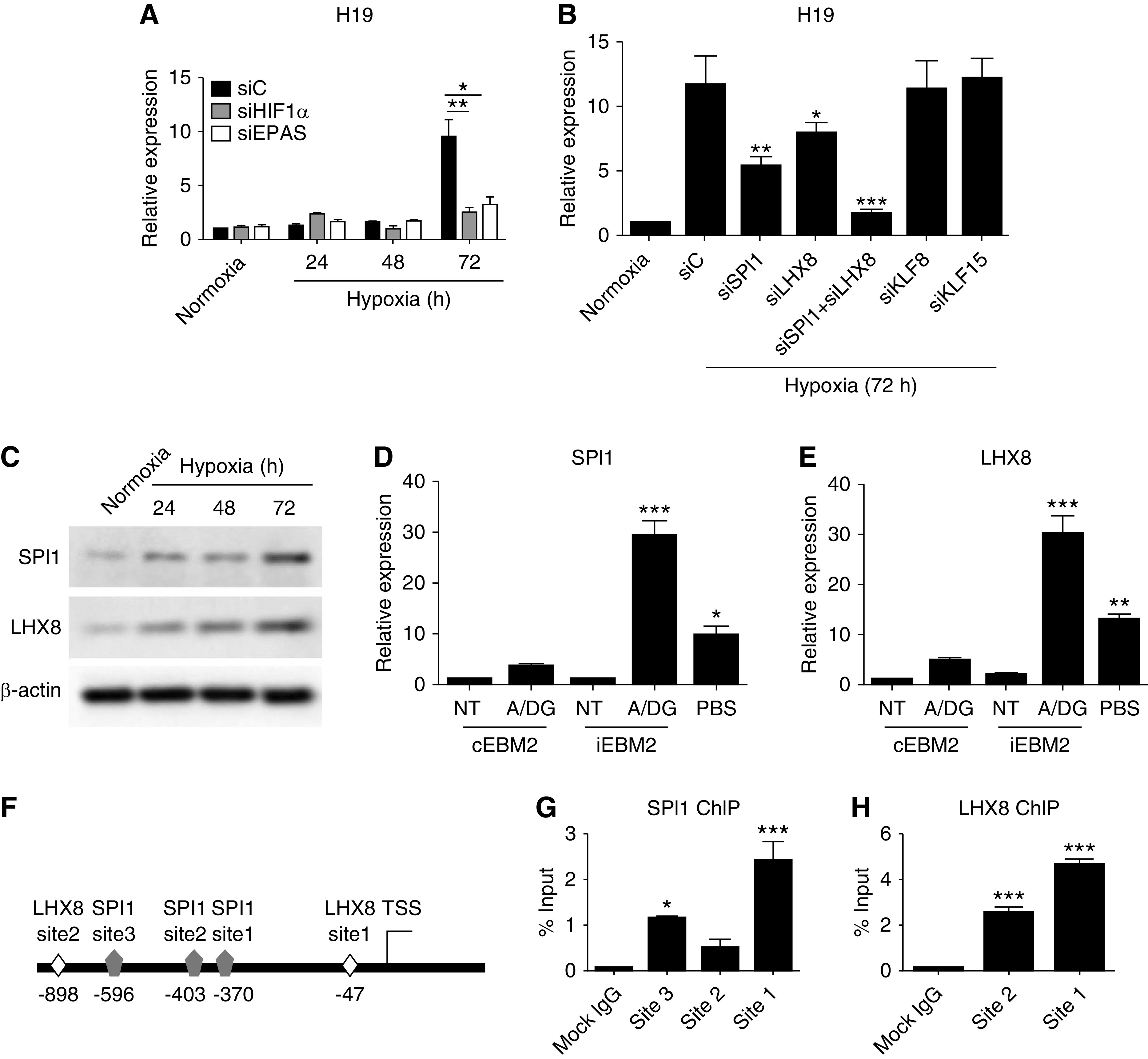
H19 is transcriptionally regulated by HIF1α, EPAS, SPI1, and LHX8. (A) Expression of H19 in response to HIF1α and/or EPAS siRNA knockdown over a 72-hour hypoxia time course. (B) H19 expression after transcription factor (SPI1, LHX8, KLF8, and KLF15) knockdown and 72 hours of hypoxia compared with normoxia. (C) Protein expression of SPI1 and LHX8 over a 72-hour hypoxia time course (n=4). (D and E) SPI1 and LHX8 induction in response to serum starvation in combination with ATP depletion or to serum starvation alone (n=4). (F) Schematic representation of SPI1 and LHX8 binding sites in the H19 promoter region. Amplification by qPCR after ChIP using specific primers to verify the binding of (G) SPI1 and (H) LHX8 to the promoter region of H19 in HUVECs subjected to 72 hours of hypoxia. Each bar represents the mean±SEM of duplicate cultures, and the data are from a representative experiment of six independent experiments that were performed (unless otherwise stated). *P<0.05; **P<0.01; ***P<0.001. A/DG, actinomycin A/2-deoxyglucose; NT, no treatment; siC, siRNA control; TSS, transcriptional start site.
Given the induction of expression of H19 in HUVECs at 72 hours of hypoxia (Figure 1G), additional transcription factors were searched for by assessing potential binding in a region 1000 bp upstream of the transcriptional start site of H19. As a result, 62 transcription factors were identified (Supplemental Table 1) and assessed by qPCR for potential upregulation after 3 days of hypoxia. Only four transcription factors, including SPI1 (PU.1), LIM homeobox 8 (LHX8), and Krüppel-like factors 8 and 15 (KLF8 and KLF15), matched the criteria (Supplemental Figure 2, A–D).
Simultaneous knockdown of both SPI1 and LHX8 resulted in the highest decrease of H19 expression, compared with the individual knockdown of SPI1 and LHX8 (also significantly downregulated), but knockdown of KLF8 or KLF15 had no effect (Figure 2B). The protein expression level of SPI1 and LHX8 showed a time-dependent increase under hypoxic conditions, peaking at 72 hours of hypoxia (Figure 2C). The knockdown efficiency of both SPI1 and LHX8 is shown in Supplemental Figure 2, E–H. In contrast, siRNA knockdown of HIF1α in RPTECs suppressed H19 expression at 24 hours of hypoxia, and significantly suppressed H19 expression at 72 hours of hypoxia (Supplemental Figure 3).
Interestingly, SPI1 and LHX8 were also induced after 16 hours of serum starvation and ATP depletion, in line with H19 expression (Figure 2, D and E). To validate the interaction of SPI1 and LHX8 with the H19 promoter, a ChIP assay was used. SPI1 and LHX8 putative binding sites are shown in a schematic representation (Figure 2F). Sheared chromatin, isolated from HUVECs exposed to hypoxia for 72 hours, were subjected to antibody pulldown and qPCR analysis. Specific primers that flanked each putative SPI1 and LHX8 binding site were analyzed by qPCR. Significant amplification of the SPI1 putative binding sites at −370 and −596 (Figure 2G), and LHX8 putative binding sites at −47 and −898 (Figure 2H), was detected.
The H19 promoter was cloned into firefly luciferase vector pXPG. HUVECs or RPTECs were transfected with 1 µg of plasmid and the cells were subjected to a hypoxia time course over 72 hours. Luciferase activity showed that the H19 promoter remained significantly active over 3 days of hypoxia in HUVECs, whereas the promoter activity in RPTECs was active for the first 48 hours of hypoxia (Supplemental Figure 4, A and B).
Functional Role of H19 in Endothelial Cell Biology
The effect of H19 modulation on cellular migration in HUVECs was evaluated using a modified Boyden chamber assay. Silencing of H19 slightly decreased the capacity to migrate (data not shown), whereas lentiviral overexpression of H19 significantly increased VEGF-induced cellular migration (Figure 3A). Endothelial cell proliferation in response to VEGF was significantly induced by H19 lentiviral overexpression (Figure 3B), whereas H19 silencing had minimal effects (data not shown). Lentiviral overexpression of H19 promoted VEGF-induced sprouting angiogenesis in a three-dimensional angiogenesis assay (Figure 3C). In addition, silencing of H19 reduced the expression of angiogenic marker genes, including Dll1 (Delta-like 1), Dll4 (Delta-like 4), Apelin, Notch1, and Notch4, under hypoxia (Supplemental Figure 5).
Figure 3.
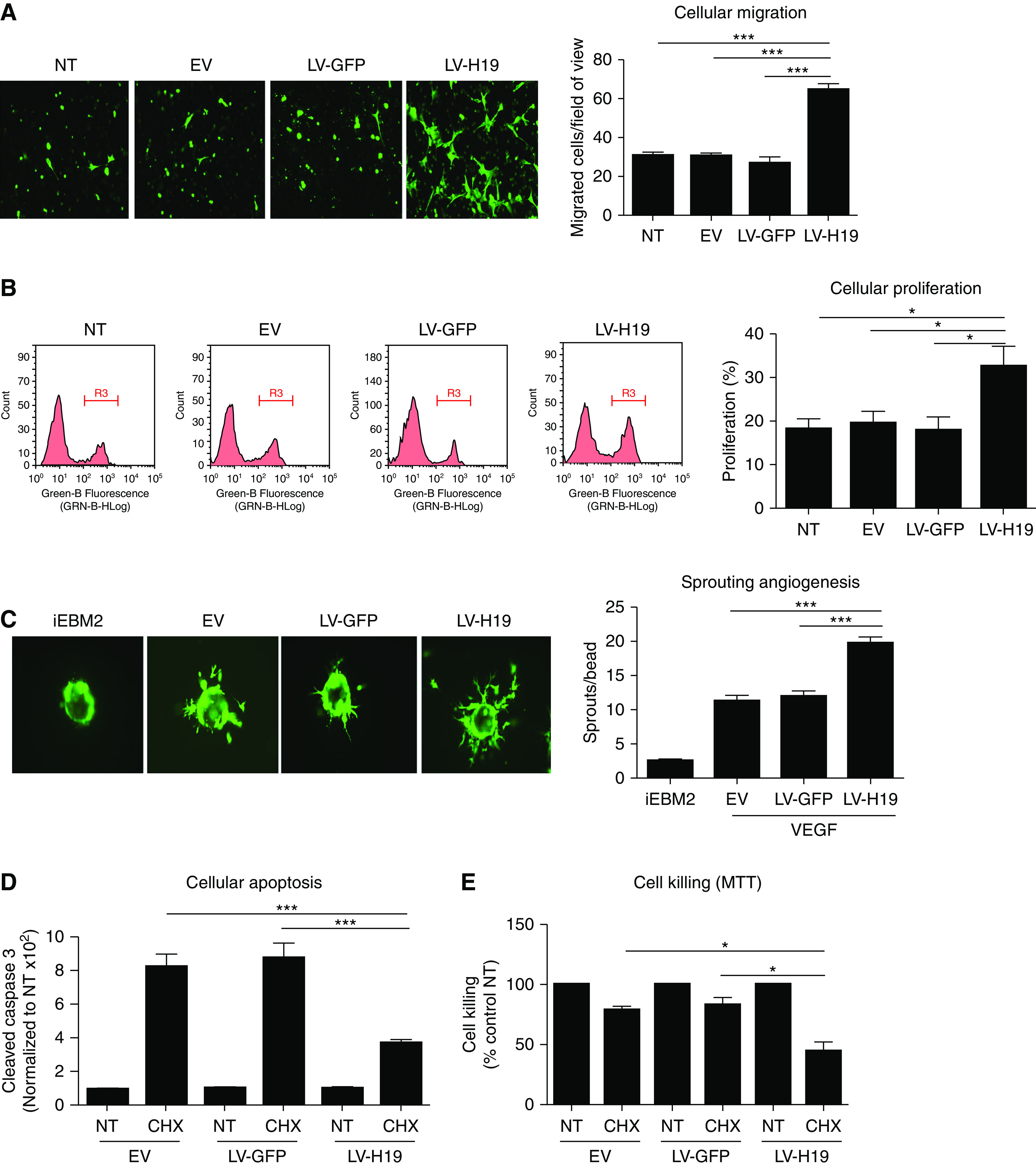
H19 increases migration, proliferation, and sprouting angiogenesis, and suppresses apoptosis in endothelial cells. (A) HUVECs overexpressing H19, GFP, empty vector, or left untreated were assayed for cellular migration in response to VEGF (50 ng/ml), using a modified Boyden chamber (8-μm pore size filters). (B) Cellular proliferation was determined using EdU cell proliferation system and flow cytometry in HUVECs treated with VEGF (50 ng/ml). (C) The effect of H19 overexpression on angiogenic potential of endothelial cells was determined using HUVECs coated on cytodex beads, and grown in fibrin gel in the presence of VEGF (50 ng/ml). (D) Cellular apoptosis was determined by cleaved caspase 3/7 in HUVECs overexpressing H19 in response to the apoptotic stimulus TNFα (10 ng/ml) and cycloheximide (25 µg/ml). (E) The effect of H19 overexpression on protection against an apoptotic stimulus in HUVECs is determined by MTT cellular toxicity assay, in response to the apoptotic stimulus TNFα (10 ng/ml) and cycloheximide (25 µg/ml). Each bar represents the mean±SEM of duplicate cultures, and the data are from a representative experiment of six independent experiments that were performed. *P<0.05; **P<0.01; ***P<0.001. CHX, cycloheximide; EV, empty vector; LV-GFP, lentivirus expressing GFP; LV-H19, lentivirus expressing H19; NT, no treatment; VEGF, vascular endothelial growth factor.
Overexpression of H19 significantly reduced apoptosis in response to an apoptotic stimulus (Figure 3D). This observation was validated by MTT cellular killing assay (Figure 3E).
The effect of H19 overexpression on signaling pathways in response to the angiogenic factor VEGF was assessed in HUVECs. Overexpression of H19 enhanced AKT and ERK phosphorylation after 5 and 10 minutes of VEGF stimulation (Figure 4, A–C). Most notably, the p38 pathway was aberrantly deregulated because it was induced after 5 minutes after VEGF treatment and remained active even after 1 hour, whereas in control cells, the p38 pathway was induced after 10 minutes of VEGF treatment and returned to basal levels by 30 minutes. Interestingly, it appears that both p38 subunits α and β are deregulated (Figure 4, A and D). In contrast, under hypoxic conditions in HUVECs, the AKT, ERK, and p38 phosphorylation was marginally increased after 72 hours of hypoxia (Supplemental Figure 6).
Figure 4.
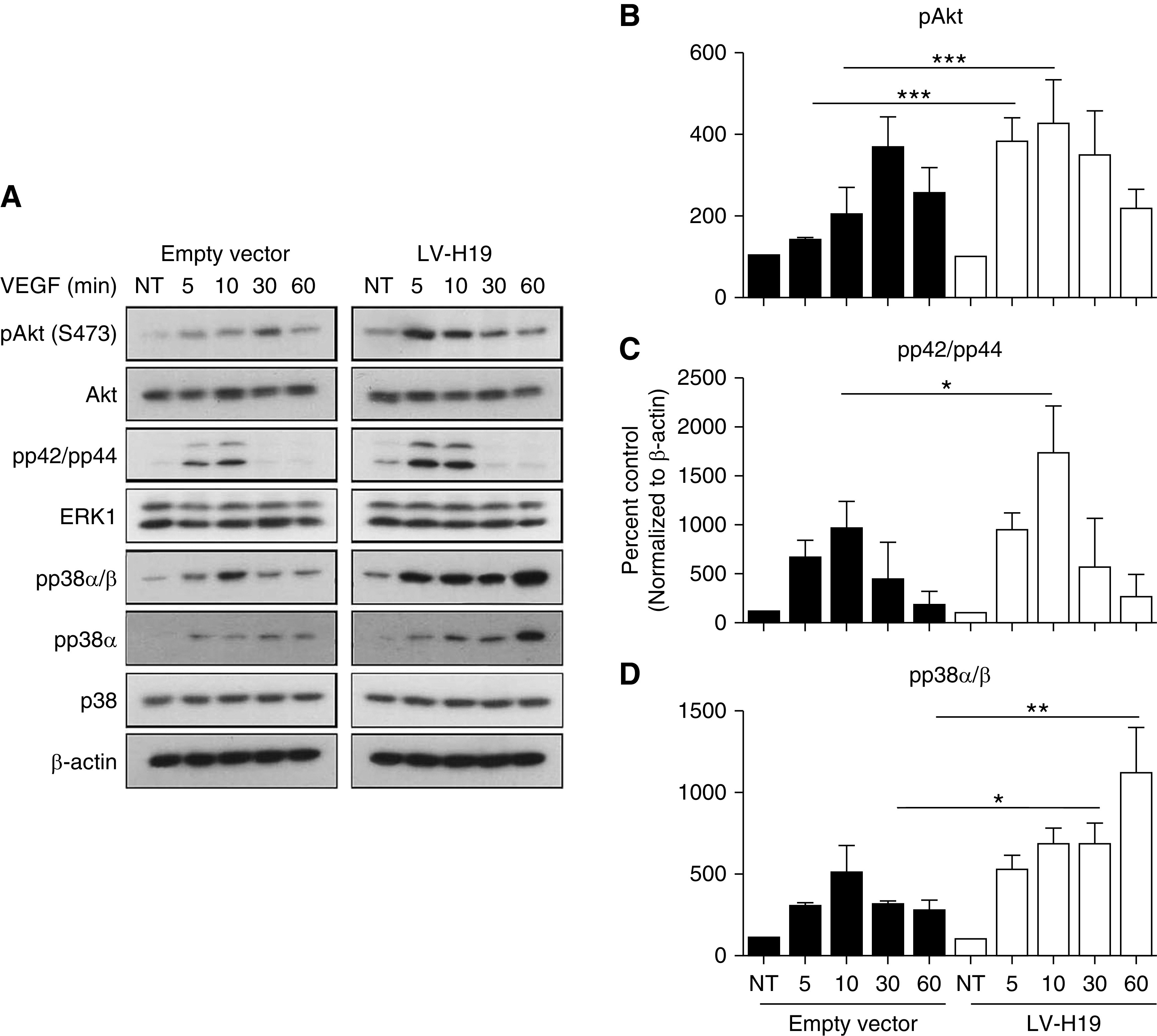
H19 promotes AKT, ERK, and p38 phosphorylation. (A) Shows representative Western blots of HUVECs transduced with a lentivirus to overexpress H19 as compared with cells transduced with an empty vector control. In both settings cells were treated with VEGF (25 ng/ml) at the indicated times. (B) Quantification of Akt phosphorylation, (C) ERK phosphorylation, and (D) p38 phosphorylation. Black bars represent cells after control virus transduction, white bars represent H19 lentivirus transduction. Each bar represents the mean±SEM of five independent experiments that were performed. *P<0.05; **P<0.01; ***P<0.001. LV-H19, lentivirus targeting lncRNA H19; NT, no treatment.
Downstream Interaction Partners of H19 in Endothelial Cells
lncRNAs are known to function as microRNA sponges in their role as competing endogenous RNA. MicroRNA binding sites in the H19 sequence were bioinformatically searched for by use of the PITA Catalogs of Predicted microRNA Targets Prediction Tool. miR-30a-5p was identified as a putative H19 target. The function of miR-30a-5p in the setting of hypoxia/ischemia in the kidney has not been described yet. To demonstrate the interaction between H19 and miR-30a-5p, H19 was cloned into pIS2 Renilla luciferase plasmid, and transfected into HUVECs in the presence of a microRNA control sequence, or with a miR-30a-5p mimic (overexpression) or miR-30a-5p inhibitor. In addition, five nucleotides in the H19 putative binding sequence CUUCC were deleted to generate a mutant plasmid. Figure 5A shows that miR-30a-5p overexpression markedly decreased luciferase activity, indicating a positive interaction between miR-30a-5p with H19. The mutant H19 sequence abrogated the effect of miR-30a-5p mimic treatment (Figure 5A).
Figure 5.
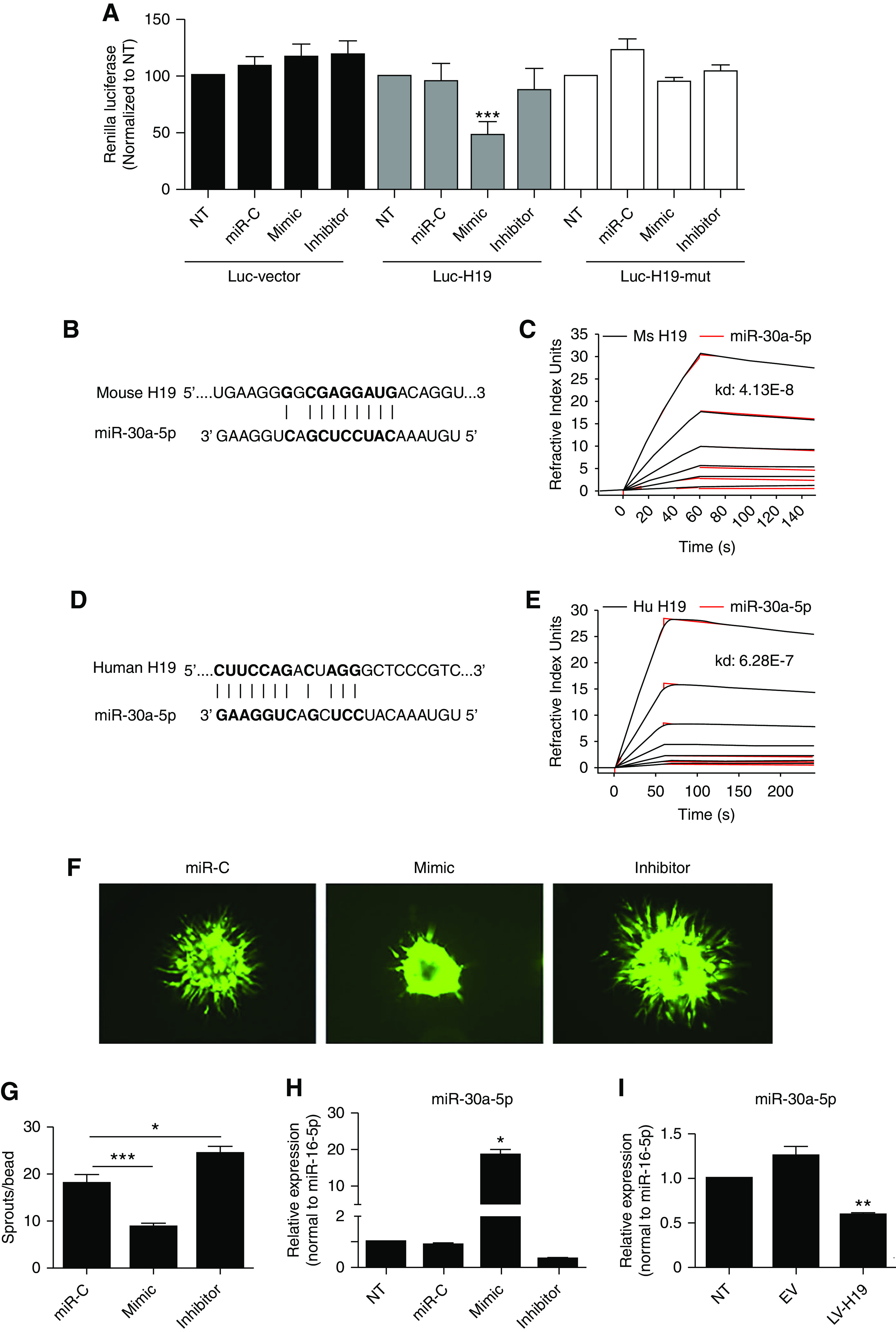
H19 sponges miR-30a-5p. (A) The potential binding of miR-30–5p with H19 was investigated using a luciferase assay. Human H19 was cloned into pIS2 Renilla luciferase vector. The mutant sequence was generated by 5- nucleotide (CUUCC) deletion, using Q5 site directed mutagenesis kit. Sequence alignment of miR-30a-5p and a binding region within (B) mouse H19 and (D) human sequence of H19. The interaction between H19 and miR-30–5p was validated using Plasmon resonance biosensor analysis (SPR) regarding the (C) mouse and (E) human sequences. The H19 probe is shown in black, and the miR-30a-5p probe is shown in red. Curves indicate strong binding between the two probes as measured by the Kd (mouse H19 miR-30a-5p Kd=4.13E−8, human H19 miR-30a-5p Kd=6.28E−7). (F) The overexpression of miR-30a-5p by the mimic sequence reduces sprouting, whereas inhibition increases sprouting activity of endothelial cells in a three-dimensional fibrin gel. (G) Quantification of results. (H) Quality control qPCR to demonstrate the increased expression of miR-30a-5p using a miR mimic and reduced expression by the specific anti-miR (or inhibitor). (I) The overexpression of H19 reduces the expression of miR-30a-5p in HUVECs. Each bar represents the mean±SEM of duplicate cultures, and the data are from a representative experiment of six independent experiments that were performed. *P<0.05; **P<0.01; ***P<0.001. EV, empty vector; LV-H19, lentivirus targeting H19; miR-C, miR control.
Furthermore, human and mouse H19 and miR-30a-5p interaction was also validated using SPR biosensor analysis. Biotinylated mouse (Figure 5, B and C) and human H19 oligonucleotides (Figure 5, D and E) were incubated with CY3-labeled miR30a-5p. As a result, the SPR analysis demonstrates a strong binding interaction between H19 and miR-30a-5p sequences (Kd values of 4.13E−8 and 6.28E−7 for mouse and human H19 sequences, respectively).
The effect of miR-30a-5p overexpression on endothelial cell sprouting activity was evaluated. miR-30a-5p overexpression significantly reduced the number of angiogenic sprouts, whereas inhibition of miR-30a-5p increased sprouting activity (Figure 5, F and G). Overexpression and silencing of miR-30a-5p was validated by qPCR (Figure 5H). Lentiviral overexpression of H19 suppressed miR-30a-5p expression in HUVECs, which is further evidence of H19 miR-30a-5p interaction (Figure 5I).
To understand the mechanism of miR-30a-5p in angiogenesis regulation, target genes that are involved in blood vessel formation, and are putative miR-30a-5p targets, were bioinformatically screened for. The bioinformatic scan revealed three potential miR-30a-5p targets that are reportedly involved in angiogenesis: (1) Autophagy-related 5 (ATG5),16 (2) Snail family transcriptional repressor 1 (SNAI1),17 and (3) Delta-like 4 (Dll4)18 (Figure 6). Western blot analysis revealed that all three targets were significantly reduced by miR-30a-5p overexpression in HUVECs, whereas miR-30a-5p inhibition significantly increased Dll4 expression, and a modest increase of ATG5 and SNAI1 was observed (Figure 6, A and B). Figure 6C shows the alignment of the miR-30a-5p sequence and putative binding sites in ATG5, Dll4, and SNAI1 3′UTR regions. To validate the interaction between miR-30a-5p and its target genes, the 3′UTR (wild-type or mutant) of ATG5, Dll4, and SNAI1 was cloned into the pIS2 Renilla luciferase vector. Interestingly, luciferase activity was significantly reduced in cells overexpressing miR-30a-5p and carrying the wild-type sequences. However, neither the miR-30a-5p inhibitory sequence nor the mutated ATG5, Dll4, and SNAI1 3′UTR sequences (deletion of TGTTT) had an effect on luciferase activity (Figure 6D), indicating that target regulation is specific to miR-30a-5p.
Figure 6.
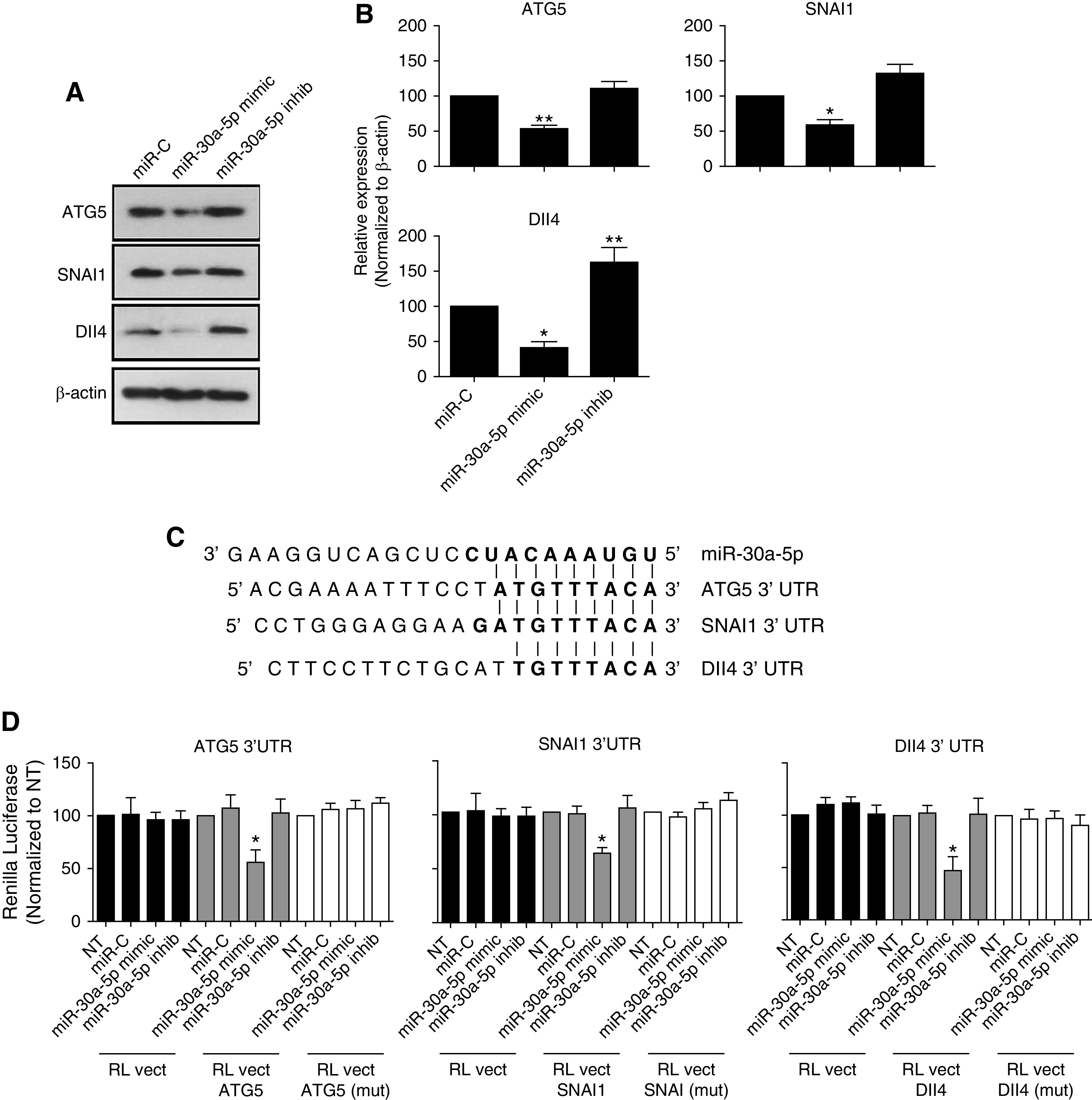
miR-30a-5p targets ATG5, SNAI1, and Dll4. (A) Representative Western blots and (B) quantification of results in HUVECs transfected with a miR-30a-5p mimic or anti-miR, and probed for Dll4, ATG5, and SNAI1. (C) Alignment of the miR-30a-5p sequence and a putative binding site in ATG5, SNAI1, and Dll4 3′UTR regions. The 3′UTR of ATG5, SNAI1, and Dll4 were cloned into the Renilla luciferase plasmid pIS2. The nucleotides sequence TGTTT was deleted by site-directed mutagenesis from ATG5, SNAI1, and Dll4 3′UTR, to generate the mutant plasmids. (D) HUVECs were transfected with the luciferase plasmids (ATG5, SNAI1, or Dll4) and either with miR-30a-5p mimic, inhibitor, or miR control sequences. Each bar represents the mean±SEM, and the data are representative of six independent experiments that were performed. *P<0.05; **P<0.01; ***P<0.001.
The effect of hypoxia on miR-30a-5p expression was assessed over a 72-hour time course. The expression decreased gradually over the hypoxia time course (Supplemental Figure 7A). The modulation of miR-30a-5p expression and its influence on angiogenic gene expression after 24 hours of hypoxia was evaluated. Supplemental Figure 7B shows efficiency of miR modulation. The mRNA expression levels of apelin, Dll1, Dll4, Kinase insert domain receptor (KDR), Notch1, and Notch4 were determined by qPCR (Supplemental Figure 7, C–H). Among those genes, unsurprisingly, only Dll4 expression was modulated by miR-30a-5p overexpression or inhibition. Hypoxic gene induction was confirmed by expression analysis of the hypoxia-sensitive gene GLUT1 (Supplemental Figure 7I).
Functional Role of H19 in Tubular Epithelial Cell Biology
A functional role of H19 in tubular epithelial cell biology could not be demonstrated in vitro. Please see the Supplemental Material for a presentation of investigated end points, including migration, proliferation, and apoptosis (Supplemental Figures 8–9). However, hypoxia in untreated cells increased migration and proliferation (Supplemental Figures 8D, 9B).
In Vivo Role of H19 on Tubular Epithelial Cell Injury, Proliferation, and Capillary Density at 24 hours of Reperfusion
The mouse H19 sequence was cloned into an AAV2 vector (AAV2-H19) and pharmacologically overexpressed in vivo, by injection of AAV2-H19 or control virus (AAV2-GFP or AAV2-CTL) directly into the renal vein at the time of clamping. AAV2-H19 delivery via the renal vein resulted in a robust and consistent overexpression of H19 in the kidney at 24 hours of reperfusion (Supplemental Figure 10, A and B). However, H19 expression was not restricted to the kidney, as the signal was also present in heart and lung tissue (Supplemental Figure 10, C–E). AAV2-H19 delivery via the renal vein resulted in a robust and consistent overexpression of H19 in the kidney at 24 hours of reperfusion (Figure 7A). A protective effect was detected in mice that were subjected to injection of AAV2-H19. Mice were injected immediately before a vascular clamp was applied to the renal pedicle. This resulted in a blunting of kidney damage, as demonstrated by KIM-1 and NGAL expression (Figure 7, B and C). Similarly, inflammatory gene expression, including TNFα (Figure 7D) and IL-1β (Figure 7E), was highly suppressed. CD31 expression as an indicator of endothelial injury was increased in mice receiving AAV2-CTL, and similar to control mice after AAV2-H19 injection (Figure 7F), suggesting a preservation of endothelial integrity. Additionally, outer medullary tubular epithelial injury amounted to 70%–80% in mice receiving AAV2-CTL (AV-GFP), whereas in mice with AAV2-H19, the percentage of injury was 40%–50% (Figure 7, G and H). Inflammatory cell influx, as assessed by CD3 and F4/80 immunofluorescence (Figure 7, I and J), and apoptosis (Figure 7K), as indicated by TUNEL staining, was significantly decreased in the AVV2-H19 group compared with the AVV2-CTL group. Moreover, a pronounced decrease in expression of adhesion molecules, particularly VCAM1 and E selectin, and to a lesser extent, ICAM1 and VE-cadherin, was seen (Supplemental Figure 11). We isolated CD31+/CD45− endothelial and aquaporin1+ proximal tubular epithelial cells after injection of AAV-H19, and compared with AAV2-CTL to verify H19 expression distribution and target regulation after virus injection. Here, we found H19 to be enriched in endothelial cells after AAV2 injection into the renal vein, in accordance with RNAscope data (Supplemental Figure 12A), whereas it was unaltered in aquaporin1+ proximal tubular cells (Supplemental Figure 12B), indicating that AAV2 injection primarily results in an endothelial phenotype. Concomitantly, miR-30a-5p was suppressed in endothelial cells, whereas it increased in proximal tubular epithelial cells (Supplemental Figure 12, C and D). Identified targets of miR-30a-5p, including Dll4, SNAI1, and ATG, were upregulated after miR-30a-5p suppression in the endothelial cell fraction (Supplemental Figure 12E, G, I), whereas they were suppressed in the proximal tubular epithelial cell fraction (Supplemental Figure 12F, H, J).
Figure 7.

AAV2-mediated H19 overexpression in vivo protects against I/R injury. (A) The overexpression of H19 in vivo was achieved through renal vein injection and validated by qPCR. Kidney injury after unilateral clamping of the left renal pedicle for 30 minutes and 24 hours of reperfusion was evaluated by qPCR and the expression of (B) KIM1, (C) NGAL, and the inflammatory markers (D) TNFα and (E) IL-1β was assessed. (F) CD31 expression indicates endothelial injury. (G) Injury in outer medulla was assessed after unilateral clamping and 24 hours of reperfusion in PAS-stained kidney sections. (H) Quantification of results. The inflammatory cells markers (I) CD3 (T-cells) and (J) F4/80 (macrophages) were analyzed by immunofluorescence and the images were captured using a confocal microscope. (K) The extent of cellular apoptosis was evaluated by TUNEL staining. Each group represents the mean±SEM of five mice used per experiment. *P<0.05; **P<0.01; ***P<0.001. AV-GFP, Adeno-associated viral vector 2 targeting a control sequence; AV-H19, Adeno-associated viral vector 2 targeting H19; Contral, contralateral control kidney; KIM1, kidney injury molecule 1; Lnc-H19, H19; NGAL, Neutrophil gelatinase-associated lipocalin; NS, nonsignificant.
In Vivo Role of H19 on Kidney Function, Capillary Density, and Apoptosis at 7 and 14 Days of Reperfusion
AAV2-mediated in vivo overexpression of H19 before unilateral clamping of the renal pedicles resulted in a preservation of outer medullary tubular epithelial cell integrity (Figure 8, A and B), and highly significant suppression of injury markers, including KIM1 and NGAL (Figure 8, C and D), on day 7 after I/R injury. Interestingly, the expression level of H19 dropped from almost 30-fold after 24 hours of virus delivery, to almost two-fold by day 7 postinjection (Figure 8E). Moreover, mice with H19 overexpression before bilateral I/R injury showed significantly reduced levels of BUN and creatinine (Figure 8, F and G) after I/R injury, indicating a preservation of kidney function. Capillary density, as assessed by endomucin staining (Figure 8H), was significantly improved on day 7 after I/R injury, whereas proliferation in the outer medulla was increased (Figure 8I) and the level of apoptosis (Figure 8J) was concomitantly reduced by AAV2-mediated H19 overexpression on day 7. Bilateral kidney clamping was performed for 15 minutes, a mild ischemia time to ensure animal survival, and kidneys were perfused for 2 weeks to determine the medium-term effect of H19 on ischemic kidney injury. Consequently, there was a slight fibrosis accumulation in the kidneys. Nonetheless, the AAV2-CTL group showed increased fibrosis levels (Supplemental Figure 13, A–C). H19 expression returned to basal levels at 2 weeks after virus delivery (Supplemental Figure 13D). Thus, the overexpression of H19 was transient. Injury marker expression was not different between the two groups (Supplemental Figure 13, E and F).
Figure 8.

AAV2-mediated H19 overexpression in vivo preserves kidney function and capillary density, promotes proliferation, and protects from apoptosis. (A and B) Injury score at 7 days of reperfusion in outer medulla of sham mice and mice receiving AAV2-H19 or AAV2-CTL in unilateral I/R injury, and quantification of results. (C and D) Renal KIM-1 and NGAL expression in mice receiving AAV2-H19 or AAV2-CTL at 7 days of reperfusion. (E) H19 expression in mice receiving AAV2-H19 or AAV2-CTL at day 7 after I/R injury. Bilateral I/R injury: both renal veins were injected with AAV2 and mice were clamped for 15 minutes and followed up for 14 days. (F and G) BUN and creatinine concentrations in mice receiving AAV2-H19 or AAV2-CTL and sham mice. (H) Endomucin expression visualizing capillary density and quantification of results in mice receiving AAV2-H19 or AAV2-CTL, and contralateral control kidneys, in unilateral I/R injury at 7 days of reperfusion. Proliferation in outer medulla as shown by (I) proliferation marker Ki67 expression, (J) TUNEL staining, and quantification of results in mice receiving AAV2-H19 or AAV2-CTL, and contralateral control kidneys, in unilateral I/R injury at 7 days of reperfusion. Each group represents the mean±SEM of five mice used per experiment. Arrows indicate proliferating (Ki67) or apoptotic nuclei (TUNEL). *P<0.05; **P<0.01; ***P<0.001. AV-GFP, Adeno-associated viral vector 2 targeting a control sequence; AV-H19, Adeno-associated viral vector 2 targeting lncRNA H19.
At 4 weeks after injection, we did not detect any increased levels of H19 in the kidney (data not shown). Moreover, long-term observation of mice after injection did not reveal an obvious change in phenotype. Mice were viable and healthy; no signs of abnormalities or tumor development were detected.
I/R Injury in Constitutive H19 Knockout Mice
H19 constitutive knockout mice were subjected to ischemia and reperfusion for 24 hours and 7 days. Outer medullary injury, kidney function, capillary density, infiltration of CD3+ and F4/80+ cells, and injury marker expression (Supplemental Figures 14 and 15) were similar on days 1 and 7 after I/R injury. The expression of miR-30a-5p increased in I/R kidneys of knockout mice after 24 hours of reperfusion, indicating a regulatory role by H19. However, miR-30a-5p expression returned to basal levels at day 7 (Supplemental Figure 16).
Discussion
Our results indicate that H19 overexpression at the time of anticipated ischemic AKI (e.g., cardiac operations with reduced cardiac output, aortic surgery with crossclamping of the aorta, kidney transplantation) may be a powerful therapeutic strategy to halt the induction of injury, and may serve as a therapeutic alternative in the future care of patients at risk for ischemic AKI. Pharmacologic modulation of H19 in renal I/R injury might result in the resolution of AKI, by influencing proregenerative and proangiogenic pathways. Small RNAs, such as microRNAs, are currently under intense investigation.5,6 Little is known about the functional role of lncRNAs. lncRNAs have been shown to regulate gene expression including the scavenging of microRNAs, as recently reviewed by our group.5 The H19 gene is located downstream of the Igf2 gene on chromosome 7 in mice and 11p15.5 in humans (ensembl.org). Its transcript, H19, has been shown to be induced during liver regeneration,3 injured rat vascular smooth muscle cells,19 severe liver injury,20 and a mouse model of hindlimb ischemia.21 Human mesenchymal stem cells stably producing prostacyclin were also found to mediate host regeneration and muscle mass gain in a paracrine manner involving H19, in hindlimb ischemia.22 In myocardial I/R injury, H19 was found to repress miR-103/107, ultimately leading to programmed necrosis.23 In addition, H19 regulates endothelial cell aging.24 These studies indicate that H19 might be part of a general mechanism of regeneration after injury. Intriguingly, the H19 RNA product is evolutionarily conserved at the nucleotide level in humans and rodents.25 Thus, findings in rodents may be directly translated into improvements in diagnosis and therapy of patients. Patients at high risk of renal I/R injury might greatly benefit from H19 overexpression before a procedure. Because we detected major effects regarding endothelial cell biology in vitro, despite modulation in cultured tubular epithelial cells showing minimal to no effects, we presume that H19 overexpression in vivo primarily preserves vascular integrity, as indicated by our results regarding capillary density at days 7 and 14. Preservation of tubular cell integrity and associated protection from acute ischemic injury is most likely secondary, and related to a stabilization of the vasculature and continued nutrient supply to the tubular epithelium. In line with our results regarding AKI, H19 has been shown to promote mucosal regeneration in intestinal inflammation. 26
We show that H19 functions as a competing endogenous RNA by sponging miR-30a-5p as a downstream effector. The interaction between H19 and different microRNA has previously been well documented. Exon 1 of H19 encodes two conserved microRNAs, miR-675–3p and miR-675–5p.27 LIN28B and let-7 interact with H19 to promote cessation of murine nephrogenesis.28 We here identified an H19 interaction with miR-30a-5p, which has not yet been described to have a role in AKI. Previously, it was shown that miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of the Warburg effect.29 Identified targets of miR-30a-5p, including Dll4, SNAI1, and ATG5, have promise with regards to I/R injury. siRNA knockdown of Dll4 impaired sprouting angiogenesis in response to VEGFC,30 and the loss of PI3K catalytic subunit p110β led to a decrease in Dll4 expression and reduction in endothelial cell sprouting in response to VEGFA.31 Previously, pretreatment with the Notch ligand Dll4 in rats subjected to renal I/R injury resulted in enhanced recovery from AKI.32 SNAI1 acts as a VEGF-induced regulator of Notch1 signaling and Dll4 expression, thereby controlling embryonic vascular development.33 Similarly, the inhibition of ATG5 by 3-methyladenine or siRNA knockdown resulted in impaired angiogenesis and tube formation capacity in bovine aortic endothelial cells.34 Endothelial-specific deletion of ATG5, or pharmacologic inhibition of autophagic flux, results in an impairment of vessel healing under injury conditions.35 These results indicate that H19/miR-30a-5p–mediated regulation of these novel targets might constitute a highly efficient model of sustained vascular integrity under ischemic stress.
We show that H19 expression is induced after 24 hours of I/R injury in vivo, whereas, under hypoxic conditions, in vitro H19 is upregulated after 72 hours in HUVECs. Endothelial cells produce 85%–90% of their energy through anaerobic glycolysis, indicating that they are well adapted to a hypoxic environment.36,37 During ischemia in vivo a complex cascade is set in motion, involving hypoxia, inflammation, and cytokine release, among other processes. As our results show, serum deprivation or ATP depletion induce an early H19 induction within 24 hours, and are therefore a clear indication of an acute H19 stress response, which might be a better reflection of the in vivo situation.
There is growing evidence that H19 is involved in tissue fibrosis in various organs.38 We observed increased (but mild) fibrosis levels in mice overexpressing H19. This reflects improved regeneration post-AKI in the AAV2-H19 group.
Given the clear protective effect of H19 overexpression on ischemic AKI, it would be expected that H19 knockout mice display exacerbated ischemic injury. However, H19 knockout mice exhibited a similar degree of injury as wild-type mice. These findings are not surprising because H19 expression in the kidney under basal conditions is hardly present. Thus, knocking out a gene that is not expressed in healthy kidney and is only reinduced after an injurious insult, would not have much impact when it is not present at all. We observed similar results in vitro by H19 silencing (no difference between control and H19 silencing).
In conclusion, we show that H19 is induced in ischemic AKI. Pharmacologic overexpression of H19 promotes regeneration of AKI. Our results may indicate that H19 could be used as a powerful therapeutic tool in the future care of patients with ischemic AKI.
Disclosures
H. Haller reports consultancy agreements with Alexion, AstraZeneca, Bayer Pharma, Boehringer, MedWiss, Phenos, and Vifor-Fresenius; honoraria from Alexion, Astra-Zeneca, Bayer Pharma, Boehringer, MedWiss, Novartis, Phenos, and Vifor-Fresenius; is a scientific advisor for, or holds membership with, Alexion, Bayer Pharma, Der Internist, and Der Nephrologe; and Speakers Bureau fees with Alexion, Amgen, AstraZeneca, Bayer Pharma, Boehringer, MedWiss, Novartis, Phenos, and Vifor-Fresenius. J.M. Lorenzen holds a patent regarding use of H19 as a therapeutic tool in ischemic AKI; and is a scientific advisor for, or holds membership with, Frontiers in (Renal) Pharmacology. R. Schmitt reports honoraria from Fresenius Medical Care and Otsuka Pharmaceutical. H. Seeger reports honoraria from Amgen, Astra-Zeneca, Bristol-Myers Squibb, Menarini AG, Mundipharma, and Vifor; and other interests/relationships with IMI Transbioline (European Union). T. Thum holds filed and licensed patents on noncoding RNAs, including H19; is shareholder and founder of Cardior Pharmaceuticals GmbH; receives support/holds advisory seats at Amicus Therapeutics, Novo Nordisk, Sanofi-Genzyme, and Takeda (all outside the field of this paper); reports patents and inventions with Cardior Pharmaceuticals and Regulus Therapeutics; is a scientific advisor for Institut Pasteur, Paris; is a member of the Editorial Board for the journals European Heart Journal, Circulation Research, Journal of Molecular and Cellular Cardiology, Atherosclerosis Thrombosis Vascular Biology, Public Library ofScience One, and Public Library of Science Genetics; and Speakers Bureau with Amicus, Genzyme, Sanofi, and Takeda. All remaining authors have nothing to disclose.
Funding
This work was supported by a National Center for Excellence in Research NCCR Kidney.CH Junior grant and Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung grant 31003A_179347 (to J.M. Lorenzen), and the Integrative Kidney Physiology and Pathophysiology 2fellowship to G. Haddad.
Supplementary Material
Acknowledgments
Dr. George Haddad performed the in vitro and in vivo analyses. Prof. Hermann Haller, Dr. Malte Kölling, Prof. Thomas Thum, Dr. Andreas D. Kistler, Prof. Thomas F. Mueller, Dr. Harald Seeger, Mr. Urs A. Wegmann, Prof. Rudolf P. Wüthrich, and Dr. Angela Dettling helped in performing the in vitro analyses. Prof. Roland Schmitt and Dr. Inga Soerensen-Zender helped in performing the animal operations. Dr. Anne Dueck and Prof. Stefan Engelhardt generated the adenoviral vectors for in vivo use. Prof. Johan M. Lorenzen conceived the idea. Dr. George Haddad and Prof. Johan M. Lorenzen wrote the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020060775/-/DCSupplemental.
Supplemental Appendix 1. Supplemental results: functional role of H19 in tubular epithelial cell biology.
Supplemental Table 1. Transcription factors that putatively bind to the H19 promoter region.
Supplemental Table 2. List of human primers.
Supplemental Table 3. List of mouse primers.
Supplemental Figure 1. Induction of HIF1α and EPAS under hypoxia.
Supplemental Figure 2. Expression of the transcription factors SPI1, LHX8, KLF8, and KLF15 under hypoxia.
Supplemental Figure 3. siRNA knockdown of HIF1α in RPTECs and the regulation of H19 expression.
Supplemental Figure 4. H19 promoter activity in HUVECs and RPTECs.
Supplemental Figure 5. Expression of angiogenic markers under hypoxia in response to H19 knockdown.
Supplemental Figure 6. Effect of a hypoxia time course on signaling pathways.
Supplemental Figure 7. Expression of miR-30a-5p under hypoxia and its modulatory effects on angiogenic gene expression.
Supplemental Figure 8. Effect of H19 overexpression in RPTECs on cellular migration.
Supplemental Figure 9. Effect of H19 overexpression or hypoxia on cellular proliferation and apoptosis in RPTECs.
Supplemental Figure 10. Expression of H19 in vivo after AAV2-mediated overexpression.
Supplemental Figure 11. Expression of adhesion molecules in mouse I/R kidney.
Supplemental Figure 12. H19, miR-30a-5p, and target gene expression in ex vivo sorted endothelial cells and tubular epithelial cells, after injection of AAV2 targeting H19 compared with AAV2-CTL.
Supplemental Figure 13. Fibrosis and injury marker gene expression on day 14 after I/R injury.
Supplemental Figure 14. Kidney injury in constitutive H19 knockout mice.
Supplemental Figure 15. Tubular injury marker expression in H19 knockout mice after unilateral I/R injury for 24 hours and 7 days.
Supplemental Figure 16. miR-30a-5p expression in wild-type and H19 knockout mice at 24 hours and7 days of reperfusion after I/R injury.
Supplemental Figure 17. Genotyping of constitutive H19 knockout mice.
References
- 1.Kelly KJ: Acute renal failure: Much more than a kidney disease. Semin Nephrol 26: 105–113, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Bon D, Chatauret N, Giraud S, Thuillier R, Favreau F, Hauet T: New strategies to optimize kidney recovery and preservation in transplantation. Nat Rev Nephrol 8: 339–347, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Lorenzen JM, Haller H, Thum T: MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat Rev Nephrol 7: 286–294, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Lorenzen JM, Kaucsar T, Schauerte C, Schmitt R, Rong S, Hübner A, et al.: MicroRNA-24 antagonism prevents renal ischemia reperfusion injury. J Am Soc Nephrol 25: 2717–2729, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lorenzen JM, Thum T: Long noncoding RNAs in kidney and cardiovascular diseases. Nat Rev Nephrol 12: 360–373, 2016 [DOI] [PubMed] [Google Scholar]
- 6.Kölling M, Genschel C, Kaucsar T, Hübner A, Rong S, Schmitt R, et al.: Hypoxia-induced long non-coding RNA Malat1 is dispensable for renal ischemia/reperfusion-injury. Sci Rep 8: 3438, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al.; Mouse Genome Sequencing Consortium: Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Cirio MC, de Groh ED, de Caestecker MP, Davidson AJ, Hukriede NA: Kidney regeneration: Common themes from the embryo to the adult. Pediatr Nephrol 29: 553–564, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanwar YS, Pan X, Lin S, Kumar A, Wada J, Haas CS, et al.: Imprinted mesodermal specific transcript (MEST) and H19 genes in renal development and diabetes. Kidney Int 63: 1658–1670, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Lorenzen JM, Schauerte C, Kielstein JT, Hübner A, Martino F, Fiedler J, et al.: Circulating long noncoding RNATapSaki is a predictor of mortality in critically ill patients with acute kidney injury. Clin Chem 61: 191–201, 2015 [DOI] [PubMed] [Google Scholar]
- 11.Ripoche MA, Kress C, Poirier F, Dandolo L: Deletion of the H19 transcription unit reveals the existence of a putative imprinting control element. Genes Dev 11: 1596–1604, 1997 [DOI] [PubMed] [Google Scholar]
- 12.Broekema M, Harmsen MC, Koerts JA, Petersen AH, van Luyn MJ, Navis G, et al.: Determinants of tubular bone marrow-derived cell engraftment after renal ischemia/reperfusion in rats. Kidney Int 68: 2572–2581, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, et al.: MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med 4: 121ra18, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakatsu MN, Davis J, Hughes CC: Optimized fibrin gel bead assay for the study of angiogenesis. J Vis Exp (3): 186, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palau W, Masante C, Ventura M, Di Primo C: Direct evidence for RNA-RNA interactions at the 3′ end of the hepatitis C virus genome using surface plasmon resonance. RNA 19: 982–991, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu Y, Yang L, Zhao M, Zhu S, Kang R, Vernon P, et al.: Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 26: 1752–1760, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J, et al.: MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer 130: 2044–2053, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Bridge G, Monteiro R, Henderson S, Emuss V, Lagos D, Georgopoulou D, et al.: The microRNA-30 family targets DLL4 to modulate endothelial cell behavior during angiogenesis. Blood 120: 5063–5072, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto Y, Nishikawa Y, Tokairin T, Omori Y, Enomoto K: Increased expression of H19 non-coding mRNA follows hepatocyte proliferation in the rat and mouse. J Hepatol 40: 808–814, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Liu C, Barbier O, Smalling R, Tsuchiya H, Lee S, et al.: Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci Rep 6: 20559, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voellenkle C, Garcia-Manteiga JM, Pedrotti S, Perfetti A, De Toma I, Da Silva D, et al.: Implication of long noncoding RNAs in the endothelial cell response to hypoxia revealed by RNA-sequencing. Sci Rep 6: 24141, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng Y, Yang Z, Terry T, Pan S, Woodside DG, Wang J, et al.: Prostacyclin-producing human mesenchymal cells target H19 lncRNA to augment endogenous progenitor function in hindlimb ischaemia. Nat Commun 7: 11276, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang JX, Zhang XJ, Li Q, Wang K, Wang Y, Jiao JQ, et al.: MicroRNA-103/107 regulate programmed necrosis and myocardial ischemia/reperfusion injury through targeting FADD. Circ Res 117: 352–363, 2015 [DOI] [PubMed] [Google Scholar]
- 24.Hofmann P, Sommer J, Theodorou K, Kirchhof L, Fischer A, Li Y, et al.: Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc Res 115: 230–242, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergström R, Whitehead J, Kurukuti S, Ohlsson R: CTCF regulates asynchronous replication of the imprinted H19/Igf2 domain. Cell Cycle 6: 450–454, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Geng H, Bu HF, Liu F, Wu L, Pfeifer K, Chou PM, et al.: In inflamed intestinal tissues and epithelial cells, interleukin 22 signaling increases expression of H19 long noncoding RNA, which promotes mucosal regeneration. Gastroenterology 155: 144–155, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dey BK, Pfeifer K, Dutta A: The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev 28: 491–501, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yermalovich AV, Osborne JK, Sousa P, Han A, Kinney MA, Chen MJ, et al.: Lin28 and let-7 regulate the timing of cessation of murine nephrogenesis. Nat Commun 10: 168, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Kang L, Zhao W, Feng Y, Liu W, Wang T, et al.: miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett 400: 89–98, 2017 [DOI] [PubMed] [Google Scholar]
- 30.Singh NK, Kotla S, Kumar R, Rao GN: Cyclic AMP response element binding protein mediates pathological retinal neovascularization via modulating DLL4-NOTCH1 signaling. EBioMedicine 2: 1767–1784, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haddad G, Zhabyeyev P, Farhan M, Zhu LF, Kassiri Z, Rayner DC, et al.: Phosphoinositide 3-kinase β mediates microvascular endothelial repair of thrombotic microangiopathy. Blood 124: 2142–2149, 2014 [DOI] [PubMed] [Google Scholar]
- 32.Gupta S, Li S, Abedin MJ, Wang L, Schneider E, Najafian B, et al.: Effect of Notch activation on the regenerative response to acute renal failure. Am J Physiol Renal Physiol 298: F209–F215, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Wu ZQ, Rowe RG, Lim KC, Lin Y, Willis A, Tang Y, et al.: A Snail1/Notch1 signalling axis controls embryonic vascular development. Nat Commun 5: 3998, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du J, Teng RJ, Guan T, Eis A, Kaul S, Konduri GG, et al.: Role of autophagy in angiogenesis in aortic endothelial cells. Am J Physiol Cell Physiol 302: C383–C391, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, et al.: Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med 19: 1281–1287, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al.: Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154: 651–663, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Zheng J: Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (review). Oncol Lett 4: 1151–1157, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang X, Ning Q: The mechanism of lncRNA H19 in fibrosis and its potential as novel therapeutic target. Mech Ageing Dev 188: 111243, 2020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.