

Significance Statement
Single-cell transcriptomes from dissociated tissues provide insights into cell type composition and gene expression, but spatial positional information is lost during tissue dissociation. Computational deconvolution of the spatial position of kidney tubule cells from whole kidney single-cell transcriptomes and reconstructed spatial and cell type–specific gene-expression patterns, from the cortex to medulla, revealed gradual changes that follow the physiological corticomedullary gradient of tissue osmolality. Quantification of osmo-responsive gene expression detected in single-cell transcriptomes identified flattened expression gradients in mice, with a genetically induced defect of medullary osmolyte retention. Hence, single-cell transcriptomes harbor previously underappreciated information on spatial gene expression and regional cellular states.
Keywords: microenvironment, spatial resolution single-cell transcriptomics, osmolality gradient, osmogenes, cell types
Visual Abstract

Abstract
Background
Single-cell transcriptomes from dissociated tissues provide insights into cell types and their gene expression and may harbor additional information on spatial position and the local microenvironment. The kidney’s cells are embedded into a gradient of increasing tissue osmolality from the cortex to the medulla, which may alter their transcriptomes and provide cues for spatial reconstruction.
Methods
Single-cell or single-nuclei mRNA sequencing of dissociated mouse kidneys and of dissected cortex, outer, and inner medulla, to represent the corticomedullary axis, was performed. Computational approaches predicted the spatial ordering of cells along the corticomedullary axis and quantitated expression levels of osmo-responsive genes. In situ hybridization validated computational predictions of spatial gene-expression patterns. The strategy was used to compare single-cell transcriptomes from wild-type mice to those of mice with a collecting duct–specific knockout of the transcription factor grainyhead-like 2 (Grhl2CD−/−), which display reduced renal medullary osmolality.
Results
Single-cell transcriptomics from dissociated kidneys provided sufficient information to approximately reconstruct the spatial position of kidney tubule cells and to predict corticomedullary gene expression. Spatial gene expression in the kidney changes gradually and osmo-responsive genes follow the physiologic corticomedullary gradient of tissue osmolality. Single-nuclei transcriptomes from Grhl2CD−/− mice indicated a flattened expression gradient of osmo-responsive genes compared with control mice, consistent with their physiologic phenotype.
Conclusions
Single-cell transcriptomics from dissociated kidneys facilitated the prediction of spatial gene expression along the corticomedullary axis and quantitation of osmotically regulated genes, allowing the prediction of a physiologic phenotype.
Recent developments in single-cell transcriptomics technologies, such as single-cell mRNA sequencing (scRNA-seq) or single-nuclei mRNA sequencing (snRNA-seq) promise breakthroughs in the understanding of cellular functions in different tissues.1–6 Although current single-cell technologies circumvent weaknesses of bulk gene expression profiling, the spatial coordinates of individual cells within tissues are lost in cell suspensions. In contrast, efforts to obtain spatially restricted transcriptomes in tissues currently lack single-cell resolution.7,8
An alternative approach to derive spatially resolved single-cell transcriptomes in suspended cells is to exploit cues to spatial origin harbored within each cell’s transcriptome.9 This approach aims to predict regional alignment of single cells on the basis of their expression similarity, and operates on the assumption that local proximity of cells implies similarities of their gene expression signatures.
The extent to which single-cell gene expression carries spatial information is likely to vary considerably across and within organs. Regional alterations of the microenvironment in a tissue may alter the gene expression in adjacent cells. Hence, cell types exposed to steep microenvironmental gradients may be particularly suitable for the reconstruction of their spatial positioning on the basis of single-cell transcriptomics. The kidneys provide an excellent candidate tissue to address this possibility, because the kidney’s tubules span large distances from kidney cortex to medulla, which are characterized by steep corticomedullary gradients of tissue osmolality and oxygen tension, resulting from coordinated transport mechanisms in kidney tubules and the vasculature,10–12 and are known to influence the gene expression of adjacent cells.13–15 Interstitial osmolalities range from the serum-isoosmolal cortex (approximately 290 mosmol/kg H2O) to the strongly hyperosmolal inner medulla (approximately 1200 mosmol/kg H2O in humans, up to 2000–4000 mosmol/kg H2O in mice).12 Tissue oxygen tension ranges from 80 mm Hg in the cortex to 10–50 mm Hg in the renal medulla.10,16–18
Here, we provide evidence that gene expression information can be utilized to infer the spatial position of individual cells within kidney dissociates, and the overall gene expression across single cells derived from a dissociated organ can help to identify gene expression patterns along the corticomedullary axis. Moreover, we show that a combination of spatial reconstruction and osmotically regulated gene expression quantitation from kidney single-cell transcriptomes facilitates an approximation of the intrarenal osmolality gradient in mice. Results from regional gene expression and spatial sorting of collecting duct principal cells (CD-PCs) can be viewed at https://shiny.mdc-berlin.de/KidneySpatial/.
Methods
Mouse Strains
Experiments were conducted using adult (3–6 months old), male, C56Bl/6N wild-type mice (dissected and whole kidney samples). For the single-nuclei whole kidney data, we used mice with a previously published collecting duct–specific (Hoxb7Cre-driven) knockout of transcription factor grainyhead-like 2 (Grhl2) and control littermates.19 These mice were on a C56Bl/6N genetic background.
Generation of scRNA-seq Data
Mouse kidneys were perfused with ice-cold PBS at 10 ml/min using a total volume of 30 ml to remove blood and cool the organ. Then, the whole organ or dissected parts of the kidney were thoroughly minced and digested using a cold protease-based protocol (protease from Bacillus licheniformis, 10 mg/ml, P5380; Sigma).20 The tissue digestion comprised four digestion steps in total, including different trituration and shaking steps of the tissue suspension (see Supplemental Material for a step-by-step protocol). The final cell suspension was fixed in 80% methanol as previously published.21 Fixed cells were stored at −80°C. Before the Dropseq run, cells were pelleted at 3000 g and resuspended in PBS/0.01% BSA + Ribolock (1U/µl) + Vanadyl Ribonucleoside Complex (VRC, S1402S, 10mM final concentration; New England Biolabs) to avoid RNA degradation, centrifuged, and resuspended again in PBS/0.01% BSA + Ribolock + VRC, and passed through a 35 µm strainer. Cells were then counted and subjected to Dropseq and library preparation, as previously published.21 Obtained libraries were sequenced (paired end) on Illumina NextSeq 500 sequencers. Digital expression matrices from raw FASTQ files were obtained using the Dropseq toolkit version 1.12.22 The inner medulla sample was pooled from three mice to obtain the needed cell number for Dropseq.
Generation of snRNA-seq Data
After mice were sacrificed, kidneys were immediately put in ice-cold PBS. A middle slice (1–2 mm) containing the cortex, outer, and inner medulla was obtained. All samples were put in 4°C RNAlater for 24 hours, then stored at −80°C. For the generation of the single-nuclei suspensions (all reactions at 4°C), specimens were thoroughly minced in nuclear lysis buffer 1 (nuclear lysis buffer [Sigma] + 1 U/µl of Ribolock + 10 mM of VRC), and homogenated using a Dounce homogenizer with pastel A (D8938–1SET; Sigma), filtered (100 µm), homogenized again (Dounce homogenizer with pastel B), filtered through a 35 µm strainer, and centrifuged (5 minutes, 500 × g). The pellet was then resuspended in nuclear lysis buffer 2 (nuclear lysis buffer + 1 U/µl of Ribolock). To remove debris from the suspension, we underlaid the suspension with lysis buffer containing 10% sucrose and 1 U/µl of Ribolock. After centrifugation (5 minutes, 500 × g), the supernatant and debris were carefully removed. Pelleted nuclei were resuspended in PBS/0.04% BSA + Ribolock (1 U/µl), filtered through a 20 µm strainer, and stained with 4'6-diamidino-2-phenylindole. The nuclei suspension was then FACS-sorted (BD FACS Aria II), counted after FACS sorting, and subjected to single-cell sequencing following the 10× genomics protocol for v2 (experiment 1) or v3.1 (experiment 2) chemistry targeting 10,000 nuclei (see Supplemental Methods for a step-by-step protocol). Obtained libraries were sequenced on Illumina HiSeq 4000 sequencers (paired end). Digital expression matrices were generated using the Cell Ranger software version 3.0.2 with –force-cells 10,000.
Single-Cell and Single-Nuclei Sequencing Data Treatment and Clustering
All datasets were imported as Seurat objects into R using Seurat version 2.3.1. Cells were filtered using a lower gene number (nGene) cutoff of 200 (500 for single-nuclei data) and an upper cutoff of 5000. Data were then log normalized and scaled according to the number of unique molecular identifiers. All single-cell and single-nuclei data samples were merged into one Seurat object, separately. Clustering was performed using highly variable genes (HVGs; Seurat parameters for FindVariableGenes: x.low.cutoff =0.0125, x.high.cutoff=Inf, y.cutoff=0.5) on all cells using Seurat’s default settings. T-distributed stochastic neighbor embedding plots were generated using 30 principal components from principal component analysis with HVGs, a perplexity of 30, and a resolution of 0.6. All other parameters were at default levels. Potential subclusters of one cell type were summarized to one cluster of the respective cell type.
Cell Type–specific HVG Selection
Cell type–specific HVGs were generated by subsetting the respective Seurat object to the respective cell types (no regional information required) and applying FindVariableGenes from the Seurat package on the remaining cells using ×.low.cutoff=0.25, x.high.cutoff=Inf, y.cutoff=1.
Randomly Chosen Genes
For regional assignment by randomly chosen genes, per cell type, a number of genes corresponding to 25% of the total cell number of the respective cell type were randomly chosen from the full expression matrix.
Computational Methods including Multinomial Classification and NovoSpaRc Sorting
Detailed information are provided in the Supplemental Methods. Multinomial classification was performed using the glmnet package version 2.0–16 and by randomly separating the cells from dissected samples of each cell type into a training (two thirds of cells) and test dataset (one third of cells).
NovoSpaRc sorting9 was performed on CD-PCs from dissected scRNA-seq data using CD-PC–specific HVGs and providing a one-dimensional space with 100 spatial positions for sorting. Spatial gene expression analyses were performed using the sdge matrix, which is the matrix obtained by multiplying the original expression matrix (dge, matrix dimensions: number of genes × number of CD-PCs, obtained from the @data slot of the respective Seurat object) and gw (matrix dimensions: number of CD-PCs × number of spatial positions), which is the matrix generated by novoSpaRc containing the probability distributions on the spatial axis of all CD-PCs. The code for spatial sorting with novoSpaRc can be downloaded at www.mdc-berlin.de/kidneyspatial.zip.
RNA In Situ Hybridization—RNAscope
The RNAscope 2.5 HD reagent kit-brown (#322300; Advanced Cell Diagnostics [ACD], Newark, CA) was used to perform chromogenic in situ hybridization on formalin-fixed paraffin embedded mouse kidney sections with probes directed against Cryab (#552741; ACD), Hsd11b2 (#494891; ACD), Scnn1g (#422091; ACD), and Fxyd4 (#820891; ACD). The probes Mm-Cryab and Mm-Fxyd4 were produced for us because they were not available in the catalog.
Kidney slices were fixed in 4% formaldehyde in PBS for 24 hours at room temperature with agitation, dehydrated in a graded series of ethanol, and embedded in paraffin. Paraffin-embedded kidney slices were cut into 5 µm sections, plated on Superfrost Plus slides, air dried overnight, baked for 1 hour at 60°C, cooled for 15 minutes, dewaxed, and air dried again. Subsequent pretreatment and RNAscope assay procedures for all probes were performed according to the “Formalin-Fixed Paraffin-Embedded (FFPE) Sample Preparation and Pretreatment” and “RNAscope 2.5 HD Detection Reagent BROWN” protocols (documents #322452 and #322310; ACD) as recommended by the manufacturer. Sections were counterstained with hematoxylin before dehydrating and applying coverslips using a xylene-based mounting medium. Images of the hybridized sections were captured on a Leica DM2000 LED bright field microscope.
Semi-Quantification of RNA In Situ Hybridizations
All image analysis and transformation steps were performed using ImageJ version 2.0.0. Original images from microscopy were transformed into gray value images. Image thresholds were then adjusted such that unstained areas showed no signal. For each of the indicated regions, five collecting ducts were quantified by manually encircling the collecting epithelium where visible in the respective area, and obtaining the average signal intensity from ImageJ’s measuring tool. Values were then normalized to the average expression of the region with highest signal intensity as indicated.
RNA Sequencing Data
Paired-end RNA sequencing (2×150 bp) was performed on (in each case) five samples from the cortex, outer, and inner medulla of adult male C56bl/6N mice using the mRNA-sequencing services of Novogene (https://en.novogene.com/next-generation-sequencing-services/gene-regulation/mrna-sequencing-service/). Samples were treated according to the provided company guidelines and libraries were sequenced (150 bp, paired end) on Illumina Novaseq 6000. Provided FASTQ files were aligned using STAR and the mm9 genome, reads were then counted using featureCounts23 with -p -t exon -O -g gene_id -s 0. Raw count values were then transformed to transcript per million values. To account for large differences between the cortex, outer medulla, and inner medulla in CD-PC abundance, we normalized RNA-seq transcript per million values from the cortex, outer, and inner medulla for each gene and CD-PC content (see Supplemental Methods).
Results
Single-Cell Transcriptomics from Whole Kidneys and Dissected Kidney Regions Allow Identification of Cell Types and Their Zonal Distribution
We prepared scRNA-seq datasets from whole wild-type male mouse kidneys (n=2, C57Bl/6N; 3–6 months old) and approximate regions representing the kidney cortex, outer medulla, and inner medulla, which were prepared by manual dissection under a dissecting microscope without safety margins to represent the entire corticomedullary axis (Figure 1A, Supplemental Figure 1). Each tissue was cold-digested20 to obtain single-cell suspensions, which were then subjected to scRNA-seq using the Dropseq method.22 This yielded gene expression profiles of 5675 cells derived from whole kidney suspensions (unknown regional origin), of 3046 cells derived from the cortex, 2392 cells derived from the outer medulla, and 889 cells derived from the inner medulla. On average, 456 unique genes and 772 unique transcripts were identified per cell. To achieve an unbiased cell type identification on the basis of transcriptomes, we used t-distributed stochastic neighbor embedding on the combined dataset and identified ten cell types (Figure 1B, Supplemental Table 1). Seven cell types represented the major tubular epithelial cell types of the kidney: proximal tubules (PT), thin limbs (TL), thick ascending limbs (TAL), distal convoluted tubules, connecting tubules, CD-PCs, and collecting duct intercalated cells (CD-ICs). The remaining three cell types represented podocytes, immune cells, and interstitial cells (Supplemental Figure 2). We then assessed the contribution of major cell types to cells derived from regions approximately representing the cortex, outer medulla, and inner medulla (Figure 1C). Consistent with the published literature, PT and TAL resided primarily in the regions representing the cortex and outer medulla, whereas TL resided in the outer and inner medulla, and CD-PCs and CD-ICs resided across all three compartments. Small subsets of PT and TAL cells in the inner medulla sample, and of distal convoluted tubules and connecting tubule cells in the outer medulla sample, might indicate some carryover of cells between the cortex, outer, and inner medulla during the dissection, but this was not critical for the purposes of this study.
Figure 1.
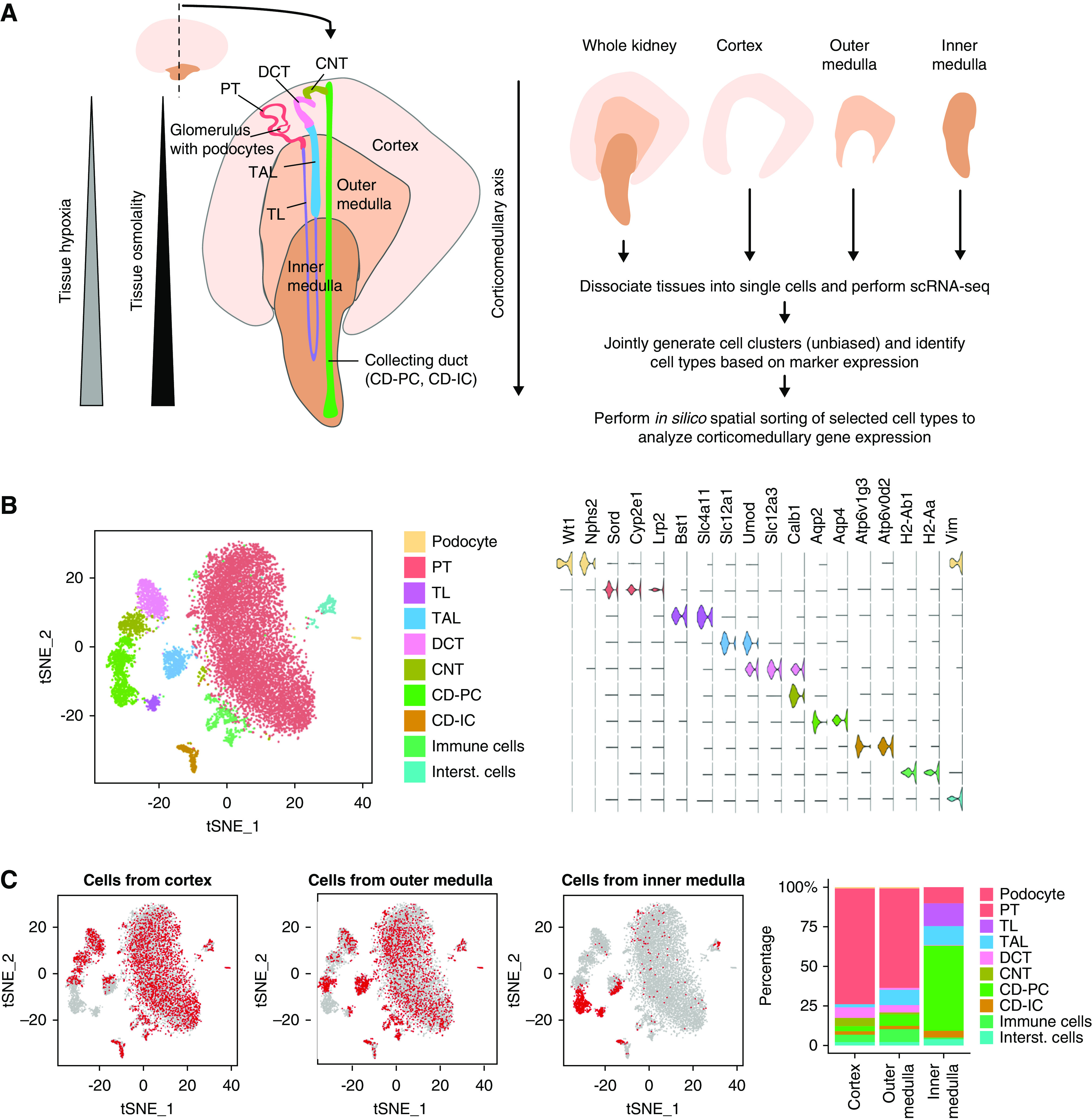
Regional distribution of cell types in the mouse kidney. (A) Schematic structure of the mouse kidney and experimental strategy. Schematically depicted is a middle slice from a mouse kidney showing the main regions of the kidney (cortex, outer, and inner medulla) with one nephron (TL; TAL; distal convoluted tubule; connecting tubule; CD-PCs/ICs). Note that only some tubular cell types (PT; TL; TAL; CD-PC; CD-IC) are present in multiple regions of the kidney. Along the corticomedullary axis, kidneys show increasing tissue osmolality and increasing tissue hypoxia from cortex toward the inner medulla. (B) Unbiased identification of kidney cell types. Joint t-distributed stochastic neighbor embedding (t-SNE) plot of scRNA-seq data from cells derived from wild-type adult male mouse whole kidneys (n=2) and dissected cortex, outer medulla, and inner medulla. Major cell types are color coded as indicated. Violin plots show expression levels (arbitrary units) of marker genes used in cell type identification. (C) Contribution of the cortex, outer, and inner medulla to kidney cell types. Left panels: using the t-SNE plot from (B), cells derived from the cortex, outer medulla, and inner medulla are labeled in red as indicated, whereas all remaining cells are depicted in gray. Right panel: contribution of cell types to cortex, outer medulla and inner medulla was quantitated and indicated as percentages of all cells from the respective zone.
Identification of HVGs within Region-Spanning Cell Types
We hypothesized that genes of highly variable expression within the major cell types of the kidney might be suitable for deriving spatial information. We focused on the five region-spanning kidney tubule cell types (PT, TL, TAL, CD-PC, CD-IC), and identified genes that displayed highly variable expression levels (HVGs) within each given cell type on the basis of an expression cutoff and a dispersion cutoff (Figure 2A, Supplemental Table 2). Most HVGs were unique to one of the five cell types (Figure 2B). We analyzed HVG expression levels in cells of known cortical, outer medullary, or inner medullary origin, and produced heatmaps reflecting their expression levels in these zones (Figure 2C). This suggested the zonal origin of cells was an important driver of variability of gene expression, especially within TL, CD-PCs, and CD-ICs. Accordingly, the calculated contribution of zonal origin to total variance for each HVG was significantly higher in TL, CD-PCs, and CD-ICs when compared with PT and TAL (Figure 2D). This was consistent with the known exposure of TL, CD-PCs, and CD-ICs to particularly steep gradients of osmolality and oxygen tension.
Figure 2.
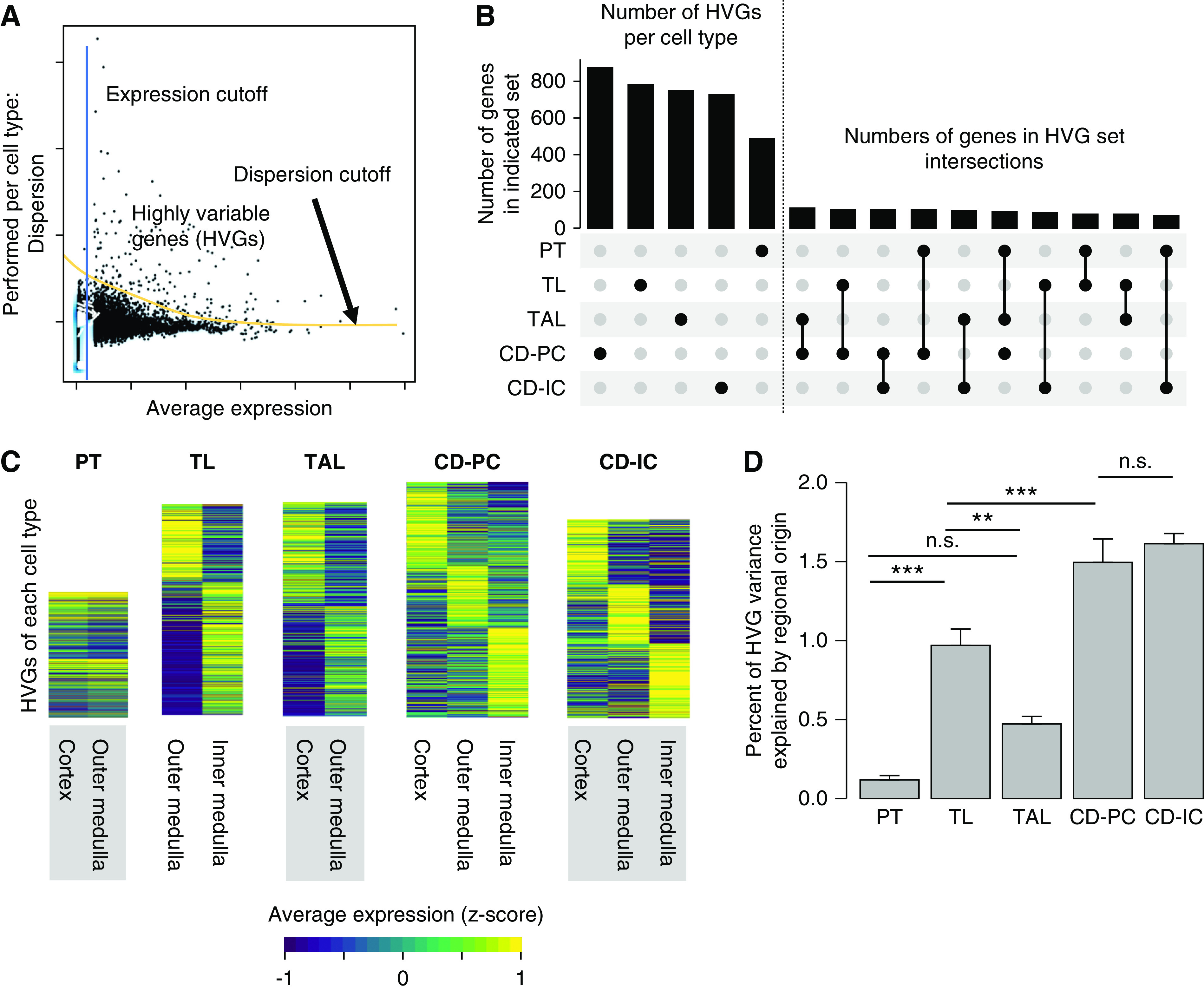
HVGs within region-spanning tubular cell types harbor spatial information. (A) HVGs are generated per cell type by applying a minimum expression and dispersion cutoff. The picture depicts a scheme for the derivation of HVGs by applying mean expression and dispersion cutoffs. This is done separately for all region-spanning cell types (PT, TL, TAL, CD-PC, CD-IC). Individual dots represent single genes. (B) Most HVGs are cell-type specific. Upset plot for HVGs. There are usually hundreds of HVGs per cell type (left of dotted line, cell type represented by black dot in lower panel), which only show very limited overlap between cell types (right of dotted line, intersections as indicated in the lower panel). (C) HVGs show regional expression profile. Shown are heatmaps of HVGs for the five region-spanning tubule cell types. Gene expression was z-score normalized over all five cell types on a per-gene basis and HVGs were fold-change sorted per cell type. (D) Regional origin contributes differently to HVG variance in region-spanning cell types. Bar plot showing the percentage of HVG variance explained by regional origin (cortex, outer, or inner medulla; mean+SEM, one-way ANOVA; P value: *<0.05, **<0.01, ***<0.001).
De novo Spatial Sorting of CD-PCs Based on Transcriptomes Identifies Continuous Transitions of Gene Expression States along the Corticomedullary Axis
To assess the general potential of HVGs to assign spatial cellular origin (cortex versus outer medulla versus inner medulla) within each of the five cell types, we trained a multinomial model to assign the zonal origin of each cell on the basis of HVG expression (Supplemental Figure 3). For this purpose, cells from each type were randomly separated into discovery set (two thirds of cells per cell type) and a validation set (one third of cells per cell type) for each run of the model. The accuracy of the spatial assignment to the cortex, outer medulla, and inner medulla was >80% for CD-PC and TL when using cell type-specific HVG sets. In contrast, the accuracy was lower in other cell types and when random genes were used instead of HVGs (Supplemental Figure 3).
We next aimed to achieve de novo spatial sorting of cells along the corticomedullary axis on the basis of their HVG transcriptomes. We decided to focus on CD-PCs, because these cells cover the entire space from the cortex to inner medulla and were present at high abundance within our dataset. In addition, CD-PCs are believed to form different subtypes on the basis of their corticomedullary location, referred to as cortical CD-PC, outer medullary CD-PC (OMCD), inner medullary CD-PC 1 (IMCD1), IMCD2, and IMCD3.5,24,25 However, it is unknown whether these subtypes truly represent distinct cell types or whether they reflect regional snapshots from a continuum of functional cell states along the corticomedullary axis. We hypothesized that the latter was true, and the corticomedullary order of CD-PCs could be derived on the basis of a gradual transition of their transcriptomes. To test this, we utilized the recently published De Novo Spatial Reconstruction of Single-Cell Gene Expression (novoSpaRc)9 tool for spatial reconstruction within scRNA-seq datasets on the basis of transcriptome similarity. NovoSpaRc enables in silico spatial sorting of cells on the basis of defined sets of ordering genes and a defined number of locations as input (Figure 3A). The algorithm assigns each cell a probability distribution on the provided space, which in our case of CD-PCs was one dimensional (CD-PCs on the corticomedullary axis). For simplicity, we selected 100 locations (altering the number of locations did not substantially change the outcome, Supplemental Figure 4) and used CD-PCs from dissected samples (753 cells, known origin) and the gene set of 872 HVGs specific to CD-PCs to instruct spatial sorting (Figure 3A). To validate the spatial ordering, we first analyzed the known origin of CD-PCs from the dissected cortex, outer medulla, and inner medulla along the 100 spatial positions (Figure 3B). Results were consistent with a gradual corticomedullary distribution between position 1 (outermost cortex) and 100 (innermost medulla). We repeated this analysis using a second dataset from mouse kidney and dissected zones, which was recently reported by Ransick et al.5 and observed virtually identical results (Supplemental Figure 5A). We next analyzed the expression of known anchor genes5,26,27 for cortical CD-PC, OMCD, IMCD1, IMCD2, and IMCD3 along spatial positions (Figure 3C), which was consistent with the known distribution of CD-PC subtypes along the corticomedullary axis, although we did observe a partial overlap between OMCD and IMCD1, and between IMCD2 and IMCD3. Again, repeating the analysis on a second independent dataset5 confirmed the result (Supplemental Figure 6B).
Figure 3.
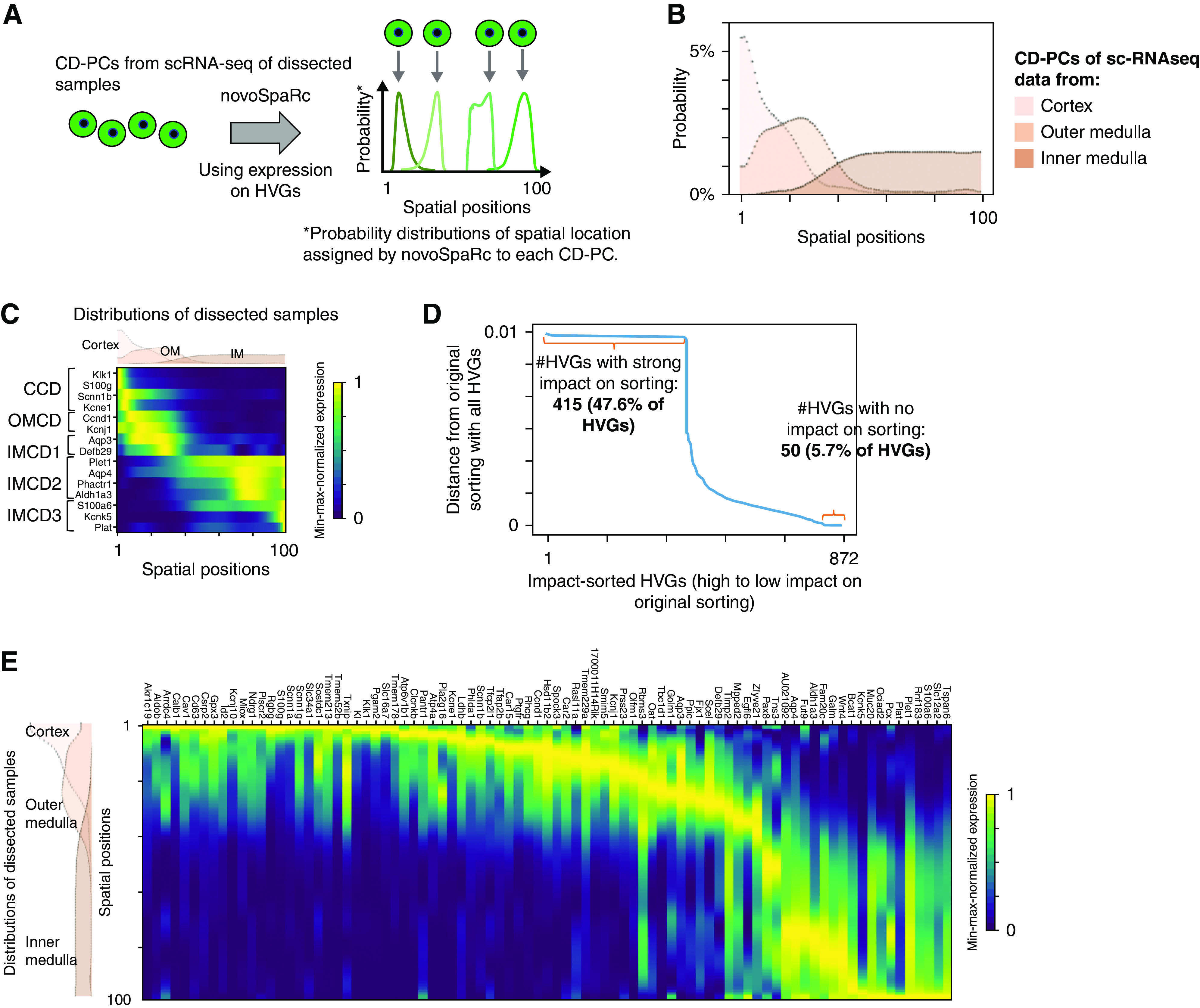
De novo spatial sorting of CD-PCs identifies a continuous transition of gene expression states along the corticomedullary axis. (A) NovoSpaRc assigns each cell a probability distribution for its spatial location on a given space on the basis of gene expression. Scheme for spatial ordering of CD-PCs using novoSpaRc and HVGs. Four hypothetical CD-PCs are shown. Each cell is assigned an individual spatial probability distribution on the basis of HVG expression. The location of maximal probability is indicated by an arrow. In our case, the given space for novoSpaRc to place cells was one dimensional with 100 spatial positions. (B) NovoSpaRc sorting of CD-PCs using HVGs correctly sorts cells derived from the cortex, outer medulla, and inner medulla. Results of joint sorting of CD-PCs from dissected samples (cortex, outer medulla, inner medulla) using 872 HVGs (CD-PC–specific HVGs). The panel shows the joint probability distribution functions for spatial location of all cortical, outer, and inner medullary CD-PCs along the spatial axis, separately, on the provided 100 spatial positions (positions 1–100). (C) NovoSpaRc sorting of CD-PCs correctly predicts known marker gene expression along the corticomedullary axis. Spatial expression analysis from in silico predictions of known CD-PC sub-cell type marker genes produced an expected distribution. The spatial probability density functions for the cortex, outer medulla, and inner medulla as shown in (B) are depicted above the heatmap. (D) NovoSpaRc sorting of CD-PCs depends on most of the HVGs. To analyze the effect of each individual HVG on sorting of CD-PCs from dissected scRNA-seq data, novoSpaRc sorting was performed iteratively by excluding one HVG at a time. We then compared the resulting sorting matrix (probability distributions for each cell) to the original sorting matrix, which was generated using the full set of HVGs. Distance is the Euclidean distance between the two resulting sorting matrices. Genes (872 CD-PC HVGs) were sorted according to the effect on sorting upon exclusion. (E) Spatial gene expression analysis of sorted CD-PCs predicts smooth transition of gene expression from cortical to inner medullary CD-PCs. Heatmaps show expression of regionally restricted genes sorted by their location of maximum expression in our scRNA-seq data from dissected samples. Genes were selected by expression thresholds and maximum-to-background ratio (Supplemental Methods). The displayed genes show a comparable spatial expression pattern in an independent second dataset5 (Supplemental Figure 5C). Gene expression values were min-max normalized per gene.
We next investigated the effect of each HVG on novoSpaRc’s sorting of CD-PCs in more detail. To estimate the effect of every single HVG, we iteratively repeated the sorting by consecutively excluding one HVG in each iteration. We then determined the Euclidean distance between the original sorting, using the full set of HVGs, and the sorting generated by excluding one HVG (Figure 3D). Interestingly, only 50 HVGs (6%) had no effect on sorting, whereas as many as 415 HVGs (48%) had a strong effect on the sorting order. This result suggests that spatial sorting of CD-PCs using HVGs was driven by a large proportion of HVGs used.
Given this information, we reasoned that selection of a larger number of location-specific markers among HVGs might allow us to reconstruct a high-resolution spatial map of CD-PCs along the corticomedullary axis. To achieve this, we selected location-specific marker genes that displayed (1) a single regional peak of expression along the corticomedullary axis, (2) a high total expression level, and (3) a high peak level to background ratio (Supplemental Methods). Using the independent mouse kidney scRNA-seq dataset from Ransick et al.5 as a discovery dataset, we identified a list of 104 predicted location-specific marker genes. In total, 85 of these genes (82%) could be validated in our own dataset, having comparable spatially restricted peak expression domains (Supplemental Methods). Heatmaps of these 85 genes along the 100 spatial positions indicated a smooth transition of cell states along the corticomedullary axis (Figure 3E, Supplemental Figure 5C). In contrast, we did not observe clusters of cell-type markers along the corticomedullary axis, as would be expected when having a limited number of distinct cell types. Our results thus indicate that cellular subtypes of CD-PCs continuously transition into one another, instead of forming distinct sharp gene-expression boundaries.
Spatial Gene-Expression Analysis Reveals Genes with Increasing or Decreasing Expression along the Corticomedullary Axis in CD-PCs
Given gene expression in CD-PCs displayed a gradual transition along the corticomedullary axis and to obtain molecular insights into the basis of this transition, we next focused on genes that displayed monotonously increasing or decreasing expression along the corticomedullary axis.
For this purpose, each gene’s expression profile along the corticomedullary axis was fitted by linear regression using a least-squares approach. Using cutoff values for the slope (75th percentile of all genes with positive or negative slope, respectively) and a correlation P value (<5e−10) and correlation coefficient (absolute value >0.6), we obtained 334 genes with negative regression coefficients (decreasing gene expression from the cortex to inner medulla) and 704 genes with positive regression coefficients (increasing gene expression from the cortex to inner medulla) (Figure 4A, Supplemental Tables 3 and 4).
Figure 4.
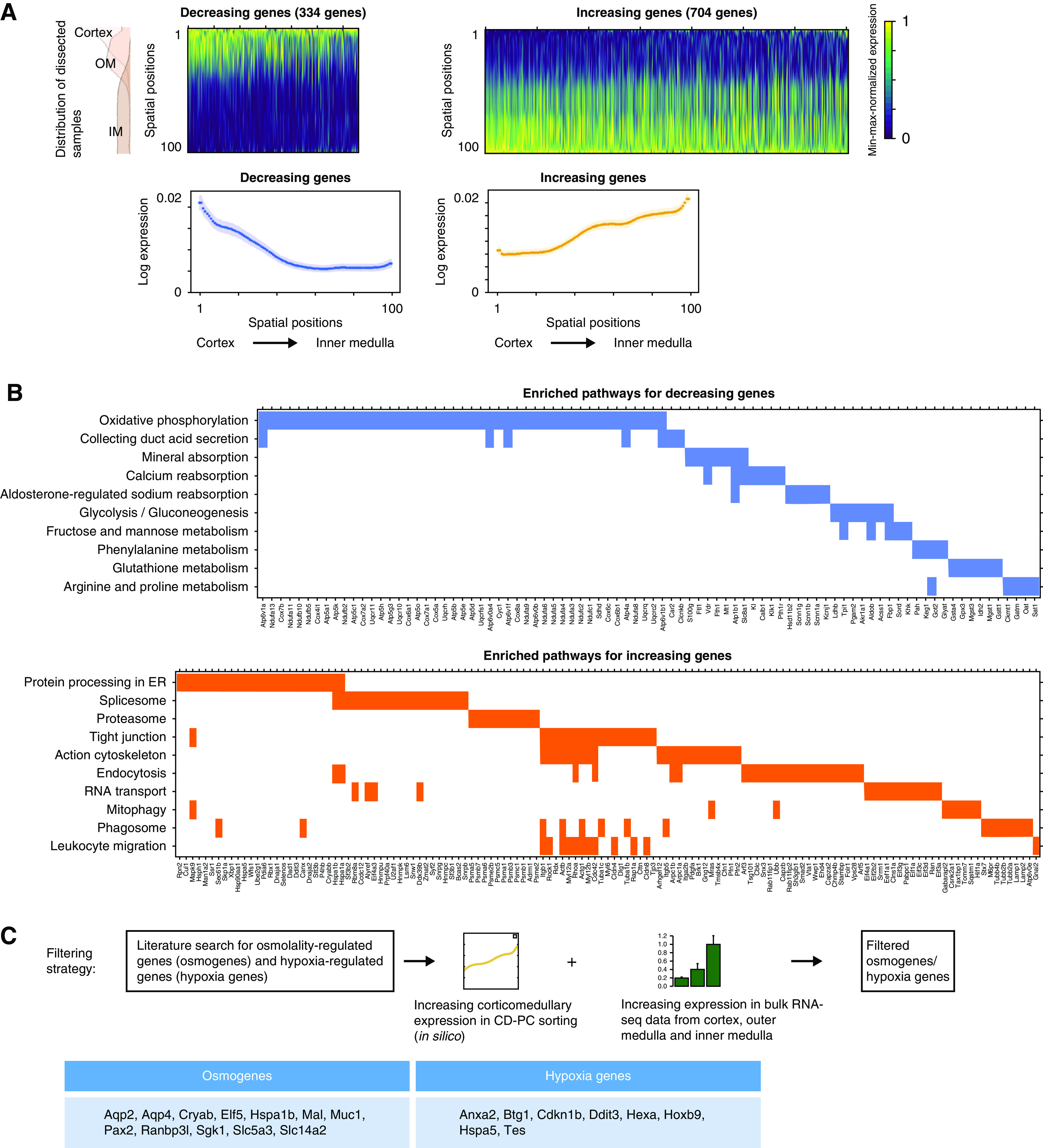
Multiple pathways show expression gradients along the corticomedullary axis in CD-PCs. (A) In silico predictions identify multiple genes with an increasing or decreasing corticomedullary expression gradient in CD-PCs. The upper panels show heatmaps and average expression plots of 704 genes with increasing expression and 334 genes with decreasing expression along the corticomedullary axis in CD-PCs. Genes were identified by linear regression of their spatial gene expression from in silico sorting. Genes are sorted from left to right by linear regression P value for each heatmap. Gene expression was min-max normalized per gene. The lower panels plot average gene expression values (log expression) along the corticomedullary axis of the genes depicted in the corresponding heatmaps above. Shades around average expression plots in lower panels represent 95% confidence intervals. (B) Multiple pathways are enriched for genes with increasing or decreasing corticomedullary expression gradients. Selected significantly enriched pathways from pathway enrichment analysis for genes depicted in (A) (decreasing or increasing genes, Supplemental Tables 3 and 4). (C) Corticomedullary gene expression analysis from in silico sorting of CD-PCs enables the identification of osmotically regulated genes (osmogenes) and hypoxia-regulated genes (hypoxia genes) that follow the known corticomedullary changes of cellular microenvironments (osmolality, hypoxia) of the kidney. Depicted is a scheme for the robust identification of osmogenes and hypoxia genes that show an increased spatial expression from cortex to the inner medulla. This would reflect the increasing corticomedullary tissue osmolality and hypoxia in the kidneys (Figure 1A) and make such genes candidate genes for physiologic phenotyping in scRNA-seq data. For this, we selected published osmogenes (Supplemental Table 5) and hypoxia genes that showed an increasing corticomedullary expression gradient in CD-PCs from our in silico sorting using novoSpaRc. We furthermore performed bulk RNA sequencing of dissected kidney regions (cortex, outer, and inner medulla, n=5 for each region) and demanded to additionally see an increasing corticomedullary expression of these genes in bulk data (Supplemental Figure 6). The lower table depicts osmogenes and hypoxia genes that fulfilled the criteria of this filtering process. ER, endoplasmatic reticulum.
Pathway enrichment analysis using Enrichr28 with the Kyoto Encyclopedia of Genes and Genomes 2019 mouse dataset revealed that genes with decreasing corticomedullary expression gradient in CD-PCs were associated with pathways such as oxidative phosphorylation and glycolysis, which may reflect the ample energy supply in the cortex and decreasing substrate availability toward the medulla. In addition, these genes were associated with energy-demanding active processes, such as amino acid metabolism and aldosterone-mediated sodium reabsorption (Figure 4B, Supplemental Table 3).
In contrast, genes with an increasing corticomedullary expression gradient were enriched for mediators of resilience against an increasingly hypoxic and hyperosmolar microenvironment in the inner medulla, and were enriched in pathways involved in cell adhesion (tight junction) and in molecular chaperones and heat shock proteins (Figure 4B, Supplemental Table 4).
We further explored the idea that genes with increasing expression from the cortex to inner medulla may contain osmotically regulated and hypoxia-regulated genes. We first conducted a literature search to generate a knowledge-based candidate list of 90 genes upregulated by surrounding tissue osmolarity (osmogenes) (Supplemental Table 5).15,29–36 A total of 14 osmogenes were represented within genes with increasing corticomedullary expression (P<0.001 for overrepresentation), but only two osmogenes among genes with decreasing corticomedullary expression (P=0.34 for overrepresentation). Among these osmogenes, 12 genes also displayed a significantly increasing expression in bulk RNA preparations from the dissected cortex, outer medulla and inner medulla (by RNA-seq from n=5 male adult C56bl/6N mice) (Figure 4C, Supplemental Figure 6). We conducted the same filtering process for hypoxia-associated genes using the hallmark hypoxia gene list from the Broad Institute (Figure 4C, Supplemental Figure 6), resulting in eight filtered hypoxia-associated genes showing increasing corticomedullary expression (Figure 4C, Supplemental Figure 6).
To validate in silico gene expression predictions, we performed RNAscope in situ hybridizations for selected abundant CD-PC genes. We stained for Scnn1g, an ENaC channel subunit, and Hsd11b2, a gene encoding for a protein preventing activation of the mineralocorticoid receptor by glucocorticoids (Figure 5A). Both genes displayed strongly decreasing corticomedullary expression gradients on the basis of in silico predictions. In contrast, Cryab, a gene encoding for heat shock protein and molecular chaperone Alpha-crystalline B chain, was among the genes with the most pronounced increasing corticomedullary expression gradient (Figure 5B). To also assess in silico predictions for more complex gene expression patterns, we stained for Fxyd4, encoding a regulatory subunit of the sodium potassium ATPase (Figure 5B). Semiquantitation of the in situ hybridization signal along the corticomedullary axis was largely consistent with the in silico predictions that were on the basis of scRNA-seq (Figure 5).
Figure 5.

In situ hybridizations of abundant CD-PC genes validate predictions from in silico sorting. (A and B) In silico predictions coincide with in situ staining for genes with increasing, decreasing, or complex corticomedullary expression pattern. Shown are RNAscope in situ hybridizations of selected abundant CD-PC genes with decreasing (Scnn1g, Hsd11b2), increasing (Cryab), or complex (Fxyd4) gene expression gradients from in silico predictions. The dotted arrow in the upper left in situ staining represents the corticomedullary axis. In silico predictions along the corticomedullary axis are plotted in yellow (left panels below in situ staining). For regional orientation, the spatial probability density functions for cortex, outer medulla, and inner medulla from sorting of dissected samples (Figure 3B) are depicted in the panels below the in silico predictions. Semi-quantification of in situ staining is depicted in the right panels next to the in silico predictions. For this, RNAscope staining was quantified (mean±SD) in two cortical regions (cortex split in half, resulting in an outer region and a region closer to the outer medulla), two outer medullary regions (corresponding to the outer and inner stripes of the outer medulla) and three inner medullary regions (inner medulla split in thirds). Five collecting ducts per region were quantified, values were normalized to the mean value of the region with maximum staining intensity. Scale bar: 1 mm.
Phenotyping of the Intrarenal Osmolality Gradient Based on Single-Cell Transcriptomics
We next hypothesized that quantitation of osmogene expression along the corticomedullary axis based on single-cell transcriptomics may allow prediction of intrarenal osmolality gradients. We had previously shown that mice which lack the transcription factor Grhl2 in collecting ducts (Grhl2CD−/− mice) have a flattened intrarenal corticomedullary osmolality gradient, characterized by a decreased osmolality in the renal medulla (1598 mosmol/kg H2O compared with 2254 mosmol/kg H2O in controls).19 Because our previous experiments indicated that cells from the kidney medulla were underrepresented in dissociated whole kidney cell suspensions, we performed snRNA-seq on kidney slices representing the cortex, outer medulla, and inner medulla (n=2, Figure 6A). We then identified CD-PCs on the basis of unbiased clustering and marker gene expression (1347 and 898 CD-PCs in control and Grhl2CD−/− samples, respectively, Supplemental Figure 7A) and aligned them to spatial positions along the corticomedullary axis on the basis of the spatial sorting of our dissected single-cell data and HVG expression (Figure 6A). An analysis of abundance of CD-PCs from the cortex, outer medulla, and inner medulla revealed this approach successfully overcame the underrepresentation of medullary CD-PCs in samples from dissociated whole kidney single-cell suspensions (Supplemental Figure 7B).
Figure 6.
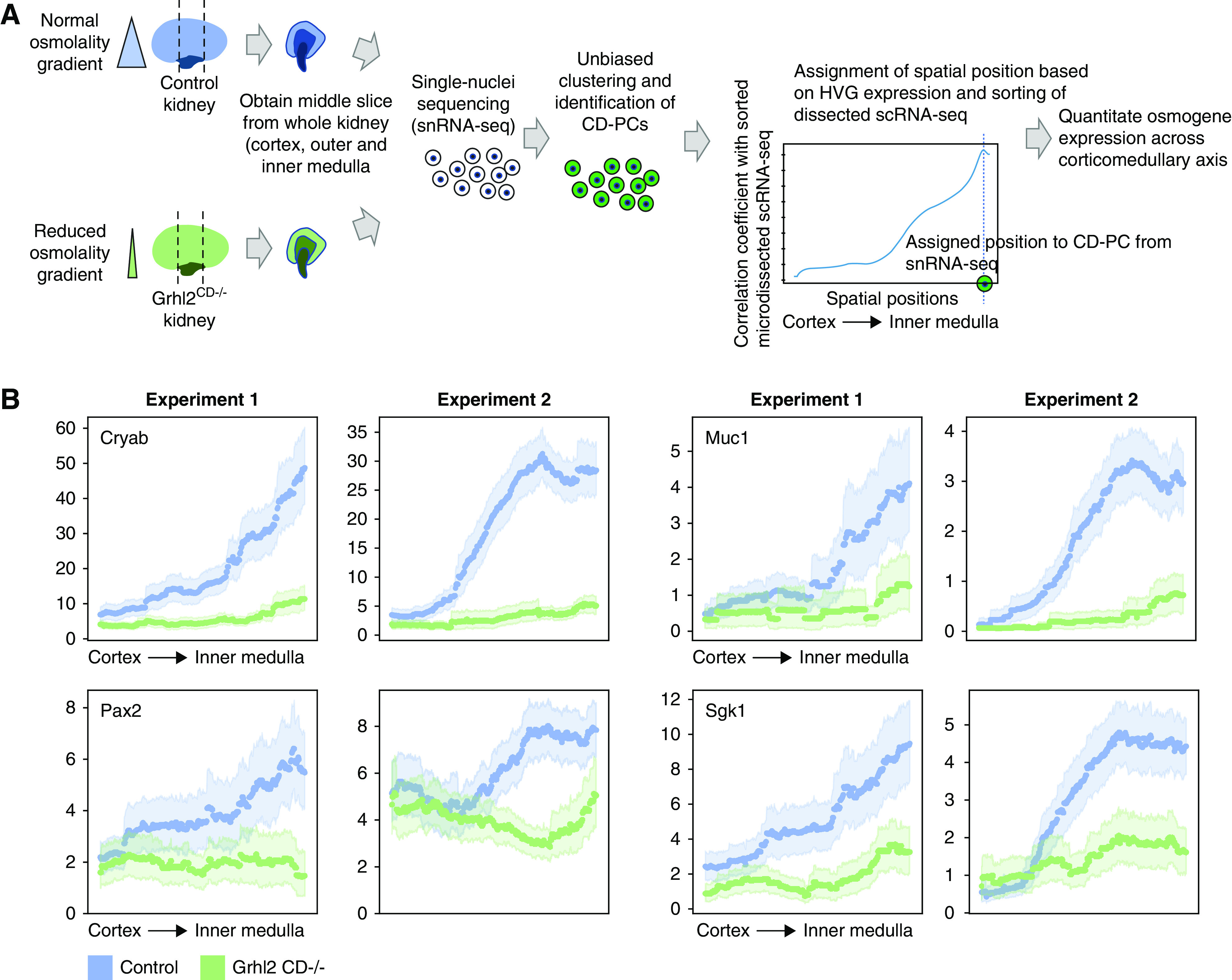
In silico spatial sorting enables physiologic phenotyping of the renal osmolality gradient in whole kidney samples. (A) Spatial sorting of CD-PCs from whole kidney snRNA-seq data. Depicted is the experimental strategy to assign spatial positions to CD-PCs in whole kidney snRNA-seq data. We chose to perform snRNA-seq to overcome the underrepresentation of medullary cells in whole kidney scRNA-seq. To further enrich for medullary cells, we obtained a middle slice containing the cortex, outer medulla, and inner medulla from each kidney of Grhl2CD−/− mice (decreased intrarenal osmolality gradient) and control mice (normal intrarenal osmolality gradient). Single-nuclei transcriptomes were obtained, and CD-PCs were identified on the basis of unbiased clustering and marker gene expression. CD-PCs from snRNA-seq were assigned regional positions by correlating HVG expression of each nucleus with HVG expression along the corticomedullary axis of sorted dissected samples (sorting performed with 500 positions, Figure 3B and C). We assigned the spatial position with maximum correlation as the new spatial position of the respective nucleus. This enabled the analysis of osmogene expression along the corticomedullary axis in the whole kidney snRNA-seq data. (B) Regional expression of selected osmogenes on the basis of snRNA-seq correctly predicts the reduced intrarenal osmolality gradient in Grhl2CD−/− mice. Depicted is a sliding average over 150 spatial positions for each whole kidney snRNA-seq sample (dotted lines with 95% confidence intervals indicated by light shades). Results are shown for two independent biologic replicates (experiment 1 and experiment 2). Expression values are library-normalized raw counts. Decreased osmogene expression in the medulla of Grhl2CD−/− kidneys reflects the known reduced medullary osmolality in these mice.19
Next, we used the osmogene set identified in Figure 4C and selected a subset of osmogenes that were expressed in >10% of CD-PCs from single-nuclei data. Using the described spatial ordering of CD-PCs from single-nuclei data, we were able to analyze expression levels of these osmogenes in CD-PCs along the corticomedullary axis comparing control and Grhl2CD−/− mice (Figure 6B, Supplemental Figure 8). This analysis predicted reduced osmogene expression toward the medulla in Grhl2CD−/−, which paralleled the degree of reduction in medullary osmolality. This demonstrated that a change in cellular microenvironment, namely, reduced medullary osmolality, could be accurately predicted on the basis of whole kidney single-nuclei transcriptomes. In principle, this approach can be applied to any existing mouse CD-PC single-cell transcriptome dataset.
Discussion
Key Findings
Our data indicate single-cell transcriptomes from dissociated kidneys harbor information that facilitates in silico prediction of spatial gene expression along the corticomedullary axis. The accuracy of spatial assignment of gene expression depended on the cell type and was highest for cell types that are exposed to a wide spectrum of microenvironmental conditions such as CD-PCs, TAL, or TL cells. We achieved a meaningful de novo reconstruction of CD-PC gene expression along the corticomedullary axis. Results were validated by analysis of established marker genes, by bulk RNA-seq of the cortex, outer, and inner medulla, by in situ hybridization of selected genes confirming the scRNA-seq–based predictions of gene expression, and by comparison with the known spatial origin of cells from the cortex, outer, and inner medulla. Genes with increasing or decreasing expression along the corticomedullary axis were enriched for functional categories related to changes of the microenvironment, including osmolality-regulated genes.
In addition, we developed a novel approach for regional quantitation of osmo-responsive gene expression on the basis of single-cell transcriptomes. This approach facilitated gene expression–based prediction of the physiologic osmolality gradient of the kidney and its alteration in Grhl2CD−/− kidneys. Notably, both spatial sorting on the basis of HVG expression and osmogene expression readouts can be obtained post hoc on existing single-cell transcriptomic datasets on the basis of code that we supply in the Supplemental Methods and the online code.
Biologic Implications
Our approach enabled us to gain novel insights into the cellular biology of the kidney. By analyzing genes with local spatial expression maxima, we observed a smooth transition from cortical to IMCD-PCs along the corticomedullary axis rather than sharp boundaries of gene expression. This observation was unexpected given it was previously assumed that at least cortical, outer medullary, and inner medullary CD-PCs represent distinct cell types.5,27,37 Previous studies have identified morphologic and functional differences within major cell types of the kidney between the cortex, outer, and inner medulla,38,39 including in CD-PCs.25,37,40 However, how these findings correlate with regional transcriptome-wide gene expression states is unknown, because previous studies had to rely on single markers instead of marker combinations. Our findings underline the complexity of distinguishing “cell types” versus “cell states” when cells are exposed to changing environmental conditions. We suggest that a rather smooth transition of gene expression profiles from cortical to medullary CD-PCs would be consistent with a model where microenvironmental gradients, such as osmotic gradients, would drive cell-state transitions. Obviously, this idea would require further validation, which may become feasible with the advent of novel spatial transcriptomic techniques (see below).
Our results highlight that categorization-based methods that group cells into different types might miss the functional relevance of continuous cell state transitions. Previous studies had mainly focused on such continuous cell-state transitions in contexts of development and differentiation,41–43 but our study highlights that they do occur in the adult kidney and may be relevant in additional biologic systems. In future studies, it will be also important to investigate gradually changing gene expression in disease conditions.
Technical Advances
In this study, we used both scRNA-seq and snRNA-seq approaches. scRNA will capture the full cellular mRNA content, but requires tissue dissociation by enzyme-based approaches, which may introduce transcriptional stress responses and alter transcriptomes in additional ways. In addition, tissue dissociation appears to be more efficient in cortex versus medulla, thereby introducing bias. In contrast, snRNA-seq does not require tissue dissociation and can be applied on frozen samples, thereby eliminating dissociation-induced transcriptional responses, and carries less of a dissociation bias, as was recently reported by others.44,45 However, due to the low medullary versus cortical mass and the physiologic abundances of cell types, whole kidney single-cell datasets will still display a large overrepresentation of proximal tubule cells and fewer cells representing the distal nephron and collecting duct. We overcame this problem by applying snRNA-seq to a central kidney block that represents the cortex, outer, and inner medulla, and thereby skews cellular abundances toward medullary cell types. This approach may be useful in future experimental settings, because it saves resources and still allows an investigation of cells along the entire corticomedullary axis.
Our study highlights the potential spatial and physiologic information harbored within single-cell transcriptomes of dissociated organs. Yet, the validation approach with in situ hybridization is time and resource consuming, limiting the number of genes that can be selected for validation. Efforts are ongoing to establish spatial transcriptomics using slides with fixed, oligo-covered beads carrying regional identifier barcodes.7,8,46,47 These approaches provide transcriptome-wide expression data at a scale of approximately 10 µm,8 but they are limited in sequencing depth. Hence, a promising perspective lies in the combination of in silico prediction-based approaches developed herein with genome-wide validation by regional transcriptomics.
Study Limitations
Analyses of single-cell data naturally depend on the number of cells, genes, and transcripts identified per cell. The number of cells, genes, and transcripts detected in our Dropseq dataset is relatively low when compared with newer platforms,1,5 and represents a limitation of our study. However, we were able to validate our findings by independent methods, including a second dataset with a higher transcriptional depth.5 This indicates the approaches developed and applied here successfully attenuate the sparseness of transcriptional data to some extent.
It must be noted that not all single-cell datasets appear to preserve the transcriptional responses of cells to the osmotic gradient that we describe here. We achieved successful spatial reconstruction of previously reported scRNA-seq datasets that used cold-protease digestion at 4°C or single-nuclear RNA-seq approaches using frozen samples.5,20,44
Our approach to spatial sorting in silico depends on the assumption that spatial proximity will translate into similarity in gene expression. Although our approach proved to be robust in the healthy kidney tissues used herein, it may not retain validity when focal variability becomes relevant, which may be the case in disease settings such as ischemia or focal inflammation.48 In addition, whereas the spatial sorting approach introduced should generally be feasible for different kidney cell types, its output quality may vary by the number of cells available, by the corticomedullary distribution pattern of the cell type, and by additional unknown factors (e.g., spatial sorting of CD-PCs showed better results than sorting of CD-ICs).
We used Grhl2CD−/− mice as a model of a flattened osmotic gradient, because we had previously shown these mice display a reduced medullary osmolality.19 We cannot completely rule out that changes of osmotically regulated gene expression in the collecting duct may be directly affected by Grhl2 deletion in these cells. In a previous study, osmotically regulated genes reported herein were not shown to be direct transcriptional targets of Grhl2,49 and we had previously provided evidence that downregulation of one osmotically regulated gene, Aqp2, was due to changes of tissue osmolality rather than direct transcriptional responses to Grhl2 deletion.19 In addition, we found downregulation of osmotically regulated genes in Grhl2 CD−/− mice not only in CD-PC, but also in TAL, TL, and endothelial cells (data not shown), supporting the idea that osmolality, rather than Grhl2-mediated gene regulation, drives these responses.
Disclosures
K.-U. Eckardt reports receiving research funding from Amgen, AstraZeneca, Bayer, Fresenius, Genzyme, Shire, and Vifor, and honoraria from Akebia, Astellas, Boehringer Ingelheim, Bayer, Genzyme, Retrophin, and Vifor. K. Schmidt-Ott reports consultancy fees from BioPorto Diagnostics; license revenue related to the use of a neutrophil gelatinase-associated lipocalin assay via Columbia University; research funding from FAST BioMedical, for being a principal investigator of the EMPAKT-CHF trial (NCT03808948), and Quark Pharmaceuticals, for being the site principal investigator for QRK309 trial (NCT03510897); and being an editorial board member for Kidney International, outside the submitted work. M. Bleich reports being a scientific advisor or a member of Pflügers Archiv—European Journal of Physiology, Kidney BP Research, JASN, and Acta Physiologica Sinica. N. Rajewsky reports being a coordinator and member of LifeTime; a member of the Organizing Committee Human Cell Atlas; an advisory member of the editorial board of the Journal of Molecular Systems Biology; a founding member of Integrative Research Institute for the Life Sciences (Humboldt University, Max Delbrueck Center for Molecular Medicine, Charité University Medicine, Berlin); a member of EMBO, Leopoldina; and a member of the scientific advisory boards of Istituto Nazionale Genetica Moleculare (Milan, Italy) and of the Medical Research Council Clinical Science Centre (London, UK). All remaining authors have nothing to disclose.
Funding
K. Schmidt-Ott was supported by the German Research Foundation (DFG) Collaborative Research Grant – Project ID 394046635 – SFB 1365 (C06), DFG Research Training Group GRK 2318 (B4), DFG Research Unit FOR 2841 (P02), and the Urological Research Foundation (Berlin, Germany). C. Hinze was supported by the Berlin Institute of Health Charité Clinician Scientist Program. N. Karaiskos was supported by DFG grant GZ KA 5006/1-1. A. Boltengagen was supported by funding from the Gottfried Wilhelm Leibniz Prize of N. Rajewsky from the DFG.
Data Sharing Statement
Regional gene expression data in all kidney cell types and in silico prediction of spatial gene expression patterns in CD-PCs are can be searched and viewed at https://shiny.mdc-berlin.de/KidneySpatial/.
All processed and raw data are available on GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145690.
Supplementary Material
Acknowledgments
We thank Tatjana Luganskaja for excellent technical support. Christian Hinze, Nikolaus Rajewsky, and Kai M. Schmidt-Ott conceived the study; Kai M. Schmidt-Ott and Nikolaus Rajewsky provided funding; Christian Hinze, Christine Kocks, and Kai M. Schmidt-Ott planned experiments; S. Steven Potter and Andrew S. Potter helped establish cold-digestion protocols (scRNA-seq); Christine Kocks supervised and Christine Hinze and Anastasiya Boltengagen carried out scRNA-seq and snRNA-seq; Katharina Walentin performed RNA in situs; Klea Redo extracted RNA for bulk RNA-sequencing; Christine Hinze, Nikos Karaiskos, Christine Kocks, Nikolaus Rajewsky, Kai-Uwe Eckardt, and Kai M. Schmidt-Ott analyzed data; Nina Himmerkus and Markus Bleich assisted with CD-PC gene expression and cell-type analysis; Christine Hinze designed (multinomial model) and performed bioinformatics analyses, supervised by Nikos Karaiskos (novoSpaRc); Christine Hinze and Kai M. Schmidt-Ott wrote the paper with critical input from Nikos Karaiskos, Christine Kocks, Kai-Uwe Eckardt, Nina Himmerkus, Markus Bleich, and Nikolaus Rajewsky.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020070930/-/DCSupplemental.
Supplemental Figure 1. Preparation of dissected samples from whole kidneys.
Supplemental Figure 2. Clustering of interstitial cells from baseline kidney samples.
Supplemental Figure 3. Correct regional assignment of kidney tubule cells on the basis of their transcriptomes is cell-type dependent.
Supplemental Figure 4. Comparison of spatial sorting with different numbers of provided positions.
Supplemental Figure 5. Validation of spatial assignments.
Supplemental Figure 6. Osmogene and hypoxia gene filtering.
Supplemental Figure 7. Whole kidney snRNA-seq data and spatial alignment of whole kidney snRNA-seq and scRNA-seq data.
Supplemental Figure 8. Spatial expression of filtered osmogenes.
Supplemental Table 1. Cell type-specific marker genes.
Supplemental Table 2. Cell type-specific highly variable genes.
Supplemental Tables 3 and 4. Pathway enrichment analysis results and gene lists for genes with corticomedullary increasing and decreasing expression gradients.
Supplemental Table 5. Osmogenes identified by literature research as indicated.
References
- 1.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al.: Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, et al.: The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A 116: 19619–19625, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu H, Humphreys BD: Single cell sequencing and kidney organoids generated from pluripotent stem cells. Clin J Am Soc Nephrol 15: 550–556, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindström NO, Guo J, Kim AD, Tran T, Guo Q, De Sena Brandine G, et al.: Conserved and divergent features of mesenchymal progenitor cell types within the cortical nephrogenic niche of the human and mouse kidney. J Am Soc Nephrol 29: 806–824, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ransick A, Lindström NO, Liu J, Zhu Q, Guo J, Alvarado GF, et al.: Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev Cell 51: 399–413.e7, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al.: Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178: 714–730.e22, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maniatis S, Äijö T, Vickovic S, Braine C, Kang K, Mollbrink A, et al.: Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 364: 89–93, 2019 [DOI] [PubMed] [Google Scholar]
- 8.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al.: Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363: 1463–1467, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nitzan M, Karaiskos N, Friedman N, Rajewsky N: Gene expression cartography. Nature 576: 132–137, 2019 [DOI] [PubMed] [Google Scholar]
- 10.Lee CJ, Gardiner BS, Evans RG, Smith DW: A model of oxygen transport in the rat renal medulla. Am J Physiol Renal Physiol 315: F1787–F1811, 2018 [DOI] [PubMed] [Google Scholar]
- 11.Cowley AW Jr.: Role of the renal medulla in volume and arterial pressure regulation. Am J Physiol 273: R1–R15, 1997 [DOI] [PubMed] [Google Scholar]
- 12.Sands JM, Layton HE: The physiology of urinary concentration: An update. Semin Nephrol 29: 178–195, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurbel S, Dodig K, Radić R: The osmotic gradient in kidney medulla: A retold story. Adv Physiol Educ 26: 278–281, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Knepper MA: Measurement of osmolality in kidney slices using vapor pressure osmometry. Kidney Int 21: 653–655, 1982 [DOI] [PubMed] [Google Scholar]
- 15.Schulze Blasum B, Schröter R, Neugebauer U, Hofschröer V, Pavenstädt H, Ciarimboli G, et al.: The kidney-specific expression of genes can be modulated by the extracellular osmolality. FASEB J 30: 3588–3597, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Brezis M, Heyman SN, Dinour D, Epstein FH, Rosen S: Role of nitric oxide in renal medullary oxygenation. Studies in isolated and intact rat kidneys. J Clin Invest 88: 390–395, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leichtweiss HP, Lübbers DW, Weiss C, Baumgärtl H, Reschke W: The oxygen supply of the rat kidney: Measurements of int4arenal pO2. Pflugers Arch 309: 328–349, 1969 [DOI] [PubMed] [Google Scholar]
- 18.Evans RG, Smith DW, Lee C-J, Ngo JP, Gardiner BS: What Makes the Kidney Susceptible to Hypoxia? Anat Rec 303: 2544–2552, 2020 [DOI] [PubMed] [Google Scholar]
- 19.Hinze C, Ruffert J, Walentin K, Himmerkus N, Nikpey E, Tenstad O, et al.: GRHL2 is required for collecting duct epithelial barrier function and renal osmoregulation. J Am Soc Nephrol 29: 857–868, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adam M, Potter AS, Potter SS: Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: A molecular atlas of kidney development. Development 144: 3625–3632, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alles J, Karaiskos N, Praktiknjo SD, Grosswendt S, Wahle P, Ruffault PL, et al.: Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biol 15: 44, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al.: Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–1214, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao Y, Smyth GK, Shi W: featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930, 2014 [DOI] [PubMed] [Google Scholar]
- 24.Sun AM, Liu Y, Centracchio J, Dworkin LD: Expression of Na+/H+ exchanger isoforms in inner segment of inner medullary collecting duct. J Membr Biol 164: 293–300, 1998 [DOI] [PubMed] [Google Scholar]
- 25.Clapp WL, Madsen KM, Verlander JW, Tisher CC: Morphologic heterogeneity along the rat inner medullary collecting duct. Lab Invest 60: 219–230, 1989 [PubMed] [Google Scholar]
- 26.Clark JZ, Chen L, Chou CL, Jung HJ, Lee JW, Knepper MA: Representation and relative abundance of cell-type selective markers in whole-kidney RNA-Seq data. Kidney Int 95: 787–796, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JW, Chou CL, Knepper MA: Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al.: Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brocker C, Thompson DC, Vasiliou V: The role of hyperosmotic stress in inflammation and disease. Biomol Concepts 3: 345–364, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berl T: How do kidney cells adapt to survive in hypertonic inner medulla? Trans Am Clin Climatol Assoc 120: 389–401, 2009 [PMC free article] [PubMed] [Google Scholar]
- 31.Berry MR, Mathews RJ, Ferdinand JR, Jing C, Loudon KW, Wlodek E, et al.: Renal sodium gradient orchestrates a dynamic antibacterial defense zone. Cell 170: 860–874.e19, 2017 [DOI] [PubMed] [Google Scholar]
- 32.Ho SN: Intracellular water homeostasis and the mammalian cellular osmotic stress response. J Cell Physiol 206: 9–15, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Siroky BJ, Kleene NK, Kleene SJ, Varnell CD Jr., Comer RG, Liu J, et al.: Primary cilia regulate the osmotic stress response of renal epithelial cells through TRPM3. Am J Physiol Renal Physiol 312: F791–F805, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein CS, Yancey PH, Martins I, Sigmund RD, Stokes JB, Davidson BL: Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am J Physiol Cell Physiol 298: C1388–C1400, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aboudehen K, Noureddine L, Cobo-Stark P, Avdulov S, Farahani S, Gearhart MD, et al.: Hepatocyte nuclear factor-1β regulates urinary concentration and response to hypertonicity. J Am Soc Nephrol 28: 2887–2900, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai Q, Dmitrieva NI, Ferraris JD, Brooks HL, van Balkom BW, Burg M: Pax2 expression occurs in renal medullary epithelial cells in vivo and in cell culture, is osmoregulated, and promotes osmotic tolerance. Proc Natl Acad Sci U S A 102: 503–508, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madsen KM, Tisher CC: Structural-functional relationships along the distal nephron. Am J Physiol 250: F1–F15, 1986 [DOI] [PubMed] [Google Scholar]
- 38.Barrett JM, Kriz W, Kaissling B, de Rouffignac C: The ultrastructure of the nephrons of the desert rodent (Psammomys obesus) kidney. I. Thin limb of Henle of short-looped nephrons. Am J Anat 151: 487–497, 1978 [DOI] [PubMed] [Google Scholar]
- 39.Kriz W: Structural organization of the renal medulla: Comparative and functional aspects. Am J Physiol 241: R3–R16, 1981 [DOI] [PubMed] [Google Scholar]
- 40.Madsen KM, Clapp WL, Verlander JW: Structure and function of the inner medullary collecting duct. Kidney Int 34: 441–454, 1988 [DOI] [PubMed] [Google Scholar]
- 41.Haghverdi L, Buettner F, Theis FJ: Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31: 2989–2998, 2015 [DOI] [PubMed] [Google Scholar]
- 42.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al.: The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32: 381–386, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bendall SC, Davis KL, Amir AD, Tadmor MD, Simonds EF, Chen TJ, et al.: Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157: 714–725, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, Kirita Y, Donnelly EL, Humphreys BD: Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 30: 23–32, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, et al.: A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun 10: 2832, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thrane K, Eriksson H, Maaskola J, Hansson J, Lundeberg J: Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res 78: 5970–5979, 2018 [DOI] [PubMed] [Google Scholar]
- 47.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al.: Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353: 78–82, 2016 [DOI] [PubMed] [Google Scholar]
- 48.Vigolo E, Markó L, Hinze C, Müller DN, Schmidt-Ullrich R, Schmidt-Ott KM: Canonical BMP signaling in tubular cells mediates recovery after acute kidney injury. Kidney Int 95: 108–122, 2019 [DOI] [PubMed] [Google Scholar]
- 49.Aue A, Hinze C, Walentin K, Ruffert J, Yurtdas Y, Werth M, et al.: A grainyhead-like 2/ovo-like 2 pathway regulates renal epithelial barrier function and lumen expansion. J Am Soc Nephrol 26: 2704–2715, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.