

Abstract
Quantitative polymerase chain reactions (qPCRs) are commonly employed to enumerate genes of interest among particular biological samples. Insertion of PCR amplicons into plasmid DNA is a mainstay for creation of known quantities of target sequences to standardize quantitative PCRs. Typically, one amplicon is inserted into one plasmid construct, the plasmid is then amplified, purified, serially diluted, and then quantified to be used to enumerate target sequences in unknown samples. As qPCR is often used to detect multiple amplicons simultaneously, individual qPCR standards are often desired to be normalized one to another. Here we report a single plasmid containing 8 amplicons which can be used to quantify several different strains of SIV and HIV, cell number equivalents for humans and nonhuman primates, T cell receptor excision circles, and bacterial 16S DNA. This FRugally Optimized DNA Octomer (FRODO) plasmid was created and standardized to quantify all 8 PCR amplicons.
Keywords: HIV, SIV, nonhuman primate, qPCR
INTRODUCTION
Commonly in immunological and microbiological studies, quantitative PCR is used to enumerate numbers of viral, bacterial or immunological genes in a biological sample. Individual DNA standards are used to construct standard curves of threshold cycle values for known quantities of DNA to which amplification of genes of interest in unknown samples can be compared. This method typically requires the generation, purification, quantification, storage and maintenance of individual DNA standards for each gene analyte. In this article, we describe a method to quantify eight microbiological analytes simultaneously using a novel qPCR standard DNA plasmid, FRODO. We provide a simple method to extract DNA from primary human or non-human primate (NHP) cells for quantification of viral, bacterial, and cell-number equivalent genes by qPCR, as well as a method to prepare the novel FRODO plasmid.
CAUTION: Follow appropriate regulations and guidelines for working with human- or NHP-derived products or infectious agents. This includes proper biosafety practices and approval of the appropriate Institutional Review Board.
STRATEGIC PLANNING
The workflow in this article begins with Basic Protocol 1 in which total genomic DNA is extracted from a biological sample to be analyzed. Target DNA in the sample extract is then quantified in Basic Protocol 2 using a DNA standard prepared in Support Protocol 1.
The starting material for DNA extraction in Basic Protocol 1 is a dry cell pellet of 5×101 to 5×105 cells. This dry cell pellet can be prepared from a single cell suspension by centrifuging 5 minutes at ≥ 500 × g, then aspirating the supernatant carefully to avoid disturbing the cell pellet. The dry cell pellet can be processed immediately as described in Basic Protocol 1 or frozen at −80°C until use. Often, it is desired to quantify target DNA from a select population of cells commonly isolated by fluorescence-activated cell sorting (FACS). Sorted cells can be pelleted and the supernatant aspirated as mentioned previously. Alternatively, single-cell suspensions from blood, tissue, cell culture, or previously frozen cells could be prepared as dry cell pellets. Approximate cell numbers from single-cell suspensions to ensure an appropriate number of cells is used. When analyzing multiple samples, it is not necessary for each sample to have equal numbers of cells as the quantitative PCR will be normalized for loading amount using cell-number equivalent genes. This procedure is appropriate for live cells, cells fixed with paraformaldehyde, and cells permeabilized followed by intracellular antigen staining and then fixed with paraformaldehyde.
The starting material for Support Protocol 1 is five dry cell pellets of 5×101, 5×102, 5×103, 5×104, and 5×105 as well as a stock solution of the FRODO standard plasmid. To obtain the cell pellets, human or NHP cells are counted and sorted into tubes by a flow cytometry cell sorting instrument, then centrifuged and media aspirated as described above. A stock solution of the FRODO standard plasmid is available from the authors upon request.
BASIC PROTOCOL 1
TOTAL GENOMIC DNA EXTRACTION FROM PRIMARY CELLS.
This protocol describes preparation of cell lysates from human and NHP cells for quantification of genomic DNA as well as HIV, SIV, or bacterial DNA present in cells by qPCR. The starting material is a dry cell pellet (see Strategic Planning) that is lysed in buffer with proteinase K. The resulting lysate can be used immediately in downstream qPCR assays or frozen for long term storage.
Materials
Between 5×101 to 5×105 human or old world primate cells, pelleted and supernatant removed
10 mM Trizma hydrochloride (TRIS), pH 7.4 (Sigma #T2663–1L)
Proteinase K, recombinant PCR Grade, 18.5 mg/mL (Roche #03115887001)
1.5 mL screw-cap microcentrifuge tubes (Sarstedt #72.692.005)
One or two heat blocks such as ThermoMixer F1.5 (Eppendorf #5384000020)
Benchtop microcentrifuge such as Centrifuge 5424 (Eppendorf #022620428)
Preheat heat block to 56 °C. If two heat blocks are available, preheat the second heat block to 95 °C.
Prepare a working solution of proteinase K diluted 1:100 in 10 mM TRIS HCl. Each sample will require 50 μL of working solution.
Resuspend the dry cell pellet in 50 μL of TRIS/proteinase K working solution making sure to mix thoroughly by pipetting.
-
Incubate tubes on a heat block for at least 1 hour at 55°C with gentle shaking (ca. 350 rpm).
When lysing larger quantities of cells, the lysis reaction may need to proceed at 55°C for longer than 1 hour.
-
Heat-inactivate the proteinase K by incubating tubes on a heat block for 10 minutes at 95 °C.
Incubating at 95 °C for longer than 10 minutes may result in loss of DNA.
-
Use cell lysates immediately in downstream qPCR reactions (see Basic Protocol 2).
Cell lysates can be stored on ice or at 4°C temporarily (< 12 hours) and should be kept at −20 °C or −80 °C for longer term storage.
BASIC PROTOCOL 2
QUANTITATIVE PCR FOR VIRAL, BACTERIAL, CELL NUMBER EQUIVALENTS
This protocol describes the real-time quantitative PCR setup and analysis with standard curves based on serial dilutions of up to eight target analytes in human or NHP primate cell lysates. The protocol uses a single plasmid DNA standard (FRODO) of known copy number containing primer and probe binding sites for each of the following amplicons: T cell receptor excision circles (TREC), HIVgag, SIVmac239, bacterial 16S rRNA, SIVsm, SIVagm, albumin, and CCR5. For quantification, a standard curve is generated from threshold cycle numbers (Ct) of serial dilutions of the FRODO plasmid. The standard curve best fit line is used to calculate DNA copy numbers from Ct values resulting from amplification of target analytes in cell lysates as prepared in Basic Protocol 1. For preparation of the DNA standard plasmid, FRODO, see Support Protocol 1.
Materials
Sample lysates as prepared in Basic Protocol 1
100 μM primers and probes (see Table 1 for sequences)
TaqMan Gene Expression MasterMix (Applied Biosystems #4369016)
FRODO plasmid standard dilution series (see Support Protocol 1)
Molecular biology grade water (Invitrogen #AM9937)
1.5 mL microcentrifuge tubes
MicroAmp Fast Optical 96-Well Reaction Plate (Applied Biosystems #4346906)
MicroAmp Optical Adhesive Film (Applied Biosystems #4311971)
Benchtop centrifuge with plate adapters such as Centrifuge 5430 (Eppendorf #022620568)
StepOnePlus Real-Time PCR System with StepOne Software (Applied Biosystems #272007793)
Table 1.
Primer and probe sequences of analytes included in FRODO.
| Human TREC (D. C. Douek et al., 2000) | |
| Forward | 5’- CACATCCCTTTCAACCATGCT -3’ |
| Reverse | 5’- GCCAGCTGCAGGGTTTAGG -3’ |
| Probe | 5’- FAM - ACACCTCTGGTTTTTGTAAAGGTGCCCACT -BHQ -3’ |
| Monkey TREC (Sodora et al., 2002) | |
| Forward | 5’- CACATCCCTTTCAACCATGCT -3’ |
| Reverse | 5’- GCCAGCTGCAGGGTTTAGG -3’ |
| Probe | 5’- FAM - ACGCCTCTGGTTTTTGTAAAGGTGCTCACT -BHQ -3’ |
| HIV gag (D.C. Douek et al., 2002) | |
| Forward | 5’- GGTGCGAGAGCGTCAGTATTAAG -3’ |
| Reverse | 5’- AGCTCCCTGCTTGCCCATA -3’ |
| Probe | 5’- FAM - AAAATTCGGTTAAGGCCAGGGGGAAAGAA -BHQ -3’ |
| SIVmac239 (Mattapallil et al., 2005) | |
| Forward | 5’- GTCTGCGTCAT(T/C)TGGTGCATTC -3’ |
| Reverse | 5’- CACTAG(C/T)TGTCTCTGCACTAT(A/G)TGTTTTG -3’ |
| Probe | 5’- FAM - CTTC(A/G)TCAGT(C/T)TGTTTCACTTTCTCTTCTGCG -BHQ -3’ |
| Human Albumin (D.C. Douek et al., 2002) | |
| Forward | 5’- TGCATGAGAAAACGCCAGTAA -3’ |
| Reverse | 5’- ATGGTCGCCTGTTCACCAA -3’ |
| Probe | 5’- FAM - TGACAGAGTCACCAAATGCTGCACAGAA -BHQ -3’ |
| Monkey Albumin (Mattapallil et al., 2005) | |
| Forward | 5’- TGCATGAGAAAACGCCAGTAA -3’ |
| Reverse | 5’- ATGGTCGCCTGTTCACCAA -3’ |
| Probe | 5’- FAM - AGAAAGTCACCAAATGCTGCACGGAATC -BHQ -3’ |
| 16S (1369–1492) (Suzuki, Taylor, & DeLong, 2000) | |
| Forward | 5’- CGGTGAATACGTTCYCGG -3’ |
| Reverse | 5’- GGWTACCTTGTTACGACTT -3’ |
| Probe | 5’- FAM - CTTGTACACACCGCCCGTC -BHQ -3’ |
| SIVsm (Brenchley et al., 2012) | |
| Forward | 5’- GGCAGGAAAATCCCTAGCAG -3’ |
| Reverse | 5’- GCCCTTACTGCCTTCACTCA -3’ |
| Probe | 5’- FAM - AGTCCCTGTTCRGGCGCCAA -BHQ -3’ |
| SIVagm (Beaumier et al., 2009) | |
| Forward | 5’- GTCCAGTCTCAGCATTTACTTG -3’ |
| Reverse | 5’- CGGGCATTGAGGTTTTTCAC -3’ |
| Probe | 5’- FAM - CAGATGTTGAAGCTGACCATTTGGG -BHQ -3’ |
| CCR5 (Venneti et al., 2008) | |
| Forward | 5’- CCAGAAGAGCTGAGACATCC -3’ |
| Reverse | 5’- CTAAYAGGCCAAGCAGCTGAG -3’ |
| Probe | 5’- FAM - TTCCCCTACAAGAAACTCTCCCCGGTAAGTA -BHQ -3’ |
| AmpR (Sastry, Johnson, Hobson, Smucker, & Cornetta, 2002) | |
| Forward | 5’- GTTGCCATTGCTACAGGCATC -3’ |
| Reverse | 5’- ACTCGCCTTGATCGTTGGG -3’ |
| Probe | 5’- FAM - ACGCTCGTCGTTTGGTATGGCTTCATTC -BHQ -3’ |
Prepare 12.5 μM working solutions of forward and reverse primer sets. These can be mixed in advance and stored at −20 °C.
-
Prepare 5 μM working solutions of each DNA probe. This may also be stored at −20 °C when made in advance.
Protect from light. When exposed to light, probes will lose fluorescent signal and decrease sensitivity in the qPCR assay.
-
For each target amplicon to be analyzed, prepare a master mix allowing for 20 μL per reaction as indicated in Table 2.
To allow for pipetting error, include excess in the calculation.
Map the layout of samples and standards in a 96-well qPCR plate, running triplicate (duplicate is fine when the experimenter is confident in their pipetting accuracy) reactions for each sample or standard. Include a no-template control (NTC) of molecular biology grade water for each amplicon master mix. See Figure 1.
Aliquot 20 μL of appropriate master mix into each well, depositing the liquid at the bottom of the well.
-
Add 5 μL of sample, NTC, or standards to wells, pipetting liquid onto the side of the well.
Addition of the standards at a bench away from the PCR bench is recommended to avoid contamination of stock PCR reagents and plasticware.
Seal the qPCR plate with MicroAmp adhesive film.
Quick spin the plate in a benchtop centrifuge to bring all reagents to the bottom of the wells and remove any bubbles.
- Transfer plate to StepOnePlus thermocycler. In the StepOne software assign targets and samples. Run the PCR at standard temperature ramp speed with the following cycle conditions:
- Step 1: 95°C for 10 min
- Step 2: 95°C for 15 sec
- Step 3: 60°C for 1 min
- Step 4: Repeat steps 2–3 for 45 cycles
In StepOne software, assign copy numbers for FRODO standard to each dilution and export results.
Table 2.
Preparation of master mix
| Component | Initial concentration | Final concentration | μL per reaction |
|---|---|---|---|
| AB TaqMan MasterMix | 2× | 1× | 10 |
| Forward primer | 12.5 μM | 0.5 μM | 1 |
| Reverse primer | 12.5 μM | 0.5 μM | 1 |
| Probe | 5 μM | 0.1 μM | 1 |
| Nuclease free water | 7 |
Figure 1.
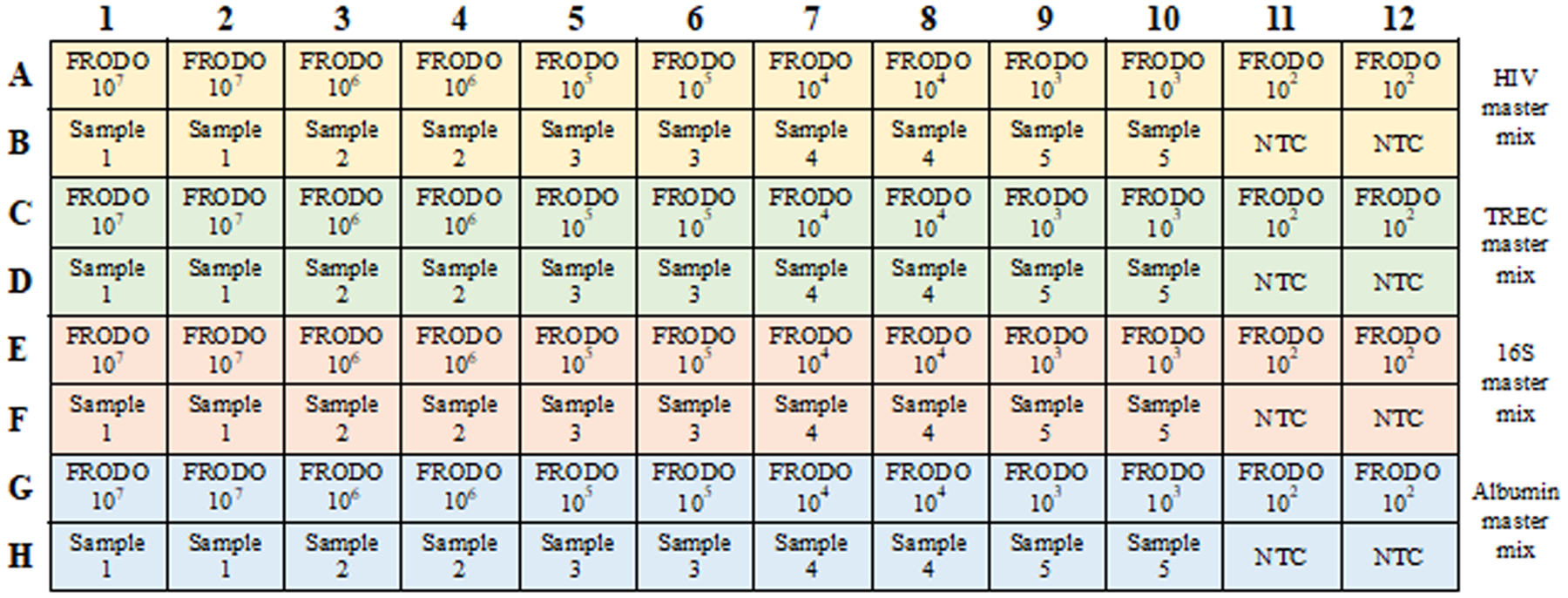
Example PCR plate layout mapping samples and FRODO standards with master mixes for each target analyte (HIV: yellow, TREC: green, 16S: red, albumin: blue)
SUPPORT PROTOCOL 1.
PURIFICATION, QUANTIFICATION, AND STORAGE OF FRODO STANDARD PLASMID DNA
This protocol describes the production and storage of the FRODO plasmid DNA standard from a plasmid preparation to standard dilution series for use in the standard curve quantification of microbiological analytes in Basic Protocol 2 (see Figure 2). This protocol begins with a stock solution of FRODO, patent application number 63/128,392. FRODO is then further purified and diluted, and the plasmid copy number is standardized against known cell numbers for use as a qPCR standard. Cell pellets containing known numbers of cells can be obtained by isolating human or non-human primate cells into tubes containing 5×101, 5×102, 5×103, 5×104, and 5×105 cells. For this protocol, it is convenient to use easily acquired cells such as peripheral blood mononuclear cells (PBMCs) though any human or old world NHP cell type from a single-cell suspension is sufficient. Dry cell pellets of 50, 500, 5000, 50000 and 500000 cells should be prepared by centrifuging for 5 minutes at 500 × g and removing the supernatant as described in the Strategic Planning section.
Figure 2.
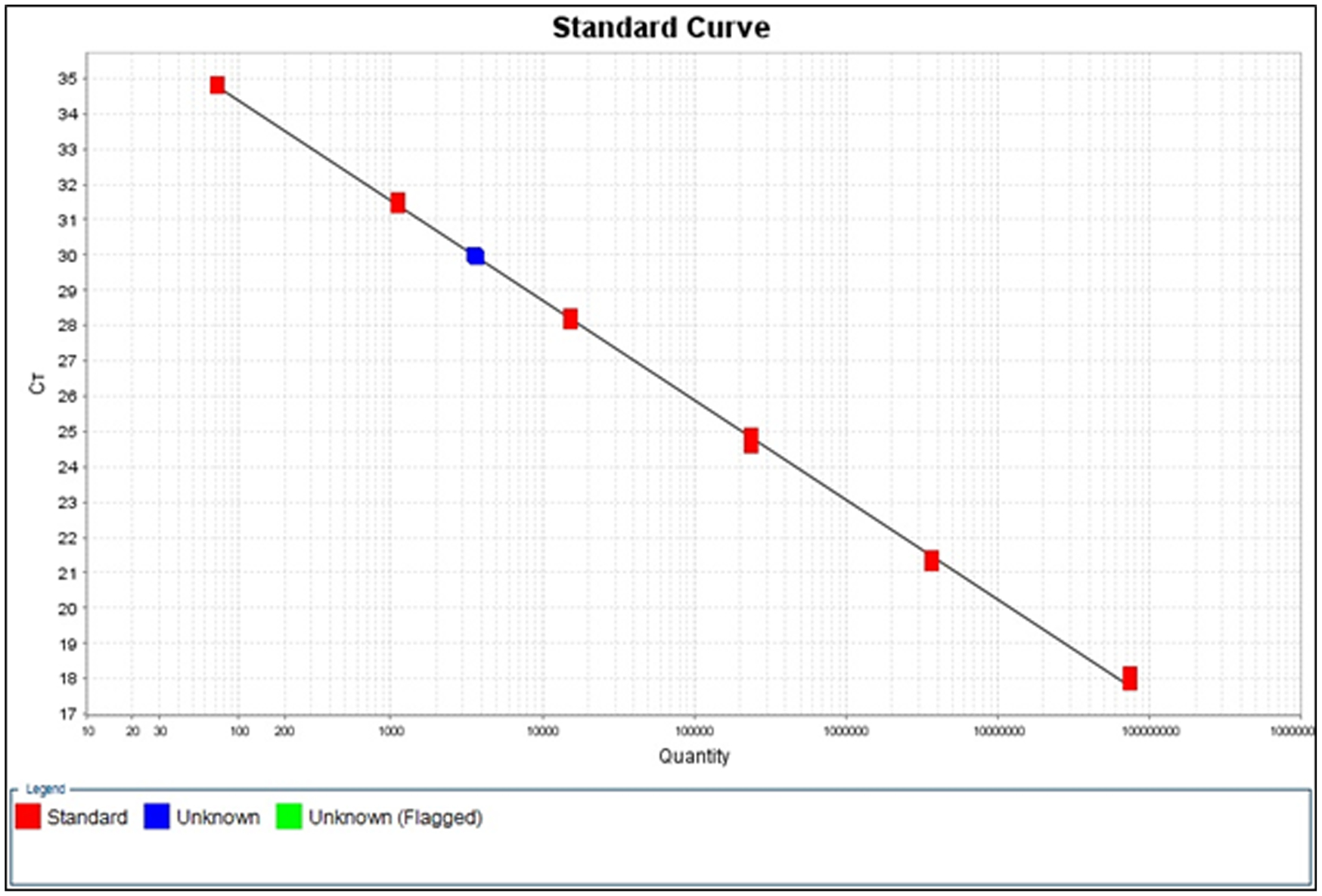
Example standard curve produced in the StepOne software for SIV in an unknown sample of cells isolated from an SIV-infected rhesus macaque.
Materials
20 μg FRODO plasmid, 200 ng/μL in water (Patent application number 63/128,392)
Molecular biology grade water (Invitrogen, AM9937)
QIAprep Spin Miniprep Kit (Qiagen, 27106)
Yeast tRNAs, 10 mg/mL (Invitrogen, AM7119)
Dry cell pellets of 5×101, 5×102, 5×103, 5×104, and 5×105 sorted human or NHP cells
1.5 mL microcentrifuge tubes
Benchtop microcentrifuge such as Centrifuge 5424 (Eppendorf #022620428)
MicroAmp Fast Optical 96-Well Reaction Plate (Applied Biosystems #4346906)
MicroAmp Optical Adhesive Film (Applied Biosystems #4311971)
Benchtop centrifuge with plate adapters such as Centrifuge 5430 (Eppendorf #022620568)
StepOnePlus Real-Time PCR System with StepOne Software (Applied Biosystems #272007793)
Synthesis and Purification
-
1
A map of FRODO is shown in Figure 3 and the complete plasmid sequence is available upon request.
-
2For use as a qPCR standard, further purify an aliquot (~20 μg) of the plasmid megaprep using a modified QIAprep Spin Miniprep Kit protocol as follows:
- Dilute 20 μg of FRODO plasmid megaprep in 500 μL Buffer PB.
- Load diluted FRODO onto DNA-binding column.
- Spin at 13,000 × g for 1 min. Discard supernatant.
- Apply 750 μL Buffer PE to column to wash.
- Spin at 13,000 × g for 1 min. Discard supernatant.
- Spin again at 13,000 × g for 1 min to get rid of excess ethanol.
- Transfer spin column to 1.5 mL collection tube being careful to avoid transferring residual Buffer PE.
- Apply 50 μL molecular biology grade water to center of column. Incubate for 1 min at room temperature, then spin at 13,000 × g to elute DNA.
- Repeat elution step with 50 μL water to increase yield. Pool eluates for 100 μL total.
- Add 500 μL Buffer PB to eluate and repeat steps b-i four more times for a total of five runs through the spin column.
- Quantify plasmid and assess purity using Denovix or Nanodrop spectrophotometer, blanking with water.
-
3
Prepare a serial dilution of purified plasmid DNA from 10−1 to 10 −12 in molecular biology grade water. Mix each solution thoroughly and change pipet tips between transfers.
The final qPCR standard dilutions should contain approximately 10 – 107 copies of DNA. After quantifying FRODO copies for each dilution from 10−1 through 10−12 in the next steps, you will select 6 to 8 dilutions that are at appropriate concentrations to use as standards and may discard the rest.
-
4
Add yeast tRNAs to a final concentration of 1 μg/mL to each dilution.
-
5
Aliquot each dilution of FRODO into 55 μL aliquots and freeze at −80°C for long term storage.
If time allows, proceed to step 6 to standardize FRODO immediately after addition of yeast tRNAs to determine which dilutions in the series are appropriate to use as qPCR standards before aliquoting and freezing.
Figure 3.
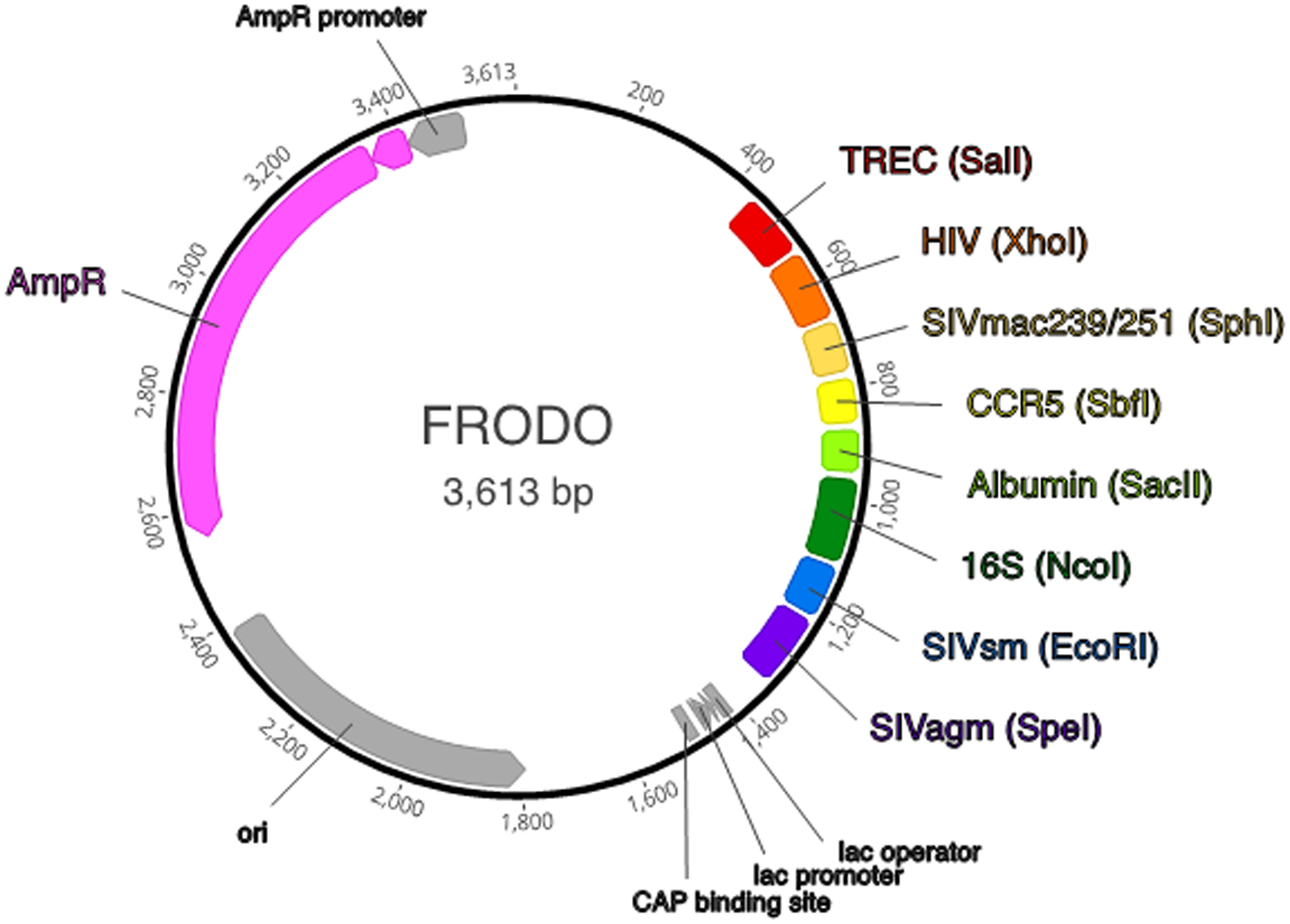
Map of FRODO plasmid indicating DNA sequences for quantification, flanking restriction enzymes, and functional plasmid components (created in Geneious Prime).
Standardizing FRODO qPCR standard
-
6
Prepare DNA extracts from dry cell pellets of 5×101, 5×102, 5×103, 5×104, and 5×105 cells as described in Basic Protocol 1.
-
7
Prepare qPCR master mix as described in Basic Protocol 2 (Preparation of Master Mix) using primers and probes for albumin (Table 2). Prepare enough master mix for triplicate reactions of each FRODO dilution, cellular DNA extracts, and no-template controls (NTC) (66 reactions).
-
8
Map FRODO dilutions, cellular DNA extracts, and no-template controls in a 96-well plate format as shown in the example in Figure 4. Aliquot 20 μL of master mix into each well, depositing the liquid at the bottom of the well.
-
9
Add 5 μL of FRODO, NTC, or cell extracts to appropriate wells, pipetting liquid onto the side of the well.
-
10
As in Basic Protocol 2, seal the qPCR plate with optical clear plate sealer, quick spin the plate and run the PCR on the StepOnePlus thermocycler using the same cycling conditions.
-
11
Calculate expected copy numbers of albumin DNA in 5 μL for each cell lysate and assign calculated values to each of the cell extract samples in the StepOne software. Export the results.
Assume each cell contains 2 copies of the albumin gene, so the expected DNA copy number for albumin in the whole sample is 2× the number of cells in the sample. Adjust this value to reflect DNA copies per 5 μL used in each PCR reaction.
-
12
For use as a qPCR standard to quantify DNA in unknown samples, retain dilutions of FRODO containing approximately 10 – 107 DNA copies per 5 μL. Store FRODO dilutions at −80°C in sufficient aliquots to reduce freeze-thaw cycles.
Figure 4.
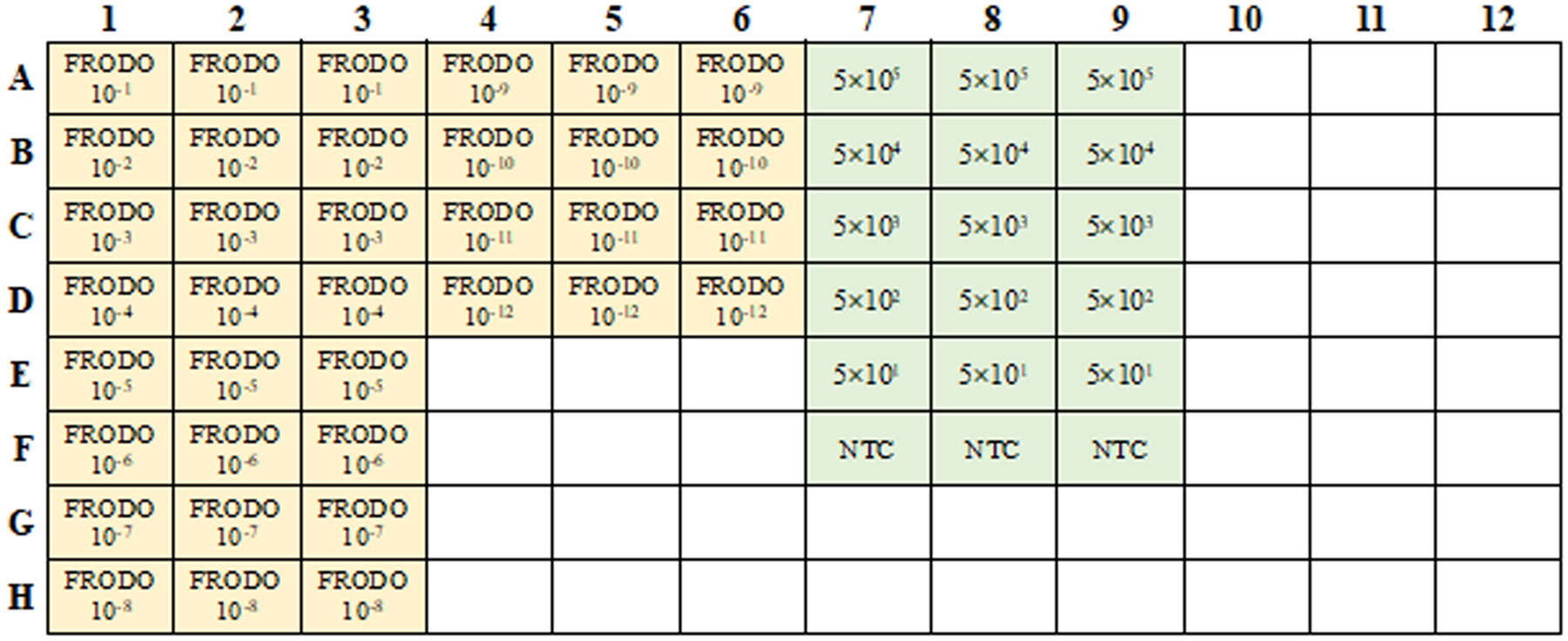
Example PCR plate layout for standardizing FRODO mapping FRODO dilutions and cell lysates of approximated cell numbers as determined by cell sorter or counting. (FRODO: yellow, cell lysates: green).
COMMENTARY
Background information
Quantitative, real time, PCR is a very commonly used technology in immunology, bacteriology, and virology. This technique can be used to enumerate numbers of viruses, bacterial taxa, and immunological genes of interest in biological samples from in vitro and ex vivo experiments. This approach is used to determine the numbers of HIV or SIV DNA copies per cell of interest after viral infection both in vitro and in vivo (Beaumier et al., 2009; Brenchley et al., 2004; Chomont et al., 2009; Lai et al., 2019; Mattapallil et al., 2005). These experiments involve measuring quantities of viral DNA relative to the number of host cells, determined by copy numbers of targeted host genes, via quantitative PCR. Moreover, qPCR is often used to measure thymic output and immunological recovery after immunological insult by measuring the numbers of rearranged T cell receptor excision circles (TREC, generated by rearrangement of T cell receptor genes, resulting in excised, circular, DNA) (D. Douek, 2004; D. C. Douek et al., 2001; McFarland, Douek, Koup, & Picker, 2000; Muraro et al., 2005). Analysis of TREC numbers has also been used to calculate the numbers of cell divisions which occur in vivo among populations of T cells (Brenchley et al., 2004; Tanaskovic, Fernandez, Price, Lee, & French, 2010). Finally, levels of DNA encoding for bacterial 16S ribosomal RNA are used to determine levels of bacterial DNA present in biological samples (Jiang et al., 2009; Klase et al., 2015).
To yield absolute quantities of gene copies, serial dilutions of known quantities of these amplicons are simultaneously analyzed with unknown samples and standard curves of cycle threshold versus analyte quantity are created (D. C. Douek et al., 1998). These known levels of amplicons are generally created by ligating amplicons into plasmid DNA followed by transformation of competent bacteria, amplification of the plasmid, and subsequent purification and quantification. Historically, one plasmid contains one amplicon of interest and laboratories which perform multiple qPCR analyses must maintain multiple plasmid standards. Here we describe creation, standardization, and use of one plasmid construct which includes 8 amplicons commonly measured in microbiology studies: SIVmac239/251, SIVsm, SIVagm, HIV-1, bacterial 16S DNA, TREC, albumin, and CCR5 (albumin and/or CCR5 can be used to determine human and NHP cell number equivalents in each PCR reaction). To synthesize the plasmid, DNA sequences of target analytes containing primer and probe binding sites were joined sequentially with unique sequences of restriction enzyme cut sites between each analyte. This conglomerate DNA sequence was synthesized by a commercial vendor and delivered in a cloning vector with a selectable marker. The plasmid has been grown up and purified in milligram quantities and the sequence has been validated to ensure sequence accuracy during synthesis and preparation. Additionally, amplification of the amplicons included in FRODO has been verified with the primer and probe sequences presented in Table 1 (Figure 5)
Figure 5.
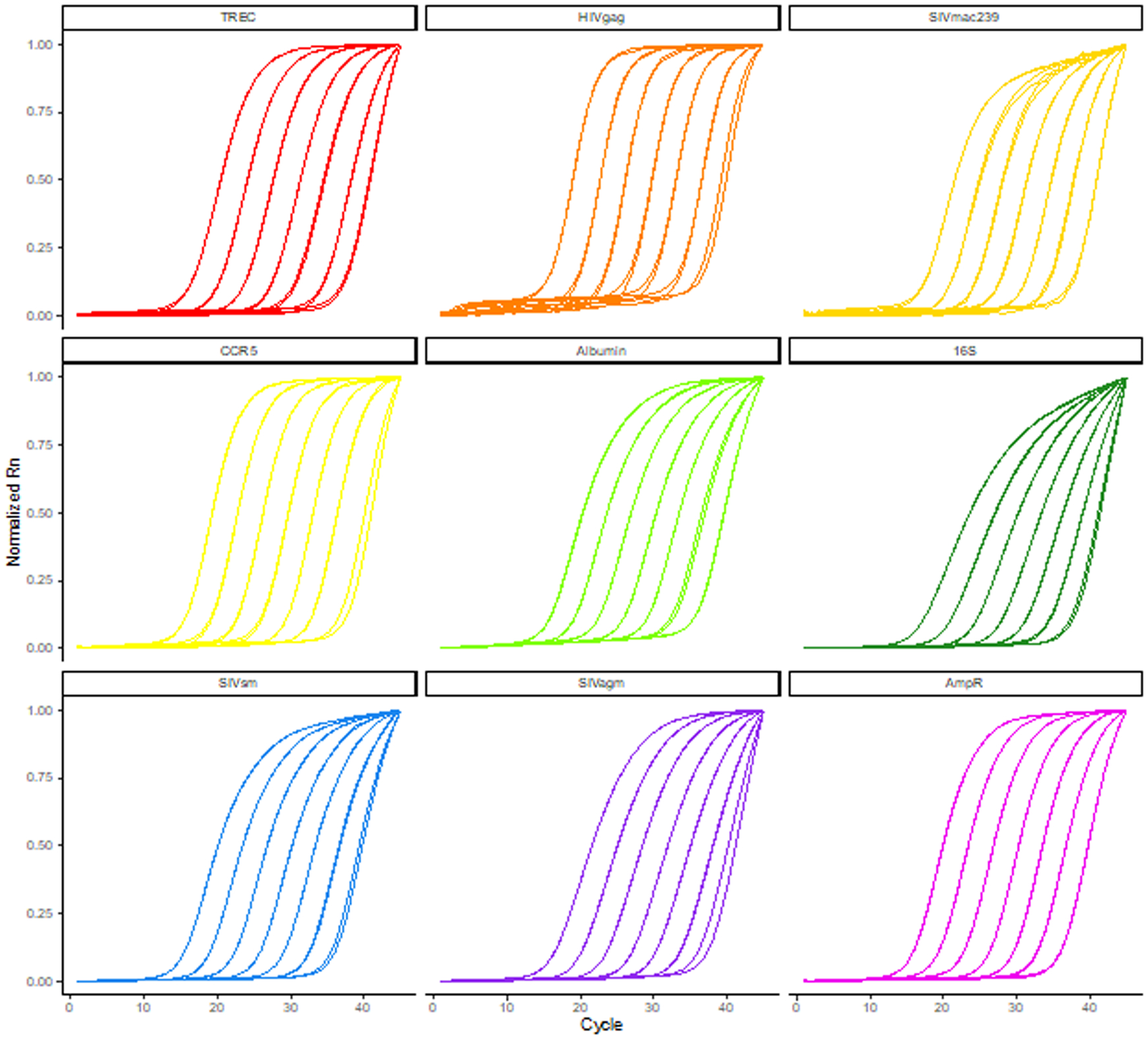
TaqMan qPCR amplification curves of FRODO plasmid dilutions with TREC, HIVgag, SIVmac239, CCR5, albumin, 16S, SIVsm, SIVagm, and AmpR primers and probes. Amplification curves were normalized by feature scaling reporter values from 0 to 1 for each reaction.
Understanding Results
Basic Protocol 1
The lysate is ready to use in downstream PCRs immediately after heat inactivation. Cell lysates may be clear to slightly cloudy depending on the number of cells lysed. If the solution is very cloudy or contains visible clumps, the lysis may be incomplete and should not be used in Basic Protocol 2 (see Critical Parameters and Troubleshooting).
Basic Protocol 2
For each target analyte, the StepOne software produces a plot of Ct versus quantity of the FRODO standard and overlays the unknown sample onto the standard curve (Figure 2). The software will calculate DNA copy numbers of target analytes in the unknown sample and report the values in the “Results” section of the data report (Table 3).
Table 3.
Results produced by StepOne software indicating Ct and quantities of target analytes calculated from the FRODO standard curve for an unknown sample of cells isolated from an SIV-infected rhesus macaque.
| Sample Name | Target Name | Cт | Quantity | Quantity Mean | Quantity SD |
|---|---|---|---|---|---|
| Unknown | TREC | 31.6437168 | 1498.48389 | 1429.29749 | 102.462669 |
| Unknown | TREC | 31.661478 | 1477.82202 | 1429.29749 | 102.462669 |
| Unknown | TREC | 31.8141289 | 1311.58679 | 1429.29749 | 102.462669 |
| Unknown | ALBUMIN | 22.7621479 | 484740.469 | 414556.969 | 67084.5 |
| Unknown | ALBUMIN | 22.9730549 | 407855.938 | 414556.969 | 67084.5 |
| Unknown | ALBUMIN | 23.1561375 | 351074.438 | 414556.969 | 67084.5 |
| Unknown | SIV | 29.9425945 | 3686.41992 | 3592.7854 | 97.2789917 |
| Unknown | SIV | 29.9717598 | 3599.70508 | 3592.7854 | 97.2789917 |
| Unknown | SIV | 30.0088978 | 3492.23145 | 3592.7854 | 97.2789917 |
| Unknown | CCR5 | 23.5239086 | 227975.000 | 217399.203 | 10119.501 |
| Unknown | CCR5 | 23.587841 | 216414.656 | 217399.203 | 10119.501 |
| Unknown | CCR5 | 23.6376972 | 207807.969 | 217399.203 | 10119.501 |
Alternatively, a computing software can be used to plot Ct values versus log copy numbers for each dilution of the FRODO standard. A standard curve is calculated using least squares regression. DNA copy numbers of target analytes in the unknown sample are calculated from the regression equation as shown below.
For improved accuracy, the Ct of the unknown sample should fall within the range of the FRODO dilutions to avoid extrapolating beyond the limits of the standard curve. If the target analyte Ct value is too high, consider repeating the qPCR assay using a 10-fold or 100-fold dilution of the sample lysate in molecular biology grade water. If the Ct value of the cell number equivalents (albumin or CCR5) falls below the Ct of the lowest standard or is not detected, consider repeating the assay with a greater number of starting cells in Basic Protocol 1. Additionally, starting with fewer cells will decrease the likelihood of detecting more rare DNA targets such as TREC or viral DNA.
Quantifying both viral DNA and cell number equivalents such as albumin or CCR5 allows the investigator to calculate the number of HIV or SIV DNA copies per cell in a sample. For this calculation, determine the copy number of each HIV or SIV DNA in the sample as well as the copy number of CCR5 or albumin DNA in the sample. Dividing the CCR5 or albumin DNA copy number by 2 gives the cell number equivalent in the sample. The ratio of HIV or SIV DNA copy number per cell number equivalents can be used to assess viral load or efficacy of antiviral therapy, for example. Similarly, quantity of TREC per cell number equivalent can be calculated to measure thymic output and immunological recovery after insult.
Support Protocol 1
The StepOne software will calculate DNA copy numbers of albumin in the FRODO dilutions (equivalent to plasmid copy number) and report the values in the “Results” section of the data report. These values reflect the DNA quantity per 5 μL reaction. Alternatively, a computing software such as Microsoft Excel can be used for the calculation as demonstrated in Basic Protocol 2. Biological samples from in vitro or ex vivo experiments analyzed by qPCR consist of hundreds to millions of cells. Therefore, we recommend retaining FRODO dilutions that contain approximately 10 to 107 plasmid copies per 5 μL reaction for use as qPCR standards for future analyses.
Critical Parameters and Troubleshooting
Some steps in the protocol may need to be adapted depending on the cell number in unknown samples. Very high cell number may result in incomplete lysis and the resulting Ct values and calculated copy numbers may underestimate of the amount of target analytes present in the sample. Additionally, because cell lysates are used as PCR templates, inhibitor molecules may be present in higher quantities than in a purified DNA sample. For samples with cell numbers >500,000 consider lysing in a larger volume of proteinase K + 10 mM TRIS or allowing the lysis reaction to proceed at 55°C for longer than 1 hour.
Because the FRODO plasmid serves as a standard for quantitation of unknown samples, it is important to accurately quantify the DNA copy number of the FRODO plasmid dilutions. In Support Protocol 1, we quantify the FRODO dilution series by comparing amplification of cell-number equivalents (e.g. albumin) between FRODO and samples of known cell number. Investigators may also standardize FRODO against existing PCR DNA standards for any of the analytes included in the FRODO plasmid to further validate plasmid copy numbers. When quantifying FRODO, verify that calculated copy numbers for each dilution approximate the expected values for a 10-fold serial dilution. This should correlate with a 3.3-point increase in threshold cycle value between dilutions. If the calculated copy number or cycle threshold values of the dilution series do not follow the expected pattern, redo the 10-fold serial dilution making sure to thoroughly mix at each step and avoid contamination by changing pipet tips for each transfer.
DNA will degrade during prolonged storage and multiple freeze-thaw cycles. To reduce loss of DNA in the FRODO standards, yeast tRNAs are added to each dilution of FRODO after purification and before freezing. We have observed consistent amplification of FRODO dilutions through at least five freeze-thaw cycles with this protocol (data not shown). If after long-term storage the investigator notices variable threshold cycle values in the FRODO standards, it is recommended to purify, dilute, and standardize a new FRODO dilution series for use as a qPCR standard (Support Protocol 1, start with step 3).
Time Considerations
Starting from freshly sorted or frozen cells, the lysis procedure in Basic Protocol 1 takes approximately 1 hour and 30 minutes, including 1 hour and 10 minutes of incubation and an ample 20 minutes for centrifugation and pipetting. The set up and run time for the qPCR reaction in Basic Protocol 2 takes approximately 3 hours, with 1 hour to prepare primer and probe dilutions and master mixes as well as loading the plate and a runtime of about 2 hours in the StepOnePlus thermocycler. The master mixes can be prepared and loaded onto the qPCR plate during the cell lysis incubation steps to reduce overall protocol time. Data from the qPCR run is available and can be analyzed immediately.
ACKNOWLEDGMENTS
Funding for this study was provided in part by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH. The content of this publication does not necessarily reflect the views or policies of the US Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US government.
LITERATURE CITED
- Beaumier CM, Harris LD, Goldstein S, Klatt NR, Whitted S, McGinty J, … Brenchley JM (2009). CD4 downregulation by memory CD4+ T cells in vivo renders African green monkeys resistant to progressive SIVagm infection. Nat Med, 15(8), 879–885. doi:nm.1970 [pii] 10.1038/nm.1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Hill BJ, Ambrozak DR, Price DA, Guenaga FJ, Casazza JP, … Koup RA (2004). T-Cell Subsets That Harbor Human Immunodeficiency Virus (HIV) In Vivo: Implications for HIV Pathogenesis. J Virol, 78(3), 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Vinton C, Tabb B, Hao XP, Connick E, Paiardini M, … Estes JD (2012). Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood, 120(20), 4172–4181. doi: 10.1182/blood-2012-06-437608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, … Sekaly RP (2009). HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med, 15(8), 893–900. doi:nm.1972 [pii] 10.1038/nm.1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek D (2004). Thymic output and HIV infection: on the right TREC. Immunity, 21(6), 744–745. doi: 10.1016/j.immuni.2004.11.005 [DOI] [PubMed] [Google Scholar]
- Douek DC, Betts MR, Hill BJ, Little SJ, Lempicki R, Metcalf JA, … Koup RA (2001). Evidence for increased T cell turnover and decreased thymic output in HIV infection. J Immunol, 167(11), 6663–6668. [DOI] [PubMed] [Google Scholar]
- Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, … Koup RA (2002). HIV Preferentially Infects HIV-Specific CD4+ T-cells. Nature, 417(6884), 95–98. [DOI] [PubMed] [Google Scholar]
- Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, … Koup RA (1998). Changes in thymic function with age and during the treatment of HIV infection. Nature, 396(6712), 690–695. [DOI] [PubMed] [Google Scholar]
- Douek DC, Vescio RA, Betts MR, Brenchley JM, Hill BJ, Zhang L, … Koup RA (2000). Assessment of thymic output in adults after haematopoietic stem-cell transplantation and prediction of T-cell reconstitution. Lancet, 355(9218), 1875–1881. [DOI] [PubMed] [Google Scholar]
- Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, … Brenchley JM (2009). Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-Treated HIV infection. J Infect Dis, 199(8), 1177–1185. doi: 10.1086/597476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klase Z, Ortiz A, Deleage C, Mudd JC, Quinones M, Schwartzman E, … Brenchley JM (2015). Dysbiotic bacteria translocate in progressive SIV infection. Mucosal Immunol, 8(5), 1009–1020. doi: 10.1038/mi.2014.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai SH, Starke CE, Flynn JK, Vinton CL, Ortiz AM, Mudd JC, & Brenchley JM (2019). Simian Immunodeficiency Virus Infects Functionally Polarized Memory CD4 T Cells Equivalently In Vivo. J Virol, 93(9). doi: 10.1128/JVI.02163-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, & Roederer M (2005). Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature, 434(7037), 1093–1097. [DOI] [PubMed] [Google Scholar]
- McFarland RD, Douek DC, Koup RA, & Picker LJ (2000). Identification of a human recent thymic emigrant phenotype. Proc Natl Acad Sci U S A, 97(8), 4215–4220. doi: 10.1073/pnas.070061597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraro PA, Douek DC, Packer A, Chung K, Guenaga FJ, Cassiani-Ingoni R, … Martin R (2005). Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med, 201(5), 805–816. doi: 10.1084/jem.20041679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry L, Johnson T, Hobson MJ, Smucker B, & Cornetta K (2002). Titering lentiviral vectors: comparison of DNA, RNA and marker expression methods. Gene Ther, 9(17), 1155–1162. doi: 10.1038/sj.gt.3301731 [DOI] [PubMed] [Google Scholar]
- Sodora DL, Milush JM, Ware F, Wozniakowski A, Montgomery L, McClure HM, … Koup RA (2002). Decreased levels of recent thymic emigrants in peripheral blood of simian immunodeficiency virus-infected macaques correlate with alterations within the thymus. J Virol, 76(19), 9981–9990. doi: 10.1128/jvi.76.19.9981-9990.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MT, Taylor LT, & DeLong EF (2000). Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5’-nuclease assays. Appl Environ Microbiol, 66(11), 4605–4614. doi: 10.1128/aem.66.11.4605-4614.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaskovic S, Fernandez S, Price P, Lee S, & French MA (2010). CD31 (PECAM-1) is a marker of recent thymic emigrants among CD4+ T-cells, but not CD8+ T-cells or gammadelta T-cells, in HIV patients responding to ART. Immunol Cell Biol, 88(3), 321–327. doi: 10.1038/icb.2009.108 [DOI] [PubMed] [Google Scholar]
- Venneti S, Bonneh-Barkay D, Lopresti BJ, Bissel SJ, Wang G, Mathis CA, … Wiley CA (2008). Longitudinal in vivo positron emission tomography imaging of infected and activated brain macrophages in a macaque model of human immunodeficiency virus encephalitis correlates with central and peripheral markers of encephalitis and areas of synaptic degeneration. Am J Pathol, 172(6), 1603–1616. doi: 10.2353/ajpath.2008.070967 [DOI] [PMC free article] [PubMed] [Google Scholar]