

Abstract
Alpha-synuclein pathology is associated with dopaminergic neuronal loss in the substantia nigra (SN) of Parkinson's patients. Working across human and mouse models, we investigated mechanisms by which the accumulation of soluble α-synuclein oligomers leads to neurodegeneration. Biochemical analysis of the midbrain of α-synuclein overexpressing BAC-transgenic male and female mice revealed age- and region-dependent mitochondrial dysfunction and accumulation of damaged proteins downstream of the RE1 Silencing Transcription Factor (REST). Vulnerable SN dopaminergic neurons displayed low REST levels compared with neighboring protected SN GABAergic neurons, which correlated with the accumulation of α-synuclein oligomers and disrupted mitochondrial morphology. Consistent with a protective role, REST levels were reduced in patient induced pluripotent stem cell-derived dopaminergic neurons carrying the SNCA-Triplication mutation, which accumulated α-synuclein oligomers and mitochondrial damage, and displayed REST target gene dysregulation. Furthermore, CRISPR-mediated REST KO induced mitochondrial dysfunction and impaired mitophagy in vitro. Conversely, REST overexpression attenuated mitochondrial toxicity and mitochondrial morphology disruption through the transcription factor PGC-1α. Finally, decreased α-synuclein oligomer accumulation and mitochondrial dysfunction in mice correlated with nuclear REST and PGC-1α in protected SN GABAergic neurons compared with vulnerable dopaminergic neurons. Our findings show that increased levels of α-synuclein oligomers cause dopaminergic neuronal-specific dysfunction through mitochondrial toxicity, which can be attenuated by REST in an early model of Parkinsonian pathology. These findings highlight REST as a mediator of dopaminergic vulnerability in PD.
SIGNIFICANCE STATEMENT Understanding early Parkinsonian pathophysiology through studies of advanced preclinical models is fundamental to the translation of disease-modifying therapies. Here we show disease-relevant levels of α-synuclein expression in mice leads to accumulation of α-synuclein oligomers in the absence of overt aggregation, and mitochondrial dysfunction in dopaminergic neurons lacking the RE1 Silencing Transcription Factor. Our findings identify the mechanism of action of RE1 Silencing Transcription Factor and PGC-1α as mediators of dopaminergic vulnerability in α-synuclein BAC-transgenic mice and induced pluripotent stem cell-derived dopaminergic cultures, highlighting their potential as therapeutic targets.
Keywords: alpha-synuclein, neurodegeneration, neuroprotection, oligomers, Parkinson's, REST
Introduction
Parkinson's disease (PD) is a complex neurodegenerative disorder affecting ∼1% of the population over 60 years old (Samii et al., 2004). Symptoms include rigidity and bradykinesia, which are caused by the loss of dopaminergic neurons of the SNc, in addition to a range of nonmotor symptoms. The underlying cause of PD is unknown, although age is the biggest risk-factor for developing the disease (Reeve et al., 2012). While the histologic hallmark of PD remains the presence of α-synuclein-positive Lewy bodies in the brain of patients (Wakabayashi et al., 2007), it is unclear whether these structures are damaging or protecting. Recent studies suggest that α-synuclein oligomers might play an important role in the disease (Roberts et al., 2015; Bengoa-Vergniory et al., 2017). α-Synuclein oligomers have been shown to promote complex I deficiencies (Miller et al., 2009), mitochondrial swelling and fragmentation (Luth et al., 2014; Plotegher et al., 2014), ER stress (Colla et al., 2012), and synaptic dysfunction (Prots et al., 2013). α-Synuclein oligomers can be cleared by the lysosomal system or by chaperone-mediated autophagy, and can accumulate if lysosomes are dysfunctional (H. J. Lee et al., 2004; Hunn et al., 2019; Ho et al., 2020).
Although we have learned much about the mechanisms and pathways of PD from rodents (Vingill et al., 2018), the toxin (Heikkila et al., 1984; Rozas et al., 1998), α-synuclein virus-injected, early transgenic (Kirik et al., 2002; Fleming et al., 2004), and α-synuclein preformed fibril (Luk et al., 2012) mouse models of PD often fail to recapitulate the progressive age-dependent neurodegeneration characteristic of sporadic disease. We have previously generated the “humanized” BAC-transgenic SNCA-OVX mouse line expressing the complete SNCA human genomic locus from a BAC insert on an endogenous mouse Snca−/− KO background (Janezic et al., 2013). The SNCA-OVX mice exhibit physiological spatial and temporal expression of α-synuclein with a 2- to 3-fold increase of α-synuclein protein. These mice demonstrate age-dependent dopaminergic neuronal loss and onset of motor dysfunction (Janezic et al., 2013; Dodson et al., 2016).
Mitochondrial dysfunction is a hallmark of PD demonstrated by genetic mutations in the mitophagy regulators PINK1 and Parkin and the induction of PD in humans and animal models by complex I inhibitors MPTP/MPP+ and rotenone (Ryan et al., 2015; Bogetofte et al., 2019). The preferential vulnerability of SNc dopaminergic neurons to mitochondrial dysfunction likely results from several biological factors, including their extensive axonal arbor and reliance on Ca2+ channel pacemaking flux, which together increase metabolic demand, mitochondrial ROS generation, and oxidative post-translational protein modification (Ryan et al., 2015). This has highlighted mitochondrial biogenesis master-regulators, such as peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) as potential therapeutic targets for PD (Zheng et al., 2010). However, the interplay between α-synuclein aggregation, mitochondrial dysfunction, and oxidative stress in dopaminergic neurons in PD remains elusive.
RE1 Silencing Transcription Factor (REST), also known as Neuron-Restrictive Silencing factor, is a transcriptional repressor controlling neuronal gene expression widely expressed in the immature brain and progressively downregulated with age and maturation (Buckley et al., 2010). REST has been associated with a number of neurodegenerative diseases, although its role as a mediator or protective factor remains complex. REST has been identified as a promoter of neurodegeneration in Huntington's disease (Zuccato and Cattaneo, 2007), and also identified as neuroprotective factor lost from the nucleus of neurons in postmortem tissue from patients with Alzheimer's disease (AD), frontotemporal dementia, and dementia with Lewy bodies (Lu et al., 2014). Conditional KO of REST in mice induces vulnerability to ethanol or MPTP-induced neurodegeneration. Conversely, overexpression of REST induces TH expression in vitro and protects cells from oxidative and manganese-induced stress, which supports a neuroprotective role for this transcriptional repressor (Tegeder et al., 2006; Cai et al., 2011; Lu et al., 2014; Pajarillo et al., 2020).
In this study, we have integrated human and mouse models of PD to identify a range of dysregulated pathways, including mitochondrial dysfunction, which contribute to α-synuclein oligomer-induced age-dependent neurodegeneration. Furthermore, we show that this dysfunction is specific to dopaminergic neurons of the SNc and is regulated by the absence of the transcription factor REST. Finally, we demonstrate that REST is protective in PD in vitro models through PGC-1α, and that absence of REST renders SNc neurons vulnerable to α-synuclein insult in vivo.
Materials and Methods
Study design
Sample size was determined according to the experimental paradigm. Any animals showing any welfare issues were taken off the study; otherwise, animals remained in the sample. For neuropathological examination, 4 or 5 animals were bred per condition to avoid n < 3 after aging. For induced pluripotent stem cell (iPSC) culture, 3 or 4 independent control lines (or clones in the case of SNCA-Triplication) were used to ensure n ≥ 3. For biochemical analysis of recombinant or biological material, experiments were performed on >3 independent samples.
Treatments and genotypes were handled blindly by coding of the different groups into numbers wherever analysis was not automated. Samples were blinded by taping over identifiers, to avoid bias when sampling. For cell imaging, 3-9 independent FOVs were analyzed per well, and 1-2 wells were analyzed per condition and line. For tissue analysis 2-4 independent fields (i.e., at least 100 µm apart to avoid resampling) of anterior SN were analyzed, and for puncta counts associated with TH+ cells, 25 neurons were sampled across the SNc/r.
Antibodies used in this study are detailed in Table 1.
Table 1.
Antibodies used in this study
| Antigen | Host species | Supplier; product number | Dilution used |
|---|---|---|---|
| 3-Nitrotyrosine | Rabbit polyclonal | Abcam; Ab42789 | 1:500 |
| α-Synuclein [MJFR1] | Rabbit polyclonal | Abcam; Ab138501 | 1:1000 |
| BIP | Rabbit polyclonal | Abcam; ab21685 | 1:1000 |
| COX IV | Rabbit monoclonal | Abcam; ab140643 | 1:500 |
| DRP-1 phospho Ser616 | Rabbit monoclonal | Cell Signaling Technology; 4494 | 1:500 |
| DRP | Rabbit monoclonal | Cell Signaling Technology Technology; 8570S | 1:1000 |
| LC3 | Rabbit polyclonal | Sigma Millipore; 7543 | 1:1000 |
| NRF2 | Rabbit polyclonal | Abcam; ab31163 | 1:1000 |
| PGC-1α | Rabbit polyclonal | Santa Cruz Biotechnology; sc-13067) | 1:200 |
| TOM20 (WB) | Rabbit polyclonal | Santa Cruz Biotechnology; sc-11021 | 1:5000 |
| TH | Rabbit polyclonal | Millipore; AB152 | 1:1000 |
| COX IV | Rabbit monoclonal | Abcam; ab140643 | 1:500 |
| TOM20 (IF, IHC) | Rabbit | Abcam; ab220822 | 1:250 |
| LAMP1 (IF) | Mouse | Santa Cruz Biotechnology; 20011 | 1:100 |
| LC3 (IF) | Rabbit | Cell Signaling Technology; 2775 | 1:200 |
| PGC-1α (IHC) | Rabbit | Cell Signaling Technology; 2178 | 1:100 |
| TH (IHC) | Rabbit | Millipore; ab152 | 1:500 |
| TH (IHC) | Chicken | Abcam; ab76442 | 1:250 |
| NEFH (IHC) | Mouse | Biolegend; 801701 | 1:500 |
| REST (IHC) | Rabbit | Millipore; 07–579 | 1:250 |
| α-synuclein (PLA) | Mouse | Abcam; ab1903 | 1:2000 |
| α-synuclein-TOM20 (PLA) | Rabbit | Abcam; ab220822 | 1:250 |
| α-synuclein-TOM20 (PLA) | Mouse | Abcam; ab1903 | 1:250 |
| Lamin A/C | Rabbit | Genscript; A01455 | 1:1000 |
Animals
WT mice (C57BL/6J) bred in house or were purchased from Charles River Laboratories. SNCA-OVX mice generated in our laboratory were used in this research project, as previously described (Janezic et al., 2013). These transgenic mice express human WT α-synuclein from the complete human SNCA locus at disease-relevant levels on a Snca−/− mouse background. Male and female mice were killed using cervical dislocation for dissection or pentobarbitone 20% (w/v) (100 μl or 20 mg per mouse, i.p.) and transcardially perfused with PBS, pH 7.4, followed by PFA 4% for tissue embedding. The total number of TH-expressing cells was assessed by stereology as previously described (Janezic et al., 2013). All procedures were conducted in accordance with the UK Animals (Scientific Procedures) Act of 1986 and approved by the Animal Welfare and Ethical Review Bodies at the Department of Physiology, Anatomy and Genetics, University of Oxford.
α-Synuclein proximity ligation assay (AS-PLA)
Oligomeric α-synuclein was detected by PLA, as previously described (Roberts et al., 2015) for brightfield preparations. For PLA coimmunofluorescence, brains were postfixed in 4% PFA for 24 h at 4°C, stored in 70% ethanol at 4°C until samples were paraffin-embedded, and 5 μm sections were cut. AS-PLA experiments were conducted on brain sections, using Duolink kits (Sigma Millipore). The α-synuclein antibody chosen for the AS-PLA probes was anti-α-synuclein 4D6 (Abcam). Sections were dewaxed in xylene and Histo-Clear, peroxidase reaction blocked in 10% H2O2 for 15 min at room temperature, and antigen retrieved in citrate buffer, pH 6 (Abcam 93678), after which brain sections were blocked in 10% normal goat serum (containing 1 m glycine TBS and 0.1% Triton X-100) and incubated for 1 h in primary antibodies. Sections were then washed with TBS containing 0.05% Tween-20 (TBS-T) and incubated for 1 h in the dark with AlexaFluor secondary antibodies (Invitrogen). All samples were washed in TBS-T and incubated in Duolink block solution for 1 h at 37°C, followed by overnight incubation with the conjugates diluted in Duolink PLA diluent (1:100) at 4°C and subsequent PLA steps as described previously (Roberts et al., 2015).
Cellular fractionation
The midbrain was dissected from one hemisphere of 18-month-old WT or SNCA-OVX mice. Tissue was lysed in 1 ml ice-cold Buffer A (250 mm sucrose, 2 mm HEPES, 0.1 mm EGTA, pH 7.4) using a glass homogenizer at 4°C. The lysate was centrifuged at 600 × g, 10 min, 4°C. The supernatant was retained, and the process was repeated on the pellet. The supernatants were pooled and centrifuged at 600 × g for 5 min, and the resulting supernatant was centrifuged at 11,000 × g for 10 min at 4°C. The resulting supernatant (organelle-depleted fraction) was stored at −80°C until analysis. The pellet (organelle-enriched fraction) was washed 3× in Buffer B (25 mm potassium phosphate, 5 mm MgCl, pH 7.2), aliquoted, and stored at −80°C. Protein was quantified by BCA assay.
Measurement of mitochondrial complex I activity
Midbrain was homogenized for 3 × 5 s in 500 µl 200 mm sodium PB, pH 7.2, using a Tissue Tearor (BioSpec Products). Homogenate was centrifuged at 800 × g for 5 min at 4°C. The supernatant was aspirated, and protein concentration was measured using BCA assay (Sigma Millipore). Mitochondrial complex I activity was measured by monitoring NADH oxidation at 340 nm, as previously described (Long et al., 2009). Briefly, sample (50 µg) was incubated in MCI buffer (200 mm sodium PB, pH 7.2, 5 mm MgCl2, 2.5 mg/ml BSA, 10 mm NADH, 1 mm KCN, and 1 mg/ml antimycin A) with or without the addition of rotenone (100 μm; final volume 125 µl). CoenzymeQ1 (65 μm) was added and absorbance at 340 nm was monitored at 30°C for 15 min. Complex I activity was calculated as the rotenone-inhabitable NADH oxidation.
Mitochondrial ROS production
Basal mitochondrial ROS production was measured in mitochondrial extracted from whole brain based on a previously published protocol (Quinlan et al., 2013). Mitochondria (15 µg) were incubated in buffer containing 120 mm KCl, 5 mm HEPES, 5 mm KH2PO4, 2.5 mm MgCl2, 1 mm EGTA, 0.3% (w/v) BSA, pH 7.0 supplemented with 1 U HRP (Sigma Millipore), 0.25 mm AmplexRed (Sigma Millipore) at 37°C. Fluorescence (ex 540 nm, em 590 nm) was monitored for 3 min to obtain unstimulated ROS production, before the addition of NADH and monitored for a further 10 min. ROS production was measured in triplicate by time-dependent increase in fluorescence over a 5 min period (N = 3). Fluorescence changes were used to calculate equivalent H2O2 production by the addition of 2.5-20 μm H2O2 to the assay and construction of a standard curve.
Identification of nitrated proteins
The presence and abundance of nitrated proteins were quantified in midbrain lysates using an anti-3-nitrotyrosine antibody. Identical samples were subjected to SDS-PAGE followed by Coomassie R-250 staining; bands corresponding to proteins with increased nitration were excised and subjected to liquid chromatography tandem mass spectrometry (LC-MS/MS).
Nitration site modeling
Human GAPDH atomic model (pdb id. 4WNC) was analyzed using Pymol software (The PyMOL Molecular Graphics System, version 2.0, Schrödinger). Four monomers of the GAPDH complex are represented in colored ribbons, and the relative position of the side chain of the nitrated residue (W313) is depicted.
Dimethyl labeling
Mitochondrial enriched/depleted fractions (100 µg protein) were incubated in 50 mm tetraethylammonium bromide and 8 m urea for 30 min. Proteins were reduced using 10 mm tris(2-carboxyethyl)phosphine for 1 h and then alkylated using 2-chloroacetamide for 30 min. Samples were diluted 1 in 4 with 25 mm tetraethylammonium bromide and incubated with trypsin 20:1 (5 µg) for 16 h at 37°C. Samples were dimethyl labeled as previously described (Boersema et al., 2009); briefly, 8 µl of 4% (v/v) CH2O (light) or CD2O (medium) was added to samples. Samples were briefly mixed and incubated with 8 µl of 600 mm NABH3CN for 1 h. The reaction was quenched with 32 µl cold 1% (v/v) ammonia solution. The samples were mixed and acidified to pH 3 with 5% (v/v) formic acid. Light and medium labeled samples were mixed at a ratio of 1:1. Samples were dried and desalted before mass spectrometry.
LC-MS/MS analysis
Peptides were resuspended in 5% formic acid and 5% DMSO. They were separated on an Ultimate 3000 UHPLC system (Thermo Fisher Scientific) and electrosprayed directly into an QExactive mass spectrometer (Thermo Fisher Scientific) through an EASY-Spray nano-electrospray ion source (Thermo Fisher Scientific). The peptides were trapped on a C18 PepMap100 precolumn (300 µm i.d. × 5 mm, 100 Å, Thermo Fisher Scientific) using Solvent A (0.1% formic acid in water) at a pressure of 500 bar. The peptides were separated on an in-house packed analytical column (75 µm i.d. packed with ReproSil-Pur 120 C18-AQ, 1.9 µm, 120 Å, Maisch) using a linear gradient (length: 120 min, 15%-35% Solvent B [0.1% formic acid in acetonitrile], flow rate: 200 nl/min). The raw data were acquired on the mass spectrometer in a data-dependent mode. Full scan MS spectra were acquired in the Orbitrap (scan range 350-1500 m/z, resolution 70 000, AGC target 3e6, maximum injection time 50 ms). After the MS scans, the 20 most intense peaks were selected for HCD fragmentation at 30% of normalized collision energy. HCD spectra were also acquired in the Orbitrap (resolution 17,500, AGC target 5e4, maximum injection time 120 ms) with first fixed mass at 180 m/z. Charge exclusion was selected for unassigned and 1+ ions. The dynamic exclusion set to 20 s.
Analysis of quantitative mass spectrometry data
Results were searched against the Uniprot Mouse database with a maximum of two missed cleavages. Protein identification was filtered using the Mascot significance threshold set at p < 0.05. Precursor mass tolerance was 10 ppm, and fragment mass tolerance was 0.02 Da. For protein abundance quantification, dynamic modifications to peptides were listed as oxidation (M), dimethyl (N-term, K; light label) and dimethyl 2H(4) (N-term, K; medium label). Carbamidomethyl modification of C residues was considered a static modification. Peptide quantitation and protein identification were performed on triplicate LC-MS/MS analyses (technical replicates) using Proteome Discoverer version 1.4 (Thermo Fisher Scientific). Peptides were filtered using a Mascot peptide ion score of 25, a false discovery rate of 1%, and peptides showing a >10-fold change were excluded from the quantitation. Additionally, proteins were only quantified if two peptides were quantified. Protein L/M ratios were normalized to the median L/M ratio for each replicate. Proteins were required to be quantified in at least three biological replicates to be included in the analysis, and proteins demonstrating a coefficient of variation >60% for the mean M/L ratio were excluded. Mitochondrial enrichment was calculated using MitoCarta 2.0 (Broad Institute; www.broadinstitute.org/mitocarta).
Pathway analysis
Proteins upregulated or downregulated by >1 SD (11.9% of the dataset) were used for further pathway analysis using Ingenuity Pathway Analysis software (QIAGEN). Data were analyzed using the Ingenuity Knowledge Base (genes only) in July 2015, using the following settings: only molecules and/or relationships experimentally observed interactions in Human, Mouse or Rat with observations in the following regions/cell types considered Substantia Nigra, Cerebral Cortex, Dorsal Root Ganglion Trigeminal Ganglion, Spinal Cord, Granule Cell Layer, Granule Cells, Subventricular Zone, Nucleus Accumbens, Hippocampus, Cerebellum, Neurons not otherwise specified, Other Nervous System, Medulla Oblongata, Pituitary Gland, Nervous System not otherwise specified, White Matter, Choroid Plexus, Amygdala, Corpus Callosum, Hypothalamus, Brain, Ventricular Zone, Parietal Lobe, Pyramidal Neurons, Cortical Neurons, Gray Matter, Putamen, Thalamus, Purkinje Cells, Caudate Nucleus, Sciatic Nerve, Striatum, Olfactory Bulb, Brainstem, Cerebral Ventricles, Other Neurons, Astrocytes, SK-N-SH, Other Neuroblastoma Cell Lines, Neuroblastoma Cell Lines not otherwise specified. Upstream regulator networks were created using upstream regulators with an activation Z score >1.
Immunohistochemistry
Sections (5 μm) were dewaxed in xylene and Histo-Clear, peroxidase reaction blocked in 0.3% H2O2 for 15 min at room temperature, and antigen retrieved in citrate buffer, pH 6 (Abcam 93678), after which brain sections were blocked in 10% normal goat serum (containing 1 m glycine TBS and 0.1% Triton X-100) and incubated overnight at 4°C in primary antibodies. The next day, sections were washed in TBS-T and incubated with appropriate AlexaFluor secondary antibodies diluted for 1 h at room temperature. For nuclear staining, sections were incubated in DAPI (1:1000, Invitrogen) for 5 min at room temperature. Sections were mounted with FluorSave mounting medium (Calbiochem) before being visualized with an Evos FL auto imaging system (Invitrogen).
Lentiviral vectors and plasmid construction
TLCV2 (Addgene #87360) was digested using the restriction enzyme BsmBI, dephosphorylated, and gel-purified. Protospacer sequences were appended in the form of double-stranded oligonucleotides with complementary sticky ends to the vector backbone (REST sgRNA1 oligo forward 5′- CACCGGATTAGTATTGTAGCCGCAG, REST sgRNA1 oligo reverse 5′-AAACCTGCGGCTACAATACTAATCC, Scrambled control forward Oligo 5′- CACCGGCACTACCAGAGCTAACTCA, Scrambled control reverse Oligo AAACTGAGTTAGCTCTGGTAGTGCC).
Lentiviral particles were produced by transfecting 5 μg of TLCV2 constructs together with packaging components (3.75 μg psPAX2 Addgene #12260; and 1.5 μg pMD2.G Addgene #12259) into human embryonic kidney cells (HEK293T) that were plated 24 h before transfection at the seeding density of 4 × 106 cells/T75 flask. Transfection in each flask was performed in the presence of 30 µl of Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer's instructions. Viral supernatant was harvested 48 h after transfection, passed through a 0.45 μm filter (Millipore), and frozen at −80°C until further use.
Lentiviral transduction
At 24 h before transduction, 2.5 × 105 SH-SY5Y cells were seeded in 2 ml of complete medium into 6-well plates; 900 μl of HEK293T supernatant containing the viral vector was used to transduce cells in the presence of polybrene (8 μg/ml, Millipore). A full media change was performed 24 h after transduction. Puromycin selection (1 μg/ml, Millipore) was started 48 h after transduction, and then gradually increased to 2 μg/ml for the consequent cell passages. Cas9-GFP expression was induced with doxycycline (1 μg/ml) and confirmed with fluorescence microscopy (EVOS). Cells were subsequently maintained in 1 µg/ml puromycin.
Cell culture
SH-SY5Y cells were seeded in a black, flat-bottom 96-well plate (Greiner) at a density of 10,000 cells/well in DMEM/F12, 10% FBS, 1% L-glutamine, 1% penicillin-streptomycin, and kept at 37°C and 5% CO2 at all times. The day after seeding, cells were transiently transfected with EF1a-REST CMV-MCherry or CMV-MCherry constructs using OptiMEM, Lipofectamine 2000, and Plus Reagent (Thermo Fisher Scientific), as previously described (Keane et al., 2015). After transfection, cells were kept in OptiMEM and starved through serum deprivation. At 24 h later, cells were treated with 1 mm MPP+ (Sigma Millipore) and incubated for another 24 h, before running downstream assays.
To-pro+ (Invitrogen) was added 1:1000 to the medium to count dead cells. For mitochondrial and lysosome, autophagosome, and autolysosome analysis, cells were fixed in 4% PFA and evaluated after immunofluorescence. Immunofluorescence was performed as described above for immunohistochemistry starting at block. TOM20 was used to visualize mitochondria, while Lamp1 and LC3 were used for lysosomal, autophagosome, and autolysosome detection. The Opera Phenix confocal microscope (PerkinElmer) was used to image cells, and ImageJ and Cell Profiler were used for image quantification.
For nuclear fractionation, NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific) were used according to the manufacturer's instructions.
Assessment of mitophagy
Mitophagy in SH-SY5Y cells was detected using the Mitophagy Detection Kit (Dojindo Molecular Technologies) according to the manufacturer's instructions. In brief, cells were seeded in a black, flat-bottom µCLEAR 96-well plate (Greiner) at a density of 20,000 cells/well. The following day, cells were washed twice with serum-free culture medium DMEM/F12 (1:1 1×, Invitrogen) before incubation with Mtphagy Dye (100 nm) at 37°C for 30 min. Cells were then washed twice in serum-free DMEM/F12 culture medium before incubation in DMEM/F12 (1:1 1×, Invitrogen) supplemented with 10% FBS (Invitrogen), 1% L-glutamine (2 mm, Invitrogen), 1% penicillin-streptomycin (Invitrogen), puromycin (1 μg/ml), and doxycycline (1 μg/ml) at 37°C for 6 h in the presence of CCCP, oligomycin, or in HBSS alone. Images were captured (×63, water objective) using the Opera Phenix high-content screening system (PerkinElmer). Throughout imaging, live cells were maintained at 37°C and 1 bar CO2 (5.0%). Three confocal channels were utilized for the selective laser-induced excitation of DAPI (NucBlue; 358/461 nm), GFP (488/509 nm), and Mtphagy dye (561/650 nm). The digital phase-contrast channel (brightfield) was also used to define the borders of individual cells within the culture. GFP intensity was measured in every defined cell, and GFP-expressing (GFP+) cells were then identified. Cells below this intensity threshold were defined as GFP– cells. Finally, Mtphagy spots were then identified in both GFP+ and GFP– cells. All cells within the previously defined populations that had ≥3 Mtphagy spots were classed as a cell undergoing mitophagy.
Statistical analysis
Differences between two groups were assessed using a one-tailed Mann–Whitney test. When more groups were present, one- or two-way ANOVAs were used; a detailed description provided for each analysis can be found in the figure legends.
Results
α-Synuclein overexpression leads to an accumulation of oligomers, dysfunctional mitochondria, and oxidative protein damage
We have previously shown that SNCA-OVX BAC-transgenic mice develop age-dependent neurodegeneration and a motor phenotype compared with mice expressing physiological levels of α-synuclein (Janezic et al., 2013). SNCA-OVX mice express 2-3 times more α-synuclein than nontransgenic WT mice (Extended Data Fig. 1-1a), show loss of TH-expressing neurons (Extended Data Fig. 1-1b–d), and accumulate high levels of early-stage oligomeric α-synuclein pathology with age in the midbrain, but not in other brain regions, as revealed by the AS-PLA (Extended Data Fig. 1-1e–q). This highlights these transgenic animals as a model of early pathology in which oligomeric pathology accumulates, as we have previously shown in postmortem human PD brain (Roberts et al., 2015).
To identify the pathways responsible for the changes associated with age-dependent neurodegeneration and accumulation of α-synuclein oligomers in the midbrain of SNCA-OVX mice, we performed LC-MS/MS proteomics on fractionated midbrain lysates from 18-month-old SNCA-OVX mice and control WT mice and identified differentially regulated proteins (Fig. 1a; Extended Data Fig. 1-2a). Interestingly, a number of neurofilament polypeptides (known to be key components of Lewy bodies) were among the most upregulated proteins in SNCA-OVX mice (Extended Data Fig. 1-2b).
Figure 1.
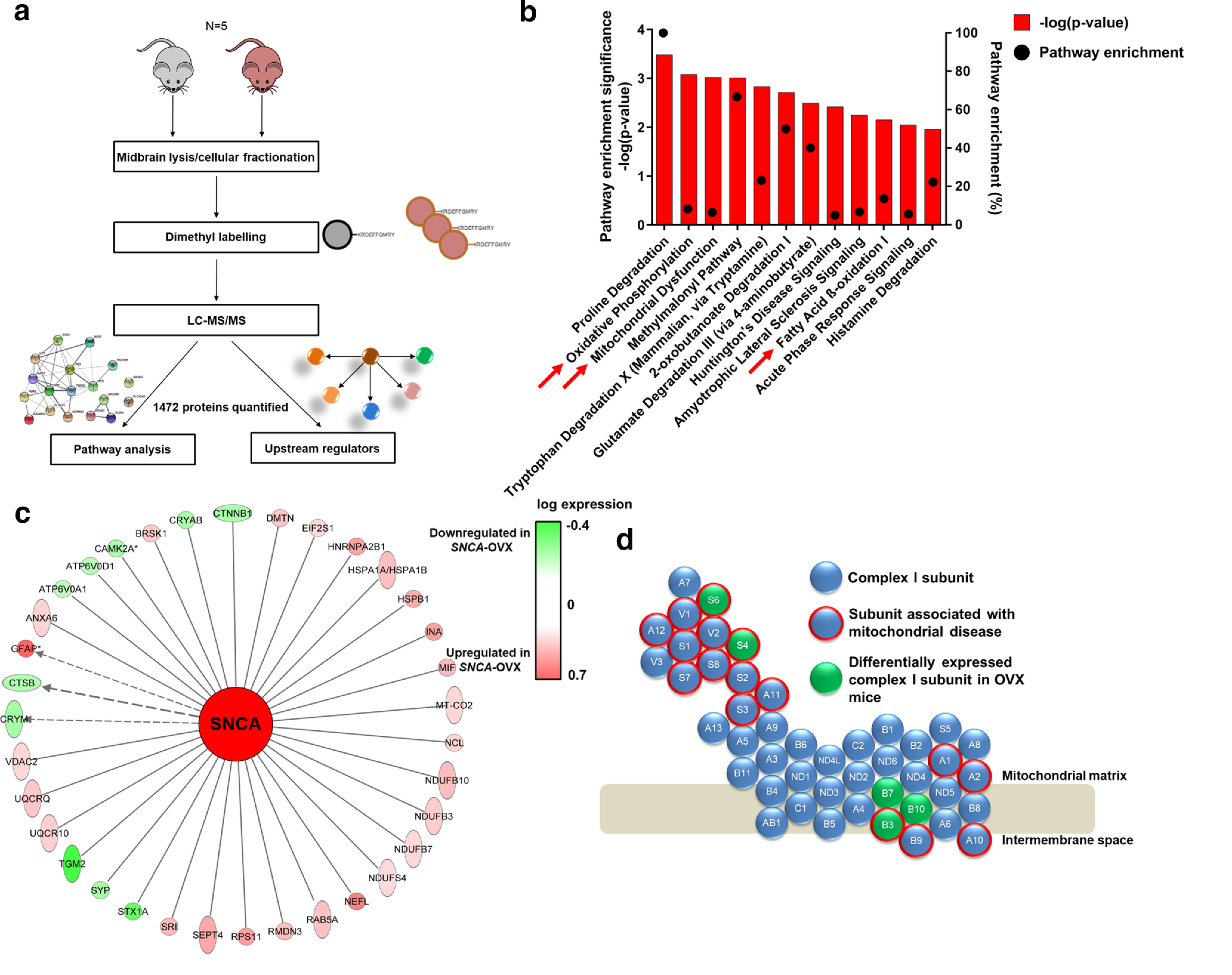
Proteomic analysis of SNCA-OVX midbrain demonstrates that mitochondrial dysfunction is a key factor in α-synuclein-induced neurodegeneration. a, Schematic representation of proteomics workflow. b, Pathway enrichment of dysregulated proteins in 18-month-old SNCA-OVX mice showing pathway enrichment (%) and enrichment significance (–log(p value)). c, Schematic representation on dysregulated mitochondrial complex I subunits caused by α-synuclein overexpression. Complex I subunits perturbed in SNCA-OVX mice (green) and subunits associated with mitochondrial diseases (red outline). d, Dysregulated proteins in SNCA-OVX mice, which are associated with altered α-synuclein levels. Upregulated/downregulated proteins are red/green, respectively, based on their magnitude of upregulation/downregulation. For dopaminergic cell and oligomeric analysis, see Extended Data Figure 1-1. For extended proteomic descriptors and validation, see Extended Data Figure 1-2.
a) Relative α-synuclein expression in midbrain lysates from WT and SNCA-OVX mice. b) Assessment of total and TH-cell counts in mouse nigra as assessed by stereology. c&d) levels of TH protein in midbrain of c) 3 month and d) 18-month-old mice as assessed by western blot (N = 3). e-g) α-synuclein oligomers are increased in the SNc of aged SNCA-OVX mice. α-Synuclein oligomers were detected by PLA staining in the SNc of α-synuclein KO, human α-synuclein (hα-syn) or SNCA-OVX mice at f) 3 or g) 24 months (p < 0.05, one-way ANOVA, Holm-Sidak; N = 3) h-q) No change in α-synuclein oligomers in the brain regions from 3 or 24-month-old SNCA-OVX and control mice (p > 0.05, one-way ANOVA, Holm-Sidak; N = 3). Download Figure 1-1, TIF file (1.1MB, tif) .
a) Protein abundance distribution in dimethyl labeling proteomics dataset (M/L ratio) dotted lines (black) indicate cutoff of proteins taken forward into pathway analysis. b) Top up and down regulated proteins identified in SNCA-OVX mice by proteomics. c) Percentage of mitochondrial subunits of each mitochondrial complex quantified by proteomics. d) Midbrain REST target gene expression analysis by qPCR of aged Wt and SNCA-OVX animals. Download Figure 1-2, TIF file (961.3KB, tif) .
A significant number of differentially regulated proteins were found to be enriched in several pathways, including oxidative phosphorylation, mitochondrial dysfunction, and β-oxidation of fatty acids (Fig. 1b; Table 2). Additionally, 16.8% of all dysregulated proteins were mitochondrial, including five mitochondrial complex I subunits, three of which are associated with mitochondrial diseases (Fig. 1c; Extended Data Fig. 1-2c,d). Furthermore, 35 dysregulated proteins were associated with altered α-synuclein levels, of which 29% are mitochondrial proteins (Fig. 1d), suggesting a direct role for increased α-synuclein in modulation of mitochondrial function in SNCA-OVX mice.
Table 2.
Biological pathways significantly enriched in differentially regulated proteins from SNCA-OVX mice
| Pathway name | Pathway enrichment –log (p) | Ratio of pathway constituents altered | Altered pathway components in dataset |
|---|---|---|---|
| Proline degradation | 3.48E + 00 | 1.00E + 00 | ALDH4A1, LOC102724788/PRODH |
| Oxidative phosphorylation | 3.08E + 00 | 8.33E-02 | NDUFB7, NDUFS6, MT-CO2, UQCRQ, NDUFB3, NDUFB10, NDUFS4 |
| Mitochondrial dysfunction | 3.02E + 00 | 6.52E-02 | NDUFB7, NDUFS6, NDUFAF2, MT-CO2, UQCRQ, NDUFB3, NDUFB10, VdAC2, NDUFS4 |
| Methylmalonyl pathway | 3.01E + 00 | 6.67E-01 | PCCA, MUT |
| Tryptophan degradation X (mammalian, via tryptamine) | 2.83E + 00 | 2.31E-01 | ALDH4A1, ALDH3A2, DDC |
| 2-oxobutanoate degradation I | 2.71E + 00 | 5.00E-01 | PCCA, MUT |
| Glutamate degradation III (via 4-aminobutyrate) | 2.50E + 00 | 4.00E-01 | GAD2, SUCLG2 |
| Huntington's disease signaling | 2.42E + 00 | 4.98E-02 | TGM2, GNB4, CAPNS1, CLTB, HSPA1A/HSPA1B, PLCB1, CAPN2, GNG3, STX1A, HSPA2 |
| Amyotrophic lateral sclerosis signaling | 2.25E + 00 | 6.74E-02 | PRPH, RAB5A, CAPNS1, NEFL, NEFH, CAPN2 |
| Fatty acid β-oxidation I | 2.15E + 00 | 1.36E-01 | ECI2, SLC27A1, ACADM |
| Acute phase response signaling | 2.05E + 00 | 5.47E-02 | HPX, C3, APOA1, TF, CP, SERPINA3, KRAS |
| Histamine degradation | 1.96E + 00 | 2.22E-01 | ALDH4A1, ALDH3A2 |
To validate these proteomic findings, consistent with mitochondrial dysfunction and oxidative stress playing a role in Parkinsonian pathology, we investigated mitochondrial dysfunction in the SNCA-OVX mouse midbrain caused by α-synuclein overexpression and oligomerization. Mitochondrial complex I activity, an established deficit reported in PD patients, was decreased by >50% in the midbrain of aged SNCA-OVX mice compared with nontransgenic control WT mice (Fig. 2a) but not in young SNCA-OVX mice nor in the cortex or whole brain homogenates of aged SNCA-OVX mice, indicating age and region specificity of this dysfunction (Fig. 2b; Extended Data Fig. 2-1a,b).
Figure 2.
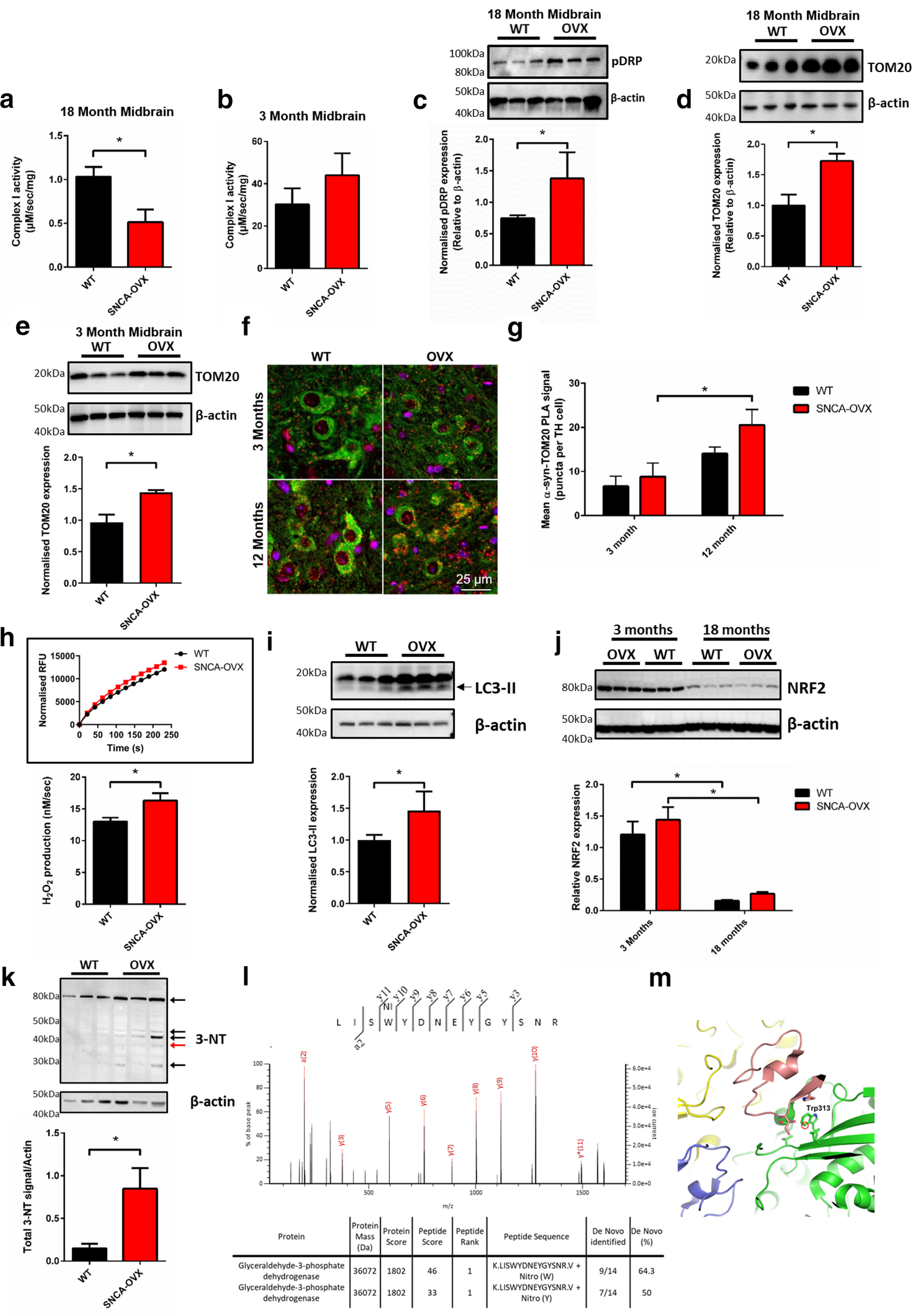
α-Synuclein overexpression induces age- and region-specific mitochondrial dysfunction and oxidative stress. a, b, Mitochondrial complex I activity is decreased in the aged midbrain of SNCA-OVX mice expressing α-synuclein. Mitochondrial complex I activity in (a) 18-month-old and (b) 3-month-old midbrain (N = 3). c, Increased midbrain levels of pS616 Drp1. Increased levels of TOM20 in (d) 18-month-old and (e) 3-month-old midbrain by Western blot (N = 3). f, g, Increased α-synuclein-TOM20 PLA signal (red puncta) in TH neurons (green). For AS-PLA puncta and mitochondrial analysis, 25 cells per neuronal cell type and animal were analyzed blindly. h, Increased mitochondrial H2O2 production in mitochondria isolated from 18-month-old mice (N = 3). i, Increased levels of autophagosome formation (LC3B lipidation; lower band) in 18-month-old midbrain. j, Decreased midbrain levels of NRF2 protein in aged (18-month-old) mice (N = 3). k, Increased midbrain levels of 3-NT containing proteins in 18-month-old midbrain lysate (N = 3). l, LC-MS/MS spectrum of excised 3-NT containing band (red arrow) showing de novo sequencing of GAPDH peptide LISWYDNEYGYSNR with summary table showing Mascot scoring de novo sequencing results. m, Molecular modeling of nitrated residue in GAPDH active site, based on PBD ID 1ZNQ. *p < 0.05 Mann–Whitney test (one-tailed) or two-way ANOVA Sidak. For extended analysis of midbrain and cortical expression levels, see Extended Data Figure 2-1. (a) Mann–Whitney p = 0.0357; (c) Mann–Whitney p = 0.05; (d) Mann–Whitney p = 0.05; (e) Mann–Whitney p = 0.05; (g) two-way ANOVA, Sidak (F(1,9) = 13.92) p = 0.042; (h) Mann–Whitney p = 0.05; (i) Mann–Whitney p = 0.05; (j) two-way ANOVA, Sidak (F(1,8) = 59.82) p < 0.001; (k) Mann–Whitney p = 0.05.
a&b) Mitochondrial complex I activity in a) 18-month cortex and b) whole brain lysates from aged SNCA-OVX mice. c) TOM20 and d) Non-phosphorylated DRP protein levels in aged midbrain, normalized to B-actin. e) Phosphorylated DRP levels in 3-month-old mouse midbrain, normalized to B-actin. f) COX IV protein levels in aged midbrain normalized to B-actin (N = 3). × g&h) Levels of lipidated LC3 (LC3-II) in g) midbrain lysates from 3-month-old mice (N = 3) and h) cortex lysates from 18-month-old mice (N = 4) normalized to β-actin levels. i-k) Levels of BIP in SNCA-OVX mice i&j) Midbrain lysates from i) 18-month-old, j) 3-month-old midbrain and k) 18-month-old cortex from OVX mice normalized to β-actin levels (N = 3). l) Quantification 3-NT band excised for LC-MS/MS analysis. Band indicated by red arrow in Figure 1k. Download Figure 2-1, TIF file (974.8KB, tif) .
Consistent with complex I dysfunction, an increase in the active (phosphorylated) form, but not in total levels, of the mitochondrial fission regulator DRP1 (Phospho S616) was observed in midbrain lysates from aged, but not young, SNCA-OVX mice compared with age-matched WT mice (Fig. 2c; Extended Data Fig. 2-1c,d), indicating increased mitochondrial fragmentation in response to α-synuclein overexpression. Furthermore, we observed increased levels of the mitochondrial outer membrane protein TOM20, but not the inner membrane protein COX IV, in the midbrain of aged SNCA-OVX mice but not young mice or in the cortex of aged mice (Fig. 2d,e; Extended Data Fig. 2-1e,f), indicating age- and region-specific impaired mitochondrial quality control in aged SNCA-OVX animals. We also observed an increase in pathogenic α-synuclein-TOM20 interactions in dopaminergic neurons of SNCA-OVX animals (Fig. 2f,g), suggesting a mechanism for mitochondrial dysfunction observed in SNCA overexpression models and PD patients (Di Maio et al., 2016; Zambon et al., 2019). Mitochondria isolated from the midbrain of aged SNCA-OVX mice also demonstrated increased ROS production (Fig. 2h). This increase in dysfunctional mitochondria was associated with increased LC3 lipidation, indicating increased autophagosome formation in an age- and region-specific manner (Fig. 2i; Extended Data Fig. 2-1g,h). Levels of the ER protein BIP were also increased indicating enhanced ER stress in aged, but not young, mice (Extended Data Fig. 2-1i–k).
To understand how ROS generated from dysfunctional mitochondria contributes to age-dependent neurodegeneration in SNCA-OVX mice, we measured levels of the antioxidant regulator NRF2 in young and aged midbrain from SNCA-OVX and control mice. A large decrease (>70%) was observed in the levels of NRF2 in whole-brain lysates of SNCA-OVX and control animals between the ages of 3 and 18 months (Fig. 2j). Given this decrease was independent of α-synuclein overexpression, decreases in cellular NRF2 appear to be a result of aging.
To investigate whether this age-dependent decrease in NRF2 potentiates oxidative/nitrative damage of proteins in SNCA-OVX mice, we measured protein nitration, a stable end product of oxidative/nitrative stress and a known modulator of α-synuclein aggregation (Matsuda et al., 2010; Burai et al., 2015). We observed an increase in nitrated proteins in aged SNCA-OVX mice (Fig. 2k; Extended Data Fig. 1-2l). Among the proteins identified, the glycolysis regulator GAPDH was found to be nitrated on the tryptophan residue of the peptide LISWYCNEYGENN (Fig. 2l). This nitrated tryptophan residue resides near the catalytic site of GAPDH in a hydrophobic site, and nitration of this residue has previously been demonstrated to be increased during oxidative insult decreasing GAPDH activity (Fig. 2m) (Jenkins and Tanner, 2006; Palamalai and Miyagi, 2010; Bailey et al., 2011). The impaired function of GAPDH caused by oxidative modification may further contribute to increased vulnerability of cells with high metabolic demand, such as dopaminergic neurons.
Together, these data demonstrate the presence of age-dependent mitochondrial dysfunction in the midbrain of SNCA-OVX mice in response to α-synuclein oligomer accumulation, suggesting that mitochondrial dysfunction may be a key contributor to the age- and region-dependent neurodegeneration observed in this model.
Proteomic analysis reveals a region-specific dysfunction of REST in the midbrain of SNCA-OVX mice
To further understand the mechanisms responsible for mitochondrial dysfunction and cell death induced by α-synuclein overexpression, we sought to identify the upstream regulators responsible for the observed changes of protein levels in the midbrains of SNCA-OVX mice. This analysis identified a number of proteins associated with neurodegeneration, including REST, also known as Neuron-Restrictive Silencing Factor (Fig. 3a; Table 3). Based on expression profiles of downstream targets that have a strong correlation with REST regulation, REST was predicted to be inhibited in α-synuclein-overexpressing mice (Fig. 3b). We therefore performed qRT-PCR to establish whether the dysregulated REST targets identified by proteomics were also perturbed at the mRNA level. We found that these targets were dysregulated and followed the same direction in expression change as detected by proteomics (Extended Data Fig. 1-2c), further suggesting REST as a contributing factor in proteomic disruption in SNCA-OVX mice.
Figure 3.
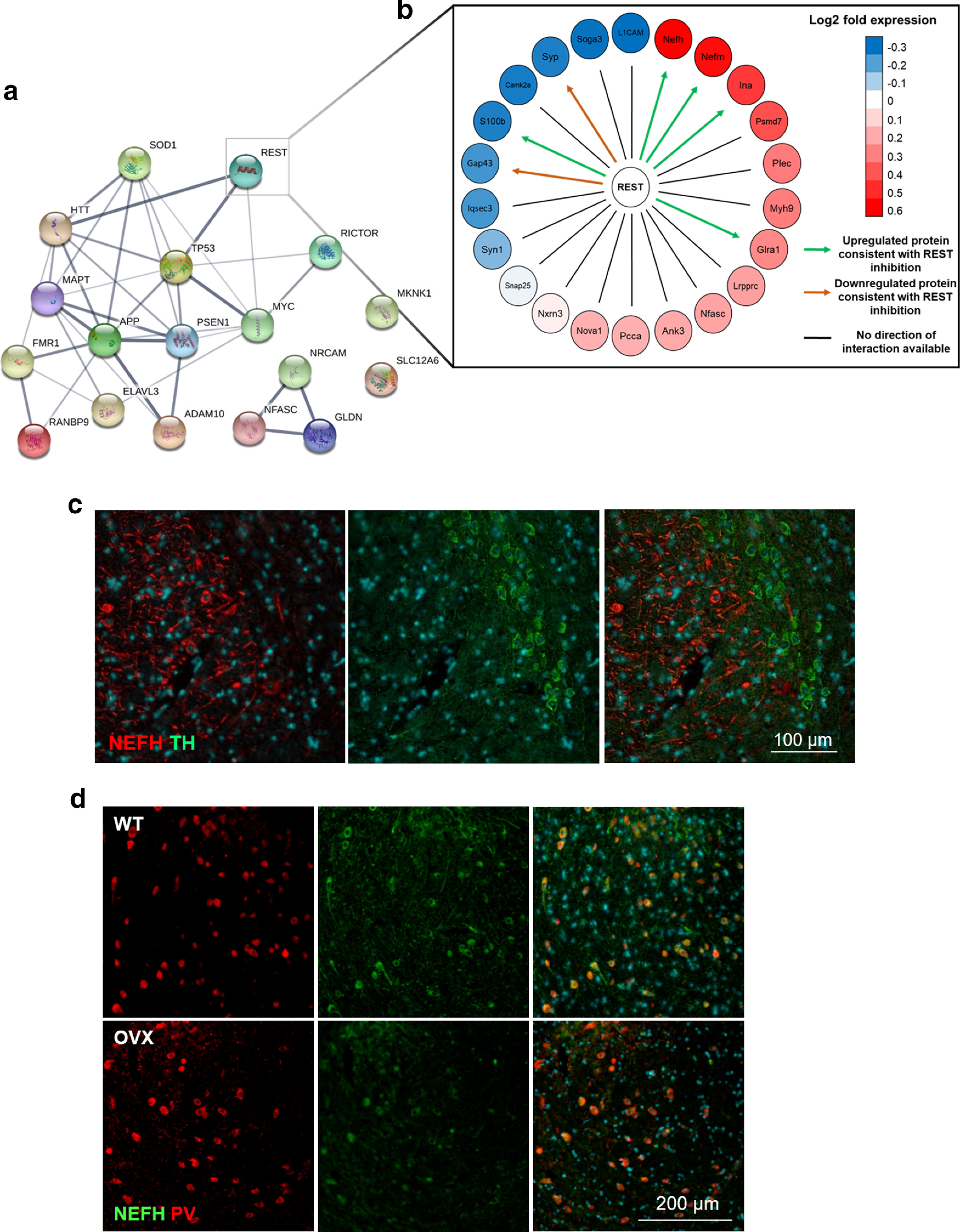
Downstream targets of REST are dysregulated in SNCA-OVX mice and reside in distinct regions of the midbrain. a, Protein-protein interaction network of upstream regulators in SNCA-OVX mice (p < 0.05). b, Directed REST interaction network showing the upregulation and downregulation of REST target proteins in SNCA-OVX mice; downregulated (blue) and upregulated (red) proteins and predicted effect of REST on downstream targets (arrows). Green arrows indicate protein regulation consistent with REST inhibition. Orange arrows indicate protein level changes inconsistent with REST inhibition. Black lines indicate proteins without directionality assigned. c, d, Immunofluorescence of mouse midbrain demonstrating (c) location of TH (green) in the SNc and NEFH (red) in the SNr and (d) colabeling of NEFH (green) and parvalbumin (PV; red) cells in 3-month-old WT (or SNCA-OVX) mouse midbrain.
Table 3.
Significant upstream regulators identified in SNCA-OVX mice (supplementary Excel sheet)
| Upstream regulator | Pathway enrichment –log (p) |
|---|---|
| MAPT | −6.627 |
| FMR1 | −6.235 |
| SOD1 | −6.168 |
| APP | −5.587 |
| GLDN | −5.266 |
| NRCAM | −5.266 |
| PSEN1 | −5.095 |
| TP53 | −4.833 |
| REST | −4.481 |
| ADAM10 | −4.382 |
| NFASC | −3.745 |
| HTT | −3.638 |
| MKNK1 | −3.603 |
| MYC | −3.545 |
| RICTOR | −3.327 |
| RANBP9 | −3.037 |
| SLC12A6 | −3.037 |
| ELAVL3 | −3.037 |
Given that we observed upregulation of neurofilament proteins in SNCA-OVX mice from the proteomic analysis of the midbrain (Extended Data Fig. 1-2b), and that REST expression negatively regulates all three neurofilament polypeptide levels (Bruce et al., 2004; Ching and Liem, 2009), we performed midbrain staining for neurofilament heavy chain (NEFH) and found a neuronal population of NEFH-labeled cells in the SNr, which distinctly segregated from TH+ dopaminergic neurons of the SNc (Fig. 3c). These SNr cells were determined to be parvalbumin+ (indicating a GABAergic origin) (C. R. Lee and Tepper, 2007; Zhou and Lee, 2011) by colocalization of NEFH (98.62 ± 1.38% in WT and 100 ± 1.85% in SNCA-OVX; Fig. 3d).
The identification of REST as an upstream regulator of proteomic dysfunction in aged SNCA-OVX mice implicates the involvement of REST in the age- and region-dependent α-synuclein oligomer formation and bioenergetic dysfunction in SNCA-OVX mice.
α-Synuclein oligomers induce mitochondrial dysfunction in dopaminergic neurons lacking nuclear REST
Given that the GABAergic neurons in the striatum and SNr are less affected by neuronal dysfunction and loss in PD than the dopaminergic neurons of the SNc (Giguère et al., 2018), we investigated the role of REST in neuronal vulnerability in PD. We observed a significant difference in REST localization between dopaminergic SNc and GABAergic SNr neurons, with REST present in the nucleus of resistant NEFH+ GABAergic SNr neurons, but completely absent in the nucleus of vulnerable dopaminergic SNc neurons at 3 and 12 months of age in SNCA-OVX and WT mice (Fig. 4a–c). Importantly, GABAergic SNr neurons were able to upregulate REST in response to α-synuclein overexpression, whereas dopaminergic neurons of the SNc were not able to respond to the insult, suggesting a lack of ability to mount a protective response against α-synuclein overexpression in SNc dopaminergic neurons.
Figure 4.
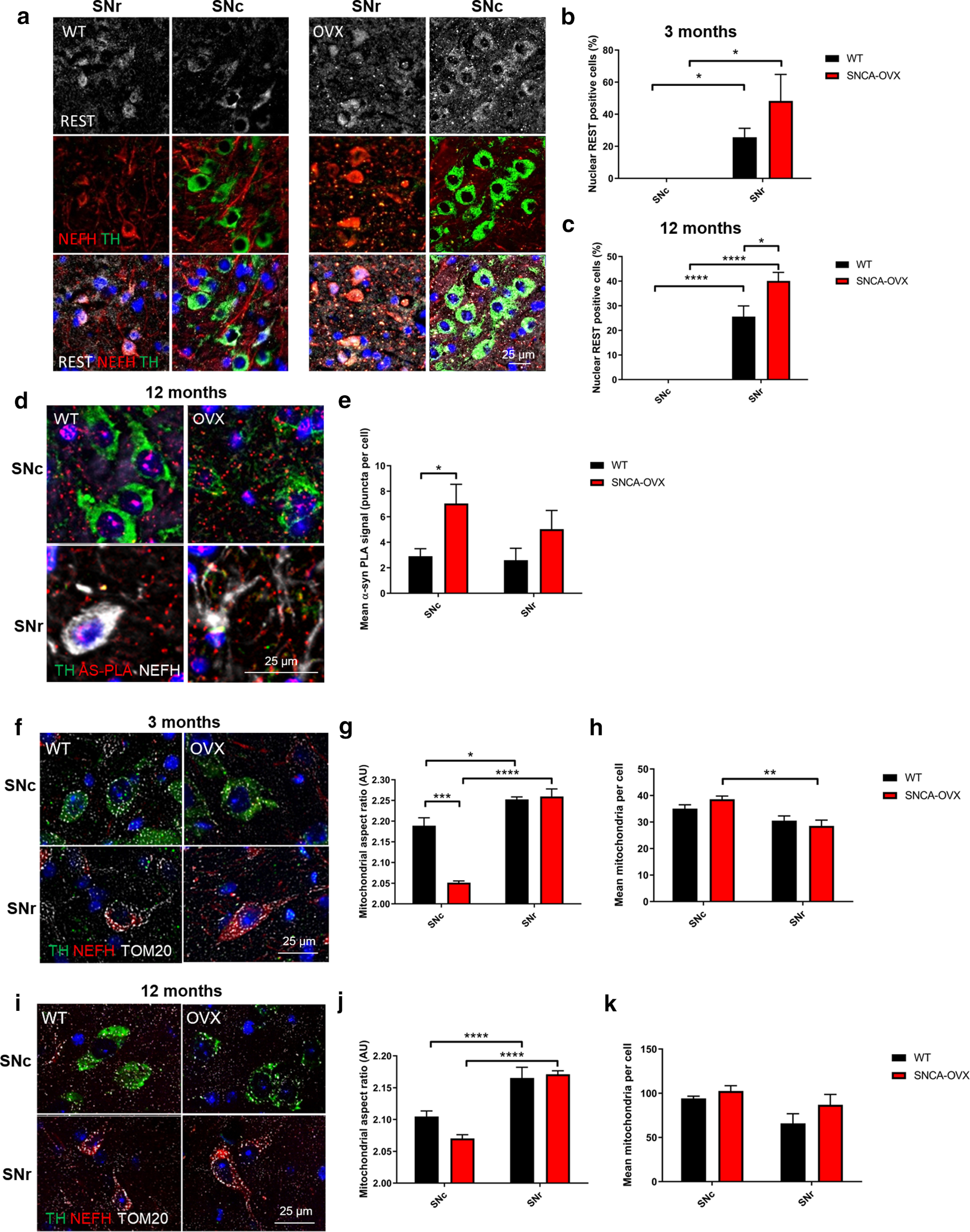
Absence of nuclear REST correlates with increased vulnerability of neurons to α-synuclein aggregation and mitochondrial dysfunction. a–c, Nuclear REST is absent from dopaminergic, but not GABAergic, cells. Quantification of nuclear REST (gray), staining in NEFH (red) and TH (green) cells in WT, and SNCA-OVX mouse midbrain at (b) 3 months and (c) 12 months. d, e, α-Synuclein oligomers are increased in dopaminergic neurons of SNCA-OVX mice. α-Synuclein PLA staining (red) in TH (green) and NEFH (gray) cells in WT and SNCA-OVX mouse midbrain. f–k, Mitochondria fragmentation in dopaminergic neurons is exacerbated by α-synuclein. Representative images of TOM20 (gray) staining in TH (green) and NEFH (red) in (f–h) 3-month-old and (i–k) 12-month-old WT and SNCA-OVX midbrain. g, j, Mitochondrial AR. h, k, Average mitochondrial count per cell. For AS-PLA puncta and mitochondrial analysis, 25 cells per neuronal cell type and animal were analyzed. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; two-way ANOVA Sidak. For extended mitochondrial morphology analysis over time, see Extended Data Figure 4-1. (b) Two-way ANOVA, Sidak (F(1,14) = 21.78) p = 0.0004; (c) two-way ANOVA, Sidak (F(1,12) = 139.1) p < 0.0001; (e) two-way ANOVA, Sidak (F(1,10) = 8.974) p = 0.0134; (g) two-way ANOVA, Sidak (F(1,8) = 98.5) p < 0.0001, (F(1,8) = 22.85) p = 0.014; (h) two-way ANOVA, Sidak (F(1,8) = 18.65) p = 0.0026; (j) two-way ANOVA, Sidak (F(1,12) = 62.44) p < 0.0001.
Mitochondrial morphology, as assessed by TOM20 staining, in aging mice demonstrating a lack of mitochondrial network in 18-month-old tissue. Download Figure 4-1, TIF file (1.1MB, tif) .
These observations, together with the accumulation of α-synuclein oligomers in the SNc of SNCA-OVX animals, prompted us to investigate whether vulnerable dopaminergic cells of the SNc accumulated more oligomeric α-synuclein than protected GABAergic neurons of the SNr. Coimmunofluorescence analysis in combination with AS-PLA revealed that α-synuclein overexpression resulted in a significant accumulation of α-synuclein in TH+ neurons of the SNc in SNCA-OVX animals (p < 0.05) but not in the SNr GABAergic neurons (p > 0.05) (Fig. 4d,e).
We next assessed mitochondrial morphology in both TH+ (dopaminergic) and NEFH+ (GABAergic) neurons in both 3- and 12-month-old WT and SNCA-OVX mice to test the correlation of mitochondrial dysfunction with age-dependent neurodegeneration. In 3-month-old SNCA-OVX animals, mitochondrial aspect ratio (AR) was reduced and mitochondrial count increased in TH+ versus NEFH+ neurons, indicating increased mitochondrial fragmentation in dopaminergic neurons versus GABAergic neurons (Fig. 4f–h). A further increase in fragmentation was observed when comparing TH+ neurons in SNCA-OVX and control mice, indicating that α-synuclein overexpression potentiates mitochondrial dysfunction specifically in dopaminergic neurons. At 12 months, the mitochondrial AR was reduced, while the total number of mitochondria was increased as an effect of aging (Fig. 4i–k). Interestingly, the mitochondrial morphology of protected GABAergic neurons was still preserved compared with that of neighboring, vulnerable dopaminergic neurons. Excessive mitochondrial fragmentation was observed in 18-month-old mice alongside neuronal loss preventing quantification (Extended Data Fig. 4-1).
These results point to distinct cell populations residing in the SNc and SNr with respect to REST expression. The vulnerable SNc dopaminergic neurons have increased α-synuclein accumulation and oligomerization, disrupted mitochondrial morphology, and no detectable nuclear REST, whereas the resistant SNr GABAergic neurons have readily detectable nuclear REST levels, a lower α-synuclein oligomer burden, and preserved mitochondrial morphology. As REST has been shown to be protective in response to manganese insult (Pajarillo et al., 2020), we assessed whether upregulation of REST in GABAergic SNr neurons is responsible for neuroprotection from toward α-synuclein-induced mitochondrial damage.
Loss of REST causes mitochondrial dysfunction and impaired mitophagy
REST has been previously shown to play a protective role in AD and in response to manganese toxicity (Lu et al., 2014; Pajarillo et al., 2020). Given our data suggesting REST as a key mediator of neuronal health in SNCA-OVX mice, we assessed REST target gene expression in iPSC-derived dopaminergic neuronal cultures from PD patients carrying the SNCA-Triplication mutation, previously shown by two independent studies to demonstrate mitochondrial dysfunction and accumulation of α-synuclein oligomers (Devine et al., 2011; Zambon et al., 2019). We observed dysregulation of REST target genes in both transcriptomic datasets (Devine et al., 2011; Zambon et al., 2019), suggesting REST inhibition (Fig. 5a; Extended Data Fig. 5-1a). Furthermore, we found that REST protein expression was reduced in dopaminergic neuronal cultures carrying the SNCA-Triplication (Fig. 5b). This suggests that even early α-synuclein aggregates inhibit and/or downregulate REST in a negative feedback loop, resulting in the accumulation of mitochondrial damage, producing ROS and further α-synuclein oligomerization.
Figure 5.
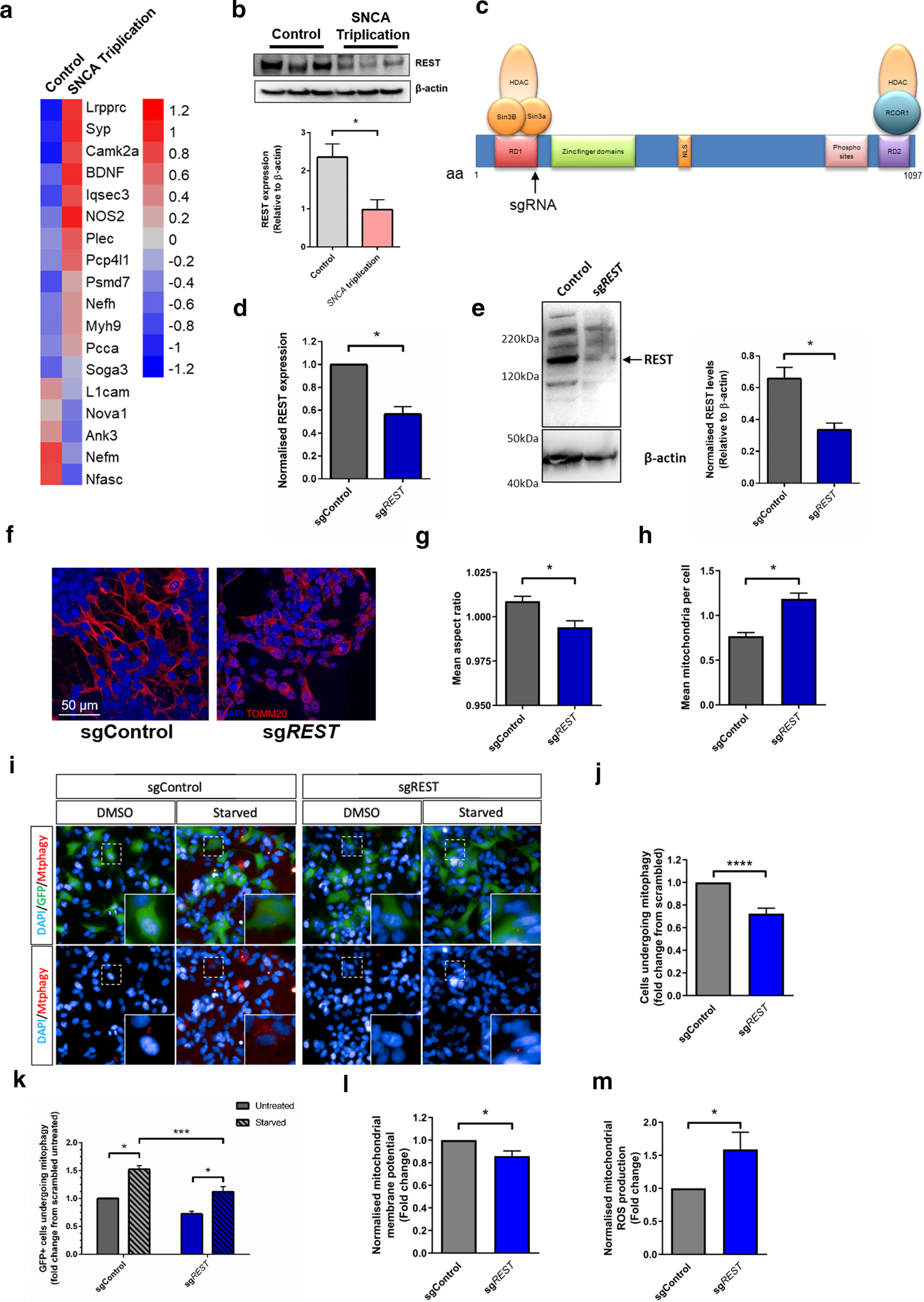
Inhibition of REST is observed in SNCA-Triplication neurons and causes mitochondrial and mitophagy defects. a, Anticorrelation of REST target gene expression in iPSC-derived dopaminergic neuronal cultures from a SNCA-Triplication patient (GDS4156) (Devine et al., 2011). b, Decreased REST protein expression levels in iPSC-derived dopaminergic neuronal cultures from a SNCA-Triplication patient (n = 3). c, Schematic representation of REST gene showing sgRNA targeting site. Confirmation of (d) REST transcript knockdown relative to HPRT expression and (e) REST protein reduced relative to β-actin after Cas9-mediated KO in a polyclonal cell population (n = 3). f–h, Representative immunofluorescence images of TOM20 (red) showing dysfunctional mitochondrial morphology in starved polyclonal REST KO lines showing (g) mean mitochondrial count per cell and (h) AR (n = 3). i–k, REST KO induced mitophagy deficits. i, Representative images of mitophagy events in SH-SY5Y cells showing mtphagy staining (red puncta) in cells expressing Cas9 (green) j, Quantification of basal mitophagy in cells expressing Cas9 and sgRNA (GFP+) or GFP– cells. k, Quantification of mitophagy in starved GFP+ cells expressing REST-targeting or scrambled guide. l, Decreased mitochondrial membrane potential in polyclonal REST KO cells assessed using JC-10, under basal conditions (n = 4). m, Increased mitochondrial ROS production under basal conditions, in polyclonal REST KO cells (n = 6). For extended analysis and assay validation, see Extended Data Figure 5-1. (b) Mann–Whitney p = 0.05; (d) Mann–Whitney p = 0.05; (e) Mann–Whitney p = 0.0089; (g) Mann–Whitney p = 0.0154; (h) Mann–Whitney p = 0.0026; (j) Mann–Whitney p < 0.0001; (k) two-way ANOVA, Sidak (F(1,20) = 29.76) < 0.0001, (F(1,20) = 54.18) < 0.0001; (l) Mann–Whitney p = 0.0143; (m) Mann–Whitney p = 0.0234. *p < 0.05; ***p < 0.001; ****p < 0.0001.
Relative expression of REST target genes in iPSC-derived dopaminergic neuronal cultures from patients with α-synuclein triplication a) dataset from Zambon et al., 2019; (Zambon et al., 2019). b-d) Basal TOM20 morphology in Cas9 expressing SH-SY5Y cells expressing sgRNA targeting REST or a scrambled sgRNA showing c) Mitochondrial AR and d) mitochondrial number. e) Images of mitophagy (as assessed by mtphagy dye; red) in Cas9 expressing SH-SY5Y cells (green) expressing sgRNA targeting REST or a scrambled sgRNA (images duplicated from Figure 5i for comparison). f-g) Quantification of mitophagy in Cas9(GFP) expressing cells as assessed by mtphagy in f) Oligomycin-treated cells and g) CCCP-treated cells. h) ATP levels in polyclonal REST KO cells (n = 4). i&j) Mitochondrial ROS production in polyclonal REST KO cells under basal and rotenone-stimulated cells (n = 6). Download Figure 5-1, TIF file (2.7MB, tif) .
To understand if the mitochondrial dysfunction observed in dopaminergic neurons of the SNc in SNCA-OVX mice and in SNCA-Triplication iPSC-derived dopaminergic neurons associated with increased α-synuclein levels could be directly attributed to inhibition of REST, we performed CRISPR-mediated KO of REST in SH-SY5Y cells, generating a polyclonal cell population with reduced REST expression (Fig. 5c–e). Consistent with our observations in both animal and iPSC-derived neuronal models, a decrease in REST expression was associated with mitochondrial fragmentation under basal conditions (Extended Data Fig. 5-1b–d) and in response to starvation (Fig. 5f–h), indicating REST regulates mitochondrial health. In addition, mitophagy was decreased in cells with reduced REST expression under basal conditions and also impaired after stimulation of mitophagy by starvation, CCCP or oligomycin (Fig. 5i–k; Extended Data Fig. 5-1e–g). Furthermore, decreased REST expression resulted in significantly decreased mitochondrial membrane potential (Fig. 5l) without overt changes in ATP production (Extended Data Fig. 5-1h) and increased ROS production under basal conditions (Fig. 5m; Extended Data Fig. 5-1i,j). Together, these data suggest that loss of nuclear REST drives mitochondrial dysfunction and impaired mitophagy.
REST exerts a mitoprotective role through PGC-1α in vitro and in vivo
To assess the potential neuroprotective role of REST, we modeled mitochondrial complex I dysfunction, as observed in SNCA-OVX mice, using the complex I inhibitor MPP+. Overexpression of REST in SH-SY5Y cells reduced cell death in response to MPP+ complex I dysfunction, indicating that REST is protective in vitro (Fig. 6a). Basal mitochondrial morphology was also improved by REST overexpression, as shown by an increase in mitochondrial AR and a decrease in number of mitochondria per cell (Fig. 6b–d), indicating that REST improves mitochondrial network integrity. These differences confirm earlier work in TH+ versus NEFH+ cells, showing that REST plays a neuroprotective role through preserving mitochondrial morphology.
Figure 6.
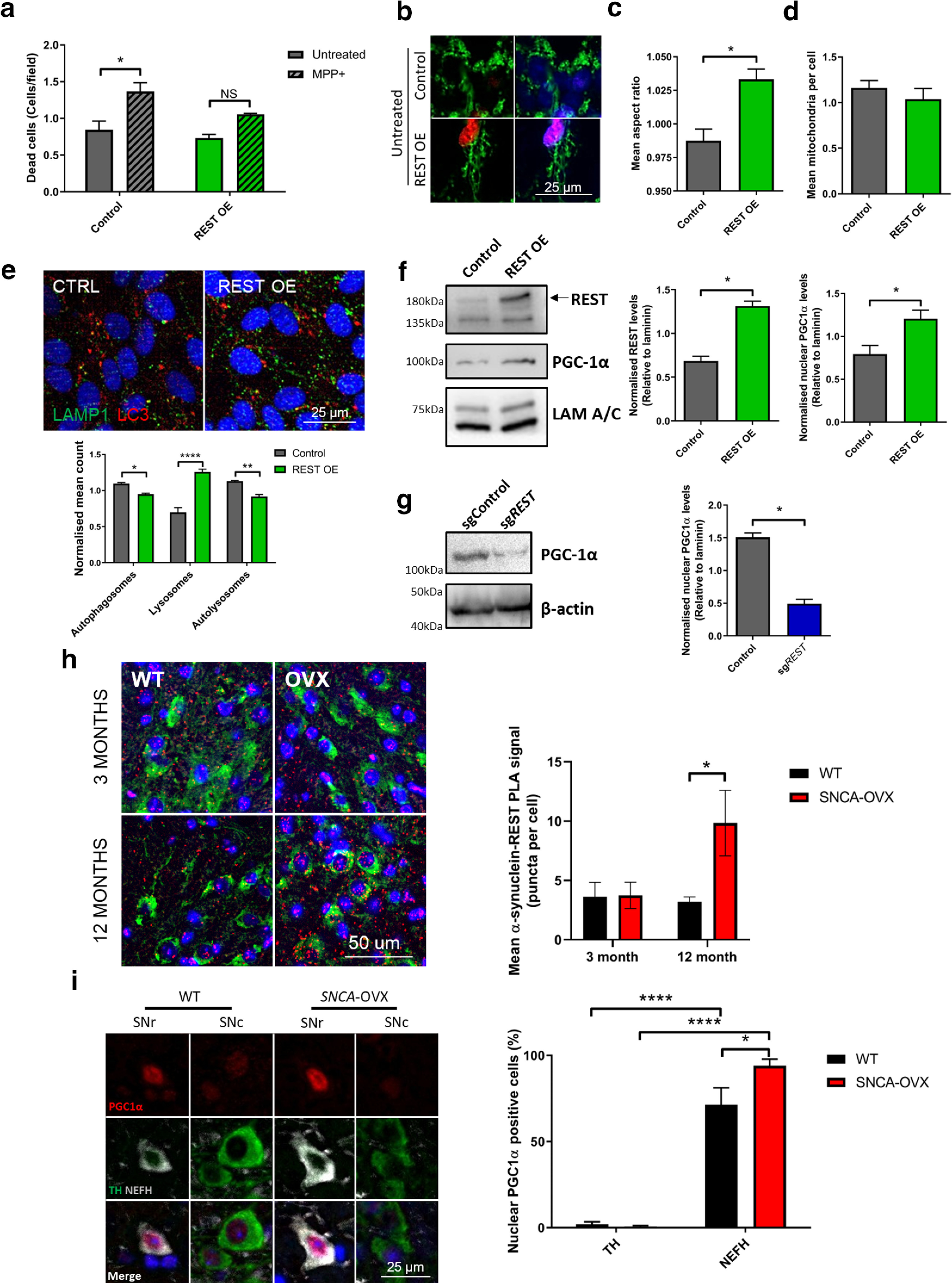
Overexpression of REST rescues cells from mitochondrial dysfunction through PGC-1α. a, REST overexpression inhibits MPP+-induced cell death as assessed by TO-PRO staining (N = 3). b, c, REST overexpression decreases mitochondrial fragmentation (N = 3). d, Autophagosome and lysosome content in REST-overexpressing cells. Quantification of LC3 puncta (red), LAMP1 puncta (green), or autolysosomes (yellow) (N = 3). e, f, Nuclear localization of PGC-1α after REST modulation. g, REST KO decreases PGC-1α in a polyclonal cell population. Polyclonal SH-SY5Y cells carrying control or sgREST were blotted for PGC-1α and β-actin (N = 3). h, Increased REST-α-syn interactions in aged mice, as assessed by PLA. i, PGC-1α is localized to the nucleus in NEFH, but not TH cells in SNCA-OVX mice. Nuclear localization of PGC-1α quantified in TH- or NEFH-positive cells in the midbrain of 12-month-old SNCA-OVX mice. *p < 0.05; **p < 0.01; ****p < 0.0001; two-way ANOVA Sidak. For further REST-α-syn interaction analysis and final model, see Extended Data Figure 6-1. (a) Two-way ANOVA, Sidak (F(1,8) = 23.35) p = 0.0013; (c) Mann–Whitney p = 0.085; (e) two-way ANOVA, Sidak (F(1,12) = 5.766) p = 0.0334; (f) Mann–Whitney p = 0.0006, p = 0.0213; (g) Mann–Whitney p = 0.0002; (h) two-way ANOVA, Sidak (F(1,13) = 3.211) p = 0.049; (i) two-way ANOVA, Sidak (F(1,12) = 236.2) p < 0.0001. NS = non significant.
a-d) PLA analysis of α-synuclein OE and MPP+ treatment in SHSY5Y cells. Cells were transfected with either empty or α-synuclein OE plasmids and treated with MPP+ 24 h post-transfection. Cells were analyzed by PLA (a-syn-PLA or REST-a-syn-PLA as indicated) at 4 DIV. e) Identification of preferential-vulnerability factors in dopaminergic neurons of the SNPc including lack of neuroprotective, nuclear REST. Download Figure 6-1, file (2.2MB, tif) .
We next tested the impact of REST overexpression on lysosomal and autophagic flux in starved cells. REST overexpression resulted in an increase in LAMP1 lysosomal puncta and a decrease in LC3+ autophagic vesicles (Fig. 6e), as well as a decrease in LAMP/LC3 double-positive autolysosomes, suggesting that REST overexpression could increase the efficiency of cargo degradation via upregulating lysosomal biogenesis.
We examined whether REST was able to stimulate the expression of the mitochondria biogenesis regulator/oxidative response transcription factor PGC-1α as a mechanism to elicit a neuroprotective role. We detected a significant upregulation of PGC-1α on REST overexpression, indicating that REST is able to elevate PGC-1α levels in the nucleus (Fig. 6f). Conversely, CRISPR-mediated reduction of REST levels resulted in decreased PGC-1α levels (Fig. 6g), suggesting that REST mediates its neuroprotective effect through PGC-1α upregulation.
Together with our previous findings and those of Kawamura et al. (2019), we hypothesized that α-synuclein oligomers might interact with REST. We therefore examined REST-α-synuclein interactions using a REST-AS-PLA. α-Synuclein overexpression in SH-SY5Y cells led to oligomer formation as measured by α-synuclein-PLA and also to increased REST-α-synuclein interactions as measured by REST-AS-PLA. In contrast, MPP+ treatment did not affect oligomer formation or REST-α-syn interactions in SH-S5Y5 cells (Extended Data Fig. 6-1a–d). This indicates that α-synuclein oligomerization, and not mitochondrial dysfunction, drives REST dysregulation. We then tested whether REST interacted with α-synuclein in WT or SNCA-OVX mice at 3 months (when no oligomers are present) or at 12 months (when increased oligomers are present). Interestingly, REST-AS-PLA was only upregulated in the dopaminergic neurons of aged SNCA-OVX animals, which indicated that α-syn oligomers are likely required for this interaction (Fig. 6h).
It has been previously demonstrated that loss of PGC-1α leads to an accumulation of α-synuclein oligomers (Eschbach et al., 2015). We therefore tested whether resistant nondopaminergic midbrain neuronal populations with increased REST expression and decreased α-synuclein oligomer burden exhibited enhanced nuclear PGC-1α expression. GABAergic neurons in the SNr expressed ∼10-fold more PGC-1α in their nucleus than dopaminergic neurons of the neighboring SNc (Fig. 6i). This difference was enhanced in SNCA-OVX mice, indicating that GABAergic, but not dopaminergic, neurons protect themselves from increased α-synuclein through activation of REST and PGC-1α.
Together, these results highlight that SNc dopaminergic neurons are made preferentially more vulnerable to insults by the absence of nuclear REST in a mouse model exemplifying early α-synuclein oligomer-driven Parkinsonian pathology (Fig. 7). In contrast, protected neurons in the SNr upregulate nuclear REST and PGC-1α in response to increased α-synuclein oligomer burden, reducing mitochondrial damage and increasing autophagic flux. Physiologically relevant increases in α-synculein inhibit REST and enhance the vulnerability of dopaminergic neurons, resulting in the accumulation of α-synuclein oligomers leading to mitchondrial damage, ROS production, and cell death. Overall, these data suggest that REST is a contributing neuroprotective factor protecting against mitochondrial dysfunction driven by α-synuclein oligomers but lacking in the nucleus of those neurons vulnerable in PD.
Figure 7.

REST regulates dopaminergic vulnerability through mitochondrial health and α-synuclein aggregation. Dopaminergic neurons of the SNc demonstrate decreased nuclear REST and an accumulation of α-synuclein oligomers, mitochondrial dysfunction, and oxidative stress. This is significantly exacerbated in SNCA-OVX mice resulting in preferential vulnerability compared with adjacent neurons in the SNr. In contrast, neurons of the SNr demonstrate increased nuclear REST and upregulation of PGC-1α and are protected from both α-synuclein oligomers and mitochondrial dysfunction.
Discussion
In the present study, we sought to explain the mechanisms by which the accumulation of soluble α-synuclein oligomers leads to bioenergetic dysfunction in SNCA-OVX mice. Using a proteomic approach, we identified mitochondrial dysfunction and mitochondrial ROS production as a prominent pathway dysregulated in SNCA-OVX mice occurring in conjunction with the inhibition of REST. We demonstrate that dopaminergic neurons of the SNc, unlike neighboring GABAergic neurons of the SNr, accumulate α-synuclein oligomers and show mitochondrial dysfunction in the absence of overt aggregation, suggesting that α-synuclein oligomers are a key driver of dopaminergic neuronal loss through mitochondrial dysfunction. Crucially, we show the absence of nuclear REST was a prominent feature of SNc neurons, whereas nuclear REST was present in the protected SNr neurons and upregulated during neurodegeneration in the SNr. Furthermore, REST was inhibited in SNCA-OVX mice and in iPSC-derived dopaminergic neurons from SNCA-Triplication PD patients (Fig. 7). Through CRISPR-KO and overexpression, we showed that REST protects cells from mitochondrial dysfunction and α-synuclein toxicity in vitro through the action of PGC-1α.
Together, these data demonstrate that α-synuclein overexpression leads to α-synuclein oligomerization and mitochondrial dysfunction. Furthermore, α-synuclein oligomers and dysfunctional mitochondria accumulate in vulnerable dopaminergic neurons of the SNc of SNCA-OVX transgenic animals, but not protected GABAergic neurons of the SNr, which inversely correlates with nuclear REST and PGC-1α. These observations highlight that lack of nuclear REST contributes to susceptibility, rendering dopaminergic neurons susceptible to mitochondrial or α-synuclein insult.
α-Synuclein oligomers have been shown to affect numerous cellular processes, such as mitochondrial function, ER stress, proteasome degradation, autophagy, and synaptic and membrane function. Oligomers have been shown to cause mitochondrial fragmentation (Plotegher et al., 2017) and to block lysosomal degradation in vitro (H. J. Lee et al., 2004). Thus, the accumulation of this α-synuclein species could potentially saturate clearance mechanisms in dopaminergic neurons, which have a high metabolic demand, relying heavily on mitochondrial respiration. The mitochondrial dysfunction observed in SNCA-OVX mice would be predicted to reduce mitochondrial ATP production and increase ROS production, as we observed. The excess production of mitochondrial ROS contributes to oxidative stress and damage of proteins and DNA, as observed in postmortem PD patients and exemplified in this study by the modification of GAPDH.
In addition, we found that α-synuclein binds TOM20 in the SNc of SNCA-OVX mice, consistent with work in postmortem human PD brain (Di Maio et al., 2016). Oligomers of α-synuclein have been shown to cause complex I dysfunction (Luth et al., 2014) and interact with a number of outer membrane proteins, including ATP synthase and the permeability transition pore (Ludtmann et al., 2018). These could be alternative or synergistic mechanisms for the α-synuclein oligomer-induced mitochondrial damage observed in SNCA-OVX animals.
Dopaminergic neurons of the SNc have specific anatomic and metabolic features that contribute to their early degeneration in PD. They have highly complex axonal arbors, which result in high energetic demand, exhibit damage associated with dopamine cytotoxicity, and have high levels of endogenous α-synuclein, which together increase ROS, α-synuclein aggregation, and toxicity (Burbulla et al., 2017; Plotegher et al., 2017; Surmeier, 2018; Alegre-Abarrategui et al., 2019). Here, we propose another contributing factor, the absence of a neuroprotective response mediated through REST. The inability of SNc dopaminergic neurons to upregulate nuclear REST, perhaps because of sequestration of REST by α-synuclein in the cytoplasm (Kawamura et al., 2019), represents a new contributing factor for preferential vulnerability of SNc dopaminergic neurons (Extended Data Fig. 6-1e). Given our observation of increased α-synuclein oligomerization in cells lacking REST, we propose a positive feedback loop whereby lack of nuclear REST causes increased mitochondrial dysfunction and increased α-synuclein oligomerization, which in turn sequesters REST in the cytoplasm.
While studies in AD, frontotemporal dementia, PD, and dementia with Lewy bodies have shown neuroprotective effects of REST (Lu et al., 2014; Kawamura et al., 2019), studies in Huntington's disease have shown that REST expression could lead to detrimental effects in the striatum (Buckley et al., 2010). These contradictory effects could be explained by dosage effects and by the nuclear role of REST. In the nucleus, REST acts as a master regulator, in part by repressing neuronal genes, which could cause neuroprotective effects by reducing neuronal activity-related stressors. However, high and sustained REST-induced repression of neuronal genes could result in detrimental effects by inhibiting critical neuronal functions, such as expression of BDNF (Zuccato and Cattaneo, 2007). A new role for REST in AD was reported recently by Meyer et al. (2019) who showed that REST was reduced in AD iPSC-derived neural progenitors, and that this resulted in increased neurogenesis and depletion of the progenitor pool. This highlights the need for careful evaluation of adult neurogenesis when considering any therapeutic interventions targeted to REST.
Kawamura et al. (2019) have shown that REST is sequestered in Lewy bodies in PD patients, inhibiting the entry of REST into the nucleus and therefore impairing REST function, providing independent validation of our observations in patients. Given the lack of Lewy body formation in our model, it could be interesting to speculate that lower-grade aggregates, such as oligomers, could also bind REST, inhibiting its DNA binding function and neuroprotective effects. Furthermore, Pajarillo et al. (2020) have shown that REST is neuroprotective against manganese-induced toxicity in immortalized cell lines. These observations by Kawamura et al. (2019) and Pajarillo et al. (2020), together with our own data here, suggest REST as a key mediator of α-synuclein, mitochondrial dysfunction and dopaminergic neuron vulnerability in PD.
Several mitochondrial and mitophagy-related processes are disrupted in PD (Pan et al., 2008; Ryan et al., 2015). We have shown that REST is able to protect cells from mitochondrial dysfunction induced by MPP+ and that REST improves basal mitochondrial morphology, as well as lysosomal and autophagic flux. Furthermore, we found that loss of REST resulted in decreased mitochondrial membrane potential and increased mitochondrial ROS production, consistent with mitochondrial uncoupling and the observed mitochondrial fragmentation. However, given the lack of significant change in cellular ATP levels, this suggests increased, but less efficient, mitochondrial oxygen consumption or compensation through increased glycolysis in cells lacking REST. Confirmation of which of these mechanisms are responsible for bioenergetic remodeling and a potential role for PGC-1α are further avenues for understanding the causal effects of REST. We found that REST expression upregulates nuclear PGC-1α, a master regulator of mitochondrial biogenesis. Two single nucleotide polymorphisms in the PGC-1a locus have been demonstrated to be linked to PD risk and age of onset (Clark et al., 2011). It has been shown previously that PGC-1α expression is protective for dopaminergic neurons in an MPTP mouse model (Mudò et al., 2012). Moreover, it has also been shown that downregulation of PGC-1α results in α-synuclein oligomeric accumulation (Ciron et al., 2015), suggesting that the absence of PGC-1α in the nucleus of dopaminergic neurons could contribute to the accumulation of α-synuclein oligomers. In addition, REST was able to upregulate autophagic flux in vitro, which is in agreement with the lower oligomeric content of SNr REST+ PGC-1α+ cells, suggesting that REST expression leads to the upregulation of a variety of neuroprotective mechanisms, including PPARγ. Our work highlights early mitochondrial pathology in an α-synuclein-overexpressing animal model that accumulates α-synuclein oligomers, which are key players in Parkinsonian neuropathology. Furthermore, we show a key role of REST in regulating dopaminergic preferential vulnerability through mitochondrial health and α-synuclein-aggregation in advanced Parkinsonian disease models. These data suggest REST, or downstream factors such as PGC-1α, as therapeutic targets in PD.
Footnotes
This work was supported Parkinson's UK Monument Trust Discovery Award J-1403. N.B.-V. and R.W.-M. were supported by the Medical Research Council UK Network of Centres of Excellence in Neurodegeneration MR/P007058/1. N.B.-V. was supported by an Oxford-BMS Fellowship. We thank Dr. Guillermo Abascal-Palacios for modeling of the nitration site on GAPDH.
The authors declare no competing financial interests.
References
- Alegre-Abarrategui J, Brimblecombe KR, Roberts RF, Velentza-Almpani E, Tilley BS, Bengoa-Vergniory N, Proukakis C (2019) Selective vulnerability in α-synucleinopathies. Acta Neuropathol 138:681–704. 10.1007/s00401-019-02010-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CE, Hammers DW, Deford JH, Dimayuga VL, Amaning JK, Farrar R, Papaconstantinou J (2011) Ischemia-reperfusion enhances GAPDH nitration in aging skeletal muscle. Aging (Albany NY) 3:1003–1017. 10.18632/aging.100394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2017) Alphα-synuclein oligomers: a new hope. Acta Neuropathol 134:819–838. 10.1007/s00401-017-1755-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4:484–494. 10.1038/nprot.2009.21 [DOI] [PubMed] [Google Scholar]
- Bogetofte H, Jensen P, Ryding M, Schmidt SI, Okarmus J, Ritter L, Worm CS, Hohnholt MC, Azevedo C, Roybon L, Bak LK, Waagepetersen H, Ryan BJ, Wade-Martins R, Larsen MR, Meyer M (2019) PARK2 mutation causes metabolic disturbances and impaired survival of human iPSC-derived neurons. Front Cell Neurosci 13:297. 10.3389/fncel.2019.00297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Göttgens B, Buckley NJ (2004) Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci USA 101:10458–10463. 10.1073/pnas.0401827101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NJ, Johnson R, Zuccato C, Bithell A, Cattaneo E (2010) The role of REST in transcriptional and epigenetic dysregulation in Huntington's disease. Neurobiol Dis 39:28–39. 10.1016/j.nbd.2010.02.003 [DOI] [PubMed] [Google Scholar]
- Burai R, Ait-Bouziad N, Chiki A, Lashuel HA (2015) Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson's disease via protein semisynthesis and mutagenesis. J Am Chem Soc 137:5041–5052. 10.1021/ja5131726 [DOI] [PubMed] [Google Scholar]
- Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Krüger R, Surmeier DJ, Krainc D (2017) Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science 357:1255–1261. 10.1126/science.aam9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Bian M, Liu M, Sheng Z, Suo H, Wang Z, Huang F, Fei J (2011) Ethanol-induced neurodegeneration in NRSF/REST neuronal conditional knockout mice. Neuroscience 181:196–205. 10.1016/j.neuroscience.2011.02.059 [DOI] [PubMed] [Google Scholar]
- Ching GY, Liem RK (2009) RE1 silencing transcription factor is involved in regulating neuron-specific expression of α-internexin and neurofilament genes. J Neurochem 109:1610–1623. 10.1111/j.1471-4159.2009.06052.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciron C, Zheng L, Bobela W, Knott GW, Leone TC, Kelly DP, Schneider BL (2015) PGC-1alpha activity in nigral dopamine neurons determines vulnerability to alphα-synuclein. Acta Neuropathol Commun 3:16. 10.1186/s40478-015-0200-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Reddy S, Zheng K, Betensky RA, Simon DK (2011) Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson's disease. BMC Med Genet 12:69. 10.1186/1471-2350-12-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK (2012) Accumulation of toxic alphα-synuclein oligomer within endoplasmic reticulum occurs in alphα-synucleinopathy in vivo. J Neurosci 32:3301–3305. 10.1523/JNEUROSCI.5368-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T (2011) Parkinson's disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun 2:440. 10.1038/ncomms1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, Hastings TG, Greenamyre JT (2016) α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med 8:342ra78. 10.1126/scitranslmed.aaf3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson PD, Dreyer JK, Jennings KA, Syed EC, Wade-Martins R, Cragg SJ, Bolam JP, Magill PJ (2016) Representation of spontaneous movement by dopaminergic neurons is cell-type selective and disrupted in parkinsonism. Proc Natl Acad Sci USA 113:E2180–E2188. 10.1073/pnas.1515941113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschbach J, von Einem B, Muller K, Bayer H, Scheffold A, Morrison BE, Rudolph KL, Thal DR, Witting A, Weydt P, Otto M, Fauler M, Liss B, McLean PJ, Spada AR, Ludolph AC, Weishaupt JH, Danzer KM (2015) Mutual exacerbation of peroxisome proliferator-activated receptor gamma coactivator 1alpha deregulation and alphα-synuclein oligomerization. Ann Neurol 77:15–32. 10.1002/ana.24294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF (2004) Early and progressive sensorimotor anomalies in mice overexpressing wild-type human α-synuclein. J Neurosci 24:9434–9440. 10.1523/JNEUROSCI.3080-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguère N, Burke Nanni S, Trudeau LE (2018) On cell loss and selective vulnerability of neuronal populations in Parkinson's disease. Front Neurol 9:455. 10.3389/fneur.2018.00455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila RE, Manzino L, Cabbat FS, Duvoisin RC (1984) Protection against the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine by monoamine oxidase inhibitors. Nature 311:467–469. 10.1038/311467a0 [DOI] [PubMed] [Google Scholar]
- Ho PW, Leung CT, Liu H, Pang SY, Lam CS, Xian J, Li L, Kung MH, Ramsden DB, Ho SL (2020) Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 16:347–370. 10.1080/15548627.2019.1603545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunn BH, Vingill S, Threlfell S, Alegre-Abarrategui J, Magdelyns M, Deltheil T, Bengoa-Vergniory N, Oliver PL, Cioroch M, Doig NM, Bannerman DM, Cragg SJ, Wade-Martins R (2019) Impairment of macroautophagy in dopamine neurons has opposing effects on parkinsonian pathology and behavior. Cell Rep 29:920–931.e927. 10.1016/j.celrep.2019.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B, Deltheil T, Kosillo P, Cioroch M, Wagner K, Ansorge O, Bannerman DM, Bolam JP, Magill PJ, Cragg SJ, Wade-Martins R (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci USA 110:4016–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins JL, Tanner JJ (2006) High-resolution structure of human D-glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr D Biol Crystallogr 62:290–301. 10.1107/S0907444905042289 [DOI] [PubMed] [Google Scholar]
- Kawamura M, Sato S, Matsumoto G, Fukuda T, Shiba-Fukushima K, Noda S, Takanashi M, Mori N, Hattori N (2019) Loss of nuclear REST/NRSF in aged-dopaminergic neurons in Parkinson's disease patients. Neurosci Lett 699:59–63. 10.1016/j.neulet.2019.01.042 [DOI] [PubMed] [Google Scholar]
- Keane H, Ryan BJ, Jackson B, Whitmore A, Wade-Martins R (2015) Protein-protein interaction networks identify targets which rescue the MPP+ cellular model of Parkinson's disease. Sci Rep 5:17004. 10.1038/srep17004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A (2002) Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J Neurosci 22:2780–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Tepper JM (2007) Morphological and physiological properties of parvalbumin- and calretinin-containing gamma-aminobutyric acidergic neurons in the substantia nigra. J Comp Neurol 500:958–972. 10.1002/cne.21220 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ (2004) Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 24:1888–1896. 10.1523/JNEUROSCI.3809-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Ma J, Luo C, Mo X, Sun L, Zang W, Liu J (2009) Comparison of two methods for assaying complex I activity in mitochondria isolated from rat liver, brain and heart. Life Sci 85:276–280. 10.1016/j.lfs.2009.05.019 [DOI] [PubMed] [Google Scholar]
- Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, Bennett DA, Colaiacovo MP, Yankner BA (2014) REST and stress resistance in ageing and Alzheimer's disease. Nature 507:448–454. 10.1038/nature13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtmann MH, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B, Al-Menhali AS, Ranasinghe RT, Whiten DR, Yapom R, Dolt KS, Devine MJ, Gissen P, Kunath T, Jaganjac M, Pavlov EV, et al. (2018) α-Synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson's disease. Nat Commun 9:2293. 10.1038/s41467-018-04422-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ (2014) Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem 289:21490–21507. 10.1074/jbc.M113.545749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–221. 10.1083/jcb.200910140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Feldman HM, Lu T, Drake D, Lim ET, Ling KH, Bishop NA, Pan Y, Seo J, Lin YT, Su SC, Church GM, Tsai LH, Yankner BA (2019) REST and neural gene network dysregulation in iPSC models of Alzheimer's disease. Cell Rep 26:1112–1127.e1119. 10.1016/j.celrep.2019.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R, James-Kracke M, Sun G, Sun A (2009) Oxidative and inflammatory pathways in Parkinson's disease. Neurochem Res 34:55–65. 10.1007/s11064-008-9656-2 [DOI] [PubMed] [Google Scholar]
- Mudò G, Mäkelä J, Liberto VD, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Mälkiä A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D (2012) Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci 69:1153–1165. 10.1007/s00018-011-0850-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajarillo E, Rizor A, Son DS, Aschner M, Lee E (2020) The transcription factor REST up-regulates tyrosine hydroxylase and antiapoptotic genes and protects dopaminergic neurons against manganese toxicity. J Biol Chem 295:3040–3054. 10.1074/jbc.RA119.011446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamalai V, Miyagi M (2010) Mechanism of glyceraldehyde-3-phosphate dehydrogenase inactivation by tyrosine nitration. Protein Sci 19:255–262. 10.1002/pro.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, Kondo S, Le W, Jankovic J (2008) The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain 131:1969–1978. 10.1093/brain/awm318 [DOI] [PubMed] [Google Scholar]
- Plotegher N, Gratton E, Bubacco L (2014) Number and Brightness analysis of alphα-synuclein oligomerization and the associated mitochondrial morphology alterations in live cells. Biochim Biophys Acta 1840:2014–2024. 10.1016/j.bbagen.2014.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher N, Berti G, Ferrari E, Tessari I, Zanetti M, Lunelli L, Greggio E, Bisaglia M, Veronesi M, Girotto S, Dalla Serra M, Perego C, Casella L, Bubacco L (2017) DOPAL derived alphα-synuclein oligomers impair synaptic vesicles physiological function. Sci Rep 7:40699. 10.1038/srep40699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prots I, Veber V, Brey S, Campioni S, Buder K, Riek R, Bohm KJ, Winner B (2013) α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem 288:21742–21754. 10.1074/jbc.M113.451815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan CL, Perevoschikova IV, Goncalves RL, Hey-Mogensen M, Brand MD (2013) The determination and analysis of site-specific rates of mitochondrial reactive oxygen species production. Methods Enzymol 526:189–217. 10.1016/B978-0-12-405883-5.00012-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Park TK, Jaros E, Campbell GR, Lax NZ, Hepplewhite PD, Krishnan KJ, Elson JL, Morris CM, McKeith IG, Turnbull DM (2012) Relationship between mitochondria and α-synuclein: a study of single substantia nigra neurons. Arch Neurol 69:385–393. 10.1001/archneurol.2011.2675 [DOI] [PubMed] [Google Scholar]
- Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2015) Direct visualization of alphα-synuclein oligomers reveals previously undetected pathology in Parkinson's disease brain. Brain 138:1642–1657. 10.1093/brain/awv040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas G, López-Martín E, Guerra MJ, Labandeira-García JL (1998) The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J Neurosci Methods 83:165–175. 10.1016/s0165-0270(98)00078-8 [DOI] [PubMed] [Google Scholar]
- Ryan BJ, Hoek S, Fon EA, Wade-Martins R (2015) Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci 40:200–210. 10.1016/j.tibs.2015.02.003 [DOI] [PubMed] [Google Scholar]
- Samii A, Nutt JG, Ransom BR (2004) Parkinson's disease. Lancet 363:1783–1793. 10.1016/S0140-6736(04)16305-8 [DOI] [PubMed] [Google Scholar]
- Surmeier DJ (2018) Determinants of dopaminergic neuron loss in Parkinson's disease. FEBS J 285:3657–3668. 10.1111/febs.14607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegeder I, Costigan M, Griffin RS, Abele A, Belfer I, Schmidt H, Ehnert C, Nejim J, Marian C, Scholz J, Wu T, Allchorne A, Diatchenko L, Binshtok AM, Goldman D, Adolph J, Sama S, Atlas SJ, Carlezon WA, Parsegian A, et al. (2006) GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med 12:1269–1277. 10.1038/nm1490 [DOI] [PubMed] [Google Scholar]
- Vingill S, Connor-Robson N, Wade-Martins R (2018) Are rodent models of Parkinson's disease behaving as they should? Behav Brain Res 352:133–141. 10.1016/j.bbr.2017.10.021 [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Tanji K, Mori F, Takahashi H (2007) The Lewy body in Parkinson's disease: molecules implicated in the formation and degradation of alphα-synuclein aggregates. Neuropathology 27:494–506. 10.1111/j.1440-1789.2007.00803.x [DOI] [PubMed] [Google Scholar]
- Zambon F, Cherubini M, Fernandes HJ, Lang C, Ryan BJ, Volpato V, Bengoa-Vergniory N, Vingill S, Attar M, Booth HD, Haenseler W, Vowles J, Bowden R, Webber C, Cowley SA, Wade-Martins R (2019) Cellular alphα-synuclein pathology is associated with bioenergetic dysfunction in Parkinson's iPSC-derived dopamine neurons. Hum Mol Genet 28:2001–2013. 10.1093/hmg/ddz038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grünblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wüllner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, et al. (2010) PGC-1alpha, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med 2:52ra73. 10.1126/scitranslmed.3001059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Lee CR (2011) Intrinsic and integrative properties of substantia nigra pars reticulata neurons. Neuroscience 198:69–94. 10.1016/j.neuroscience.2011.07.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E (2007) Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol 81:294–330. 10.1016/j.pneurobio.2007.01.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a) Relative α-synuclein expression in midbrain lysates from WT and SNCA-OVX mice. b) Assessment of total and TH-cell counts in mouse nigra as assessed by stereology. c&d) levels of TH protein in midbrain of c) 3 month and d) 18-month-old mice as assessed by western blot (N = 3). e-g) α-synuclein oligomers are increased in the SNc of aged SNCA-OVX mice. α-Synuclein oligomers were detected by PLA staining in the SNc of α-synuclein KO, human α-synuclein (hα-syn) or SNCA-OVX mice at f) 3 or g) 24 months (p < 0.05, one-way ANOVA, Holm-Sidak; N = 3) h-q) No change in α-synuclein oligomers in the brain regions from 3 or 24-month-old SNCA-OVX and control mice (p > 0.05, one-way ANOVA, Holm-Sidak; N = 3). Download Figure 1-1, TIF file (1.1MB, tif) .
a) Protein abundance distribution in dimethyl labeling proteomics dataset (M/L ratio) dotted lines (black) indicate cutoff of proteins taken forward into pathway analysis. b) Top up and down regulated proteins identified in SNCA-OVX mice by proteomics. c) Percentage of mitochondrial subunits of each mitochondrial complex quantified by proteomics. d) Midbrain REST target gene expression analysis by qPCR of aged Wt and SNCA-OVX animals. Download Figure 1-2, TIF file (961.3KB, tif) .
a&b) Mitochondrial complex I activity in a) 18-month cortex and b) whole brain lysates from aged SNCA-OVX mice. c) TOM20 and d) Non-phosphorylated DRP protein levels in aged midbrain, normalized to B-actin. e) Phosphorylated DRP levels in 3-month-old mouse midbrain, normalized to B-actin. f) COX IV protein levels in aged midbrain normalized to B-actin (N = 3). × g&h) Levels of lipidated LC3 (LC3-II) in g) midbrain lysates from 3-month-old mice (N = 3) and h) cortex lysates from 18-month-old mice (N = 4) normalized to β-actin levels. i-k) Levels of BIP in SNCA-OVX mice i&j) Midbrain lysates from i) 18-month-old, j) 3-month-old midbrain and k) 18-month-old cortex from OVX mice normalized to β-actin levels (N = 3). l) Quantification 3-NT band excised for LC-MS/MS analysis. Band indicated by red arrow in Figure 1k. Download Figure 2-1, TIF file (974.8KB, tif) .
Mitochondrial morphology, as assessed by TOM20 staining, in aging mice demonstrating a lack of mitochondrial network in 18-month-old tissue. Download Figure 4-1, TIF file (1.1MB, tif) .
Relative expression of REST target genes in iPSC-derived dopaminergic neuronal cultures from patients with α-synuclein triplication a) dataset from Zambon et al., 2019; (Zambon et al., 2019). b-d) Basal TOM20 morphology in Cas9 expressing SH-SY5Y cells expressing sgRNA targeting REST or a scrambled sgRNA showing c) Mitochondrial AR and d) mitochondrial number. e) Images of mitophagy (as assessed by mtphagy dye; red) in Cas9 expressing SH-SY5Y cells (green) expressing sgRNA targeting REST or a scrambled sgRNA (images duplicated from Figure 5i for comparison). f-g) Quantification of mitophagy in Cas9(GFP) expressing cells as assessed by mtphagy in f) Oligomycin-treated cells and g) CCCP-treated cells. h) ATP levels in polyclonal REST KO cells (n = 4). i&j) Mitochondrial ROS production in polyclonal REST KO cells under basal and rotenone-stimulated cells (n = 6). Download Figure 5-1, TIF file (2.7MB, tif) .
a-d) PLA analysis of α-synuclein OE and MPP+ treatment in SHSY5Y cells. Cells were transfected with either empty or α-synuclein OE plasmids and treated with MPP+ 24 h post-transfection. Cells were analyzed by PLA (a-syn-PLA or REST-a-syn-PLA as indicated) at 4 DIV. e) Identification of preferential-vulnerability factors in dopaminergic neurons of the SNPc including lack of neuroprotective, nuclear REST. Download Figure 6-1, file (2.2MB, tif) .