

SUMMARY
Neural network dysfunction may play an important role in Alzheimer’s disease (AD). Neuronal circuits vulnerable to AD are also affected in human amyloid precursor protein (hAPP) transgenic mice. hAPP mice with high levels of amyloid-β peptides in the brain develop AD-like abnormalities, including cognitive deficits and depletions of calcium-related proteins in the dentate gyrus, a region critically involved in learning and memory. Here we report that hAPP mice have spontaneous non-convulsive seizure activity in cortical and hippocampal networks, which is associated with GABAergic sprouting, enhanced synaptic inhibition, and synaptic plasticity deficits in the dentate gyrus. Many Aβ-induced neuronal alterations could be simulated in nontransgenic mice by excitotoxin challenge, and prevented in hAPP mice by blocking overexcitation. Aberrant increases in network excitability and compensatory inhibitory mechanisms in the hippocampus may contribute to Aβ-induced neurological deficits in hAPP mice and, possibly, also in humans with AD.
INTRODUCTION
The hippocampus plays key roles in learning and memory (Burgess et al., 2002; Frankland and Bontempi, 2005; Morris et al., 1982; O’Keefe et al., 1975) and is a prime target of Alzheimer’s disease (AD), which causes progressive memory impairments (Blennow et al., 2006). Although much is known about the neural circuits and molecular pathways required for normal hippocampal functions, the processes by which AD disables this brain region remain to be defined. We recently postulated that destabilization of neuronal network activity may contribute to cognitive impairments associated with AD, and hypothesized that such destabilization may result, at least in part, from aberrant alterations in neuronal network activity and resulting compensatory responses (Palop et al., 2006).
A key molecule that may perturb network activity in AD is the amyloid-β (Aβ) peptide, which is derived from the amyloid precursor protein (APP) by proteolytic cleavage. The excessive accumulation of pathogenic Aβ assemblies in the brain appears to play a causal role in AD (Glenner and Wong, 1984; Tanzi and Bertram, 2005; Walsh and Selkoe, 2004). In strong support of this notion, neuronal expression of human APP (hAPP) and Aβ in transgenic mice elicits several AD-like abnormalities, including amyloid plaques, neuritic dystrophy, aberrant sprouting of axon terminals, functional and structural synaptic deficits, impairments in learning and memory, and other behavioral alterations (Chin et al., 2005; Chin et al., 2004; Games et al., 1995; Götz et al., 2004; Kobayashi and Chen, 2005; Palop et al., 2005; Palop et al., 2003).
The relevance of these transgenic mouse models to the human condition is further underlined by the fact that their impairments do not affect the entire brain but primarily those regions that are particularly vulnerable to AD, including the hippocampus. Aβ-dependent deficits in hippocampal learning and memory correlate well with alterations in calcium- and synaptic activity-related proteins in granule cells of the dentate gyrus (Chin et al., 2005; Palop et al., 2005; Palop et al., 2003), a brain region critically involved in learning and memory (Singer et al., 2005). These functionally relevant molecular alterations include reductions in the calcium-binding protein calbindin-D28K, the immediate-early gene products Arc and Fos, the dendritic spine actin-binding protein α-actinin-II, and the phosphorylation states of the NMDA receptor subunit NR2B and the MAP kinases ERK1/2, as well as increases in the α7 nicotinic acetylcholine receptor and striatal-enriched phosphatase. The levels of these molecules are affected to different degrees in different familial AD-mutant hAPP (hAPPFAD) mice but to very similar degrees in any given hAPPFAD mouse, suggesting a common underlying mechanism (Chin et al., 2005; Palop et al., 2005; Palop et al., 2003).
Notably, several of these dentate alterations have also been observed in humans with AD (Palop et al., 2003), humans with epilepsy (Nägerl et al., 2000), and animal models with chronic aberrant increases in excitatory neuronal activity (overexcitation) (Marksteiner et al., 1990; Peng and Houser, 2005; Tonder et al., 1994a; Tonder et al., 1994b). These findings suggest that the dentate alterations of hAPPFAD mice and humans with AD may be caused, at least in part, by aberrant increases in neuronal activity, consistent with the increased risk of seizure activity in sporadic and familial AD and with the excitotoxicity hypothesis of neurodegenerative disorders (for review, Amatniek et al., 2006; Mark et al., 1995; Mattson et al., 1993; Olney, 1995; Rothman and Olney, 1995; Snider et al., 2005). However, Aβ decreases glutamatergic synaptic transmission, at least at certain synapses, presumably through a mechanism that involves the endocytosis of AMPA receptors and subsequent collapse of dendritic spines (Hsia et al., 1999; Hsieh et al., 2006; Kamenetz et al., 2003; Shankar et al., 2007; Walsh et al., 2002). Therefore, the depletion of calcium- and synaptic activity-related proteins in the dentate gyrus of hAPPFAD mice may also be caused by the suppression of overall network activity. Because Aβ might act differentially on neuronal subtypes and brain regions, it is unclear whether the net effect of Aβ on specific networks is excitatory, inhibitory, or combines elements of both.
To explore whether Aβ-dependent neuronal alterations associated with cognitive deficits are more closely related to overexcitation or suppression of neuronal activity in cortical and hippocampal networks, we analyzed four lines of transgenic mice expressing FAD-mutant or wildtype (WT) hAPP. Compared with nontransgenic (NTG) mice and with hAPPWT transgenic mice, hAPPFAD mice with high levels of Aβ showed anatomical and biochemical alterations in the dentate gyrus that are indicative of aberrant excitatory neuronal activity. Electroencephalographic (EEG) recordings in hAPPFAD mice revealed intermittent epileptiform cortical and hippocampal synchronous discharges and generalized non-convulsive seizure activity. These abnormalities were associated with a prominent remodeling of inhibitory circuits, increased synaptic inhibition, and synaptic plasticity deficits in the dentate gyrus. Many of these alterations could be simulated in nontransgenic (NTG) mice by systemic injection with kainate, and prevented in both hAPPFAD mice and kainate-treated NTG mice by removal of Tau, a genetic manipulation that protects against excitotoxin-induced seizure activity (Roberson et al., 2007). Our data demonstrate that hAPP/Aβ induces aberrant excitatory neuronal network activity in vivo and triggers compensatory inhibitory mechanisms in hippocampal circuits. Both the aberrant excitatory neuronal activity and the compensatory inhibitory mechanisms may limit the capacity for synaptic plasticity and contribute to AD-related network dysfunction.
RESULTS
Remodeling of neuronal circuits in the dentate gyrus
Axonal sprouting of GABAergic interneurons and alterations in the expression of neuropeptide Y (NPY) are sensitive indicators of imbalances between excitatory and inhibitory neuronal activities in the hippocampus (for review, Vezzani et al., 1999). Compared with NTG controls, hAPPFAD mice from the high expresser J20 line (hAPP-J20 mice) showed ectopic expression of NPY in the molecular layer of the dentate gyrus and in the axons of granule cells (mossy fibers) (Fig. 1A). Double-labeling for NPY and calbindin revealed that these alterations were associated with depletions of calbindin in granule cells (Fig. 1B–D), which correlate with deficits in learning and memory in this model and are also seen in humans with AD (Palop et al., 2003). Compared with NTG controls, hAPP-J20 mice showed a marked increase in NPY mRNA levels in granule cells (Fig. 1E), suggesting that the ectopic presence of NPY in mossy fibers was caused by increased NPY gene expression in these neurons. In contrast, the NPY-positive sprouting in the molecular layer more likely emanated from hilar NPY- and somatostatin (SOM)-positive GABAergic interneurons (Bakst et al., 1986; Freund and Buzsáki, 1996; Vezzani et al., 1999), since these fibers were also immunoreactive for somatostatin (Fig. 1F, G) and failed to stain with the Timm method, which detects the high levels of zinc present in mossy fibers (not shown). Because NPY- and SOM-positive GABAergic hilar interneurons inhibit the dendrites of granule cells (Freund and Buzsáki, 1996), the inhibitory sprouting would be expected to increase inhibition of granule cells. Dentate mRNA levels for NPY-Y1 and -Y2 receptors were determined by quantitative fluorogenic RT-PCR. Compared with NTG mice, Y1 receptors, which exert weak excitatory activity of NPY (Vezzani et al., 1999), were decreased in hAPP-J20, whereas Y2 receptors, which mediate potent inhibitory activity of NPY (Vezzani et al., 1999), were increased (Fig. 1H).
Figure 1. Remodeling of inhibitory circuits and alterations in the expression of NPY and its receptors in the dentate gyrus of hAPP-J20 mice.

(A) Brain sections were immunoperoxidase-stained for NPY. Compared with NTG controls, hAPP-J20 mice had an increase in NPY expression in the molecular layer of the dentate gyrus (arrow) and in the mossy fibers (arrowhead). (B) Confocal microscopic imaging of sections double-labeled for calbindin (red) and NPY (green) demonstrated sprouting of NPY axons in the molecular layer (arrow), ectopic expression of NPY in mossy fibers (arrowheads), and severe depletion of calbindin (CB) in granule cells of an hAPP-J20 mouse. Images represent the regions indicated by squares in panel A. (C–D) Calbindin depletions correlated with NPY increases in the dentate gyrus (C) and mossy fibers (D) in hAPP-J20 mice. (E) In situ hybridization revealed ectopic expression of NPY mRNA in granule cells (arrow) of an hAPP-J20 mouse (bottom panel). (F) Compared with NTG mice, hAPP-J20 mice had sprouting of somatostatin (SOM)-positive axons in the molecular layer (top panels), which was quantitated by densitometry (bottom panel). (G) Axonal sprouts in the molecular layer of hAPP-J20 mice were double-labeled for NPY (green) and SOM (red), indicating that they originated from hilar GABAergic cells. (H) NPY receptors in the dentate gyrus were also altered in hAPP-J20 mice, as determined by quantitative fluorogenic RT-PCR: Y1 receptors were decreased, whereas Y2 receptors were increased compared with NTG controls. Numbers in bars indicate number of mice per group. *P<0.05, **P<0.01, ***P<0.001 vs. NTG by Student’s t test.
To exclude the possibility that the NPY alterations in hAPP-J20 mice resulted from hAPP overexpression or from a transgene insertion effect, we analyzed additional hAPPWT (hAPP-I5) and hAPPFAD (hAPP-ARC48 and hAPP-J9/FYN) transgenic lines. (Cheng et al., 2004; Cheng et al., 2007; Chin et al., 2005; Chin et al., 2004; Mucke et al., 2000). NPY expression in hAPP-I5 mice, which have similar hAPP levels as hAPP-J20 mice but much lower levels of Aβ, was indistinguishable from that in NTG controls (Supplemental Fig. 1A), excluding a direct effect of hAPP overexpression on these alterations. The two additional hAPPFAD lines showed prominent increases in NPY expression (Supplemental Fig. 1B–E), demonstrating that this alteration is not restricted to hAPP-J20 mice.
Ectopic expression of NPY in mossy fibers has previously been identified in aged hAPPFAD mice, and was ascribed to late amyloid deposition (Diez et al., 2003; Diez et al., 2000). However, we found that the magnitude of NPY elevation was similar in the three independent hAPPFAD lines, even though they have vastly different plaque loads (Supplemental Fig. 1F and Cheng et al., 2004; Chin et al., 2005; Palop et al., 2003). Thus, it is very likely that NPY and calbindin alterations in hAPPFAD mice are unrelated to amyloid deposition.
Circuit alterations that distinguish hAPPFAD mice from epilepsy models
The GABAergic sprouting and NPY alterations we identified in different hAPPFAD lines have also been observed in animal models with chronic epilepsy and aberrant increases in neuronal activity (for review, Vezzani et al., 1999). We therefore examined whether hAPP-J20 mice resemble epilepsy models also with respect to circuit alterations that are widely presumed to promote epileptogenesis: recurrent excitatory sprouting of collateral mossy fibers in the inner molecular layer and loss of inhibitory interneurons in the hilus (Cobos et al., 2005; de Lanerolle et al., 1989; Mathern et al., 1995). We found no evidence for recurrent mossy fiber sprouting in the inner molecular layer of hAPP-J20 mice either by NPY immunostaining (Fig. 1 A, B) or by Timm staining (Fig. 2A). Instead, there was an increase of collateral mossy fibers crossing the granule cell layer (Fig. 2A, B). Double-labeling for collateral mossy fibers (Timm) and markers of GABAergic basket cells (parvalbumin or NPY) revealed that the Timm-positive sprouting was specifically and heavily decorating somas and proximal dendrites of basket cells (Fig. 2C). Because the GABAergic basket cells inhibit the granule cells (Freund and Buzsáki, 1996), increased excitatory mossy fiber innervation of basket cells would be expected to enhance inhibition of granule cells. In a similar vein, quantification of the number of somatostatin-positive interneurons in the hilus throughout the rostrocaudal extent of the hippocampus showed no significant difference between NTG and hAPP-J20 mice (Fig. 2D, E). Thus, the dentate gyrus of hAPP-J20 mice exhibits circuit alterations that are widely presumed to enhance inhibition but lacks those that promote hippocampal epileptogenesis, suggesting an incomplete mechanistic overlap with models of epilepsy.
Figure 2. Aberrant innervation of inhibitory basket cells by glutamatergic mossy fiber collaterals.

Brain sections from hAPP-J20 and NTG mice were stained with the Timm method to detect glutamatergic synaptic vesicles in collateral mossy fibers, and/or immunostained for the GABAergic basket cell markers parvalbumin (PV) or NPY. (A) Sprouting of mossy fiber collaterals in the dentate gyrus was evident in hAPP-J20 mice but not in the NTG controls. Note the increase of Timm-positive axons in the granular layer (arrow) and the Timm-positive clusters in the inner molecular layer (arrowhead) of the hAPP-J20 mouse. (B) Quantitation of Timm-positive collateral mossy fibers in the granular layer of hAPP-J20 and NTG mice. (C) Collateral mossy fibers outlined specific cells in the granular layer of hAPP-J20 mice (left). Double-labeling for Timm/PV (center) or Timm/NPY (right) identified these cells as basket cells. (D) Somatostatin (SOM)-positive GABAergic interneurons (arrow) in the hilus of the dentate gyrus in an hAPP-J20 mouse and a NTG control. (E) Quantitation of SOM-positive interneurons in the hilus. Data represent the mean (± SEM) number of cells at different rostrocaudal levels. **P<0.01 vs. NTG by Student’s t test.
Increased inhibition in granule cells of hAPP-J20 mice
In models of epilepsy, synaptic inhibition in the dentate gyrus is compromised primarily due to the loss of hilar NPY- and SOM-positive GABAergic cells (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005). To test whether the remodeling of inhibitory hippocampal circuits in hAPP-J20 mice is associated with increased inhibition in granule cells, we performed whole-cell voltage-clamp recordings to isolate miniature inhibitory postsynaptic currents (mIPSCs). We found that the frequency of mIPSCs recorded from dentate granule cells was significantly increased in hAPP-J20 mice relative to NTG controls (Fig. 3A–C). Although mIPSC amplitudes were not significantly different between NTG and hAPP-J20 mice when all events were averaged (data not shown), the cumulative probability plot demonstrated that large-amplitude events were enhanced in hAPP-J20 mice (Fig. 3D). When the largest 25% of events were averaged, there was a significant difference in mIPSC amplitude between NTG and hAPP-J20 mice (Fig. 3E). The increases in mIPSC frequency and large-amplitude events observed in granule cells of hAPP-J20 mice suggest an increase in the number of functional GABAergic synapses onto the granule cells and may reflect enhanced presynaptic GABA release, enhanced postsynaptic GABA sensitivity, or both.
Figure 3. Increase in mIPSC frequency and large-amplitude mIPSCs in dentate gyrus slices from hAPP-J20 mice.
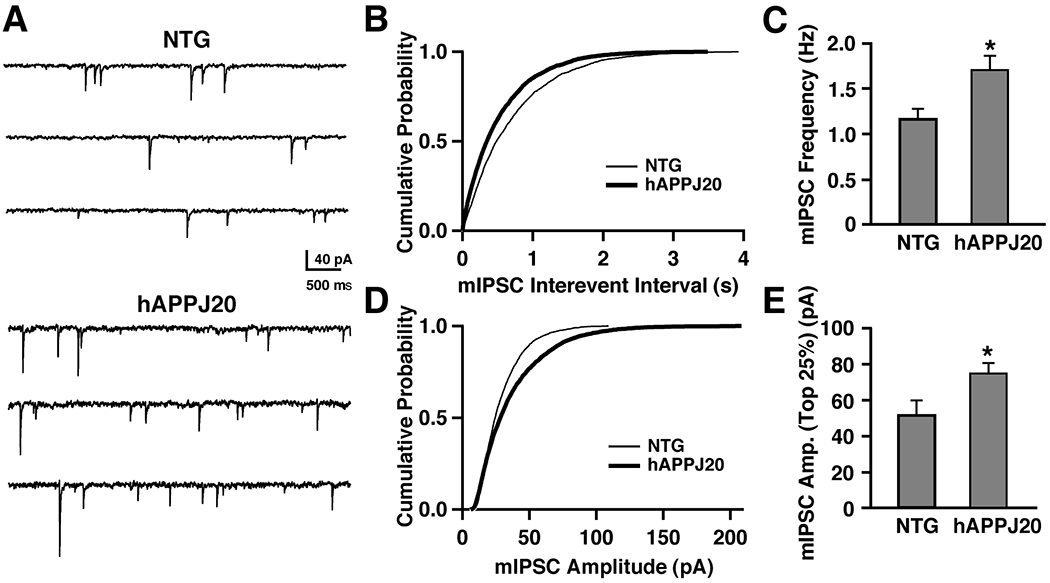
(A) Representative traces of mIPSCs recorded from NTG (top) and hAPP-J20 (bottom) mice. (B) Average cumulative probability plot of mIPSC interevent intervals for NTG (thin line, n = 7 cells, 3 mice) and hAPP-J20 (thick line, n = 10 cells, 3 mice) mice shows decreased interevent intervals in hAPP-J20 mice. (C) mIPSC frequency was significantly increased in hAPP-J20 mice (NTG, n=7 cells, 3 mice; hAPP-J20, n=10 cells, 3 mice). (D) Average cumulative probability plot of mIPSC amplitudes recorded from NTG (thin line, n = 7 cells, 3 mice) and hAPP-J20 (thick line, n = 10 cells, 3 mice) mice shows enhanced number of large-amplitude mIPSCs. (E) The largest 25% of mIPSCs were increased in amplitude in the hAPP-J20 mice. (n=10 cells, 3 mice) relative to NTG controls (n=7 cells, 3 mice). *P<0.05 by Student’s t test.
Pharmacological induction of neuronal overexcitation in NTG mice triggers molecular and anatomical alterations resembling those observed in untreated hAPP-J20 mice
The circuit alterations we identified in the dentate gyrus of hAPPFAD mice may be typical responses of hippocampal networks to aberrant increases in neuronal activity. To begin to address this possibility, we used the excitatory amino acid kainate to elicit aberrant increases in neuronal activity in NTG mice. Three days after receiving an IP injection of kainate or saline, NTG mice were analyzed for a number of neuronal alterations known to be affected in untreated hAPP-J20 mice and hAPP-J9/FYN mice (this study and Chin et al., 2005; Palop et al., 2005; Palop et al., 2003). Kainate-treated NTG mice showed dose-dependent increases in NPY and depletions of Arc-positive granule cells in the dentate gyrus (Fig. 4A–D). Levels of STEP, α7 nAchR, and calbindin (Fig. 4E) were also altered in the same direction as in untreated hAPP-J20 and hAPP-J9/FYN mice (Chin et al., 2005). Thus, a single excitotoxin challenge was sufficient to trigger in NTG mice many of the alterations observed in the dentate gyrus of hAPPFAD mice at baseline.
Figure 4. Pharmacological induction of neuronal overexcitation in NTG mice triggers molecular and anatomical alterations resembling those observed in untreated hAPPFAD mice.
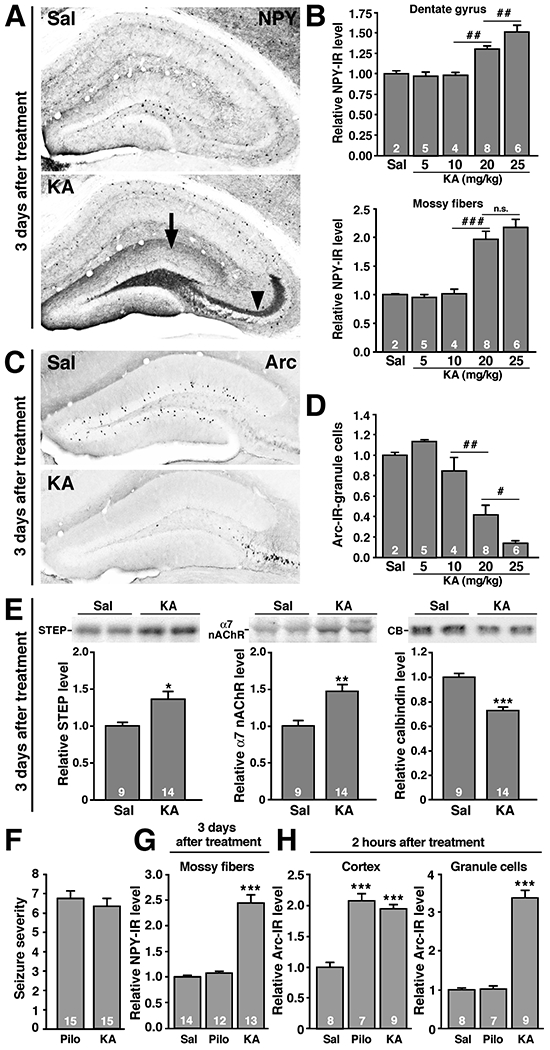
(A-E) NTG mice were injected IP with saline or kainate (KA) (5, 10, 20, or 25 mg/kg) and analyzed 3 days later. (A) Representative photomicrographs depicting KA-induced sprouting of NPY-positive terminals in the molecular layer of the dentate gyrus (arrow) and ectopic expression of NPY in the mossy fibers (arrowhead). (B) Quantitation of KA-induced increases in NPY expression demonstrating dose-dependence. (C) Typical KA-induced reduction of Arc expression in granule cells of the dentate gyrus. (D) This effect was also dose-dependent. (E) Representative western blots of dentate lysates and quantitations of signals, illustrating KA-induced changes in the levels of STEP (left), α7 nAchR (center), and calbindin (right). Each lane represents an individual mouse. (F-H) NTG mice were injected IP with pilocarpine (Pilo) (250 mg/kg) or kainate (25 mg/kg) and euthanized 2 h or 3 days later for analysis of NPY and Arc expression. (F) At these concentrations, both drugs elicited similar overall seizure severity within 20 min after their injections. (G) By 3 days after the injection, KA, but not pilocarpine, increased NPY expression in mossy fibers. (H) By 2 h after the injection, both drugs elicited comparable increases in Arc expression in the neocortex, but only KA elicited marked Arc expression in granule cells. #P<0.05, ##P<0.01, ##P<0.01 by ANOVA and contrasts test. *P<0.05, **P<0.01, ***P<0.001 vs. Sal by Student’s t test or Tukey-Kramer test.
Interestingly, systemic injection of kainate (25 mg/kg) or of the muscarinic receptor agonist pilocarpine (250 mg/kg) elicited tonic-clonic seizure activity of comparable severity (Fig. 4F) in NTG mice, but only kainate led to an increase in NPY expression (Fig. 4G), suggesting that seizure activity per se was not sufficient to alter NPY expression. To assess whether particular patterns of neuronal activation may be required to increase NPY expression, we analyzed Arc expression 2 h after the treatments. Although both kainate and pilocarpine acutely increased neuronal Arc expression in the neocortex, only kainate increased Arc expression in granule cells (Fig. 4H). Thus, overexcitation of granule cells may be required to trigger the aberrant NPY expression in the dentate gyrus.
Tau reduction prevents kainate- and hAPP/Aβ-induced changes in NPY
Genetic reduction of the microtubule-binding protein tau diminished the susceptibility of hAPP-J20 and NTG mice to seizures induced by kainate or the GABAA receptor antagonist pentylenetetrazole (PTZ), and ameliorated Aβ-dependent cognitive deficits in hAPP-J20 mice (Roberson et al., 2007). We therefore examined whether the kainate- or Aβ-induced alterations described above could also be prevented by tau reduction. After intraperitoneal (IP) injection with kainate (17 mg/kg), seizures were less severe in Tau−/− mice than in Tau+/+ mice (Fig. 5A), as described (Roberson et al., 2007). Three days after the injection, brains were analyzed for NPY expression. Tau removal effectively prevented kainate-induced NPY increases in the dentate gyrus and mossy fibers (Fig. 5B). In untreated hAPP-J20 mice, tau reduction dose-dependently prevented NPY increases (Fig. 5C–E) and calbindin depletions (Fig. 5F, G). The observations that Aβ and excitotoxins elicit similar molecular and anatomical alterations, and that these alterations can be prevented by the same genetic manipulation suggest mechanistic overlap and support the hypothesis that Aβ promotes neuronal overexcitation.
Figure 5. Tau reduction prevents KA- and hAPP/Aβ-induced changes in NPY.

(A-B) Tau−/− and Tau+/+ mice were injected IP with saline or 17 mg/kg of kainate (KA), and brains were analyzed 3 days later. (A) Compared with Tau+/+ mice, Tau−/− mice displayed less KA-induced freezing. (B) In Tau+/+ mice, but not in Tau−/− mice, KA induced robust increases in NPY in the dentate gyrus (left) and the mossy fibers (right). (C-G) Brain sections from hAPP-J20 mice with or without Tau expression were immunostained for calbindin or NPY; immunoreactivity was quantified by densitometry. (C) Increased NPY expression was prominent in hAPP-J20/Tau+/+ mice, but absent in hAPP-J20/Tau−/− mice. (D, E) Quantitation of NPY-IR in the molecular layer (D) (hAPP x Tau interaction, P < 0.02; ***P < 0.001, **P < 0.01 vs. groups without hAPP) and mossy fibers (E) (hAPP x Tau interaction, P < 0.02; **P < 0.01 vs. groups without hAPP). (F) Calbindin depletion in the dentate gyrus was observed in hAPP-J20/Tau+/+ mice, but not in hAPP-J20/Tau−/− mice. (G) Quantitation of calbindin immunoreactivity (CB-IR) in the molecular layer (hAPP x Tau interaction, P < 0.0001; ***P < 0.001 vs. groups without hAPP.)
Aβ-dependent alterations in the dentate gyrus are associated with increased seizure activity after inhibition of GABAA receptors
To test this hypothesis more directly, we assessed the propensity of hAPPFAD mice and NTG controls to develop seizures after systemic challenge with PTZ. After IP injection with PTZ, hAPP-J20 mice had earlier and more severe seizures than NTG controls (Fig. 6A). Seizure-induced death was more frequent in hAPP-J20 mice than NTG controls (Fig. 6A). hAPP-ARC48 mice and hAPP-J9/FYN mice were also more susceptible to PTZ-induced seizures than controls (Fig. 6B, C). These findings are consistent with results obtained in another line of hAPP mice (Del Vecchio et al., 2004), and suggest that chronic exposure to high levels of Aβ sensitizes at least some neuronal networks to overexcitation.
Figure 6. Aβ-dependent alterations in the dentate gyrus are associated with increased susceptibility of hAPPFAD mice to seizures induced by a GABAA antagonist.

Mice were injected IP with PTZ (40 or 80 mg/kg). Brain tissues were collected 20 min thereafter, or earlier if they developed fatal seizures. Behavior was video-recorded, and seizure severity was scored off-line. (A) Compared with NTG controls, hAPP-J20 mice had shorter latencies to reach a given seizure severity (left), greater overall seizure severity (center), and more seizure-associated deaths (right). (B) hAPP-ARC48 mice and hAPP-J9/FYN mice also had increased overall seizures severity compared with littermate controls. (C) hAPP-J9/FYN mice also had a greater incidence of seizure-associated death than the control groups. (D) All NTG mice treated with 80 mg/kg of PTZ developed fatal seizure activity. (E-G) Protein levels of NPY (E), calbindin (F), and Fos (G) in the dentate gyrus were not altered by fatal seizures. (H-K) Compared with hAPP-J20 mice that survived the PTZ injection, hAPP-J20 mice that developed fatal seizures had higher levels of NPY in the dentate gyrus and mossy fibers (H), lower dentate calbindin levels (I), and lower numbers of granule cells expressing Fos (J) or Arc (K). *P<0.05, **P<0.01, ***P<0.001 vs. NTG by Student’s t test or Tukey-Kramer test. #P<0.05, ##P<0.01 by Fisher’s exact test.
Interestingly, 50% of the hAPP-J20 mice developed fatal status epilepticus after PTZ administration, whereas the other 50% did not (Fig. 6A, right panel). To test whether this difference was related to molecular alterations in the dentate gyrus, we first confirmed that the levels of the proteins we wanted to analyze would not be affected by acute fatal seizure activity during the 20 min duration of this experiment. NPY, calbindin, and Fos levels in the dentate gyrus were not altered in NTG mice that died acutely from a PTZ-induced seizure, or that survived for 20 min after PTZ-induced seizures (Fig. 6D–G), suggesting that the levels of these proteins reflect the baseline situation even when analyzed after acute seizures. hAPP-J20 mice that developed fatal seizures after PTZ injection had greater increases in NPY and more severe depletions of calbindin, Fos, and Arc than hAPP-J20 mice with less severe seizures and NTG controls (Fig. 6H–K). Thus, the magnitude of these molecular alterations likely reflects the severity of Aβ-induced increases in network excitability.
In vivo EEG recordings revealed hyperexcitability and non-convulsive seizure activity in hAPP-J20 mice
To more directly test whether aberrant neuronal activity is present in hAPP-J20 mice, we performed prolonged videoEEG monitoring of six freely behaving hAPP-J20 adult mice (aged 3-7 months). In all hAPP-J20 mice, the cortical background activity showed very frequent (5-50/min), generalized, sharp, synchronous discharges in all cortical electrodes throughout the monitoring period that were never seen in NTG controls (Fig. 7A). When depth electrodes were bilaterally implanted in the hippocampus of two hAPP-J20 mice, similar high amplitude discharges could occasionally be recorded even unilaterally (Fig. 7B). These results demonstrate hippocampal hyperexcitability and suggest the involvement of hippocampal networks in the aberrant generalized neocortical synchronization.
Figure 7. Aberrant synchronous neuronal network activity and spontaneous non-convulsive seizures in hAPP-J20 mice.
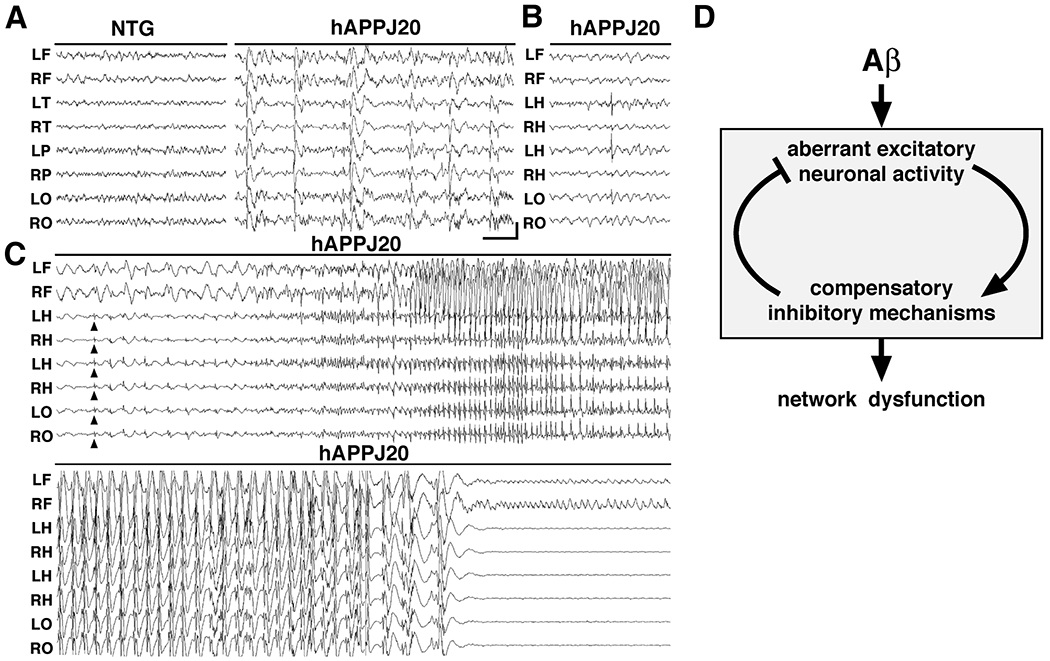
Chronic cortical and hippocampal EEG recordings were performed in freely moving, untreated hAPP-J20 mice and NTG controls. L, left; R, right; F, frontal; T, temporal; P, parietal; O, posterior-parietal; and H, hippocampal, indicate the position of recording electrodes. (A) In contrast to NTG mice, which showed normal EEG activity (left), hAPP-J20 mice exhibited frequent (5–50/min) generalized cortical epileptiform (interictal) spike discharges (right). (B) Bilateral depth electrode recordings from the hippocampus detected occasional discharges of isolated hippocampal origin. In this example, a prominent left-sided discharge remains focal with minimal spread to the neocortex. (C) Initiation (upper panel) and termination (lower panel) of a cortical seizure in an hAPP-J20 mouse. Concurrent bilateral hippocampal depth electrode recordings show abnormal synchronization beginning as spike discharges (arrowheads) in the hippocampus and overlying posterior-parietal neocortex (LO-RO). Synchronous seizure activity then generalizes in the neocortex without behavioral signs of convulsive activity, followed by profound electroencephalographic postictal depression. Calibration: 1 sec and 400 mV. (D) Model of Aβ-induced network dysfunction. We propose that high levels of Aβ induce aberrant excitatory neuronal activity, which triggers compensatory inhibitory mechanisms to counteract overexcitation. Both aberrant excitatory neuronal activity and compensatory inhibitory mechanisms may contribute to AD-related network dysfunction.
In contrast to NTG controls, hAPP-J20 mice also exhibited intermittent non-convulsive electroencephalographic seizures. This activity typically started with generalized spike and slow-wave discharges, which gradually increased in frequency to a rapid spike discharge and then decelerated to slow (1-2/second) spike-wave rhythms, followed by an abrupt termination of the ictal event with pronounced cortical EEG depression (Fig. 7C). During these electroencephalographic seizures, the mice were immobile, showing no visible myoclonic activity. When the postictal EEG depression ended, they resumed normal exploratory behaviors (data not shown).
Aberrant Arc expression in granule cells of hAPPFAD mice
To assess whether spontaneous episodes of neuronal overexcitation could also be detected by gene expression imaging (Link et al., 1995; Lyford et al., 1995), we analyzed Arc expression in a large number of untreated mice to broadly profile neuronal activity patterns. In the majority of hAPP-J20 mice, Arc expression in granule cells was much lower than in NTG controls (Fig. 8A, B), consistent with previous findings (Palop et al., 2005). However, 13 of 151 untreated hAPP-J20 mice (8.6%) had markedly increased Arc expression in granule cells (Fig. 8C). This abnormal pattern of Arc expression was not found in any of the 135 NTG controls and likely reflects a recent episode of abnormal overexcitation (Link et al., 1995; Lyford et al., 1995; Palop et al., 2006). hAPP-J20 mice with increased Arc expression had even higher NPY levels and lower calbindin levels than hAPP-J20 mice with reduced Arc expression (Fig. 8D, E).
Figure 8. Alterations in Arc expression and in NMDA and AMPA receptors in the dentate gyrus of hAPP-J20.

Brain sections from 151 untreated hAPP-J20 mice and 142 NTG controls were immunostained for Arc, calbindin, and NPY. (A-C) Photomicrographs of the dentate gyrus showing three distinct representative patterns of Arc expression we identified. (A) Normal pattern of Arc expression found in virtually all NTG mice. (B) Typical reduction of Arc expression observed in the majority of hAPP-J20 mice. (C) Roughly 9% of hAPP-J20 mice showed abnormally increased Arc expression in the granule cells, suggestive of recent seizure activity (Lyford et al., 1995). (D-E) Calbindin depletions (D) and NPY increases (E) were even more severe in hAPP-J20 mice with excessive Arc expression than in hAPP-J20 mice with reduced Arc expression. (F-G) Representative western blots of dentate lysates and quantitations of signals illustrating reduced levels of tyrosine-phosphorylated NR2B (pY1472), but not total NR2B levels (F), and reduced levels of AMPA receptor subunits GluR1 and GluR2 (G) in hAPP-J20 mice compared to NTG controls. Each lane represents an individual mouse. **P<0.01, ***P<0.001 by Student’s t test.
Depletions of NMDA and AMPA receptors in the dentate gyrus of hAPP-J20 mice
Alterations in NMDA and AMPA glutamate receptors have been linked to Aβ-induced impairments in synaptic plasticity and the collapse of dendritic spines (Hsieh et al., 2006; Kamenetz et al., 2003; Shankar et al., 2007). To test whether hAPP-J20 mice exhibit alterations in glutamate receptors, we examined the phosphorylation state of tyrosine residue 1472 of the NR2B subunit of NMDARs, which modulates calcium conductance, is associated with LTP induction, and controls internalization of the receptor (reviewed in Salter and Kalia, 2004). Levels of tyrosine-phosphorylated NR2B in the dentate gyrus were lower in hAPP-J20 mice than in NTG controls, whereas total levels of NR2B were unchanged (Fig. 8F, and Palop et al., 2005). We also examined AMPAR subunits GluR1 and GluR2, whose synaptic localization is decreased by Aβ (Almeida et al., 2005; Hsieh et al., 2006; Snyder et al., 2005). Dentate levels of GluR1 and GluR2 were lower in hAPP-J20 mice than in NTG controls (Fig. 8G).
Short- and long-term plasticity impairments in the dentate gyrus of hAPP-J20 mice
As illustrated by this and previous studies (Chin et al., 2005; Palop et al., 2005; Palop et al., 2003), neuronal circuits in the dentate gyrus are exquisitely vulnerable to Aβ-induced molecular and anatomical alterations. To further assess how these alterations may relate to synaptic dysfunction, we performed field EPSPs recordings in hippocampal slices from the dentate gyrus and CA1 region. Perforant pathway to granule cell synapses displayed severe impairments in long-term potentiation (LTP) and alterations in paired-pulse modification (Fig. 9A, B), but normal synaptic transmission strength (Fig. 9C), consistent with results obtained in other hAPP models (Chapman et al., 1999). Analysis of the Schaffer collateral to CA1 pyramidal cell synapse revealed an opposite pattern of deficits, consistent with our original observations in the CA1 region of two other hAPPFAD lines (H6 and J9) (Hsia et al., 1999) and subsequent findings by others (Fitzjohn et al., 2001). LTP and paired-pulse facilitation were normal (Fig. 9D, E), whereas synaptic transmission strength was reduced (Fig. 9F). Thus, marked brain region-specific deficits also exist at the electrophysiological level, and the dentate gyrus appears to be particularly vulnerable to impairments in both short- and long-term plasticity.
Figure 9. Region-specific electrophysiological alterations in hippocampal slices from hAPP-J20 mice.
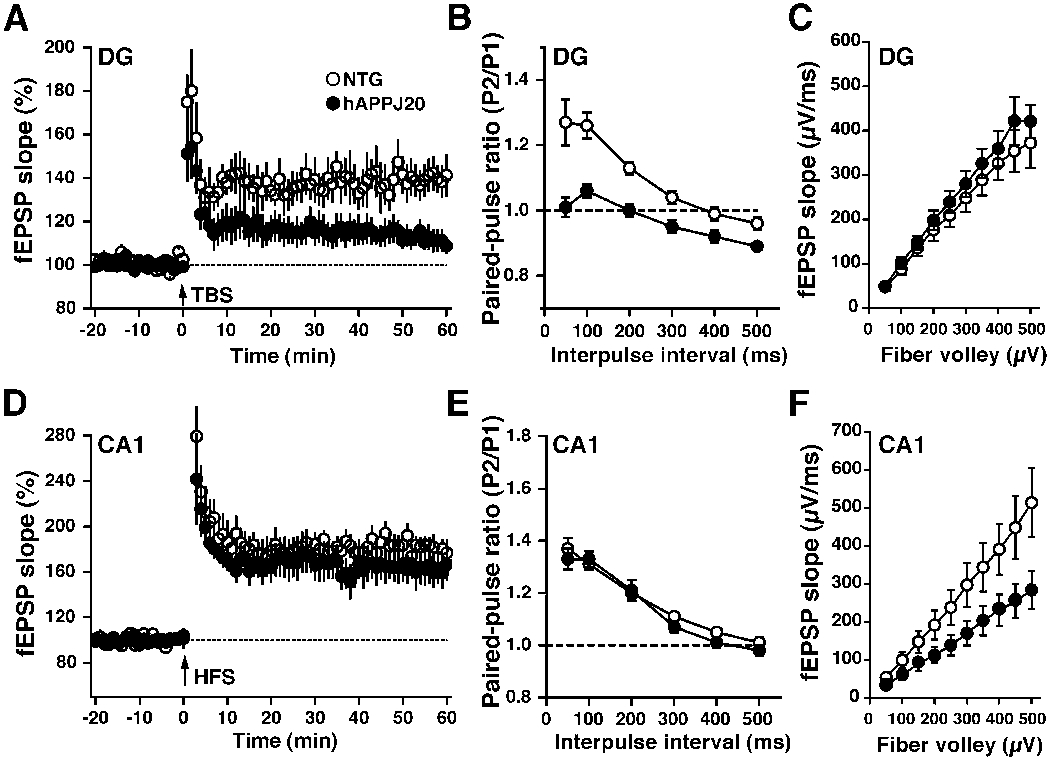
(A) LTP at the medial perforant pathway synapse within the dentate gyrus (DG) was significantly depressed in hAPP-J20 mice (6 slices, 6 mice) compared with NTG mice (9 slices, 8 mice) (P<0.01 by repeated measures ANOVA on data collected from minutes 51-60). (B) At the medial or lateral (not shown) perforant pathway, paired-pulse ratio, a common measure of presynaptic function, was significantly different in hAPP-J20 mice (15 slices, 7 mice) compared with NTG mice (15 slices, 8 mice) (P<0.01 by Student’s t test at all interpulse intervals). (C) The slope of the input-output relationship, a measure of synaptic transmission, along the medial or lateral (not shown) perforant pathway was similar in hAPP-J20 (14 slices, 7 mice) and NTG (12 slices, 7 mice) mice. (D) LTP at the Schaffer collateral synapse within the CA1 region (CA1) was similar in hAPP-J20 (5 slices, 5 mice) and NTG (8 slices, 6 mice) mice. (E) Paired-pulse ratio at this synapse was similar in hAPP-J20 (14 slices, 6 mice) and NTG (16 slices, 7 mice) mice. (F) In contrast, synaptic transmission along the Schaffer collateral synapse was significantly less in hAPP-J20 mice (14 slices, 5 mice) than in NTG mice (11 slices, 5 mice) (P<0.01 by ANCOVA). TBS, theta-burst stimulation; HFS, high frequency stimulation.
DISCUSSION
Our study demonstrates that hAPPFAD mice with high Aβ levels have spontaneous non-convulsive seizure activity in cortical and hippocampal networks, indicating that the net effect of Aβ on these networks is excitatory. AD patients also have a higher incidence of seizures than reference populations (Amatniek et al., 2006; Hauser et al., 1986; Hesdorffer et al., 1996; Lozsadi and Larner, 2006; Mendez and Lim, 2003). Interestingly, the risk of epileptic activity is particularly high in AD patients with early-onset dementia and during the earlier stages of the disease, reaching an 87-fold increase in seizure incidence compared with an age-matched control population (Amatniek et al., 2006; Mendez et al., 1994). Thus, aberrant neuronal overexcitation may play an important role not only in hAPPFAD mouse models, but also in the pathogenesis of dementia in sporadic AD. It is interesting in this regard that the relationship between seizures and AD is even tighter in autosomal dominant early-onset FAD. Pedigrees with epilepsy have been identified in FAD linked to mutations in presenilin-1, presenilin-2, and APP (Edwards-Lee et al., 2005; Marcon et al., 2004; Snider et al., 2005), as well as in FAD linked to duplications of APP (Cabrejo et al., 2006). Over 30 different mutations in presenilin-1 are associated with seizures (Larner and Doran, 2006), and 56% of FAD patients with APP duplications have seizures (Cabrejo et al., 2006). Our results suggest that the increased epileptic activity in sporadic and autosomal dominant AD may be caused by Aβ-induced increases in network excitability. Although Aβ may increase the incidence of seizures, clinically evident convulsive seizure activity is not a prominent feature in AD. Notably, our hAPPFAD mice had frequent electroencephalographic seizures that were not accompanied by obvious tonic-clonic movements, raising the question of whether the extent of subclinical epileptic activity in AD may be underestimated.
Generalized epileptic activity can trigger downstream cellular and circuit alterations in hippocampal circuits (Fig. 7D and Supplemental Fig. 2) (Marksteiner et al., 1990; Tonder et al., 1994b; Vezzani et al., 1999). Some of these alterations have been characterized as compensatory inhibitory mechanisms in different epileptic models, since they can ameliorate neuronal overexcitation. hAPPFAD mice exhibited a variety of such alterations in the dentate gyrus, including altered levels of NPY receptors, ectopic NPY expression, GABAergic sprouting, and increased synaptic inhibition. NPY-Y1 receptors, which were decreased in hAPPFAD mice, typically reside on dendrites and mediate weak excitatory effects of NPY (Vezzani et al., 1999). NPY-Y1 receptor antagonists had anticonvulsive effects in rat seizure models (Gariboldi et al., 1998). NPY-Y2 receptors, which were increased in hAPPFAD mice, typically reside on presynaptic terminals and mediate strong presynaptic inhibition of glutamate release via inactivation of voltage-gated calcium channels (Qian et al., 1997; Schwarzer et al., 1998). This inhibitory presynaptic mechanism of glutamate release would likely be augmented by the ectopic expression of NPY in mossy fibers (Klapstein and Colmers, 1993; Vezzani and Sperk, 2004). NPY and particularly NPY-Y2 receptor agonists potently suppress seizure activity in hippocampal slices in vitro and in experimental animals in vivo (Baraban et al., 1997; Bijak, 1995; Colmers et al., 1987; Smialowska et al., 1996; Vezzani et al., 1999). Viral vector-mediated NPY expression in the dentate gyrus of NTG rats inhibited seizure activity induced by intrahippocampal kainate injection (Richichi et al., 2004). We also found prominent GABAergic sprouting and increases in mIPSC frequency and large amplitude events in the dentate gyrus of hAPPFAD mice, indicating that GABAergic function is enhanced in this brain region. Thus, most of the alterations we identified in the dentate gyrus of hAPPFAD mice likely serve to counteract Aβ-induced neuronal overexcitation.
The tenuous balance between excitation and inhibition in hAPPFAD mice was highlighted by our analysis of neuronal Arc expression. Although Arc was reduced in most hAPPFAD mice with high levels of Aβ, consistent with previous results (Palop et al., 2005), a small fraction of hAPPFAD mice had markedly increased Arc expression in the dentate gyrus, possibly reflecting recent break-through seizure activity resulting from severe neuronal overexcitation and/or relative inadequacies in synaptic inhibition. Thus, the activity of granule cells in hAPPFAD mice, and possibly also in humans with AD, may fluctuate between aberrant increases and postictal or compensatory suppression.
In the current study, a single systemic injection of kainate was sufficient to trigger prominent NPY/SOM-positive sprouting and ectopic NPY expression in a distribution very similar to that observed in untreated hAPPFAD mice. Similar alterations have been observed after electroconvulsive seizures, electrical stimulation of the entorhinal cortex, pharmacological stimulation of NMDA receptors or metabotropic glutamate receptors, or inhibition of GABA receptors (Marksteiner et al., 1990; Tonder et al., 1994b), making it likely that aberrant increases in excitatory neuronal activity are the common underlying mechanism. It is interesting in this regard that kainate injection also replicated several other Aβ-induced dentate alterations, including increases in the levels of STEP and α7 nAChRs, and reductions in calbindin and Arc (Chin et al., 2005; Palop et al., 2005; Palop et al., 2003), suggesting that the pathogenesis of these alterations in hAPPFAD mice may also involve aberrant increases in neuronal activity.
Interestingly, hAPPFAD mice resemble models of chronic temporal lobe epilepsy only in some respects, but not in others. In contrast to chronic epilepsy models (de Lanerolle et al., 1989; Nadler, 2003; Tu et al., 2006), hAPPFAD mice showed no loss of NPY/SOM-positive GABAergic interneurons and no recurrent sprouting of excitatory collateral mossy fibers in the inner molecular layer. Instead, hAPPFAD mice showed sprouting of collateral mossy fibers onto GABAergic basket cells. The circuit alterations in the chronic epilepsy models would be expected to promote hippocampal epileptogenesis, whereas those in hAPPFAD mice should counteract it. Consistent with this interpretation, synaptic inhibition in granule cells is largely compromised in models of epilepsy (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005), but was significantly enhanced in hAPPFAD mice. This difference may help explain why hAPPFAD mice had primarily non-convulsive seizures and why escalation of epileptic activity into convulsive seizures is relatively infrequent in both hAPPFAD mice and humans with AD.
While the chronic activation of inhibitory mechanisms can limit excitotoxic injury, it may also interfere with processes required for learning and memory and other neural functions (Fig. 7D). Consistent with this idea, increased inhibition of granule cells in a model of Down’s syndrome caused LTP deficits in the dentate gyrus, as suggested by the prevention of such deficits by treatment with the GABAA antagonist picrotoxin (Kleschevnikov et al., 2004). We found that increased inhibition and GABAergic remodeling was also associated with deficits in short- and long-term plasticity in the dentate gyrus of hAPPFAD mice. However, LTP deficits were not normalized by picrotoxin in hAPPFAD slices, suggesting additional pathogenic mechanisms. Since depletion of calbindin (Molinari et al., 1996), reduction of Arc expression (Guzowski et al., 2000), and an increase of inhibition (Kleschevnikov et al., 2004) can cause LTP deficits, and such alterations are present in hAPPFAD mice (this study and Palop et al., 2005; Palop et al., 2003), it is likely that LTP impairments in hAPPFAD mice have a multifactorial origin. The increased inhibition of granule cells may underlie or contribute to the reduced expression of immediate-early genes, such as Arc and Fos, in the dentate gyrus of hAPPFAD mice (Chin et al., 2005; Lee et al., 2004; Palop et al., 2005; Palop et al., 2003). It is interesting in this regard that GABAergic sprouting correlated with reductions in Arc (not shown) and calbindin in granule cells of hAPPFAD mice, which correlate well with deficits in learning and memory (Palop et al., 2005; Palop et al., 2003). Because the experimental reduction of calbindin, Fos, or Arc in otherwise healthy rodents elicits neuronal deficits and memory impairments (Guzowski et al., 2000; He et al., 2002; Molinari et al., 1996; Plath et al., 2006; Tzingounis and Nicoll, 2006), the depletion of these factors could further contribute to granule cell dysfunction and behavioral deficits in hAPPFAD mice.
Notably, hAPPFAD mice showed impairments in both synaptic glutamatergic transmission and levels of NMDAR and AMPAR. Specifically, we showed reduced LTP and paired-pulse modification, reduced phosphorylation of the NMDAR subunit NR2B, and reduced levels of the AMPAR subunits GluR1 and GluR2 in the dentate gyrus of hAPPFAD mice. These data are consistent with the growing literature suggesting that Aβ induces impairments in synaptic glutamatergic transmission and retraction of excitatory dendritic spines (Hsia et al., 1999; Hsieh et al., 2006; Kamenetz et al., 2003; Palop et al., 2005; Shankar et al., 2007; Walsh et al., 2002). Therefore, aberrant increases in neuronal activity in hippocampal and cortical networks can coexist with impaired glutamatergic transmission in hAPPFAD mice. Although the exact relationship between these two phenomena needs to be further elucidated, several non-mutually exclusive possibilities could explain their coexistence. First, depressed glutamatergic transmission could be a synaptic compensatory mechanism against overexcitation. Second, inhibitory interneurons could be more susceptible to the suppressive effects of Aβ on glutamatergic synaptic transmission than excitatory principal neurons, leading to an overall increase in neuronal excitability. Third, cortical or subcortical regions that control neuronal excitability on a broad scale could be particularly susceptible to Aβ-induced impairments of glutamatergic synaptic transmission, inreasing overall network excitability.
In conclusion, our findings suggest that Aβ triggers intermittent aberrant excitatory neuronal activity in the cortex and hippocampus, resulting in a prominent remodeling of inhibitory circuits and increased inhibition of granule cells. Thus, cognitive deficits in hAPPFAD mice, and perhaps also in humans with AD, may result from the combination of neuronal overexcitation and the subsequent development of compensatory inhibitory mechanisms that reduce overexcitation but end up constraining the functional agility of specific excitatory circuits. Studies are needed to determine whether blocking Aβ-induced neuronal overexcitation can prevent the activation of inhibitory pathways as well as the development of AD-related neurological deficits.
MATERIALS AND METHODS
TG mice.
We studied 4- to 7-month-old heterozygous TG and NTG mice from lines J20, J9, ARC48, I5, and N8, as well as J9/FYN doubly transgenic mice and hAPP-J20/Tau−/− mice. Lines J9 and J20 express hAPP carrying the Swedish and Indiana FAD mutations; line ARC48 expresses hAPP carrying the Swedish, Indiana, and Arctic FAD mutations; line I5 expresses wildtype hAPP; and line N8 overexpresses wildtype mouse Fyn (Cheng et al., 2004; Chin et al., 2005; Chin et al., 2004; Dawson et al., 2001; Mucke et al., 2000; Roberson et al., 2007). See Supplemental Methods for more information.
Drugs.
Pentylenetetrazole (PTZ) (Sigma, St. Louis, MO), kainate (Sigma), and pilocarpine (Tocris) were dissolved in PBS at 5, 1.8, and 20 mg/ml respectively, and injected intraperitoneally at the doses indicated in the Results section.
Seizure severity score.
After drug administration (PTZ, pilocarpine, or kainate), each mouse was placed in a new cage and its behavior was videorecorded for 20 min or manually recorded for 3 hours for Tau-manipulated mice treated with kainate. An investigator blinded to the genotype and treatment of the mice quantified the time course and severity of seizures according to published scales (Loscher et al., 1991; Racine, 1972). Seizure severity scores were 0 = normal exploratory behavior; 1 = immobility, 2 = generalized spasm, tremble, or twitch, 3 = tail extension, 4 = forelimb clonus, 5 = generalized clonic activity, 6 = bouncing or running seizures, 7 = full tonic extension, 8 = death.
Immunohistochemistry.
Tissue preparation and immunohistochemistry were performed as described (Palop et al., 2005; Palop et al., 2003). Primary antibodies used included: rabbit anti-Arc (1:8,000; a gift from S. Chowdhury and P. F. Worley, Johns Hopkins University School of Medicine, Baltimore, MD), rabbit anti-calbindin (1:15,000 for DAB; Swant, Bellinzona, Switzerland), mouse anti-calbindin (1:1,000 for fluorescence; Swant), rabbit anti-Fos (1:10,000; Ab-5, Oncogene, San Diego, CA), rabbit anti-neuropeptide Y (1:8,000 for DAB, 1:4,000 for fluorescent; ImmunoStar, Hudson, WI), rabbit anti-parvalbumin (1:5,000; Swant), rat anti-somatostatin (1:200 for DAB, 1:50 for fluorescence; Chemicon, Temecula, CA), or mouse biotinylated anti-Aβ (1:400, 3D6; Elan Pharmaceuticals, South San Francisco, CA). Primary antibodies were detected with biotinylated goat anti-rabbit (1:200; Vector Laboratories, Burlingame, CA) or goat anti-rat (1:200; Vector Laboratories), or with fluorescein-labeled donkey anti-rabbit (1:300, Jackson ImmunoResearch, West Grove, PA), fluorescein-labeled goat anti-rabbit (1:150; Vector Laboratories), Alexa Fluor 594 tyramide-labeled goat anti-rat (1:300; Molecular Probes, Carlsbad, CA), or Texas red-labeled donkey anti-mouse (1:300, Jackson ImmunoResearch).
Timm staining.
Sodium selenite (Sigma) was dissolved in PBS (10 mg/ml) and injected IP at a dose of 20 mg/kg. Mice were sacrificed 1 h after the injection. Sliding microtome floating sections were processed for the detection of vesicular zinc with a slightly modified Timm method. See Supplemental Methods for details.
Quantitative analysis of brain sections.
Calbindin, Arc, and Fos were quantified as described (Palop et al., 2005; Palop et al., 2003), and NPY was quantified similarly with a slight modification (see Supplemental Methods for details).
In situ hybridization and RT-PCR.
For NPY in situ hybridization, antisense and sense cRNA probes were generated from a linearized plasmid (IMAGE:5683102) containing full-length NPY cDNA (561bp) with T7 and T3 polymerase (Promega, Madison, WI) and premixed RNA-labeling nucleotide mixes containing digoxigenin (Roche Molecular Biochemicals, Palo Alto, CA). For quantitative fluorogenic RT-PCR analysis of mRNAs encoding NPY-Y1 or -Y2 receptors, the following primers were used: NPY-Y1, 5′-CAGTGAGACCAAGCGAATCAAC-3′, 5′-CTGGTGGTTCCAGTCGAACA −3′; NPY-Y2, 5′-TGGGCCAGGGCACACTAC-5′, 5′-TCACCTGCACCTCGACCA-3′. In situ hybridization and RT-PCR were performed as described (Palop et al., 2005).
Western blot analysis.
Microdissections and western blot analysis was performed as described (Chin et al., 2005; Palop et al., 2005). The following antibodies were used and detected with species-appropriate horseradish peroxidase-conjugated secondary antibodies: anti-α7 nAChR (1:1,000; mouse monoclonal, Covance, Princeton, NJ), anti-calbindin (1:15,000; rabbit polyclonal, Swant), anti-GluR1 and anti-GluR2 (1:20,000; rabbit polyclonals, Chemicon), anti-pY1472 (1:1,000; rabbit polyclonal, Chemicon), anti-NR2B (1:10,000 rabbit polyclonal, Chemicon), and anti-STEP (1:5,000; mouse monoclonal, Novus Biologicals, Littleton, CO). Bands were visualized by ECL and quantitated densitometrically with ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Electrophysiology.
Transverse hippocampal slices (300 μm thick for mIPSC; 350 μm thick for fEPSP) were prepared from 3-4-month-old NTG and hAPP-J20 mice. For more details see Supplemental Methods.
mIPSC recordings:
NBQX (2 μM), D-AP5 (25 μM), and TTX (1 μM) were added to the external solution to isolate miniature IPSCs. All recordings were performed at room temperature. Whole-cell voltage-clamp recordings were obtained from visualized hippocampal dentate gyrus granule cells using IR-DIC video microscopy. Glass electrodes (3-4 MΩ) were filled with a solution containing (in mM): 140 CsCl, 10 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, adjusted to pH 7.3 with CsOH. Access resistance and leak currents were monitored continuously and experiments were rejected if these parameters changed by more than 15% during the experiment.
Voltage-clamp recordings were performed using a Multiclamp 700B amplifier (Molecular Devices); data was filtered at 2 kHz and digitized at 10 kHz. Acquisition and analysis were performed using custom Igor Pro software.
fEPSP recordings:
Bipolar stimulating electrodes consisted of a glass microelectrode (10 ~ 20 μm OD) filled with bathing solution or 1 M NaCl and 25 mM HEPES (pH = 7.3). Stimulating electrodes were placed in the middle of the outer or middle molecular layer of the dentate gyrus or in the middle of the stratum radiatum. Field potential recordings were obtained using a MultiClamp 700A amplifier (Axon Instruments, Inc, Union City, California) controlled by Axon’s pClamp 9. Recording pipettes had resistances of 2-4 MΩ when filled with 1 M NaCl and 25 mM HEPES (pH = 7.3). The bathing solution contained 100 μM picrotoxin and 20 μM bicuculline to block inhibitory transmission and was perfused at 35°C at approximately 2 ml/min. LTP was induced by high frequency stimulation (HFS, 100 pulses at 100 Hz, four times in 20 s intervals) in CA1 or by theta-burst stimulation (TBS, 10 bursts at 5 Hz, repeated 10 times in 15 s intervals. Each burst consisted of four pulses of 100 Hz) in the dentate gyrus. For data collection and analyses see Supplemental Methods.
Chronic Electroencephalographic (EEG) Recordings.
hAPP-J20 and NTG mice were implanted for chronic videoEEG monitoring after anesthesia with Avertin (1.25% tribromoethanol/amyl alcohol solution, injected IP at 0.02 ml/g). Teflon-coated silver wire electrodes (0.005 inch diameter) attached to a microminiature connector were implanted bilaterally into the subdural space over frontal, central, parietal, and occipital cortices. Simultaneous depth recordings were also obtained from the hippocampal formation in two hAPP-J20 mice. Digital EEG activity was monitored daily for up to two weeks during prolonged overnight and random 2 hr sample recordings (Stellate Systems, Harmonie software version 5.0b). Recordings of similar durations were made in NTG and hAPP-J20 mice, and were reviewed by an investigator unaware of the genotypes of the mice. A video camera was used to monitor behavior during the EEG recording periods. All recordings were carried out at least 24 h after surgery on mice freely moving in the test cage.
Statistical analysis.
Statistical analyses were performed with SPSS 10.0 (SPSS, Chicago, IL). Unless indicated otherwise, differences between two means were assessed by unpaired, two-tailed Student’s t test and differences among multiple means by ANOVA and Tukey–Kramer post hoc test. Differences between expected and observed frequencies were assessed by Fisher’s exact test. Correlations were examined by simple regression analysis. Null hypotheses were rejected at the 0.05 level.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by a fellowship from the McBean Foundation (J.J.P.) and by National Institutes of Health Grants AG023501, AG011385, and NS041787 to L.M., NS29709 and HD24064 to J.L.N., NS39074 and AG022074 to S.F., NS54811 to E.D.R., and a facilities grant (RR 018928) from the National Center for Research Resources. We thank M. Vitek and H. Dawson for Tau−/− mice, R. Malenka for use of equipment; S. Chowdhury and P. F. Worley for the Arc antibody; W. Morishita for advice on slice preparations; C. McCulloch for statistical advice; F. Yan, X. Wang, and H. Solanoy for technical support; G. Howard and S. Ordway for editorial review; and D. Murray McPherson for administrative assistance.
REFERENCES
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, and Gouras GK (2005). β-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20, 187–198. [DOI] [PubMed] [Google Scholar]
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, and Stern Y (2006). Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 47, 867–872. [DOI] [PubMed] [Google Scholar]
- Bakst I, Avendano C, Morrison JH, and Amaral DG (1986). An experimental analysis of the origins of somatostatin-like immunoreactivity in the dentate gyrus of the rat. J Neurosci 6, 1452–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Hollopeter G, Erickson JC, Schwartzkroin PA, and Palmiter RD (1997). Knock-out mice reveal a critical antiepileptic role for neuropeptide Y. J Neurosci 17, 8927–8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijak M (1995). Inhibitory effect of neuropeptide y on epileptiform activity in the frontal cortex and hippocampus in vitro. Pol J Pharmacol 47, 461–463. [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, and Zetterberg H (2006). Alzheimer’s disease. Lancet 368, 387–403. [DOI] [PubMed] [Google Scholar]
- Burgess N, Maguire EA, and O’Keefe J (2002). The human hippocampus and spatial and episodic memory. Neuron 35, 625–641. [DOI] [PubMed] [Google Scholar]
- Cabrejo L, Guyant-Maréchal L, Laquerriére A, Vercelletto M, De La Fourniére F, Thomas-Antérion C, Verny C, Letournel F, Pasquier F, Vital A , et al. (2006). Phenotype associated with APP duplication in five families. Brain 129, 2966–2976. [DOI] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TVP, Hyman BT , et al. (1999). Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2, 271–276. [DOI] [PubMed] [Google Scholar]
- Cheng I, Palop J, Esposito L, Bien-Ly N, Yan F, and Mucke L (2004). Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nat Med 10, 1190–1192. [DOI] [PubMed] [Google Scholar]
- Cheng I, Scearce-Levie K, Legleiter J, Palop J, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe K, Muchowski P, and Mucke L (2007). Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem, In press. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, and Mucke L (2005). Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci 25, 9694–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu G-Q, Kojima N, Masliah E, and Mucke L (2004). Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci 24, 4692–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, and Rubenstein JL (2005). Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci 8, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Colmers WF, Lukowiak K, and Pittman QJ (1987). Presynaptic action of neuropeptide Y in area CA1 of the rat hippocampal slice. J Physiol 383, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, and Vitek MP (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci 114, 1179–1187. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, and Spencer DD (1989). Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res 495, 387–395. [DOI] [PubMed] [Google Scholar]
- Del Vecchio RA, Gold LH, Novick SJ, Wong G, and Hyde LA (2004). Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci Lett 367, 164–167. [DOI] [PubMed] [Google Scholar]
- Diez M, Danner S, Frey P, Sommer B, Staufenbiel M, Wiederhold KH, and Hokfelt T (2003). Neuropeptide alterations in the hippocampal formation and cortex of transgenic mice overexpressing β-amyloid precursor protein (APP) with the Swedish double mutation (APP23). Neurobiol Dis 14, 579–594. [DOI] [PubMed] [Google Scholar]
- Diez M, Koistinaho J, Kahn K, Games D, and Hökfelt T (2000). Neuropeptides in hippocampus and cortex in transgenic mice overexpressing V717F β-amyloid precursor protein - Initial observations. Neuroscience 100, 259–286. [DOI] [PubMed] [Google Scholar]
- Edwards-Lee T, Ringman JM, Chung J, Werner J, Morgan A, St George Hyslop P, Thompson P, Dutton R, Mlikotic A, Rogaeva E, and Hardy J (2005). An African American family with early-onset Alzheimer disease and an APP (T714I) mutation. Neurology 64, 377–379. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, and Seabrook GR (2001). Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci 21, 4691–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, and Bontempi B (2005). The organization of recent and remote memories. Nat Rev Neurosci 6, 119–130. [DOI] [PubMed] [Google Scholar]
- Freund TF, and Buzsáki G (1996). Interneurons of the hippocampus. Hippocampus 6, 347–470. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F , et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373, 523–527. [DOI] [PubMed] [Google Scholar]
- Gariboldi M, Conti M, Cavaleri D, Samanin R, and Vezzani A (1998). Anticonvulsant properties of BIBP3226, a non-peptide selective antagonist at neuropeptide Y Y1 receptors. Eur J Neurosci 10, 757–759. [DOI] [PubMed] [Google Scholar]
- Glenner GG, and Wong CW (1984). Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120, 885–890. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel DW Jr., Morris JC, Growdon JH, and Hyman BT (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 16, 4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, and Chen F (2004). Transgenic animal models of Alzheimer’s disease and related disorders: Histopathology, behavior and therapy. Mol Psychiatry 9, 664–683. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, and Barnes CA (2000). Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci 20, 3993–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser WA, Morris ML, Heston LL, and Anderson VE (1986). Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 36, 1226–1230. [DOI] [PubMed] [Google Scholar]
- He J, Yamada K, and Nabeshima T (2002). A role of Fos expression in the CA3 region of the hippocampus in spatial memory formation in rats. Neuropsychopharmacology 26, 259–268. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Hauser WA, Annegers JF, Kokmen E, and Rocca WA (1996). Dementia and adult-onset unprovoked seizures. Neurology 46, 727–730. [DOI] [PubMed] [Google Scholar]
- Hsia A, Masliah E, McConlogue L, Yu G, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, and Mucke L (1999). Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA 96, 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, and Malinow R (2006). AMPAR Removal Underlies Aβ-Induced Synaptic Depression and Dendritic Spine Loss. Neuron 52, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, and Malinow R (2003). APP processing and synaptic function. Neuron 37, 925–937. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, and Colmers WF (1993). On the sites of presynaptic inhibition by neuropeptide Y in rat hippocampus in vitro. Hippocampus 3, 103–111. [DOI] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, and Mobley WC (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci 24, 8153–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi DT, and Chen KS (2005). Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer’s Disease. Genes Brain and Behav 4, 173–196. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, and Buckmaster PS (2003). Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci 23, 2440–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopniczky Z, Dobo E, Borbely S, Vilagi I, Detari L, Krisztin-Peva B, Bagosi A, Molnar E, and Mihaly A (2005). Lateral entorhinal cortex lesions rearrange afferents, glutamate receptors, increase seizure latency and suppress seizure-induced c-fos expression in the hippocampus of adult rat. J Neurochem 95, 111–124. [DOI] [PubMed] [Google Scholar]
- Larner AJ, and Doran M (2006). Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin-1 gene. J Neurol 253, 139–158. [DOI] [PubMed] [Google Scholar]
- Lee KW, Lee SH, Kim H, Song JS, Yang SD, Paik SG, and Han PL (2004). Progressive cognitive impairment and anxiety induction in the absence of plaque deposition in C57BL/6 inbred mice expressing transgenic amyloid precursor protein. J Neurosci Res 76, 572–580. [DOI] [PubMed] [Google Scholar]
- Link W, Koniezko U, Kauselmann G, Krug M, Schwanke B, Frey U, and Kuhl D (1995). Somatodendritic exprsesion of an immediate early gene is regulated by synaptic activity. Proc Natl Acad Sci USA 92, 5734–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, Honack D, Fassbender CP, and Nolting B (1991). The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res 8, 171–189. [DOI] [PubMed] [Google Scholar]
- Lozsadi DA, and Larner AJ (2006). Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer’s disease. Dement Geriatr Cogn Disord 22, 121–124. [DOI] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, and Worley PF (1995). Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 14, 433–445. [DOI] [PubMed] [Google Scholar]
- Marcon G, Giaccone G, Cupidi C, Balestrieri M, Beltrami CA, Finato N, Bergonzi P, Sorbi S, Bugiani O, and Tagliavini F (2004). Neuropathological and clinical phenotype of an Italian Alzheimer family with M239V mutation of presenilin 2 gene. J Neuropathol Exp Neurol 63, 199–209. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Ashford JW, Goodman Y, and Mattson MP (1995). Anticonvulsants attenuate amyloid β-peptide neurotoxicity, Ca2+ deregulation, and cytoskeletal pathology. Neurobiol Aging 16, 187–198. [DOI] [PubMed] [Google Scholar]
- Marksteiner J, Ortler M, Bellmann R, and Sperk G (1990). Neuropeptide Y biosynthesis is markedly induced in mossy fibers during temporal lobe epilepsy of the rat. Neurosci Lett 112, 143–148. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Pretorius JK, and Leite JP (1995). Reactive synaptogenesis and neuron densities for neuropeptide Y, somatostatin, and glutamate decarboxylase immunoreactivity in the epileptogenic human fascia dentata. J Neurosci 15, 3990–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, and Rydel RE (1993). β-amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer’s disease. TINS 16, 409–414. [DOI] [PubMed] [Google Scholar]
- Mendez M, and Lim G (2003). Seizures in elderly patients with dementia: Epidemiology and management. Drugs Aging 20, 791–803. [DOI] [PubMed] [Google Scholar]
- Mendez MF, Catanzaro P, Doss RC, R AR, and Frey WH 2nd (1994). Seizures in Alzheimer’s disease: Clinicopathologic study. J Geriatr Psychiatry Neurol 7, 230–233. [DOI] [PubMed] [Google Scholar]
- Molinari S, Battini R, Ferrari S, Pozzi L, Killcross AS, Robbins TW, Jouvenceau A, Billard J-M, Dutar P, Lamour Y , et al. (1996). Deficits in memory and hippocampal long-term potentiation in mice with reduced calbindin D28K expression. Proc Natl Acad Sci USA 93, 8028–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, and O’Keefe J (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297, 681–683. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, and McConlogue L (2000). High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci 20, 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JV (2003). The recurrent mossy fiber pathway of the epileptic brain. Neurochem Res 28, 1649–1658. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Mody I, Jeub M, Lie AA, Elger CE, and Beck H (2000). Surviving granule cells of the sclerotic human hippocampus have reduced Ca2+ influx because of a loss of calbindin-D28K in temporal lobe epilepsy. J Neurosci 20, 1831–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe J, Nadel L, Keightley S, and Kill D (1975). Fornix lesions selectively abolish place learning in the rat. Exp Neurol 48, 152–166. [DOI] [PubMed] [Google Scholar]
- Olney JW (1995). NMDA receptor hypofunction, excitotoxicity, and Alzheimer’s disease. NeurobiolAging 16, 459–461. [Google Scholar]
- Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu G-Q, and Mucke L (2005). Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J Neurosci 25, 9686–9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, and Mucke L (2006). A network dysfunction perspective on neurodegenerative diseases. Nature 443, 768–773. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, and Mucke L (2003). Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci USA 100, 9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, and Houser CR (2005). Temporal patterns of fos expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. J Neurosci 25, 7210–7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, et al. (2006). Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444. [DOI] [PubMed] [Google Scholar]
- Qian J, Colmers WF, and Saggau P (1997). Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: Modulation of presynaptic Ca2+ entry. J Neurosci 17, 8169–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Richichi C, Lin EJ, Stefanin D, Colella D, Ravizza T, Grignaschi G, Veglianese P, Sperk G, During MJ, and Vezzani A (2004). Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci 24, 3051–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, and Mucke L (2007). Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Rothman SM, and Olney JW (1995). Excitotoxicity and the NMDA receptor - still lethal after eight years. Trends Neurosci 18, 57–58. [DOI] [PubMed] [Google Scholar]
- Salter MW, and Kalia LV (2004). Src kinases: A hub for NMDA receptor regulation. Nat Rev Neurosci 5, 317–328. [DOI] [PubMed] [Google Scholar]
- Schwarzer C, Kofler N, and Sperk G (1998). Up-regulation of neuropeptide Y-Y2 receptors in an animal model of temporal lobe epilepsy. Mol Pharmacol 53, 6–13. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, and Sabatini BL (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27, 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao LR, and Dudek FE (2005). Changes in mIPSCs and sIPSCs after kainate treatment: evidence for loss of inhibitory input to dentate granule cells and possible compensatory responses. J Neurophysiol 94, 952–960. [DOI] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, and Masliah E (2005). Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci 8, 1343–1349. [DOI] [PubMed] [Google Scholar]
- Smialowska M, Bijak M, Sopala M, and Tokarski K (1996). Inhibitory effect of NPY on the picrotoxin-induced activity in the hippocampus: a behavioural and electrophysiological study. Neuropeptides 30, 7–12. [DOI] [PubMed] [Google Scholar]
- Snider BJ, Norton J, Coats MA, Chakraverty S, Hou CE, Jervis R, Lendon CL, Goate AM, McKeel DW Jr., and Morris JC (2005). Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch Neurol 62, 1821–1830. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, and Greengard P (2005). Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci 8, 1051–1058. [DOI] [PubMed] [Google Scholar]
- Tanzi R, and Bertram L (2005). Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 120, 545–555. [DOI] [PubMed] [Google Scholar]
- Tonder N, Kragh J, Bolwig T, and Zimmer J (1994a). Transient decrease in calbindin immunoreactivity of the rat fascia dentata granule cells after repeated electroconvulsive shocks. Hippocampus 4, 79–83. [DOI] [PubMed] [Google Scholar]
- Tonder N, Kragh J, Finsen BR, Bolwig TG, and Zimmer J (1994b). Kindling induces transient changes in neuronal expression of somatostatin, neuropeptide Y, and calbindin in adult rat hippocampus and fascia dentata. Epilepsia 35, 1299–1308. [DOI] [PubMed] [Google Scholar]
- Tu B, Jiao Y, Herzog H, and Nadler JV (2006). Neuropeptide Y regulates recurrent mossy fiber synaptic transmission less effectively in mice than in rats: Correlation with Y2 receptor plasticity. Neuroscience 143, 1085–1094. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, and Nicoll RA (2006). Arc/Arg3.1: Linking gene expression to synaptic plasticity and memory. Neuron 52, 403–407. [DOI] [PubMed] [Google Scholar]
- Vezzani A, and Sperk G (2004). Overexpression of NPY and Y2 receptors in epileptic brain tissue: An endogenous neuroprotective mechanism in temporal lobe epilepsy? Neuropeptides 38, 245–252. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Sperk G, and Colmers WF (1999). Neuropeptide Y: Emerging evidence for a functional role in seizure modulation. Trends Neurosci 22, 25–30. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, and Selkoe DJ (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Walsh DM, and Selkoe DJ (2004). Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44, 181–193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.