

Supplemental Digital Content is available in the text.
Keywords: chromatin, hormone, human development, progesterone, sexual maturation, transcription factor
Abstract
Background:
Despite in-depth knowledge of the molecular mechanisms controlling embryonic heart development, little is known about the signals governing postnatal maturation of the human heart.
Methods:
Single-nucleus RNA sequencing of 54 140 nuclei from 9 human donors was used to profile transcriptional changes in diverse cardiac cell types during maturation from fetal stages to adulthood. Bulk RNA sequencing and the Assay for Transposase-Accessible Chromatin using sequencing were used to further validate transcriptional changes and to profile alterations in the chromatin accessibility landscape in purified cardiomyocyte nuclei from 21 human donors. Functional validation studies of sex steroids implicated in cardiac maturation were performed in human pluripotent stem cell–derived cardiac organoids and mice.
Results:
Our data identify the progesterone receptor as a key mediator of sex-dependent transcriptional programs during cardiomyocyte maturation. Functional validation studies in human cardiac organoids and mice demonstrate that the progesterone receptor drives sex-specific metabolic programs and maturation of cardiac contractile properties.
Conclusions:
These data provide a blueprint for understanding human heart maturation in both sexes and reveal an important role for the progesterone receptor in human heart development.
Clinical Perspective.
What Is New?
Recent advances in single-cell sequencing are providing an unprecedented view of cellular heterogeneity in the healthy and diseased human heart.
We performed single nucleus RNA sequencing to capture transcriptional changes across multiple cardiac cell populations during human heart development from fetal stages to adulthood.
Our data reveal sex-specific transcriptional mechanisms governing maturation of multiple cell types in the heart, including a previously unrecognized role for the progesterone receptor in human cardiomyocyte maturation.
What Are the Clinical Implications?
Despite well-documented sex differences in cardiovascular physiology and disease outcomes, the transcriptional mechanisms driving the emergence of sex-specific human cardiac biology remain largely unknown.
Our findings uncover sex-specific functions of the progesterone receptor in the regulation of cardiomyocyte metabolism in males and females, thus raising the possibility that progesterone signaling could be therapeutically targeted to modify sexually dimorphic disease outcomes in the future.
The potential direct effects of progesterone on the heart should be considered with respect to cardiovascular adaptations during pregnancy and in women taking oral contraceptives or hormone replacement therapy.
Heart development begins in the embryo and continues into adulthood to establish the mature gene expression programs that sustain cardiac output throughout life. The human heart undergoes profound changes in many facets of cardiac physiology after birth, including increased contractility and shifts in metabolic substrate use to adapt to the increased oxygen perfusion and growth demands of the postnatal environment.1,2 Although the molecular mechanisms governing embryonic development in vertebrates have been intensively interrogated, little is known about the mechanisms controlling postnatal maturation and the acquisition of adult cell identity in mammals.1 Although recent studies have begun to provide a transcriptional framework for cardiac development and disease at the single-cell level,3–7 the mechanisms regulating postnatal heart maturation in humans have not been defined. Moreover, recent single-cell transcriptomic studies have identified sexually dimorphic gene expression programs in adult mice8 and humans,7 suggesting that cardiac identity and cellularity are dependent on biological sex and regulated by gonadal hormones.9 However, despite well-documented sex differences in cardiovascular physiology and disease outcomes,10,11 the transcriptional mechanisms governing the emergence of sex-specific gene expression programs during human heart development are unknown.
Methods
Please refer to the Data Supplement Methods and Tables I–IV for a full description of experimental procedures.
Human Ethics and Tissue Collection
Human ethics for cardiac tissue biobanking and human pluripotent stem cell research was approved by The Royal Children’s Hospital Melbourne Human Research Ethics Committee (approval numbers: HREC 38192, HREC 33001A, HREC 37172A, HREC 36358A), The University of Queensland Institutional Human Research Ethics Committee (SBS/2014000329), QIMR Berghofer Human Research Ethics Committee (P2385), and The University of Sydney Human Research Ethics Committee (HREC 2016/923). Fetal tissues were procured from Advanced Bioscience Resources (CA). Informed consent was obtained from all human subjects, and studies were conducted in accordance with the approved protocols and guidelines from the National Health and Medical Research Council of Australia. All human heart specimens (apical region of left ventricle) were obtained from donor hearts without any clinical evidence of cardiovascular disease and stored in liquid nitrogen (Table V in the Data Supplement).
Animal Ethics and Tissue Collection
Animal experiments were conducted in accordance with the relevant codes of practice for the care and use of animals for scientific purposes as stipulated by the National Health and Medical Research Council of Australia and conducted with approval from the Animal Ethics Committees of the Alfred Medical Research and Education Precinct (E/1706/2017/B) and The University of Adelaide (M 2018-122). For neonatal recombinant adeno-associated viral vector (AAV) injection experiments, C57BL/6J mice were maintained in a 12-hour light:dark cycle at a temperature of 22±1 °C and provided ad libitum access to food and water during the study duration. At postnatal day 1, male and female C57BL/6J mice received a single intraperitoneal injection of AAV serotype 6 vectors containing either the longest isoform of the full-length human progesterone receptor (PGR) sequence NM_000926.4 (AAV_PGR) or a negative control AAV lacking a sequence insert in the multiple cloning site (AAV_Con), under the control of the human cytomegalovirus promoter. All animals were treated with 1×1012 vector genomes resuspended in a total volume of 50 μL of PBS. Neonates were weaned between 21 and 28 days of age. PRKO (Pgrtm1Bwo knockout) mice were obtained from The Jackson Laboratory and maintained at The University of Adelaide under a 12-hour light:dark cycle at a temperature of 22±1 °C with ad libitum access to food and water during the study duration. For both AAV and PRKO mice studies, male and female adult mice were humanely euthanized by cervical dislocation at postnatal day 56, and the heart was rapidly excised. Atrial tissues were removed from ventricles (septum intact), and ventricles were collected and snap-frozen in liquid nitrogen for storage at –80 °C until processing.
Cardiac Nuclei Isolation
Cardiac nuclei were isolated as described previously,12 with minor modifications. For single-nucleus RNA sequencing (RNA-seq [snRNA-seq]), ≈20 to 100 mg of human left ventricular tissue were used per sample, whereas ≈0.5 to 1.0 g of tissue was required for bulk RNA-seq of purified cardiomyocyte nuclei (mouse and human).12 All steps were conducted on ice.
SnRNA-Seq Library Preparation and Sequencing
Isolated nuclei were stained with Hoechst 33342 (Thermo Fisher Scientific) before sorting on an Influx cell sorter (Becton Dickinson) with a 70-µmol/L nozzle and 60 psi pressure setting. Sorted nuclei were counted on a hemocytometer to calculate nuclei density and then loaded onto the Chromium Controller (10X Genomics) for gel bead emulsion formation with ≈10 000 nuclei loaded per sample for library preparation. After gel bead emulsion formation, library preparation was conducted according to the manufacturer’s recommended protocol using the Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit version 3.1. Libraries were sequenced on the NovaSeq 6000 (Illumina) at 50 000 reads per nuclei resolution.
PCM1 Cardiomyocyte Nuclei Enrichment, RNA-Seq Library Preparation, and Sequencing
PCM1 nuclear enrichment, RNA extraction, library preparation, and sequencing were performed as described previously with minor modifications.12,13 Full details are provided in the Data Supplement.
PCM1+ Cardiomyocyte Bulk Assay for Transposase-Accessible Chromatin Sequencing Library Preparation and Sequencing
After PCM1 nuclei sorting and pelleting, Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) libraries were generated according to the methods described in a previous publication.14 Libraries were sequenced for 120 million paired-end reads on HiSeq2500 (Illumina).
Bioinformatics Analysis for SnRNA-Seq
The sequencing reads were mapped, processed, and counted using Cell Ranger (version 3.0.2), resulting in a table of unique molecular identifier counts for 33 939 genes for each of the 9 samples. The number of cells captured per biological replicate ranged from 1681 to 10 948, with a median of 5558. All subsequent analysis was performed using the R statistical programming language (version 3.6.0). The quality of the cells was assessed for each sample independently by examining the total number of cells, the distributions of total unique molecular identifier counts, the number of unique genes detected, and the proportions of ribosomal and mitochondrial content per cell.
For each developmental group (fetal, young, adult) we performed gene filtering, SCTransform normalization,15 data integration of the 3 biological replicates,16–18 data scaling, and graph-based clustering separately, using the R package Seurat (version 3.0.2). For gene filtering, mitochondrial and ribosomal genes were discarded, as well as genes that were not annotated. Genes that had at least 1 count in at least 20 cells were retained for further analysis, assuming a minimum cluster size of 20 cells. All genes on the X and Y chromosomes were removed before clustering and all subsequent analysis. Data integration of the biological replicates for each group was performed using canonical correlation analysis with 30 dimensions and 3000 integration anchors followed by data scaling. Clustering of the cells was performed with 20 principal components and resolution parameter set to 0.3 for fetal and young samples and 0.6 for adult samples. Taking this strategy, 22, 17, and 21 clusters were identified in the fetal, young, and adult samples, respectively.
For marker analysis, the counts were log2 transformed, adding a pseudocount of 0.5 proportional to the library size. Using the limma R package (version 3.40.2), marker genes were identified as significantly upregulated genes for each cluster using moderated t tests, accounting for the mean variance trend and using robust empirical Bayes shrinkage of the variances, followed by TREAT tests specifying a log-fold change threshold of 0.5 and false discovery rate (FDR) cutoff <0.0519-21. The marker genes, in conjunction with visualizations including violin plots, t-distributed stochastic neighbor embedding, and uniform manifold approximation, as well as projection plots, clustering trees,22 and heat maps showing expression of previously published marker genes, were used to aid in interpretation of the clusters. Seven broad cell types were defined for fetal, young, and adult samples by combining clusters together that had similar gene expression profiles. In addition, a small population of erythroid cells in the fetal samples was identified. Marker analysis at the broad cell type level was performed as described earlier for each group separately, as well as for all samples across groups, confirming the broad cell type identities. Gene set testing to test for enrichment of REACTOME gene sets (https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp) and transcription factor (TF) target sets was performed using CAMERA.23 The ENCODE (Encyclopedia of DNA Elements) TF target sets were curated as described previously.13 Significant differences in cell type composition between groups was assessed using a propeller test from the speckle R package (https://github.com/Oshlack/speckle).
Pseudobulk samples were created by summing the counts for each biological replicate for each cell type.24 The data were transformed using voom25 and differential expression analysis within each broad cell type performed using limma, testing for genes differentially expressed across the developmental trajectory, as well as sex differences within each time point. Robust empirical Bayes variance estimation was performed, and TREAT tests with a log-fold change threshold of 0.5 identified significantly differentially expressed genes for each comparison at FDR <0.05. Gene set testing of the REACTOME and ENCODE TF targets sets was performed using CAMERA.
To identify subclusters of the broad cell type lineages, cells from all 9 biological replicates were integrated for each broad cell type separately. Cells that had <500 genes expressed and <2500 total unique molecular identifier counts were discarded before data integration and clustering. Gene filtering was performed as before. The data were normalized, integrated, scaled, and clustered (resolution parameter = 0.1) as described earlier. Marker analysis and gene set testing were performed. These analysis steps were repeated for each of the 7 broad cell types. For broad cell types with fewer cells, the number of principal components used for clustering was reduced (immune cells = 15 principal components, neural cells and smooth muscle cells = 10 principal components). Propeller tests were run to identify population shifts across development within the broad cell types.
PCM1+ Cardiomyocyte Bulk RNA-Seq and ATAC-Seq Bioinformatics Analyses
PCM1+ bulk RNA-seq and ATAC-seq bioinformatic analyses were conducted as described previously with minor modifications.13 Full details are provided in the Data Supplement.
Human-Induced Pluripotent Stem Cell–Cardiomyocyte Differentiation for ATAC-Seq
The human induced pluripotent stem cell (hiPSC) lines MCRIi001-A (male) and MCRIi004-A (female) were reprogrammed by the iPSC Derivation Core Facility at the Murdoch Children’s Research Institute from the blood of healthy donors.26 Both lines were cultured as described previously.27 See the Data Supplement Methods for a full description.
Human Embryonic Stem Cell–Derived Cardiomyocyte Differentiation, Extracellular Field Potential, and Impedance Analysis
The human embryonic stem cell (hESC) line HES3 NKX2-5eGFP/w (female) was used for functional studies in 2-dimensional monolayer cultures of human cardiomyocytes.28 Cardiomyocytes were cultured in maturation media and placed on the CardioExcyte plate reader (Nanion) for calibration. At least 12 hours after calibration, baseline recordings were obtained, and the cells were then treated with the following conditions: 0.1% ethanol as a vehicle control, 1 µM hydrocortisone, 1 µM testosterone, or 10 µM progesterone. Another recording was made 48 hours after treatment, and each well was then normalized to its baseline recording before comparisons were performed between groups. All conditions were electrically stimulated at 1 Hz, and only samples that were definitively paced at 60±6 beats per minute were included for downstream analyses. See Data Supplement Methods for a full description.
Human Cardiac Organoid Differentiation and Functional Analysis
Human cardiac organoids (hCOs) were generated as described previously using H9 human embryonic stem cells (female) until day 15.27, 29, 30 Over 4 days, hCOs were exposed to different treatment conditions including 0.1% ethanol as a vehicle control, 10 µM progesterone, and 10 µM progesterone with 10 µM mifepristone with treatment refreshed every 48 hours. hCOs were measured for functional parameters such as force, rate, 50% activation time, and 50% relaxation time as per previous publications.27,29
Statistical Analysis
All nonbioinformatics statistical analyses were performed using GraphPad Prism 8.1.1. For animal experiments, sample sizes were determined using predicted size of effects from previous work with an α value of 0.05 and a β value of 0.1, and animals were randomly assigned to experimental groups. For comparisons between 2 groups, a 2-tailed unpaired Student t test was used. For multiple group comparisons, a 1-way ANOVA followed by Tukey posttest was used. For comparison between multiple groups with multiple variables, a 2-way ANOVA followed by Sidak or Tukey posttest was used. For quantitative polymerase chain reaction validation of PCM1 enrichment, nonparametric Wilcoxon matched-pairs signed-rank test was performed on individual gene markers, because the data were not normally distributed and each PCM1+ sample was coupled to its respective PCM1– control. All data are presented as mean±SEM. Detailed statistical analysis methods and sample sizes are indicated in the figure legends. For all statistical analyses, *P≤0.05 and **P≤0.001.
Data Access
All data in the main and supplemental figures are associated with raw data and are available on reasonable request. All snRNA-seq, bulk RNA-seq, and ATAC-seq raw fastq.gz files have been deposited to the Gene Expression Omnibus under accession No. GSE156707. Comprehensive bioinformatics analyses can be retrieved from the following websites:
https://bphipson.github.io/Human_Development_snRNAseq/
https://evangelynsim.github.io/Human_Development_RNAseq_bulk/
https://evangelynsim.github.io/Human_Development_ATACseq_bulk/
Results
Single-Cell Analysis Reveals Age- and Sex-Dependent Transcriptional Changes During Human Heart Development
To gain insight into the developmental mechanisms underpinning human heart maturation, we transcriptionally profiled cardiac tissue samples isolated from the left ventricle of healthy donor hearts at fetal, young, and adult stages of development (Figure 1A and Table V in the Data Supplement). Because intact cardiomyocytes cannot be readily dissociated from frozen heart tissue or sorted through microfluidic channels in conventional Drop-seq platforms for single-cell gene expression analysis,7,31 we adapted protocols for isolation of individual cardiac nuclei for downstream transcriptional profiling (Figure 1A). SnRNA-seq of 54 140 single cardiac nuclei from fetal, young, and adult hearts revealed 8 major cell types within the human left ventricle (Figure 1B and Figure I in the Data Supplement). Analysis of marker genes and gene ontology terms in these 8 clusters enabled discrimination of major cardiac cell types including cardiomyocytes, fibroblasts, endothelial cells, epicardial cells, immune cells, smooth muscle cells, and neural and erythroid cells (Figures II and III in the Data Supplement). With the exception of erythroid cells, which were only present in cardiac nuclear preparations from fetal hearts, all other major cardiac cell types were detected across all developmental time points (Figure IB in the Data Supplement). Cardiac maturation was associated with major shifts in cellular composition characterized by a significant expansion in the relative proportion of cardiac fibroblasts and immune cells in the postnatal human heart, accompanied by a significant decrease in the proportion of cardiomyocytes (Figure 1C, FDR<0.05, propeller test). As reported previously, we observed variation in individual sample contributions to snRNA-seq clusters.7,32 All fetal cardiac cell types clustered distinctly from postnatal cells (Figure 1D), with the majority of differentially expressed genes during cardiac maturation accounted for by changes in cardiomyocytes, epicardial cells, fibroblasts, and endothelial cells (Figure 1E). Statistically significant sex differences in the transcriptional profiles of all cardiac cell types were identified across all stages of development, with the majority of these differences observed in cardiomyocytes (Figure 1F). Cardiomyocyte maturation in humans was associated with repression of cell cycle genes and activation of metabolic genes involved in oxidative phosphorylation and the respiratory electron transport chain (Figure 1G), which is consistent with well-described processes regulated during cardiac maturation in mammals.13 Maturation of nonmyocyte populations was associated with increased interferon signaling capacity and responsiveness in fibroblasts, endothelial cells, immune cells, and epicardial and neural cells (Figure 1G). In addition, 37 subclusters of cardiac cells were identified across the 7 major cell types (Figures IV, V, and VI in the Data Supplement), consistent with other reports of heterogeneity and interindividual variation within human cardiac cell populations.7 For example, 7 subclusters of cardiomyocytes were identified, including mitotic cardiomyocytes (cluster 2), which were more abundant in the fetal heart compared with postnatal human hearts (Figures VIA through VIC and VII in the Data Supplement). Overall, human heart development is characterized by a maturation trajectory in all cardiac cell types, with the largest number of changes including sex differences occurring in cardiomyocytes.
Figure 1.
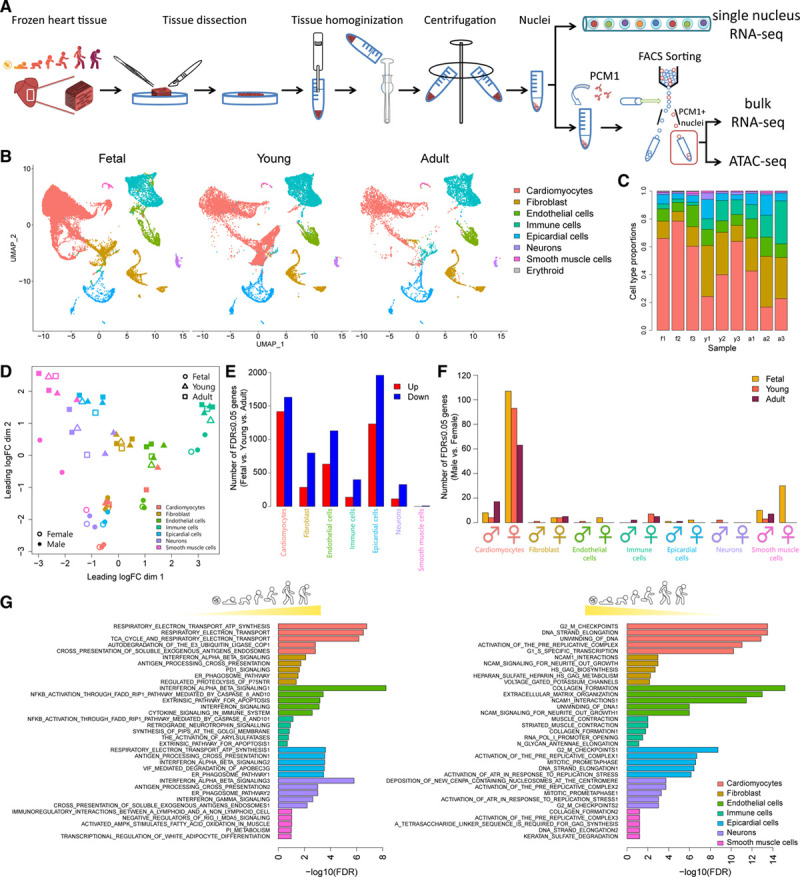
SnRNA-seq reveals age- and sex-dependent maturation of cardiac cells during human development. A, Schematic of nuclei isolation for snRNA-seq, bulk RNA-seq, and ATAC-seq. B, UMAP plot of nuclei showing distinct clusters of cardiac cell types at different stages of human heart development. C, Bar plot of cell type proportions for each biological replicate (f = fetal, y = young, a = adult, 1–3 denote biological replicate). D, MDS plot displaying pseudobulk profiles of cell types in samples. The largest projection of expression variation is shown on the x axis, and the second largest orthogonal projection is on the y axis. E, Bar plot of the number of DE genes in each cell type showing the number of DE genes that are progressively upregulated (red) or downregulated (blue) during human heart development from fetal to adult stages. F, Bar plot showing the number of DE genes between males and females across different cell types in fetal (gold), young (orange), and adult (maroon). G, GSEA reactome analysis of developmentally upregulated (left) and downregulated (right) genes for each cardiac cell type. Top 5 gene sets displayed for each cell type. ATAC-seq indicates Assay for Transposase-Accessible Chromatin using sequencing; DE, differentially expressed; GSEA, gene set enrichment analysis; MDS, multidimensional scaling; snRNA-seq, single-nucleus RNA sequencing; and UMAP, Uniform Manifold Approximation and Projection.
Cardiomyocytes Undergo Profound Transcriptional Maturation in a Sex-Dependent Manner During Human Heart Development
To further explore the degree to which sex differences contribute to the emergence of diverse cardiomyocyte transcriptional networks during cardiac maturation, we performed deep RNA sequencing of highly purified cardiomyocyte nuclei on an expanded set of human cardiac tissue samples (n=21 samples including 10 males and 11 females; Tables V and VI in the Data Supplement). Consistent with previous reports,12,13,33,34 PCM1-purified cardiomyocyte nuclei exhibited increased ploidy during cardiomyocyte maturation (Figures VIIIA and VIIIB in the Data Supplement) and were enriched for cardiomyocyte markers and depleted of nonmyocyte transcripts (Figure VIIIC in the Data Supplement). Unsupervised hierarchical clustering and principal coordinate analysis revealed distinct clusters of cardiomyocytes that could be separated on the basis of developmental stage and sex, with the majority of sex differences emerging in adulthood (Figure 2A and 2B). Consistent with our snRNA-seq analysis, cardiomyocyte maturation was associated with highly connected pathways linked to repression of cell cycle genes and activation of metabolic processes related to the tricarboxylic acid cycle and respiratory electron transport chain (Figure 2C and Figure IXA through IXD in the Data Supplement). Bulk RNA-seq of PCM1-purified cardiomyocyte nuclei identified thousands of differentially expressed genes among fetal, young, and adult cardiomyocytes (Figure 2D) with strong overlap between differentially expressed genes identified by bulk RNA-seq and snRNA-seq (Figure XA in the Data Supplement). Sex differences identified using snRNA-seq were confirmed and expanded using bulk RNA-seq of purified cardiomyocyte nuclei (Figure XB in the Data Supplement). It is interesting to note that although broad cardiomyocyte maturation programs were shared between males and females, a large subset of genes (N=1647, FDR≤0.05) exhibited sexually dimorphic developmental regulation characterized by reciprocal regulation of genes during cardiomyocyte maturation in males and females (Figure 2E). Transcriptional networks that were induced in males but repressed in females during cardiomyocyte maturation were associated with chromatin organization, histone modification, and RNA processing (Figure 2F). In contrast, transcriptional networks that were induced in females but repressed in males during cardiomyocyte maturation were involved in vesicle-mediated transport (Figure 2F). Although some sex differences were apparent between male and female cardiomyocytes at early stages of fetal development (N=35, FDR≤0.05, Figure 2G), a much larger number of differentially expressed genes (N=2818, FDR≤0.05, Figure 2G) were identified in adulthood, and these were mostly involved in the regulation of cytoskeleton organization, RNA processing, and cell adhesion (Figure 2H and Figure IXE in the Data Supplement). Sex differences at fetal stages of development were primarily attributed to X and Y chromosome–linked genes, whereas the majority of differentially expressed genes in adulthood were autosomal (see gold circles for X- and Y-linked genes in Figure 2G). These results unveil sex-specific transcriptional programs that are established during human cardiomyocyte maturation.
Figure 2.
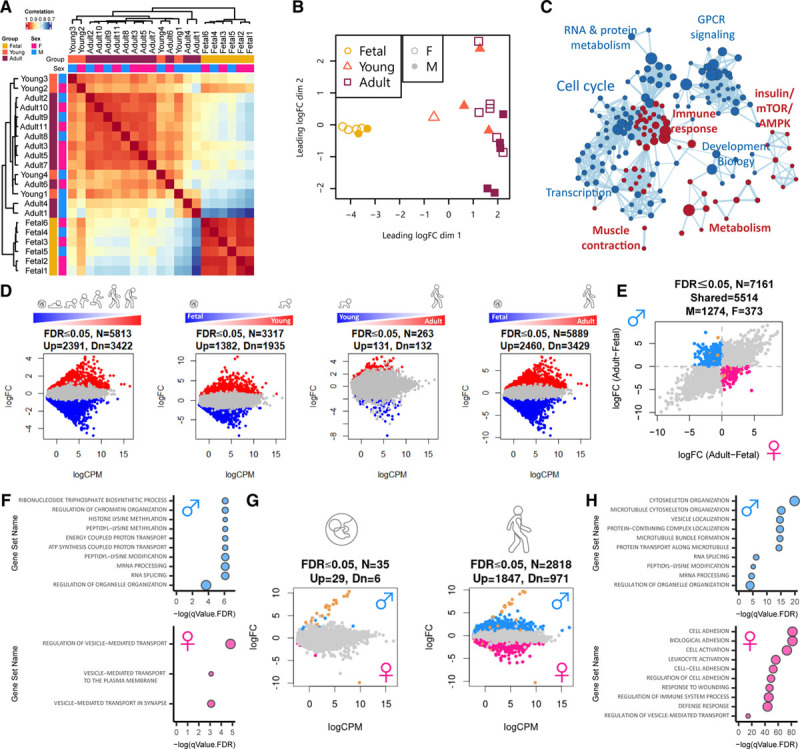
Cardiomyocytes undergo sex-specific transcriptional maturation during human development. A, Pearson correlation heat map of bulk RNA-seq showing distinct clustering of PCM1-purified cardiomyocytes from fetal versus young and adult samples. B, MDS plot illustrating the largest source of variation in the data are between fetal (gold circles) versus young (orange triangles) and adult (maroon squares) cardiomyocytes with some separation of female versus male (open versus closed) cardiomyocytes evident in adult samples. C, Cytoscape representation of GSEA reactome analysis of genes that are progressively upregulated (red) or downregulated (blue) during human cardiomyocyte maturation from fetal to adult stages. D, Mean-difference scatter plots highlighting differentially expressed genes (both sexes) for all comparisons including dynamically regulated genes (fetal>young>adult), fetal versus young, young versus adult, and fetal versus adult cardiomyocytes. Genes that are significantly upregulated (red) or downregulated (blue) are highlighted (FDR≤0.05). E, Log-fold change scatter plot of developmentally regulated genes (fetal versus adult) displaying reciprocal regulation during cardiomyocyte maturation in males and females. Genes that are upregulated in males and downregulated in females (blue), upregulated in females and downregulated in males (pink), or on chromosome X or Y (gold) are highlighted. F, Gene ontology analysis for genes that are upregulated in males and downregulated in females (blue) or upregulated in females and downregulated in males (pink). G, Mean-difference scatter plots comparing male versus female cardiomyocytes at fetal (left) and adult (right) stages. Genes that are upregulated in males (blue), upregulated in females (pink), or on chromosome X or Y (gold) are highlighted (FDR≤0.05). H, Gene ontology analysis of differentially expressed genes between adult male (blue) and female (pink) cardiomyocytes. An FDR≤0.05 using the correction procedure of Benjamini and Hochberg was used. FDR indicates false discovery rate; GSEA, gene set enrichment analysis; and MDS, multidimensional scaling.
Analysis of the Chromatin Accessibility Landscape Identifies TFs Associated With Human Cardiomyocyte Maturation
We next sought to identify potential transcriptional drivers of sex-specific maturation programs by surveying the accessible chromatin landscape of developing cardiomyocytes. Open chromatin regions were analyzed using the ATAC-seq on PCM1-purified nuclei. Fetal, young, adult, and hiPSC-derived cardiomyocytes composed of both male and female samples were profiled across all groups (n=27 samples in total, Tables V and VII in the Data Supplement). Principal coordinate analysis and unsupervised hierarchical clustering separated samples on the basis of developmental stage and sex with the majority of differences accounted for by hiPSC-derived cardiomyocytes compared with native cardiomyocytes (Figure 3A and 3B). Human cardiomyocyte maturation was associated with global shifts in chromatin accessibility across 4707 genomic regions from midgestation to adulthood (Figure 3C). The majority (>99%) of these developmental changes in chromatin accessibility emerged between midgestation (19–20 weeks) and early childhood (<4 years) with very few distinguishable peaks identified between young and adult samples (Figure 3C). The distribution of ATAC-seq peaks across genomic features was comparable among fetal, young, and adult samples (Figure 3D). However, hiPSC-derived cardiomyocytes were characterized by a greater proportion of ATAC-seq peaks across gene promoters (Figure 3D). The majority (≈70%) of differentially accessible chromatin regions among fetal, young, and adult groups occurred across intronic and intergenic regions with ≈20% situated around gene promoters and ≈10% falling inside exons and transcription termination sites (Figure 3D). The distribution of ATAC-seq peaks across autosomes was comparable between male and female cardiomyocytes at all developmental stages (Figure XIA in the Data Supplement). As expected, open chromatin regions at gene promoters were strongly correlated with active gene transcription (Figure XIB in the Data Supplement). In accordance with previously published RNA-seq data from PCM1-enriched human cardiomyocyte nuclei34 and ATAC-seq data from adult human heart tissue from ENCODE, integration of our ATAC-seq and RNA-seq data sets validated known cardiomyocyte maturation markers (Figure XIC in the Data Supplement). For example, cardiomyocyte maturation was associated with transcriptional repression and loss of chromatin accessibility around the promoters of TNNI1 and BMP5 (Figure XIC in the Data Supplement), as well as transcriptional activation and gain of accessibility around the promoters of the metabolic genes PDK4 and PFKFB2 (Figure XIC in the Data Supplement). To identify putative TFs driving cardiomyocyte maturation programs, we next surveyed accessible chromatin regions for enriched TF motifs across all groups in our data set (Figure 3E). Accessible chromatin regions in hiPSC-derived cardiomyocytes were highly enriched for pluripotency TF binding motifs (Figure 3E), suggesting that in vitro cardiac differentiation protocols do not completely remodel the genomic landscape of human pluripotent stem cells to differentiated cardiomyocytes. Comparison of fetal, young, and adult cardiomyocytes revealed a progressive loss of accessibility around TF binding sites associated with cardiac development (GATA4, TBX20) and the cell cycle (E2F1, E2F4, and TEAD; Figure 3E). In contrast, cardiomyocyte maturation was associated with a gain of accessibility around TF binding sites for steroid hormone nuclear receptors (glucocorticoid response element, androgen response element, and PGR), as well as TFs belonging to the AP1-JUN family (Figure 3E).
Figure 3.
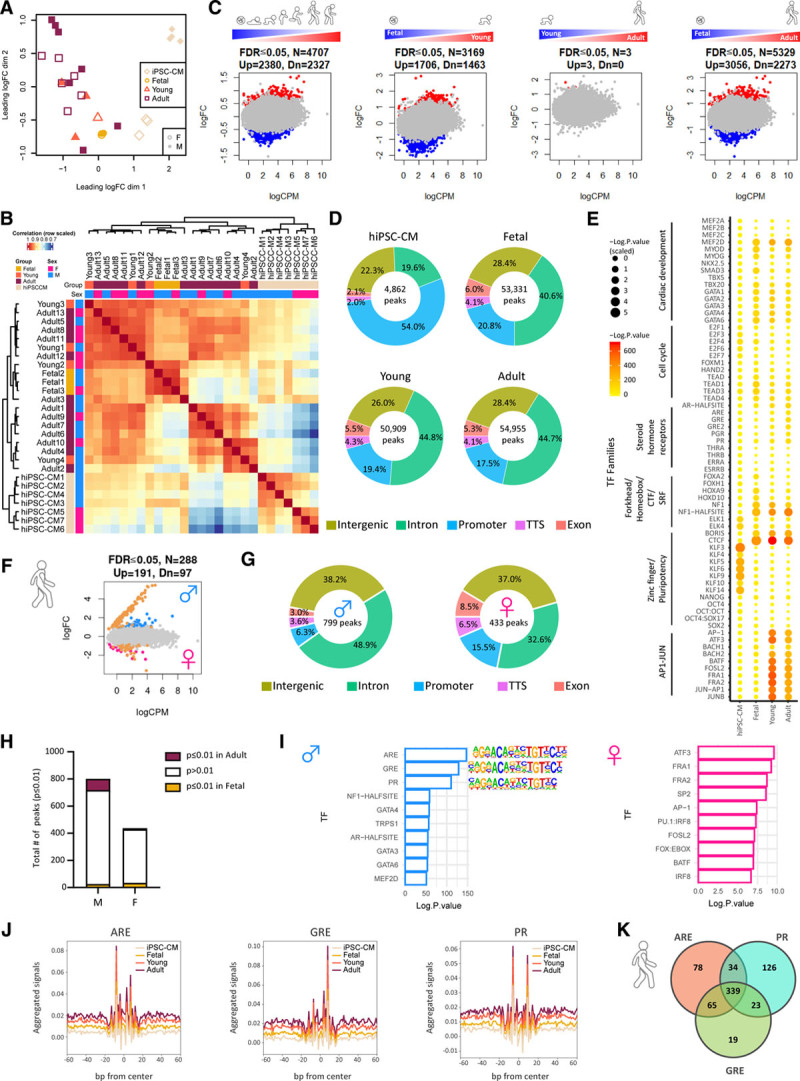
Cardiomyocyte maturation is associated with sex-specific chromatin remodeling around steroid hormone nuclear receptor motifs during human heart development. A, MDS plot shows that the largest source of variation in the ATAC-seq data are between the hiPSC-CM (apricot) and fetal (gold), young (orange), and adult (maroon) samples. Female samples are indicated with open plotting characters and male closed. B, Pearson correlation heat map of bulk ATAC-seq illustrating distinct clustering of PCM1-purified cardiomyocytes from hiPSC-CM versus fetal, young, and adult samples. C, Mean-difference scatter plots of differentially regulated open chromatin regions for all comparisons including dynamically regulated regions (fetal>young>adult), fetal versus young, young versus adult, and fetal versus adult cardiomyocytes. Regions that are more accessible (red) or less accessible (blue) are highlighted (FDR≤0.05). D, Genomic distribution of ATAC-seq peaks in each group. E, TF motif predictions for open chromatin regions in hiPSC-CM, fetal, young, and adult cardiomyocytes. Circle size and color denotes the statistical significance of enriched TFs at each specific time point. F, Mean-difference scatter plot of open chromatin regions in adult male versus female cardiomyocytes. Regions that are more accessible in females (pink), more accessible in males (blue), or on chromosome X or Y (gold) are highlighted. G, Genomic distribution of differentially regulated open chromatin regions in adult male and female cardiomyocytes. H, Bar plot showing number of developmentally regulated ATAC-seq peaks (fetal = gold, adult = maroon) for differentially open chromatin regions in adult male (M) and female (F) cardiomyocytes. I, Bar plot showing significantly enriched TF binding motifs in differentially regulated open chromatin regions between adult male (blue) and female (pink) cardiomyocytes. J, TF footprinting for ARE, GRE, and PR confirming increased binding to open chromatin regions in adult (maroon) and young (orange) cardiomyocytes versus fetal (gold) and iPSC-derived (apricot) cardiomyocytes. Venn diagram showing shared and unique transcription factor binding sites for ARE, GRE, and PR in open chromatin regions of adult cardiomyocytes compared with fetal. An FDR≤0.05 with the correction procedure of Benjamini and Hochberg was used. ARE indicates androgen response element; ATAC-seq, Assay for Transposase-Accessible Chromatin using sequencing; FDR, false discovery rate; GRE, glucocorticoid response element; hiPSC-CM, human induced pluripotent stem cell cardiomyocyte; iPSC, induced pluripotent stem cell; MDS, multidimensional scaling; PR, progesterone receptor; and TF, transcription factor.
As expected, significant differences in ATAC-seq peak distribution across the X and Y chromosomes were noted for male and female samples across all groups (Figure XIA in the Data Supplement). Moreover, hundreds of differentially accessible chromatin regions (N=288, FDR≤0.05) were identified between adult male and female cardiomyocytes, with the majority of differences linked to sex chromosomes (see gold circles for X- and Y-linked regions in Figure 3F). Adult male-specific ATAC-seq peaks were characterized by a greater proportion of peaks across intronic regions, whereas females had a greater peak distribution across gene promoters (Figure 3G). The majority of sex-specific open chromatin regions were not developmentally regulated (Figure 3H). However, open chromatin regions in adult male cardiomyocytes were highly enriched for steroid hormone nuclear receptors including androgen response element, glucocorticoid response element, and PGR, whereas accessible chromatin regions in female cardiomyocytes were highly enriched for AP1-JUN motifs (Figure 3I). To confirm the putative role of steroid hormone nuclear receptors in cardiomyocyte maturation, we analyzed the Tn5 insertion signal across all ATAC-seq peaks, which confirmed distinct TF footprints for androgen response element, glucocorticoid response element, and PGR in open chromatin regions of adult versus fetal and hiPSC-derived cardiomyocytes (Figure 3J). Moreover, interrogation of differentially open chromatin regions during cardiomyocyte maturation revealed considerable overlap between TF binding sites for the androgen response element, glucocorticoid response element, and PGR (Figure 3K). Collectively, these results suggest that steroid hormone nuclear receptors drive sex-specific transcriptional programs associated with cardiomyocyte maturation in humans.
Progesterone Receptor Controls Sex-Specific Transcriptional Programs Associated With Human Cardiomyocyte Maturation
Given the well-established functions of sex steroids and steroid hormone nuclear receptors in maturation of reproductive tissues during development,35 we subsequently focused on their potential roles as mediators of sex-specific differences arising during cardiomyocyte maturation. Androgen receptor (AR) and progesterone receptor (PGR) gene expression levels increased during cardiomyocyte maturation, although expression levels did not differ between male and female cardiomyocytes (Figure 4A and 4B). Single-cell analysis confirmed upregulation of PGR in human cardiomyocytes during postnatal maturation, whereas AR expression was below the threshold of detection in our snRNA-seq analysis (Figure XIIA in the Data Supplement). The gene encoding the glucocorticoid receptor nuclear receptor subfamily 3 group C member 1 (NR3C1), was downregulated in cardiomyocytes during cardiac maturation (Figure 4A and 4B and Figure XIIA in the Data Supplement). It is interesting to note that consistent with well-known species differences between mice and humans in the developmental regulation of sarcomeric protein isoforms during cardiomyocyte maturation,36 we also observed species differences in the developmental regulation of steroid hormone nuclear receptors (Figure 4A and Figure XIIB in the Data Supplement). In contrast with the relatively high expression of the AR and PGR in human cardiomyocytes (Figure 4A), Ar was barely detectable and Pgr was undetectable in mouse cardiomyocytes (Figure XIIB in the Data Supplement). These findings reveal potentially important species differences in the regulation of cardiomyocyte maturation between mice and humans.
Figure 4.
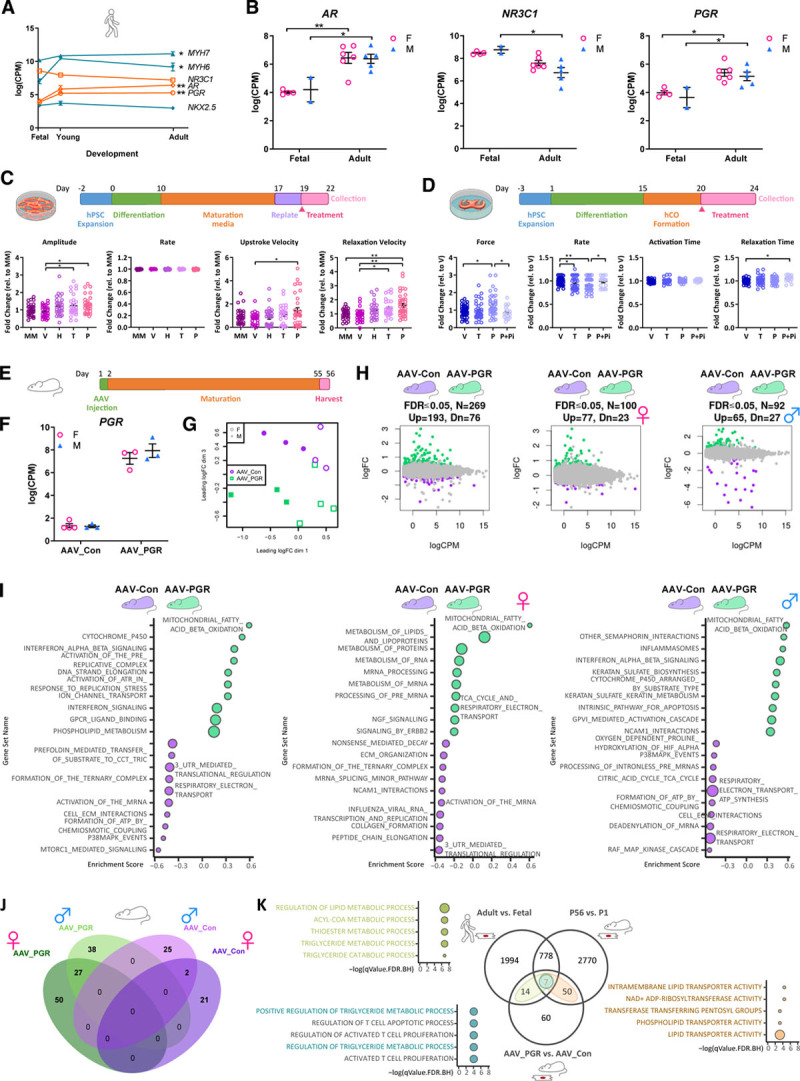
The progesterone receptor augments cardiac contractility and activates sex-specific metabolic networks associated with cardiomyocyte maturation. A, Gene expression (log CPM reads) showing developmental regulation of sarcomeric genes (MYH6, MYH7), cardiac TFs (NKX2.5), and nuclear steroid hormone receptors (AR, NR3C1, PGR) during human cardiomyocyte maturation. Data are mean±SEM. *FDR≤0.05; **FDR≤0.001. B, Gene expression (log CPM reads) for nuclear steroid hormone receptors (AR, GR, PGR) in male (blue) versus female (pink) cardiomyocytes at fetal and adult stages. Data are mean±SEM. *FDR≤0.05, **FDR≤0.001. C, Progesterone treatment increases contractile force of 2-dimensional hESC-CMs. Dot plots showing baseline-normalized functional readouts for HES3 NKX2-5eGFP/w cardiomyocytes cultured in MM and treated with vehicle (V: 0.1% ethanol), glucocorticoids (G: 1 µM hydrocortisone), testosterone (T: 1 µmol/L of testosterone), or progesterone (P: 10 µmol/L of progesterone). Data are presented as fold change for n=5 independent experiments with >27 replicates per condition. A 1-way ANOVA followed by a Tukey posttest was used. Data are mean±SEM. *P≤0.05, **P≤0.001. D, Progesterone treatment increases contractile force and decreases spontaneous beating rate of hCOs. Dot plots showing functional readouts of H9 hCOs cultured in MM and treated with vehicle (V: 0.1% ethanol), progesterone (P: 10 µmol/L of progesterone), or progesterone inhibitor (P+Pi: 10 µmol/L of progesterone + 10µM RU-486). Data are presented as fold change for n=6 independent experiments with n>19 tissues per group, respectively. Data are mean±SEM. A 1-way ANOVA followed by a Tukey posttest was used. *P≤0.05, **P≤0.001. E, Schematic overview of AAV-PGR gain-of-function experiment in mice. F, Gene expression (log CPM reads) for PGR in PCM1-purified cardiomyocytes from AAV-PGR and AAV-Con treated adult female (pink) and male (blue) mice. Data are mean±SEM. G, MDS plot shows clear separation of female (open) and male (closed) PCM1-purified cardiomyocytes (dimension 1) from control AAV-Con (green) and AAV-PGR-treated (purple) mice (dimension 3). H, Mean-difference scatter plots of AAV-PGR versus AAV-Con (combined male and female, female only, and male only). Genes that are upregulated in AAV-PGR (green) or AAV-Con (purple) are highlighted (FDR≤0.05). I, GSEA reactome analysis of differentially expressed genes between AAV-PGR (green) and AAV-Con (purple) for combined male and female, female only, and male only samples. J, Venn diagram of differentially expressed genes in male and female AAV-Con and AAV-PGR–treated cardiomyocytes. Genes that are upregulated in AAV-PGR (green), upregulated in AAV-Con (purple), upregulated in male (light shade), or upregulated in female (dark shade) are highlighted. K, Venn diagram and pathway analysis for mouse and human gene orthologs that are upregulated in 3 comparisons: 1) AAV-PGR versus AAV-Con, 2) adult (postnatal day [P] 56) versus neonatal (P1) mouse cardiomyocytes, and 3) adult versus fetal human cardiomyocytes. Genes/pathways that are shared between AAV-PGR upregulated and adult mouse upregulated are highlighted in brown, AAV-PGR upregulated and adult human upregulated are highlighted in green, and genes/pathways that are common across all 3 comparisons are highlighted in blue. An FDR≤0.05 with the correction procedure of Benjamini and Hochberg was used. AAV indicates adeno-associated viral vector; Con, control; CPM, count per million; FDR, false discovery rate; GSEA, gene set enrichment analysis; hCO, human cardiac organoid; hESC, human embryonic stem cell; MDS, multidimensional scaling; MM, maturation media; PGR, progesterone receptor; and TF, transcription factor.
To assess the potential effect of steroid hormones on human cardiomyocyte function, we screened a number of steroids for potential effects on cardiac contractility using cardiomyocytes differentiated from the HES3 NKX2-5eGFP/w human embryonic stem cell line.37 Standard 2-dimensional monolayers of human cardiomyocytes were treated with vehicle (ethanol), hydrocortisone (cortisol), testosterone, or progesterone (ligands for NR3C1, AR, and PGR, respectively), and cardiomyocyte electrophysiology and contractility were concurrently recorded using extracellular field potential and impedance measurements. Initial screening of steroid hormones revealed a positive inotropic effect of progesterone, which increased contractile amplitude, upstroke velocity, and relaxation velocity (Figure 4C). We subsequently validated this finding in 3-dimensional hCOs cultured under maturation conditions from an independent human embryonic stem cell line (H9), which confirmed a significant increase in cardiac force production in the presence of progesterone (Figure 4D). It is important to note that the positive inotropic effect of progesterone was abolished in the presence of the PGR antagonist mifepristone (RU-486; Figure 4D). The spontaneous contraction rate of hCOs was also reduced in the presence of progesterone, and this effect was abrogated in the presence of RU-486 (Figure 4D). These data indicate that PGR activation augments cardiac contractility and reduces the spontaneous contraction rate of hCOs, which are consistent with a putative role in cardiomyocyte maturation.
In contrast with human cardiomyocytes, the PGR is not expressed in adult mouse cardiomyocytes (Figure XIIB in the Data Supplement). Accordingly, RNA-seq analysis of Pcm1-purified cardiomyocyte nuclei from PRKO mice revealed that the transcriptional program of adult mouse cardiomyocytes was not affected by genetic deletion of Pgr (Figure XIIC and XIID in the Data Supplement). To evaluate the potential effects of human PGR activation on cardiomyocyte maturation in vivo, we performed a gain-of-function experiment in mice (Figure 4E). The human PGR was delivered to neonatal mice using adeno-associated virus (AAV) serotype 6, which displays very high tropism for cardiac muscle in vivo38 and results in preferential transduction of cardiomyocytes.39 Expression of the human PGR was confirmed in adult cardiomyocytes from mice treated with AAV-PGR compared with mice treated with an AAV empty vector control (Figure 4F). RNA-seq of purified adult cardiomyocytes from AAV-PGR and AAV empty vector control–treated mice demonstrated distinct clustering of samples on the basis of treatment and sex (Figure 4G). Expression of the human PGR during cardiomyocyte maturation in vivo altered the expression of 269 genes in adult cardiomyocytes (Figure 4H). Human PGR activation in vivo was associated with sex-specific activation of transcriptional networks involved in fatty acid oxidation, lipid metabolism, tricarboxylic acid cycle, and respiratory electron transport chain (Figure 4H through 4J). More than 40% of PGR-regulated genes were also induced during cardiomyocyte maturation in mice,13 and these were broadly associated with lipid transport (Figure 4K). Moreover, a subset of these genes associated with activation of lipid metabolic processes, including acyl-CoA metabolism, were also regulated during human cardiomyocyte maturation (Figure 4K). Thus, the PGR modulates human cardiomyocyte contractility and transcriptionally regulates metabolic networks associated with cardiomyocyte maturation in a sex-specific manner.
Discussion
Sex steroids play an important role in regulating cardiac physiology and cardiovascular disease outcomes. However, studies to date have almost exclusively focused on the contributions of estrogen and testosterone.10 Progesterone via PGR is traditionally associated with functions in female reproductive physiology, including essential roles in ovulation, uterine receptivity, and pregnancy maintenance.40 Our study reveals a previously unappreciated role for progesterone in human cardiomyocyte maturation in both males and females, including the establishment of sex-specific transcriptional programs associated with oxidative metabolism in cardiomyocytes. Progesterone-driven alterations in cardiomyocyte metabolism might contribute to previously described effects of progesterone on cardiac hypertrophy and protein synthesis in female rats associated with cardiac adaptations during pregnancy.41–43 It is interesting to note that progesterone levels transiently increase 20-fold in the luteal phase44 and 300-fold during pregnancy (or can be chronically elevated with progestin-based oral contraceptives). However, serum progesterone levels are comparable in males and females outside the luteal phase of the reproductive cycle,45 suggesting that progesterone may also regulate cardiac physiology in males. These findings raise the possibility that fluctuating progesterone ligand, in combination with sex-specific DNA binding of PGR in cardiomyocytes, contributes to sex-based differences in cardiovascular health. Given the widespread prevalence of cardiovascular disease in children and adults worldwide,46,47 it will be important to determine whether progesterone signaling can be therapeutically targeted to modify sexually dimorphic disease outcomes in the future. In addition, our findings reveal that human heart development is associated with profound maturation of all cardiac cell lineages from fetal development to adulthood, which is consistent with previous observations in rodents.13 It is currently unclear whether progesterone plays a similar role in postnatal maturation of other cardiac lineages, although it is tempting to speculate that PGR may also regulate development of cardiac immune cells given that it is upregulated in this lineage during human heart maturation (Figure XII in the Data Supplement). Additional studies are required to define the biological functions of progesterone during cardiac development in diverse lineages including characterization of direct PGR targets and underlying mechanisms of action. Given the species differences reported here, as well as recent studies highlighting the prevalence of species differences in the regulation of sex-specific gene expression programs,48 it will be critical to dissect such mechanistic biology in humans. However, studying human heart maturation will require ongoing refinement of organoid technologies to better reflect the complexity of diverse cell lineages in the human heart. Moreover, it is important to recognize the limitations of working with primary, nondiseased human tissue samples, which are difficult to obtain and could be influenced by unintended surgical biases, including slight variations in sampling from different anatomic regions. It is expected that our results will inform more detailed assessments of spatiotemporal variation in gene expression at the single-cell level and propel studies with larger cohorts to determine the roles of age, sex, and ancestry on normal human heart development. To this end, the key resource data sets outlined in this study provide a blueprint defining sex differences in human heart development and a framework for understanding mechanisms underpinning the acquisition of adult cardiac cell identity.
Acknowledgments
The authors thank the Australian Red Cross Blood Service for coordination of donor heart retrieval for the Sydney Heart Bank. The authors gratefully acknowledge technical support and advice for next-generation sequencing including snRNA-seq from M. Le Moing, G. D'Cunha, W.S. Lee, and S. Stephenson from the Translational Genomics Unit, Australian Genome Research Facility (AGRF) and the Murdoch Children's Research Institute (MCRI). The authors thank M. Burton and E. Jones from MCRI and G. Osborne and V. Nink from the University of Queensland for technical assistance and advice for fluorescence-activated cell sorting. The authors gratefully acknowledge technical support from H. Qian for adeno-associated virus production. The authors thank L. Ling and D. Godler from MCRI for technical assistance and use of the ViiA 7 Real-Time PCR System. The authors gratefully acknowledge K. Karavendzas and L.Qian from MCRI for technical assistance and advice on stem cell culture.The Australian National Fabrication Facility Queensland Node fabricated Heart-Dyno molds. The authors thank J. Stolper and F. Bolk for graphical assistance with figures.
C.B.S., J.E.H., and E.R.P. conceptualized the project. C.B.S., B.P., M.Z., D.A.E., A.O., A.E.-O., J.E.H., and E.R.P designed the experiments. C.B.S. and C.V. performed sample preparation for sequencing. C.B.S., B.P., M.Z., H.R., R.K.R.K., A.K., and H.K. analyzed sequencing data. C.B.S., R.J.M., K.D.A.-B., H.K.V., A.H., and J.E.H. conducted experiments with human pluripotent stem cells and cardiac organoids including data analysis. K.I.W. and P.G. generated adeno-associated viral vectors and conducted mouse experiments. D.T.D. and R.L.R. provided knockout mice and conducted mouse experiments. C.B.S. and M.t.H. performed fluorescence-activated cell sorting analysis for cardiomyocyte ploidy. C.G.d.R., S.L., A.T.P., and I.E.K. contributed to patient recruitment, consenting, and biobanking. L.M.D. provided critical insight into interpretations of sex steroid signaling in the heart. C.B.S., E.R.P., and J.E.H. analyzed and interpreted data. E.R.P., J.E.H., A.O., A.E.-O. and I.E.K. obtained funding and managed the project. C.B.S., E.R.P., and J.E.H. wrote the article. All authors reviewed the article.
Sources of Funding
The authors received grant and fellowship support from the National Health and Medical Research Council of Australia (E.R.P., J.E.H., A.O., A.E.-O., B.P., R.R.), the Australian Research Council (E.R.P., A.E.-O., L.M.D.), the Heart Foundation of Australia (E.R.P. and J.E.H.), the Stafford Fox Medical Research Foundation (E.R.P.), and the Royal Children's Hospital Foundation (E.R.P., I.E.K., D.A.E., L.M.D.). MCRI is supported by the Victorian Government's Operational Infrastructure Support Program.
Disclosures
E.R.P., J.E.H., and R.J.M. are cofounders, scientific advisors, and hold equity in Dynomics, a biotechnology company focused on the development of heart failure therapeutics. The other authors report no conflicts.
Supplemental Materials
Data Supplement Methods
Data Supplement Tables I–VII
Data Supplement Figures I–XII
References 49–72
Supplementary Material
Footnotes
The full author list is available on page 1625.
Sources of Funding, see page 1626
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.120.051921.
Contributor Information
Choon Boon Sim, Email: evangelyn.sim@mcri.edu.au.
Belinda Phipson, Email: Belinda.Phipson@petermac.org.
Mark Ziemann, Email: m.ziemann@deakin.edu.au.
Haloom Rafehi, Email: rafehi.h@wehi.edu.au.
Richard J. Mills, Email: richard.mills@qimrberghofer.edu.au.
Kevin I. Watt, Email: kevin.watt@unimelb.edu.au.
Kwaku D. Abu-Bonsrah, Email: dad.abubonsrah@mcri.edu.au.
Ravi K.R. Kalathur, Email: enzo.porrello@mcri.edu.au.
Holly K. Voges, Email: holly.voges@mcri.edu.au.
Doan T. Dinh, Email: doan.dinh@adelaide.edu.au.
Menno ter Huurne, Email: menno.terhuurne@mcri.edu.au.
Celine J. Vivien, Email: celine.vivien@mcri.edu.au.
Antony Kaspi, Email: akaspi@gmail.com.
Harikrishnan Kaipananickal, Email: Harikrishnan.Kaipananickal@monash.edu.
Alejandro Hidalgo, Email: hidala@gmail.com.
Leanne M.D. Delbridge, Email: lmd@unimelb.edu.au.
Rebecca L. Robker, Email: rebecca.robker@adelaide.edu.au.
Paul Gregorevic, Email: pgre@unimelb.edu.au.
Cristobal G. dos Remedios, Email: crisdosremedios@gmail.com.
Sean Lal, Email: sean.lal@sydney.edu.au.
Adam T. Piers, Email: adam.piers@mcri.edu.au.
Igor E. Konstantinov, Email: igorkonst@hotmail.com.
David A. Elliott, Email: david.elliott@mcri.edu.au.
Assam El-Osta, Email: sam.el-osta@monash.edu.
Alicia Oshlack, Email: Alicia.Oshlack@petermac.org.
References
- 1.Guo Y, Pu WT. Cardiomyocyte maturation: new phase in development. Circ Res. 2020;126:1086–1106. doi: 10.1161/CIRCRESAHA.119.315862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vivien CJ, Hudson JE, Porrello ER. Evolution, comparative biology and ontogeny of vertebrate heart regeneration. NPJ Regen Med. 2016;1:16012. doi: 10.1038/npjregenmed.2016.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, Hu Y, Dong J, Li Q, Wu X, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019;26:1934–1950.e5. doi: 10.1016/j.celrep.2019.01.079 [DOI] [PubMed] [Google Scholar]
- 4.DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, et al. Single-cell resolution of temporal gene expression during heart development. Dev Cell. 2016;39:480–490. doi: 10.1016/j.devcel.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lescroart F, Wang X, Lin X, Swedlund B, Gargouri S, Sànchez-Dànes A, Moignard V, Dubois C, Paulissen C, Kinston S, et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science. 2018;359:1177–1181. doi: 10.1126/science.aao4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G, Xu A, Sim S, Priest JR, Tian X, Khan T, Quertermous T, Zhou B, Tsao PS, Quake SR, et al. Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev Cell. 2016;39:491–507. doi: 10.1016/j.devcel.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC, Jr, Akkad AD, Herndon CN, Arduini A, Papangeli I, et al. Transcriptional and cellular diversity of the human heart. Circulation. 2020;142:466–482. doi: 10.1161/CIRCULATIONAHA.119.045401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, Cohen CD, Pandey R, Diep H, Vinh A, et al. High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation. 2020;142:1448–1463. doi: 10.1161/CIRCULATIONAHA.119.045115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Squiers GT, McLellan MA, Ilinykh A, Branca J, Rosenthal NA, Pinto AR. Cardiac cellularity is dependent upon biological sex and is regulated by gonadal hormones. Cardiovasc Res. 2020. doi: 10.1093/cvr/cvaa265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Regitz-Zagrosek V, Kararigas G. Mechanistic pathways of sex differences in cardiovascular disease. Physiol Rev. 2017;97:1–37. doi: 10.1152/physrev.00021.2015 [DOI] [PubMed] [Google Scholar]
- 11.Aggarwal NR, Patel HN, Mehta LS, Sanghani RM, Lundberg GP, Lewis SJ, Mendelson MA, Wood MJ, Volgman AS, Mieres JH. Sex differences in ischemic heart disease: advances, obstacles, and next steps. Circ Cardiovasc Qual Outcomes. 2018;11:e004437. doi: 10.1161/CIRCOUTCOMES.117.004437 [DOI] [PubMed] [Google Scholar]
- 12.Bergmann O, Jovinge S. Isolation of cardiomyocyte nuclei from post-mortem tissue. J Vis Exp. 2012;65:e4205. doi: 10.3791/4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE, Porrello ER. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation. 2017;136:1123–1139. doi: 10.1161/CIRCULATIONAHA.117.028252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, III, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet. 2019;20:257–272. doi: 10.1038/s41576-019-0093-7 [DOI] [PubMed] [Google Scholar]
- 19.Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann Appl Stat. 2016;10:946–963. doi: 10.1214/16-AOAS920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarthy DJ, Smyth GK. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009;25:765–771. doi: 10.1093/bioinformatics/btp053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zappia L, Oshlack A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience. 2018;7doi: 10.1093/gigascience/giy083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu D, Smyth GK. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012;40:e133. doi: 10.1093/nar/gks461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crowell HL, Soneson C, Germain PL, Calini D, Collin L, Raposo C, Malhotra D, Robinson MD. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat Commun. 2020;11:6077. doi: 10.1038/s41467-020-19894-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vlahos K, Sourris K, Mayberry R, McDonald P, Bruveris FF, Schiesser JV, Bozaoglu K, Lockhart PJ, Stanley EG, Elefanty AG. Generation of iPSC lines from peripheral blood mononuclear cells from 5 healthy adults. Stem Cell Res. 2019;34:101380. doi: 10.1016/j.scr.2018.101380 [DOI] [PubMed] [Google Scholar]
- 27.Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci U S A. 2017;114:E8372–E8381. doi: 10.1073/pnas.1707316114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson DJ, Kaplan DI, Bell KM, Koutsis K, Haynes JM, Mills RJ, Phelan DG, Qian EL, Leitoguinho AR, Arasaratnam D, et al. NKX2-5 regulates human cardiomyogenesis via a HEY2 dependent transcriptional network. Nat Commun. 2018;9:1373. doi: 10.1038/s41467-018-03714-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills RJ, Parker BL, Quaife-Ryan GA, Voges HK, Needham EJ, Bornot A, Ding M, Andersson H, Polla M, Elliott DA, et al. Drug screening in human PSC-cardiac organoids identifies pro-proliferative compounds acting via the mevalonate pathway. Cell Stem Cell. 2019;24:895–907.e6. doi: 10.1016/j.stem.2019.03.009 [DOI] [PubMed] [Google Scholar]
- 30.Voges HK, Mills RJ, Elliott DA, Parton RG, Porrello ER, Hudson JE. Development of a human cardiac organoid injury model reveals innate regenerative potential. Development. 2017;144:1118–1127. doi: 10.1242/dev.143966 [DOI] [PubMed] [Google Scholar]
- 31.Hu P, Liu J, Zhao J, Wilkins BJ, Lupino K, Wu H, Pei L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018;32:1344–1357. doi: 10.1101/gad.316802.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, et al. Cells of the adult human heart. Nature. 2020;588:466–472. doi: 10.1038/s41586-020-2797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilsbach R, Preissl S, Grüning BA, Schnick T, Burger L, Benes V, Würch A, Bönisch U, Günther S, Backofen R, et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun. 2014;5:5288. doi: 10.1038/ncomms6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Schneider P, Nührenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun. 2018;9:391. doi: 10.1038/s41467-017-02762-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wierman ME. Sex steroid effects at target tissues: mechanisms of action. Adv Physiol Educ. 2007;31:26–33. doi: 10.1152/advan.00086.2006 [DOI] [PubMed] [Google Scholar]
- 36.Lompré AM, Nadal-Ginard B, Mahdavi V. Expression of the cardiac ventricular alpha- and beta-myosin heavy chain genes is developmentally and hormonally regulated. J Biol Chem. 1984;259:6437–6446. [PubMed] [Google Scholar]
- 37.Elliott DA, Braam SR, Koutsis K, Ng ES, Jenny R, Lagerqvist EL, Biben C, Hatzistavrou T, Hirst CE, Yu QC, et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat Methods. 2011;8:1037–1040. doi: 10.1038/nmeth.1740 [DOI] [PubMed] [Google Scholar]
- 38.Weeks KL, Gao X, Du XJ, Boey EJ, Matsumoto A, Bernardo BC, Kiriazis H, Cemerlang N, Tan JW, Tham YK, et al. Phosphoinositide 3-kinase p110α is a master regulator of exercise-induced cardioprotection and PI3K gene therapy rescues cardiac dysfunction. Circ Heart Fail. 2012;5:523–534. doi: 10.1161/CIRCHEARTFAILURE.112.966622 [DOI] [PubMed] [Google Scholar]
- 39.Quaife-Ryan GA, Mills RJ, Lavers G, Voges HK, Vivien CJ, Elliott DA, Ramialison M, Hudson JE, Porrello ER. beta-Catenin drives distinct transcriptional networks in proliferative and nonproliferative cardiomyocytes. Development. 2020;147doi: 10.1242/dev.193417 [DOI] [PubMed] [Google Scholar]
- 40.Wu SP, Li R, DeMayo FJ. Progesterone receptor regulation of uterine adaptation for pregnancy. Trends Endocrinol Metab. 2018;29:481–491. doi: 10.1016/j.tem.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chung E, Yeung F, Leinwand LA. Akt and MAPK signaling mediate pregnancy-induced cardiac adaptation. J Appl Physiol (1985). 2012;112:1564–1575. doi: 10.1152/japplphysiol.00027.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung E, Yeung F, Leinwand LA. Calcineurin activity is required for cardiac remodelling in pregnancy. Cardiovasc Res. 2013;100:402–410. doi: 10.1093/cvr/cvt208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldstein J, Sites CK, Toth MJ. Progesterone stimulates cardiac muscle protein synthesis via receptor-dependent pathway. Fertil Steril. 2004;82:430–436. doi: 10.1016/j.fertnstert.2004.03.018 [DOI] [PubMed] [Google Scholar]
- 44.Stricker R, Eberhart R, Chevailler MC, Quinn FA, Bischof P, Stricker R. Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT analyzer. Clin Chem Lab Med. 2006;44:883–887. doi: 10.1515/CCLM.2006.160 [DOI] [PubMed] [Google Scholar]
- 45.Oettel M, Mukhopadhyay AK. Progesterone: the forgotten hormone in men? Aging Male. 2004;7:236–257. doi: 10.1080/13685530400004199 [DOI] [PubMed] [Google Scholar]
- 46.Rossano JW. Congenital heart disease: a global public health concern. Lancet Child Adolesc Health. 2020;4:168–169. doi: 10.1016/S2352-4642(19)30429-8 [DOI] [PubMed] [Google Scholar]
- 47.Savarese G, Lund LH. Global public health burden of heart failure. Card Fail Rev. 2017;3:7–11. doi: 10.15420/cfr.2016:25:2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naqvi S, Godfrey AK, Hughes JF, Goodheart ML, Mitchell RN, Page DC. Conservation, acquisition, and functional impact of sex-biased gene expression in mammals. Science. 2019;365:eaaw7317doi: 10.1126/science.aaw7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S, et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A. 2013;110:1446–1451. doi: 10.1073/pnas.1214608110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li M, Parker BL, Pearson E, Hunter B, Cao J, Koay YC, Guneratne O, James DE, Yang J, Lal S, et al. Core functional nodes and sex-specific pathways in human ischaemic and dilated cardiomyopathy. Nat Commun. 2020;11:2843. doi: 10.1038/s41467-020-16584-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Heesch S, Witte F, Schneider-Lunitz V, Schulz JF, Adami E, Faber AB, Kirchner M, Maatz H, Blachut S, Sandmann CL, et al. The translational landscape of the human heart. Cell. 2019;178:242–260.e29. doi: 10.1016/j.cell.2019.05.010 [DOI] [PubMed] [Google Scholar]
- 52.Jiang H, Lei R, Ding SW, Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014;15:182. doi: 10.1186/1471-2105-15-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 55.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Team RC. R: A Language and Environment for Statistical Computing. 2019. R Foundation for Statistical Computing [Google Scholar]
- 57.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57:289–300. doi: [Google Scholar]
- 58.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bardou P, Mariette J, Escudié F, Djemiel C, Klopp C. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics. 2014;15:293. doi: 10.1186/1471-2105-15-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walter W, Sánchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31:2912–2914. doi: 10.1093/bioinformatics/btv300 [DOI] [PubMed] [Google Scholar]
- 62.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amemiya HM, Kundaje A, Boyle AP. The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep. 2019;9:9354. doi: 10.1038/s41598-019-45839-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bentsen M, Goymann P, Schultheis H, Klee K, Petrova A, Wiegandt R, Fust A, Preussner J, Kuenne C, Braun T, et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat Commun. 2020;11:4267. doi: 10.1038/s41467-020-18035-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44W1W160–W165. doi: 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaspi A, Ziemann M. Mitch: multi-contrast pathway enrichment for multi-omics and single-cell profiling data. BMC Genomics. 2020;21:447. doi: 10.1186/s12864-020-06856-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hidalgo A, Glass N, Ovchinnikov D, Yang SK, Zhang X, Mazzone S, Chen C, Wolvetang E, Cooper-White J. Modelling ischemia-reperfusion injury (IRI) in vitro using metabolically matured induced pluripotent stem cell-derived cardiomyocytes. APL Bioeng. 2018;2:026102. doi: 10.1063/1.5000746 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.