

Abstract
Objective
To accurately categorize the phenotypes of individuals with collagen VI–related dystrophies (COL6-RDs) during the first years of life to predict long-term motor function and pulmonary function, to provide phenotype-specific anticipatory care, and to improve clinical trial readiness.
Methods
This retrospective, multicenter, international study analyzed the relationship of long-term motor and pulmonary function with the initial maximal motor ability achieved in individuals with COL6-RD.
Results
We studied 119 patients with COL6-RD from Spain (n = 54) and the United States (n = 65). The early maximal motor milestones of ability to rise from the floor unassisted and ability to climb 4 steps without holding onto a railing demonstrated reliability in distinguishing between 3 COL6-RD phenotypic subgroups: (1) Ullrich congenital muscular dystrophy, (2) intermediate COL6-RD, and (3) Bethlem myopathy. Long-term motor function and pulmonary function are strongly correlated with the maximal motor ability achieved during the first years of life. Maximal motor capacity can predict other disease-relevant events such as the age at loss of ambulation and the need for the initiation of nocturnal noninvasive ventilation.
Conclusion
This work proposes a prospective phenotypic classification for COL6-RDs that will enable an accurate prediction of a patient's COL6-RD phenotype during the first years of life. The ability to establish a patient's COL6-RD phenotypic classification early will enable a more accurate prognosis of future motor and pulmonary function, thus improving anticipatory clinical care, and it will be instrumental in aiding the design of future clinical trials by allowing early stratification of trial cohorts.
Dominant or recessive mutations in COL6A1, COL6A2, or COL6A3 affect the complex assembly and secretion of collagen VI (COL6), a muscle extracellular matrix protein, and result in a congenital muscular dystrophy (CMD) subtype known as the COL6-related dystrophies (COL6-RDs).1,2 The COL6-RDs encompass a continuum of overlapping clinical phenotypes ranging from Ullrich CMD (UCMD) to Bethlem myopathy (BM) with intermediate phenotypes in between. Clinical hallmarks of the COL6-RDs include muscle weakness, distal joint hyperlaxity, contractures of proximal joints, and progressive respiratory insufficiency in most patients.1,3–13 Previous attempts at categorizing COL6-RD phenotypes either lacked clearly delineated boundaries or were dependent on the timing of the loss of ambulation (LoA), which is often between 10 and 20 years of age.1,14–17 In particular, predictive phenotypic characterization in COL6-RDs has remained challenging.
In this study, we stratified the phenotypes of a large cohort of patients with COL6-RD into 3 categories according to the maximal motor capacity achieved by the age of 5 years and then correlated the proposed COL6-RD classification with long-term motor and pulmonary function. Establishing an accurate phenotypic classification of the COL6-RDs from an early age would enable clinicians to more precisely counsel patients and their families about the projected clinical course, thus enabling appropriate anticipatory care. An earlier, confident phenotype subtype classification would also aid in the design of viable, COL6-RD subtype–specific outcome measures and clinical trial stratification, thus improving clinical trial preparedness of the COL6-RD patient population as a whole for the therapeutic approaches that are currently under development.18–21
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
Data were collected in accordance with ethics guidelines of each of the institutions involved. Written informed consent and age-appropriate assent for study participation were obtained by a qualified investigator for US patients (protocol 12-N-0095, approved by the Institutional Review Board of the NIH).
Study Design and Patients
This is a retrospective, multicenter study with the aim of characterizing the long-term motor and pulmonary function in patients with COL6-RD on the basis of developmental motor milestones in early childhood. To this end, the relationship of long-term motor and pulmonary function with the maximal motor ability achieved was analyzed.
Clinical data of patients with a genetically or pathologically confirmed diagnosis of COL6-RD were reviewed at 6 different centers with neuromuscular expertise in the United States and Spain: 1 specialized clinical research center in the United States (National Institute of Neurological Disorders and Stroke, NIH, Bethesda, MD) and 5 specialized neuromuscular centers in Spain (Hospital Sant Joan de Déu, Barcelona; Hospital 12 de Octubre, Madrid; Hospital de la Santa Creu i Sant Pau, Barcelona; Hospital de Bellvitge, L'Hospitalet de Llobregat, Barcelona; and Hospital Clínic, Barcelona). Patients with pathogenic mutation/s in COL6A1, COL6A2, or COL6A3 found on direct Sanger sequencing or on next-generation sequencing, including whole exome sequencing, or evidence of significantly decreased or mislocalized COL6 on immunohistochemical studies performed on muscle biopsy or dermal fibroblasts were included in this study. Phenotypic data were collected from patient and family interviews, medical records, and patient questionnaires, with an emphasis on motor development milestones achieved, as well as relevant events such as initiation and LoA.
Clinical Information and Outcome Assessment Measures
Demographic information such as age and sex and natural history data, including age at symptom onset (by parental recollection and by verification of medical records), age at clinical diagnosis, and history of motor developmental milestones gained or lost, were collected. Other relevant medical and surgical history information was captured, with attention to pulmonary function. Methods of diagnosis (clinical phenotype, immunohistochemical studies of COL6 expression performed on muscle biopsy and/or dermal fibroblasts and/or molecular genetic confirmation) and family history of COL6-RD were collected. Genotype was also collected on all patients in whom genetic testing had been performed.
Spirometry techniques were performed according to international standards.22 Upright (seated) forced vital capacity (FVC) measurements (expressed as percent predicted) were captured in all patients. Data on the motor milestones of independent walking, rising from the floor unassisted, and climbing a minimum of 4 steps without holding onto a railing were collected for all patients. These 3 motor milestones were specifically selected following a pilot study performed in 40 patients with COL6-RD in Spain (Hospital Sant Joan de Déu, Barcelona, Spain) in which the following 6 developmental motor milestones were captured and assessed for their the ability to predict future maximal motor ability: independent sitting, independent walking, running, jumping, rising from the floor unassisted, and climbing a minimum of 4 steps without the use of a railing. Jumping was defined as being able to jump a minimum of 10 cm off the ground with both feet.23 Running was defined as a gait that demonstrates an aerial phase during which no limbs touch the ground. From this pilot study, 3 maximal motor milestones—independent walking, rising from the floor (from a supine position to a standing position) unassisted (except for potentially pushing off one's own legs/use of the Gowers' maneuver), and climbing a minimum of 4 steps without holding onto a railing—seemed to more reliably reflect the phenotype and were more objective. Therefore, they were chosen for use in collecting motor function data from the present cohort.
Motor function testing included the Motor Function Measure-32 (MFM-32),24,25 6-minute walk test (6MWT),26–31 the North Star Ambulatory Assessment (NSAA),24,32,33 and timed function tests.31 The MFM-32 scale consists of 32 items that are subdivided in 3 functional dimensions: D1, which tests standing and transfers; D2, which tests axial and proximal motor function; and D3, which tests distal motor function.24,25 This scale was performed in all of the patients. The 6MWT, NSAA, and timed function tests were performed only in ambulant patients. Motor function testing was performed by trained clinical evaluators.
Patient Phenotypes and Clinical Classification
Patients were divided into 3 COL6-RD phenotypic subgroups based on the maximal motor ability achieved at 5 years of age: achieved rising from the floor only with assistance (UCMD), achieved rising from the floor unassisted (intermediate COL6-RD), and achieved stair climbing (ascending a minimum of 4 steps without holding onto of a railing) (BM). Maximal motor ability achieved was assigned an ordinal variable because all the patients who were able to ascend 4 steps without holding onto a railing were also able to rise from the floor unassisted.
Statistical Analysis
Statistical analyses were performed with Statistical Package for the Social Sciences 24.0 (IBM SPSS Statistics 24.0, Chicago, IL). Demographic and medical characteristics of patients are summarized with descriptive statistics. Data are presented as mean ± SD or as median and range when appropriate. The Kruskal-Wallis test was used to evaluate the association between maximal motor ability achieved and function scales and the association between maximal motor ability achieved and pulmonary function. An α level of 0.01 was used as the criterion for statistical significance. A linear regression model, including the interaction between age and phenotype, was used to compare the effect of age on motor function scale scores among the 3 phenotypic groups.
We performed time-to-event analyses of LoA and initiation of noninvasive ventilation (NIV) with age (years) as the time variable and LoA or initiation of NIV as events. Median age at LoA and at initiation of NIV corresponding 95% confidence intervals were estimated (taking into account censored data) by plotting empirical Kaplan-Meier curves for patient groups defined by maximal motor ability achieved, with the above-described grouping. Cox proportional hazard models were used to estimate and compare age-related risks of both LoA and initiation of NIV. Statistical significance was also set at p < 0.01.
To examine the concurrent validity between MFM-32 total scale score, the 3 MFM-32 domains, NSAA score, 6MWT, timed test of 10-m run, ascending 4 steps, and descending 4 steps, and percent predicted FVC, Spearman rank-order correlations were performed. Correlations were designated as large when they were >0.70 or <−0.70.
Data Availability
The data not published are available from the corresponding author and will be shared with any qualified investigator on reasonable request.
Results
Demographics
Our cohort totaled 119 patients (62 male, 57 female) with COL6-RD from Spain (n = 54) and from the United States (n = 65). All of the patients had motor function scales on record. Patients ranged in age from 5 to 73 years (mean 23.2 ± 16.1 years) at the time of last clinical evaluation. Confirmed pathogenic mutations were identified in COL6A1, COL6A2, or COL6A3 in 115 patients (97%), and muscle biopsy immunohistochemical evidence of significantly decreased, mislocalized, or absent COL6 with pending molecular genetic confirmation was found in 4 patients. Motor function and pulmonary function data from 23 US patients with COL6-RD have been described previously.34
Correlation Between Early Motor Maximal Ability and Later Disease Category
Patients were separated into 3 groups according to maximal motor ability achieved by 5 years of age (table 1). (1) Thirty-eight patients (32%) achieved the ability to rise from the floor only with support and thus were categorized as having UCMD. Patients in this UCMD group were last clinically assessed at a mean age of 12 years (rang: 5–34 years, SD ± 6.7 years). (2) Thirty-five patients (29%) achieved the ability to rise from the floor unassisted (except for potentially pushing off one's legs/use of the Gowers' maneuver) but were never able to ascend 4 steps without holding onto a railing and thus were categorized as having intermediate COL6-RD. The mean age at the time of last clinical assessment for the intermediate group was 19 years (range 6–48 years, SD ± 10.5 years). (3) Forty-six patients (39%) achieved the abilities to rise from the floor unassisted and to ascend 4 steps without holding onto a railing and thus were categorized as having BM. The mean age at the time of last clinical assessment for the patients with BM was 34.4 years (range 5–73 years, SD ± 18.3 years).
Table 1.
Clinical Characteristics of COL6-RD Phenotypes

Eight of the 38 patients classified as having UCMD (21%) never achieved independent ambulation. LoA as defined by full-time wheelchair dependence in those patients who had attained independent ambulation occurred in 30 patients at the time of this study: 22 of 30 with UCMD (range 4–15 years), 8 of 35 patients with intermediate COL6-RD (range 6–12 years), and 0 of 46 patients with BM, because all remained ambulant at their last follow-up assessment. While the age ranges at LoA between those patients classified as having UCMD and those patients classified as having intermediate COL6-RD demonstrated an overlap, differences in the age at the time of LoA between the established COL6-RD subgroups were statistically significant (p < 0.001).
Those patients who achieved a higher initial maximal motor ability and thus were classified in COL6-RD subgroups associated with milder phenotypic severity obtained higher scores in the motor function scales performed at the time of last clinical assessment, regardless of their age at that time. The association between the initial maximal motor ability achieved and the scores of all the function scales assessed—MFM-32 (including each of its 3 functional domains: D1, D2, and D3), NSAA, and 6MWT—was highly significant statistically (p < 0.001) (figure 1).
Figure 1. Motor Function in Patients with COL6-RD by Phenotype.

Highly significant direct correlation is seen between the scores of all the motor function scales assessed: (A) Motor Function Measure (MFM)-32 (including each of its 3 functional domains: [B] D1, [C] D2, and [D] D3), (E) North Star Ambulatory Assessment (NSAA), and (F) 6-minute walk test (6MWT) and collagen VI–related dystrophy (COL6-RD) phenotype (based on the classification proposed here using maximal motor function achieved) (p < 0.001, Kruskal-Wallis test). Box plots show that, regardless of current age, patients whose maximal motor function achieved was climbing 4 steps without holding onto a railing (categorized as Bethlem myopathy [BM]) obtain higher scores in the motor function scales than those whose maximal motor function achieved was rising from the floor unassisted (intermediate COL6-RD) and those who did not achieve either of these 2 milestones (Ullrich congenital muscular dystrophy [UCMD]). Box plots represent medians and quartiles; whiskers signify the 5th and 95th percentiles.
We analyzed the effect of age on motor function scale scores and the effect of age on the 3 phenotypic subgroups of COL6-RD. As we expected, there was an inverse correlation between the scores of all of the function scales tested and age, both for the cohort as a whole and for each of the 3 subgroups (figure 2). Among patients belonging to the same phenotypic subgroup, the greater the age was, the lower their corresponding scores were in MFM-32, NSAA, and 6MWT assessments. A significantly sharper decline in the MFM-32 score with age was detected in the UCMD group (p = 0.03). It is notable that the patients classified in the subgroups with milder phenotypes (intermediate COL6-RD and BM) were older on average at the time of last clinical assessment and had higher motor function scale scores. Kaplan-Meier curves depicting the estimated probability of LoA by age and phenotypic group are shown in figure 3.
Figure 2. Motor Function and Age.

(A–F) An indirect relationship is seen between the scores of all the motor function scales and age, both for the entire collagen VI–related dystrophy (COL6-RD) cohort and for each of the COL6-RD phenotypes. A significantly sharper decline in the Motor Function Measure (MFM) total score in relation to age (A) was detected for those patients within the UCMD subgroup (p = 0.03). BM = Bethlem myopathy; D1 = domain 1; D2 = domain 2; D3 = domain 3; NSAA = North Star Ambulatory Assessment; 6MWT = 6-minute walk test; UCMD = Ullrich congenital muscular dystrophy.
Figure 3. LoA in Patients With COL6-RD by Phenotype.
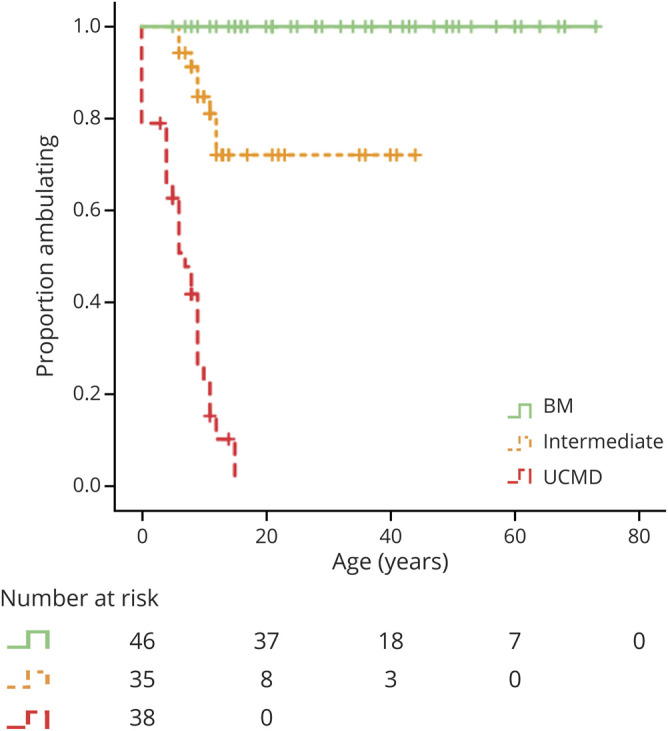
Kaplan-Meier curves depict the probability of loss of ambulation (LoA) by age and collagen VI–related dystrophy (COL6-RD) phenotypic classification BM = Bethlem myopathy; UCMD = Ullrich congenital muscular dystrophy.
The correlations between the MFM-32, the NSAA, and the 6MWT were robust and statistically significant (p < 0.001). A correlation matrix comparing the different motor function scales and pulmonary function as measured by FVC is shown in figure 4. Concurrent validity between the MFM-32 and the NSAA scales was 0.92, between the MFM-32 and the 6MWT was 0.89, and between the NSAA and the 6MWT was 0.95. The 6MWT inversely correlated significantly (p < 0.001) with the timed 10-m run (rs = −0.71) and ascend 4 steps (rs = −0.80) tests. This latter finding means that individuals who covered longer distances in 6MWT tended to run 10 m faster and ascend 4 steps faster.
Figure 4. Correlation of Motor Function and Pulmonary Function Assessments.
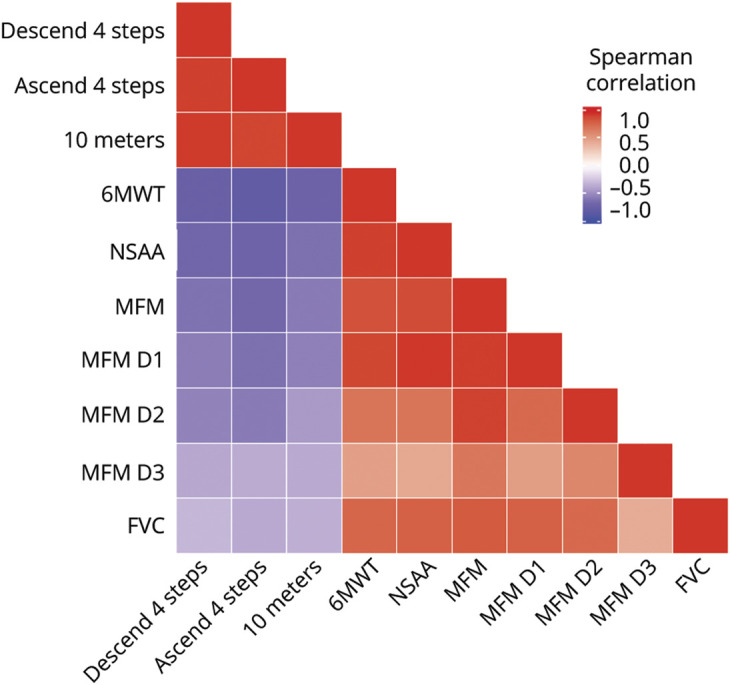
Correlation matrix compares different motor function scales and pulmonary function as measured by forced vital capacity (FVC) in our cohort of patients with collagen VI–related dystrophy. Red indicates positive correlation; purple indicates negative correlation. D1 = domain 1; D2 = domain 2; D3 = domain 3; MFM = Motor Function Measure; NSAA = North Star Ambulatory Assessment; 6MWT = 6-minute walk test.
Correlation Between Maximal Motor Capacity and Pulmonary Function
The relationship between the pulmonary function testing measurement of FVC and maximal motor capacity was highly statistically significant (p < 0.001). The distribution of FVC values demonstrated a direct relationship with severity of clinical phenotype. The relationship between age and FVC for the 3 COL6-RD subgroups was also highly significant (p < 0.001). In a similar way that the mean score of the motor function scales was associated with a sharper decline in UCMD patients, the mean FVC by age was lower among the UCMD and intermediate COL6-RD subgroups (figure 5).
Figure 5. Pulmonary Function and NIV in Patients With Col6-RD by Phenotype.

Relationship between pulmonary function and collagen VI–related dystrophy (COL6-RD) phenotype (based on the classification proposed here using maximal motor function achieved) is highly statistically significant (p < 0.001) as demonstrated by (A) box plots (representing medians and quartiles with whiskers signifying the 5th and 95th percentiles) and (B) scatterplots. (C) Kaplan-Meier curves depict the probability of initiation of noninvasive ventilation (NIV) by age and COL6-RD phenotypic classification. BM = Bethlem myopathy; FVC = forced vital capacity; UCMD = Ullrich congenital muscular dystrophy.
Nocturnal NIV in the form of bilevel positive airway pressure (BiPAP) had been initiated in a total of 33 patients at the time of this study. Of the 38 patients with UCMD, 23 (61%) had initiated NIV at the time of this study. The average age at initiation of NIV in patients with UCMD was 10.1 years (SD ± 4.9 years). Of the 35 patients with intermediate COL6-RD, 9 (26%) had started nocturnal NIV at the time of the study, with an average age at the time of NIV initiation of 15.6 years (SD ± 9.6 years). Only 1 of the 46 patients with BM was using nocturnal NIV at the time of last clinical assessment, which had been initiated at 63 years of age. Kaplan-Meier curves depicting estimated ventilation-free probability demonstrated a statistically significant (p < 0.001) difference between the 3 COL6-RD subgroups, with 50% of patients with UCMD on BiPAP by 12 years of age. Differences were also statistically significant by the age of 20 years: 79.4% of the patients with UCMD, 29.8% of the intermediate patients, and 0% of the patients with BM were using nocturnal NIV in the form of BiPAP.
Correlation Between Genotype and Disease Category Based on Early Motor Maximal Ability
Eighty-seven different mutations were identified in the 115 patients with genetically confirmed diagnosis. These mutations were distributed along the 3 COL6 genes with 41 (47%) in COL6A1, 32 (37%) in COL6A2, and 14 (16%) in COL6A3 (table e-1 available from Dryad, doi:10.5061/dryad.2v6wwpzkk). Forty-nine different dominantly acting mutations (24 in COL6A1, 15 in COL6A2, and 10 in COL6A3) and 38 recessively acting mutations (17 in COL6A1, 17 in COL6A2, and 4 in COL6A3) were detected. A total of 84 patients harbored dominantly acting mutations, while 31 patients carried recessively acting biallelic mutations: 8 in homozygosity and 23 as compound heterozygous alleles. Variants are numbered according to RefSeq transcripts NM_001848.2 for COL6A1, NM_001849.3 for COL6A2, and NM_004369.3 for COL6A3.
The most common mutations acting in a dominant-negative fashion were (1) splice-site mutations resulting in-frame triple helical (TH) exon skipping and (2) missense changes affecting glycine residues located within TH domain. Roughly one-third of cases (40 of 115) were caused by dominant exon-skipping mutations within the 5′ part of the TH domains of any of the 3 COL6 chains. Approximately another one-third of cases (37 of 115) resulted from dominant glycine substitutions in the TH domain, clustered mainly in Gly-X-Y triplets 3–20 at the 5′ part of the domain. Overall, exon-skipping mutations in the 5′ exons encoding for the TH domain resulted in phenotypes consistent with BM in 48.5% of cases, intermediate COL6-RD in 25%, and UCMD in 27.5%. Similarly, in patients with glycine substitutions within the TH domain, 38% resulted in BM, 38% in intermediate COL6-RD, and 24% in UCMD. One study proposed that glycine substitutions in the critical region including Gly-X-Y triplets 10 to 15 in the N-terminal of the TH domain tend to result in more severe phenotypes than those glycine substitutions located in the TH but outside this critical region,35 as it was confirmed in a review of 194 cases of patients with COL6-RD with glycine substitutions in the TH domain of the COL6.36 In our cohort, we also found a higher proportion of milder COL6-RD phenotypes in those patients whose glycine substitutions were located outside the critical region: 55% of patients with glycine substitutions outside the critical region showed a BM phenotype (18% intermediate COL6-RD phenotype, 27% UCMD phenotype), whereas only 25% of patients with glycine substitutions within the critical region showed the milder BM phenotype (50% intermediate COL6-RD phenotype, 25% UCMD phenotype). The third cause of autosomal dominant COL6-RD in terms of frequency (6 of 115) in our study was the recently described recurrent de novo base pair substitution in intron 11 of COL6A1 (c.930 + 189 C>T), which results in a dominantly acting splice-gain event and the insertion of a pseudoexon, disrupting the N-terminal end of the TH domain.37 The ability to rise from the floor unassisted was reported as being attained by only 1 of these 6 patients. That same patient started using a wheelchair by 6 years of age and started NIV in the form of BiPAP by 12 years of age and thus had progressed to a clinical phenotype of UCMD.
As with the substitution in intron 11 of COL6A1, other dominant mutations also showed a strong association with specific phenotypes: the 5 individuals who harbored the COL6A1 c.1056+1 G>A splicing mutation (intron 14) and the 3 with the COL6A3 c.6239 G>A glycine substitution (exon 17) invariably resulted in a BM phenotype. The recurrent COL6A1 c.877 G>A glycine substitution (exon 10) resulted in mild or intermediate phenotypes: an intermediate COL6-RD phenotype was observed in 8 of 11 individuals who harbored it, and the other 3 of 11 showed a BM phenotype. In contrast, patients harboring the same mutation while manifesting a varying range of phenotypic severity were also noted, including, for example, the mutation COL6A3 c.6210+1 G>A (intron 16), which was identified in 1 patient with BM phenotype, 1 with intermediate COL6-RD phenotype, and 2 with UCMD phenotype.
For the recessively acting mutations, some genotype-phenotype correlations were also identified. The first is that individuals who are compound heterozygous for 2 exon-skipping mutations or 2 missense mutations outside the TH domain resulted in relatively mild phenotypes: 62.5% resulted in BM (10 patients) and 37.5% in the intermediate COL6-RD subtype (6 patients), with no detected cases of UCMD. The second is that homozygous mutations that introduce premature termination codons tend to result in severe phenotypes (2 of 2 patients had an UCMD phenotype). The phenotype of those patients with a truncating mutation (premature termination codons or frameshift mutations) in compound heterozygosity with a missense or exon-skipping mutation also resulted in relatively severe phenotypes; however, the phenotypes were more heterogeneous, depending on the second mutation: 22% resulted in BM, 11% in intermediate COL6-RD, and 67% in UCMD.
Discussion
The ability to accurately categorize patients with COL6-RD into clinically relevant phenotypic subgroups from the first years of life will help in optimizing anticipatory clinical care and in assisting in the design of clinical trials for the therapeutic strategies currently in development.18–21,37,38 Natural history and cohort studies of individuals with COL6-RD have highlighted the difficulty of distinguishing between COL6-RD phenotypes, especially for those phenotypes falling between UCMD and BM and, in particular, for those phenotypes bordering established phenotypes.1,10,17 Clear clinical criteria for differentiating between COL6-RD phenotypes in such gray areas surrounding phenotypic boundaries are lacking. Furthermore, the use of motor milestones such as attaining the ability to jump and run, albeit useful, is not able to distinguish phenotypically between patients with COL6-RD during the first years of life.
The 2 motor milestones of rising from the floor unassisted and climbing 4 steps without use of a railing had been selected among the 6 motor developmental milestones previously collected as part of a pilot study of motor function milestones in patients with COL6-RD that assessed sitting unassisted, independent walking, running, jumping, rising from the floor unassisted, and climbing a minimum of 4 steps without holding onto a railing. Sitting unassisted and independent ambulation were not able to discriminate among the COL6-RD phenotypes because the vast majority of patients had achieved these milestones. The motor milestones of running and jumping were found to be prone to misinterpretation among patients and parents who participated in the pilot study. Thus, these milestones were not collected as part of this multicenter study.
Here, we demonstrate that the use of 2 early maximal motor milestones, the ability to rise from the floor unassisted and the ability to ascend 4 steps without holding onto a railing, can reliably distinguish between various COL6-RD phenotypes in a clinically meaningful manner and has the potential for accurately providing a prognosis of disease severity and progression. Our data show that long-term motor function is strongly related to pulmonary function in all patients with COL6-RD. We demonstrate that initial maximal motor capacity can predict other disease-relevant events such as the age at LoA and the need for the initiation of nocturnal NIV. This work shows that classifying patients with COL6-RD into phenotypic groups that exhibit differences in long-term motor function and pulmonary function is effective for studying COL6-RD subgroup–specific natural history and for improving clinical trial readiness for the COL6-RD population as a whole.
Some dominantly acting mutations were found to be strongly associated with specific phenotypes. Among them is the recurrent COL6A1 deep intronic mutation (c.930 + 189C>T) which demonstrates a phenotype of a relative paucity of prominent symptoms during the first 1 to 2 years of life followed by an accelerated progression to a severe UCMD phenotype. Some patients with this mutation achieved independent ambulation, but independent ambulation is subsequently lost between 6 and 9 years of age, consistent with UCMD. Given this new, consistent observation of rapid progression in phenotypic severity associated with this genotype and the potential that our proposed COL6-RD classification may not be as accurate in prospectively predicting the COL6-RD phenotype in this particular group of patients, work on defining this mutation-specific phenotype in an international cohort is underway.
Our study also confirms other previously proposed general genotype-phenotype correlations for COL6-RDs such as the severe phenotype associated with glycine substitutions located in the critical region including Gly-X-Y triplets 10 to 15 in the N-terminal region of the TH domain. We also provide some hypotheses that may be useful to predict the phenotype in those patients with recessively acting mutations.
Of note, individuals with an intermediate COL6-RD phenotype show a progressive deterioration of pulmonary function that is comparatively steeper than the decline in motor function over the same age range. Although the motor function trajectory of individuals with intermediate COL6-RD may at times be difficult to distinguish from that of individuals with BM, the pulmonary function trajectory of these 2 phenotypes is highly distinct. Indeed, the pivotal contribution of this proposed classification is its ability to anticipate a decline in pulmonary function. Given the ability of this classification to predict COL6-RD phenotypes from an early age and result in anticipatory clinical care, the risk of unrecognized or underrecognized hypoventilation, historically the leading cause of morbidity and mortality in the COL6-RDs, should be significantly decreased.
This study has some limitations. We performed a retrospective review of the clinical data that had been collected in the course of patients' clinical care; therefore, most but not all parameters were available for every patient. Furthermore, the maximal motor milestones recorded in the medical record that were not captured on video and are no longer possible to demonstrate clinically may have reduced accuracy due to recall bias. This is particularly the case in the attainment of the ability to rise from the floor, because whether this milestone was achieved without assistance is used to predict a future phenotype of intermediate COL6-RD vs UCMD and would be most accurate if documented after direct observation or review of a video recording of this milestone. For this reason, robust prospective natural history studies with the collection of maximal motor milestone data by either direct observation or video capturing are essential for validating this phenotypic classification. Prospective natural history data collection would also be useful for validating the time-to-event analyses performed in this study (displayed by Kaplan-Meier curves in figures 3 and 5).
Our study has demonstrated some evidence of overlap for age at LoA and age at the initiation of NIV between the UCMD and intermediate COL6-RD phenotypes, namely for patients whose phenotypes fall along the boundary between UCMD and intermediate COL6-RD. While an overlap in phenotypic categorization is not entirely unexpected, given that COL6-RDs manifest along a continuous phenotypic spectrum, it is important to recognize that in reference to the timing of NIV initiation, variability in clinical care between centers and patient compliance/lack of compliance may result in significant variability in the timing of NIV initiation, even when clinically indicated. It is our hope that our proposed prospective classification using early maximal motor milestones to predict future motor function and pulmonary function may prove to be an effective means for individual patient-level prognostication, which, if used across centers, could positively affect care, including the timing of the initiation of NIV. Furthermore, a prospective COL6-RD natural history study would provide the opportunity to better determine the ability of the motor milestone of rising from the floor unassisted to accurately distinguish between the intermediate and UCMD phenotypes.
We note that for patients with Duchenne muscular dystrophy in whom variability on motor function tests has historically complicated the interpretation of clinical trials, latent class trajectory modeling based on 6MWT or NSAA trajectories has proved to be useful in characterizing subgroups of patients with Duchenne muscular dystrophy.39,40 This type of analysis requires the availability of longitudinal motor function scale scores starting at early ages, which was not available for all the patients in this COL6-RD retrospective study. In a future prospective natural history study in patients with COL6-RD, the use of latent class trajectory modeling could be considered as a means of potential validation for the phenotypic classification proposed here.
Our study of a large cohort of patients with COL6-RD from Spain and the United States (n = 119) identifies 2 initial maximal motor function milestones, the ability to rise from the floor unassisted and the ability to climb 4 steps without holding onto a railing, as predictive of the corresponding COL6-RD phenotypic subgroups as follows: (1) achieved rising from the floor only with assistance (UCMD), (2) achieved rising from the floor unassisted (intermediate COL6-RD), and (3) achieved stair climbing (ascending a minimum of 4 steps without use of a railing) (BM). This classification offers a probabilistic approach for predicting the future phenotype of individual patients with COL6-RD from an early age, thus resulting in improved anticipatory care and clinical trial readiness.
Previous studies of patients with COL6-RD had categorized patient phenotypes on the basis of motor function features such as LoA or the achievement of running or jumping; however, none of those proposed phenotypic classifications allowed accurate phenotypic identification from an early age. In contrast, this proposed prospective phenotypic classification for COL6-RDs should enable clinicians to predict a patient's future COL6-RD phenotype from the first years of life. We suggest that our proposed phenotypic classification may be as definitive and relevant from a clinical research perspective as the phenotypic classification of patients with spinal muscular atrophy.41 This study adds to prior studies of COL6-RDs in demonstrating that the early maximal motor function achieved correlates with both motor function and pulmonary function long term. We believe that this classification will help clinicians in providing a more accurate prognosis of future motor function and pulmonary function, thus enabling optimized anticipatory clinical care. This proposed COL6-RD classification will also aid in the design of clinical trials by enabling the phenotypic stratification of clinical trial cohorts in patients with COL6-RD from an early age. Given the goal of making the therapeutic strategies currently in development available to patients with COL6-RD during the first years of life, the accurate prediction of phenotypic subtype from an early age will improve the clinical trial readiness of young patients with COL6-RD.
Acknowledgment
The authors thank the patients and their family members for their participation in this study. They also wish to acknowledge the contribution of Fundación Noelia (Spanish association of patients with COL6-RD) in helping to coordinate patient participation in this study.
Glossary
- BiPAP
bilevel positive airway pressure
- BM
Bethlem myopathy
- CMD
congenital muscular dystrophy
- COL6
collagen VI
- COL6-RD
COL6-related dystrophy
- FVC
forced vital capacity
- LoA
loss of ambulation
- MFM-32
Motor Function Measure-32
- NIV
noninvasive ventilation
- NSAA
North Star Ambulatory Assessment
- 6MWT
6-minute walk test
- TH
triple helical
- UCMD
Ullrich CMD
Appendix. Authors
Study Funding
This study was partially supported by the Instituto de Salud Carlos III and cofunded with European Regional Development Fund “A Way to Achieve Europe” Río Hortega Grant CM17/00054 (D.N.-d.B) and grant PI19/00122 (C.J.M) and intramural funds of the National Institute of Neurological Disorders and Stroke, NIH (C.G.B).
Disclosure
Daniel Natera-de Benito was partially supported by Instituto de Salud Carlos III, Río Hortega Grant CM17/00054. Cecilia Jimenez-Mallebrera is partially supported by Instituto de Salud Carlos III and cofunded with European Regional Development Fund “A Way to Achieve Europe” grant PI19/00122. Carsten G. Bönnemann is supported by intramural funds of the National Institute of Neurological Disorders and Stroke, NIH. The rest of authors report no disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Foley AR, Quijano-Roy S, Collins J, et al. Natural history of pulmonary function in collagen VI-related myopathies. Brain 2013;136:3625–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamandé SR, Bateman JF. Collagen VI disorders: insights on form and function in the extracellular matrix and beyond. Matrix Biol Int Soc Matrix Biol 2018;71–72:348–367. [DOI] [PubMed] [Google Scholar]
- 3.Mohassel P, Reghan Foley A, Bönnemann CG. Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol 2018;71–72:188–204. [DOI] [PubMed] [Google Scholar]
- 4.Bönnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol 2011;7:379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yonekawa T, Nishino I. Ullrich congenital muscular dystrophy: clinicopathological features, natural history and pathomechanism(s). J Neurol Neurosurg Psychiatry 2015;86:280–287. [DOI] [PubMed] [Google Scholar]
- 6.Lampe AK, Flanigan KM, Bushby KM, Hicks D. Collagen type VI-related disorders. In: GeneReviews. Seattle: University of Washington; 1993-2020. [Google Scholar]
- 7.Deconinck N, Richard P, Allamand V, et al. Bethlem myopathy: long-term follow-up identifies COL6 mutations predicting severe clinical evolution. J Neurol Neurosurg Psychiatry 2015;86:1337–1346. [DOI] [PubMed] [Google Scholar]
- 8.Graziano A, Bianco F, D´Amico A, et al. Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology 2015;84:904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sframeli M, Sarkozy A, Bertoli M, et al. Congenital muscular dystrophies in the UK population: clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul Disord 2017;27:793–803. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Park H, Park Y, Kim S, Choi Y. Clinical, pathologic, genetic features of collagen 6-related myopathy in Korea. Neuromuscul Disord 2015;25:S266. [Google Scholar]
- 11.Okada M, Kawahara G, Noguchi S, et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007;69:1035–1042. [DOI] [PubMed] [Google Scholar]
- 12.Peat RA, Smith JM, Compton AG, et al. Diagnosis and etiology of congenital muscular dystrophy. Neurology 2008;71:312–321. [DOI] [PubMed] [Google Scholar]
- 13.Clement EM, Feng L, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001-2008. Neuromuscul Disord 2012;22:522–527. [DOI] [PubMed] [Google Scholar]
- 14.Rodríguez MA, Barquero LMDR, Ortez CI, et al. Differences in adipose tissue and lean mass distribution in patients with collagen VI related myopathies are associated with disease severity and physical ability. Front Aging Neurosci 2017;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cruz S, Figueroa-Bonaparte S, Gallardo E, et al. Bethlem myopathy phenotypes and follow up: description of 8 patients at the mildest end of the spectrum. J Neuromuscul Dis 2016;3:267–274. [DOI] [PubMed] [Google Scholar]
- 16.Toni S, Morandi R, Busacchi M, et al. Nutritional status evaluation in patients affected by Bethlem myopathy and Ullrich congenital muscular dystrophy. Front Aging Neurosci 2014;6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Briñas L, Richard P, Quijano-Roy S, et al. Early onset collagen VI myopathies: genetic and clinical correlations. Ann Neurol 2010;68:511–520. [DOI] [PubMed] [Google Scholar]
- 18.Marrosu E, Ala P, Muntoni F, Zhou H. Gapmer antisense oligonucleotides suppress the mutant allele of COL6A3 and restore functional protein in Ullrich muscular dystrophy. Mol Ther Nucleic Acids 2017;8:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gualandi F, Manzati E, Sabatelli P, et al. Antisense-induced messenger depletion corrects a COL6A2 dominant mutation in Ullrich myopathy. Hum Gene Ther 2012;23:1313–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolduc V, Zou Y, Ko D, Bönnemann CG. SiRNA-mediated allele-specific silencing of a COL6A3 mutation in a cellular model of dominant Ullrich muscular dystrophy. Mol Ther Nucleic Acids 2014;3:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noguchi S, Ogawa M, Kawahara G, Malicdan MC, Nishino I. Allele-specific gene silencing of mutant mRNA restores cellular function in Ullrich congenital muscular dystrophy fibroblasts. Mol Ther Nucleic Acids 2014;3:e171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J 2005;26:319–338. [DOI] [PubMed] [Google Scholar]
- 23.Natera-de Benito D, Alarcon M, Ortez C, et al. Clinical and genetic characterization of collagen VI-related myopathies: difficulties in phenotypic characterization in the first years of life. Neuromuscul Disord 2017;27:S105. [Google Scholar]
- 24.Meilleur KG, Jain MS, Hynan LS, et al. Results of a two-year pilot study of clinical outcome measures in collagen VI- and laminin alpha2-related congenital muscular dystrophies. Neuromuscul Disord 2015;25:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bérard C, Payan C, Hodgkinson I, Fermanian J. A motor function measure scale for neuromuscular diseases: construction and validation study. Neuromuscul Disord 2005;15:463–470. [DOI] [PubMed] [Google Scholar]
- 26.Pane M, Mazzone ES, Sormani MP, et al. 6 Minute walk test in Duchenne MD patients with different mutations: 12 month changes. PLoS One 2014;9:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mcdonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle and Nerve 2013;48:343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li AM, Yin J, Yu CCW, et al. The six-minute walk test in healthy children: reliability and validity. Eur Respir J 2005;25:1057–1060. [DOI] [PubMed] [Google Scholar]
- 29.Geiger R, Strasak A, Treml B, et al. Six-minute walk test in children and adolescents. J Pediatr 2007;150:395–399. [DOI] [PubMed] [Google Scholar]
- 30.Gibbons WJ, Fruchter N, Sloan S, Levy RD. Reference values for a multiple repetition 6-minute walk test in healthy adults older than 20 years. J Cardiopulm Rehabil 2001;21:87–93. [DOI] [PubMed] [Google Scholar]
- 31.Li AM, Yin J, Au JT, et al. Standard reference for the six-minute-walk test in healthy children aged 7 to 16 years. Am J Respir Crit Care Med 2007;176:174–180. [DOI] [PubMed] [Google Scholar]
- 32.Mazzone E, Martinelli D, Berardinelli A, et al. North Star Ambulatory Assessment, 6-minute walk test and timed items in ambulant boys with Duchenne muscular dystrophy. Neuromuscul Disord 2010;20:712–716. [DOI] [PubMed] [Google Scholar]
- 33.Tiziano FD, Lomastro R, Di Pietro L, et al. Clinical and molecular cross-sectional study of a cohort of adult type III spinal muscular atrophy patients: clues from a biomarker study. Eur J Hum Genet 2013;21:630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain MS, Meilleur K, Kim E, et al. Longitudinal changes in clinical outcome measures in COL6-related dystrophies and LAMA2-related dystrophies. Neurology 2019;93:e1932–e1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pace RA, Peat RA, Baker NL, et al. Collagen VI glycine mutations: perturbed assembly and a spectrum of clinical severity. Ann Neurol 2008;64:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butterfield RJ, Foley AR, Dastgir J, et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen vi myopathies. Hum Mutat 2013;34:1558–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolduc V, Foley AR, Solomon-Degefa H, et al. A recurrent COL6A1 pseudoexon insertion causes muscular dystrophy and is effectively targeted by splice-correction therapies. JCI Insight 2019;4:e124403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguti S, Bolduc V, Ala P, et al. Exon-skipping oligonucleotides restore functional collagen VI by correcting a common COL6A1 mutation in Ullrich CMD. Mol Ther Nucleic Acids 2020;21:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mercuri E, Signorovitch JE, Swallow E, et al. Categorizing natural history trajectories of ambulatory function measured by the 6-minute walk distance in patients with Duchenne muscular dystrophy. Neuromuscul Disord 2016;26:575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muntoni F, Domingos J, Manzur AY, et al. Categorising trajectories and individual item changes of the North Star Ambulatory Assessment in patients with Duchenne muscular dystrophy. PLoS One 2019;14:e0221097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerres K, Schöneborn SR. Natural history in proximal spinal muscular atrophy: clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol 1995;52:518–523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data not published are available from the corresponding author and will be shared with any qualified investigator on reasonable request.