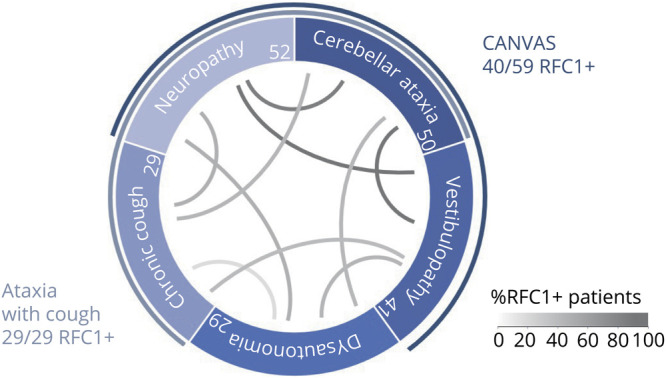
Andreas Traschütz
Andreas Traschütz, MD, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Andrea Cortese
Andrea Cortese, MD, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Selina Reich
Selina Reich, MSc
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Natalia Dominik
Natalia Dominik, MSc
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Jennifer Faber
Jennifer Faber, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Heike Jacobi
Heike Jacobi, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Annette M Hartmann
Annette M Hartmann, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Dan Rujescu
Dan Rujescu, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Solveig Montaut
Solveig Montaut, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Andoni Echaniz-Laguna
Andoni Echaniz-Laguna, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Sevda Erer
Sevda Erer, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Valerie Cornelia Schütz
Valerie Cornelia Schütz, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Alexander A Tarnutzer
Alexander A Tarnutzer, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Marc Sturm
Marc Sturm, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Tobias B Haack
Tobias B Haack, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Nadège Vaucamps-Diedhiou
Nadège Vaucamps-Diedhiou, MSc
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Helene Puccio
Helene Puccio, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Ludger Schöls
Ludger Schöls, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Thomas Klockgether
Thomas Klockgether, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Bart P van de Warrenburg
Bart P van de Warrenburg, MD, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Martin Paucar
Martin Paucar, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Dagmar Timmann
Dagmar Timmann, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Ralf-Dieter Hilgers
Ralf-Dieter Hilgers, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Jose Gazulla
Jose Gazulla, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Michael Strupp
Michael Strupp, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
German Moris
German Moris, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Alessandro Filla
Alessandro Filla, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Henry Houlden
Henry Houlden, MD, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Mathieu Anheim
Mathieu Anheim, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Jon Infante
Jon Infante, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
A Nazli Basak
A Nazli Basak, PhD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,
Matthis Synofzik
Matthis Synofzik, MD
1From the Department of Neurodegenerative Diseases (A.T., S.R., L.S., M. Synofzik), Hertie-Institute for Clinical Brain Research and Center of Neurology, and German Center for Neurodegenerative Diseases (DZNE) (A.T., S.R., L.S., M. Synofzik), University of Tübingen, Germany; MRC Centre for Neuromuscular Diseases (A.C., N.D., H.H.), Department of Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, UCL Queen Square Institute of Neurology, London, UK; Department of Brain and Behaviour Sciences (A.C.), University Pavia, Italy; Department of Neurology (J.F., T.K.), University Hospital Bonn; German Center for Neurodegenerative Diseases (DZNE) (J.F., H.J., T.K.), Bonn; Department of Neurology (H.J.), University Hospital of Heidelberg; Department of Psychiatry, Psychotherapy and Psychosomatics (A.M.H., D.R.), University of Halle, Germany; Département de Neurologie (S.M., M.A.), Hôpital de Hautepierre, Hôpitaux Universitaires de Strasbourg; Department of Neurology (A.E.-L.), APHP, CHU de Bicêtre; French National Reference Center for Rare Neuropathies (NNERF) (A.E.-L.); Inserm U1195 and Paris-Sud University (A.E.-L.), Le Kremlin Bicêtre, France; Medical Faculty (S.E.), Department of Neurology, Uludag University, Bursa, Turkey; University of Zurich (V.C.S., A.A.T.); Department of Neurology (V.C.S., A.A.T.), University Hospital Zurich, Switzerland; Institute of Medical Genetics and Applied Genomics (M. Sturm, T.B.H.) and Center for Rare Diseases (T.B.H.), University of Tübingen, Germany; Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (N.V.-D., H.P.); INSERM (N.V.-D., H.P.), U1258; CNRS (N.V.-D., H.P.), UMR7104, Illkirch; Université de Strasbourg (H.P.), France; Department of Neurology (B.P.v.d.W.), Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, the Netherlands; Department of Neurology (M.P.), Karolinska University Hospital; Department of Clinical Neuroscience (M.P.), Karolinska Institute, Stockholm, Sweden; Department of Neurology (D.T.), Essen University Hospital, University of Duisburg-Essen, Essen; Department of Medical Statistics (R.-D.H.), RWTH Aachen University, Germany; Department of Neurology (J.G.), Hospital Universitario Miguel Servet. Zaragoza, Spain; Department of Neurology (M. Strupp), University Hospital, and German Center for Vertigo and Balance Disorders (M.Strupp), Ludwig Maximilians University, Munich, Germany; Neurology Service (G.M.), Hospital Unversitario Central de Asturias (HUCA), SESPA, Oviedo, Spain; Department of Neurosciences and Reproductive and Odontostomatological Sciences (A.F.), Federico II University Naples, Italy; Institute of Genetics and Molecular and Cellular Biology (M.A.), INSERM-U964/CNRS-UMR7104, University of Strasbourg, Illkirch; Strasbourg Federation of Translational Medicine (M.A.), University of Strasbourg, Strasbourg, France; Service of Neurology (J.I.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, “Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED),” Santander, Spain; and Suna and Inan Kıraç Foundation (A.N.B.), Neurodegeneration Research Laboratory, KUTTAM, Koç University School of Medicine, Istanbul, Turkey.
1,✉;
on behalf of the RFC1 Study Group1