

Abstract
Objective
To assess the frequency of biologically defined Alzheimer disease (AD) in relation to age, sex, APOE ε4, and clinical diagnosis in a prospective cohort study evaluated with amyloid-PET and tau-PET.
Methods
We assessed cognitively unimpaired (CU) elderly (n = 166), patients with amnestic mild cognitive impairment (n = 77), and patients with probable AD dementia (n = 62) who underwent evaluation by dementia specialists and neuropsychologists in addition to amyloid-PET with [18F]AZD4694 and tau-PET with [18F]MK6240. Individuals were grouped according to their AD biomarker profile. Positive predictive value for biologically defined AD was assessed in relation to clinical diagnosis. Frequency of AD biomarker profiles was assessed using logistic regressions with odds ratios (ORs) and 95% confidence intervals (CIs).
Results
The clinical diagnosis of probable AD dementia demonstrated good agreement with biologically defined AD (positive predictive value 85.2%). A total of 7.88% of CU were positive for both amyloid-PET and tau-PET. Frequency of biologically defined AD increased with age (OR 1.14; p < 0.0001) and frequency of APOE ε4 allele carriers (single ε4: OR 3.82; p < 0.0001; double ε4: OR 17.55, p < 0.0001).
Conclusion
Whereas we observed strong, but not complete, agreement between clinically defined probable AD dementia and biomarker positivity for both β-amyloid and tau, we also observed that biologically defined AD was not rare in CU elderly. Abnormal tau-PET was almost exclusively observed in individuals with abnormal amyloid-PET. Our results highlight that even in tertiary care memory clinics, detailed evaluation by dementia specialists systematically underestimates the frequency of biologically defined AD and related entities.
Classification of Evidence
This study provides Class I evidence that biologically defined AD (abnormal amyloid PET and tau PET) was observed in 85.2% of people with clinically defined AD and 7.88% of CU elderly.
Definitive diagnosis of Alzheimer disease (AD) requires the presence of β-amyloid (Aβ) plaques and tau neurofibrillary tangles upon postmortem examination.1,2 Diagnosis of probable AD (pAD), a clinical syndrome characterized by progressive amnestic multidomain cognitive impairment resulting in dementia,3,4 shows varying degrees of sensitivity and specificity for the core neuropathologic features of AD.5,6 Whereas the pAD clinical syndrome displays good but imperfect concordance with neuropathology, significant aggregation of Aβ plaques and tau neurofibrillary tangles is sometimes observed in the brains of cognitively unimpaired (CU) elderly individuals at autopsy,7,8 or in individuals with more subtle cognitive impairment who do not exhibit dementia.9–11
The recent biological research framework for AD extends the neuropathologic definition of AD to living humans with the use of in vivo biomarkers of Aβ and tau.12 Therefore, it is now possible to identify biologically defined AD in individuals without evidence of cognitive impairment. Correspondingly, it is also now feasible to provide estimates of discordance between traditional clinical definitions of pAD dementia and of biologically defined AD in living humans. Here, we assess the frequency of biologically defined AD entities in relation to age, sex, and APOE ε4, as well as the rates of concordance between clinical and biological definitions of AD in a prospective cohort study.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
This study's protocol was approved by McGill University's Institutional Review Board. Informed written consent was obtained from all participants.
Participants
All individuals in this study were part of the Translational Biomarkers in Aging and Dementia (TRIAD) cohort,13 a longitudinal imaging and biofluid cohort study of aging and AD. Participants were recruited through advertisements in the community, newspaper advertisements, word of mouth, and referrals from the McGill Centre for Studies in Aging. Evaluations of participants included a review of their medical history and an interview with the participant and his or her study partner followed by a neurologic examination by a dementia specialist and a neuropsychological examination. Participants were given a diagnosis of CU (defined as not mild cognitive impairment [MCI] or dementia),14 single or multi-domain amnestic MCI,15 or probable Alzheimer disease (pAD)4 using established criteria. All participants were evaluated at the McGill Center for Studies in Aging, a tertiary care memory clinic specializing in the diagnosis and clinical management of neurodegenerative diseases. The pAD group included individuals with a typical amnestic phenotype3,4 as well as some individuals with established atypical AD syndromes: language-predominant, visuospatial-predominant, and behavioral/dysexecutive-predominant. Four individuals with pAD included in this study had autosomal dominant AD. Clinical diagnoses were always made blinded to PET results (patients only participate in research after they have been evaluated by the study physician and have a diagnosis); in other words, AD was first clinically defined before comparing with biologically defined AD. Individuals with dementia due to a suspected etiology other than AD (n = 49) were not included in the study. Other exclusion criteria were imaging contraindications, active substance abuse, active psychiatric disorder, and untreated medical conditions. See figure 1 for a flowchart of this study's selection process.
Figure 1. Study Flowchart.
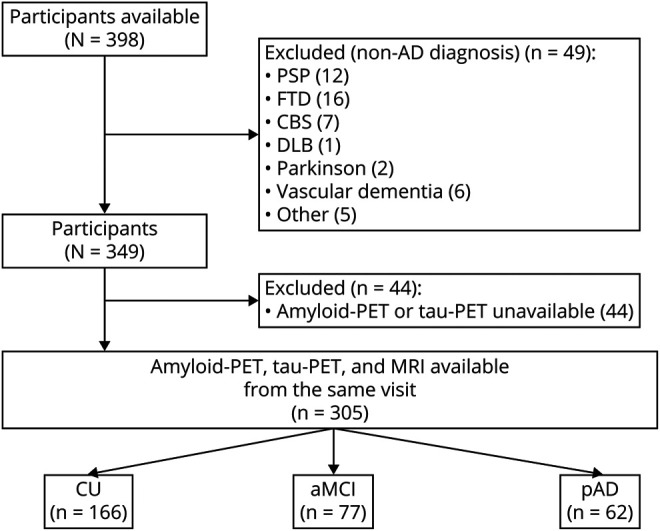
aMCI = amnestic mild cognitive impairment; CBS = corticobasal syndrome; CU = cognitively unimpaired; DLB = dementia with Lewy bodies; FTD = frontotemporal dementia; pAD = probable Alzheimer disease; PSP = progressive supranuclear palsy.
We assessed CU elderly (n = 166), patients with single or multidomain amnestic MCI (n = 77), and patients with pAD dementia (n = 62) who were evaluated with Aβ PET with [18F]AZD4694, tau PET with [18F]MK6240, anatomical MRI, and genotyping for APOE ε4. Consistent with the biological AD research framework from the National Institute of Aging–Alzheimer's Association,12 study participants without objective cognitive dysfunction who reported subjective memory complaints were analyzed together with CU individuals. All participants had clinical assessments including Clinical Dementia Rating (CDR), Mini-Mental State Examination (MMSE), and cerebrovascular disease risk using the Hachinski Ischemic Scale.
PET Image Acquisition and Processing
PET image acquisition and processing has been described previously.13 [18F]MK6240 standardized uptake value ratio (SUVR) images were normalized using the inferior cerebellar gray matter as a reference region and [18F]AZD4694 SUVR maps were generated using the cerebellar gray matter as a reference region. A global neocortical [18F]AZD4694 SUVR was estimated for each study participant by averaging the [18F]AZD4694 SUVR from the precuneus, prefrontal, orbitofrontal, temporal, parietal, anterior, and posterior cingulate cortices. [18F]MK6240 SUVRs were calculated in a Braak stage I–II region of interest (ROI) comprising the entorhinal cortex and hippocampus16,17 as well as a temporal meta-ROI comprising the entorhinal cortex, hippocampus, fusiform, parahippocampal, inferior temporal, and middle temporal cortices.18
MRI Acquisition and Processing
Structural MRI data were acquired at the Montreal Neurologic Institute for all participants on a 3T Siemens Magnetom using a standard head coil. The MRI protocol also included a fluid-attenuated inversion recovery (FLAIR) sequence, which was employed to quantify white matter hyperintensity (WMH) volume. Cortical thickness measures were determined using Freesurfer (v6.0). We computed the AD-signature meta-ROI based on the weighted average of the bilateral entorhinal, inferior temporal, middle temporal, and fusiform cortices. WMH volume was quantified using the lesion prediction algorithm as implemented in the LST toolbox version 2.0.15 in SPM, which calculated a probability of WMH at each voxel. All segmented WMH images were examined visually. A binary WMH image was then defined for WMH probability >0.5.
Determination of AD Biomarker Abnormality
Amyloid-PET positivity was determined using an [18F]AZD4694 SUVR threshold of 1.55 validated using the following methodologic approaches: (1) Gaussian mixture modeling, (2) ROC analyses contrasting CU elderly with pAD dementia, (3) ROC analyses contrasting visually positive/negative amyloid-PET scans, (4) comparison with Aβ positivity determined by CSF biomarkers, and (5) comparisons with young adults (age <25).19 These approaches converged at an [18F]AZD4694 SUVR threshold of 1.55.
To determine tau-PET positivity, we created thresholds based on the means and SDs of a population of 30 amyloid-negative CU young adults scanned with [18F]MK6240 (age <25; clinical and demographic data provided in supplementary table 1, data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr). In primary analyses, a participant was considered positive for tau-PET if he or she surpassed the mean + 2 SDs from the CU young population in a temporal meta-ROI comprising the hippocampus, entorhinal cortex, fusiform, parahippocampal, inferior temporal, and middle temporal cortices.18 This resulted in an [18F]MK6240 SUVR threshold of 1.24. We chose to select CU young individuals as the reference group because large-scale postmortem examinations report frequent neurofibrillary tangle pathology in CU elderly,20,21 a finding also observed in multiple [18F]Flortaucipir PET studies.22 Therefore, taking the mean and SD from a population of CU elderly individuals may lack sensitivity to detect neurofibrillary changes.18
Because dichotomous classification of continuous variables into positive/negative categories inevitably presents with a number of conceptual and analytical idiosyncrasies, we conducted sensitivity analyses in which we defined tau positivity based on Braak I–II regions, where positivity was determined using means and 2 SDs from a sample of 30 CU young adults23 (Braak I–II ROI mean [18F]MK6240 SUVR in young adults 0.79; SD 0.14; cutoff 1.1). Braak I–II regions were selected because these regions are characterized by early tau accumulation compared to other brain regions.20,21 We employed the entorhinal cortex and hippocampus as Braak I–II ROIs given that [18F]MK6240 is not susceptible to off-target binding to the choroid plexus.24 A contingency table illustrating the proportion of the sample labeled as positive according to both the temporal meta-ROI and Braak I-II ROIs, as well as representative discordant cases, is displayed in supplementary figure 1 (data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr).
For the study's primary hypotheses, individuals were grouped into 1 of 4 AD biomarker categories based on their AD biomarker abnormality: A−T−: negative AD biomarkers; A+T−: Alzheimer pathologic change; A−T+: non-Alzheimer pathologic change; A+T+: biological AD.12,14,25 Because our primary hypothesis was to determine frequency of AD pathology, we did not stratify individuals based on neurodegeneration biomarkers as neurodegeneration is neither sensitive nor specific to AD (it does not define AD biologically among other neurodegenerative diseases). However, we repeated all analyses using the additional stratification based on neurodegeneration (N) biomarkers in secondary analyses. In these analyses, an individual was considered positive for neurodegeneration if cortical thickness in AD signature regions (bilateral entorhinal, inferior temporal, middle temporal, and fusiform cortices) fell below 2.77. This threshold was determined based the least distance from (0,1) point to the ROC curve contrasting Aβ− CU elderly with cognitively impaired individuals, providing the best tradeoff between sensitivity and specificity between these groups.
Statistical Analyses
Baseline demographics were evaluated using multiple t tests and χ2 tests using R version 3.5.3 (r-project.org/). We employed logistic regression to assess the relationships between AD spectrum clinical entities in relation to age, sex, and APOE ε4 status, with relative frequencies computed with odds ratios (ORs) and corresponding 95% confidence intervals (CIs).26 We repeated APOE ε4 analyses excluding the 4 cases with autosomal dominant AD. Rates of concordance between clinically diagnosed pAD dementia and biologically defined AD are reported and statistically assessed with χ2 contingency analyses. Rates of concordance between clinically defined and biologically defined AD stratified by age are reported for the CU elderly and individuals with pAD dementia. Rates of A−T+ biomarker profiles as estimated by the temporal meta-ROI and Braak I–II ROIs were compared using χ2 contingency analyses. The evidence provided in this study is Class I because of the prospective data collection, inclusion of a broad spectrum of individuals suspected of having the disease, and determination of disease status made independently of the test results. We carried out model diagnostics using the car package in R to determine the presence of multicollinearity. We calculated the variance inflation factor (VIF), a measure of the extent to which variance in regression coefficients is inflated due to multicollinearity.
Data Availability
Anonymized data and documentation from this study can be made available to qualified investigators on reasonable request. Such arrangements are subject to standard data sharing agreements.
Results
Demographic and clinical information is presented in table 1. At the group level, participants with clinically diagnosed pAD dementia had higher neocortical [18F]AZD4694 SUVR, higher [18F]MK6240 SUVR in Braak I–II regions, higher [18F]MK6240 SUVR in the temporal meta-ROI, and lower MMSE scores compared to CU elderly. Individuals with amnestic MCI (aMCI) also had higher neocortical [18F]AZD4694 SUVR and higher [18F]MK6240 SUVR in Braak I–II regions, and higher [18F]MK6240 SUVR in the temporal meta-ROI, than CU elderly. APOE ε4 carriership was highest in the AD dementia group. The aMCI and pAD dementia groups had significantly lower cortical thickness and higher WMH volume than healthy controls. Amyloid-PET and tau-PET biomarkers in each AD phenotype are presented in supplementary table 2 (data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr). Across all diagnostic groups, age was significantly associated with both cortical thickness (β estimate −0.007, SE 0.001, p < 0.0001) and WMH volume (β estimate 0.35, SE 0.07, p < 0.0001). VIFs for all variables were below 2, indicating that problematic levels of multicollinearity are very unlikely to be present in our analyses. Full model statistics for all frequency analyses are summarized in supplementary tables 3–6 (data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr).
Table 1.
Demographic and Key Characteristics of the Sample

Associations Between AD Biomarker Status and Clinical Impairment
Figure 2 represents the association between biologically defined AD spectrum entities and severity of clinical impairment as indexed by CDR score. Frequency of AD biomarkers increased corresponding to increasing clinical impairment. CU individuals (CDR 0) were most likely to be AD biomarker–negative (72.3%), although 8.1% of individuals were positive for both amyloid- and tau-PET. Individuals with a CDR of 0.5 displayed heterogeneous patterns of AD biomarkers, with 34.6% being biomarker-negative and under 47.4% being positive for both amyloid-PET and tau-PET. Individuals with a CDR of 1 were most likely to be positive for both amyloid-PET and tau-PET biomarkers (84.2%). Finally, in a tertiary care memory clinic setting, 100% of individuals with AD dementia and a CDR of 2 were positive for both amyloid- and tau-PET biomarkers. No individuals with severe dementia (CDR 3) were recruited. Associations between A/T/N status and CDR are reported in supplementary figure 2 (data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr).
Figure 2. Associations Between Alzheimer Disease (AD) Biomarker Status and Clinical Impairment.

AD biomarker status according to Clinical Dementia Rating (CDR). Each plot represents individuals grouped according to CDR, with bar colors corresponding to AD biomarker status. Individuals with CDR 0 were the most likely to be AD biomarker–negative (72.3%). Despite lower frequency, 8.1% of cognitively unimpaired individuals were positive for both amyloid- and tau-PET biomarkers. Individuals with a CDR of 0.5 displayed heterogeneous patterns of AD biomarkers, with under 40% being biomarker-negative and under 50% being positive for both amyloid- and tau-PET. Individuals with a CDR of 1 were most likely to be positive for both amyloid- and tau-PET biomarkers (84.2%). Finally, 100% of individuals with AD dementia and a CDR of 2 were positive for both amyloid- and tau-PET biomarkers. A−T− = amyloid-negative/tau-negative (normal AD biomarkers); A+T− = amyloid-positive/tau-negative (AD pathologic change); A−T+ = amyloid-negative/tau-positive (non-AD pathologic change); A+T+ = amyloid-positive/tau positive (biological AD).
Frequency of Biologically Defined AD Spectrum Entities Stratified by Age
We next assessed the frequency of biologically defined AD spectrum entities with respect to age. As a specialized memory clinic, the McGill Center for Studies in Aging sees a disproportionately high level of patients with early-onset AD (familial as well as sporadic), who by definition have both Aβ and tau positivity at a young age (symptom onset <65 years old). Because these individuals are rare but constitute a relatively large proportion of the patients with AD in our cohort, we display results without the patients with early-onset AD in figure 3A and with early-onset AD in figure 3B. When excluding individuals with early-onset AD, we observed that younger individuals were more likely to be AD biomarker–negative (β estimate −0.11, SE 0.02, p < 0.00001). Conversely, the frequency of biologically defined AD rose with age (β estimate 1.3, SE 0.03, p < 0.00001). Similarly, the frequency of AD pathologic change increased with age (β estimate 0.05, SE 0.02, p = 0.03). When investigating tau positivity as assessed with the temporal meta-ROI, we did not observe increases in the frequency of non-AD pathologic change (A−T+) with age (β estimate 0.06, SE 0.06, p = 0.3). When including patients with early-onset AD, as expected, we observed that the frequency of biologically defined AD in individuals under age 65 increased substantially. Otherwise, the same age-related trends remained. Associations between age and A/T/N status are reported in supplementary figure 4A (data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr).
Figure 3. Frequency of Biologically Defined Alzheimer Disease (AD) Spectrum Entities Stratified by Age.

Frequency of AD biomarkers rises with age. Individuals are grouped by AD biomarker status, with colored bars representing each age group. (A) Tau positivity defined in the temporal meta–region of interest. Individuals below the age of 65 were the most likely to be AD biomarker–negative (p < 0.0001). Correspondingly, the likelihood of being AD biomarker–negative decreased with each age group. In contrast, the frequency of biologically defined AD (A+T+) increased with age (p < 0.0001). There was no statistically significant association between non-AD pathologic change and age (p = 0.3). (B) When defining tau positivity using Braak I–II regions, we observed a higher frequency of the A−T+ biomarker profile until age 75. We also observed higher frequency of the A+T+ biomarker profile. Individuals with early-onset AD are excluded from this figure and presented in supplementary figure 3 (available at doi.org/10.5061/dryad.69p8cz8zr).
Association Among APOE ε4, Sex, and Biologically Defined AD Spectrum Entities
Figure 4 displays the relative frequency of APOE ε4 status (ε4 noncarrier/ε4 heterozygous/ε4 homozygous) in relation to the 4 biologically defined AD spectrum entities. APOE ε4 displayed a gene–dose association with biologically defined AD as indexed by amyloid-PET and tau-PET positivity (APOE ε4 heterozygosity: OR 3.82, 95% CI 2.19–6.71, p < 0.0001; APOE ε4 homozygosity: OR 17.55, 95% CI 5.25–80.2, p < 0.0001). Notably, no APOE ε4 homozygotes were found in the A−T+ category. Figure 5 represents the relative frequency of AD spectrum entities as stratified by sex. We did not observe significant differences in the frequency of AD spectrum entities between men and women based on dichotomous cutoffs. Associations between APOE ε4 status, sex, and A/T/N status are reported in supplementary figure 4B and C (data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr).
Figure 4. Association Between APOE4 and Biologically Defined Alzheimer Disease (AD) Spectrum Entities.

Relative frequency of APOE ε4 status (ε4 noncarrier/ε4 heterozygous/ε4 homozygous) in relation to the 4 biologically defined AD spectrum entities. APOE ε4 displayed a gene–dose association with both amyloid-β and tau-PET positivity. Zero percent APOE ε4 of homozygotes were A−T+. When excluding the 4 autosomal dominant AD cases, we observed a slightly lower frequency (17% vs 19%) of A+T+ in individuals who were APOE ε4 noncarriers.
Figure 5. Biologically Defined Alzheimer Disease (AD) Spectrum Entities Stratified by Sex.
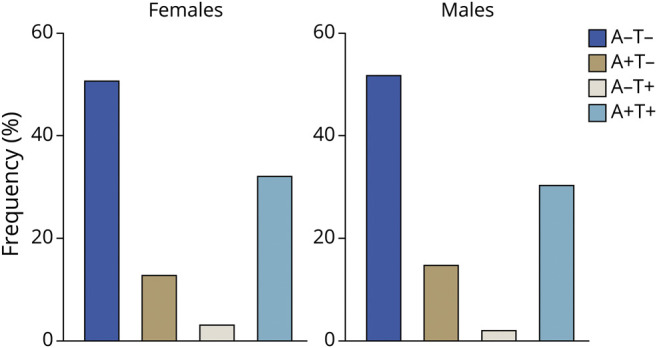
Relative frequency of AD spectrum entities as stratified by sex. We did not observe statistically significant differences in the frequency of AD spectrum entities between men and women based on dichotomous cutoffs.
Concordance of Biologically vs Clinically Defined AD
Rates of agreement between biologically defined and clinically defined AD are presented in figure 6. We evaluated 62 participants with pAD according to established guidelines.3,4 Of these 62 participants, 52 were positive for both amyloid-PET and tau-PET (positive predictive value 85.2%, p < 0.0001), indicating good concordance between clinical phenotype and presence of both Aβ and tau. Of the remaining participants with pAD, 4 (6.6%) participants had Alzheimer pathologic change (A+T−) and 3 individuals (5%) had non-Alzheimer pathologic change (A−T+). Two (3.2%) participants with pAD did not display abnormal AD biomarkers (A−T−). Of the 166 CU elderly participants, 13 (7.8%) had biologically defined AD (positive for both amyloid-PET and tau-PET) (negative predictive value 92%). A total of 28 (16.9%) CU elderly individuals had AD pathologic change (A+T−) and 3 (1.2%) had non-Alzheimer pathologic change (A−T+). A total of 121 (73.3%) CU elderly individuals did not display abnormal AD biomarkers (figure 6). Of the 77 participants with single or multidomain amnestic MCI, 29 (38.1%) had biologically defined AD. A total of 10 (13.2%) had AD pathologic change (A+T−) and 3 (3.9%) had non-AD pathologic change. A total of 34 individuals (44.7%) did not display abnormal AD biomarkers. When assessing agreement between clinically and biologically defined AD in CU elderly, we observed that rates of concordance dropped with advancing age (β estimate 0.15, SE 0.06, p = 0.014; supplementary figure 5, data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr), indicating higher rates of biologically defined AD with advanced age. In the pAD dementia group, there was no statistically significant association between patient age and clinical–biological discordance (β estimate −0.005, SE 0.05, p = 0.96). Associations between clinical diagnosis and A/T/N status are reported in supplementary figure 6 (data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr).
Figure 6. Concordance Between Biologically and Clinically Defined Alzheimer Disease (AD).

Whereas the majority of cognitively unimpaired (CU) participants had negative AD biomarkers, approximately 8% had biologically defined AD. Conversely, whereas the majority of patients with pAD dementia were positive for both amyloid-β and tau-PET, there was imperfect agreement, with 15% of patients with pAD dementia being amyloid-PET–negative but tau-PET–positive (5%), amyloid-PET–positive but tau-PET–negative (6.5%), or both amyloid-PET and tau-PET–negative (3.3%). Amyloid-PET positivity was more compatible with normal cognition than tau-PET positivity. A−T− = amyloid-negative/tau-negative (normal AD biomarkers); A+T− = amyloid-positive/tau-negative (AD pathologic change); A−T+ = amyloid-negative/tau-positive (non-AD pathologic change); A+T+ = amyloid-positive/tau positive (biological AD).
Sensitivity Analyses
Due to the relatively recent emergence of tau-PET and the corresponding novelty of defining positive/negative cutoffs based on tau-PET SUVR, we performed sensitivity analyses using Braak I–II ROIs, considered to be regions of early tau accumulation.20,21 While the general pattern of results remained the same, some notable differences emerged: in individuals with CDR 0 and CDR 0.5, tau positivity was higher than when using the temporal meta-ROI, likely reflecting the characteristic early tau aggregation in these regions (supplementary figure 7, data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr). When measuring tau-PET in Braak I–II ROIs we again did not observe increased frequency of non-AD pathologic change with age (p = 0.98). While we observed relationships between age and positivity in Braak I–II ROIs (p < 0.0001), these relationships no longer existed when including Aβ positivity as a covariate (p = 0.54) (figure 3). Where no sex differences were observed when employing the temporal meta-ROI, we observed differences in the frequency of Braak I–II tau positivity, such that women were more likely than mento have biologically defined AD (β estimate 0.75, SE 0.36, p = 0.03; OR 0.47, 95% CI 0.23–0.93) (supplementary figure 8, data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr). The same pattern of relationships remained between APOE ε4 and AD biomarkers. Finally, a similar pattern of concordance was observed between clinical diagnosis of AD and Aβ and tau-PET positivity (positive predictive value 85%). However, more CU elderly individuals were labeled as being A+T+ (11.9%). Overall, we observed a significantly larger proportion of individuals with the A−T+ biomarker profile when assessing tau-PET positivity using Braak I–II ROIs (47 A−T+ individuals) compared to the temporal meta-ROI (9 A−T+ individuals); χ2 = 30.1, p < 0.0001 (supplementary figure 9, data available from Dryad, doi.org/10.5061/dryad.69p8cz8zr).
Discussion
We investigated the frequency of biologically defined AD spectrum entities in relation to age, sex, and APOE ε4, as well as the rates of discordance between clinically and biologically defined AD. We found good, but not perfect, agreement between clinically defined pAD and biomarker evidence for both Aβ and tau. A number of conclusions can be reached. First, elevated tau-PET was rarely observed in the absence of elevated amyloid-PET. Second, increasing clinical impairment was associated with increased likelihood of positivity of both AD biomarkers. Elevated amyloid-PET uptake was more compatible with normal cognition than elevated tau-PET; tau-PET positivity was more frequently associated with clinical impairment. Finally, biologically defined AD was observed in approximately 8% of CU elderly participants, indicating that even in centers specializing in the diagnosis and clinical management of neurodegenerative diseases, clinical phenotyping and detailed neuropsychological evaluation systematically underestimate the frequency of detectable AD.
In our study, most but not all (85.2%) individuals diagnosed with pAD had biologically defined AD. Because multiple neuropathologic degenerative processes can result in the same clinical phenotype, the pAD clinical syndrome lacked specificity for biologically defined AD.27 Argyrophilic grain disease,28 limbic-predominant age-related TDP-43 encephalopathy,29 hippocampal sclerosis, and neurofibrillary tangle predominant dementia,30 among others, can all present with progressive amnestic impairment that mirrors the pAD phenotype. Without biomarkers specific for these neurodegenerative processes, we cannot determine the cause of cognitive impairment in individuals with the pAD clinical syndrome who do not have biologically defined AD. While biomarkers of neurodegeneration are generally not considered specific in differentiating among neurodegenerative diseases, recent studies using [18F]FDG-PET in tau-negative individuals with progressive amnestic impairment have provided evidence that the topography of temporal [18F]FDG-PET is associated with hippocampal sclerosis at autopsy.31 Recent studies have provided evidence that negative amyloid-PET and tau-PET scans may be useful in ruling out AD as the cause of progressive amnestic impairment.32 Furthermore, it is conceivable that neurofibrillary tangle predominant dementia30 may be identified based on the A−/T+ biomarker profile in individuals with dementia, though it may also be necessary to rule out other pathologies. There remains a need for biomarkers specific to other neuropathologies in order to determine the etiology (etiologies) of non-AD cognitive impairment in vivo. The heterogeneity of pathologic insults (either together or in isolation) that can be associated with the same phenotype highlights the limitations of clinical phenotyping even in specialized centers.
The limitations in using clinical phenotyping to identify biologically defined AD are exacerbated when examining individuals with only mild cognitive symptoms, despite the fact that the disease process may have been taking place for several years.33,34 For CU elderly and aMCI groups, the concordance between clinically and biologically defined AD dropped substantially. Approximately 36% of individuals with aMCI had biologically defined AD; nearly 50% were AD biomarker–negative. While this presents a lower frequency than some autopsy studies of MCI, autopsy studies are typically conducted on much older individuals,10,11 who are more likely to harbor AD pathology. The same is true of autopsy studies of CU individuals, who are also more likely to display AD pathology with advanced age.8 Indeed, in our study, advanced age was strongly associated with the presence of biologically defined AD in CU individuals. Biologically defined AD in CU elderly was far from rare: in our study, estimates ranged from 7% to 12%, depending on the brain regions used to determine tau positivity. Early Braak regions resulted in higher levels of tau positivity. While by definition CU elderly individuals do not demonstrate overt cognitive impairment, tau pathology in early Braak regions is associated with memory dysfunction,35 neurodegeneration,36 as well as longitudinal cognitive decline.37 Thus, identification of individuals at greatest risk for cognitive decline may be aided by the quantification of AD neuropathology using PET.
Tau abnormality as determined by PET occurred almost exclusively in the presence of amyloid-PET positivity. However, the reverse was not the case: a significant portion of individuals across clinical presentations displayed amyloid-PET positivity in the absence of tau-PET positivity. Though our data are cross-sectional and correspondingly cannot infer a causal link between Aβ and tau, these findings can be interpreted in the context of longitudinal studies of autosomal dominant34 and sporadic33 AD in which Aβ abnormality precedes tau abnormality as assessed with CSF by several years. Taken together, our study supports a model of AD in which amyloid-PET abnormality is an early marker of a protracted disease process, while tau-PET abnormality is more proximal to the clinical impairment typically associated with pAD dementia.
Despite the fact that almost all individuals with positive tau-PET had a positive amyloid-PET scan, a non-negligible portion of participants in our study (∼3–6%) with significant tau pathology did not display abnormal Aβ deposition. While these findings are in agreement with postmortem studies reporting tau neurofibrillary tangle pathology in aging20 and dementia30 in the absence of Aβ, it will be crucial to determine the factors underlying neurofibrillary tangle aggregation in amyloid-negative individuals and to what extent these represent a pathologic process distinct from AD.38 It is also important to remember that an amyloid-negative PET scan does not signify the absence of cerebral Aβ39; it is conceivable that specific vulnerability factors in some individuals may require lower (i.e., subthreshold) concentrations of cerebral Aβ to unleash significant tau aggregation across the neocortex. However, in our sample, significant tau aggregation in medial temporal (Braak I–II) regions was more common than significant tau aggregation in the larger temporal meta-ROI in individuals without elevated amyloid-PET uptake (∼29% of Aβ− individuals) and may be an indication of primary age-related tauopathy (PART).40 Whereas the temporal meta-ROI may have lower sensitivity to detect PART, it is unclear to what extent medial temporal tauopathy represents the AD pathologic process. Therefore, dichotomization of tau-PET status using the temporal meta-ROI may have higher specificity for the AD pathologic process.
A surprising finding in our study was the frequency of Alzheimer pathologic change (A+T−) in cognitively impaired individuals with aMCI or pAD dementia. Given that widely accepted models of AD consider abnormal amyloid-PET to be insufficient for causing cognitive impairment,12,33,34 more research is needed to identify the pathologic processes related to cognitive impairment in these individuals. In our study, approximately 14% of aMCI participants and 10% of pAD dementia participants were amyloid-positive but tau-negative. This finding has implications for the utility of amyloid-PET in the diagnosis and clinical management of individuals with aMCI and dementia41: a positive amyloid-PET scan does not have 100% sensitivity or specificity for biologically defined AD, which requires concurrent tau abnormality. In other words, although a negative amyloid-PET scan is useful in ruling out AD in the differential diagnosis of individuals with cognitive impairment,42 its inherent limitation of identifying 1 of 2 core AD pathologic features suggest that a positive tau-PET scan43 may also be needed for ruling in AD.
Our study extends many findings from recent large-scale studies of AD biomarkers in relation to age, sex, APOE ε4, and clinical impairment to a specialized memory clinic setting. Our finding of higher rates of biologically defined AD in CU elderly is consistent with a recent population-based study in which the prevalence of biologically defined AD exceeded the prevalence of clinically defined AD across all age groups.25 Because of the recruited nature of the TRIAD cohort and the relatively young age of the participants, the 7%–12% of CU elderly individuals with biologically defined AD reported in this study cannot be interpreted as a prevalence estimate. However, recent population-based studies report that the prevalence of biologically defined AD is strongly associated with age, with prevalence estimates of approximately 10% at age 70 and 33% at age 85.25 Our observation that the frequency of AD biomarker abnormality (as well as A+/T+/N+ status) increases with age in CU individuals is consistent with prior work44 and highlights the therapeutic potential for disease-modifying interventions for AD in the preclinical phase of the disease. We observed a slightly higher frequency of the A−T+ biomarker profile than reported in a multicenter tau imaging study43 and a population-based study using CSF biomarkers.45 It is not clear whether this discrepancy is due to specific properties of our study population, thresholds for determination of positivity,46 or the use of a tau-PET ligand with sub-nanomolar affinity for neurofibrillary tangles.47 However, in line with previous studies, the vast majority of individuals with abnormal tau-PET levels had abnormal amyloid-PET levels. Our study also provides evidence that estimation of tau pathology in individuals with atypical AD phenotypes, who often have regional differences in tau-PET uptake, is feasible using commonly employed meta-ROIs. Accurate estimation of the prevalence of biologically defined AD in the general population as well as in clinical populations is of great importance as prevalence estimates of biologically defined AD mirror the prevalence of clinically defined pAD dementia 15 years later.25
Our study was designed to estimate the frequency of biologically defined AD entities in a memory clinic and prospective cohort study and cannot estimate a true prevalence of biologically defined AD, for which population-based studies with random sampling are necessary. The TRIAD cohort constitutes a group of individuals motivated to participate in research on aging and dementia; CU elderly individuals who participate in a study on brain aging may be motivated by experiencing subjective memory concerns. For this reason, we use the term frequency and not prevalence. The frequency of APOE ε4 in individuals with dementia was slightly lower in our sample compared to multicenter studies,48 which could be related to the relative distributions of AD biomarker profiles reported in this study. Furthermore, whereas our study identifies a substantial number of CU elderly individuals with biologically defined AD, it is important to remember that these individuals were identified based on a detection threshold of PET imaging. Postmortem studies report higher rates of Aβ and tau neurofibrillary tangle aggregation in CU elderly individuals than reported in our study,20 although the younger age of our sample in comparison to other cohorts is also a factor. Despite the limitations of in vivo measurements, quantification of Aβ plaques and tau neurofibrillary tangles was nonetheless more sensitive than clinical phenotyping and neuropsychological evaluation in the detection of biologically defined AD in CU elderly. Correspondingly, our study highlights that the number of individuals who could potentially benefit from disease-modifying therapies for AD, should they become available, exceed the current estimates of clinically defined AD. This is an encouraging finding in light of the frequent failures of disease-modifying trials in individuals in the dementia phase of AD.
Our study has limitations. Because of the recruited nature of the cohort, our results cannot be interpreted as prevalence estimates. A crucial limitation is that thresholds for abnormality are invariably bound by tradeoffs in sensitivity vs specificity.6,49 We attempted to circumvent this limitation by conducting sensitivity analyses. Although converging evidence from postmortem and in vivo studies suggests that tau aggregation appears early in Braak stage I–II regions, it remains unclear to what extent tau pathology in these regions represents the AD pathologic process vs other factors such as aging16 or genetic factors.13 Overall, more studies are needed to determine optimal thresholds for tau positivity, including the possibility of more than one threshold to capture the substantial variability in both topography and magnitude of cortical tau deposition. Although other groups have studied the prevalence of AD biomarkers45,48 in various clinically defined groups, an advantage of our study is the direct comparison between clinically and biologically defined AD within the same individuals.
Acknowledgment
The authors thank the study participants; the staff of the McGill Center for Studies in Aging for their role in data collection; and Dean Jolly, Alexey Kostikov, Monica Samoila-Lactatus, Karen Ross, Mehdi Boudjemeline, and Sandy Li for their assistance with radiochemistry production.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- aMCI
amnestic mild cognitive impairment
- CDR
Clinical Dementia Rating
- CI
confidence interval
- CN
cognitively normal
- CU
cognitively unimpaired
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- OR
odds ratio
- pAD
probable Alzheimer disease
- PART
primary age-related tauopathy
- ROI
region of interest
- SUVR
standardized uptake value ratio
- TRIAD
Translational Biomarkers in Aging and Dementia
- VIF
variance inflation factor
- WMH
white matter hyperintensity
Appendix. Authors
Footnotes
Study Funding
Study funded by McGill University Faculty of Medicine, Alzheimer's Society of Canada, Alzheimer's Association, Weston Brain Institute, and Canadian Institutes of Health Research (CIHR).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/Nhttps://n.neurology.org/lookup/doi/10.1212/WNL.0000000000011416 for full disclosures.
References
- 1.Ball M, Braak H, Coleman P, et al. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol Aging 1997;18 (suppl 1):S1–S2. [PubMed] [Google Scholar]
- 2.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939. [DOI] [PubMed] [Google Scholar]
- 4.McKhann G, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim A, Tsuang D, Kukull W, et al. Clinico-neuropathological correlation of Alzheimer's disease in a community-based case series. J Am Geriatr Soc 1999;47:564–569. [DOI] [PubMed] [Google Scholar]
- 6.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer disease centers, 2005-2010. J Neuropathol Exp Neurol 2012;71:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology 2000;55:370–376. [DOI] [PubMed] [Google Scholar]
- 8.Knopman DS, Parisi JE, Salviati A, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 2003;62:1087–1095. [DOI] [PubMed] [Google Scholar]
- 9.Abner EL, Kryscio RJ, Schmitt FA, et al. Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 2017;81:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 2006;63:665–672. [DOI] [PubMed] [Google Scholar]
- 11.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009;66:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jack CR, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Therriault J, Benedet AL, Pascoal TA, et al. Association of apolipoprotein e ϵ4 with medial temporal tau independent of amyloid-β. JAMA Neurol 2020;77:470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jack CR, Wiste HJ, Botha H, et al. The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain 2019;142:3230–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004:183–194. [DOI] [PubMed] [Google Scholar]
- 16.Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron 2016;89:971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Therriault J, Benedet AL, Pascoal TA, et al. APOE ε4 potentiates the relationship between amyloid-β and tau pathologies. Mol Psychiatry Epub 2020 Mar 11. [DOI] [PMC free article] [PubMed]
- 18.Jack CR, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement 2017;13:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Therriault J, Benedet AL, Pascoal TA, et al. Determining amyloid-B positivity using [18F]AZD4694 PET imaging. J Nucl Med Epub 2020 Jul 31. [DOI] [PubMed]
- 20.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997;18:377–379. [DOI] [PubMed] [Google Scholar]
- 21.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 22.Schöll M, Maass A, Mattsson N, et al. Biomarkers for tau pathology. Mol Cell Neurosci 2019;97:18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowe VJ, Wiste HJ, Senjem ML, et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer's dementia. Brain 2018;141:271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hostetler ED, Walji AM, Zeng Z, et al. Preclinical characterization of 18F-MK-6240, a promising PET tracer for in vivo quantification of human neurofibrillary tangles. J Nucl Med 2016;57:1599–1606. [DOI] [PubMed] [Google Scholar]
- 25.Jack CR, Therneau TM, Weigand SD, et al. Prevalence of biologically vs clinically defined Alzheimer spectrum entities using the National Institute on aging-Alzheimer’s association research framework. JAMA Neurol 2019;76:1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Therriault J, Ng KP, Pascoal TA, et al. Anosognosia predicts default mode network hypometabolism and clinical progression to dementia. Neurology 2018;90:e932–e939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knopman DS, Petersen RC, Jack CR. A brief history of “Alzheimer disease”: multiple meanings separated by a common name. Neurology 2019;92:1053–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrer I, Santpere G, Van Leeuwen FW. Argyrophilic grain disease. Brain 2008;131:1416–1432. [DOI] [PubMed] [Google Scholar]
- 29.Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019;142:1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bancher C, Jellinger KA. Neurofibrillary tangle predominant form of senile dementia of Alzheimer type: a rare subtype in very old subjects. Acta Neuropathol 1994;88:565–570. [DOI] [PubMed] [Google Scholar]
- 31.Botha H, Mantyh WG, Murray ME, et al. FDG-PET in tau-negative amnestic dementia resembles that of autopsy-proven hippocampal sclerosis. Brain 2018;141:1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Botha H, Mantyh WG, Graff-Radford J, et al. Tau-negative amnestic dementia masquerading as Alzheimer disease dementia. Neurology 2018;90:e940–e946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowe VJ, Bruinsma TJ, Wiste HJ, et al. Cross-sectional associations of tau-PET signal with cognition in cognitively unimpaired adults. Neurology 2019;93:e29–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ossenkoppele R, Smith R, Ohlsson T, et al. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology 2019;92:e601–e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Betthauser TJ, Koscik RL, Jonaitis EM, et al. Amyloid and tau imaging biomarkers explain cognitive decline from late middle-age. Brain 2019;143:320–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci 2020;21:21–35. [DOI] [PubMed] [Google Scholar]
- 39.Bischof GN, Jacobs HIL. Subthreshold amyloid and its biological and clinical meaning: long way ahead. Neurology 2019;93:72–79. [DOI] [PubMed] [Google Scholar]
- 40.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rabinovici GD, Gatsonis C, Apgar C, et al. Association of amyloid positron emission tomography with subsequent change in clinical management among Medicare beneficiaries with mild cognitive impairment or dementia. JAMA 2019;321:1286–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the amyloid imaging Task Force, the society of nuclear medicine and molecular imaging, and the Alzheimer's association. Alzheimers Dement 2013;54:476–490. [DOI] [PubMed] [Google Scholar]
- 43.Ossenkoppele R, Rabinovici GD, Smith R, et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 2018;320:1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jack CR, Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. Lancet Neurol 2017;16:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kern S, Zetterberg H, Kern J, et al. Prevalence of preclinical Alzheimer disease: comparison of current classification systems. Neurology 2018;90:E1682–E1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattsson-Carlgren N, Leuzy A, Janelidze S, et al. The implications of different approaches to define AT(N) in Alzheimer disease. Neurology 2020;24:e2233–e2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pascoal TA, Shin M, Kang MS, et al. In vivo quantification of neurofibrillary tangles with [18F]MK-6240. Alzheimer's Res Ther; 2018;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ossenkoppele R, Jansen WJ, Rabinovici GD, et al. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 2015;313:1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McSweeney M, Pichet Binette A, Meyer PF, et al. Intermediate flortaucipir uptake is associated with Aβ-PET and CSF-tau in asymptomatic adults. Neurology Epub 2020 Feb 3. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data and documentation from this study can be made available to qualified investigators on reasonable request. Such arrangements are subject to standard data sharing agreements.