

SUMMARY
Although neutralizing monoclonal antibodies (mAbs) against epitopes within the alphavirus E2 protein can protect against infection, the functional significance of non-neutralizing mAbs is poorly understood. Here, we evaluate the activity of 13 non-neutralizing mAbs against Mayaro virus (MAYV), an emerging arthritogenic alphavirus. These mAbs bind to the MAYV virion and surface of infected cells but fail to neutralize infection in cell culture. Mapping studies identify six mAb binding groups that localize to discrete epitopes within or adjacent to the A domain of the E2 glycoprotein. Remarkably, passive transfer of non-neutralizing mAbs protects against MAYV infection and disease in mice, and their efficacy requires Fc effector functions. Monocytes mediate the protection of non-neutralizing mAbs in vivo, as Fcγ-receptor-expressing myeloid cells facilitate the binding, uptake, and clearance of MAYV without antibody-dependent enhancement of infection. Humoral protection against alphaviruses likely reflects contributions from non-neutralizing antibodies through Fc-dependent mechanisms that accelerate viral clearance.
Graphical abstract
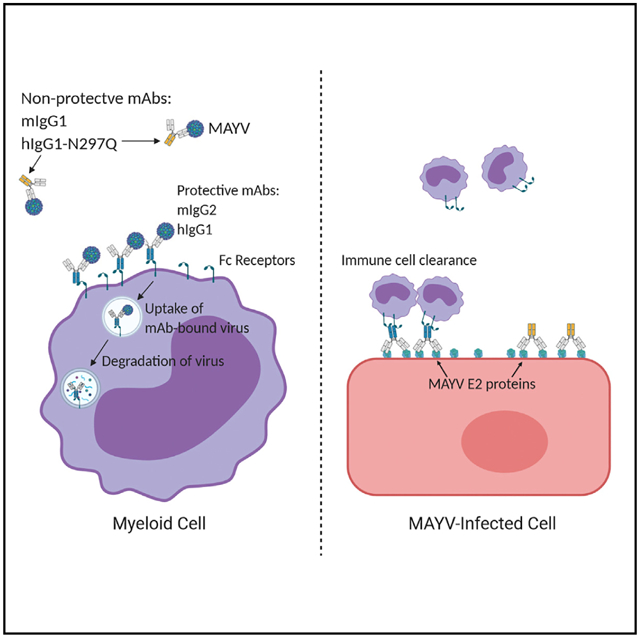
In brief
Earnest et al. characterize the protective antibody response against Mayaro virus, an emerging arthritogenic alphavirus. They find that antibody-mediated protection in mice does not require virus neutralization but can rely on Fc effector functions associated with myeloid cells.
INTRODUCTION
Alphaviruses are mosquito-transmitted, positive-sense RNA viruses in the Togaviridae family and are classified into groups based on genetic relatedness and disease potential. Encephalitic alphaviruses, including Eastern, Western, and Venezuelan equine encephalitic viruses (EEEV, WEEV, and VEEV, respectively), infect neuronal cells, which can lead to encephalitis and death. Arthritogenic alphaviruses, including chikungunya (CHIKV), Ross River (RRV), O’nyong-nyong (ONNV), and Mayaro (MAYV) viruses, infect joint-associated tissues and cause acute and chronic musculoskeletal disease. MAYV was described in 1954 in Trinidad (Causey and Maroja, 1957) and circulates in the Caribbean Islands and South America (Azevedo et al., 2009; Causey and Maroja, 1957; Pinheiro et al., 1981). MAYV infection causes an acute febrile illness that can progress to acute and chronic arthritis, much like CHIKV (Halsey et al., 2013). Due to serum cross-reactivity and overlapping epidemiology with CHIKV, MAYV infections may be more prevalent than previously appreciated (Hozé et al., 2020). Currently, no treatments are approved for any alphavirus infection.
The alphavirus virion is comprised of a single ~11.4-kb RNA genome encapsidated in a nucleocapsid core and surrounded by a host-derived membrane. The genome encodes for four non-structural proteins, namely, nsP1–4, which mediate viral translation, viral replication, and host subversion and evasion (Rupp et al., 2015); and six structural proteins, namely, capsid, E3, E2, 6K, transframe (TF), and E1. The viral glycoproteins are cleaved from a structural polyprotein precursor and form the heterodimer p62(E3-E2)-E1. p62 is cleaved by furin proteases in the trans-Golgi network (Heidner et al., 1996), and the mature E2 and E1 proteins transit to the surface of the cell where they may still associate with E3 (Uchime et al., 2013; Yap et al., 2017). Virion morphogenesis occurs at the plasma membrane, and the mature virion displays trimers of E2-E1 heterodimers assembled into higher order spikes. The virus is released into the extracellular space by budding (Carleton et al., 1997). Because E1 and E2 proteins are exposed on virions and the surface of infected cells, they are the targets of the host antibody response.
Antibody-mediated protection from alphavirus infection occurs through several mechanisms, including neutralization by inhibiting virus attachment, entry, fusion, and/or egress from host cells (Earnest et al., 2019; Fox et al., 2015; Jin and Simmons, 2019). Indeed, neutralizing monoclonal antibodies (mAbs) that bind the B domain of MAYV E2 prevent virus-cell membrane fusion or viral egress (Earnest et al., 2019). The efficacy of anti-alphavirus neutralizing antibodies in vivo also is modulated by Fc effector functions (Earnest et al., 2019; Fox et al., 2019). Antibodies that bind to viral proteins on the surface of infected cells may facilitate complement deposition and/or innate immune cell targeting (Bournazos et al., 2015; Lu et al., 2018).
Little is known about the functional significance of non-neutralizing antibodies in the context of infection and immunity of alphaviruses or other families of enveloped viruses. Here, we isolated a panel of murine mAbs against the MAYV E2 protein of the IgG2c subclass with no measurable neutralizing activity in vitro. These mAbs bound virions in a capture ELISA and mapped to six distinct epitopes within or proximal to the A domain of the E2 of MAYV. Remarkably, the majority of non-neutralizing mAbs conferred protection against arthritis in immunocompetent mice and prevented lethal challenge in immunodeficient mice. Protection in vivo was immunoglobulin G (IgG) subclass and Fcγ receptor (FcγR) dependent and required the presence of monocytes. Mechanism of action studies showed that non-neutralizing mAbs can enhance the binding, uptake, and clearance of MAYV on FcγR-expressing myeloid cells. Our results indicate that direct neutralization is not required for antibody-mediated protection, as Fc effector functions and monocytes can promote antibody-dependent control of alphavirus infection.
RESULTS
Isolation of non-neutralizing anti-MAYV mAbs
C57BL/6J mice were inoculated with the MAYV strain CH and boosted twice with recombinant MAYV E2 ectodomain protein (amino acids 1–340) in Freund’s incomplete adjuvant. Three days after the second booster immunization, we performed a splenocyte-myeloma fusion to generate hybridomas (Earnest et al., 2019; Pal et al., 2013). We isolated 114 mAbs that bound to the MAYV E2 protein by ELISA. To identify mAbs that more broadly recognized MAYV, we also assessed binding to Vero cells infected with the heterologous MAYV strain BeH407 (96% amino acid identity in E2-E1) by flow cytometry. We successfully cloned 73 hybridomas with these features. Because previous studies demonstrated that IgG2a/c isotypes exhibited superior protective activity against alphaviruses (Earnest et al., 2019; Pal et al., 2013), we isotyped the clones and selected 13 IgG2c mAbs for further characterization.
The 13 mAbs were purified and evaluated for neutralizing activity by using focus reduction neutralization tests (FRNTs) in Vero and C2C12 myoblast cells. Serial dilutions of mAb were mixed with 102 focus-forming units (FFUs) of MAYV-BeH407 before incubation with the two target cells. In contrast to a previously described neutralizing mAb (MAY-117; Earnest et al., 2019), none of the 13 mAbs showed measurable inhibitory activity in either Vero or C2C12 cells even at concentrations of 50 μg/ml (Figures 1A and 1B).
Figure 1. Binding of non-neutralizing anti-MAYV mAbs to virions and recombinant E2 protein.
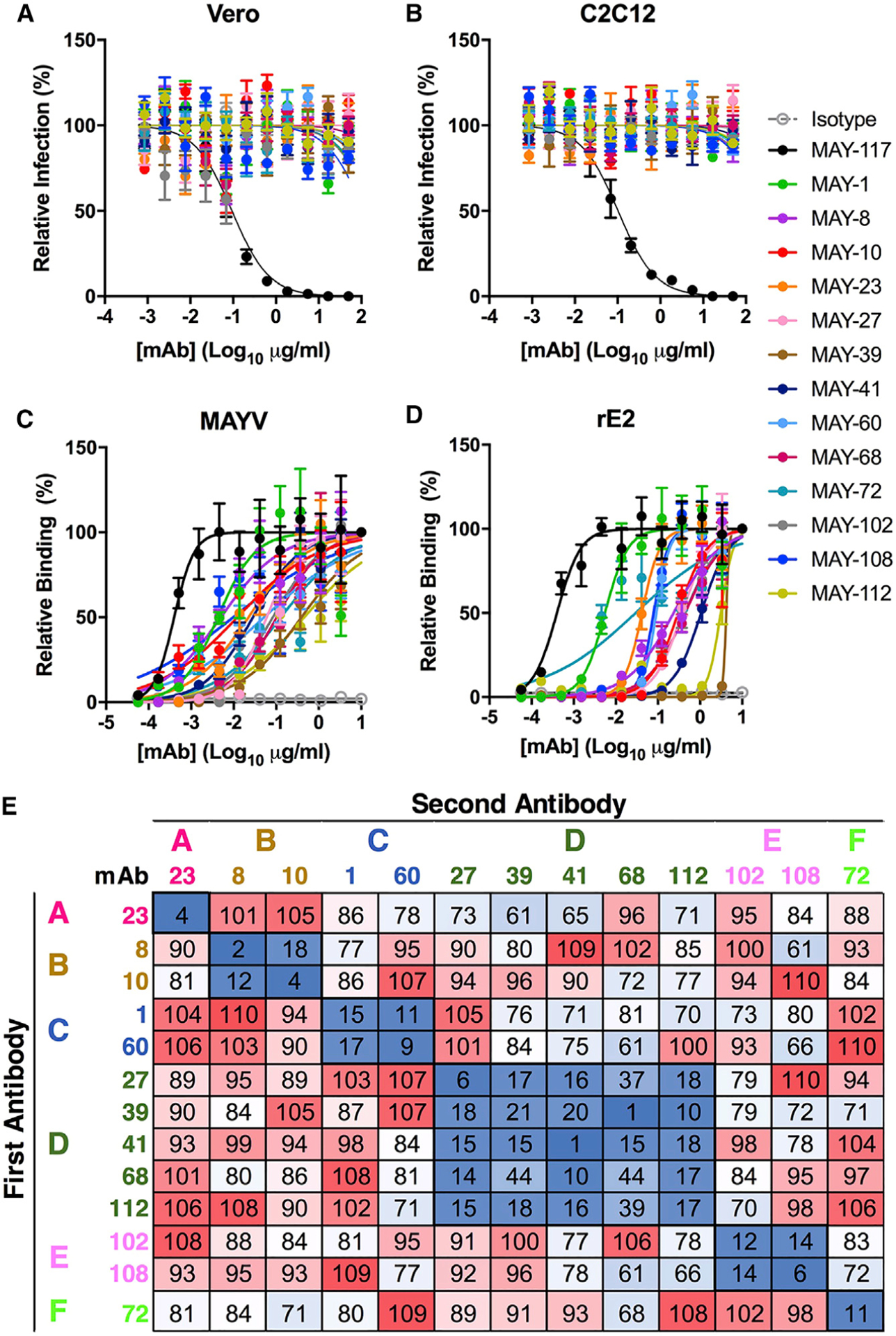
(A and B) Anti-MAYV mAbs were tested for neutralization of MAYV on Vero (A) and C2C12 myoblast (B) cells. Serial dilutions of the indicated mAbs were incubated with 102 FFU of MAYV-BeH407 and then added to the indicated cells. Viral foci are plotted relative to a no mAb control. The neutralizing mAb MAY-117 was used as a positive control, and an irrelevant mIgG2c mAb was used as a negative isotype control (mean and SD of two experiments performed in triplicate).
(C and D) Binding to MAYV virions (C) or recombinant MAYV E2 protein (D) by ELISA. Virions were captured with a humanized mAb to MAYV, and recombinant MAYV E2 protein was bound directly to microtiter plates. Bound murine mAbs were detected with an horseradish peroxidase (HRP)-conjugated secondary antibody. Data are expressed as OD values relative to the 10-μg/ml sample (mean and SD of two experiments performed in triplicate).
(E) MAYV mAbs were competed for binding to MAYV (strain BeH407) by ELISA. Virus was captured on plates using a humanized anti-MAYV mAb. Captured virion was incubated with 10 μg/ml of the indicated mAb (first antibody). Antibody-virus complexes were incubated with 10 ng/ml of the indicated mAb labeled with biotin (second antibody). Binding was detected using streptavidin HRP and is indicated by color from high (red) to low (blue). Data are presented relative to a control with no first antibody and are representative of two experiments.
We next evaluated these mAbs for binding to MAYV virions (Figure 1C) and recombinant E2 protein (Figure 1D). Half-maximal binding (EC50) to virions was measured by ELISA after MAYV was captured with an anti-MAYV mAb containing a human Fc region. A neutralizing anti-MAYV E2 B domain mAb (MAY-117) (Earnest et al., 2019) was used for comparison. All 13 mAbs bound to intact virions with EC50 values ranging from 4 to 379 ng/ml, with 9 of 13 having EC50 values less than 100 ng/ml (Table 1). The EC50 values, however, were 10-fold higher than those for MAY-117 (0.5 ng/ml). The maximal binding values (Bmax) generally were consistent among the mAbs, with the majority showing optical density (OD) values between 1.7 and 2.8. However, MAY-39 and MAY-112 had maximal OD values of <1, which suggests that fewer mAbs can bind the virus at saturation (Table 1; Figure S1A). ELISA-based competition binding studies for MAYV virions revealed six distinct groups (Figure 1E). The majority of anti-MAYV mAbs also bound the recombinant E2 protein in an ELISA, although the EC50 values generally were higher than that observed for virion binding, suggesting less avid binding to recombinant protein than intact virions (Table 1). Furthermore, MAY-39 and MAY-112 did not bind appreciably to solid phase recombinant E2 protein by ELISA. The disparities in binding to recombinant protein versus intact virion may reflect differences in epitope presentation or engagement of a quaternary epitope between or across E2 proteins within a spike, which is present exclusively on the virion, as seen with anti-CHIKV (Fox et al., 2015) and anti-RRV (Powell et al., 2020) mAbs. As all of the non-neutralizing mAbs bound E2 after western blotting when we used both non-reducing and reducing conditions, they likely do not recognize conformational epitopes (Figure S1B). To acquire more quantitative data, we measured monovalent binding affinities to the MAYV E2 protein by biolayer interferometry (BLI) (Table 1; Figure S1C). The measured kinetic binding constant (KD) rate for the mAbs varied by more than 100-fold from 4 to 548 nM, with the affinity of binding correlating inversely with the half-life (t1/2). MAY-10, MAY-108, MAY-8, and MAY-23 showed the highest affinities with KD values of <19 nM and t1/2 values over 130 s. MAY-60, MAY-68, and MAY-112 showed no appreciable binding.
Table 1.
Binding affinities of anti-MAYV mAbs to virions and recombinant E2 protein
| mAb | EC50, virion (ng/ml) | EC50, E2 (ng/ml) | Bmax virion (OD) | KD (kinetic) (nM) | t1/2 (s) |
|---|---|---|---|---|---|
| 117 (Earnest et al., 2019) | 0.4 (0.3–0.5) | 0.4 (0.3–0.5) | 2.84 | 9.2 | 264.1 |
| 10 | 12 (8–17) | 302 (274–334) | 1.66 | 14.0 | 231.8 |
| 108 | 10 (5–23) | 82 (72–95) | 2.65 | 4.1 | 213.9 |
| 8 | 5 (3–7) | 195 (157–242) | 1.22 | 16.6 | 136.3 |
| 23 | 21 (14–31) | 41 (36–45) | 2.04 | 18.4 | 142.6 |
| 102 | 83 (61–113) | 461 (406–523) | 2.06 | 211.3 | 61.5 |
| 60 | 45 (28–75) | 84 (77–92) | 1.90 | ND | ND |
| 1 | 4 (2–6) | 5 (4–6) | 2.79 | 160.0 | 14.1 |
| 41 | 25 (19–33) | 863 (773–997) | 1.99 | 47.3 | 92.5 |
| 27 | 56 (41–75) | 344 (317–374) | 1.72 | 110.5 | 12.6 |
| 39 | 332 (238–463) | >1,000 | 0.80 | 547.6 | 74.5 |
| 68 | 100 (71–138) | 383 (333–440) | 2.00 | ND | ND |
| 72 | 134 (79–225) | 33 (16–68) | 2.04 | 165.6 | 14.9 |
| 112 | 379 (230–637) | >1,000 | 0.88 | ND | ND |
Anti-MAYV mAbs were measured for binding to intact MAYV virions by capture ELISA and recombinant MAYV E2 protein by ELISA and BLI. Effective concentration of 50% binding (EC50) was calculated from the OD values with serial dilutions of the indicated mAb. Bmax was measured as the highest OD value observed in the virion capture ELISA. BLI was performed by binding biotinylated mAbs to streptavidin-coated pins and flowing over recombinant E2 protein. The equilibrium binding constant (KD) and half-life (t1/2) of antibody binding were measured. The previously characterized (Earnest et al., 2019) positive-control MAY-117 mAb is listed first for comparison, and subsequently, mAbs are arranged from most to least protective. MAY-10, MAY-108, MAY-8, MAY-23, and MAY-102 mAbs had ≥50% protection in the lethal challenge model. Data are the mean of two independent experiments, and 95% confidence intervals are in parentheses. ND, not determined due to poor binding.
Epitope mapping of anti-MAYV mAbs
Although our ELISA, BLI, and western blotting data establish that the non-neutralizing mAbs recognize the MAYV E2 glycoprotein, we observed no binding to the recombinant E2 B domain (J.T.E. and M.S.D., unpublished data). To test whether the mAbs bound to regions in the A domain, we performed alanine scanning mutagenesis in the context of expression of the structural polyprotein (C-E3-E2-6K-E1). Based on the CHIKV pE2-E1 structure (PDB: 3N42), we introduced alanine substitutions in predicted solvent-exposed amino acids of the A domain and the β-ribbon arch connecting the A and B domains (residues 1–172) of the MAYV E2 protein. When an alanine was present in the viral sequence, we substituted a serine residue. 293T cells were transfected with wild-type (WT) or individual mutant plasmids, and mAb binding was measured by flow cytometry. An oligoclonal pool of the 13 mAbs as well as MAY-117, an anti-E2 B domain mAb, was used to control for mutant protein expression. Key interaction residues were defined when mAb binding to a given mutant was ≤25% after normalization of binding to cells expressing the WT plasmid (Table S1). The 13 mAbs mapped to 6 different sites within or near the A domain (Figure 2A), which correlated with the following competition groups (Figure 1E): group A, residues 27–29 (Figure S2A); group B, residues 57–61 (Figure S2B); group C, residues 72–77 (Figure S2C); group D, residues 81–86 (Figure S2D); group E, residues 159–163 (Figure S2E); and group F, residues 168–173 (Figure S2F). Group D mAbs, which had the largest number in our panel, map to an epitope facing the inside of the E2-E1 trimer (Figures 2B and 2C); mutation of these residues resulted in loss of binding of MAY-27, MAY-39, MAY-41, MAY-68, and MAY-112. Two mAb groups map to sites adjacent to the A domain in the β-ribbon arch connecting the A and B domains of E2 (159–163, group E, MAY-102 and MAY-108; and 168–173, group F, MAY-72). MAY-23, the lone member of group A, localizes to a β strand adjacent to the N-terminal linker on the outer face of the E2-E1 trimeric complex (Figure 2C). The group B (MAY-8 and MAY-10) and C (MAY-1 and MAY-60) mAbs map to two structurally adjacent regions near the top of the spike complex (Figures 2B and 2C). As non-neutralizing anti-MAYV mAbs recognize denatured forms of E2 (Figure S1B) and their mapping residues cluster together, these antibodies likely preferentially engage linear peptide epitopes.
Figure 2. Mapping of mAbs to MAYV E2 protein.
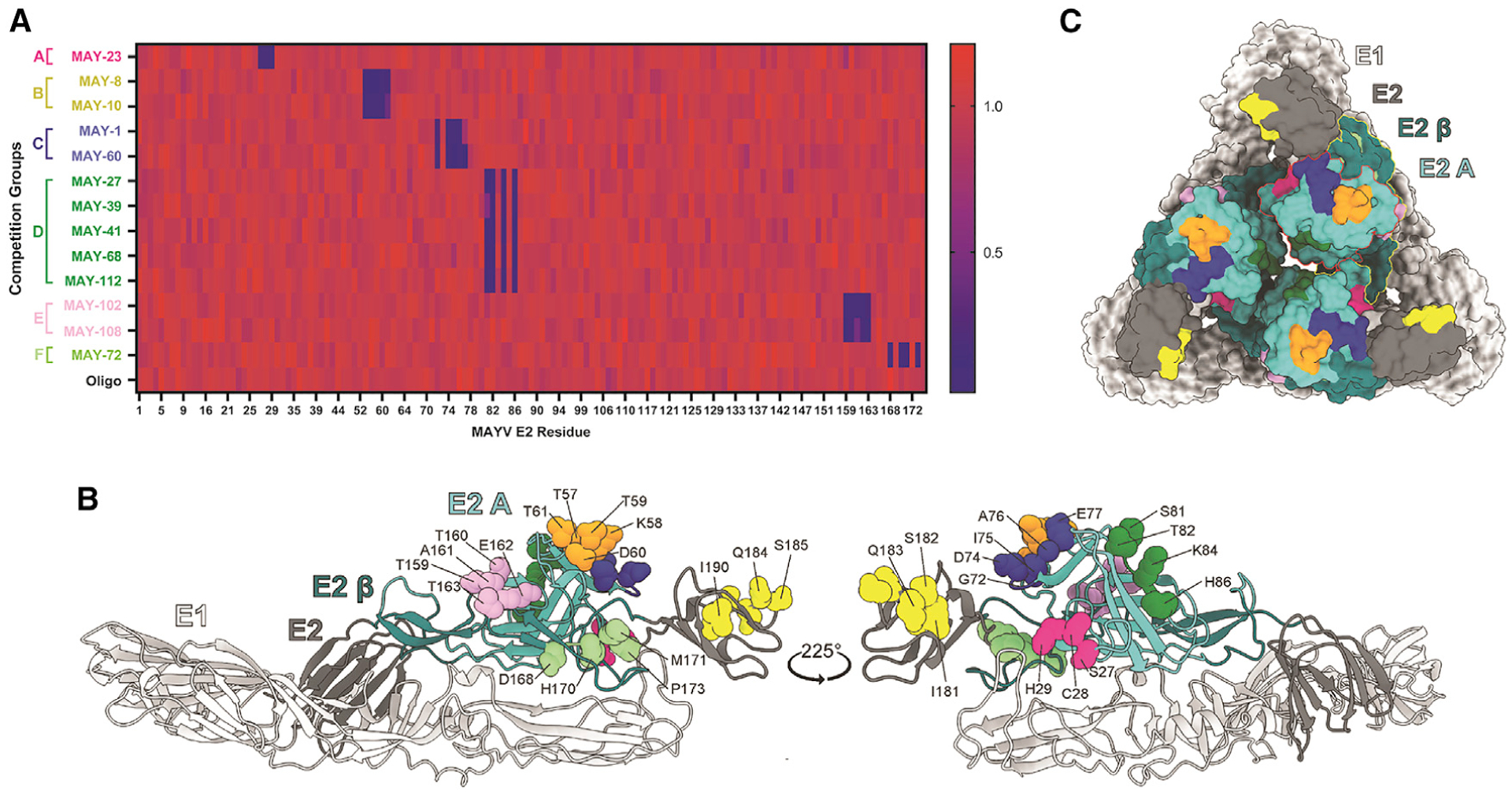
(A) Heatmap of relative binding of anti-MAYV mAbs to MAYV-E2 A domain mutants. 293T cells were transfected with a C-E3-E2–6K-E1 plasmid containing alanine mutations in the A domain of E2. Binding of the indicated mAbs were measured by flow cytometry; the full dataset is shown in Table S1 and Figure S2. Relative binding compared to an oligoclonal control is indicated by color from high (red) to low (blue).
(B and C) Residues required for mAb engagement are depicted as balls and sticks on a ribbon diagram of the predicted structure of MAYV E2-E1 monomer generated using Phyre2 (B) and are highlighted on the monomers arranged as a trimeric spike (C). The E1 and E2 glycoproteins are light and dark gray, respectively. Within E2, domain A is cyan, the β-ribbon is dark cyan. In the surface representation (C), the A domain and β-ribbon regions are outlined in red and yellow, respectively. Competition groups are color coded as follows: group A, dark pink (residues 27–29); group B, orange (57–61); group C, blue (72–77); group D, dark green (81–86); group E, lavender (159–163); group F, light green (168–173); and the anti-B domain control mAb MAY-117, bright yellow (181–186). See also Table S1 and Figure S2.
Protection against lethal MAYV challenge by mAbs
We tested the 13 non-neutralizing mAbs in a lethal MAYV challenge model in 4-week-old female C57BL/6J mice. Because MAYV does not cause mortality in immunocompetent mice, we treated animals with 100 μg of an anti-Ifnar1 mAb (MAR-5A3; Sheehan et al., 2006) to transiently immunosuppress them. Mice given anti-Ifnar1 mAb succumbed to subcutaneous inoculation of 103 FFU of MAYV-BeH407 between 3 and 5 days post-infection (dpi) (Figure 3). To assess for protection as prophylaxis in this model, mice were treated with a single 100-μg (5 mg/kg) dose of individual anti-MAYV mAbs 1 day before virus inoculation. We observed a range of protective activity of the mAbs, as follows: MAY-10 and MAY-108 conferred 100% protection (Figure 3A); MAY-23, MAY-8, and MAY-102 protected ≥50% of mice from lethality; and MAY-1, MAY-39, MAY-60, MAY-68, MAY-72, and MAY-112 exhibited less protective activity (30%–40%) (Figure 3B). MAY-27 and MAY-41 had no significant protective activity. Notably, the in vivo activity of the non-neutralizing antibodies generally correlated with binding affinity, with the most protective mAbs having lower KD and higher t1/2 values (Table 1). Moreover, the most protective mAbs map to one of two epitopes, as follows: group B mAbs localize to an epitope at the top of the spike trimer (MAY-10 and MAY-8; Figures 2B and 2C), and group E mAbs engage an epitope in the linker region between the A and B domains on the side of the spike trimer (MAY-23, MAY-102, and MAY-108; Figures 2B and 2C). These results suggest both binding affinity and epitope location facilitate optimal protection by non-neutralizing mAbs.
Figure 3. Antibody protection against lethal MAYV challenge.
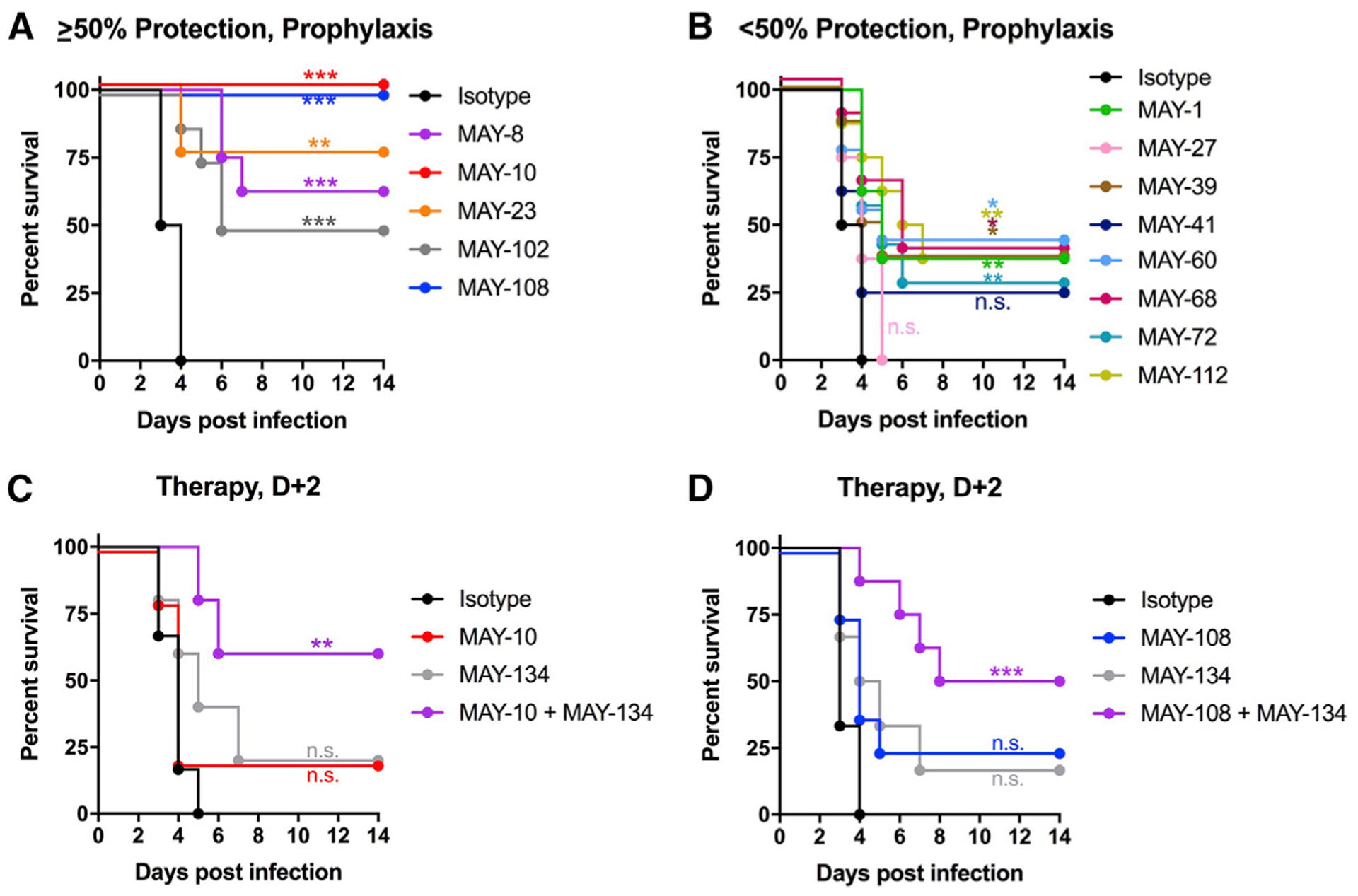
Four-week-old C57BL/6J female mice were treated with 100 μg of anti-Ifnar1 mAb 1 day before subcutaneous inoculation of MAYV-BeH407.
(A and B) A single 100-μg dose of anti-MAYV mAbs was administered by intraperitoneal injection 1 day before virus inoculation. The mAbs exhibited a range of activity with some showing >50% protection (A) and others <50% (B). Data are from two experiments.
(C and D) Combination therapy of an anti-MAYV E2 B domain mAb (MAY-134) and anti-MAYV E2 A domain mAbs. C57BL6/J mice were treated with 100 μg of anti-Ifnar1 mAb 1 day before subcutaneous virus inoculation. (C) Two days after infection, mice were treated with 200 μg of MAY-10 or MAY-134 or 100 μg each of MAY-10 and MAY-134. (D) Two days after infection, mice were treated with 200 μg of MAY-108 or MAY-134 or 100 μg each of MAY-108 and MAY-134 (two experiments, n = 8; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; log-rank test with Bonferroni correction compared to isotype control mAb). See also Figure S3.
We previously described a protective neutralizing anti-MAYV mAb (MAY-134) that maps to the B domain of E2 (Earnest et al., 2019). Because MAY-134 is protective and does not compete for binding with either MAY-10 or MAY-108 (Figure S3), we tested whether combinations of neutralizing (MAY-134) and non-neutralizing (MAY-10 or MAY-108) mAbs could enhance therapeutic efficacy in our lethal challenge mouse model. We administered 200 μg (10 mg/kg) of either MAY-10 (Figure 3C), MAY-108 (Figure 3D), or MAY-134 (Figures 3C and 3D) or 100 μg each of MAY-10 or MAY-108 and MAY-134 2 days after mice were inoculated with 103 FFU of MAYV-BeH407. Although we observed no statistically significant protection from lethal challenge in mice treated with any single mAb, combination therapy with MAY-10 and MAY-134 or MAY-108 and MAY-134 protected 60% and 50% of mice, respectively (Figures 3C and 3D). Thus, non-neutralizing A domain and neutralizing B domain mAbs together provide greater protection as post-exposure therapy than either mAb alone.
Antibody protection in vivo requires Fc effector functions
We hypothesized that protection by non-neutralizing mAb might require Fc effector functions. To test this idea, we repeated passive transfer experiments in anti-Ifnar1-mAb-treated C57BL/6J mice lacking the common signaling gamma (γ) chain and expression of activating FcγR (FcγR−/−). These mice were treated with a single 100-μg dose of MAY-10 or MAY-108 1 day prior to subcutaneous inoculation with MAYV-BeH407. Remarkably, both isotype control and anti-MAYV (MAY-10 or MAY-108)-treated FcγR−/− mice uniformly succumbed by 4 dpi (Figure 4A), which contrasts with results seen in congenic WT C57BL/6J mice (Figure 3A).
Figure 4. Protection by non-neutralizing mAbs is Fc dependent.

(A) Four-week-old C57BL/6J FcγR−/− male and female mice were administered 100 μg of MAY-10 or MAY-108 1 day before subcutaneous inoculation of MAYV-BeH407 (two experiments, n = 6).
(B) Isotype-switched mAb binding to recombinant murine FcγRI, FcγRIII, and FcγRIV. MAY-10 or MAY-108 of the indicated isotype were added to plates coated with FcγRs. Binding data: two-way ANOVA with Sidak’s post-test, compared to mIgG2c isotype mAb.
(C–F) MAY-10 (C) and MAY-108 (E) were isotype switched from murine IgG2c to murine IgG1, human IgG1, or human IgG1-N297Q. Each antibody variant was tested for binding to captured MAYV by ELISA. For protection studies (D and F), 100 μg of the indicated mAb was administered to 4-week-old C57BL/6J mice 1 day before subcutaneous inoculation with MAYV (two experiments, n = 6; log-rank test with Bonferroni correction compared to isotype control mAb). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To corroborate the role of Fc effector function in the protective activity of non-neutralizing anti-MAYV mAbs, we engineered isotype-switched versions of MAY-10 and MAY-108. Murine IgG2c (mIgG2c) antibodies bind mouse Fc gamma receptor (FcγR)I and FcγRIV with high and moderate affinity, whereas murine IgG1 (mIgG1) binds less avidly to these receptors (Mancardi et al., 2008). Furthermore, human IgG1 (hIgG1) binds strongly to these murine FcγRs in a manner similar to mIgG2c (Dekkers et al., 2017; Earnest et al., 2019). We cloned the variable regions of MAY-10 and MAY-108 into antibody expression constructs with Fc regions of mIgG1 or hIgG1. Additionally, we introduced an N297Q mutation in the Fc region of the hIgG1 construct to remove an N-linked glycan that is necessary for Fc-FcγR interactions (Tao and Morrison, 1989).
We first tested whether the isotype-switched MAY-10 and MAY-108 had different binding patterns to recombinant murine FcγRs (I, III, and IV) by ELISA. As expected, mIgG1 mAbs showed less binding to all of the tested FcγRs (Figure 4B). hIgG1 mAbs showed slightly lower binding, but it was more similar to mIgG2c than mIgG1. The N297Q variant lost ~90% of binding activity compared to mIgG2c. We confirmed that the isotype-switched MAY-10 and MAY-108 mAbs bound MAYV virions similarly (Figures 4C and 4E), and thus, altering Fc interactions did not affect antibody-antigen binding. We administered a single 100-μg dose of the isotype-switched mAbs to anti-Ifnar1-mAb-treated WT C57BL/6J mice 1 day before subcutaneous inoculation of MAYV. As expected, mIgG2c MAY-10 and MAY-108 protected mice from lethal challenge. Similarly, hIgG1 mAbs protected 90% (MAY-10) and 100% (MAY-108) of mice from mortality. In contrast, the mIgG1 and aglycosyl hIgG1-N297Q forms of MAY-10 and MAY-108 lost activity (Figures 4D and 4F). Collectively, these experiments establish that protection against lethal challenge by non-neutralizing anti-MAYV mAbs requires Fc-effector-function-dependent activity.
Protection against MAYV-induced musculoskeletal disease by mAbs
We assessed the activity of our two most protective non-neutralizing mAbs in a more physiologically relevant model of MAYV-induced disease. Subcutaneous inoculation of MAYV in the foot of immunocompetent C57BL/6J mice results in joint swelling and musculoskeletal disease (Earnest et al., 2019). Viral infection is first observed in the ipsilateral foot, ankle, and calf muscle before disseminating to the contralateral extremity. Similarly, foot swelling occurs first in the ipsilateral ankle and later in the contralateral ankle (Earnest et al., 2019). We treated C57BL6/J mice with a single 100-μg dose of MAY-10, MAY-108, or an isotype control mAb before subcutaneous inoculation of MAYV in the foot. At 1 and 7 dpi, animals were euthanized and perfused extensively with PBS. The ipsilateral ankle and calf muscle, contralateral ankle and calf muscle, and spleen were harvested, and viral RNA levels were measured using quantitative reverse-transcriptase PCR (qRT-PCR) with probes targeting the 5′ untranslated region of MAYV (Waggoner et al., 2018). At 1 dpi, we observed a >1,000-fold reduction in MAVY RNA in the ipsilateral ankles of both MAY-10- and MAY-108-treated mice compared to the isotype control mAb (Figure 5A). Moreover, we observed dissemination of MAYV to the ipsilateral calf muscle, the contralateral leg, and the spleen in isotype mAb treated mice, but there was no detectable viral RNA in mice treated with the non-neutralizing mAbs MAY-10 or MAY-108 (Figures 5B–5E). Remarkably, at 7 dpi, MAY-10- and MAY-108-treated mice had cleared viral RNA levels from the ipsilateral foot, mice showed no infection of the contralateral extremity, and only one animal had detectable viral RNA in the spleen (Figures 5F–5J).
Figure 5. Antibodies clear MAYV, prevent viral dissemination, and protect against musculoskeletal disease.
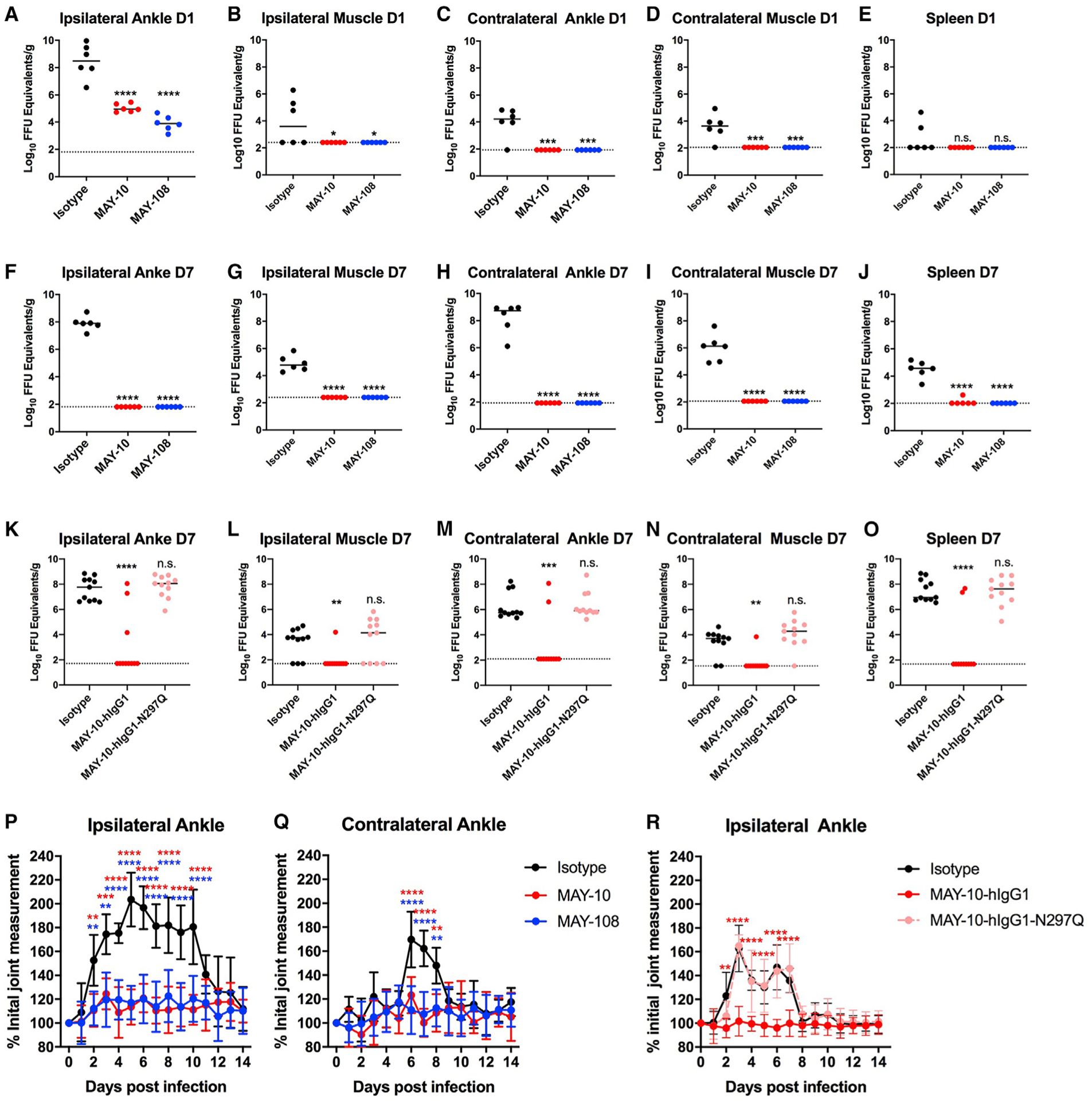
(A–J) Tissue titers of MAYV at 1 (A–E) or 7 (F–J) dpi. Four-week-old C57BL/6J mice treated with 100 μg of MAY-10, MAY-108, or an isotype control mAb 1 day before subcutaneous inoculation with MAYV-BeH407. At the indicated days, the ipsilateral ankle (A and F), ipsilateral calf muscle (B and G), contralateral ankle (C and H), contralateral calf muscle (D and I), and spleen (E and J) were harvested, and viral RNA was measured (two experiments; n = 6, one-way ANOVA with Dunnett’s post-test).
(K–O) Tissue titers of mice treated with 100 μg of MAY-10-hIgG1 or MAY-hIgG1-N297Q 1 day before infection. MAYV titers in the ipsilateral ankle (K) and calf muscle (L), the contralateral ankle (M) and calf muscle (N), and the spleen (O) were measured at 7 dpi (three experiments; n = 11, one-way ANOVA with Dunnett’s post-test).
(P–R) Four-week-old C57BL/6J mice were treated with MAY-10 or MAY-108 and infected as described above. Ipsilateral (P) and contralateral (Q) ankle joints were measured using digital calipers. (R) Ipsilateral ankle swelling was measured in mice treated with MAY-10-hIgG1 or MAY-10-hIgG1-N297Q (mean and SD of two experiments; n = 10, two-way ANOVA with Tukey’s post-test). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Figure S4.
We next tested whether antibody effector functions were required to limit viral infection and control musculoskeletal disease in immunocompetent mice by treating WT C57BL/6J mice with MAY-10-hIgG1 or aglycosyl MAY-10-hIgG1-N297Q 1 day before MAYV infection. At 7 dpi, the humanized version of MAY-10 protected mice from virus infection (Figures 5K–5O) to a similar extent as the parental mouse mAbs (Figures 5F–5J), with decreases in all tissues measured when compared to isotype control mAb. However, MAY-10-hIgG1-N297Q did not provide virological protection in this model. We also observed Fc-effector-function-dependent decreases in the inflammatory cytokines (tumor necrosis factor α [TNF-α], CXCL1, CXCL9, CXCL10, CCL3, CCL4, CCL5, and CSF1) in the ipsilateral ankles of MAY-10-hIgG1-treated animals at 7 dpi (Figure S4). These data indicate that the effector functions of non-neutralizing mAbs are required for efficient viral clearance and reduction of inflammation in joint-associated tissues of infected animals.
To determine if the mAbs protect against MAYV-mediated musculoskeletal disease, we measured ankle swelling. We observed substantial swelling in the ipsilateral ankle from 2–10 dpi (Figure 5K) and the contralateral ankle from 6–8 dpi (Figure 5L) in mice treated with an isotype control mAb, whereas animals treated with MAY-10 or MAY-108 showed no ankle swelling. Antibody-mediated protection was Fc effector function dependent, as MAY-10-hIgG1, but not MAY-10-hIgG1-N297Q, limited swelling in this model (Figure 5R). Thus, even without neutralizing activity, anti-MAYV mAbs can prevent dissemination, clear infection, and limit musculoskeletal disease in immunocompetent mice.
Myeloid-cell-dependent protection of non-neutralizing mAbs
Because non-neutralizing mAbs control MAYV infection in an Fc-effector-function-dependent manner, we hypothesized that specific immune cells bearing FcγRs mediate this protection (Bournazos et al., 2015; Lu et al., 2018). Previous studies have shown that monocytes and natural killer (NK) cells mediate antibody-dependent antiviral protection in vivo by antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), or antibody-dependent virus opsonization (Lu et al., 2018). To determine the cell type responsible for antibody-dependent protection against MAYV, we depleted monocytes or NK cells with specific mAbs (anti-CCR2 and anti-NK1.1, respectively) beginning 1 day before infection using the anti-Ifnar1-mAb-treated immunocompromised lethal challenge mouse model. As expected, we observed complete protection against mortality in mice treated with MAY-10 or MAY-108 in the absence of immune-cell-depleting antibody (Figures 6A and 6B). However, in the presence of anti-CCR2 mAb treatment and monocyte depletion (Figure S5A), MAY-10 and MAY-108 protection decreased to 40% and 30%, respectively. When similar depletion experiments were performed with anti-NK1.1 mAb to deplete NK cells (Figure S5B), we saw no change in protective activity in MAY-10 or MAY-108 (Figure 6B). These data suggest that CCR2+ monocytes are principally responsible for the protection conferred by non-neutralizing anti-MAYV mAbs.
Figure 6. Protection by non-neutralizing mAbs depends on monocytes.
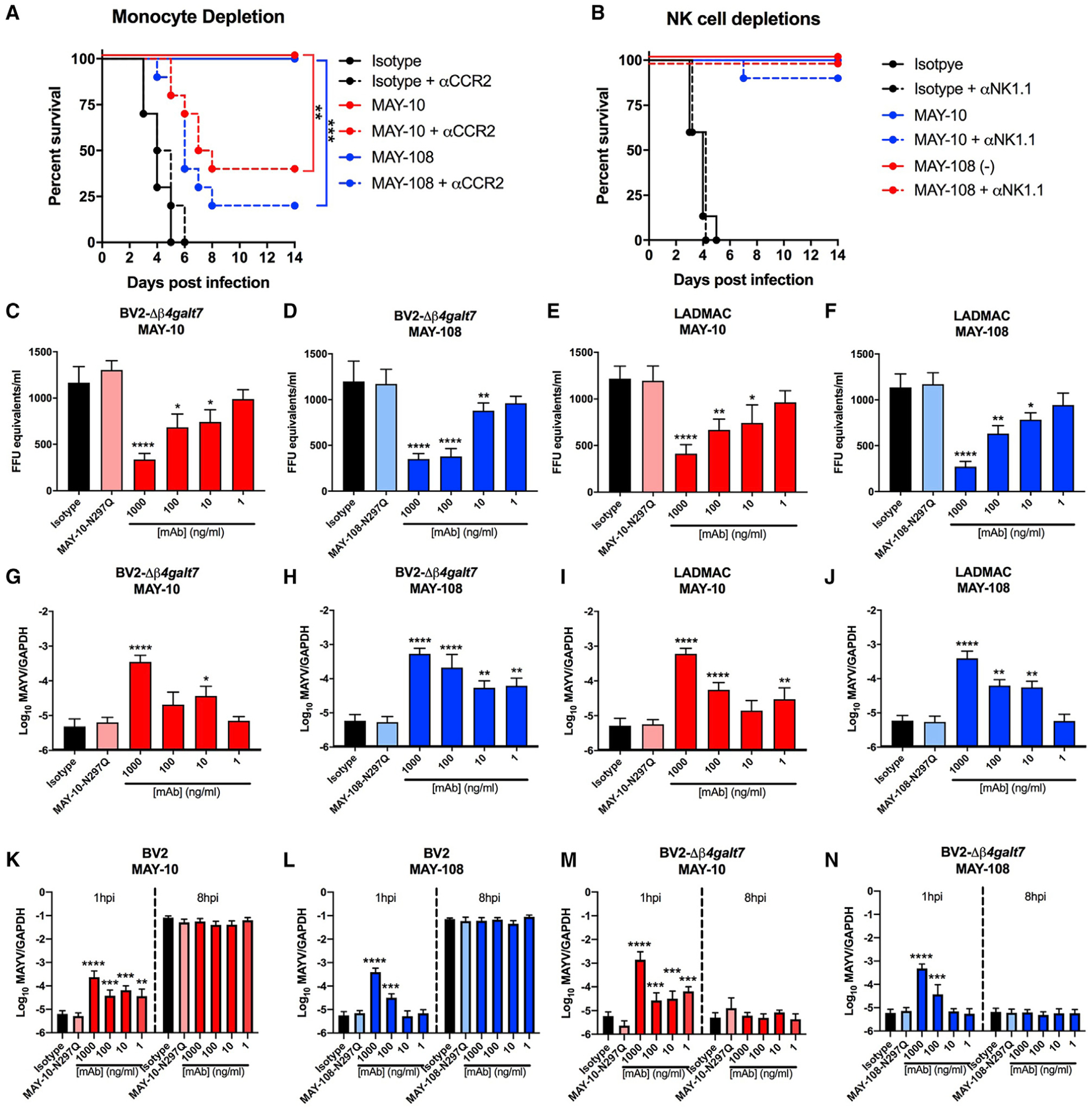
(A) Four-week-old C57BL6/J mice were treated with 100 μg of anti-Ifnar1 mAb and 100 μg of MAY-10, MAY-108, or isotype control mAb 1 day before infection with MAYV-BeH407. Indicated mice also were treated with 25 μg of an anti-CCR2 mAb at 1 day before infection and every other day after (two experiments, n = 10; log-rank test with Bonferroni correction).
(B) NK cells were depleted by treating mice with 200 μg mAb of anti-NK1.1 mAb 1 day before infection and every other day after. Mice were treated with anti-Ifnar1 mAb and MAY-10, MAY-108, or isotype control mAb as above (two experiments, n = 10; log-rank test with Bonferroni correction).
(C–J) Antibody-mediated binding of hIgG1 variants of MAYV to BV2-Δβ4galt7 (C, D, G, and H) or LADMAC (E, F, I, and J) cells. Virus binding to cells was measured indirectly by the depletion of MAYV from supernatants (C–F) or by direct binding and/or internalization of target cells (G–J). For measuring supernatants, MAYV was incubated with the indicated amount of hIgG1 variants of MAY-10 (C and E) or MAY-108 (D and F) for 1 h at 37°C before adding to the indicated cell for 30 min at 37°C. Viral RNA from supernatants was measured. Isotype-matched antibodies and hIgG1-N297Q mAb variants served as negative controls. To measure virus binding and internalization, BV2-Δβ4galt7 or LADMAC cells were inoculated with MAYV that had been pre-incubated with MAY-10 (G and I) or MAY-108 (H and J). After incubating for 30 min at 37°C, cells were washed with PBS and lysed, and viral RNA was measured.
(K–N) Antibody-dependent infection assays. Serial dilutions of hIgG1 mAbs were pre-incubated with MAYV before being added to permissive BV2 cells (K and L) or non-permissive BV2-Δβ4galt7 cells (M and N). Binding and internalization into cells was measured as above (G–H) at 1 or 8 hpi after removal of unbound virus (mean and SD of three experiments performed in triplicate; one-way ANOVA with Dunnett’s post-test compared to the isotype mAb control). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Figures S5–S8.
We evaluated how monocytes might contribute to antibody-mediated protection. To assess whether non-neutralizing anti-MAYV mAbs promote opsonization of free virions and clearance by monocytes, we performed in vitro binding assays in the folllowing two murine myeloid cell lines: microglial-derived BV2 cells (Figures 6C, 6D, 6G, and 6H) and bone-marrow-derived monocytic LADMAC cells (Figures 6E, 6F, 6I, and 6J). Although LADMAC cells are essentially non-permissive for MAYV infection unless the Mxra8 receptor is ectopically expressed (Zhang et al., 2018), BV2 cells can be infected at low levels because they express heparin sulfate (HS) as an attachment factor. To minimize the effects of HS on MAYV binding and infection of BV2 cells, we used BV2 cells lacking β−1,4-galactosyltransferase 7 (BV2-Δβ4galt7) (Ma et al., 2020), a key enzyme required for glycosaminoglycan synthesis. Flow cytometry analysis showed that BV2 cells express high levels of FcγRI, FcγRII, FcγRIII, and FcγRIV on their surface, whereas LADMAC cells express FcγRI, FcγRII, and FcγRIV and at lower levels (Figure S6).
We performed two sets of experiments, namely, an antibody-dependent virus depletion assay from the supernatant (Figures 6C–6F) and cell binding/internalization assays (Figures 6G–6J). For the virus depletion assay, 103 FFU of MAYV was pre-incubated with serial dilutions of the hIgG1 variant of MAY-10, MAY-108, or isotype control mAb before being added to BV2-Δβ4galt7 or LADMAC cells. After a 30-min incubation at 37°C, the supernatant containing unbound virus was collected, and after virion lysis, viral RNA was measured by qRT-PCR. Viral RNA levels were compared with a standard curve generated from known quantities of infectious MAYV to determine viral equivalents per ml. Notably, less viral RNA remained in the supernatant after treatment with MAY-10 (Figures 6C and 6E) and MAY-108 (Figures 6D and 6F) than that with the isotype control mAb. Viral clearance from the supernatant occurred dose dependently and required Fc effector functions, as it was not observed with the aglycosyl hIgG1-N297Q variants of MAY-10 and MAY-108. We observed similar results with the mouse IgG2c versions of both MAY-10 and MAY-108 (Figures S7A–S7D). These data suggest that the Fc region of anti-MAYV mAbs promotes clearance of MAYV virions from the inoculum, presumably by binding FcγR on the myeloid cells.
To test this hypothesis directly, we performed cell binding and internalization assays (Figures 6G–6J, S6, and S8). MAYV was incubated with MAY-10, MAY-108, or isotype control mAb for 30 min at 37°C. Antibody-virion complexes then were added to BV2-Δβ4galt7 or LADMAC cells and incubated for 1 h at 37°C. Cells then were rinsed thoroughly with PBS and lysed, and attached and/or internalized viral RNA was measured by qRT-PCR. Pre-treatment of MAYV with MAY-10 and MAY-108 significantly increased the level of cell-associated viral RNA compared to the isotype control mAb. Antibody-dependent binding and/or internalization of MAYV virions by BV2-Δβ4galt7 or LADMAC cells occurred dose dependently and required a functional Fc region, as no increase was observed with the aglycosyl N297Q variants of MAY-10 and MAY-108. For the highest concentrations of MAY-10 and MAY-108, we observed a >100-fold increase in cell-associated viral RNA compared to the isotype control mAb. Similar data were observed using mouse IgG2c versions of MAYV antibodies (Figures S7E–S7H). Treatment of BV2-Δβ4galt7 or LADMAC cells at 1 h after 37°C incubation with proteinase K and RNase A, to remove bound but not internalized virus, revealed that a significant fraction of opsonized MAYV transited into the cells (Figure S8). Collectively, these data suggest that antibody-dependent binding to MAYV in myeloid cells was dependent on Fc-FcγR interactions, resulted in enhanced cell binding and uptake, and was not due to virion cross-linking and aggregation, as seen for some anti-alphavirus antibodies (Zhou et al., 2020).
We next evaluated if MAYV association with target FcγR-expressing myeloid cells resulted in abortive or productive (and possibly even antibody enhanced) infection. We used both permissive WT BV2 (Figures 6K and 6L) and non-permissive BV2-Δβ4galt7 (Figures 6M and 6N) cells to track viral infection in the presence and absence of non-neutralizing mAbs. MAYV was incubated with serial dilutions of MAY-10 (Figures 6K and 6M) or MAY-108 (Figures 6L and 6N) to form antibody-antigen complexes. These complexes were added to target cells for a 1-h incubation at 37°C, and the cells then were rinsed to remove unbound virus. The cells were lysed either immediately after rinsing (1 h post-infection [hpi]) or after another 7 h-incubation at 37°C (8 hpi), and cell-associated viral RNA was measured by qRT-PCR. As expected, we observed an initial increase in cell-associated viral RNA at 1 hpi when the virions were pre-treated with the mIgG2c but not N297Q forms of MAY-10 and MAY-108. For the WT BV2 cells (Figures 6K and 6L), we observed a ~10,000-fold increase in viral RNA at 8 hpi compared to 1 hpi with the isotype-mAb-treated virions, which indicates that MAYV replicated in the BV2 cells. However, there was no difference in viral RNA levels at 8 hpi between anti-MAYV and isotype mAb treatments, indicating that the greater level of virus binding and internalization at 1 hpi caused by non-neutralizing mAbs MAY-10 and MAY-108 did not result in enhanced infection in WT BV2 cells. To determine if mAb-induced virion binding and internalization caused enhancement of infection of non-permissive myeloid cells (as seen with dengue virus [Brandt et al., 1982; Halstead et al., 1980]), we repeated experiments in the non-permissive BV2-Δβ4galt cells (Figures 6M and 6N). We observed the expected increase in cell-associated viral RNA at 1 hpi, and clearance was observed at 8 hpi with no evidence of productive infection. We observed similar results by using mIgG2c versions of the anti-MAYV mAbs (Figures S7I–S7L). To determine if BV2 cells are even capable of antibody-dependent enhancement (ADE), we repeated the experiments with Zika virus (ZIKV), a flavivirus whose infection is enhanced in myeloid cells by cross-reactive, non-neutralizing antibodies (Castanha et al., 2017; Dejnirattisai et al., 2016). We incubated a mouse-adapted ZIKV virus (Gorman et al., 2018) with serial dilutions of the mAb E60, a poorly neutralizing mAb that binds the conserved fusion loop of the flavivirus E protein (Oliphant et al., 2006). Despite not observing ADE with MAYV, we observed robust ADE with ZIKV and the E60 mAb in BV2 and LADMAC cells (Figures S7M–S7O), suggesting the outcome of antibody engagement in cells expressing FcγRs is virus specific. In the case of MAYV, non-neutralizing mAbs facilitate the interaction of virions with myeloid cells that results in abortive infection and clearance.
DISCUSSION
Previous studies have analyzed the importance of neutralizing antibody responses in protecting against alphavirus infection. These studies highlighted both the effects of virus neutralizing activity (Earnest et al., 2019; Fox et al., 2015; Jin et al., 2015, 2018; Martins et al., 2019; Pal et al., 2013) and Fc effector functions (Earnest et al., 2019; Fox et al., 2019) for optimal in vivo activity. However, they could not fully gauge the protective activity of Fc effector functions because the antibodies were inherently neutralizing. Here, we identified a panel of 13 anti-MAYV mAbs that bind avidly to virions and infected cells yet exhibit no detectable neutralizing activity against the virus in Vero and C2C12 cells. Passive transfer of non-neutralizing mAbs conferred significant protection in vivo that was completely dependent on Fc effector interactions, as determined using IgG subclass switch variants and N297Q variants lacking the ability to engage FcγRs.
The mIgG2c mAbs that we characterized exhibited a range of protective ability in the lethal MAYV challenge model. The reasons for these differences may be due to several factors. We observed variable binding to MAYV virions in a capture ELISA experiment and to recombinant E2 protein by using ELISA and BLI experiments. The mAbs that bound most avidly to virions and recombinant E2 proteins showed the greatest protective activity in vivo. Indeed, MAY-10 and MAY-108, our most protective mAbs, had KD values that were 100-fold lower and had substantially longer binding t1/2 than many poorly protecting mAbs. A higher level of virus binding likely enhances the Fc-dependent clearance by myeloid cells. Furthermore, the most protective mAbs map to two epitopes within or proximal to the A domain, as follows: one near the amino terminus on the top of the spike trimeric complex (group B), and a second in the β-ribbon region on the outer face of the E2-E1 spike (group E). Possibly, the mAbs binding these regions of the E2 protein are more accessible for engagement by FcγRs on monocytes, enabling virus clearance and protection. The orientation of mAb binding to the virion also could affect presentation of the Fc region to FcγRs, as was seen with anti-dengue virus mAbs (Renner et al., 2018).
Our non-neutralizing antibodies bound to six distinct regions of the MAYV E2 A domain and the β-ribbon region between the A and B domains. In comparison, other mAbs against arthitogenic alphavirues (e.g., CHIKV) that map to epitopes within the A domain can be potently neutralizing. The anti-CHIKV human mAbs 1H12 and 3N23 are potently inhibitory in cell culture and yet share interaction residues with group B and C mAbs from our panel (Smith et al., 2015). Similarly, the highly neutralizing anti-CHIKV mAbs 4J21 and 5M16 (Long et al., 2015) bind amino acids shared by group B and F mAbs as well as others throughout the A and B domains. The distinguishing feature of these neutralizing mAbs is their ability to bind residues within multiple E2 domains (e.g., A and B) or across different E2 proteins, whereas the majority of the non-neutralizing mAbs appear to recognize linear determinants. Engagement of tertiary and quaternary epitopes within and across E2 may be required for alphavirus neutralization. Indeed, structural studies have demonstrated cross-linking of multiple distinct domains in E2 by potently neutralizing mAbs (Fox et al., 2015; Powell et al., 2020).
We found that monocytes were necessary for antibody-effector-mediated protection from MAYV infection. Our in vitro studies suggest a possible mechanistic basis for at least part of the inhibitory activity. Non-neutralizing anti-MAYV mAbs bind virus and facilitate clearance by Fc-dependent internalization and destruction in myeloid cells. This abortive infection mechanism explains how non-neutralizing mAbs could prevent dissemination from the site of inoculation but might not explain how antibodies effectively clear MAYV-infected cells. Additionally, monocytes might clear virus from infected cells by ADCP (Bournazos et al., 2015; Lu et al., 2018) because the viral E1 and E2 structural proteins are displayed on the plasma membrane surface prior to virion morphogenesis and budding and can be recognized by antibodies. Alternatively, the enhanced entry of virus into myeloid cells in an antibody- and Fc-dependent manner could promote antigen cross-presentation and accelerated CD8+-T-cell-mediated clearance (Bournazos et al., 2020a; Platzer et al., 2014). ADCC by NK cells did not appear to have a dominant role in protection by non-neutralizing mAbs, as depletion did not impact survival.
Under certain circumstances, monocytes themselves can be infected by alphaviruses (Her et al., 2010; Winkler et al., 2020), which may be a potential mechanism for viral dissemination. Our studies indicate that the enhanced virus binding and internalization of MAYV facilitated by non-neutralizing mAbs in myeloid cell lines does not result in enhanced infection in vitro or in vivo. This result contrasts with flaviviruses for which non-or poorly neutralizing antibodies can promote infection of FcγR-expressing myeloid cells through ADE (Bournazos et al., 2020b; Halstead et al., 2010; Rey et al., 2018), which is believed to result in severe disease during secondary dengue infection (Halstead, 1988). In comparison, ADE and pathogenic antibodies that enhance myeloid cell infections are not believed to contribute to alphavirus pathogenesis, although one study reported higher viremia and worse arthritis in mice in the setting of passive transfer of an anti-CHIKV mAb (Lum et al., 2018). Clearly, studies that examine the effects of in vivo passive transfer of antibodies with no, weak, or potently neutralizing activity with multiple alphaviruses are needed to establish the generaliz-ability of our findings.
Antibody-dependent protection against alphaviruses is facilitated by two main functions, as follows: Fab-mediated virus neutralization and Fc-dependent effector functions. Data from this study and others (Earnest et al., 2019; Fox et al., 2015; Jin and Simmons, 2019; Pal et al., 2013) suggest that neutralization and Fc effector functions can control alphavirus infections through a range of mechanisms. Analyzing the polyclonal antibody response to infection by MAYV or other alphaviruses in the context of natural infection or immunization (Choi et al., 2019; Weise et al., 2014) to determine the relative amounts of neutralizing and non-neutralizing antibodies may provide insight as to which functions ultimately are most important for controlling infection.
The efficacy of a protective antibody response to alphavirus infection is determined by the location of antibody binding on intact virions and structural proteins displayed on the surface of infected cells, the inherent neutralizing ability and mechanism (blockade of attachment, entry, fusion, or egress), and likely the accessibility of the Fc region of antibodies to engage FcγR and mediate effector functions. Because accelerated virus clearance might mitigate the development of chronic musculoskeletal disease, designing vaccines and analyzing antibody responses in the context of effector function responses may be important. Although many neutralizing anti-alphavirus mAbs have been described that bind the A and B domain of E2, our study shows that non-neutralizing mAbs recognizing epitopes within or near the A domain also can prevent or clear virus from infected hosts by optimal effector functions. The six epitopes we identified are also highly conserved across the 73 complete MAYV genomes annotated in public databases. Amino acid interaction residues from mAbs in groups A, D, and F are entirely conserved, whereas there are a small number of single amino acid substitutions in binding residues from mAbs in group B (9/73 strains with amino acid substitution at T55), C (2/73 strains with amino acid substitution at D74), or E (1/73 strains with amino acid substitution at A161). Further analysis of human antibody responses to natural infections, in both patients who have cleared virus and those with persistent disease, may provide insight into the contribution of Fc effector functions to protection and disease pathogenesis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael S. Diamond (diamond@wusm.wustl.edu).
Materials availability
All requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact author. This includes mice, antibodies, viruses, and proteins. All reagents will be made available on request after completion of a Materials Transfer Agreement.
Data and code availability
All data supporting the findings of this study are available within the paper and are available from the corresponding author upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Vero, HEK293T, and C2C12 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml of penicillin, 100 μg/ml of streptomycin, 1X MEM non-essential amino acids, 1 mM sodium pyruvate, 2 mM L-Glutamine and 10 mM HEPES pH 7.3. Hybridomas were cultured in Isocove’s modified Eagle medium (IMDM) supplemented with 20% FBS, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 1 mM sodium pyruvate. Expi293 cells were maintained in Expi293 medium (GIBCO). BV2-Δβ4galt7 cells were produced previously (Ma et al., 2020). BV2, BV2-Δβ4galt7, and LADMAC cells were maintained in DMEM supplemented with 5% FBS, 100 U/ml of penicillin, 100 μg/ml of streptomycin, 1X MEM non-essential amino acids, and 10 mM HEPES pH 7.3.
Viruses
MAYV (strain BeH407) was obtained from the World Reference Center for Emerging Viruses and Arboviruses (K. Plante, and S. Weaver, University of Texas Medical Branch) and passaged in Vero cells from lyophilized stocks. Recombinant viruses were produced after linearization of a prS2 vector containing cDNA from MAYV (strain CH) (Weise et al., 2014) that was provided by S. Weaver (University of Texas Medical Branch). After in vitro transcription with mMESSAGE mMACHINE SP6 transcription kit (Invitrogen) and transfection into BHK-21 cells, p0 virus stocks were harvested and passaged once (p1) in Vero cells. Virus titers were determined by focus forming assay (Fox et al., 2015). Mouse adapted Zika-Dakar virus (Gorman et al., 2018) was propagated in Vero cells.
Mouse experiments
All animal experiments and procedures were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (Assurance number A3381–01). Injections were performed under anesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering.
WT C57BL/6J male mice were purchased from Jackson Laboratories. Common γ-chain deficient (FcRγ−/−) C57BL/6 mice were obtained commercially (Taconic), backcrossed using speed congenics to C57BL/6J mice, and bred at the Washington University School of Medicine Animal Facility. FcRγ−/− experiments were performed with both male and female mice. Unless otherwise indicated, four-week-old mice were used in all experiments. Anti-MAYV mAbs were administered by intraperitoneal injection at specified times before or after inoculation in the left footpad with 103 FFU of MAYV in Hank’s Balanced Salt Solution (HBSS) supplemented with 1% heat-in-activated FBS. Foot swelling was monitored via measurements (width × height) using digital calipers. Tissues were harvested after perfusion with 40 mL of PBS and titered by qRT-PCR using RNA isolated from viral stocks as a standard curve to determine FFU equivalents. For lethal challenge experiments, mice were administered via intraperitoneal injection a single 100 μg dose of anti-Ifnar1 mAb MAR1–5A3 (Sheehan et al., 2006) (BioXCell) one day before infection. Monocyte depletion experiments were performed by administering 25 μg of anti-CCR2 (clone MC-21) at day −1 and every other day subsequently. NK cell depletion experiments were performed by administering 200 μg of NK1.1 (BioXCell clone PK136) at day −1, and every other day subsequently.
METHOD DETAILS
Protein expression and purification
The MAYV (strain TRVL4675) E2 ectodomain (residues 1–340) was cloned into the pET21a expression vector and expressed in BL21 (DE3) E. coli cells. Protein production was induced using 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), where E2 partitioned into the inclusion body fraction and was refolded using an oxidative refolding protocol (Nelson et al., 2014). Briefly, 10 mL of solubilized inclusion body was injected at 1ml/h into a 1 l volume of arginine refolding buffer (400 mM L-arginine, 100 mM Tris [pH 8.5], 5 mM reduced glutathione, 0.5 mM oxidized glutathione, and 0.2 mM PMSF), and then allowed to stir slowly overnight at 4°C. The refolded protein was filtered, concentrated using a 30 kDa cutoff stirred cell concentrator (EMD Millipore), and purified by HiLoad 16/600 Superdex 75 size exclusion chromatography (GE Healthcare).
mAb generation
Ten week-old female C57BL/6J mice were inoculated with 103 FFU of MAYV-CH. Mice were boosted with 100 μg of recombinant MAYV E2 protein mixed 1:1 with Freund’s Incomplete adjuvant at 14, 28, and 42 days after initial infection. Spleens were harvested at 45 dpi and fused with P3X63 Ag.8.6.5.3 mouse myeloma cells as described previously (Pal et al., 2013). Hybridoma supernatants were screened for antibodies that bound to recombinant MAYV E2 in an ELISA and/or to MAYV (strain BeH407)-infected cells by flow cytometry. Neat hybridoma supernatants were screened for neutralization of MAYV-CH using a FRNT (described below). Selected mAbs were isotyped by ELISA and purified by protein A affinity chromatography (Thermo).
ELISA
For the virion capture ELISA, a humanized mAb specific for MAYV (MAY-134) (Earnest et al., 2019) was adsorbed to Maxisorp Immunocapture ELISA plates (Thermo) in a sodium bicarbonate buffer pH 9.3 overnight at 4°C. Wells were washed with PBS and blocked with blocking buffer (PBS + 5% BSA [Sigma]) for 1 h at 37°C. Blocking buffer was removed and replaced with 103 FFU/well of MAYV-BeH407 diluted in blocking buffer and incubated at 37°C for 1 h. Unbound virus was washed away with PBS and serial dilutions of anti-MAYV mAbs, diluted in blocking buffer, were added to the wells and incubated for 1 h at 4°C. Unbound mAb was washed away with PBS, and wells were incubated with an HRP conjugated goat anti-mouse Fc antibody for 1 h at 4°C. Plates were washed and developed with TMB one-step substrate (Thermo) for 10 minutes. The reaction was stopped with 1 N H2SO4, and absorbance was measured at 450 nm. For the mAb competition binding ELISA, virus was captured to plates as above and incubated with 10 μg/ml of the indicated primary mAbs. Unbound mAbs were rinsed away, and wells were incubated with 10 ng/ml of the secondary mAbs labeled with NHS-Biotin (Thermo). After washing, biotinylated mAbs were detected using a streptavidin-HRP secondary (Vector Laboratories). For the E2 protein ELISA, 50 ng/well of bacterially produced recombinant MAYV E2 ectodomain was adsorbed to plates as above. Plates were washed with ELISA wash buffer (PBS + 0.05% Tween 20) and incubated with serial dilutions of anti-MAYV mAbs diluted in blocking buffer. MAbs were detected using secondary reagents and OD was measured as described above.
Focus reduction neutralization tests (FRNT)
Anti-MAYV mAbs were diluted serially and incubated with 102 FFU of MAYV-BeH407 for 1 h at 37°C in triplicate wells. Virus-mAb mixtures were incubated on Vero or C2C12 cells for 60 min at 37°C before being overlaid with 1% methylcellulose in minimal essential medium (MEM) supplemented with 10 mM HEPES pH 7.3, 100 U/ml of penicillin, 100 μg/ml of streptomycin, 2 mM L-glutamine, and 2% FBS. Eighteen hours after virus inoculation, cells were fixed with 1% paraformaldehyde (PFA) in PBS. Cells then were washed and overlaid with 1 μg/ml of biotinylated MAY-118 (Earnest et al., 2019) for 2 h. Cells were washed and overlaid with streptavidin-HRP for 1 h. Foci of infection were detected using TrueBlue substrate (KPL) and counted using a Biospot plate reader (Cellular Technology). Wells containing virus incubated with mAbs were compared to wells treated with virus containing no mAb.
Western blotting
Recombinant MAYV-E2 protein was mixed with LDS buffer (Life Technologies) in the presence (reducing) or absence (non-reducing) of 20 mM dithiothreitol. After heating at 70°C for 10 min, samples were electrophoresed using a 4%–12% Bis-Tris gel (Life Technologies). Proteins were transferred to nitrocellulose membranes using an iBlot2 Dry Blotting System (Life Technologies). Membranes were blocked with 5% BSA, cut into strips, and probed with the indicated mAbs. Unbound mAb was rinsed away, and the mAbs were detected with an anti-mouse HRP-conjugated secondary antibody (Vector Laboratories). Blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Life Technologies).
Biolayer interferometry
The binding affinity of purified recombinant MAYV E2 ectodomain protein to MAYV mAbs was evaluated at 25°C using an Octet-Red96 device (Pall ForteBio). 100 μg of each mAb was mixed with biotin (EZ-Link-NHS-PEG4-Biotin, Thermo Fisher) at a molar ratio of 20:1 biotin:protein and incubated at room temperature for 30 min. The unreacted biotin was removed by passage through a de-salting column (5 mL Zeba Spin 7 kDa molecular weight cut-off, Thermo Fisher). The biotinylated-mAbs were loaded onto streptavidin biosensor pins (ForteBio) until saturation, typically 10 μg/ml for 2 min, in 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, 0.005% P20 surfactant, and 1% BSA. The pins were equilibrated in binding buffer alone before being plunged into wells containing various concentrations of MAYV E2, then being placed back into binding buffer to allow for dissociation. Real-time data were analyzed using BIAevaluation 3.1 (GE Healthcare). Kinetic profiles and steady-state equilibrium concentration curves were fitted using a global 1:1 binding algorithm with drifting baseline.
Alanine scanning mutagenesis
A pcDNA3.1(+) plasmid expressing a codon-optimized MAYV (strain TRVL4675) structural polyprotein (C, E3, E2, 6K, and E1 genes) was synthesized and mutated by GenScript. Alanine scanning mutagenesis was performed on amino acids in the A domain of the E2 protein (residues 1–173) that were predicted to be solvent exposed. Plasmids were transfected into HEK293T cells using Lipofectamine 3000 (Thermo Fisher). Eighteen hours later, cells were chilled to 4°C, washed with PBS, and incubated with anti-MAYV mAbs (10 μg/ml) in PBS with 2% FBS for 1 h at 4°C. An oligoclonal mixture of the 13 mAbs as well as an anti-B domain mAb (MAY-117) was used as a control for mutant E2 protein expression. Anti-MAYV mAb binding was detected using Alexa Fluor 647 conjugated goat anti-mouse IgG (Thermo Fisher) diluted 1:1000. After 1 h, cells were washed, fixed with 1% PFA in PBS, and analyzed by flow cytometry using a MACSQuant Analyzer (Miltenyi Biotec). Using previously described criteria (Smith et al., 2015), critical residues were defined as those with ≤25% binding to an individual mAb but ≥ 75% binding to an oligoclonal pool of anti-MAYV mAbs as determined by flow cytometry.
Isotype switching of mAbs
MAY-10 and MAY-108 variable regions were sequenced and cloned using previously described methods (Ho et al., 2016). Total RNA was isolated from hybridomas and cDNA was produced using random hexamers and Oligod(T)20 using a SuperScript IV First Strand Synthesis kit (Invitrogen). Heavy and light chain variable regions were amplified and sequenced using mouse-specific primer sets (Ho et al., 2016). Allele-specific primers were used to amplify variable regions and append Gibson assembly sequences to the 5′ and 3′ ends. The variable regions then were cloned into plasmids containing the constant regions of human IgG1 (pAbVec-hIgG1) or mouse IgG1 (pAbVec-mIgG1) or the appropriate kappa chain (pAbVec-hIgKappa or pAbVec-mIgKappa) using NEBuilder (New England Bio-labs). The human IgG1-N297Q vector was produced by site directed mutagenesis of the human IgG1 vector using a Phusion site directed mutagenesis kit. Antibodies were produced by co-transfecting Expi293 cells with an appropriate heavy and kappa chain plasmid using Hype5 transfection reagent (Oz Biosciences). Four days after transfection, supernatant was collected and mAbs were purified on a Pierce protein A agarose column (Thermo).
Cytokine analysis
Ankle homogenates were incubated with Triton X-100 (1% final concentration) for 1 h at room temperature to inactivate MAYV. Homogenates then were analyzed for cytokines and chemokines by Eve Technologies Corporation (Calgary, AB, Canada) using their Mouse Cytokine Array/Chemokine Array 31-Plex platform.
Flow cytometry
For cell depletion experiments, whole blood was harvested from mice and mixed with 5 mM EDTA (Corning). Red blood cells were lysed with ACK lysis buffer at room temperature before being washed and chilled in PBS + 5% FCS. Monocytes were stained with CD45 BUV395 (BD Biosciences clone 30-F11), CD11b PerCP-Cy5.5 (BioLegend clone M1/70), Ly6B FITC (Abcam clone 7/4), Ly6G PE-Cy7 (BioLegend clone 6D5), Ly6C Pacific Blue (BioLegend clone HK1.4) and MHC class II A700 (BioLegend clone M5/114.15.2). NK cells were stained with CD45 BUV95, CD3 APC-Cy-7 (BioLegend clone 145–2C11), CD19 BV605 (BioLegend clone 6D5), and NK1.1 Pe-Cy7 (BioLegend clone PK136). Viability was determined through exclusion of a fixable viability dye (e506;eBiosciences). Samples were fixed and processed on a BD-Fortessa X20. For FcγR expression experiments BV2, LADMAC, and Vero cells were stained with one of the following: CD64 APC (BioLegend clone X54–5/7.1), CD32b APC (Invitrogen clone AT130–2), CD16 FITC (BioLegend clone 221–3A4), CD16.2 APC (BioLegend clone 9E9). Cells were analyzed on a MACSQuant analyzer (Miltenyi). All FACS data were analyzed by FlowJo v. 10.7.
Antibody-induced depletion of MAYV from cell supernatants
MAYV was treated with 100 μg/ml of RNase A (Thermo #EN0531) for 30 min at 37°C to degrade unencapsidated RNA. RNase was inactivated by incubating samples with 100 U of RiboLock RNase inhibitor (Thermo #E00381) at 37°C for 15 minutes. 103 FFU of RNase-treated virus was incubated with serial dilutions of the indicated mAb for 30 min at 37°C. Virus-mAb complexes were placed on cells and incubated for 30 min at 37°C. The virus inoculum then was removed and viral RNA was isolated using a MagMax Viral Isolation Kit (Applied Biosystems). MAYV RNA was quantified using a Taqman RNA-to-Ct 1-step kit (Thermo Fisher) and a 5′ UTR and nsp1 specific primer/probe set (Waggoner et al., 2018) along with a standard curve of MAYV stock virus.
Antibody-induced virus binding/internalization assay
Target cells were counted, and the indicated mAbs were incubated for 30 min at 37°C with MAYV at a multiplicity of infection (MOI) of 10. Virus-antibody complexes were added to cells and incubated at 37°C for 1 h. Cells were washed six times with ice cold PBS and lysed in RLT buffer (QIAGEN). Viral RNA was isolated using a MagMax Viral Isolation Kit. MAYV RNA and GAPDH RNA was quantified using a Taqman RNA-to-Ct 1-step kit with either anti-MAYV primers or anti-mouse Gapdh primer/probe. Data are expressed as amount of MAYV RNA relative to GAPDH RNA. For internalization assays, the same protocol was followed, but after rinsing mono-layers after 1 h incubation at 37°C, cells were treated with 100 μg/ml of proteinase K (Invitrogen) for 15 min at 37°C. Proteinase K was rinsed away, and cells were treated with 100 μg/ml of RNase A for 30 min at 37°C. Cells were lysed, and viral RNA was detected from cellular lysates as described above.
Antibody dependent enhancement assays
For MAYV, target cells were counted and the indicated mAbs were incubated for 30 min at 37°C with MAYV at a MOI of 10. Virus-antibody complexes were added to cells and incubated at 37°C for 1 h. Unabsorbed virus was removed by washing cells 6X with 37°C DMEM. Cells were incubated at 37°C in DMEM + 2% FBS for 7 additional hours. Cells were lysed and viral RNA was quantified as described above.
For ZIKV, target cells were counted and either MAY-10 or the cross-reactive anti-E mAb E60 (Oliphant et al., 2006) were pre-incubated with ZIKV for 30 min at 37°C. Virus-antibody complexes were added to the cells at an MOI of 50 and incubated at 37°C for 2 h. Unabsorbed virus was removed by rinsing cells six times with DMEM. Cells were incubated at 37°C in DMEM + 2% FBS for 16 h. Cells were lysed and viral RNA was quantified using a published primer/probe set (White et al., 2018).
MAYV E2-E1 structure depiction
Structural figures were created using UCSF ChimeraX (Goddard et al., 2018). The predicted structure of the MAYV E2-E1 monomer was generated as previously described (Earnest et al., 2019). To depict the envelope proteins as a trimeric spike, the predicted monomers were superimposed onto the model of the CHIKV E2-E1 spike (PDB: 6NK5) using the matchmaker command in UCSF ChimeraX (Goddard et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was assigned with P values < 0.05 using GraphPad Prism version 7.0. The specific test for each dataset is indicated in respective figure legends and was selected based on the number of comparison groups and variance of the data. For foot swelling analysis, significance was determined by a two-way ANOVA with Tukey’s post-test (more than two groups) or Sidak’s post-test (between two groups). Viral burden data were analyzed by a one-way ANOVA with a Dunnett’s post-test. Survival curve analysis was analyzed by the log rank test. A Bonferroni correction was used depending on the number of comparison groups.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MAY-1, anti-MAYV mAb | This Paper | N/A |
| MAY-8, anti-MAYV mAb | This Paper | N/A |
| MAY-8-mIgG1, anti-MAYV mAb | This Paper | N/A |
| MAY-8-hIgG1, anti-MAYV mAb | This Paper | N/A |
| MAY-8-hIgG1-N297Q, anti-MAYV mAb | This Paper | N/A |
| MAY-10, anti-MAYV mAb | This Paper | N/A |
| MAY-23, anti-MAYV mAb | This Paper | N/A |
| MAY-27, anti-MAYV mAb | This Paper | N/A |
| MAY-39, anti-MAYV mAb | This Paper | N/A |
| MAY-41, anti-MAYV mAb | This Paper | N/A |
| MAY-59, anti-MAYV mAb | This Paper | N/A |
| MAY-60, anti-MAYV mAb | This Paper | N/A |
| MAY-68, anti-MAYV mAb | This Paper | N/A |
| MAY-72, anti-MAYV mAb | This Paper | N/A |
| MAY-102, anti-MAYV mAb | This Paper | N/A |
| MAY-108, anti-MAYV mAb | This Paper | N/A |
| MAY-108-mIgG1, anti-MAYV mAb | This Paper | N/A |
| MAY-108-hIgG1, anti-MAYV mAb | This Paper | N/A |
| MAY-108-hIgG1-N297Q, anti-MAYV mAb | This Paper | N/A |
| MAY-112, anti-MAYV mAb | This Paper | N/A |
| MAY-117, anti-MAYV mAb | Earnest et al., 2019 | N/A |
| MAY-117-hIgG1, anti-MAYV mAb | Earnest et al., 2019 | N/A |
| E60, anti-Zika mAb | Oliphant et al., 2006 | N/A |
| MAR1-5A3, anti-Ifnar1 mAb | Leinco | Cat #: BP024; RRID:AB_2491621 |
| MC-21, anti-CCR2 mAb | (Mack et al., 2001) | N/A |
| InVivomAb anti-mouse NK1.1 | Bio X Cell | Cat # BE0036; RRID:AB_1107737 |
| Goat anti-mouse IgG, human ads-HRP | Southern Biotech | Cat # 1030-05; RRID:AB_2619742 |
| Goat anti-mouse IgG, human ads-BIOT | Southern Biotech | Cat # 1030-08; RRID:AB_2794296 |
| Goat anti-mouse IgG, human ads-AlexaFluor 647 | Invitrogen | Cat # A-21236; RRID:AB_2535805 |
| Goat anti-human IgG, HRP | Thermo Fisher | Cat # 62-8420; RRID:AB_2533962 |
| Streptavidin-HRP | Vector Laboratories | Cat # SA-5004; RRID:AB_2336509 |
| BUV95 anti-CD45 | BD BioSciences | Cat # 564279; RRID:AB_2651134 |
| Fixable Aqua dead cell stain | Invitrogen | Cat # L34965 |
| PerCP-Cy5.5 anti-CD11b | BioLegend | Cat # 101207; RRID: AB_312790 |
| FITC anti-Ly6B | Abcam | Cat # ab53457; clone: 7/4 |
| PE-Cy7 anti-Ly6G | BioLegend | Cat # 115511; RRID: AB_313646 |
| Pacific Blue anti-Ly6C | BioLegend | Cat # 128015; RRID: AB_1732087 |
| AlexaFluor 700 anti-MHC class II | BioLegend | Cat # 107621; RRID: AB_493726 |
| APC-Cy-7 anti-CD3 | BioLegend | Cat # 100329; RRID: AB_1877171 |
| BV605 anti-CD19 | BioLegend | Cat # 115539; RRID: AB_11203538 |
| PE-Cy7 anti-NK1.1 | BioLegend | Cat # 108713; RRID: AB_389363 |
| APC anti-CD64 | BioLegend | Cat # 139305; RRID: AB_11219205 |
| APC anti-CD32b | Invitrogen | Cat # 17-0321-82; RRID: AB_2573142 |
| APC anti-CD16.2 | BioLegend | Cat # 149505; RRID: AB_2565812 |
| Virus and bacterial strains | ||
| MAYV-CH | Weise et al., 2014 | N/A |
| MAYV-BeH407 | World Reference Center for Emerging Viruses and Arboviruses, The University of Texas Medical Branch | N/A |
| Zika-Dakar-MA | Gorman et al., 2018 | N/A |
| Experimental models: cell lines | ||
| Vero E6 | ATCC | CRL-1586; RRID:CVCL_0574 |
| C2C12 | ATCC | CRL-1772; RRID: CVCL_0188 |
| HEK293T | ATCC | CRL-3216; RRID:CVCL_0063 |
| Expi293F | Invitrogen | Cat # A14527; RRID: CVCL_D615 |
| BV2 | Ma et al., 2020 | N/A |
| BV2-β4galt7 | Ma et al., 2020 | N/A |
| LADMAC | ATCC | CRL-2420; RRID:CVCL_2550 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Cat # 000664; RRID: IMSR_JAX:000664 |
| Mouse: C57BL/6 FcRγ−/− | Taconic | Cat # 583 |
| Oligonucleotides | ||
| MAYV-rtPCR-F: 5′-AAGCTCTTCCTCTGCATTGC-3′ | Waggoner et al., 2018 | N/A |
| MAYV-rtPCR-R: 5′-TGCTGGAAACGCTCTCTGTA-3′ | Waggoner et al., 2018 | N/A |
| MAYV-rtPCR-Probe: 5′-/56-FAM/-GCCGAGAG/ZEN/CC CGTTTTTAAAATCAC/3IABkFQ-3′ | Waggoner et al., 2018 | N/A |
| Zika-rtPCR-F: 5′-CCACCAATGTTCTC TTGCAGACATATTG-3′ | White et al., 2018 | N/A |
| Zika-rtPCR-R: 5′-TTCGGACAGCCGT TGTCCAACACAAG-3′ | White et al., 2018 | N/A |
| Zika-rtPCR-Probe: 5′-/56-FAM/AGCCTACCT/Zen/TGA CAAGCAGTC/3IABkFQ/−3′ | White et al., 2018 | N/A |
| Gapdh TaqMan Primer/Probe set | IDT | Mm.PT.39a.1 |
| Recombinant DNA | ||
| pET21a-MAYV-TRVL4675-rE2 protein | Earnest et al., 2019 | N/A |
| pCDNA3.1-MAYV-Polyprotein (including alanine scan mutants in E2 residues 1–174) | This Paper | N/A |
| pAbVec-mIgG1 | Ho et al., 2016 | N/A |
| pAbVec-mIgKappa | Ho et al., 2016 | N/A |
| pAbVec-hIgG1 | Ho et al., 2016 | N/A |
| pAbVec-hIgG1-N297Q | Earnest et al., 2019 | N/A |
| pAbVec-hIgKappa | Ho et al., 2016 | N/A |
| Software and algorithms | ||
| FlowJo | FlowJo, LLC | v10 |
| GraphPad Prism | GraphPad | v 8.2.1 |
| BIAevaluation | GE Healthcare | v 3.1 |
| UCSF ChimeraX | RBVI | v 1.1 |
Highlights.
Anti-MAYV antibodies protect against infection without neutralizing the virus
Protection by non-neutralizing anti-MAYV antibodies requires Fc effector function
Anti-MAYV antibody Fc effector functions require monocytes to mediate protection
Non-neutralizing anti-MAYV antibodies promote abortive infection in myeloid cells
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 AI141436, R01 AI114816, and U19 AI142790 and contracts AI201800001, 75N93019C00062, and HHSN272201700060C. We thank Michelle Noll for mouse husbandry and Ted Pierson for comments on the manuscript. BioRender software was used to generate some of the images.
DECLARATION OF INTERESTS
M.S.D. is a consultant for Inbios, Vir Biotechnology, NGM Biopharmaceuticals, and the Carnival Corporation and on the Scientific Advisory Board of Moderna and Immunome. The Diamond laboratory has received sponsored research agreements from Moderna, Vir Biotechnology, and Emergent BioSolutions.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108962.
REFERENCES
- Azevedo RS, Silva EV, Carvalho VL, Rodrigues SG, Nunes-Neto JP, Monteiro H, Peixoto VS, Chiang JO, Nunes MR, and Vasconcelos PF (2009). Mayaro fever virus, Brazilian Amazon. Emerg. Infect. Dis 15, 1830–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S, DiLillo DJ, and Ravetch JV (2015). The role of Fc-FcγR interactions in IgG-mediated microbial neutralization. J. Exp. Med 212, 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S, Corti D, Virgin HW, and Ravetch JV (2020a). Fc-optimized antibodies elicit CD8 immunity to viral respiratory infection. Nature 588, 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S, Gupta A, and Ravetch JV (2020b). The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol 20, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt WE, McCown JM, Gentry MK, and Russell PK (1982). Infection enhancement of dengue type 2 virus in the U-937 human monocyte cell line by antibodies to flavivirus cross-reactive determinants. Infect. Immun 36, 1036–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleton M, Lee H, Mulvey M, and Brown DT (1997). Role of glycoprotein PE2 in formation and maturation of the Sindbis virus spike. J. Virol 71, 1558–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanha PMS, Nascimento EJM, Braga C, Cordeiro MT, de Carvalho OV, de Mendonça LR, Azevedo EAN, França RFO, Dhalia R, and Marques ETA (2017). Dengue Virus-Specific Antibodies Enhance Brazilian Zika Virus Infection. J. Infect. Dis 215, 781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causey OR, and Maroja OM (1957). Mayaro virus: a new human disease agent. III. Investigation of an epidemic of acute febrile illness on the river Guama in Pará, Brazil, and isolation of Mayaro virus as causative agent. Am. J. Trop. Med. Hyg 6, 1017–1023. [PubMed] [Google Scholar]
- Choi H, Kudchodkar SB, Reuschel EL, Asija K, Borole P, Ho M, Wojtak K, Reed C, Ramos S, Bopp NE, et al. (2019). Protective immunity by an engineered DNA vaccine for Mayaro virus. PLoS Negl. Trop. Dis 13, e0007042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, Sakuntabhai A, Cao-Lormeau V-M, Malasit P, Rey FA, et al. (2016). Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat. Immunol 17, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers G, Bentlage AEH, Stegmann TC, Howie HL, Lissenberg-Thunnissen S, Zimring J, Rispens T, and Vidarsson G (2017). Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs 9, 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnest JT, Basore K, Roy V, Bailey AL, Wang D, Alter G, Fremont DH, and Diamond MS (2019). Neutralizing antibodies against Mayaro virus require Fc effector functions for protective activity. J. Exp. Med 216, 2282–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MKS, Fong RH, Kahle KM, Smit JM, Jin J, Simmons G, et al. (2015). Broadly Neutralizing Alphavirus Antibodies Bind an Epitope on E2 and Inhibit Entry and Egress. Cell 163, 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JM, Roy V, Gunn BM, Huang L, Edeling MA, Mack M, Fremont DH, Doranz BJ, Johnson S, Alter G, and Diamond MS (2019). Optimal therapeutic activity of monoclonal antibodies against chikungunya virus requires Fc-FcγR interaction on monocytes. Sci. Immunol 4, eaav5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman MJ, Caine EA, Zaitsev K, Begley MC, Weger-Lucarelli J, Uccellini MB, Tripathi S, Morrison J, Yount BL, Dinnon KH III., et al. (2018). An Immunocompetent Mouse Model of Zika Virus Infection. Cell Host Microbe 23, 672–685.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halsey ES, Siles C, Guevara C, Vilcarromero S, Jhonston EJ, Ramal C, Aguilar PV, and Ampuero JS (2013). Mayaro virus infection, Amazon Basin region, Peru, 2010–2013. Emerg. Infect. Dis 19, 1839–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead SB (1988). Pathogenesis of dengue: challenges to molecular biology. Science 239, 476–481. [DOI] [PubMed] [Google Scholar]
- Halstead SB, Porterfield JS, and O’Rourke EJ (1980). Enhancement of dengue virus infection in monocytes by flavivirus antisera. Am. J. Trop. Med. Hyg 29, 638–642. [DOI] [PubMed] [Google Scholar]
- Halstead SB, Mahalingam S, Marovich MA, Ubol S, and Mosser DM (2010). Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect. Dis 10, 712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidner HW, Knott TA, and Johnston RE (1996). Differential processing of sindbis virus glycoprotein PE2 in cultured vertebrate and arthropod cells. J. Virol 70, 2069–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her Z, Malleret B, Chan M, Ong EKS, Wong S-C, Kwek DJC, Tolou H, Lin RTP, Tambyah PA, Rénia L, and Ng LF (2010). Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J. Immunol 184, 5903–5913. [DOI] [PubMed] [Google Scholar]
- Ho IY, Bunker JJ, Erickson SA, Neu KE, Huang M, Cortese M, Pulendran B, and Wilson PC (2016). Refined protocol for generating monoclonal antibodies from single human and murine B cells. J. Immunol. Methods 438, 67–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hozé N, Salje H, Rousset D, Fritzell C, Vanhomwegen J, Bailly S, Najm M, Enfissi A, Manuguerra J-C, Flamand C, and Cauchemez S (2020). Reconstructing Mayaro virus circulation in French Guiana shows frequent spillovers. Nat. Commun 11, 2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, and Simmons G (2019). Antiviral Functions of Monoclonal Antibodies against Chikungunya Virus. Viruses 11, 305–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Liss NM, Chen DH, Liao M, Fox JM, Shimak RM, Fong RH, Chafets D, Bakkour S, Keating S, et al. (2015). Neutralizing Monoclonal Antibodies Block Chikungunya Virus Entry and Release by Targeting an Epitope Critical to Viral Pathogenesis. Cell Rep. 13, 2553–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Galaz-Montoya JG, Sherman MB, Sun SY, Goldsmith CS, O’Toole ET, Ackerman L, Carlson LA, Weaver SC, Chiu W, and Simmons G (2018). Neutralizing Antibodies Inhibit Chikungunya Virus Budding at the Plasma Membrane. Cell Host Microbe 24, 417–428.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Fong RH, Austin SK, Chen Z, Klose T, Fokine A, Liu Y, Porta J, Sapparapu G, Akahata W, et al. (2015). Cryo-EM structures elucidate neutralizing mechanisms of anti-chikungunya human monoclonal antibodies with therapeutic activity. Proc. Natl. Acad. Sci. USA 112, 13898–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LL, Suscovich TJ, Fortune SM, and Alter G (2018). Beyond binding: antibody effector functions in infectious diseases. Nat. Rev. Immunol 18, 46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum F-M, Couderc T, Chia B-S, Ong R-Y, Her Z, Chow A, Leo Y-S, Kam Y-W, Rénia L, Lecuit M, and Ng LFP (2018). Antibody-mediated enhancement aggravates chikungunya virus infection and disease severity. Sci. Rep 8, 1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Kim AS, Kafai NM, Earnest JT, Shah AP, Case JB, Basore K, Gilliland TC, Sun C, Nelson CA, et al. (2020). LDLRAD3 is a receptor for Venezuelan equine encephalitis virus. Nature 588, 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachý J, Brühl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, and Schlöndorff D (2001). Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J. Immunol 166, 4697–4704. [DOI] [PubMed] [Google Scholar]
- Mancardi DA, Iannascoli B, Hoos S, England P, Daëron M, and Bruhns P (2008). FcgammaRIV is a mouse IgE receptor that resembles macrophage FcepsilonRI in humans and promotes IgE-induced lung inflammation. J. Clin. Invest 118, 3738–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins KA, Gregory MK, Valdez SM, Sprague TR, Encinales L, Pacheco N, Cure C, Porras-Ramirez A, Rico-Mendoza A, Chang A, et al. (2019). Neutralizing Antibodies from Convalescent Chikungunya Virus Patients Can Cross-Neutralize Mayaro and Una Viruses. Am. J. Trop. Med. Hyg 100, 1541–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CA, Lee CA, and Fremont DH (2014). Oxidative refolding from inclusion bodies. Methods Mol. Biol 1140, 145–157. [DOI] [PubMed] [Google Scholar]
- Oliphant T, Nybakken GE, Engle M, Xu Q, Nelson CA, Sukupolvi-Petty S, Marri A, Lachmi B-E, Olshevsky U, Fremont DH, et al. (2006). Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J. Virol 80, 12149–12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MK, et al. (2013). Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 9, e1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro FP, Freitas RB, Travassos da Rosa JF, Gabbay YB, Mello WA, and LeDuc JW (1981). An outbreak of Mayaro virus disease in Belterra, Brazil. I. Clinical and virological findings. Am. J. Trop. Med. Hyg 30, 674–681. [DOI] [PubMed] [Google Scholar]
- Platzer B, Stout M, and Fiebiger E (2014). Antigen cross-presentation of immune complexes. Front. Immunol 5, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell LA, Miller A, Fox JM, Kose N, Klose T, Kim AS, Bombardi R, Tennekoon RN, Dharshan de Silva A, Carnahan RH, et al. (2020). Human mAbs Broadly Protect against Arthritogenic Alphaviruses by Recognizing Conserved Elements of the Mxra8 Receptor-Binding Site. Cell Host Microbe 28, 699–711.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Flanagan A, Dejnirattisai W, Puttikhunt C, Kasinrerk W, Supasa P, Wongwiwat W, Chawansuntati K, Duangchinda T, Cowper A, et al. (2018). Characterization of a potent and highly unusual minimally enhancing antibody directed against dengue virus. Nat. Immunol 19, 1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FA, Stiasny K, Vaney MC, Dellarole M, and Heinz FX (2018). The bright and the dark side of human antibody responses to flaviviruses: lessons for vaccine design. EMBO Rep. 19, 206–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp JC, Sokoloski KJ, Gebhart NN, and Hardy RW (2015). Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol 96, 2483–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan KC, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, and Schreiber RD (2006). Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res 26, 804–819. [DOI] [PubMed] [Google Scholar]
- Smith SA, Silva LA, Fox JM, Flyak AI, Kose N, Sapparapu G, Khomandiak S, Ashbrook AW, Kahle KM, Fong RH, et al. (2015). Isolation and Characterization of Broad and Ultrapotent Human Monoclonal Antibodies with Therapeutic Activity against Chikungunya Virus. Cell Host Microbe 18, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao MH, and Morrison SL (1989). Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol 143, 2595–2601. [PubMed] [Google Scholar]
- Uchime O, Fields W, and Kielian M (2013). The role of E3 in pH protection during alphavirus assembly and exit. J. Virol 87, 10255–10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner JJ, Rojas A, Mohamed-Hadley A, de Guillen YA, and Pinsky BA (2018). Real-time RT-PCR for Mayaro virus detection in plasma and urine. J. Clin. Virol 98, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise WJ, Hermance ME, Forrester N, Adams AP, Langsjoen R, Gorchakov R, Wang E, Alcorn MDH, Tsetsarkin K, and Weaver SC (2014). A novel live-attenuated vaccine candidate for mayaro Fever. PLoS Negl. Trop. Dis 8, e2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Xiong S, Malvin NP, Khoury-Hanold W, Heuckeroth RO, Stappenbeck TS, and Diamond MS (2018). Intestinal Dysmotility Syndromes following Systemic Infection by Flaviviruses. Cell 175, 1198–1212.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler ES, Shrihari S, Hykes BL Jr., Handley SA, Andhey PS, Huang YS, Swain A, Droit L, Chebrolu KK, Mack M, et al. (2020). The Intestinal Microbiome Restricts Alphavirus Infection and Dissemination through a Bile Acid-Type I IFN Signaling Axis. Cell 182, 901–918.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap ML, Klose T, Urakami A, Hasan SS, Akahata W, and Rossmann MG (2017). Structural studies of Chikungunya virus maturation. Proc. Natl. Acad. Sci. USA 114, 13703–13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, Poddar S, et al. (2018). Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou QF, Fox JM, Earnest JT, Ng T-S, Kim AS, Fibriansah G, Kostyuchenko VA, Shi J, Shu B, Diamond MS, and Lok SM (2020). Structural basis of Chikungunya virus inhibition by monoclonal antibodies. Proc. Natl. Acad. Sci. USA 117, 27637–27645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and are available from the corresponding author upon request.