

Abstract
Enterovirus D68 (EV-D68) is an RNA virus that causes respiratory illnesses mainly in children. In severe cases, it can lead to neurological complications such as acute flaccid myelitis (AFM). EV-D68 belongs to the enterovirus genera of the Picornaviridae family, which also includes many other significant human pathogens such as poliovirus, enterovirus A71 and rhinovirus. There are currently no vaccines or antivirals against EV-D68. In this review, we present the current understanding of the link between EV-D68 and AFM, the mechanism of viral replication, and recent progress in developing EV-D68 antivirals by targeting various viral proteins and host factors that are essential for viral replication. The future directions of EV-D68 antiviral drug discovery and the criteria for drugs to reach clinical trials are also discussed.
Keywords: enterovirus D68, acute flaccid myelitis, antiviral
Graphical Abstract

Enterovirus D68 (EV-D68) is an RNA virus that mainly causes respiratory illnesses in children, but in rare cases also leads to neurological complications such as acute flaccid myelitis (AFM). There are currently no vaccines or antivirals against EV-D68. In this review, we discuss recent progress in developing EV-D68 antivirals by targeting different stages of viral replication cycle. In addition, we provide insights regarding future directions of EV-D68 antiviral drug discovery and the criteria for drugs to reach clinical trials.
Enteroviruses include poliovirus and non-polio enteroviruses such as enterovirus A71 (EV-A71), enterovirus D68 (EV-D68), coxsackievirus, echovirus, and rhinovirus. They are non-enveloped, single-stranded, positive-sense RNA viruses that belong to the enterovirus genera of the Picornaviridae family. EV-D68 mainly infects children and causes flu-like symptoms. EV-D68 has two unique features that distinguish itself from other enteroviruses. First, instead of transmitting through the fecal-oral route like many other enteroviruses (poliovirus, echovirus, coxsackievirus), EV-D68 mainly transmits through airways. The molecular explanation underlying this phenotype is that EV-D68 is acid labile, therefore it cannot survive in the acidic environment of the gastrointestinal tract. It has been shown that low pH can trigger uncoating of viral capsids of EV-D68.1 Second, EV-D68 prefers lower growth temperature at 33°C, while other enteroviruses such as echovirus and coxsackievirus prefer 37°C. Overall, EV-D68 is more closely related with rhinovirus. Historically, EV-D68 was misclassified as rhinovirus 87 prior to genetic sequencing.
EV-D68 emerged as public health threat when its first outbreak in 2014 coincided with increasing cases of acute flaccid myelitis (AFM), a neurological complication that resembles poliovirus infection. AFM is defined as acute onset of paralysis, accompanied by lesions in the grey matter of the spinal cord shown by magnetic resonance imaging. Although AFM is a severe disease, it remains rare. Majority of EV-D68 infection only leads to mild respiratory illness, and a small fraction will progress to AFM.2 Poliovirus is a well-known cause of AFM and is associated with poliomyelitis. A number of non-polio viruses have also been reported to be associated with AFM including West Nile virus, Japanese encephalitis virus, adenovirus, and echovirus.3
EV-D68 and AFM
Poliomyelitis was nearly eliminated through vaccination with only limited cases reported in three countries – Pakistan, Afghanistan, and Nigeria. However, neurological infections caused by non-polio enteroviruses, including EV-D68 and EV-A71 haven risen continuously in recent years. EV-D68 was first isolated in 1962 and rarely caused any significant problems until the first outbreak in 2014. The rise in AFM cases led to the speculation that contemporary EV-D68 viruses might have evolved to become more pathogenic than historic viruses such as the Fermon strain isolated in 1962. This is not surprising as EV-D68 and poliovirus actually belong to the same genera of enterovirus family. Indeed, sequencing of the contemporary EV-D68 viruses revealed 12 substitutions in both the coding and non-coding regions that showed similar sequences identity with neurotropic poliovirus and EV-A71,4 suggesting EV-D68 viruses have evolved overtime to become neurotropic. To fulfill Koch’s postulations that EV-D68 is the causative agent of AFM, Tyler et al. infected Swiss Webster (SW) mice with several EV-D68 isolates from the 2014 U.S. outbreak and were able to reproduce flaccid paralysis symptoms.5 In a follow-up study, they further showed that contemporary EV-D68 strains isolated during the 2014 outbreak such as US/MO/47 were able to infect and replicate in neuronal cell lines such as SH-SY5Y as well as primary human neuron cultures.6 In contrast, EV-D68 strains isolated prior to the 2014 outbreak such as VR1197 (similar to the prototypical Fermon strain isolated in 1962) and US/TN (isolated in 2012), failed to grow in these neuronal cells. Furthermore, among the three viruses tested, only contemporary EV-D68 strain US/MO/47 was able to cause paralysis in neonatal mice following intramuscular injection as shown by the lack of movement in one or more limbs. Increasing viral titers were also detected in the spinal cord and muscle after US/MO/47 infection. In addition, mouse models developed by several other groups were also able to reproduce the paralysis symptoms when mice were infected with contemporary EV-D68 viruses.7, 8 Other than the neurological mouse model, the respiratory mouse model for EV-D68 was also reported.9 More information on the development of mouse models of EV-D68 infection can be found in recent reviews.10–12
In contrast to the conclusion that neurotropism is only a recent acquired phenotype, Rosenfeld et al. recently reported that both historic and contemporary EV-D68 isolates were able to replicate in organotypic mouse brain slice cultures as well as induced human astrocytes.13 All viruses except the 952 isolate also replicated in neurons derived from human induced pluripotent stem cells. There appears to be no clear difference between historic strains and contemporary strains in terms of their ability to infect neuronal cells. Thus, they concluded that neurotropism is not a recently acquired phenotype. The controversy raised by these two studies might have resulted from different cell types used for viral infection: Brown et al. used human neuroblastoma-derived neuronal cell line SH-SY5Y as a cell culture model, which Rosenfeld et al. argued is not suitable for studying neurotropism as this cell line might lose the neuronal characteristics upon passage. Instead, Rosenfeld et al. chose stem cell induced neurons, including cortical neurons and astrocytes, as well as mice organotypic brain slice cultures, all of which have been widely used to study other neurotropic viruses such as ZIKA virus and measles virus.14 However, in Brown et al.’s study, aside from SH-SY5Y cells, they also showed that only contemporary EV-D68 strain US/MO/47, and not the historic strains US/TN and VR1197, was able to replicate in primary human fetal brain-derived neurons,6 raising further questions regarding the neurotropism.
The mechanism by which EV-D68 leads to neurological complications in humans is still under investigation. Clinically, EV-D68 virus was rarely detected in the patients’ blood, cerebrospinal spinal fluid, or spinal cord samples.15 Only one sample of cerebrospinal fluid from AFM patients tested positive for EV-D68 in 2018.15, 16 Absence of viral RNA in AFM might be due to the timing of sampling or the extremely low levels of viral genome in the CSF. This raised the questions of how viruses spread in the body and whether viruses penetrate blood-brain barrier or blood-spinal cord barrier. To address this question, a recent study took an alternative approach by identifying EV-D68 antibodies in the CSF samples from AFM patients instead of detecting viral genome directly.17 Specifically, peptide microarrays consisting of 160,000 unique peptide fragments from the capsid proteins of enteroviruses were used to isolate binding antibodies from the CSF samples. Encouragingly, antibodies reacting specifically to a 22 amino acid peptide fragment derived from the EV-D68 VP1 capsid protein were identified in 6 out of 11 CSF samples and in 8 out of 11 serum samples. In contrast, no reactive antibody against this peptide was detected in 26 control samples. Although the authors could not completely rule out the possibility of the CSF antibodies originating from the blood, this finding strongly suggests the presence of virus in the CSF at some point during viral infection. Another independent study using a similar strategy came up with a similar conclusion stating that there is a plausible correlation between EV-D68 infection and AFM.16
EV-D68 REPLICATION CYCLE
EV-D68 virion contains a single positive-sense RNA genome surrounded by an icosahedral capsid made of viral capsid proteins VP1-VP4 with VP1-VP3 displayed on the surface and VP4 sitting underneath.18 It is among the smallest viruses with a diameter of ~ 30 nm. Each virion contains 60 copies of the capsid proteins VP1-VP4. Inside the virion is a single-stranded, positive-sense viral RNA that is approximately 7400 bases long. The viral RNA contains three segments: a 5’ untranslated region (UTR), a single open reading frame (ORF), and a 3’-UTR. Upon binding to host cell receptors (sialic acid, ICAM-5, or others), virion enters cell through endocytosis (Figure 1).19, 20 The low pH in the endosome triggers viral uncoating and releasing of viral RNA.1 The host cell ribosome then binds to an internal ribosomal entry site (IRES) within the 5’-UTR and initiates translation. Similar to HIV and HCV viruses, the protein-coding region within the ORF is first translated into a single viral polypeptide in which all viral proteins are linked together. Next, two viral proteases, 2A and 3C, that are within this polypeptide self-cleave the polypeptide into individual viral proteins – four capsid proteins VP1-VP4 and seven non-structural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D). Viral genome replication starts with transcription of the genomic (+)-RNA to the (−)-RNA by the viral RNA-dependent RNA polymerase (3Dpol). The (−)-RNA then serves as a template for the synthesis of new (+)-RNA. Replication of enteroviruses typically involves membranous replication organelles, and several viral proteins (2C and 3A) and host factors (ACBD3, PI4KB and OSBP) are involved in this membrane remodeling process.21, 22 Finally, capsid proteins, together with viral genomic RNA, self-assemble and exit the host cell as progeny virions either through exocytosis or lytic release.23
Figure 1.

Viral replication cycle of EV-D68. EV-D68 enters a cell via receptor-mediated endocytosis. The low pH in the endosome triggers uncoating of the viral capsid proteins and the releasing of the viral RNA. The (+)-RNA is translated into a single viral polypeptide, which is subsequently cleaved by two viral proteases 2A and 3C into individual viral proteins. Next, viral 3D polymerase mediates the synthesis of (+)-RNA via (−)-RNA in the replication organelle. Finally, the progeny virion is released from the infected cell.
ANTIVIRAL DRUG DISCOVERY STRATEGIES
Theoretically, compounds targeting any step during the viral replication process could be developed as antivirals. The compounds can act on either viral proteins or host factors that are essential for viral replication. It is generally more feasible to develop direct-acting antivirals than host-targeting antivirals due to off-target issues. In reality, maraviroc, an HIV CCR5 co-receptor antagonist, is the only specifically designed host-target antiviral.24 Other known host-targeting antivirals include interferons and ribavirin; however their mechanism of action is not well understood and multiple mechanisms of action might be involved.
The goal of this review is to summarize the recent progress and discuss the future directions in developing EV-D68 antivirals. We will mainly focus on drug candidates that have a defined mechanism of action. The following section is organized based on individual drug targets.
There are three general approaches used to discover antivirals for EV-D68. The first approach is to test EV-D68 against existing antivirals that have antiviral activity against similar viruses such as poliovirus, EV-A71, CVB3, or rhinovirus. The rationale is that viruses in the same family generally share a certain degree of sequence similarity; therefore, antivirals targeting conserved viral proteins might have broad-spectrum antiviral activity against viruses in the same family. Examples of compounds identified through this approach include pleconaril and rupintrivir. The second approach involves screening of chemical libraries against EV-D68 in phenotypic viral cytopathic effect assay. The mechanisms of action by the positive hits are then elucidated through either resistance selection or various cellular and biochemical assays. The third approach involves target-based design and screening of inhibitors targeting certain viral proteins from EV-D68.
CAPSID INHIBITORS (VP1-VP4)
The VP1, VP2, and VP3 subunits from EV-D68 assemble and form an icosahedral shell with VP4 located in the inner surface of the viral capsid (Figure 2A). Pleconaril (1) was originally developed as a capsid inhibitor for rhinovirus, and it was later found to be active against EV-D68 (Figure 2B). Pleconaril (1) failed in the clinical trial for the treatment of rhinovirus-induced common colds due to a lack of efficacy.25 The X-ray crystal structure of EV-D68 virion in complex with pleconaril (1) was solved at 2.32 Å resolution (PDB: 4WM7) (Figure 2C).18 Pleconaril (1) fits in a hydrophobic drug-binding pocket in the canyon region located at the VP1 protein and protects the virion against thermal inactivation. It inhibits EV-D68 replication in cell culture with single-digit to submicromolar potency as shown by multiple studies (Table 1),26–28 and it is often used as a positive control compound in antiviral assays as well as in mechanistic studies. Pleconaril (1) is active against multiple EV-D68 strains tested. However, it was not active against the close-related EV-A71 virus.28, 29 Resistance to pleconaril (1) was mapped to VP1-V81A (V69A in the 4WM7 numbering), a residue that locates right above the benzene ring of pleconaril (1). Mutating a bulky valine to a smaller alanine is likely to diminish the hydrophobic interactions between VP1 and pleconaril (1), leading to drug resistance. However, the fitness of the VP1-V69A mutant virus was not studied, and it is not clear whether this drug resistant mutation might be a potential concern or not. Other capsid inhibitors tested against EV-D68 include vapendavir (2), pirodavir (3), and pocapavir (4) (Figure 2B), all of which were originally designed as rhinovirus antivirals. The antiviral potency of vapendavir (2) against EV-D68 was not conclusive. In one study, vapendavir inhibited multiple EV-D68 strains but only one with single to submicromolar potency,26and in another study, it was not active against the four EV-D68 strains tested (EC50 > 10 μM) (Table 1).27 Pirodavir (3) was less active against EV-D68 and had a low selectivity index, and it was not active against the historic strain rhinovirus 87 (EC50 > 19 μM). Pocapavir (4) was not active against EV-D68.27 No mechanistic studies have been performed to elucidate the mechanism of action of these capsid inhibitors, but they presumably bind to the same binding pocket as pleconaril (1). From these drug-repurposing results, it can be concluded that although the capsid proteins from EV-D68 share many similarities with other enterovirus such as EV-A71, rhinovirus and poliovirus, not all previously discovered enterovirus capsid inhibitors are active against EV-D68. Main reason for this is due to the VP1 sequence divergence in the capsid inhibitor drug-binding site.18
Figure 2.
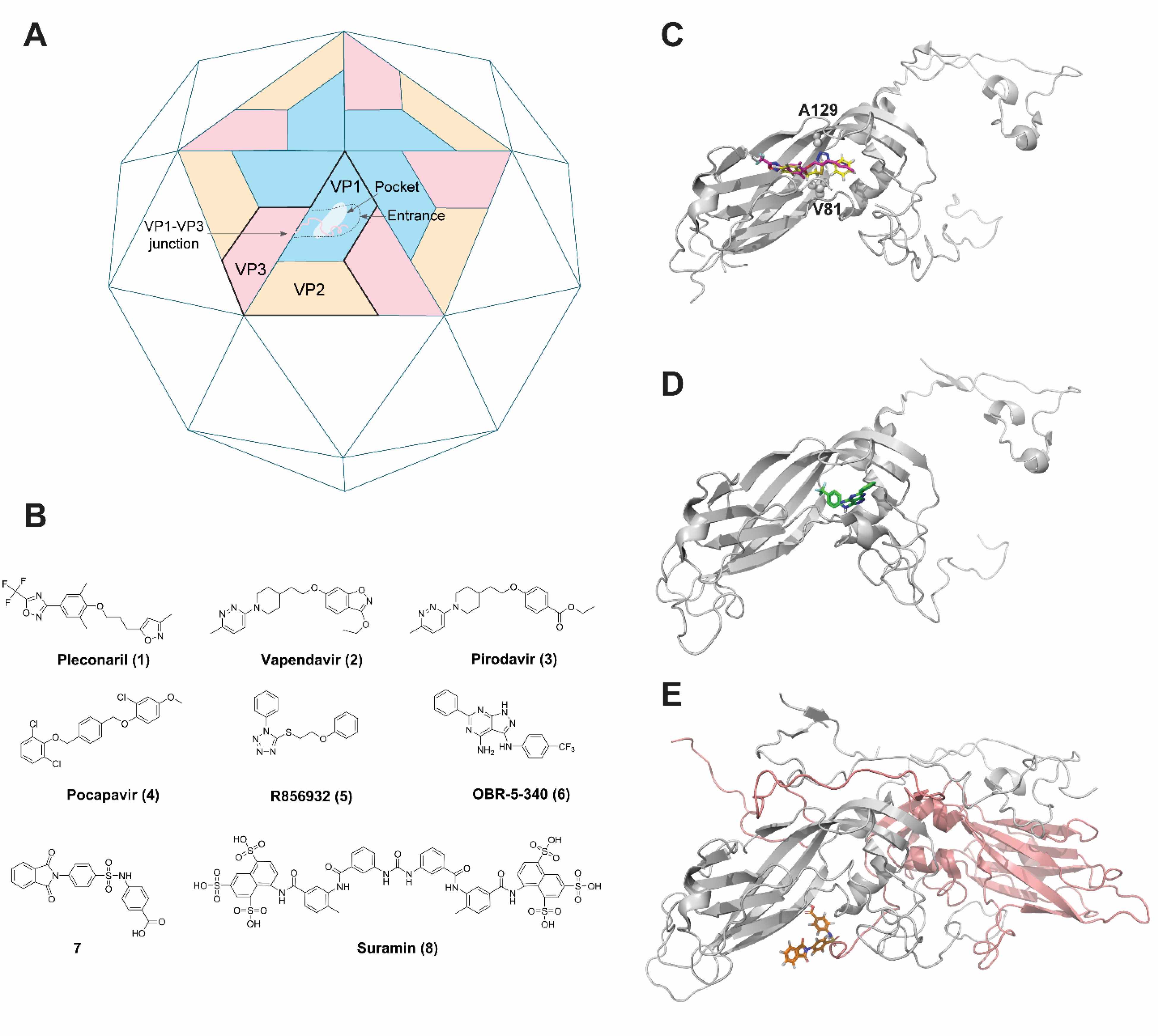
EV-D68 capsid proteins and capsid inhibitors. (A). Cartoon representation of the EV-D68 capsid shell and three different drug-binding sites. (B). Structures of capsid inhibitors. Note that not all listed compounds are active against EV-D68. (C). X-ray crystal structure of EV-D68 VP1 in complex with pleconaril (1) (PDB: 4WM7). R856932 (5) binds to the same pocket and the docking pose was overlaid with pleconaril (1). (D). Cryo-electron microscopy (Cryo-EM) structure of RV-B5 VP1 in complex with OBR-5–340 (6) (PDB: 6SK5). (E). Cryo-EM structure of CV-B3 VP1-VP3 in complex with compound 7. VP1 and VP3 are colored in grey and orange, respectively.
Table 1.
Antiviral activity of selected compounds on the in vitro replication of EV-D68 viruses
| Inhibitor | EV-D68 potency | Inhibitor | EV-D68 potency |
|---|---|---|---|
| Capsid and entry inhibitors | 2A protease inhibitors | ||
| Pleconaril (1) | EC50 = 0.081 to 0.83 μM26 EC50 = 0.38 to 6.11 μM27 EC50 = 0.13 μM28 |
Telaprevir (9) | EC50 = 0.4 to 1.9 μM40 |
| Vapendavir (2) | EC50 = 2.4 to 9.6 μM26 EC50 = 7 to 13 μM28 |
3C protease inhibitors | |
| Pirodavir (3) | EC50 = 2.4 to 9.6 μM26 EC50 = 7 to 13 μM28 |
Rupintrivir (10) | EC50 = 0.0019 to 0.0030 μM26 EC50 = 0.0015 to 0.0046 μM27 EC50 = 0.0022 to 0.0046 μM28 |
| Pocapavir (4) | EC50 > 10 μM27 | AG7404 (11) | EC50 = 0.0035 to 0.027 μM27 |
| R856932 (5) | EC50 = 0.46 to 4.39 μM29 | 15 | EC50 = 0.04 μM41 |
| OBR-5–340 (6) | Not tested38 | IRES inhibitors | |
| 7 | EC50 > 296 μM39 | Emetine (27) | EC50 = 0.0187 μM42 |
| DAS181 | EC50 = 0.0012 to 0.114 μM27 | 3D polymerase inhibitors | |
| Suramin (8) | No inhibition43 | FNC (34) | EC50 = 1.548 nM44 |
| 2C inhibitors | GPC-N114 (38) | EC50 = 1.44 μM45 | |
| Guanidine (16) | EC50 = 80 to 91 μM28 EC50 = 219.4 μM46 |
Host-targeting antivirals | |
| Fluoxetine (17) | (s)-fluoxetine EC50 = 0.67 μM (r)-fluoxetine Not active47 |
Enviroxime (40) | EC50 = 0.19 to 0.45 μM26 EC50 = 0.27 to 0.31 μM28 |
| Pirlindole mesylate (18) | EC50 = 9.24 μM48 EC50 = 8.4 μM46 |
Itraconazole (41) | EC50 = 0.32 μM49 EC50 > 10 μM27 |
| Formoterol (19) | EC50 > 10 μM27 EC50 = 0.51 μM48 EC50 = 24.7 μM46 |
||
| Zuclopenthixol (20) | Not tested | ||
| Dibucaine (21) | EC50 = 3.03 μM48 EC50 = 5.3 μM46 |
||
When evaluated against EV-D68 in both the respiratory model and the neurological model of infection in AG129 mice, pleconaril (1) and pirodavir (3) had no effect in reducing lung virus titers and improving survival.7 Although this result was somewhat disappointing, it should be noted that the mouse of EV-D68 infection was still in its early development stage, and the lack of in vivo efficacy in this study does not imply EV-D68 capsid inhibitors should be terminated. It is reasonable to speculate that the failure was not due to the in vivo pharmacokinetic properties of pleconaril (1), as this compound was active in the in vivo mouse model of rhinovirus infection as well as coxsackievirus infection.30 It was also evaluated in human clinical trials for common colds.31, 32 The in vivo antiviral efficacy of pleconaril (1) deserved to be re-tested in other relevant EV-D68 mouse models when possible, and the drug formulation, dose, and dosing frequency all need to be systematically evaluated.
A recent study from our group identified a tetrazole compound R856932 (5) that inhibited multiple EV-D68 strains with single-digit to submicromolar potency.29 Mechanistic studies showed that R856932 (5) had an overlapping drug-binding site as pleconaril (Figure 2C). A mutation VP1-A129V led to drug resistance against R856932 (5). However, VP1-A129V mutant virus was shown to have reduced fitness of replication compared to wild-type (WT) EV-D68 as shown by the competition growth assay. Therefore, R856932 (5) might represent a novel capsid inhibitor for further development as an EV-D68 antiviral.
For review of capsid inhibitors targeting enteroviruses in general, please refer to recent reviews.33, 34 A number of capsid inhibitors have been reported to inhibit EV-A71, CVB3 or poliovirus in either in vitro cell culture model or in vivo mouse model studies.35–37 However, their antiviral activity has not been evaluated against EV-D68.
Overall, existing results suggest VP1 is a validated antiviral drug target for EV-D68. Other than the hydrophobic pleconaril drug-binding pocket, there are two additional drug-binding pockets that might be worth pursuing. One is located at the entrance of the hydrophobic canyon pocket of VP1 (Figures 2A & 2D). A recent study from Wald et al suggested that designing inhibitors targeting this entrance region might be feasible.38 Cryo-EM structure showed that a pyrazolopyrimidine compound OBR-5–340 (6) was located at the entrance of the VP1 capsid protein of rhinovirus-B5 (RV-B5). OBR-5–340 (6) potently inhibited RV-B5 and did not show cross-resistance with pleconaril (1) since it did not have an overlapping binding site with pleconaril (1). However, it is yet unknown whether EV-D68 is sensitive towards OBR-5–340 (6). Another potential capsid drug-binding pocket is located at the VP1-VP3 junction, and this pocket appears to be conserved among coxsackieviruses (Figure 2E).39 However, when tested against EV-D68, the most active compound 7 (numbered 17 in the original paper) did not show inhibition. Nevertheless, it is reasonable to speculate that the same drug-binding pocket located at the VP1-VP3 junction in EV-D68 might be similarly targeted by small molecules. Since the high-resolution structure of EV-D68 capsid protein is available (PDB: 4WM7), both rational design and high-throughput screening can be applied in searching for EV-D68 capsid inhibitors with a novel mechanism of action. Of particular interest are the capsid inhibitors that have a novel mechanism of action and a high genetic barrier to drug resistance. Although capsid inhibitors are unlikely to be used as monotherapy, due to high mutation rates, they are nevertheless valuable candidates for combination therapy.
INHIBITORS TARGETING/MIMICKING THE CELL SURFACE SIALIC ACID RECEPTORS
Several studies have shown that the EV-D68 capsid proteins bind to the sialic acid receptor on the host cell surface, suggesting that EV-D68 uses sialic acid-containing proteins/lipids for cell entry.50 The X-ray crystal structure of sialic acid bound to EV-D68 further strengthened this claim.20 DAS181, a sialidase enzyme that cleaves sialic acid, inhibited the replication of both historic and contemporary EV-D68 strains in RD cells with nanomolar potency.27 However, a recent study showed that the interaction between sialic acid and EV-D68 capsid protein is both strain and cell type specific, and the neurotropism is independent of the sialic acid receptor.13 In other words, the in vitro antiviral activity of DAS181 against EV-D68 in RD cells might be cell-type dependent. Therefore, entry inhibitors designed as decoy receptors should be evaluated against multiple EV-D68 strains in multiple cell lines to ensure their physiological relevance.51 Indeed, suramin (8) (Figure 2B), a naphthalenetrisulfonic acid compound, binds to the positively charged region in the 5-fold axis of the EV-A71 capsid proteins and showed potent antiviral activity against EV-A71 and several coxsackievirus, but not against the closely related polioviruses and EV-D68.43 However, suramin is a well-known promiscuous compound that may represent a Pan-Assay Interference compound (PAIN),52 therefore lacking translational potential.
It has also been shown that EV-D68 virus can enter cell through sialic acid-independent mechanism via binging to cellular receptors such as intercellular adhesion molecule 5 (ICAM-5/telencephalin).19, 50 Soluble ICAM-5 fragments inhibited EV-D68 virus replication in multiple permissive cells. However, no small molecule has been reported to block ICAM-5-depenent EV-D68 entry. Interestingly, sialic acid-independent EV-D68 strains also bind to sialic acid-containing human erythrocytes, suggesting these viruses might use multiple receptors for cell entry.50
2A PROTEASE INHIBITORS
EV-D68 2A protease has long been speculated as a viral cysteine protease based on sequence alignment. However, its protease activity had not been demonstrated in vitro until recently.40 It turned out that the 2A protein itself could not be efficiently expressed in E. coli. Nevertheless, our group was able to express this protein with a cleavable SUMO tag. The protease activity of the tag-free 2A protein was shown by the fluorescence resonance energy transfer (FRET) assay. 2A protease specifically cleaved a peptide substrate corresponding to the VP1–2A junction, but not the 3C protease substrate. Subsequently, a FRET-based high-throughput screening was applied to identify 2A protease inhibitors. Telaprevir (9) (Figure 3A), an FDA-approved HCV NS3/NS4A serine protease inhibitor, inhibited EV-D68 2A protease with an IC50 value of 0.2 μM. To profile the specificity, telaprevir (1) was tested against two closely related viral cysteine proteases, the EV-D68 3C protease and the EV-A71 2A protease. It was found that telaprevir did not show comparable inhibition against these two proteases, suggesting that the inhibition of EV-D68 2A protease by telaprevir (9) is specific. Enzyme kinetic studies suggest that telaprevir (9) forms a nearly irreversible complex with EV-D68 2Apro, and this conclusion was further supported by the dialysis experiment. In cell culture, telaprevir (9) inhibited multiple contemporary EV-D68 strains with single-digit to submicromolar potency in multiple cell lines, including RD, A549, HEK293, and neuronal cells A172. Serial viral passage experiment identified one mutation— 2A-N84T that confers resistance in both the enzymatic assay and the cellular antiviral assay. In thermal shift assay, telaprevir binds and stabilizes the WT 2Apro, but not the 2A-N84T mutant protein. In the docking model, telaprevir (9) was positioned at the active site with its ketone in close proximity with the catalytic cysteine (Figure 3C). Collectively, this result showed that EV-D68 2A is a validated antiviral drug target, and that telaprevir (9) is a specific EV-D68 2A protease inhibitor.
Figure 3.

EV-D68 2A and 3C protease inhibitors and their mechanism of action. (A). Structures of enterovirus 2A and 3C protease inhibitors. Note that not all compounds have been tested against EV-D68 2A and 3C protease. (B). Mechanisms of action of compounds 13 and GC376 (14). (C). Docking model of telaprevir (9) with EV-D68 2A protease. (D). X-ray crystal structure of rupintrivir (10) in complex with EV-A71 3C protease (PDB: 3SJO).
Although the discovery of telaprevir (9) as a potent antiviral for EV-D68 is encouraging, caution should be taken in repurposing telaprevir (9) as an antiviral for EV-D68. Telaprevir (9) is no longer on the market as a HCV drug because newer generations of HCV NS3/NS4A protease inhibitors were introduced, and Vertex announced the discontinuation of telaprevir (9). Telaprevir (9) also carries a black box warning of potential severe skin reactions, which makes it undesired for pediatric patients. As telaprevir (9) was not specifically developed for EV-D68 2Apro, it is expected that re-designing telaprevir (9) through either structure-based approach or structure-activity relationship studies might further improve its potency and selectivity index against EV-D68 2Apro. In this regard, solving the high-resolution structure of the 2A protease will help facilitate this process. As for now, only the EV-A71 2Apro is available,53 and the EV-D68 2Apro has not been solved yet. In addition, large-scale high-throughput screening might identify novel 2A protease inhibitors for further development.
3C PROTEASE INHIBITORS
Viral 3C or 3C-like proteases (3Cpro and 3CLpro, respectively) have been extensively studied as antiviral drug targets for picornaviruses, noroviruses, and coronaviruses.54 The 3Cpro is more conserved than the 2Apro among various viral families. Therefore, viral 3Cpro and 3CLpro inhibitors are expected to have broad-spectrum antiviral activity against multiple viruses. The X-ray crystal structures of several viral 3Cpro and 3CLpro proteases including EV-D68 3Cpro and EV-A71 3Cpro with and without inhibitor bound have also been solved, paving the way for structure-based drug design.55 Viral 3Cpro and 3CLpro are cysteine proteases that adopt a typical chymotrypsin-like fold with a catalytic triad of Cys, His, and Glu (or Asp). One of the reasons that viral 3Cpro and 3CLpro are attractive antiviral drug targets is because these proteases prefer a glutamine residue at the P1 position on the substrate, a unique feature that is not shared among majority of the host proteases.56 Therefore, it is feasible to design highly selective and specific 3Cpro and 3CLpro inhibitors as antivirals without potential side effects. Among the reported viral 3Cpro and 3CLpro inhibitors, one of the most advanced compounds is rupintrivir (AG7088) (10) (Figure 3A), a peptide-mimetic with an α,β-unsaturated ester warhead. Another similar analog that was advanced to human clinical trial was AG7404 (11) (Figure 3A), which had improved oral bioavailability compared to rupintrivir (10). Rupintrivir (10) and AG7404 (11) act as irreversible inhibitors against viral 3Cpro and 3CLpro by forming a covalent bond with the active side cysteine (PDB: 3SJO) (Figure 3D).57 Rupintrivir (10) was originally developed as an antiviral against the human rhinovirus 3C protease, but dropped out of clinical trials due to a lack of efficacy in naturally acquired rhinovirus infections. Given the sequence similarities between the 3Cpro from rhinovirus and EV-D68, further studies showed that rupintrivir (10) was highly active against EV-D68 with nanomolar potency in cytopathic effect assay.27, 28 Several other viral 3Cpro and 3CLpro inhibitors have also been reported in literature showing varying degrees of potency in the enzymatic assay and cellular antiviral assay against EV-D6855, 58. The majority of them follow the similar design of rupintrivir (10) – a peptidomimetic sequence with a covalent warhead.41 Rupintrivir (10) was tested in the EV-D68 respiratory and neurological infection mouse models, but failed to show significant efficacy compared to the control group.7 In contrast, rupintrivir (10) was active in protecting suckling mice from EV-A71 infection induced limb paralysis.59 As rupintrivir (10) is well-tolerated and safe in humans,60 the lack of efficacy against EV-D68 might be due to the specific mouse model used, rather than the poor pharmacokinetic properties of the drug. Thus, research on viral 3Cpro and 3CLpro inhibitors should not be discouraged by this particular study.
A non-peptidomimetic small molecule DC07090 (12) (Figure 3A) was identified as a non-covalent inhibitor of EV-A71 3Cpro through virtual screening and showed moderate antiviral activity against EV-A71 (EC50 = 22.09 ± 1.07 μM).61 However, this compound has not been tested against EV-D68.
The majority of the cysteine protease inhibitors feature a peptide mimetic sequence and a reactive warhead, which are designed to mimic the substrate and covalently label the catalytic cysteine residue, respectively. Although many of the reported 3C inhibitors have potent enzymatic inhibition as well as cellular antiviral activity, very few of them have demonstrated in vivo antiviral efficacy in animal model studies. Up to now, the FDA has not approved a single cysteine protease inhibitor. One caveat of covalent inhibitors, especially 3C protease inhibitors, is their poor selectivity and pharmacokinetic properties. Reactive warheads such as aldehyde and α,β-unsaturated ester tend to react non-specifically with free thiols from glutathione or cysteine residues from host proteins or proteases, leading to reduced antiviral efficacy and toxic side effects.62 To balance the selectivity and potency of 3C protease inhibitors, novel warheads are explored. For example, compound 13 (numbered 30 in the original paper) featuring N-disubstituted cyanoacrylamide had not only potent enzymatic inhibition (Ki = 9.3 nM against EV-A71 3C) and cellular antiviral activity (EC50 = 51 nM against EV-A71),63 but also promising selectivity against three mammalian proteases, including the serine protease chymotrypsin and the two cysteine proteases cathepsin K and calpain-1. The proposed mechanism of action is that the α-proton in the enzyme-inhibitor adduct was sterically shielded from deprotonation, resulting in slow off-rate and long residence time (Figure 3B). Follow up studies should be planned to investigate whether the in vitro potency and selectivity of this series of compounds could be translated into in vivo antiviral activity and selectivity in animal models. Another 3C inhibitor worth highlighting is GC376 (14), which is being developed as a veterinary medicine to treat cats with feline infectious peritonitis (FIP). FIP virus is a coronavirus that similarly encodes a 3CLpro. GC376 (14) was designed to target the 3CLpro and had potent antiviral activity in cell culture against a panel of picornaviruses, noroviruses, and coronaviruses, including EV-A71 and rhinoviruses.54, 64 More importantly, cats infected with feline infectious peritonitis (FIP) showed full recovery after treatment with GC376 (14),65 suggesting that GC376 (14) is well tolerated and had favorable in vivo PK properties. GC376 (14) is the bisulfite adduct of the aldehyde and is slowly reverted to aldehyde in solution (Figure 3B). It acts as a reversible inhibitor of the 3C protease.66 GC376 (14) has not been tested against EV-D68, but if it is active in the cell culture, it will be a promising candidate for the in vivo mouse model study. A third class of promising 3C protease inhibitors are the ones containing α-ketoamide as a warhead such as compound 15 (numbered 10 in the original paper) (Figure 3A).41 Compound 15 inhibited EV-D68 with high potency in cell culture (EC50 = 0.04 μM). The translational potential of α-ketoamide is conferred by two FDA-approved drugs, telaprevir (9) and boceprevir. Therefore, continuous development of 3C inhibitors with α-ketoamide warhead will likely yield clinical candidates. Overall, a fine balance between the reactivity of the cysteine-reactive warhead and the substrate mimic sequence should be fine-tuned to achieve high potency and selectivity simultaneously. In addition, more efforts should focus on optimizing the in vitro and in vivo pharmacokinetic properties of these 3Cpro and 3CLpro inhibitors and advancing them to the in vivo mouse model study.
2C INHIBITORS
2C is approximately 328 amino acid protein with an N-terminal membrane-binding domain that spans residues 1–116, followed by an ATPase domain between 116 and 266 that resemble the AAA+ superfamily of ATPases. Residues that span amino acids 266 to 299 contain a zinc finger domain with three cysteine residues that coordinate the ion. The C-terminal domain forms a 29 amino-acid long α-helix that is suggested to play a role in forming 2C dimers or higher order oligomers, possibly hexamers.67, 68 The functions of the viral 2C protein are still under investigation and are not fully understood. The known functions of 2C protein include but are not limited to RNA binding and replication, membrane rearrangement, encapsidation of the viral genome, and progeny viral assembly.12, 22 The 2C protein was also reported to interact with multiple host proteins.69 Although the investigation of the precise roles of 2C protein in the viral life cycle is still ongoing, the multifunctional roles of 2C protein render it a prominent antiviral drug target. Structural diverse compounds have been reported as EV-D68 antivirals by targeting the viral 2C protein. Among all the 2C inhibitors, guanidine (16) and fluoxetine (17) were the most extensively studied ones (Figure 4). Guanidine (16) inhibited multiple EV-D68 strains in cell culture with close to 100 μM potency.28, 46 In addition, guanidine (16) also inhibited EV-A71 virus replication. The antiviral mechanism of action of guanidine (16) was studied using EV-A71 virus and mutations in 2C protein confer drug resistance.70 Surprisingly, despite the weak in vitro antiviral potency of guanidine (EC50 > 50 μM), it was the only small molecule that showed in vivo antiviral efficacy in the EV-D68 respiratory and neurological infection models.7 Another 2C inhibitor that moved to human clinical trials for EV-D68 infection is fluoxetine (Prozac) (17). Fluoxetine (17) is an FDA approved antidepressant drug; therefore it is well positioned for the treatment of EV-D68 induced AFM.71 Fluoxetine (17) inhibited multiple EV-D68 strains with single-digit or submicromolar potency in cell culture.27, 46, 72 In addition, it also inhibited CVB3, EV-A71, but was not active against poliovirus and rhinovirus.72, 73 The antiviral mechanism of action of fluoxetine (17) was studied using the CVB3 virus, and mutations located at the 2C protein (A224V, I227V, and A229V) conferred drug resistance. However, fluoxetine (17) did not show antiviral efficacy in the EV-D68 infection mouse models.7, 74 When tested in patients with AFM, fluoxetine (17) also failed to improve clinical symptoms, although it was well-tolerated.75 It was not clear the reasons behind the failure. Nevertheless, one immunocompromised pediatric patient with chronic enteroviral encephalitis did show stabilization and improvement following fluoxetine (17) treatment,76 emphasizing the need to continuously develop more potent and specific 2C inhibitors for the treatment of EV-D68 infection.
Figure 4.

Structures of enterovirus viral 2C inhibitors. Note that not all compounds have been tested against EV-D68.
Other 2C inhibitors discovered through drug repurposing approach that are active against EV-D68 in vitro include pirlindole mesylate (18), formoterol (19), zuclopenthixol (20), and dibucaine (21) with single-digit to double-digit micromolar potency (Figure 4).27, 46, 48 To be effective in clinic, the antiviral efficacy and selectivity index of these compounds need to be significantly improved. In this regard, a recent structure-activity relationship study based on dibucaine (21) led to the discovery of several quinoline analogs such as compound 22 with nanomolar potency and a high selectivity index (SI > 500).46 Other synthetic small molecules that were reported as enterovirus 2C inhibitors include TBZE-029 (23) 72, 77 HBB (24), MRL-1237 (25), and pyrazolopyridine (26) (Figure 4).78, 79 However their antiviral potency against EV-D68 has not been tested.
It remains intriguing that structural diverse compounds inhibit enterovirus replication by targeting the same viral 2C protein, albeit at different locations.67 Sequence alignment of various members of the enterovirus family showed ~60% amino acid sequence identity, suggesting EV-D68 2C protein might function similarly as other enteroviral 2C proteins.67, 68 The structure of EV-D68 2C protein has not been solved yet, but the existing drug resistance mutations, coupled with the X-ray crystal structures of the 2C proteins from poliovirus 67 and EV-A7168, might offer insights regarding the mechanism of action of EV-D68 2C inhibitors.
The pharmacology of 2C inhibitors needs to be further delineated. Specifically, do these molecules directly bind to 2C protein or do they inhibit the interactions between 2C protein and viral RNA or host protein? How do these inhibitors affect the ATPase and helicase activities of 2C protein? In this regard, high-resolution structures of EV-D68 2C protein with and without inhibitors are highly desired to elucidate the mechanism of action. Of particular highlight is a recent study investigating the antiviral mechanism of action of fluoxetine against CVB3. Fluoxetine (17) was shown to bind to CVB3 2C protein in a stereospecific manner with the (S)-enantiomer significantly more potent than the (R)-enantiomer.47
INTERNAL RIBOSOME ENTRY SITE (IRES) INHIBITORS
IRES is located at the 5’-UTR of the viral genome and is approximately 450 nucleotides long. IRES directs the translation of viral polyprotein in a cap-independent manner mediated by the interactions between the multiple stem-loop structures located within IRES element and the host IRES trans-acting factors (ITAFs).69 Enteroviruses contain type I IRES.80 This unique translation mechanism is distinct from the canonical eukaryotic cap-dependent translation, suggesting that it might be feasible to selectivity perturb the IRES-directed viral translation to achieve antiviral activity.
In a recent study, emetine (27) was shown to inhibit multiple enteroviruses including EV-A71, CV-A16, CV-B1, EV-D68, and Echov-6 viruses with low submicromolar potency (Figure 5).42 Significantly, when tested in the in vivo EV-A71 infected mouse model, emetine treated mice showed 100% survival protection when dosed as low as 0.2 mg/kg twice a day. In addition, viral loads in several organs of the infected mice including front and hind limbs, brain, and spleen were significantly lower in the emetine-treated group than the vehicle-treated control group. Given the promising in vitro results of emetine against EV-D68, coupled with the in vivo antiviral efficacy against EV-A71, it might be worthwhile to test emetine in the EV-D68 infected mouse models.
Figure 5.

Structures of IRES inhibitors. Note that not all compounds have been tested against EV-D68.
Other reported IRES inhibitors include idarubicin (28),81 apigenin (29) (Figure 5).82 However, their antiviral activity against EV-D68 has not yet been confirmed.
3D POLYMEREASE (3Dpol) INHIBITORS
The 3Dpol of EV-D68 is a RNA-dependent RNA polymerase that mediates viral genome replication within the replication organelles. EV-D68 3Dpol resembles a right hand with finger, thumb, and palm motifs. The palm holds the RNA template and is involved in catalysis through the coordination of magnesium ions. Fingers and thumb motifs coordinate the RNA strand and NTPs. The mechanism of action of antivirals targeting the 3Dpol may be through inhibition of transcript polymerization or through lethal mutagenesis. The X-ray crystal structure of the EV-D68 3Dpol was solved (PDB: 5XE0) (Figure 6A), paving the way for structure-based drug design.83 Ribavirin (30) showed weak antiviral activity against EV-D68 (EC50 > 100 μM).28 Another nucleoside drug tested against EV-D68 was favipiravir (T-705) (31), which similarly showed weak antiviral activity (EC50 > 50 μM) and was not active against all EV-D68 strains tested.26 Screening existing nucleoside/nucleotide drugs against EV-D68 might yield additional 3Dpol inhibitors. For example, NITD008 (32), a failed clinical candidate for the treatment of dengue virus infection due to toxicity, showed promising in vitro and in vivo antiviral activity against EV-A71 infection.84 Gemcitabine (33) also showed potent inhibition against EV-A17 and CVB3 in cell culture,85 as well as poliovirus.86 However, it is not known whether NITD008 (32) or gemcitabine (33) is active against EV-D68.
Figure 6.

Structures of EV-D68 3Dpol and viral polymerase inhibitors. (A). X-ray crystal structure of EV-D68 3Dpol in complex with GTP (PDB: 5XE0). (B). Structures of enterovirus polymerase inhibitors. Note that not all compounds have been tested against EV-D68 virus.
A recent study showed that a cytidine analog 2’-deoxy-2’-β-fluoro-4’-azidocytidine, also known as azvudine or FNC (34) (Figure 6B), had broad-spectrum antiviral activity against multiple enteroviruses including EV-A71, coxsackievirus A16 (CV-A16), CV-A6, CV-B3, and EV-D68 with EC50 values at the nanomolar range.44 The mechanism of action of FNC (34) was confirmed in the in vitro 3Dpol assay as well as the isothermal titration calorimetry (ITC) assays. Encouragingly, FNC (34) had promising in vivo antiviral efficacy and protected neonatal mice against lethal challenges with EV-A71 and CV-A16 viruses and reduced the viral load in several organs. FNC (34) was originally developed as a HIV drug and is currently in clinical trial phase II in China. Therefore, FNC (34) represents a promising candidate for EV-D68.
Several non-nucleoside compounds were also reported as enterovirus antivirals by targeting the 3Dpol, including aurintricarboxylic acid (35),87 amiloride (36),88 BPR-3P0128 (37),89 GPC-N114 (38),45 and DTrip-22 (39) (Figure 6B).90 However, the exact mechanism of action remains uncharacterized. One exception is GPC-N114 (38), which had broad-spectrum antiviral activity against enteroviruses with EC50 values in the single-digit and submicromolar range, including EV-D68 (EC50 = 1.44 ± 0.13 μM). GPC-N114 (38) has a novel mechanism of action by targeting the RNA template-primer site in the core of 3Dpol. The mechanism of action was characterized by X-ray crystallography, in vitro viral polymerase assay, and mutagenesis studies. However, this compound lacks drug-like properties and was not advanced to animal model studies.91 Nevertheless, the identification of this novel drug-binding pocket in 3Dpol opens the possibility to develop second-generation inhibitors with favorable pharmacokinetic properties.
The advantages of targeting viral 3Dpol include high potency, high genetic barrier to drug resistance, and broad-spectrum antiviral activity. However, the concerns that need to be addressed with nucleoside/nucleotide analogues are off-target side effects including immunosuppression and cytotoxicity.
HOST-TARGETING ANTIVIRALS
Enterovirus genome replication occurs at the membranous replication organelles, and several host proteins as well as viral proteins are known to participate in this process, including host acyl-CoA-binding domain-containing protein 3 (ACBD3), phosphatidylinositol-4-kinase IIIβ (PI4KIIIβ), oxysterol-binding protein (OSBP), and viral 2BC, 3A, 2C and 3D proteins (Figure 7A). However, the exact composition of the replication organelle and the mechanism of coordination among these proteins remain elusive. Nevertheless, these replication organelle-associated host proteins have been shown to be relevant antiviral drug targets.
Figure 7.

Replication organelles and the structures of host-targeting antivirals. (A). Cartoon representation of replication organelle-associated host factors and viral proteins. Note that the exact order of interactions is still under investigation. (B) Structures of host-targeting antivirals.
Enviroxime (40) inhibits multiple EV-D68 and EV-A71 strains with single-digit micromolar to submicromolar potency in cell culture (Figure 7B).26, 28 Enviroxime (40) was advanced to clinical trials for treatment of human rhinovirus-associated common cold, but was dropped out of phase II due to toxicity issues.92 The mechanism of action of enviroxime (40) was studied using resistance selection with rhinovirus, enterovirus, and poliovirus, and in all cases, resistance mutations were mapped to the viral 3A protein.93 Interestingly, enviroxime (40) inhibits the enzymatic activity of PI4KIIIβ in vitro.94 Therefore, although resistant mutants were mapped to the viral 3A protein, the 3A protein is not the direct target of enviroxime (40), but rather the host protein PI4KIIIβ. It was shown that enterovirus 3A mediates the interaction between host PI4KIIIβ and ACBD4 protein.95 Several other enviroxime-like inhibitors have also been reported as potent antivirals against enteroviruses,96, 97 but none of them have been tested in animal models.
Itraconazole (41) inhibits EV-D68 with an EC50 value of 0.32 μM.49 However, the antiviral potency appears to be not conclusive, as another study showed that itraconazole (41) was not active against multiple EV-D68 strains in cell culture (EC50 > 10 μM).27 The mechanism of action of itraconazole (41) was proposed to be mediated through OSBP.98 Similarly, OSW-1 (42), a specific OSBP antagonist, also had antiviral activity against multiple enteroviruses. Similar to enviroxime (40), resistant mutants against intraconazole were mapped to the viral 3A protein,49 suggesting that OSBP might interact with 3A directly or indirectly within the replication organelle.
It is noted that PI4KIIIβ inhibitors and OSBP antagonists are also often referred to as 3A inhibitors. For host-targeting antivirals, the in vitro potency may or may not correlate with the in vivo efficacy, therefore in vivo study should be planned when available for further target validation.
A recent genome-wide CRISPR screen identified the actin histidine methyltransferase SET domain containing 3 (SETD3) as an essential host factor for the replication of multiple enteroviruses including EV-A71 and EV-D68.99 It was shown that SETD3 specifically interacts with the viral 2A protease, and 2A mutants that abolish the interaction with SETD3 have compromised RNA replication. This study suggest that SETD3–2A interaction might be a novel host-targeting antiviral drug target for EV-D68.
PERSPECTIVES OF EV-D68 ANTIVIRAL DRUG DISCOVERY
Although AFM is a severe and devastating disease, it remains rare, and only a small fraction of the EV-D68 infected children progressed to neurological complications. Notably, in the majority of the AFM cases, infected children had fever and respiratory illness prior to limb weakness.100 Therefore, due to the limited therapeutic window, early diagnosis is the key for therapeutic intervention, and drug treatment should be initiated at the respiratory infection phase to prevent CNS invasion by the viruses. Indeed, EV-D68 is the most frequently detected virus in respiratory samples among the 233 patients with confirmed AFM in 2018, highlighting the feasibility of early diagnosis.
Drug discovery targeting EV-D68 is still at its infancy. The compounds presented above are promising starting points for further optimization, but they are still far from reaching human clinical trials. The only small molecule that has in vivo antiviral efficacy against EV-D68 is guanidine, whose mechanism of action remains elusive. To develop EV-D68 antivirals with potential clinical impact, the following features should be taken into consideration: 1) The in vitro antiviral potency EC50 of EV-D68 antivirals should be at the submicromolar range and ideally nanomolar range. A high potency is a pre-requisite to ensure that the in vivo drug concentration can exceed the in vitro EC90 value. A high selectivity index/therapeutic window is also desired since the majority of the patient population are children. 2) The drug candidates should have favorable in vitro and in vivo pharmacokinetic properties, including solubility, membrane permeability, stability, and minimal drug-drug interactions. 3) Blood-brain barrier (BBB) penetrating antivirals might be needed for the late-stage treatment of EV-D68 infection in which viruses already invade the central nervous system. Nevertheless, BBB-impermeable antivirals are also highly desired for early intervention to achieve maximum clinical benefits. 4) EV-D68 is an RNA virus with high mutation rates, and combination therapy might be necessary to suppress drug resistance. Although capsid inhibitors and protease inhibitors typically have low genetic barrier to drug resistance, they are valuable candidates for combination therapy.
Although drug discovery targeting EV-D68 is still in the early stage, we have accumulated a significant amount of knowledge and expertise from studying related enteroviruses including poliovirus, rhinovirus, coxsackievirus, and EV-A71. Given the similarities between EV-D68 and other enteroviruses (EV-A71, rhinovirus, coxsackievirus, and poliovirus) in terms of their viral protein sequence and replication mechanism, it is worthwhile to test existing EV-A71 antivirals,34, 101 coxsackievirus antivirals,78, 102 rhinovirus antivirals, and poliovirus antivirals against EV-D68, especially the ones that already have proven in vivo antiviral efficacy in animal models. Priority should be given to molecules that have proven in vivo antiviral efficacy against other enteroviruses. In parallel, with the high-resolution structures of various viral proteins, structure-based drug design and target-based virtual screening are now feasible.
Highly potent and selective enterovirus antivirals will not only serve as drug candidates for translational research, but also as valuable chemical probes to help understand the mechanism of viral replication and viral-host interactions. The progress made by the EV-D68 scientific community over the past 6 years is exciting, and developing effective treatments of EV-D68 infection is promising and shows great potential in the future.
Acknowledgements
This research is supported by the NIH grants (AI119187, AI144887, and AI147325), the Young Investigator Award grant from the Arizona Biomedical Research Centre to J.W (ADHS18-198859).
REFERENCES
- 1.Liu Y, Sheng J, van Vliet ALW, Buda G, van Kuppeveld FJM, and Rossmann MG (2018) Molecular basis for the acid-initiated uncoating of human enterovirus D68, Proc. Natl. Acad. Sci. U. S. A 115, E12209–E12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morens DM, Folkers GK, and Fauci AS (2019) Acute Flaccid Myelitis: Something Old and Something New, mBio 10, e00521–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilsdorf JR (2019) Acute Flaccid Myelitis: Lessons From Polio, J Pediatric Infect Dis Soc 8, 550–553. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Cao J, Zhang S, Lee AJ, Sun GY, Larsen CN, Zhao HT, Gu ZP, He S, Klem EB, and Scheuermann R0H (2016) Genetic changes found in a distinct clade of Enterovirus D68 associated with paralysis during the 2014 outbreak, Virus Evol 2, vew015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hixon AM, Yu G, Leser JS, Yagi S, Clarke P, Chiu CY, and Tyler KL (2017) A mouse model of paralytic myelitis caused by enterovirus D68, PLoS Pathog 13, e1006199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown DM, Hixon AM, Oldfield LM, Zhang Y, Novotny M, Wang W, Das SR, Shabman RS, Tyler KL, and Scheuermann RH (2018) Contemporary Circulating Enterovirus D68 Strains Have Acquired the Capacity for Viral Entry and Replication in Human Neuronal Cells, mBio 9, e01954–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurst BL, Evans WJ, Smee DF, Van Wettere AJ, and Tarbet EB (2019) Evaluation of antiviral therapies in respiratory and neurological disease models of Enterovirus D68 infection in mice, Virology 526, 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun SY, Bian LL, Gao F, Du RX, Hu YL, Fu Y, Su Y, Wu X, Mao QY, and Liang ZL (2019) A neonatal mouse model of Enterovirus D68 infection induces both interstitial pneumonia and acute flaccid myelitis, Antiviral Res 161, 108–115. [DOI] [PubMed] [Google Scholar]
- 9.Evans WJ, Hurst BL, Peterson CJ, Van Wettere AJ, Day CW, Smee DF, and Tarbet EB (2019) Development of a respiratory disease model for enterovirus D68 in 4-week-old mice for evaluation of antiviral therapies, Antiviral Res 162, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hixon AM, Frost J, Rudy MJ, Messacar K, Clarke P, and Tyler KL (2019) Understanding Enterovirus D68-Induced Neurologic Disease: A Basic Science Review, Viruses 11, E821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun J, Hu XY, and Yu XF (2019) Current Understanding of Human Enterovirus D68, Viruses 11, E490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baggen J, Thibaut HJ, Strating J, and van Kuppeveld FJM (2018) The life cycle of non-polio enteroviruses and how to target it, Nat Rev Microbiol 16, 368–381. [DOI] [PubMed] [Google Scholar]
- 13.Rosenfeld AB, Warren AL, and Racaniello VR (2019) Neurotropism of Enterovirus D68 Isolates Is Independent of Sialic Acid and Is Not a Recently Acquired Phenotype, mBio 10, e02370–02319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenfeld AB, Doobin DJ, Warren AL, Racaniello VR, and Vallee RB (2017) Replication of early and recent Zika virus isolates throughout mouse brain development, Proc. Natl. Acad. Sci. U. S. A 114, 12273–12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Messacar K, Asturias EJ, Hixon AM, Van Leer-Buter C, Niesters HGM, Tyler KL, Abzug MJ, and Dominguez SR (2018) Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality, Lancet Infect Dis 18, e239–e247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra N, Ng TFF, Marine RL, Jain K, Ng J, Thakkar R, Caciula A, Price A, Garcia JA, Burns JC, Thakur KT, Hetzler KL, Routh JA, Konopka-Anstadt JL, Nix WA, Tokarz R, Briese T, Oberste MS, and Lipkin WI (2019) Antibodies to Enteroviruses in Cerebrospinal Fluid of Patients with Acute Flaccid Myelitis, mBio 10, e01903–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schubert RD, Hawes IA, Ramachandran PS, Ramesh A, Crawford ED, Pak JE, Wu W, Cheung CK, O’Donovan BD, Tato CM, Lyden A, Tan M, Sit R, Sowa GA, Sample HA, Zorn KC, Banerji D, Khan LM, Bove R, Hauser SL, Gelfand AA, Johnson-Kerner BL, Nash K, Krishnamoorthy KS, Chitnis T, Ding JZ, McMillan HJ, Chiu CY, Briggs B, Glaser CA, Yen C, Chu V, Wadford DA, Dominguez SR, Ng TFF, Marine RL, Lopez AS, Nix WA, Soldatos A, Gorman MP, Benson L, Messacar K, Konopka-Anstadt JL, Oberste MS, DeRisi JL, and Wilson MR (2019) Pan-viral serology implicates enteroviruses in acute flaccid myelitis, Nature Med 25, 1748–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Sheng J, Fokine A, Meng G, Shin WH, Long F, Kuhn RJ, Kihara D, and Rossmann MG (2015) Structure and inhibition of EV-D68, a virus that causes respiratory illness in children, Science 347, 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei W, Guo HR, Chang JL, Yu YZ, Liu GC, Zhang NN, Willard SH, Zheng S, and Yu XF (2016) ICAM-5/Telencephalin Is a Functional Entry Receptor for Enterovirus D68, Cell Host Microbe 20, 631–641. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Sheng J, Baggen J, Meng G, Xiao C, Thibaut HJ, van Kuppeveld FJ, and Rossmann MG (2015) Sialic acid-dependent cell entry of human enterovirus D68, Nat Commun 6, 8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melia CE, Peddie CJ, de Jong AWM, Snijder EJ, Collinson LM, Koster AJ, van der Schaar HM, van Kuppeveld FJM, and Barcena M (2019) Origins of Enterovirus Replication Organelles Established by Whole-Cell Electron Microscopy, mBio 10, e00951–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Linden L, Wolthers KC, and van Kuppeveld FJM (2015) Replication and Inhibitors of Enteroviruses and Parechoviruses, Viruses-Basel 7, 4529–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corona AK, Saulsbery HM, Velazquez AFC, and Jackson WT (2018) Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit, Cell Rep 22, 3304–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Clercq E, and Li GD (2016) Approved Antiviral Drugs over the Past 50 Years, Clin Microbiol Rev 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senior K (2002) FDA panel rejects common cold treatment, Lancet Infect Dis 2, 264. [DOI] [PubMed] [Google Scholar]
- 26.Sun L, Meijer A, Froeyen M, Zhang L, Thibaut HJ, Baggen J, George S, Vernachio J, van Kuppeveld FJ, Leyssen P, Hilgenfeld R, Neyts J, and Delang L (2015) Antiviral Activity of Broad-Spectrum and Enterovirus-Specific Inhibitors against Clinical Isolates of Enterovirus D68, Antimicrobial Agents Chemother 59, 7782–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhoden E, Zhang M, Nix WA, and Oberste MS (2015) In Vitro Efficacy of Antiviral Compounds against Enterovirus D68, Antimicrob Agents Chemother 59, 7779–7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smee DF, Evans WJ, Nicolaou KC, Tarbet EB, and Day CW (2016) Susceptibilities of enterovirus D68, enterovirus 71, and rhinovirus 87 strains to various antiviral compounds, Antiviral Res 131, 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma C, Hu Y, Zhang J, Musharrafieh R, and Wang J (2019) A Novel Capsid Binding Inhibitor Displays Potent Antiviral Activity against Enterovirus D68, ACS Infect Dis 5, 1952–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pevear DC, Tull TM, Seipel ME, and Groarke JM (1999) Activity of pleconaril against enteroviruses, Antimicrob Agents Chemother 43, 2109–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayden FG, Herrington DT, Coats TL, Kim K, Cooper EC, Villano SA, Liu S, Hudson S, Pevear DC, Collett M, McKinlay M, and Pleconaril Respiratory Infection Study, G. (2003) Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: results of 2 double-blind, randomized, placebo-controlled trials, Clin Infect Dis 36, 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pevear DC, Hayden FG, Demenczuk TM, Barone LR, McKinlay MA, and Collett MS (2005) Relationship of pleconaril susceptibility and clinical outcomes in treatment of common colds caused by rhinoviruses, Antimicrob Agents Chemother 49, 4492–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egorova A, Ekins S, Schmidtke M, and Makarov V (2019) Back to the future: Advances in development of broad-spectrum capsid-binding inhibitors of enteroviruses, Eur J Med Chem 178, 606–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim ZQ, Ng QY, Ng JWQ, Mahendran V, and Alonso S (2019) Recent progress and challenges in drug development to fight hand, foot and mouth disease, Expert Opin Drug Discov, 1–13. [DOI] [PubMed] [Google Scholar]
- 35.Zhang M, Wang Y, He W, Sun Y, Guo Y, Zhong W, Gao Q, Liao M, Wang X, Cai Y, Guo Y, and Rao Z (2020) Design, Synthesis, and Evaluation of Novel Enterovirus 71 Inhibitors as Therapeutic Drug Leads for the Treatment of Human Hand, Foot, and Mouth Disease, J Med Chem 63, 1233–1244. [DOI] [PubMed] [Google Scholar]
- 36.Madia VN, Messore A, Pescatori L, Saccoliti F, Tudino V, De Leo A, Scipionet L, Fiore L, Rhoden E, Manetti F, Oberste MS, Di Santo R, and Costi R (2019) In Vitro Antiviral Activity of New Oxazoline Derivatives as Potent Poliovirus Inhibitors, J Med Chem 62, 798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho JY, Chern JH, Hsieh CF, Liu ST, Liu CJ, Wang YS, Kuo TW, Hsu SJ, Yeh TK, Shih SR, Hsieh PW, Chiu CH, and Horng JT (2016) In vitro and in vivo studies of a potent capsid-binding inhibitor of enterovirus 71, J Antimicrob Chemother 71, 1922–1932. [DOI] [PubMed] [Google Scholar]
- 38.Wald J, Pasin M, Richter M, Walther C, Mathai N, Kirchmair J, Makarov VA, Goessweiner-Mohr N, Marlovits TC, Zanella I, Real-Hohn A, Verdaguer N, Blaas D, and Schmidtke M (2019) Cryo-EM structure of pleconaril-resistant rhinovirus-B5 complexed to the antiviral OBR-5–340 reveals unexpected binding site, Proc. Natl. Acad. Sci. U. S. A 116, 19109–19115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdelnabi R, Geraets JA, Ma Y, Mirabelli C, Flatt JW, Domanska A, Delang L, Jochmans D, Kumar TA, Jayaprakash V, Sinha BN, Leyssen P, Butcher SJ, and Neyts J (2019) A novel druggable interprotomer pocket in the capsid of rhino- and enteroviruses, PLoS Biol 17, e3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musharrafieh R, Ma C, Zhang J, Hu Y, Diesing JM, Marty MT, and Wang J (2019) Validating Enterovirus D68–2A(pro) as an Antiviral Drug Target and the Discovery of Telaprevir as a Potent D68–2A(pro) Inhibitor, J Virol 93, e02221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim Y, Kankanamalage AC, Damalanka VC, Weerawarna PM, Groutas WC, and Chang KO (2016) Potent inhibition of enterovirus D68 and human rhinoviruses by dipeptidyl aldehydes and alpha-ketoamides, Antiviral Res 125, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Q, Li S, Du L, Chen S, Gao J, Cai Y, Xu Z, Zhao Z, Lan K, and Wu S (2020) Emetine protects mice from enterovirus infection by inhibiting viral translation, Antiviral Res 173, 104650. [DOI] [PubMed] [Google Scholar]
- 43.Ren P, Zheng Y, Wang W, Hong L, Delpeyroux F, Arenzana-Seisdedos F, and Altmeyer R (2017) Suramin interacts with the positively charged region surrounding the 5-fold axis of the EV-A71 capsid and inhibits multiple enterovirus A, Sci Rep 7, 42902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu N, Yang J, Zheng B, Zhang Y, Cao Y, Huan C, Wang S, Chang J, and Zhang W (2020) The pyrimidine analog FNC potently inhibits the replication of multiple enteroviruses, J Virol 94, e00204–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Linden L, Vives-Adrian L, Selisko B, Ferrer-Orta C, Liu X, Lanke K, Ulferts R, De Palma AM, Tanchis F, Goris N, Lefebvre D, De Clercq K, Leyssen P, Lacroix C, Purstinger G, Coutard B, Canard B, Boehr DD, Arnold JJ, Cameron CE, Verdaguer N, Neyts J, and van Kuppeveld FJ (2015) The RNA template channel of the RNA-dependent RNA polymerase as a target for development of antiviral therapy of multiple genera within a virus family, PLoS Pathog 11, e1004733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Musharrafieh R, Zhang J, Tuohy P, Kitamura N, Bellampalli SS, Hu Y, Khanna R, and Wang J (2019) Discovery of Quinoline Analogues as Potent Antivirals against Enterovirus D68 (EV-D68), J Med Chem 62, 4074–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bauer L, Manganaro R, Zonsics B, Strating J, El Kazzi P, Lorenzo Lopez M, Ulferts R, van Hoey C, Mate MJ, Langer T, Coutard B, Brancale A, and van Kuppeveld FJM (2019) Fluoxetine Inhibits Enterovirus Replication by Targeting the Viral 2C Protein in a Stereospecific Manner, ACS Infect Dis 5, 1609–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ulferts R, de Boer SM, van der Linden L, Bauer L, Lyoo HR, Mate MJ, Lichiere J, Canard B, Lelieveld D, Omta W, Egan D, Coutard B, and van Kuppeveld FJM (2016) Screening of a Library of FDA-Approved Drugs Identifies Several Enterovirus Replication Inhibitors That Target Viral Protein 2C, Antimicrob Agents Chemother 60, 2627–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao Q, Yuan S, Zhang C, Wang Y, Wang Y, He G, Zhang S, Altmeyer R, and Zou G (2015) Discovery of itraconazole with broad-spectrum in vitro antienterovirus activity that targets nonstructural protein 3A, Antimicrob Agents Chemother 59, 2654–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baggen J, Thibaut HJ, Staring J, Jae LT, Liu Y, Guo H, Slager JJ, de Bruin JW, van Vliet AL, Blomen VA, Overduin P, Sheng J, de Haan CA, de Vries E, Meijer A, Rossmann MG, Brummelkamp TR, and van Kuppeveld FJ (2016) Enterovirus D68 receptor requirements unveiled by haploid genetics, Proc. Natl. Acad. Sci. U. S. A 113, 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Earley DF, Bailly B, Maggioni A, Kundur AR, Thomson RJ, Chang CW, and von Itzstein M (2019) Efficient Blocking of Enterovirus 71 Infection by Heparan Sulfate Analogues Acting as Decoy Receptors, ACS Infect Dis 5, 1708–1717. [DOI] [PubMed] [Google Scholar]
- 52.Wiedemar N, Hauser DA, and Maser P (2020) 100 Years of Suramin, Antimicrob Agents Chemother 64, e01168–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai Q, Yameen M, Liu W, Gao Z, Li Y, Peng X, Cai Y, Wu C, Zheng Q, Li J, and Lin T (2013) Conformational plasticity of the 2A proteinase from enterovirus 71, J Virol 87, 7348–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim Y, Lovell S, Tiew KC, Mandadapu SR, Alliston KR, Battaile KP, Groutas WC, and Chang KO (2012) Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses, J Virol 86, 11754–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan J, George S, Kusov Y, Perbandt M, Anemuller S, Mesters JR, Norder H, Coutard B, Lacroix C, Leyssen P, Neyts J, and Hilgenfeld R (2013) 3C protease of enterovirus 68: structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses, J Virol 87, 4339–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang KO, Kim Y, Lovell S, Rathnayake AD, and Groutas WC (2019) Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors, Viruses 11, E197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu G, Qi J, Chen Z, Xu X, Gao F, Lin D, Qian W, Liu H, Jiang H, Yan J, and Gao GF (2011) Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti-hand, foot, and mouth disease virus drug design, J Virol 85, 10319–10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang L, Lin D, Kusov Y, Nian Y, Ma Q, Wang J, von Brunn A, Leyssen P, Lanko K, Neyts J, de Wilde A, Snijder EJ, Liu H, and Hilgenfeld R (2020) alpha-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment, J Med Chem asap. [DOI] [PubMed] [Google Scholar]
- 59.Zhang XN, Song ZG, Qin BY, Zhang XL, Chen LX, Hu YW, and Yuan ZH (2013) Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: Evaluation of antiviral efficacy in a murine infection model, Antiviral Res 97, 264–269. [DOI] [PubMed] [Google Scholar]
- 60.Patick AK, Brothers MA, Maldonado F, Binford S, Maldonado O, Fuhrman S, Petersen A, Smith GJ 3rd, Zalman LS, Burns-Naas LA, and Tran JQ (2005) In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease, Antimicrob Agents Chemother 49, 2267–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma GH, Ye Y, Zhang D, Xu X, Si P, Peng JL, Xiao YL, Cao RY, Yin YL, Chen J, Zhao LX, Zhou Y, Zhong W, Liu H, Luo XM, Chen LL, and Shen X (2016) Identification and biochemical characterization of DC07090 as a novel potent small molecule inhibitor against human enterovirus 71 3C protease by structure-based virtual screening, Eur J Med Chem 124, 981–991. [DOI] [PubMed] [Google Scholar]
- 62.Siklos M, BenAissa M, and Thatcher GRJ (2015) Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors, Acta Pharmaceutica Sinica B 5, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma Y, Li L, He S, Shang C, Sun Y, Liu N, Meek TD, Wang Y, and Shang L (2019) Application of Dually Activated Michael Acceptor to the Rational Design of Reversible Covalent Inhibitor for Enterovirus 71 3C Protease, J Med Chem 62, 6146–6162. [DOI] [PubMed] [Google Scholar]
- 64.Kim Y, Shivanna V, Narayanan S, Prior AM, Weerasekara S, Hua DH, Kankanamalage AC, Groutas WC, and Chang KO (2015) Broad-spectrum inhibitors against 3C-like proteases of feline coronaviruses and feline caliciviruses, J Virol 89, 4942–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim Y, Liu H, Galasiti Kankanamalage AC, Weerasekara S, Hua DH, Groutas WC, Chang KO, and Pedersen NC (2016) Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor, PLoS Pathog 12, e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim Y, Mandadapu SR, Groutas WC, and Chang KO (2013) Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease, Antiviral Res 97, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guan HX, Tian J, Zhang C, Qin B, and Cui S (2018) Crystal structure of a soluble fragment of poliovirus 2C(ATPase), Plos Pathogens 14, e1007304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guan H, Tian J, Qin B, Wojdyla JA, Wang B, Zhao Z, Wang M, and Cui S (2017) Crystal structure of 2C helicase from enterovirus 71, Sci Adv 3, e1602573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shih SR, Stollar V, and Li ML (2011) Host factors in enterovirus 71 replication, J Virol 85, 9658–9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sadeghipour S, Bek EJ, and McMinn PC (2012) Selection and characterisation of guanidine-resistant mutants of human enterovirus 71, Virus Res 169, 72–79. [DOI] [PubMed] [Google Scholar]
- 71.Tyler KL (2015) Rationale for the evaluation of fluoxetine in the treatment of enterovirus D68-associated acute flaccid myelitis, JAMA Neurol 72, 493–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ulferts R, van der Linden L, Thibaut HJ, Lanke KH, Leyssen P, Coutard B, De Palma AM, Canard B, Neyts J, and van Kuppeveld FJ (2013) Selective serotonin reuptake inhibitor fluoxetine inhibits replication of human enteroviruses B and D by targeting viral protein 2C, Antimicrob Agents Chemother 57, 1952–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuo J, Quinn KK, Kye S, Cooper P, Damoiseaux R, and Krogstad P (2012) Fluoxetine Is a Potent Inhibitor of Coxsackievirus Replication, Antimicrob Agents Chemother 56, 4838–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hixon AM, Clarke P, and Tyler KL (2017) Evaluating Treatment Efficacy in a Mouse Model of Enterovirus D68-Associated Paralytic Myelitis, J Infect Dis 216, 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Messacar K, Sillau S, Hopkins SE, Otten C, Wilson-Murphy M, Wong B, Santoro JD, Treister A, Bains HK, Torres A, Zabrocki L, Glanternik JR, Hurst AL, Martin JA, Schreiner T, Makhani N, DeBiasi RL, Kruer MC, Tremoulet AH, Van Haren K, Desai J, Benson LA, Gorman MP, Abzug MJ, Tyler KL, and Dominguez SR (2019) Safety, tolerability, and efficacy of fluoxetine as an antiviral for acute flaccid myelitis, Neurology 92, e2118–e2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gofshteyn J, Cardenas AM, and Bearden D (2016) Treatment of Chronic Enterovirus Encephalitis With Fluoxetine in a Patient With X-Linked Agammaglobulinemia, Pediatr Neurol 64, 94–98. [DOI] [PubMed] [Google Scholar]
- 77.De Palma AM, Heggermont W, Lanke K, Coutard B, Bergmann M, Monforte AM, Canard B, De Clercq E, Chimirri A, Purstinger G, Rohayem J, van Kuppeveld F, and Neyts J (2008) The thiazolobenzimidazole TBZE-029 inhibits enterovirus replication by targeting a short region immediately downstream from motif C in the nonstructural protein 2C, J Virol 82, 4720–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zuo J, Kye S, Quinn KK, Cooper P, Damoiseaux R, and Krogstad P (2015) Discovery of Structurally Diverse Small-Molecule Compounds with Broad Antiviral Activity against Enteroviruses, Antimicrob Agents Chemother 60, 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xing Y, Zuo J, Krogstad P, and Jung ME (2018) Synthesis and Structure-Activity Relationship (SAR) Studies of Novel Pyrazolopyridine Derivatives as Inhibitors of Enterovirus Replication, J Med Chem 61, 1688–1703. [DOI] [PubMed] [Google Scholar]
- 80.Furuse Y, Chaimongkol N, Okamoto M, and Oshitani H (2019) Evolutionary and Functional Diversity of the 5’ Untranslated Region of Enterovirus D68: Increased Activity of the Internal Ribosome Entry Site of Viral Strains during the 2010s, Viruses 11, E626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hou HY, Lu WW, Wu KY, Lin CW, and Kung SH (2016) Idarubicin is a broad-spectrum enterovirus replication inhibitor that selectively targets the virus internal ribosomal entry site, J Gen Virol 97, 1122–1133. [DOI] [PubMed] [Google Scholar]
- 82.Lv X, Qiu M, Chen D, Zheng N, Jin Y, and Wu Z (2014) Apigenin inhibits enterovirus 71 replication through suppressing viral IRES activity and modulating cellular JNK pathway, Antiviral Res 109, 30–41. [DOI] [PubMed] [Google Scholar]
- 83.Wang C, Wang C, Li Q, Wang Z, and Xie W (2017) Crystal Structure and Thermostability Characterization of Enterovirus D68 3D(pol), J Virol 91, e00876–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deng CL, Yeo H, Ye HQ, Liu SQ, Shang BD, Gong P, Alonso S, Shi PY, and Zhang B (2014) Inhibition of enterovirus 71 by adenosine analog NITD008, J Virol 88, 11915–11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kang H, Kim C, Kim DE, Song JH, Choi M, Choi K, Kang M, Lee K, Kim HS, Shin JS, Kim J, Han SB, Lee MY, Lee SU, Lee CK, Kim M, Ko HJ, van Kuppeveld FJ, and Cho S (2015) Synergistic antiviral activity of gemcitabine and ribavirin against enteroviruses, Antiviral Res 124, 1–10. [DOI] [PubMed] [Google Scholar]
- 86.Zhang Z, Yang E, Hu C, Cheng H, Chen CY, Huang D, Wang R, Zhao Y, Rong L, Vignuzzi M, Shen H, Shen L, and Chen ZW (2017) Cell-Based High-Throughput Screening Assay Identifies 2’,2’-Difluoro-2’-deoxycytidine Gemcitabine as a Potential Antipoliovirus Agent, ACS Infect Dis 3, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hung HC, Chen TC, Fang MY, Yen KJ, Shih SR, Hsu JT, and Tseng CP (2010) Inhibition of enterovirus 71 replication and the viral 3D polymerase by aurintricarboxylic acid, J Antimicrob Chemother 65, 676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gazina EV, Smidansky ED, Holien JK, Harrison DN, Cromer BA, Arnold JJ, Parker MW, Cameron CE, and Petrou S (2011) Amiloride Is a Competitive Inhibitor of Coxsackievirus B3 RNA Polymerase, J Virol 85, 10364–10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Velu AB, Chen GW, Hsieh PT, Horng JT, Hsu JT, Hsieh HP, Chen TC, Weng KF, and Shih SR (2014) BPR-3P0128 inhibits RNA-dependent RNA polymerase elongation and VPg uridylylation activities of Enterovirus 71, Antiviral Res 112, 18–25. [DOI] [PubMed] [Google Scholar]
- 90.Chen TC, Chang HY, Lin PF, Chern JH, Hsu JT, Chang CY, and Shih SR (2009) Novel antiviral agent DTriP-22 targets RNA-dependent RNA polymerase of enterovirus 71, Antimicrob Agents Chemother 53, 2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bauer L, Lyoo H, van der Schaar HM, Strating JR, and van Kuppeveld FJ (2017) Direct-acting antivirals and host-targeting strategies to combat enterovirus infections, Curr Opin Virol 24, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Phillpotts RJ, Jones RW, Delong DC, Reed SE, Wallace J, and Tyrrell DA (1981) The activity of enviroxime against rhinovirus infection in man, Lancet 1, 1342–1344. [DOI] [PubMed] [Google Scholar]
- 93.Heinz BA, and Vance LM (1996) Sequence determinants of 3A-mediated resistance to enviroxime in rhinoviruses and enteroviruses, J Virol 70, 4854–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van der Schaar HM, van der Linden L, Lanke KHW, Strating JRPM, Purstinger G, de Vries E, de Haan CAM, Neyts J, and van Kuppeveld FJM (2012) Coxsackievirus mutants that can bypass host factor PI4KIII beta and the need for high levels of PI4P lipids for replication, Cell Res 22, 1576–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xiao X, Lei X, Zhang Z, Ma Y, Qi J, Wu C, Xiao Y, Li L, He B, and Wang J (2017) Enterovirus 3A Facilitates Viral Replication by Promoting Phosphatidylinositol 4-Kinase IIIbeta-ACBD3 Interaction, J Virol 91, e00791–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, and Shimizu H (2011) Phosphatidylinositol 4-Kinase III Beta Is a Target of Enviroxime-Like Compounds for Antipoliovirus Activity, J Virol 85, 2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.De Palma AM, Thibaut HJ, van der Linden L, Lanke K, Heggermont W, Ireland S, Andrews R, Arimilli M, Al-Tel TH, De Clercq E, van Kuppeveld F, and Neyts J (2009) Mutations in the nonstructural protein 3A confer resistance to the novel enterovirus replication inhibitor TTP-8307, Antimicrob Agents Chemother 53, 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Strating JR, van der Linden L, Albulescu L, Bigay J, Arita M, Delang L, Leyssen P, van der Schaar HM, Lanke KH, Thibaut HJ, Ulferts R, Drin G, Schlinck N, Wubbolts RW, Sever N, Head SA, Liu JO, Beachy PA, De Matteis MA, Shair MD, Olkkonen VM, Neyts J, and van Kuppeveld FJ (2015) Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein, Cell Rep 10, 600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Diep J, Ooi YS, Wilkinson AW, Peters CE, Foy E, Johnson JR, Zengel J, Ding S, Weng KF, Laufman O, Jang G, Xu J, Young T, Verschueren E, Kobluk KJ, Elias JE, Sarnow P, Greenberg HB, Huttenhain R, Nagamine CM, Andino R, Krogan NJ, Gozani O, and Carette JE (2019) Enterovirus pathogenesis requires the host methyltransferase SETD3, Nat Microbiol 4, 2523–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Helfferich J, Knoester M, Van Leer-Buter CC, Neuteboom RF, Meiners LC, Niesters HG, and Brouwer OF (2019) Acute flaccid myelitis and enterovirus D68: lessons from the past and present, Eur J Pediatr 178, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lin JY, Kung YA, and Shih SR (2019) Antivirals and vaccines for Enterovirus A71, J Biomed Sci 26, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Garmaroudi FS, Marchant D, Hendry R, Luo HL, Yang DC, Ye X, Shi JY, and McManus BM (2015) Coxsackievirus B3 replication and pathogenesis, Future Microbiol 10, 629–652. [DOI] [PubMed] [Google Scholar]