

Abstract
This Phase II study evaluated analgesia, abuse liability, and cognitive performance of hydromorphone and oral delta-9-tetrahydrocannabinol (THC; dronabinol) using a within-subject, double-blind, randomized, placebo-controlled, human laboratory trial. Healthy adults (N = 29) with no history of drug use disorder received combinations of placebo, hydromorphone (4 mg; oral), and dronabinol (2.5 mg, 5.0 mg, 10 mg; oral). Primary outcomes were quantitative sensory testing (QST) measures of acute (thermal, pressure pain; thermal, punctate probe temporal summation; cold pressor; conditioned pain modulation) and chronic pain (capsaicin 10% topical cream with thermal rekindling), measures of drug abuse liability, cognitive functioning, and adverse events. Subgroup analyses were conducted within opioid-responders (endorsed >20 on a Drug Effect visual analog scale during the hydromorphone-only condition) and nonresponders. A consistent dose-effect relationship of dronabinol on hydromorphone across all measures was not observed. Analgesia only improved in the hydromorphone + dronabinol 2.5 mg condition. Hydromorphone + dronabinol 2.5 mg showed the lowest and hydromorphone+dronabinol 5 mg showed the highest risk for abuse. Hydromorphone+dronabinol 10 mg produced a high rate of dysphoric effects, and hydromorphone+dronabinol 5 mg and hydromorphone + dronabinol 10 mg produced AEs. Subgroup analyses showed subjective effects and abuse risk was increased among opioid responders and largely absent among nonresponders. Overall, only hydromorphone+dronabinol 2.5 mg modestly enhanced hydromorphone-based analgesia and hydromorphone + dronabinol 5 mg and 10 mg increased risk for abuse and AEs. These data can help inform opioid-sparing efforts in clinical pain populations. Demonstration that potential opioid effects varied as a function of participant opioid sensitivity (e.g., responder status) is a novel finding that warrants additional research.
Subject terms: Experimental models of disease, Outcomes research
Introduction
Opioid use disorder (OUD) is a significant public health problem that is reducing the average life expectancy in the United States. [1, 2] Taking high doses of opioids increases risk for OUD [3–5], and there is interest in identifying medications that can be co-administered with low-dose opioids to produce acceptable levels of analgesia while reducing OUD risk [6–9].
The opioid-sparing properties of cannabinoids were first acknowledged in 1889 [10], and are evident within preclinical studies that report high potency cannabinoids produce robust and meaningful shifts in opioid-induced analgesia [11, 12]. Patient surveys [13–16] and epidemiological evidence reports that reductions in opioid-reliance and consequences following medicinal cannabis exposure [17, 18] suggested these preclinical outcomes might generalize into human clinical samples, and this collective information was the basis upon which several states added OUD as a qualifying condition for medicinal cannabis prescribing [19].
Such legislative approvals are likely premature; human empirical evaluation of cannabis for opioid-sparing is sparse and the ability to generalize from preclinical studies is tenuous since the cannabinoids examined have potencies that can cause psychiatric adverse events in humans [20, 21]. More recent epidemiological reports partially refute earlier studies by suggesting that cannabinoid access does not improve opioid-related morbidity and mortality [22] and empirical examinations of opioid-cannabis interactions in humans have found that oral delta-9-tetrahydrocannabinol (THC; dronabinol) reduced rather than improved analgesia from oxycodone and morphine and also showed increased risk for abuse [23–25]. Smoked cannabis has been shown to enhance oxycodone-based analgesia but may also have increased risk for abuse [26].
These prior empirical evaluations have focused on acute rather than chronic pain models, despite evidence that cannabinoids may be most efficacious for centralized pain conditions [27]. They also did not account for whether participants were able to discriminate between an opioid and placebo, a recognized concern for opioid administration studies [28]. The current Phase II randomized, controlled, within-subject clinical trial (NCT02901275) extends prior research by examining whether dronabinol enhanced the effects of a low dose of hydromorphone. Hydromorphone was selected because it is less impacted by P450 enzymes than other opioids, which reduces the likelihood that individual participant outcomes will be differentially impacted by variability in metabolic status [29]. Although oral hydromorphone dosing has not been directly compared to heroin exposure, direct comparisons of intravenous and subcutaneously-delivered hydromorphone and heroin have confirmed similarity in effects between the two drugs that support the use of hydromorphone as a laboratory surrogate for heroin [30, 31]. Primary outcomes were measures of acute and chronic pain as assessed using quantitative sensory testing (QST), standardized measures of abuse liability, and cognitive performance evaluated within the entire sample and as a function of opioid sensitivity.
Methods
Participants
Healthy individuals were enrolled between 02/2017 and 03/2020. Full eligibility criteria and a CONSORT flowchart are provided in supplemental materials. The study was approved by the Johns Hopkins University School of Medicine Institutional Review Board and all participants provided informed consent to participate. The trial was registered on clinicaltrials.gov as NCT02901275.
Study design
Eligibility was established via self-report and semi-structured observer measures, blood and urine samples that tested for recent exposure to substances (i.e., opioids, buprenorphine, methadone, amphetamine, benzodiazepines, cocaine, and THC), pregnancy, and medical eligibility, breath samples to check for recent alcohol exposure, and an ECG with a history and physical. To ensure rigorous blinding and reduce bias in responses, neither participants nor research staff were informed about the specific medications under investigation [32].
Study sessions
Participants completed five outpatient laboratory sessions scheduled a minimum 7 days apart. Sessions began around 08:00 and lasted 8 h. Transportation to and from the session was provided by the study. Breath alcohol and urine toxicology testing were used to verify participants had not recently used alcohol or illicit drugs and were not pregnant, and a standardized breakfast was provided. Baseline ratings for all study measures were collected, study drugs were co-administered at 10:00, and study assessments were obtained repeatedly for 6 h post-dosing.
Study measures
QST measures
Laboratory models of acute and chronic pain were used to assess analgesia [33]. A QST assessment was conducted during screening to familiarize participants with the process and during sessions at baseline and each hour post-drug administration for 4 h. Upper limits were imposed for all tests to ensure safety and participant discontinuation of tests was operationalized as pain tolerance. Acute measures included thermal and pressure pain, temporal summation of pain using thermal and punctate probe stimuli, cold pressor testing, and conditioned pain modulation (CPM) [34–36]. Capsaicin (10% topical cream) was used to model chronic pain by rekindling the treated area with a thermal stimulus at each assessment [34, 36–41]. A full description of QST methods is provided as Supplementary Material.
Acute pain outcomes
Threshold response primary outcomes were detection limit and tolerance for thermal (heat pain threshold [HPTh] and tolerance [HPTo]) and pressure pain (threshold; PPTh). Thermal temporal summation (TTS) was the change of pain ratings from the first heat stimulus and the maximum pain rating after a train of thermal stimuli. Mechanical temporal summation (MTS) was the peak pain following a 10-stimuli train divided by pain from a single stimulus. Cold pressor pain was evaluated using time to first pain (threshold) and time to hand withdrawal (tolerance) following hand immersion in a cold-water bath (5 °C). CPM was evaluated by repeating the cold pressor task for 20 s before delivering concurrent pressure pain or mechanical temporal summation tasks in random order, after which initial PPTh and peak pain during MTS, respectively, were subtracted from the value collected following single cold pressor hand immersion.
Chronic pain outcomes
Chronic pain primary outcomes were HPTh and MTS following initial 30-min exposure of capsaicin 10% topical cream and rekindling the capsaicin-affected area using a thermal stimulus at each assessment.
Global QST outcomes
Three Z-score standardized global QST outcomes were derived from the full QST testing battery. For Central Sensitization (the average of MTS, TTS, CPM, and after-sensation Z-scores) and General Sensitivity (the average of PPTh, HPTh, HPTo, and time to cold pressor latency), higher values represent greater sensitization/sensitivity. Nociceptive profile was derived from temporal summation and CPM, with negative and positive values denoting antinociception and pronociceptive properties, respectively.
Participant-reported effects
Consistent with FDA guidance [42], participant drug effect ratings were collected at baseline and each hour post-dose for 6-h using the following visual analog scales (VAS; 0–100): Drug Effect, Good Effect, Bad Effect, High, Like the Way I Feel, and Nausea. Primary outcomes were peak ratings post-dose per session.
Measures of abuse liability
Abuse liability was operationalized as a post-drug rating of ≥60 on the VAS scale “High” [42]. Participants also reported whether they enjoyed study medications, the monetary amount they would pay for this medication, and the likelihood they would take the medication again (0 “not at all” to 5 “extremely”).
Cognitive performance
Cognitive performance was assessed using three tasks [43–45]. The Digit Symbol Substitution Task is a measure of psychomotor ability in which participants used a keypad to replicate observed patterns on a computer screen; the primary outcome was proportion correct. The Paced Serial Addition Task is a measure of working memory in which participants add sequentially presented integers together in rapid sequence; the primary outcomes were mean reaction time and percent correct. A circular light test of fine motor movement required participants to repeat visual patterns displayed on a board for a 60-s period; the primary outcome was maximum correct number correct.
Adverse events
Adverse events (AEs) were prompted throughout the session by asking participants at every data collection time point whether they were experiencing any side effects of the study medications. Any reported event was documented and classified according to severity (i.e., mild, moderate, severe) and relatedness (i.e., unrelated, possibly, probably, definitely). A change in severity was documented as a new AE. Primary outcomes were the total number of AEs, number of AEs (collapsed across possible, probable, and definite relation) rated as mild, moderate, or severe, and the percent of participants who experienced ≥1 AE during a session.
Study medications
Study medications were oral hydromorphone (4 mg, Sky Pharma), dronabinol (2.5 mg, 5 mg, 10 mg; Akorn), and placebo. Medications were over-encapsulated using size 00 gelcaps to blind dose condition to participants and staff. To model real-world settings, doses were not adjusted for weight and medications were consumed at the same time. The study included two control conditions: placebo+placebo (placebo) and hydromorphone 4 mg + placebo (hydromorphone + placebo), and three experimental conditions: hydromorphone 4 mg + dronabinol 2.5 mg, hydromorphone 4 mg + dronabinol 5 mg, and hydromorphone 4 mg + dronabinol 10 mg. All doses were selected because they were within the range approved by the FDA for prescribing and could therefore be prescribed clinically. Lower doses were also considered preferable for both tolerability and to avoid ceiling effects that might obscure evidence of drug interactions. Hydromorphone 4 mg was observed in a parallel (unpublished) trial to produce a low level of analgesia on an identical battery of QST tasks and was well-tolerated by most participants. Dronabinol doses were selected to be linear, starting with the lowest prescribable dose (2.5 mg) and based upon prior experience from our group that a 20 mg dronabinol dose produced a high rate of AEs in persons with no recent history of THC exposure. The first drug session was always the hydromorphone+placebo condition to ensure participants safely tolerated hydromorphone before receiving the combination. Participants (N = 5) who experienced elevated negative opioid agonist signs (e.g., vomiting) during this session were discontinued from the study; only study completers are reported here. Thereafter, session order was randomized by a research pharmacist who had no participant interaction using a random number sequence, with the exception that no sequences scheduled hydromorphone + dronabinol 10 mg first.
Data analyses
The study hypothesis was that combining dronabinol with hydromorphone would dose-dependently increase analgesia on measures of acute and chronic pain while also increasing abuse liability and/or cognitive impairment relative to both placebo and hydromorphone + placebo conditions. A power analysis based upon a prior evaluation of oxycodone + smoked cannabis effects on cold pressor tolerance and threshold tests [26] determined a sample size of 15 would be sufficient to detect large effects (Cohen’s d = 1.154) of drug condition. A sample of N = 30 was planned to support subgroup comparisons though due to human subjects restrictions, the study closed at the onset of the COVID-19 pandemic with a final enrollment of N = 29.
Demographic variables are presented descriptively below and in Supplementary Table 1. Primary outcomes were evaluated using mixed-effects models for continuous variables and generalized estimating equations for dichotomous outcomes. Partial eta square (η2) was derived for all outcomes to identify small (0.01), medium (0.06), and large (0.14) effect sizes and Tukey test post-hoc comparisons were conducted. Analyses were next replicated with (1) data converted to a change from baseline and (2) body mass index (BMI) as a covariate; in both cases, results did not meaningfully change so only primary data are reported. Participants were then classified as opioid responders or nonresponders. An opioid responder was defined as having >20-point difference between baseline and post-drug administration on the Drug Effects VAS scale during the hydromorphone + placebo session [46]. Next, analyses were repeated adding responder status as a covariate, and then sensitivity analyses of main effects were conducted within the opioid responder and nonresponder subgroups following the analytic plan described above. All data were analyzed using SAS version 9.4 with alpha set at 0.05.
Results
Participant demographics
Participants (N = 29) were 52% female, mean (SD) 30.4 (9.2) years of age, and had a mean BMI of 25.6 (4.8). The sample was 48% Caucasian, 41% African American, 10% Asian and 7% were of Hispanic origin. Additional characteristics are presented in Supplementary Table 1.
QST outcomes
Acute Pain Measures
There was limited evidence of dronabinol enhancement of hydromorphone on QST measures (Table 1, Fig. 1). A significant main effect was evident on HPTh (F(4,112)=2.81, p = 0.029, η2 = 0.05), and HPTo (F(4,112) = 3.03, p = 0.020, η2 = 0.05); in both cases only the hydromorphone+dronabinol 2.5 mg condition (p’s < 0.02) increased analgesia greater than hydromorphone+placebo (but not placebo + placebo). A main effect on TTS (F(4,112) = 2.87, p = 0.026, η2 = 0.05) was evident wherein the hydromorphone+dronabinol 10 mg condition increased analgesia relative to placebo + placebo (p = 0.018), but not the hydromorphone+placebo condition. Hydromorphone+dronabinol 10 mg also reduced the pain rating at time of first pain during the cold pressor task (F(4,112) = 3, p = 0.021, η2 = 0.05) significantly more than placebo+placebo (p = 0.041) but not hydromorpone+placebo, though actual changes in cold pressor tolerance and threshold were not significant. No CPM measures showed significant main effects.
Table 1.
Primary outcomes among full sample (N = 29).
| Placebo | Hydromorphone 4 mg | P value (partial eta2) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dronabinol 0 mg | Dronabinol 2.5 mg | Dronabinol 5 mg | Dronabinol 10 mg | ||||||||
| Mean/% | SD | Mean/% | SD | Mean/% | SD | Mean/% | SD | Mean/% | SD | ||
| Quantitative Sensory Testing | |||||||||||
| Acute Pain Model | |||||||||||
| Pressure Pain, threshold (0–1200) | 586.2 | 256.9 | 637.2 | 256.8 | 630.9 | 250.3 | 673.0 | 242.3 | 652.0 | 227.0 | NS |
| Thermal Pain | |||||||||||
| Threshold | 44.7 | 3.1 | 44.0a | 2.6 | 45.6b | 1.9 | 44.4 | 3.0 | 45.0 | 3.2 | 0.029 (0.04) |
| Tolerance | 48.5 | 1.2 | 48.1a | 1.2 | 48.9b | 1.1 | 48.5 | 1.5 | 48.8 | 1.4 | 0.02 (0.05) |
| Temporal Summation | |||||||||||
| Mechanical | 6.8 | 12.8 | 5.5 | 8.2 | 6.8 | 11.3 | 5.9 | 7.7 | 5.4 | 7.6 | NS |
| Thermal | 11.9 | 15.0 | 17.4a | 17.9 | 9.9 | 10.0 | 11.3 | 10.8 | 8.4b | 8.5 | 0.026 (0.05) |
| Cold Pressor | |||||||||||
| Threshold (time in seconds) | 16.0 | 14.5 | 14.5 | 9.9 | 15.9 | 17.0 | 18.8 | 27.7 | 14.0 | 10.9 | NS |
| Threshold severity rating (0–100) | 55.3a | 22.0 | 54.3 | 19.7 | 48.7 | 23.9 | 48.8 | 20.2 | 46.0b | 17.8 | 0.02 (0.05) |
| Tolerance (time in seconds) | 84.5 | 99.3 | 87.0 | 96.7 | 85.0 | 95.8 | 99.2 | 107.3 | 86.0 | 87.8 | NS |
| Chronic Pain Model | |||||||||||
| Capsaicin, Heat-Pain Threshold | 44.2 | 3.4 | 43.3 | 3.5 | 44.4 | 2.7 | 44.4 | 3.1 | 44.2 | 3.1 | NS |
| Capsaicin, MTS | 4.5 | 6.3 | 5.5a | 6.6 | 5.1 | 4.5 | 10.8 | 19.6 | 8.2b | 18.6 | NS |
| Conditioned Pain Modulation | |||||||||||
| MTS | 14.7a | 26.4 | 11.1 | 15.0 | 20.4b | 23.1 | 18.3 | 26.8 | 16.7 | 26.3 | NS |
| Pressure Pain Threshold, peak difference | 155.7 | 135.3 | 153.2 | 146.0 | 145.1 | 126.9 | 150.3 | 125.0 | 114.0 | 127.8 | NS |
| Global Measures | |||||||||||
| Central Sensitization (z-score) | 0.14 | 0.42 | 0.32a | 0.49 | 0.10b* | 0.36 | 0.19 | 0.48 | 0.28 | 0.71 | 0.03 (0.05) |
| General Sensitivity (z-score) | 0.39 | 0.65 | 0.52 | 0.73 | 0.48b | 1.86 | 0.21 | 0.86 | 0.27 | 0.71 | NS |
| Nociceptive Profile (z-score) | −0.34 | 1.42 | 0.14 | 1.30 | 0.55 | 1.40 | 0.07 | 1.25 | 0.28 | 1.41 | NS |
| Participant Ratings (0–100) | |||||||||||
| Drug Effects | 12.9a | 21.5 | 39.2b | 27.4 | 47.6b | 32.2 | 50.3b | 34.5 | 49.1b | 34.3 | <0.0001 (0.30) |
| Good Effects | 8.2a | 15.2 | 22.1 | 23.3 | 33.2b | 29.6 | 28.2b | 30.1 | 19.7 | 30.2 | <0.001 (0.12) |
| Bad Effects | 8.4a | 17.9 | 13.0c | 22.4 | 17.4 | 23.3 | 23.1b | 27.4 | 28.3b,d | 28.0 | 0.001 (0.10) |
| High | 2.1a | 6.0 | 17.0b | 19.5 | 23.9b | 31.8 | 30.5b | 30.9 | 25.6b | 30.5 | <0.0001 (0.19) |
| Like the Way I Feel | 30.4 | 33.8 | 42.0a | 25.6 | 36.5 | 33.5 | 38.4 | 33.0 | 26.6b | 31.9 | 0.019 (0.06) |
| Nausea | 0.9a | 1.9 | 8.3 | 16.1 | 7.9 | 16.4 | 15.8b | 22.5 | 17.2b | 22.7 | <0.0001 (0.13) |
| Abuse Potential Measures | |||||||||||
| Enjoyed medication (%) | 10.3a | 31.0b | 34.5b | 41.4b | 17.2b | <0.0001 (0.23) | |||||
| Would take medication again (%) | 27.6 | 34.5 | 31.0 | 51.7 | 24.1 | 0.043 (0.04) | |||||
| ≥60 on “High” rating scale (%) | 0 | 3.4 | 20.7 | 20.7 | 17.2 | 0.032 (0.21) | |||||
| Willingness to pay for medication ($) | 4.7 | 18.6 | 4.1 | 6.2 | 3.2 | 6.7 | 6.9 | 8.9 | 2.6 | 4.2 | NS |
| Cognitive Testing | |||||||||||
| Circular lights, max per minute | 48.6a | 10.6 | 44.1b,c | 11.0 | 47.1 | 11.7 | 48.2d | 10.3 | 49.6d | 10.90 | <0.0001 (0.17) |
| DSST, proportion correct | 0.91 | 0.92 | 0.95 | 0.95 | 0.92 | NS | |||||
| PASAT, Mean Reaction Time Correct (s) | 1207.2 | 271.0 | 1214.0 | 441.2 | 1312.7 | 179.9 | 1236.4 | 278.9 | 1256.7 | 248.50 | NS |
| PASAT, Correct (%) | 78 | 73.3 | 78.9 | 79.1 | 75.5 | NS | |||||
Outcomes represent mean peak ratings or percent participants for each condition (N = 29).
Hydro hydromorphone, SD standard deviation, TTS thermal temporal summation, MMT mechanical temporal summation, DSST Digit Symbol Substitution Task, PASAT Paced Auditory Serial Addiction Task.
Subscripts denote significant differences in posthoc comparisons. Differences exist between a and b, c and d.
Partial eta2 effect sizes provided for significant results: small (0.01), medium (0.06), large (0.14).
Fig. 1. Quantitative Sensory Testing (QST) Outcomes.
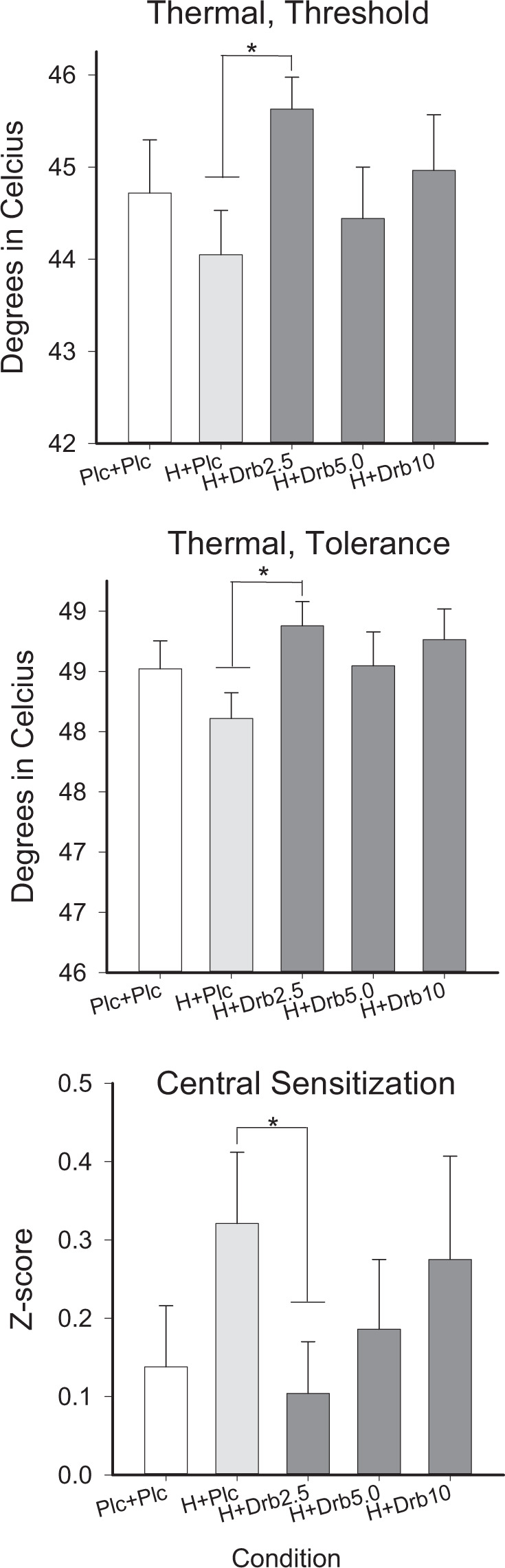
Data show results from thermal threshold (top) and tolerance (middle) test, as well as Central Sensitization (bottom) global QST measure, as a function of study condition (x-axis). Medication conditions were Placebo + Placebo (Plc + Plc), oral hydromorphone 4 mg + placebo (H + Plc), and hydromorphone 4 mg combined with oral dronabinol 2.5 mg (Drb2.5 mg), 5 mg (Drb5 mg), and 10 mg (Drb10 mg) doses. * represents differences p < 0.05 and error bars represent SEM.
Chronic pain measures
No main effects of peak chronic pain measures reached significance.
Global measures
A main effect of Central Sensitization (F(4,112)=2.7, p = 0.034, η2 = 0.05) revealed the hydromorphone+dronabinol 2.5 mg condition (p = 0.053) reduced sensitization when compared to hydromorphone + placebo but not placebo+placebo (Fig. 3). Peak Nociceptive Profile and Global Sensitivity ratings were not significantly different across doses.
Fig. 3. Measures of abuse liability.
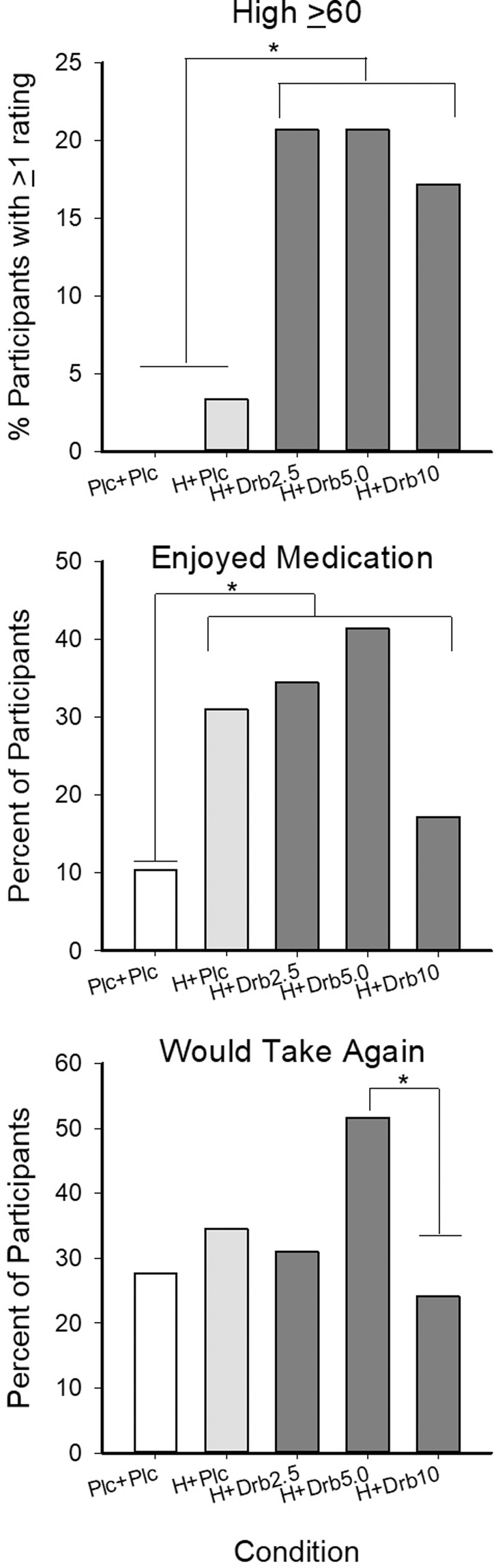
Data present percent of participants rating their feeling of High on a visual analog scale (VAS) 60 or higher at least once during a session (top), the percent of participants who reported enjoying their medication (collapsed across ratings of a little, moderately, quite a bit, and very much; middle), and percent of participants who indicated willingness to take the medication combination again (bottom). Medication conditions were Placebo+Placebo (Plc + Plc), oral hydromorphone 4 mg + placebo (H + Plc), and hydromorphone 4 mg combined with oral dronabinol 2.5 mg (Drb2.5 mg), 5 mg (Drb5 mg), and 10 mg (Drb10 mg) doses. * represents differences p < .05.
Participant ratings
Drug conditions generally produced subjective ratings that were significantly different from placebo+placebo but not hydromorphone + placebo (Table 1, Fig. 2). All active drug conditions increased ratings of Drug Effects (F(4,112)=16.65, p < 0.001, η2 = 0.30) significantly more than placebo + placebo (p’s < 0.001), but no dronabinol dose increased Drug Effect ratings relative to hydromorphone + placebo. Ratings for Good Effects (F(4,112) = 5.86, p < 0.001, η2 = 0.12) were significantly higher than placebo+placebo in the lower dronabinol dose conditions (hydromorphone+dronabinol 2.5 mg [p < 0.001] and hydromorphone+dronabinol 5 mg [p = 0.004]), but not different significantly from hydromorphone+placebo. Bad Effect ratings (F(4,112) = 5.14, p < 0.001, η2 = 0.10) were significantly higher than placebo+placebo in the hydromorphone+dronabinol 5 mg condition (p = 0.029) and significantly higher placebo + placebo (p = 0.002), hydromorphone+placebo (p = 0.001), and hydromorphone+dronabinol 2.5 mg (p = 0.012) in the hydromorphone+dronabinol 10 mg condition. Nausea (F(4,112) = 6.47, p < 0.001, η2 = 0.13) was also significantly higher than placebo+placebo for both the hydromorphone+dronabinol 5 mg (p = 0.001) and hydromorphone+dronabinol 10 mg (p < 0.001) conditions, but not the hydromorphone+placebo condition. The hydromorphone + dronabinol 10 mg condition produced significantly lower ratings of Like the Way I Feel relative to hydromorphone+placebo (p = 0.022), but not placebo + placebo. Finally, all active drug conditions produced ratings of High (F(4,112) = 9.36, p < 0.0001, η2 = 0.19) that were significantly greater than placebo+placebo (p’s < 0.05), though dronabinol did not increase ratings beyond those observed in the hydromorphone+placebo condition.
Fig. 2. Participant ratings.

Data present results of visual analog scale (VAS) 0–100 ratings on Drug Effect (left, upper), Good Effect (right, upper), Bad Effect (left, lower), and High (right, lower) for the entire sample (black bars) as well as persons categorized as Opioid Responders (light gray) and Opioid Nonresponders (dark gray). Opioid responders achieved a >20 point difference between baseline and active drug during the Hydromorphone 4 mg + placebo condition. Medication conditions were Placebo + Placebo (Plc + Plc), oral hydromorphone 4 mg + placebo (H + Plc), and hydromorphone 4 mg combined with oral dronabinol 2.5 (Drb2.5 mg), 5 mg (Drb5 mg), and 10 (Drb10 mg) doses. Error bars represent SEM.
Abuse liability measures
Participants enjoyed study medications more in all active drug conditions as compared to placebo+placebo (F (4,112) = 11.59, p < 0.001; η2 = 0.23, Table 1, Fig. 3) though dronabinol did not increase ratings relative to hydromorphone+placebo. Dronabinol increased the percent of participants rating High ≥60 when compared to both placebo+placebo and hydromorphone + placebo (X2(4) = 10.5, p = 0.032, η2 = 0.21). Participants reported the least interest in taking the hydromorphone+dronabinol 10 mg combination again (F (4,112) = 2.56, p = 0.043, η2 = 0.04) (Table 1, Fig. 3). No significant main effect of drug condition was observed on the amount of money participants were willing to pay for the drug.
Cognitive testing
A significant main effect was observed for peak accuracy on the circular lights task (F (4,1122) = 6.82, p < 0.001, η2 = 0.14). Hydromorphone + placebo significantly decreased accuracy as compared to placebo + placebo (p = 0.001), and hydromorphone+dronabinol 5 mg (p = 0.004) and hydromorphone + dronabinol 10 mg (p < 0.001) significantly increased accuracy compared to hydromorphone+placebo. No differences were observed on other cognitive performance assessments.
Adverse events (AEs)
AEs were documented in 72 (49.7%) sessions and were experienced by 21 (72.4%) participants, ranging from 1 to 11 AEs within a single session. No serious AEs occurred. A significant main effect of drug condition was observed for the number of AEs rated as mild (F(4,112) = 3.28, p = 0.014, η2 = 0.06) and moderate (F(4,112) = 3.47, p = 0.010, η2 = 0.06). Hydromorphone + dronabinol 5 mg (p = 0.038) and hydromorphone+dronabinol 10 mg (p = 0.010) both produced significantly more mild AEs than placebo + placebo, but did not differ significantly from hydromorphone+placebo. Hydromorphone+dronabinol 10 mg also produced more moderate AEs than placebo + placebo (p = 0.044), but not hydromorphone + placebo. The main effect of total AEs (independent of severity) was significant (F(4,112) = 3.74, p = 0.007, η2 = 0.07). Significantly more AEs occurred during the hydromorphone+dronabinol 5 mg (p = 0.015) and hydromorphone+dronabinol 10 mg (p = 0.017) conditions relative to the placebo+placebo condition, with no difference observed relative to hydromorphone+placebo. The percent of participants experiencing any AE was also significant (X2(4)=10.100, p = 0.039, η2 = 0.20) and revealed that AEs were more likely to occur in all active drug conditions when compared to placebo+placebo (p’s < 0.01), but that dronabinol did not increase the rate of AEs relative to hydromorphone+placebo. One participant who did not complete the study experienced an acute panic attack during the hydromorphoned+dronabinol 10 mg condition.
Responder analysis
When opioid-responder status was added as a covariate to statistical tests, subjective rating of High was the only outcome for which a significant main effect of dose condition remained. Ratings of High in the hydromorphone+placebo and all hydromorphone+dronabinol conditions were greater than placebo+placebo. A sensitivity analyses conducted just within the opioid-responder subsample (N = 20; 69%) showed that the hydromorphone+placebo condition increased subjective drug effect ratings, increased abuse liability ratings, and decreased cognitive performance compared with placebo+placebo, but that the addition of dronabinol to hydromorphone did produce results that differed significantly from hydromorphone+placebo (Table 2, Fig. 3). However, responders did not consistently show greater analgesic efficacy across QST assessments. Greater analgesia was observed on the cold pressor (p = 0.002, η2 = 0.13) and TTS (p = 0.024, η2 = 0.07) outcomes in the hydromorphone+placebo and hydromorphone+dronabinol conditions compared with placebo+placebo, but no additive effect of hydromorphone+dronabinol combination was observed. No other QST measures differed by dose condition in the responder group. A sensitivity analysis within the nonresponder sample (N = 9; 31%) showed that subjective Drug Effects (p = 0.05, η2 = 0.13) were significantly greater than placebo+placebo in all active drug conditions, but only the highest (10 mg) dose of dronabinol increased abuse liability (“Enjoyed Medication”) compared with placebo+placebo (F(4,32) = 2.99, p = 0.03, η2 = 0.15). Heat pain threshold [HPTh] (F(4,32) = 3.1, p = 0.03, η2 = 0.16) increased in the hydromorphone+placebo and hydromorphone+dronabinol dose conditions compared with placebo+placebo among opioid non-responders; an effect not observed in the total study sample.
Table 2.
Responder analyses.
| Placebo | Hydromorphone 4 mg | P value (partial eta2) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dronabinol 0 mg | Dronabinol 2.5 mg | Dronabinol 5 mg | Dronabinol 10 mg | ||||||||
| Mean/% | SD | Mean/% | SD | Mean/% | SD | Mean/% | SD | Mean/% | SD | ||
| Opioid Responders (n = 20) | |||||||||||
| Participant Ratings (0–100) | |||||||||||
| Drug Effects | 10.0a | 10.1 | 51.1b | 22.9 | 55.7b | 29.4 | 55.7b | 29.3 | 56.9b | 31.7 | <0.001 (0.45) |
| Good Effects | 8.1a | 7.9 | 23.2 | 12.4 | 35.1b | 20.7 | 27.2b | 19.9 | 22.0 | 22.7 | 0.001 (0.14) |
| Bad Effects | 5.4a | 6.5 | 17.2 | 17.3 | 19.5 | 16.4 | 27.1b | 19.4 | 31.4b | 20.8 | <0.001 (0.16) |
| High | 2.9a | 3.6 | 21.3b | 12.7 | 30.6b | 21.4 | 37.3b | 23.1 | 30.3b | 22.8 | <0.001 (0.25) |
| Like the Way I Feel | 32.6 | 29.9 | 45.7a | 24.0 | 37.6 | 26.1 | 35.4 | 26.6 | 26.8b | 25.8 | 0.05 (0.06) |
| Nausea | 0.9a | 0.9 | 10.5 | 9.4 | 8.1 | 7.9 | 16.7b | 13.0 | 17.5b | 15.5 | 0.004 (0.11) |
| Abuse Potential Measures | |||||||||||
| Enjoyed medication (%) | 10.0a | 35.0b | 35.0b | 40.0b | 20.0b | <0.0001 (0.27) | |||||
| Willingness to pay for medication ($) | 0.01a | 2.7 | 3.4 | 4.8 | 3.5 | 7.6 | 6.9b | 8.3 | 2.9 | 4.6 | 0.009 (0.11) |
| Cognitive Testing | |||||||||||
| Circular lights, max per minute | 48.6a | 10.3 | 44.2b,c | 10.0 | 47.2 | 11.4 | 47.6 | 8.8 | 50.1d | 10.2 | <0.001 (0.17) |
| PASAT, Correct (%) | 79.2a | 72.3b,c | 79.2c | 80.0 | 80.0 | 0.026 (0.07) | |||||
| Opioid Nonresponders (n = 8) | |||||||||||
| Participant Ratings (0–100) | |||||||||||
| Drug Effects | 19.2 | 26.8 | 12.9a | 18.6 | 29.3 | 28.6 | 38.4b | 29.7 | 31.9 | 28.4 | 0.047 (0.02) |
| Good Effects | 8.6 | 17.0 | 19.7 | 30.6 | 29.0 | 33.7 | 30.4 | 37.6 | 14.7 | 26.0 | NS |
| Bad Effects | 15.0 | 26.2 | 3.7 | 5.3 | 12.9 | 16.2 | 14.3 | 15.8 | 21.7 | 29.8 | NS |
| High | 0.1 | 0.3 | 7.7 | 16.4 | 9.0 | 23.5 | 15.6 | 23.5 | 15.0 | 29.4 | NS |
| Like the Way I Feel | 25.4 | 35.9 | 33.8 | 35.1 | 34.1 | 37.6 | 45.2 | 36.5 | 26.2 | 29.8 | NS |
| Nausea | 0.9 | 2.3 | 3.3 | 6.7 | 6.6 | 7.3 | 13.7 | 20.2 | 16.4 | 18 | NS |
| Abuse Potential Measures | |||||||||||
| Enjoyed medication (%) | 7.7a | 20.0 | 42.8 | 50.0 | 33.3b | 0.033 (0.15) | |||||
| Willingness to pay for medication ($) | 12.8 | 32.9 | 5.7 | 8.7 | 2.7 | 4.4 | 6.8 | 9.6 | 1.8 | 3.2 | NS |
| Cognitive Testing | |||||||||||
| Circular lights, max per minute | 48.6 | 11.1 | 44.0a | 13.9 | 47.1 | 12.9 | 49.7b | 13.6 | 48.3 | 13.3 | 0.048 (0.13) |
| PASAT, Correct (%) | 75.6 | 75.6 | 78.1 | 78.1 | 66.3 | NS | |||||
Opioid responder defined as >20 point difference on Drug Effects visual analog scale between baseline and post-dose during hydromorphone 4 mg + placebo condition. 1 participant omitted from responder analysis due to elevated ratings (>50) at baseline. Only significant outcomes are shown.
Hydro hydromorphone, SD standard deviation, PASAT Paced Auditory Serial Addiction Task, sec second.
Subscripts denote significant differences in posthoc comparisons. Differences exist between a and b, c and d.
Partial eta2 effect sizes provided for significant results: small (0.01), medium (0.06), large (0.14).
Discussion
This study used a human laboratory model to assess changes in analgesic, abuse liability, and cognitive performance when hydromorphone was co-administered with dronabinol as compared with hydromorphone+placebo or placebo+placebo. This controlled, within-subject trial provided a rigorous method for assessing opioid–cannabinoid interactions using a small sample size that was not impacted by differences in participant pain profile or severity, or lifetime history and/or current use of opioid or other medications for analgesia. Thus, this design provided an opportunity to assess for a positive signal of dronabinol on opioid-sparing potential before moving to a large-scale trial within a clinical pain population.
As expected, a low dose of hydromorphone (4 mg) alone did not produce strong effects on QST outcomes when compared with placebo. The addition of dronabinol to hydromorphone increased analgesia on some QST methods, but the magnitude of the increase was modest and a clear dose-dependent increase in the strength of the effect was not observed. Rather, the smallest (2.5 mg) dronabinol dose showed the largest signal for increasing hydromorphone analgesic efficacy and the highest (10 mg) dronabinol dose produced a hyperalgesic effect wherein pain sensitivity increased on some QST outcomes. This pattern of dronabinol increasing opioid analgesic efficacy at low doses and increasing pain sensitivity at high doses has been observed in prior studies [25, 26]. The combined hydromorphone and dronabinol conditions (particularly 5 mg and 10 mg of dronabinol) also showed greater evidence of potential for abuse and greater impairment on one cognitive task relative to placebo, while also producing significantly more AEs. Together these data suggest that dronabinol may enhance the analgesic effects of a low dose of hydromorphone, indicative of possible opioid-sparing effects, but that this effect only occurs within a narrow dose range beyond which hyperalgesia, increased risk for AEs, and abuse liability are more likely to occur.
An analysis within participants categorized as “opioid-responders” (e.g., persons who subjectively reported discriminative drug effects during the hydromorphone-placebo condition) revealed that while dronabinol did not dose-dependently change their response to hydromorphone, they did experience overall greater subjective drug effects, showed more evidence of liking the study medication and willingness to pay for the study medication, and evidenced greater cognitive impairment than what was observed among the overall sample. In contrast, a parallel analysis within opioid nonresponders found that drug-based differences in participant self-reports that had been apparent in the larger sample were no longer significant and that abuse liability was only evident at the highest dronabinol dose. Though it remains possible that the sample was not sufficiently powered to detect an effect, these data might provide initial evidence that dronabinol enhancement of opioid effects and associated risks for abuse could vary depending on whether the individual is sensitive to a given opioid dose. This may partially explain the inconsistency that exists between patient reports of the opioid-sparing potential of THC and empirical research. Although this is the first opioid–cannabinoid administration study to examine the contribution of opioid responder status on outcomes, these results are consistent with prior laboratory evaluations that have found pronounced and clinically-meaningful individual differences in response to hydromorphone, heroin, and oxycodone [31, 46]. The fact that effects were observed within subjective reports, abuse liability, and cognition, but not analgesia, suggests that opioid sensitivity may have multiple physiological and/or pharmacological mechanisms. Additional research that examines the degree to which being an opioid responder impacts analgesic efficacy and/or the opioid-sparing properties of candidate medications is warranted.
This study is limited by lack of additional hydromorphone doses to examine interactions, examination of non-oral THC, and lack of self-administration to directly assess drug reinforcement. This study also did not examine cannabis, which has additional chemical constituents that may change its impact on opioids in ways we do not understand. Study results are further limited by the fact that we relied on experimental models of clinical pain conditions, rather than studying patients with clinical pain. However, the models used here have been shown to be predictive of analgesia in chronic pain populations in prior studies, and the use of experimental pain methods eliminates time-dependent variance in pain severity in patient populations. The need to standardize the hydromorphone+placebo condition as the first drug exposure session to establish safety may also have introduced an order effect, though this was minimized by the administration of QST prior to the first session and evidence that QST measures are robust and not significantly impacted by practice effects [47–49]. In addition, the doses examined here were selected for safety and to be consistent with prescribable dose ranges, however, it remains possible that higher or lower doses of hydromorphone and/or dronabinol may yield more clinically-meaningful effects. The study also did not collect carbon monoxide assessments of recent tobacco exposure prior to study sessions so cannot evaluate the degree to which this may have impacted analgesia. Finally, given the exploratory nature of this study [50], adjustments for multiple comparisons were not made and such adjustments could influence the overall significance of the effects.
Ultimately, these data largely replicate and extend upon prior research to suggest that a low dose (2.5 mg) of orally-administered THC (dronabinol) may enhance some analgesic effects of the opioid hydromorphone, but that undesirable effects emerge quickly (5 mg) and can become very uncomfortable (10 mg), likely undermining the clinical utility of this approach. The analgesic benefit observed with the hydromorphone+2.5 mg dronabinol dose was also not very large or consistent across all QST measures assessed. Data further suggested that effects were strongest within the subgroup of participants who were sensitive to opioid effects, an effect that should be explored further. Altogether these data provide limited support for the broad advancement of dronabinol as an opioid-sparing compound, with the acknowledgement that evaluations with different cannabinoids or in clinical populations may reveal different outcomes.
Funding and disclosures
This study was supported by grant funding from the National Institute on Drug Abuse (NIDA) R01DA040644 (MPI Dunn, Campbell), R01DA042751 (MPI Dunn, Campbell), R01DA035246 (Dunn), and F32DA049393 (Mun). The investigators have no relevant conflicts of interest to disclose. In the past 3 years, KED has consulted for Grünenthal, Inc. and MindMed, received honoraria for advisory board work for Canopy Corporation and Beckley-Canopy, and served as an unpaid advisor to Peabody Pharmaceuticals. ASH receives partial salary support from Ashley Addiction Treatment. RV has been a paid consultant to Canopy Health Innovations and received honoraria for advisory board work from FSD Pharma, Greenwich Biosciences, and Present Life Corporation.
Supplementary information
Acknowledgements
The study team thanks Ian Geithner, Megan Greene, Caitlyn Grubb, Jasmyne Jardot, Haley Puddy, Sandra Okobi, Leticia Nanda, and Dr. Annie Umbricht for their assistance with study sessions and Paul Nuzzo for his assistance conducting data analyses.
Author contributions
KED, RV, and CMC designed the trial and received grant funding, all study authors contributed to daily management of the study and session completion, KED led the data analyses and drafted the first edition of the manuscript, all authors reviewed and contributed to the final study manuscript.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41386-021-01007-4.
References
- 1.Ciccarone D. The triple wave epidemic: supply and demand drivers of the US opioid overdose crisis. Int J Drug Policy. 2019;71:183–8. doi: 10.1016/j.drugpo.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dowell D, Arias E, Kochanek K, et al. Contribution of opioid-involved poisoning to the change in life expectancy in the United States, 2000–2015. JAMA. 2017;318:1065–7. doi: 10.1001/jama.2017.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah A, Hayes CJ, Martin BC. Characteristics of initial prescription episodes and likelihood of long-term opioid use—United States, 2006–2015. Morb Mortal Wkly Rep. 2017;66:265–9. doi: 10.15585/mmwr.mm6610a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Johnson P, Jeng PJ, Reid MC, Witkin LR, Schackman BR, et al. First opioid prescription and subsequent high-risk opioid use: a national study of privately insured and Medicare advantage adults. J Gen Intern Med. 2018;33:2156–62. [DOI] [PMC free article] [PubMed]
- 5.Raman SR, Bush C, Karmali RN, Greenblatt LH, Roberts AW, Skinner AC. Characteristics of new opioid use among medicare beneficiaries: identifying high-risk patterns. J Managed Care Specialty Pharm. 2019;25:966–72. doi: 10.18553/jmcp.2019.25.9.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LaPietra AM, Motov S. A country in crisis: opioid sparing solutions for acute pain management. Mol Med. 2019;116:140. [PMC free article] [PubMed] [Google Scholar]
- 7.Foll L. Opioid-sparing effects of cannabinoids: myth or reality? Prog Neuro-Psychopharmacol Biol Psychiatry. 2020:110065. [DOI] [PubMed]
- 8.Babalonis S, Walsh SL. Therapeutic potential of opioid/cannabinoid combinations in humans: review of the evidence. Eur Neuropsychopharmacol. 2020. [DOI] [PMC free article] [PubMed]
- 9.Nielsen S, Sabioni P, Trigo JM, Ware MA, Betz-Stablein BD, Murnion B, et al. Opioid-sparing effect of cannabinoids: a systematic review and meta-analysis. Neuropsychopharmacology. 2017;42:1752–65. [DOI] [PMC free article] [PubMed]
- 10.Birch EA. The use of Indian hemp in the treatment of chronic chloral and chronic opium poisoning. Lancet. 1889;1:25. [Google Scholar]
- 11.Maguire DR, Yang W, France CP. Interactions between mu-opioid receptor agonists and cannabinoid receptor agonists in rhesus monkeys: antinociception, drug discrimination, and drug self-administration. J Pharm Exp Ther. 2013;345:354–62. doi: 10.1124/jpet.113.204099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunn KE, Huhn AS, Bergeria CL, Gipson CD, Weerts EM. Non-opioid neurotransmitter systems that contribute to the opioid withdrawal syndrome: a review of preclinical and human evidence. J Pharm Exp Ther. 2019;371:422–52. doi: 10.1124/jpet.119.258004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corroon JM, Jr, Mischley LK, Sexton M. Cannabis as a substitute for prescription drugs–a cross-sectional study. J Pain Res. 2017;10:989–98. doi: 10.2147/JPR.S134330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucas P, Walsh Z, Crosby K, Callaway R, Belle-Sile L, Kay R, et al. Substituting cannabis for prescription drugs, alcohol and other substances among medical cannabis patients: The impact of contextual factors. Drug Alcohol Rev. 2016;35:326–33. doi: 10.1111/dar.12323. [DOI] [PubMed] [Google Scholar]
- 15.Bergeria CL, Huhn AS, Dunn KE. The impact of naturalistic cannabis use on self-reported opioid withdrawal. J Subst Abuse Treat. 2020:108005. [DOI] [PMC free article] [PubMed]
- 16.Lötsch J, Weyer‐Menkhoff I, Tegeder I. Current evidence of cannabinoid‐based analgesia obtained in preclinical and human experimental settings. Eur J Pain. 2018;22:471–84. doi: 10.1002/ejp.1148. [DOI] [PubMed] [Google Scholar]
- 17.Bachhuber MA, Saloner B, Cunningham CO, Barry CL. Medical cannabis laws and opioid analgesic overdose mortality in the United States, 1999–2010. JAMA Intern Med. 2014;174:1668–73. doi: 10.1001/jamainternmed.2014.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell G, Hall W, Nielsen S. What does the ecological and epidemiological evidence indicate about the potential for cannabinoids to reduce opioid use and harms? A comprehensive review. Int Rev Psychiatry. 2018;30:91–106. doi: 10.1080/09540261.2018.1509842. [DOI] [PubMed] [Google Scholar]
- 19.Humphreys K, Saitz R. Should physicians recommend replacing opioids with cannabis? JAMA. 2019;321:639–40. doi: 10.1001/jama.2019.0077. [DOI] [PubMed] [Google Scholar]
- 20.Hindley G, Beck K, Borgan F, Ginestat CE, McCutcheon R, Kleinloog D, et al. Psychiatric symptoms caused by cannabis constituents: a systematic review and meta-analysis. Lancet Psychiatry. 2020. [DOI] [PMC free article] [PubMed]
- 21.Hobbs M, Kalk NJ, Morrison PD, Stone JM. Spicing it up-synthetic cannabinoid receptor agonists and psychosis-a systematic review. Eur Neuropsychopharmacol. 2018;28:1289–304. doi: 10.1016/j.euroneuro.2018.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Shover CL, Davis CS, Gordon SC, Humphreys K. Association between medical cannabis laws and opioid overdose mortality has reversed over time. Proc Natl Acad Sci. 2019;116:12624–6. doi: 10.1073/pnas.1903434116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naef M, Curatolo M, Petersen-Felix S, Arendt-Nielsen L, Zbinden A, Brenneisen R. The analgesic effect of oral delta-9-tetrahydrocannabinol (THC), morphine, and a THC-morphine combination in healthy subjects under experimental pain conditions. Pain. 2003;105:79–88. doi: 10.1016/s0304-3959(03)00163-5. [DOI] [PubMed] [Google Scholar]
- 24.Roberts JD, Gennings C, Shih M. Synergistic affective analgesic interaction between delta-9-tetrahydrocannabinol and morphine. Eur J Pharmacol. 2006;530:54–8. doi: 10.1016/j.ejphar.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 25.Babalonis S, Lofwall MR, Sloan PA, Nuzzo PA, Fanucchi LC, Walsh SL. Cannabinoid modulation of opioid analgesia and subjective drug effects in healthy humans. Psychopharmacology. 2019;236:3341–52. doi: 10.1007/s00213-019-05293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper ZD, Bedi G, Ramesh D, Balter R, Comer SD, Haney M. Impact of co-administration of oxycodone and smoked cannabis on analgesia and abuse liability. Neuropsychopharmacology. 2018;43:2046–55. doi: 10.1038/s41386-018-0011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aviram J, Samuelly-Leichtag G. Efficacy of cannabis-based medicines for pain management: a systematic review and meta-analysis of randomized controlled trials. Pain Physician. 2017;20:E755–E96. [PubMed] [Google Scholar]
- 28.Comer SD, Zacny JP, Dworkin RH, Turk DC, Bigelow GE, Foltin RW, et al. Core outcome measures for opioid abuse liability laboratory assessment studies in humans: IMMPACT recommendations. Pain. 2012;153:2315–24. doi: 10.1016/j.pain.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Overholser BR, Foster DR. Opioid pharmacokinetic drug-drug interactions. Am J Manag Care. 2011;17:S276–S87. [PubMed] [Google Scholar]
- 30.Dunn KE, Brands B, Marsh DC, Bigelow GE. Characterizing the subjective, observer-rated, and physiological effects of hydromorphone relative to heroin in a human laboratory study. Psychopharmacology. 2018;235:971–81. doi: 10.1007/s00213-017-4814-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunn KE, Barrett FS, Brands B, Marsh DC, Bigelow GE. Individual differences in human opioid abuse potential as observed in a human laboratory study. Drug Alcohol Depend. 2019;205:107688. [DOI] [PMC free article] [PubMed]
- 32.Carter LP, Griffiths RR. Principles of laboratory assessment of drug abuse liability and implications for clinical development. Drug Alcohol Depend. 2009;105:14. doi: 10.1016/j.drugalcdep.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arendt-Nielsen L. Central sensitization in humans: assessment and pharmacology. Handb Exp Pharmacol. 2015;227:79–102. doi: 10.1007/978-3-662-46450-2_5. [DOI] [PubMed] [Google Scholar]
- 34.Campbell CM, Buenaver LF, Raja SN, Kiley KB, Swedberg LJ, Wacnik PW, et al. Dynamic pain phenotypes are associated with spinal cord stimulation-induced reduction in pain: a repeated measures observational pilot study. Pain Med. 2015;16:1349–60. doi: 10.1111/pme.12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rolke R, Baron R, Maier CA, Tolle TR, Treede DR, Beyer A, et al. Quantitative sensory testing in the German research network on neuropathic pain (DFNS): standardized protocol and reference values. Pain. 2006;123:231–43. doi: 10.1016/j.pain.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 36.Campbell CM, Buenaver LF, Finan P, Bounds SC, Redding M, McCauley L, et al. Sleep, pain catastrophizing, and central sensitization in knee osteoarthritis patients with and without insomnia. Arthritis Care Res. 2015;67:1387–96. [DOI] [PMC free article] [PubMed]
- 37.Dirks J, Petersen KL. The heat/capsaicin sensitization model: a methodologic study. J Pain. 2003;4:122–8. doi: 10.1054/jpai.2003.10. [DOI] [PubMed] [Google Scholar]
- 38.Mathiesen O, Imbimbo BP, Hilsted KL, Fabbri L, Dahl JB. CHF3381, a N-methyl-D-aspartate receptor antagonist and monoamine oxidase—a inhibitor, attenuates secondary hyperalgesia in a human pain model. J Pain. 2006;7:565–74. doi: 10.1016/j.jpain.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Treede R, Meyer RA, Raja SN, Campbell JN. Peripheral and central mechanisms of cutaneous hyperalgesia. Prog Neurobiol. 1992;38:397–421. doi: 10.1016/0301-0082(92)90027-c. [DOI] [PubMed] [Google Scholar]
- 40.Frymoyer AR, Rowbotham MC, Petersen KL. Placebo-controlled comparison of a morphine/dextromethorphan combination with morphine on experimental pain and hyperalgesia in healthy volunteers. J Pain. 2007;8:19–25. doi: 10.1016/j.jpain.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Petersen KL, Rowbotham MC. A new human experimental pain model: the heat/capsaicin sensitization model. Neuroreport. 1999;10:1511–6. doi: 10.1097/00001756-199905140-00022. [DOI] [PubMed] [Google Scholar]
- 42.US Food and Drug Administration. Assessment of abuse potential of drugs: Guidance for industry. US Department of Health and Human Services. https://www.fda.gov/downloads/drugs/guidances/ucm198650.pdf. 2017. Accessed 04 April 2021.
- 43.Herrmann ES, Cone EJ, Mitchell JM, Bigelow GE, LoDico C, Flegel R, et al. Non-smoker exposure to secondhand cannabis smoke II: effect of room ventilation on the physiological, subjective, and behavioral/cognitive effects. Drug Alcohol Depend. 2015;151:194–202. doi: 10.1016/j.drugalcdep.2015.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mintzer MZ. Effects of opioid pharmacotherapy on psychomotor and cognitive performance: a review of human laboratory studies of methadone and buprenorphine. Heroin Addiction Relat Clin Probl. 2007;9:5–24. [Google Scholar]
- 45.Mintzer MZ, Copersino ML, Stitzer ML. Opioid abuse and cognitive performance. Drug Alcohol Depend. 2005;78:225–30. doi: 10.1016/j.drugalcdep.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 46.Antoine DG, Strain EC, Tompkins DA, Bigelow GE. Opioid abusers’ ability to differentiate an opioid from placebo in laboratory challenge testing. Drug Alcohol Depend. 2013;132:369–72. doi: 10.1016/j.drugalcdep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Neill S, O’Neill L. Improving QST reliability—more raters, tests, or occasions? A multivariate generalizability study. J Pain. 2015;16:454–62. doi: 10.1016/j.jpain.2015.01.476. [DOI] [PubMed] [Google Scholar]
- 48.Kennedy DL, Kemp HI, Ridout D, Yarnitsky D, Rice AS. Reliability of conditioned pain modulation: a systematic review. Pain. 2016;157:2410. doi: 10.1097/j.pain.0000000000000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nothnagel H, Puta C, Lehmann T, Baumbach P, Menard MB, Gabriel B, et al. How stable are quantitative sensory testing measurements over time? report on 10-week reliability and agreement of results in healthy volunteers. J Pain Res. 2017;10:2067–78. doi: 10.2147/JPR.S137391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Althouse AD. Adjust for multiple comparisons? it’s not that simple. Ann Thorac Surg. 2016;101:1644–45. doi: 10.1016/j.athoracsur.2015.11.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.