

Abstract
Ageing is a risk factor for chronic diseases including cancer, cardiovascular diseases, neurodegenerative disorders, and metabolic syndrome. Among others, senescence mechanisms have become a target of huge research on the topic of the ageing process. Cellular senescence is a state of an irreversible growth arrest that occurs in response to various forms of cellular stress and is characterized by a pro-inflammatory secretory phenotype. Multiple studies showed that cellular senescence occurs in both physiological and pathophysiological conditions. Senescent cells accumulate with ageing and can contribute to age-related decline in tissue function. Obesity is a metabolic condition that can accelerate the ageing process by promoting a premature induction of the senescent state of the cells. In contrast, caloric restriction without malnutrition is currently the most effective non-genetic intervention to delay ageing, and its potential in decreasing the cellular senescent burden is suggested. Here, it will be highlighted the cellular and molecular mechanisms involved in cellular senescence and discussed some of the research that is being done about how environmental conditions such as diet can affect the accumulation of senescent cells.
Keywords: ageing, caloric restriction, caloric restriction mimetics, cellular senescence, obesity
Introduction
Ageing is an inevitable and multi-factorial biological process that promotes a progressive deterioration across multiple organ systems, resulting in tissue dysfunction.1 Therefore, age is a risk factor for chronic diseases including cancer,2 cardiovascular diseases,3 neurodegenerative disorders,4 and metabolic syndrome.5 Despite the available evidence, there is yet a lot to be known about the ageing mechanisms and the specific molecular traits involved in this process. The identification of so-called ageing hallmarks was an asset on ageing research once it allowed the delaying of multiple age-related diseases by targeting ageing process.6 Among all of the ageing hallmarks, cellular senescence mechanisms are considered the key factor in the complexity of ageing, which explains the huge research that recently is being done about this topic.7,8 Thus, this review focuses on cellular senescence process, in some of the stress mechanisms that drive this cellular state and its consequences to cell homeostasis. Also, the influence of some metabolic conditions, like obesity, in the increment of the senescent cell burden and the beneficial effects of caloric restriction (CR) and resveratrol, a caloric restriction mimetic (CRM), in the prevention of the accumulation of senescent cells will be discussed.
Cellular senescence—What is it?
Senescence is a cellular state characterized by a cell cycle division arrest in response to a variety of cellular stresses, including telomere shortening and oxidative stress, which involves chromatin remodelling and metabolic reprogramming (Fig. 1).9,10 In contrast with apoptosis, in which phagocytes are released to remove damaged cells without causing inflammation, senescent cells survive due to the stimulation of the inflammatory environment, caused by activation of senescence-associated secretory phenotype (SASP) factors.11
Figure 1.
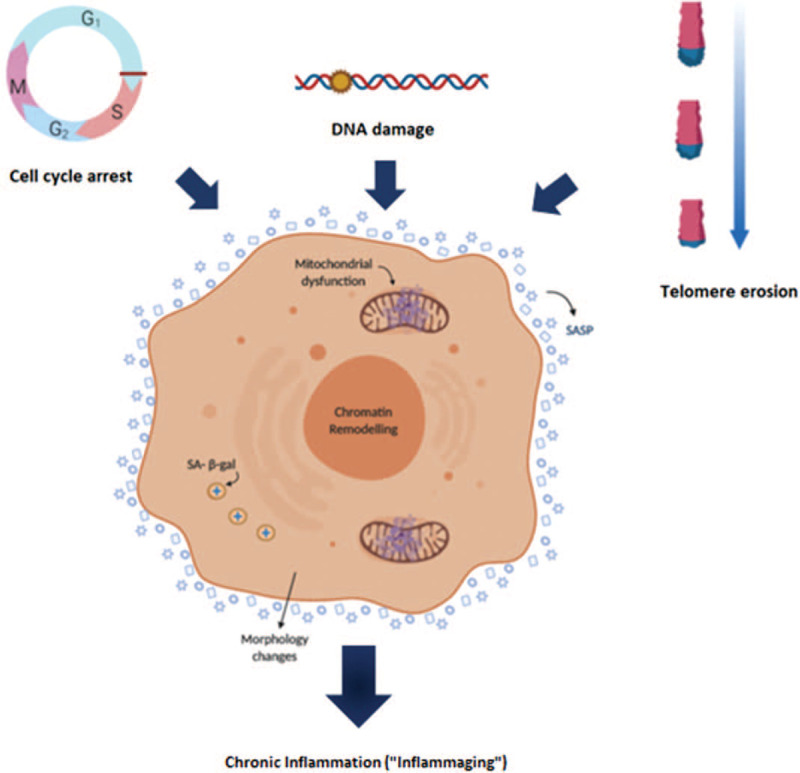
Senescence as a key factor of ageing. Telomere shortening, DNA damage by ROS, and cell cycle arrest are some of the stimuli that mediate damage during ageing. These drivers of damage are known to be involved in the cellular senescence initiation. Senescence is a cellular state characterized by some phenotypic alterations and during ageing, the increase of the cellular senescence burden causes sterile inflammation. pRB = protein of retinoblastoma; SA-b-gal = senescence-associated b-galactosidase.
Senescence was first described by Hayflick and Moorhead (1961) during serial passage of human fibroblasts. Thus, it was establish that, in normal cell cultures, replicative senescence occurs after a limited number of population doublings, the so-called Hayflick limit.12 Besides fibroblasts, cellular senescence is described in other cell types, including epithelial and endothelial cells, lymphocytes, and possibly even post-mitotic cells, like neurons or glial cells.13 Multiple studies demonstrated that cellular senescence occurs in both physiological and pathophysiological conditions. For instance, senescence can be engaged during development and is also essential for tissue remodelling.14 Also, transient induction of senescent cells is observed during wound healing and contributes to its resolution.15 Senescence can also play a protective stress response. For example, senescence is best known as a potent anticancer mechanism.16 However, with age, senescent cells can accumulate. Senescent cells limit the regeneration of metabolic tissues and through the SASP, modify tissue architecture, and cause sterile inflammation. This leads to a disruption of the metabolic homeostasis, causing age-related diseases.17
Cells undergoing cellular senescence have a stable growth arrest, are usually enlarged, and report some morphology alterations but are metabolically active.18 The most common marker to detect senescent cells in vitro is the senesce-associated β-galactosidase (SA-β-gal), which is an isoform of beta-galactosidase enzyme and whose activity is increased in lysosomes of senescent cells.19
Factors driving senescence
Oxidative stress
Reactive oxygen species (ROS), natural by-products of normal oxygen metabolism, are considered to regulate several physiological functions such as signal transduction, gene expression, and proliferation. However, high ROS content is associated with a decreased lifespan and with the rise and maintenance of the senescent phenotype.20 The balance between oxidant generation and antioxidant processes is kept in healthy tissues due to a predominance of various antioxidants but, with age, this balance is disrupted, which causes damage in macromolecules (DNA, proteins, and lipids).21
Endogenous or exogenous sources of ROS, such as UV and ionizing radiation or environmental toxins, stimulate the cellular senescent phenotype by mechanisms that involve the response to DNA damage, epigenetic regulation and tumour suppression pathway activation (mostly the ones involved in cell cycle control: p53 (cellular tumour antigen p53), p21 (cyclin-dependent kinase inhibitor 1), Rb (retinoblastoma protein)). ROS together with SASP factors release stimulate a positive feedback loop that results in increased ROS production, especially in mitochondrial, which increases intracellular ROS and contributes to the maintenance of the senescent phenotype by telomere shortening and dysfunction.22,23
Telomere shortening
Telomeres are highly repetitive DNA specialized structures located at the end of chromosomes, composed of several kilobases (kb) of simple repeats (TTAGGG)n. The biological function of telomeres is to protect the chromosomes from end-to-end fusions, harmful rearrangements, and chromosome loss.24 The length of telomeres is intimately related to the replicative ability of cells, and thus it can be used as a predictor of cellular senescence. DNA polymerases cannot synthesize DNA without a template, and the end-replication problem in telomeres is a consequence of this. This means that telomere shortening occurs in every cell cycle division.25 In youth, it is estimated that the length of the telomere is around 11 to 15 kb and about 4 to 7 kb in the elderly.26 During normal ageing, the shortening of the telomeres is controlled by the telomerase. However, under some pathological conditions, there is an imbalance between telomere shortening and the counteracting by telomerase, which results in accelerated senescence.
Tumour suppressors and cell cycle inhibitors
Nowadays, several suppressors and cell cycle inhibitors are known. Activation of p16INK4A, p53, and p21 pathways occurs during senescence and is triggered by DNA damage. Telomere shortening can trigger a permanent DNA damage response (DDR) in human cells. This event leads to the recruitment of the damage sensor ataxia telangiectasia mutated (ATM) to uncapped telomeres, resulting in the upregulation of p53 and the p53 transcriptional target p21. In this setting, p21 inhibits cyclin-dependent kinase 2 (CDK2)-mediated inactivation of RB, preventing cells to enter the S phase of the cell cycle. DNA damage by oxidative stress can also support the ATM-p53-p21 axis. Another barrier to proliferation is p16Ink4a, which prevents CDK4- and CDK6-mediated inactivation of RB, resulting in cell cycle arrest. Depending on stress or cell type, this mechanism can act either alone or in combination with the p53-p21 pathway (Fig. 2).27
Figure 2.
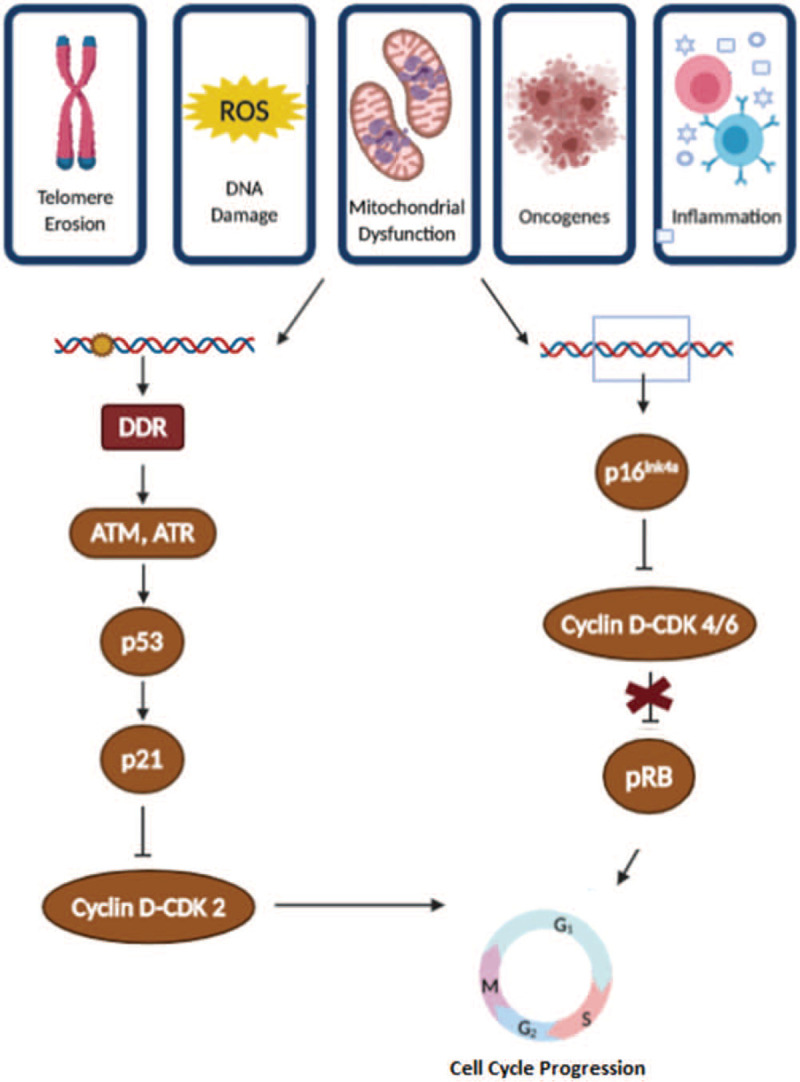
Molecular pathways involved in the senescence-associated growth arrest. A spectrum of stressors induces senescence-associated growth arrest. Cell cycle arrest is mediated through 2 main pathways, p16INK4a/pRb and p53/p21CIP1. Both collide in the activation of the pRB, which prevents cells to enter the S phase of the cycle. ATM = ataxia telangiectasia mutated; ATR = ATM and Rad3-related; CDK = cyclin-dependent kinase; DDR = DNA damage response; p16 = cyclin-dependent kinase inhibitor 16; p21 = cyclin-dependent kinase inhibitor 21; p53 = tumour suppressor p53 protein; pRB = tumour protein of the retinoblastoma.
A typical essay that is normally used to verify if cells have exited the cell cycle is the measurement of the expression levels of the CDKIs p16 and p21.28
Consequences of cellular senescence
The SASP
An important feature of senescent cells is the SASP. Despite the specific combination of secreted factors may depend on the cell type, many of the key effectors of the SASP and his regulatory mechanisms seemed to be shared. The proinflammatory nuclear factor-kB (NF-kB) and CCAAT/enhancer-binding protein beta (CEBP/ β) are the key transcriptional SASP regulators.29 DNA damage, mTOR signalling, Jak2/Stat3 pathway, and mitochondrial dysfunction can also contribute to the release of SASP factors.30,31 The SASP is mainly composed by an increased secretion of a range of factors, such as pro-inflammatory cytokines, especially interleukin-1 (IL-1), interleukin-6 (IL-6) and interleukin-8 (IL-8), chemokines (eg, MCP-1), growth factors, like insulin-growth factor (IGF), tumour necrosis factor-alpha (TNF-α) and metalloproteinases. Due to its complexity, the SASP is responsible for both positive and negative biological functions.32 The major function of the SASP is to recruit the immune system to destroy senescent cells, by stimulation of adaptive and innate immune cells.33 On the other hand, factors secreted by senescent cells can drive both differentiated cells and stem cells into the senescent phenotype, exacerbating the cellular senescence burden during ageing. Also, some of the SASP factors can modify the extracellular matrix (ECM) components and be involved in the epithelial-to-mesenchymal transition in susceptible cells. Thus, with age, the accumulation of senescent cells amplifies the SASP signal, which changes the cellular microenvironment and results in chronic inflammation (Fig. 3). This persistent chronic inflammation that occurs during ageing, also known as inflammaging, is responsible for the development of various age-related pathological conditions.34
Figure 3.
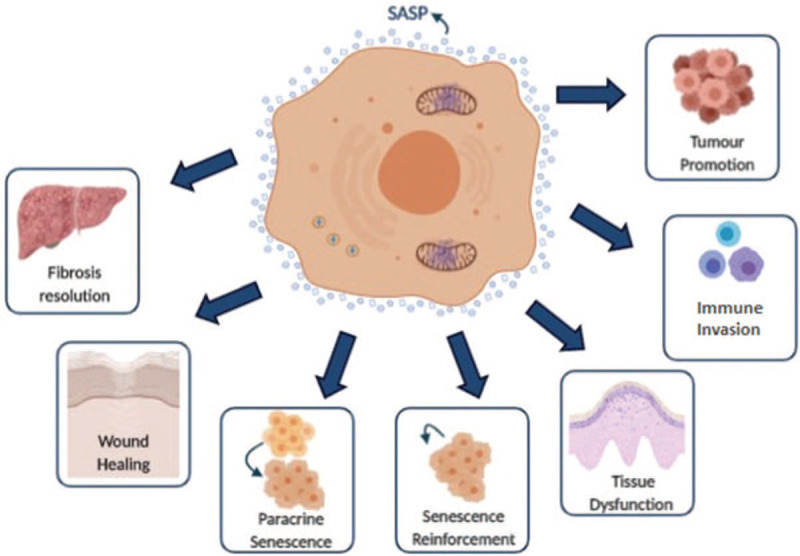
The senescence-associated secretory phenotype (SASP) as an important mediator of the pathological functions of senescence. Schematic representation of some of the consequences of the senescence cells to the organism mediated by SASP factors release.
Metabolic shift
In physiological conditions, cells maintain a balance between cell division and cell metabolism to preserve the optimal building of cell components and the cellular energy levels for cell division activity. On the other hand, when cells undergo senescence, they are in a permanently arrested state of cell growth and seem to exhibit an altered metabolism.35 Some studies report a shift to a more glycolytic state and a less energetic one, in cells under replicative senescence, in culture.36 However, in a contradictory way, it is also described that, during senescence, the AMP: ATP and ADP: ATP ratios increase, resulting in AMPK dephosphorylation and activation of its signalling pathway.31 The AMPK acts as a sensor of reduced energetic cellular state, consequently promoting catabolic pathways activation, like β-oxidation, while inhibiting biosynthetic ones. During senescence, AMPK can be activated in an ATM-dependent manner in response to genotoxic stress. This will result in direct phosphorylation of p53 and the increase of p21 transcription. Also, AMPK can inhibit Hu antigen R (HuR), which increases the stability of the p21 and p16 mRNAs, factors involved in the unleash of the cellular senescent process by increasing the activity of the pRB tumour suppressor. p53 is known to regulate cellular metabolism by inhibiting glycolysis37,38 and promoting the tricarboxylic acid (TCA) cycle, oxidative phosphorylation, and β-oxidation, yet pRB seems to promote glucose uptake and glycolysis (Fig. 4).39,40
Figure 4.
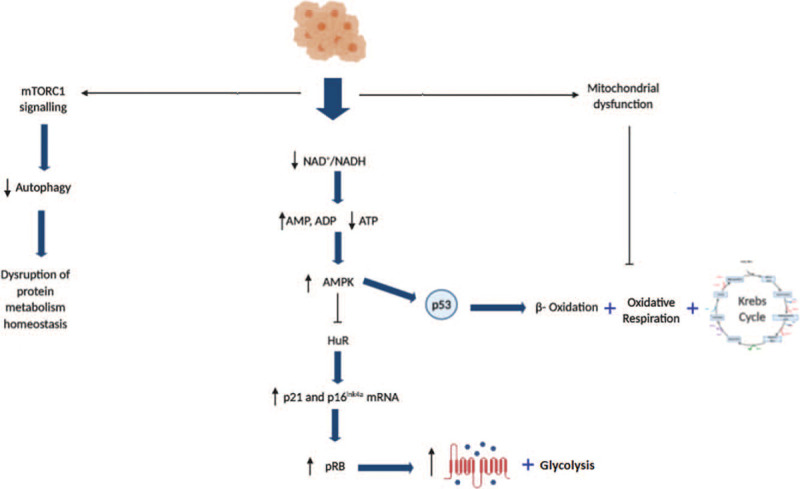
Metabolic changes in cellular senescence. Activation of the key signalling pathways that act as metabolic switches to regulate senescence. ADP = adenosine dinucleotide; AMP = adenosine mononucleotide; AMPK = AMP-activated protein kinase; ATP = adenosine trinucleotide; HuR = Hu antigen receptor; mRNA = messenger RNA; mTORC1 = mammalian target of rapamycin complex 1; NAD+ = oxidized nicotinamide adenine dinucleotide; NADH reduced nicotinamide adenine dinucleotide; p16 = cyclin-dependent kinase inhibitor 16; p21 = cyclin-dependent kinase inhibitor 21; p53 = tumour suppressor p53 protein; pRB = tumour protein of the retinoblastoma.
While there is strong evidence that correlates senescence and glycolysis, the precise mechanisms involved in this metabolic shift are unclear. One possible explanation could be related to the demands of the SASP promoting considerable macromolecular biosynthesis. Thus, it is possible that senescence cells, in a similar manner to cancer cells, enhance the glycolytic process due to its ability to provide precursors for producing protein, lipids, and other cellular components.41
Mitochondrial dysfunction and mTOR activation are also responsible for some metabolic changes in senescent cells. Mitochondrial dysfunction-associated senescence (MiDAS) activates AMPK, reducing TCA cycle activity and NAD+ content, which are both involved in the cellular senescence phenotype.42 Moreover, mTOR signalling decreases autophagy, influencing the protein metabolism homeostasis.43
Extracellular matrix remodelling
Recently, it was suggested that cells under senescence promote an altered expression of adhesion molecules (AMs) and, consequently, remodelling in the extracellular matrix (ECM). AMs are pro-inflammatory proteins involved in cell-cell/cell-matrix interactions, which are required for the inflammation process and the immune response. E-selectin, P-selectin, vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1 (ICAM-1) are the major players in this process. They are maintained at low levels in endothelial cells, in normal biological conditions, but under stress (inflammation or oxidative stress), there is an increased content of these molecules in the plasma. The high levels of soluble AMs in plasma have been used as a vascular risk factor for many cardiovascular abnormalities, including peripheral arterial disease, coronary heart disease, stroke, and other conditions that may occur with age.44
During ageing, cells lose their ability to synthesize the correct matrix components. The proportion of ECM components, like collagen fibers, fibronectin, or laminin increase with age.45 The changes that occur in the ECM are involved in the impairment of cell attachment and changes in cellular morphology. For instance, it was observed that, during the senescence process, the ECM becomes less soluble and proteolytically digestible, which can emerge as a consequence of the formation of age-related intermolecular cross-links.46
Conditions that accelerate and exacerbate cellular senescence
Obesity
Obesity is a metabolic disease caused by an unbalance between the nutrient consumption and the energy expenditure, considered to be a major risk factor for chronic diseases, including cardiovascular diseases, type 2 diabetes, cancer, osteoarthritis, and sleep apnea.47 Recently, some studies have associated the obese condition with a decrease in lifespan and it is thought that its impact in cellular mechanisms can be similar to the ageing process.48 As a matter of fact, nutrient excess may promote a premature induction of cellular senescence by accelerating intrinsic, age-related causes of the senescence process. For example, some studies have reported telomere attrition in obese adult.49 Also, increased metabolic loading is associated with mitochondrial dysfunction, causing an uncoupling in the mitochondrial respiration, impaired electron transport chain function, and increased production of ROS.50 Indeed, increased markers of ROS damage have been identified in samples of adipose tissue, cardiovascular tissue, and liver of obese humans and mice.51 In addition, markers of DNA damage, one of the stimuli that are involved in the activation of the senescent profile, have been found in obese humans with type 2 diabetes. Finally, obesity is also associated with metabolic changes, including glucose uptake and the activation of the biosynthetic pathways, which can promote the activation and maintenance of the senescent state of the cells.52
Caloric restriction as a cellular senescence modulator
Caloric restriction
CR is a well-established intervention for reducing age-associated chronic diseases and enhancing lifespan, which typically involves reducing caloric intake by 30% to 40% while maintaining adequate nutrition. The beneficial effects of CR for extending healthspan and lifespan were observed in multiple animal models, including yeast, fruit flies, worms, and rodent.53 In mice, the reduction of dietary calories by 20 up to 50% promotes a significant extension of both average and maximal lifespan. Also, many of the typical age-associated chronic diseases can be prevented or delayed. For example, the incidence of cancer is drastically reduced in animals under CR; plus similar reduction or slowing down of disease progression was observed for neurodegenerative disorders, cardiovascular diseases, and metabolic syndrome.54 There is also some interesting data reporting the benefits of CR in non-human primates. In rhesus monkeys, 2 differently designed studies showed distinct results on lifespan but similar health benefits improvements and delayed onset of ageing phenotypes.55 In humans, CR with adequate intake of vitamins and minerals seems to counteract several of the mechanisms involved in age-associated diseases. Non-obese, healthy adults following continuous CR (15%–25%) for 24 months demonstrated improvements in the quality of life and reduced body mass (mostly).56 Many aspects, including fasting insulin levels, resting energy expenditure, thyroid axis, oxidative stress, inflammation markers, and cardiometabolic risk factors were significantly reduced under CR.57–59 In obese humans, CR improves general health and has a significant impact on weight loss.60
Molecular mechanisms underlying CR effects
CR interventions are associated with several age-associated pathophysiological changes, including reduction of metabolic rate and oxidative damage, enhanced cellular turnover and protein homeostasis and improvement of chronic metabolic disorders. The specific mechanisms underlying the benefits of CR are not fully understood, however, some potential target pathways have been suggested, including the activation of AMP protein kinase (AMPK) and sirtuins (SIRTs), inhibition of insulin-like growth factor-1 (IGF-1) signalling, and the inhibition of the mechanistic target of rapamycin (mTOR) (Fig. 5).61
Figure 5.
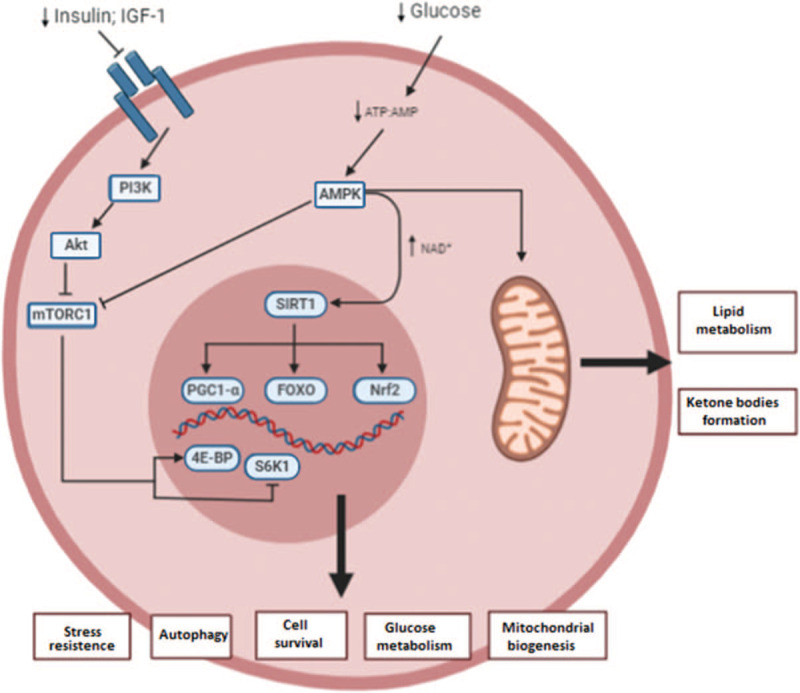
Molecular mechanisms involved in caloric restriction (CR). The decreased energy intake promoted by CR intervention involves the modulation of mainly 4 signalling pathways: AMP-activated protein kinase (AMPK), insulin-like growth factor-1 (IGF-1), mammalian target of rapamycin (mTOR), and silent mating type information regulation 2 homolog (SIRTs). AE BP = adipocyte enhancer binding protein; Akt = protein kinase B; AMP = adenosine mononucleotide; AMPK = AMP-activated protein kinase; ATP = adenosine trinucleotide; FOXO = forkhead box O; mTORC1 = mammalian target of rapamycin complex 1; NAD+ = oxidized nicotinamide adenine dinucleotide; Nrf2 = nuclear factor erythroid 2-related factor 2; PGC1-a = peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PI3K = phosphatidylinositol 3-kinase; S6K1 = ribosomal protein S6 kinase beta-1.
CR implies a decrease in the energy intake that will increase AMP to ATP ratios. This promotes AMPK activation, which triggers repair and some metabolic changes: in a short time frame, there is an increase in glycolysis and fatty acid oxidation that will be replaced by an increase in mitochondrial content and the use of mitochondrial substrates as an energy source. AMPK activation leads with Acetyl-CoA and nicotine amide adenine dinucleotide (NAD+) formation, which serve as cofactors for epigenetic modifiers and SIRTs. SIRTs deacetylate forkhead box Os (FOXOs) and peroxisome proliferator-activated receptor ϒ coactivator 1α (PGC-1α), factors respectively involved in stress resistance and mitochondrial biogenesis.62 Production of ketone bodies like β-hydroxybutyrate from β-oxidation can act as endogenous histone deacetylase (HDAC) inhibitors and its contribute to epigenetic control of gene expression, DNA repair, and genome stability, is suggested.63 Ketogenesis also promotes synaptic plasticity and neurogenesis by increasing the expression of brain-derived neurotrophic factor (BDNF).64
CR has also impact in IGF-1 pathway. After a few hours of fasting, there is a down-regulation of the IGF-1 signalling pathway, which represses the activity of mTOR and its downstream effector, the ribosomal protein S6 kinase beta-1 (S6K). This mechanism inhibits global protein synthesis and promotes the recycling of macromolecules by stimulation of autophagy. Autophagy is an important mechanism for the removal of dysfunctional organelles, amyloid, and other protein aggregates that interfere with normal cell function.65
Additionally, recent studies proved that CR promotes the expression and activity of NRF2, which induces several antioxidative and carcinogen-detoxifying enzymes.66
CR and cellular senescence
Recent studies have shown that CR reduces senescence markers in different mouse organs and human colon mucosa.67–69 The precise mechanisms involved in the delaying of cellular senescence by CR are yet poorly understood, however, that are some targets that can be distinguished. One of the main inducers of senescence is cellular damage. Thereby, it is proposed that CR can prevent the damage to occur, reducing the generation of senescent cells by 2 different mechanisms. CR can protect against cellular senescence by interfering with the source of damage (oxidative stress or inflammation), or repairing/eliminating already present damage, perhaps by increasing autophagy.57,70,71
Caloric restriction mimetics
Despite the benefits of CR in lifespan and healthspan, most people cannot follow such a severe diet program, especially in the long term. This raises the need for the development of natural or synthetic molecules that can mimic the effects of CR, without reducing food intake. Such compounds are identified as CR mimetics (CRM) and their beneficial effects were observed in both mice and humans.72–74 The potential CRM under research acts on the same signalling pathways as CR, including insulin pathway, activation of AMPK, autophagy stimulus, alpha-lipoic acid, and other antioxidants.75 In this context, the role of polyphenols can be highlighted once they are natural compounds with reported antioxidant and anti-inflammatory activity.76,77 In addition, natural polyphenols show low toxicity and the absence of high collateral effects.78
Resveratrol
Resveratrol is one of the most well-known natural polyphenols, which is particularly abundant in the peel of grapes and in red wine. Several studies report that its administration can promote longevity across species and improve some age-related parameters in mice.79 Also, resveratrol can protect against metabolic syndrome, type 2 diabetes, cancer, neurodegeneration, and cardiovascular diseases.80 Most of the benefic effects of Resveratrol are associated to its ability to induce autophagy, however, like other polyphenols, it was found that this CRM is also capable of modulating the expression of pro- and anti-apoptotic factors, eliminate free radical species, improve mitochondrial functions and avoid protein aggregation.81 Recently, the role of resveratrol in extending lifespan has been linked to an increase of SIRT1 activity, which is the same mechanism suggested for the longevity caused by CR. In a comparative study regarding the anti-ageing effect of resveratrol and CR, it is shown that both have similar activities of recovering SIRT1 mRNA levels, increasing the protein expression of FOXO 3a, AROS and HuR and decreasing p53 and DBC1 levels. Thus, it is suggested that resveratrol and CR exhibited similar anti-ageing activities both in vitro and in vivo by regulation of the SIRT1 pathway, implicating the potential of resveratrol as a CR mimetic.82
Conclusion
Normal human cells do not divide indefinitely. When cultured in vitro, cells can undergo only a finite number of divisions before entering in a nondividing state, the so-called replicative senescence. Senescence has been suggested both as contribute and a consequence of the ageing process and is involved in the development of many age-related chronic diseases. An increasing body of evidence supports that this cellular process can be triggered and exacerbated by nutrient excess.83 Besides, recent studies shown that CR can reduce senescence in the mouse liver and intestine.84 However, the precise mechanisms underlying the effect of obesity in the induction of premature cellular senescence are poorly understood and warrant further investigation. Moreover, more studies are required to understand how lowering calories intake reduces cellular senescence burden, and whether this can directly lower levels of molecules involved in the inflammation process, like interleukins, which, for instance, could also be promoted by other variables independently altered by senescence. Plus, in obesity and ageing studies, the researchers tend to focus on one specific organ or pathology type, which limits the information that may be collected about the temporal biological order of senescence induction. Thus, more in vitro studies are required especially in cellular model systems that can replicate the alterations seen during in vivo progression in the ageing process. So far, most of the in vitro studies involving senescence or data showing the influence of nutrient excess/deprivation on this process are presented in fibroblasts as a cellular model. Therefore, studies in other types of cells, such as endothelial ones, which are particularly prone to damage and dysfunction, are needed to elucidate the cellular and molecular mechanisms of cellular senescence occurring in the endothelium during ageing.
Conflicts of interest
There are no conflicts of interest regarding this paper.
References
- [1].Kirkwood T. Understanding the odd science of aging. Cell. 2005;120:437–447. [DOI] [PubMed] [Google Scholar]
- [2].De Magalhães JP. How ageing processes influence cancer. Nat Rev Cancer. 2013;13:357–365. [DOI] [PubMed] [Google Scholar]
- [3].North B, Sinclair D. The intersection between aging and cardiovascular disease. Circ Res. 2012;110:1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lardenoije R, Pishva E, Lunnon K, van den Hove D. Neuroepigenetics of aging and age-related neurodegenerative disorders. Prog Mol Biol Transl. 2018;158:49–82. [DOI] [PubMed] [Google Scholar]
- [5].Dominguez L, Barbagallo M. The biology of the metabolic syndrome and aging. Curr Opin Clini Nutr Metab Care. 2016;19:5–11. [DOI] [PubMed] [Google Scholar]
- [6].López-Otín C, Blasco M, Partridge L, Serrando M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hansson G. Mechanisms of disease: inflammation, atherosclerosis, and coronary artery disease. N Eng J Med. 2005;352:1685–1695. [DOI] [PubMed] [Google Scholar]
- [8].Mchugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kuilman T, Michaloglou C, Mooi WJ, Peeper D. The essence of senescence. Genes Dev. 2010;24:2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Childs B, Baker D, Kirkland J, Campisi J, Deursen J. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15:1139–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hayflick L. The limited in nitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. [DOI] [PubMed] [Google Scholar]
- [13].Coppé J, Patil C, Rodier F, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5:e9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Storer M, Mas A, Robert-Moreno A, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. [DOI] [PubMed] [Google Scholar]
- [15].Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jeyapalan J, Ferreira M, Sedivy J, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007;128:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Itahana K, Campisi J, Dimri G. Mechanisms of cellular senescence in human and mouse cells. Biogerontoly. 2004;5:1–10. [DOI] [PubMed] [Google Scholar]
- [19].Dimri G, Leet X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. PNAS. 1995;92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxi Med Cell Longev. 2016;2016:3565127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pole A, Dimri M, Dimri P. Oxidative stress, cellular senescence and ageing. AIMS Mol Sci. 2016;3:300–324. [Google Scholar]
- [22].Ziegler D, Wiley C, Velarde M. Mitochondrial effectors of cellular senescence: beyond the free radical the ory of aging. Aging Cell. 2015;14:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liguori I, Russo G, Curcio F, et al. Oxidative stress, aging, and diseases. Clin Interv Aging. 2018;13:757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Siderakis M, Tarsounas M. Telomere regulation and function during meiosis. Chromosome Res. 2007;15:667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sanders J, Newman A. Telomere length in epidemiology: a biomarker of aging, age-related disease, both, or neither? Epidemiol Rev. 2013;35:112–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chandrasekaran A, Sosa P, Melendez J. Redox control of senescence and age-related disease. Redox Biol. 2017;11:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Childs BG, Durik M, Baker DJ, Van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sharpless N, Sherr C. Forging a signature of in vivo senescence. Nat Publ. 2015;15:397–408. [DOI] [PubMed] [Google Scholar]
- [29].Acosta J, Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. [DOI] [PubMed] [Google Scholar]
- [30].Teissier T, Boulanger É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition rece ptor (PRR) for inflammaging. Biogerontology. 2019;20:279–301. [DOI] [PubMed] [Google Scholar]
- [31].Hernandez-segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–453. [DOI] [PubMed] [Google Scholar]
- [32].Copp J, Desprez P, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Path. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kang T, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. [DOI] [PubMed] [Google Scholar]
- [34].Franceschi C, Capri M, Monti D, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. [DOI] [PubMed] [Google Scholar]
- [35].Matsumura T, Zerrudo Z, Hayflick L. Senescent human diploid cells in culture: survival, DNA synthesis and morphology. J Gerontol. 1979;34:328–334. [DOI] [PubMed] [Google Scholar]
- [36].Zwerschke W, Mazurek S, Ockl PST, Utter EH, Eigenbrodt E. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J. 2003;376:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Green DR. Chipuk JE. p53 and metabolism: inside the TIGAR. Cell. 2006;126:30–32. [DOI] [PubMed] [Google Scholar]
- [38].Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. [DOI] [PubMed] [Google Scholar]
- [39].Wiley CD, Cam pisi J. From ancient pathways to aging cells—connecting metabolism and cellular senescence. Cell Metab. 2016;23:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Strycharz J, Drzewoski J, Szemraj J, Sliwinska A. Is p53 involved in tissue-specific insulin resistance formation? Oxi Med Cell Longev. 2017;2017:9270549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Heidenr MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Correia-melo C, Passos JF. Mitochondria: Are they causal players in cellular senescence? BBA. 2015;1847:1373–1379. [DOI] [PubMed] [Google Scholar]
- [43].Nacarelli T, Sell C. Targeting metabolism in cellular senescence, a role for intervention. Mol Cell Endocrinol. 2017;455:83–92. [DOI] [PubMed] [Google Scholar]
- [44].Zou Y, Jung KJ, Kim JW, Yu BP, Chung HY. Alteration of soluble adhesion molecules during aging and their modulation by calorie restriction. FASEB J. 2004;18:320–322. [DOI] [PubMed] [Google Scholar]
- [45].Yang KE, Kwon J, Rhim JH, et al. Differential expression of extracellular matrix proteins in senescent and young human fibroblasts: a comparative proteomics and microarray study. Mol Cells. 2011;32:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sell DR, Monnir VM. Structure elucidation of a senescence cross-link from human extracellular matrix. implication of pentoses in the aging process. J Biol Chem. 1989;264:21597–21602. [PubMed] [Google Scholar]
- [47].Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282:1523–1529. [DOI] [PubMed] [Google Scholar]
- [48].Ahima RS. Connecting obesity, aging and diabetes. Nat Med. 2009;15:996–997. [DOI] [PubMed] [Google Scholar]
- [49].Nordfjäll K, Eliasson M, Stegmayr B, Melander O, Nilsson P, Roos G. Telomere length is associated with obesity parameters but with a gender difference. Obesity. 2008;16:2682–2689. [DOI] [PubMed] [Google Scholar]
- [50].Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. [DOI] [PubMed] [Google Scholar]
- [51].Schafer MJ, Miller JD, LeBrasseur NK. Cellular senescence: implications for metabolic disease. Mol Cell Endocrinol. 2017;455:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Al-Aubaidy HA, Jelinek HF. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur J Endocrinol. 2011;164:899–904. [DOI] [PubMed] [Google Scholar]
- [53].Fontana L, Partridge L, Longo VD. Dietary restriction, growth factors and aging: from yeast to humans. Science. 2010;328:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mitchell SJ, Matute JM, Scheibye-knudsen M, et al. Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab. 2016;23:1093–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mattison JA, Colman RJ, Beasley TM, et al. Caloric restriction improves health and survival of rhesus monkeys. Nat Commun. 2017;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Martin C, Bhapkar M, Pittas A, et al. Effect of calorie restriction on mood, quality of life, sleep, and sexual function in healthy nonobese adults: the CALERIE 2 randomized clinical trial. JAMA Intern Med. 2016;176:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Il’yasova D, Fontana L, Bhapkar M, et al. Effects of 2 years of caloric restriction on oxidative status assessed by urinary F2-isoprostanes: the CALERIE 2 randomized clinical trial. Aging Cell. 2018;17:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Redman LM, Smith SR, Burton JH, Martin CK, Il’yasova D, Ravussin E. Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab. 2018;27:805–815.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Most J, Gilmore LA, Smith SR, Han H, Ravussin E, Redman LM. Significant improvement in cardiometabolic health in healthy nonobese individuals during caloric restriction-induced weight loss and weight loss maintenance. Am J Physiol Endocrinol Metab. 2018;314:E396–E405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ard JD, Gower B, Hunter G, Ritchie CS, et al. Effects of calorie restriction in obese older adults: the CROSSROADS randomized controlled trial. J Gerontol A Biol Sci. 2017;73:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Madeo F, Carmona-Gutierrez D, Hofer SJ, Kroemer G. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab. 2019;29:592–610. [DOI] [PubMed] [Google Scholar]
- [62].Cantó C, Auwerx J. Calorie restriction: is AMPK as a key sensor and effector? Physiology. 2013;26:214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Di Francesco A, Di Germanio C, Bernier M, De Cabo R. A time to fast. Science. 2018;362:770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].De Cabo R, Mattson MP. Effects of intermittent fasting on health, aging, and disease. N Engl Med. 2019;381:2541–2551. [DOI] [PubMed] [Google Scholar]
- [65].Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease mTOR in growth control. Trends Mol Med. 2013;18:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Martín-Montalvo A, Villalba JM, Navas P, De Cabo R. NRF2, cancer and calorie restriction. Oncogene. 2011;30:505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fontana L, Mitchell SE, Wang B, et al. The effects of graded caloric restriction: XII. Comparison of mouse to human impact on cellular senescence in the colon. Aging Cell. 2018;17:4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ogrodnik M, Miwa S, Tchkonia T, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yang L, Licastro D, Cava E, et al. Long-term calorie restriction enhances cellular quality-control processes in human skeletal muscle. Cell Rep. 2016;14:422–428. [DOI] [PubMed] [Google Scholar]
- [71].Meydani SN, Das SK, Pieper CF, et al. Long-term moderate calorie restriction inhibits inflammation without impairing cell-mediated immunity: A randomized controlled trial in non-obese humans. Aging (Albany NY). 2016;8:1416–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nasri H, Rafieian-Kopaei M. Metformin: current knowledge. J Res Med Sci. 2014;19:658–664. [PMC free article] [PubMed] [Google Scholar]
- [73].Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Berman AY, Motechin RA, Wiesenfeld MY, Holz MK. The therapeutic potential of resveratrol: a review of clinical trials. NPJ Precis Oncol. 2017;1:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yessenkyzy A, Saliev T, Zhanaliyeva M, et al. Polyphenols as caloric-restriction mimetics and autophagy inducers in aging research. Nutrients. 2020;12:1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Santarelli V, Neri L, Sacchetti G, Di Mattia CD, Mastrocola D, Pittia P. Response of organic and conventional apples to freezing and freezing pre-treatments: focus on polyphenols content and antioxidant activity. Food Chem. 2020;308:125570. [DOI] [PubMed] [Google Scholar]
- [77].Bermúdez-Oria A, Rodríguez-Gutiérrez G, Alaiz M, Vioque J, Girón-Calle J, Fernández-Bolaños J. Polyphenols associated to pectic polysaccharides account for most of the antiproliferative and antioxidant activities in olive extracts. J Funct Foods. 2019;62:103530. [Google Scholar]
- [78].Pallauf K, Rimbach G. Autophagy, polyphenols and healthy ageing. Ageing Res Revi. 2013;12:237–252. [DOI] [PubMed] [Google Scholar]
- [79].Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metabo. 2018;27:529–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nabavi SF, Sureda A, Dehpour AR, et al. Regulation of autophagy by polyphenols: paving the road for treatment of neurodegeneration. Biotechnol Adv. 2018;36:1768–1778. [DOI] [PubMed] [Google Scholar]
- [82].Li J, Zhang CX, Liu YM, Chen KL, Chen G. A comparative study of anti-aging properties and mechanism: resveratrol and caloric restriction. Oncotarget. 2017;8:65717–65729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Schafer MJ, White TA, Evans G, et al. Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes. 2016;65:1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang C, Maddick M, Miwa S, et al. Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging (Abany NY). 2010;2:555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]