

Abstract
Background:
Hemoglobin disorders are the leading health concern in the world including India. There is a paucity of literature on the spectrum of hemoglobin disorders in southern districts of Odisha state. This study was undertaken to elucidate the occurrence of different hemoglobin disorders in a tertiary health care facility of Odisha state, India.
Methods:
The study cases were suspected patients of all age groups advised for screening of different hemoglobin disorders. Hemoglobin disorders were screened by sickling slide test and high-performance liquid chromatography (HPLC) using the Variant-II hemoglobin testing system as per the manufacturer's guidelines.
Results:
Over 2 years, 2332 blood samples (including 1102 pediatric and 1230 adult cases) were investigated, out of which, 1380 (59.2%) of cases had abnormal hemoglobin disorders. The most common was sickle cell disorders (48.67%, 1135/2332) followed by β-thalassemia (11.32%, 264/2332). Some rare variants were detected as hemoglobin D-Punjab, hemoglobin E, hemoglobin Lepore, hereditary persistence of fetal hemoglobin, hemoglobin with high P2 window, hemoglobin with high P3 window etc, Among the cases with abnormal hemoglobin disorders, 744 (53.9%), 545 (39.5%) and, 91 (6.6%) cases were found to have the heterozygous, homozygous and, double heterozygous state. Of the 188 ante-natal cases screened, 31.4% of cases had abnormal hemoglobin variants with sickle cell disorders being the most prevalent one.
Conclusion:
Along with the high occurrence of sickle cell disorders in the study area, some other rare hemoglobin disorders are also prevalent which calls for a large community-based cohort study.
Keywords: anti-natal case, hemoglobin variant, HPLC, retention time, sickle cell, variant II
Introduction
The inherited hemoglobin disorders are autosomal recessive disorders of hemoglobin. They are categorized into 2 main groups, that is, (a) thalassemias characterized by the reduced (+) or absent (0) production of one or both of the globin chains of the hemoglobin tetramer and (b) structural hemoglobin variants resulting from single amino-acid substitutions in the α or β globin chains.1 According to the WHO report, hemoglobin disorders are originally endemic to 61% of the 229 countries around the world and 5.2% of the world population carries a significant hemoglobin variant. Again approximately 1.1% of couples are at risk for having children with a hemoglobin disorder and 2.7 per 1000 conceptions are affected worldwide.2 In India, the prevalence of hemoglobin disorders is high accounting for a significant proportion of populations with specific hemoglobin disorders. Among various hemoglobin disorders, sickle cell disorders, β-thalassemia, and α-thalassemia are highly prevalent in India. India has been positioned second worst affected country for sickle cell disorders.3 The prevalence of β-thalassemia carriers is 3% to 4% caters to around 35 to 45 million carriers in India.4 Similarly α-thalassemia is widespread in India with a prevalence of 10% to 25%, however, in certain communities, it is highly prevalent, that is, up to 80% of the population.5
After decades of research activities on the prevalence and management of different hemoglobin disorders in India, a lot of works have to be carried out to fill the knowledge gap. In India, a large number of geographical region are still there including southern districts of Odisha state, India where no significant information are available on the prevalence of various hemoglobin disorders. This tertiary care hospital-based study was carried out to elucidate the occurrence of different hemoglobin disorders in southern districts of Odisha state, India.
Methods
This observational study was carried out in Multi-Disciplinary Research Unit in association with Department of Pathology of Maharaja Krishna Chandra Gajapati Medical College, Berhampur, Odisha, India. This is a tertiary health care referral centre with 1190 beds and providing health care facilities to 13 millions people residing in southern Odisha, India (Coordinates 19.32°N 84.78°E). Patients who attended the hospital from November, 2016 to December, 2018 were considered for this study. The suspected cases were advised for the diagnosis of hemoglobin disorders from various departments of this institution and send to the Department of Pathology for screening. Some cases during family screening registered at the Department of Pathology along with the advised cases from various departments were also included in the study after taking required consent. Three milliliters of venous blood was collected in a K2 EDTA vacutainer (BD Vacutainer) from each case for testing. The complete blood count analysis was performed by using the XN-1000 auto-analyzer (Sysmex Cobe Japan) and hemoglobin fraction was analyzed by Variant-II hemoglobin testing system by using β-thalassemia short program (Bio-Rad Laboratories, Hercules, CA, USA). For both the instruments, all the possible parameters were analyzed as per the manufacturer guidelines. In detail for the analysis of hemoglobin fraction, the whole blood samples was diluted 200 times by supplied lysis buffer and were placed in the rack for the analysis. The resulted chromatogram showed the fraction of each hemoglobin variant in a sample and was analyzed by looking at the relative retention time.
The reporting of the hemoglobin disorders was carried out by looking results of slide test, hemoglobin fractions in the chromatogram, and complete blood counts analysis. The suspected cases for β-thalassemia diagnosed as a high A2 level (>3.9%) with a microcytic and hypochromic red cell pictures. Other rare hemoglobin variants were analyzed based on previously published reports. Hemoglobin Lepore was diagnosed in the A2 window with a value of 6% to 15% and eluted earlier 3.35 to 3.50 minutes compared to the A2 window.6,7 Hemoglobin E was suspected with an A2 level of more than 20%. All the diagnosis was made after review by the hematological experts. The generated data was entered in a predesigned excel format for analysis.
Results
During a study period of 2 years, 2393 blood samples (cases) were advised for diagnosis of different hemoglobin disorders at Variant-II, and finally, 2332 blood samples were analyzed. From 2332 cases, 1102 (47.26%) cases were found to be from pediatric and 1230 (52.74) cases were from the adult group. The recruitment of cases has been illustrated in Figure 1. We have noticed 8 different known windows as F, A0, A2, D, S, P1, P2 and P3) in the chromatograms with a median retention time of 1.09 minutes, 2.42 minutes, 3.64 minutes, 4.09 minutes, 4.36 minutes, 0.77 minutes, 1.34 minutes and 1.74 minutes, respectively.
Figure 1.

Recruitment of cases in the study.
After analysis, 59.2% of study cases had abnormal hemoglobin chromatograms. The most common abnormal hemoglobin disorder was found to be sickle hemoglobin variant (48.67%, 1135/2332) which was diagnosed as sickle cell trait (n = 535), homozygous sickle cell anemia (n = 517), double heterozygous state of sickle cell-β-thalassemia (n = 75), sickle cell-hemoglobin Lepore (n = 6) and sickle cell-hemoglobin E (n = 2). The second prevalent variant was β-thalassemia (11.32%, 264/2332) which was diagnosed as β-thalassemia trait, β-thalassemia major, and double heterozygous state with sickle cell, hemoglobin E and hemoglobin Lepore. Some rare hemoglobin variants were diagnosed including, hemoglobin D-Punjab, hemoglobin E, hemoglobin Lepore, hereditary persistence of fetal hemoglobin (HPFH), hemoglobin with high P2 window, hemoglobin with high P3 window, etc. Hemoglobin variants were inherited either alone as heterozygous state or with the same variant as homozygous state or with different variants as a double heterozygous state. Out of the 1380 cases with abnormal hemoglobin disorders, 744 (53.9%), 545 (39.5%), and 91 (6.6%) cases were diagnosed as heterozygous, homozygous and double heterozygous state, respectively. The detailed diagnosis of hemoglobin disorders and the number of recruited cases both in pediatric and adult populations has been shown in Table 1. The represented chromatograms of various hemoglobin disorders in the heterozygous state have been depicted in Figure 2 and represented chromatograms for both homozygous and double heterozygous states have been depicted in Figure 3.
Table 1.
Distribution of study cases on the basis of their hemoglobin disorders.
| Genotypes | Hemoglobin disorders | Pediatric population (n = 1102) number | Adult population (n = 1230) number | Total (n = 2332) number |
|---|---|---|---|---|
| Normal | Normal β globin (AA) | 404 | 514 | 918 |
| Heterozygous state | Sickle cell trait (AS) | 254 | 281 | 535 |
| β-Thalassemia trait (Aβ) | 37 | 119 | 156 | |
| Hemoglobin D-Punjab trait (AD-Punjab) | 1 | 0 | 1 | |
| Heterozygous for high P2 (AP2) | 0 | 2 | 2 | |
| Heterozygous for high P3 (AP3) | 3 | 0 | 3 | |
| Hemoglobin E trait (AE) | 2 | 9 | 11 | |
| Heterozygous for high HbF (AF) | 27 | 1 | 28 | |
| Lepore trait (AL) | 0 | 8 | 8 | |
| Homozygous state | β-Thalassemia major (ββ) | 25 | 0 | 25 |
| Sickle cell anemia (SS) | 274 | 243 | 517 | |
| Homozygous for high HbF (FF) | 2 | 1 | 3 | |
| Double heterozygous state | HbE and β-thalassemia (Eβ) | 1 | 3 | 4 |
| Sickle cell and β-thalassemia (Sβ) | 34 | 41 | 75 | |
| Sickle cell and Lepore (SL) | 1 | 5 | 6 | |
| Sickle cell and hemoglobin E (SE) | 2 | 0 | 2 | |
| Lepore and β-thalassemia (Lβ) | 1 | 3 | 4 |
The hemoglobin disorders of 34 infants (<6 mo of age) were not analyzed though they were included in the pediatric population.
Figure 2.
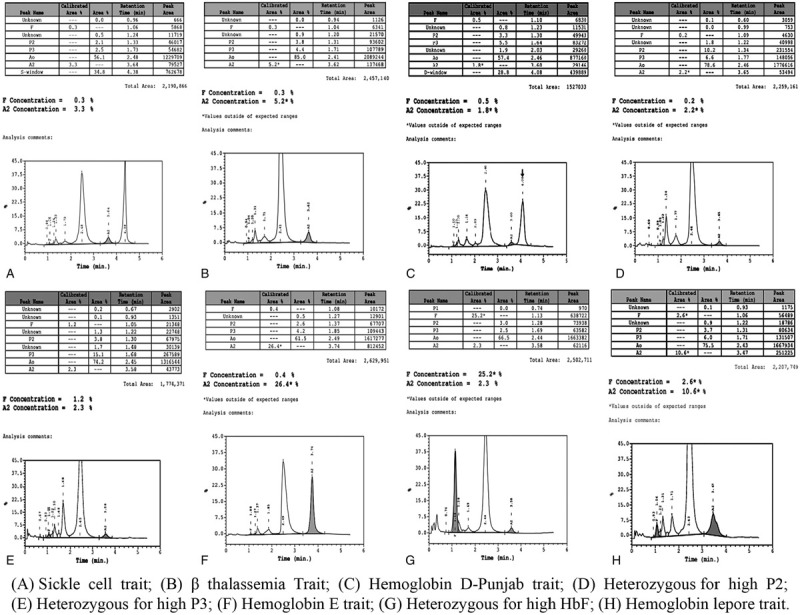
Chromatogram of various hemoglobin disorders in heterozygous state.
Figure 3.
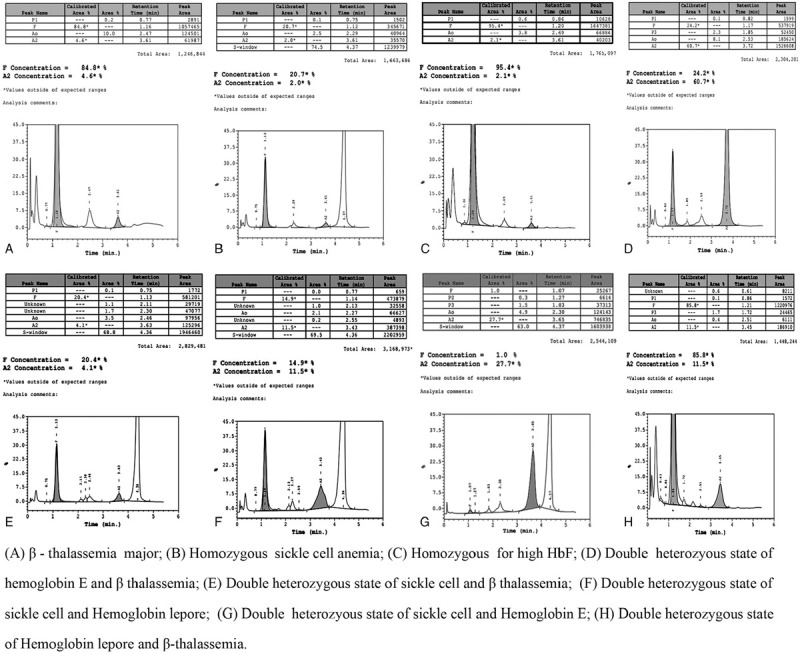
Chromatogram of various hemoglobin disorders in homozygous and double heterozygous state.
All the cases advised from the Obstetrics and Gynecology department were antenatal. Out of 188 ante-natal cases, 31.4% of cases were found to have abnormal hemoglobin variants. Among the abnormal variants, sickle cell trait, homozygous sickle cell anemia, β-thalassemia trait, sickle cell-β-thalassemia, hemoglobin Lepore trait and Lepore-β-thalassemia was diagnosed in 28 (14.9%), 12 (6.38%) and 14 (7.45%), 2 (1.06%), 2 (1.06%), and 1 (0.53%) cases, respectively.
Discussion
Out of the 2332 suspected individuals screened for hemoglobin variants, 59.2% of cases were diagnosed with abnormal hemoglobin disorders. This high occurrance of abnormal hemoglobin disorders is due to the selection biased of samples because only suspected cases were allowed to screen for hemoglobin variants from different departments of this institution. In the chromatogram, the retention time of different windows were per the manufacturer guidelines of the Instruments. Among the abnormal hemoglobin variants, the majority of cases had sickle cell variant which was eluted at a retention time of 4.22 to 4.44 minutes with a median value of 4.36 minutes. The high occurrence of sickle cell disorders in this study area was also reported from an earlier study.8 Along with sickle cell disorders, many other hemoglobin variants were also diagnosed. The second most hemoglobin disorder was found to be β-thalassemia which was diagnosed when HbA2 value was ≥3.9% with a microcytic and hypochromic red cells picture. β-thalassemia was diagnosed as β-thalassemia trait, β-thalassemia major, and co-inherited with sickle cell, hemoglobin E and hemoglobin Lepore etc. All the 25 cases diagnosed as β-thalassemia major were children which predicted the non-survivability of cases beyond the age of 14 years. A similar observation has also been noticed in a study from southern India.9 However, due to advances in red cell transfusion, introduction of new iron chelators and chelation regimes, screening for blood-borne viruses and other infections, and proper management, patients with β-thalassemia major have increased their life expectancy up to 40 years or more.10,11
In the A2 window, the other 2 hemoglobin disorders have been suspected as hemoglobin E and hemoglobin Lepore. Hemoglobin E was diagnosed in 17 cases including hemoglobin E trait in 11 cases, co-inherited with β-thalassemia in 4 cases, and co-inherited with sickle cell in 2 cases. This is the first study to report the cases with hemoglobin E in southern Odisha. Earlier studies have reported the occurrence of hemoglobin E in Odisha state specific to the western belt of Odisha.12,13 The occurrence of hemoglobin E in a region where both sickle cell disorders and β-thalassemia is highly prevalent is important because the co-inheritance of hemoglobin E with either of these 2 hemoglobin variants resulted with a significant clinical severity.12,14 Chromatogram characterized by a high A2 level (range 7%–14%) with a downward notch suspected as hemoglobin Lepore. Another identification remark for diagnosis of hemoglobin Lepore had a low retention (elution) time (3.37–3.46 minutes) compared to A2 (3.60–3.67 minutes) as similar to the earlier reports published from India.6,7 This is the first study to report the occurrence of suspected hemoglobin Lepore in the state of Odisha, India. However, the confirmation for diagnosis of hemoglobin Lepore can only be done by molecular diagnosis methods.
Hereditary persistence of fetal hemoglobin has been diagnosed in 31 cases both in heterozygous (28 cases) and homozygous state (3 cases). The diagnosis of these hemoglobin disorders revealed the possible prevalence of either HPFH or δβ-thalassemia in the study area which can be confirmed by molecular diagnosis methods. In the state of Odisha, both of these hemoglobin disorders have been diagnosed earlier from the western belt of Odisha state.15–17
In this study, a single case had a significant peak in the D window (retention time: 4.09 minutes), which was diagnosed to be the HbD-Punjab trait. The incidence of hemoglobin D-Punjab in the population of Odisha state-reported earlier.18 The occurrence of hemoglobin D-Punjab in a region where sickle cell disorders are highly prevalent is important because the co-inheritance of these 2 hemoglobin variants may result in a significant clinical severity.18–20
High P2 window was observed in 2 adult cases (20.5% and 10.2%, respectively). HbA1c which is usually increased in diabetes cases elutes in the P2 window.21 Another variant hemoglobin Hope is elutes in the P2 window but its value is generally >40%.22 High P3 window was observed in 3 pediatric cases (11.3%, 11.7%, and 19%, respectively). Several hemoglobin variants have been reported in the P3 window including both α and β globin variants.22 In India, various hemoglobin variants have been reported in the P3 window as HbJ-Meerut,23 HbJ-Oxford,24 other hemoglobin J variants9 etc. Some hemoglobin fractions that formed after post-translational modifications of adult hemoglobin also elutes in the P3 window but the value usually less than 10%. The P3 level also increases when the samples get older or more degradation of hemoglobin occurs.14,25
Among the antenatal cases, around one-third of study women had abnormal hemoglobin disorders. Majorities of them had sickle cell disorders. It has been reported that the pregnancy outcome in women with sickle cell disorders is poor compared to normal hemoglobin genotypes. In a recent retrospective study carried out in women with sickle cell disease from the state of Odisha, 25% of the pregnancies were unsuccessful.26 This needs attention for the early diagnosis and management of pregnant women with sickle cell disorders in the state.
In conclusion, this 2 years hospital based study revealed the spectrum of various abnormal hemoglobin variants among the studied patients of southern Odisha. Moreover, knowledge of common hemoglobin variants and its chromatogram patterns in a particular region will help for easy diagnosis of hemoglobin disorders and formulation of appropriate preventive measures.
Acknowledgments
The authors acknowledge laboratory technicians, Mrs Radha Rani Dora, Mrs Deepika Mishra, and Mr Subash Satapathy for their support in collections and processing of blood samples during the investigations. The authors also acknowledge to Mr Satyanarayana Mohanty, Data Entry Operator, Multi-Disciplinary Research Unit for data processing.
Author contributions
D.T., B.M., and D.P.M. design the concept; P.S., R.T., J.N., S.K.A., S.K.B., M.K.P., D.T., B.M., and D.P.M. involved in the recruitment of cases; P.P., R.T., J.N., S.K.B., M.K.P., and D.P.M. were involved in the experiments; P.S., P.P., and S.M. were involved in the data acquisition; P.P. was involved in the data analysis; P.P., N.K., and D.P.M. were involved in the manuscript preparation. All the authors have reviewed the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
References
- [1].Weatherall DJ, Clegg JB. Inherited hemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- [2].Modell B, Darlison M. Global epidemiology of hemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:417–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hockham C, Bhatt S, Colah R, et al. The spatial epidemiology of sickle-cell anemia in India. Sci Rep. 2018;8:17685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Colah RB, Italia K, Gorakshakar A. Burden of thalassemia in India: the road map for control. Pediatr Hematol-Oncol J. 2017;2:79–84. [Google Scholar]
- [5].Williams TN, Weatherall DJ. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harbor Perspect Med. 2012;2:a011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shaji RV, Edison ES, Krishnamoorthy R, Chandy M, Srivastava A. Hb Lepore in the Indian population. Hemoglobin. 2003;27:7–14. [DOI] [PubMed] [Google Scholar]
- [7].Nadkarni A, Italia K, Sawant P, Ghosh K, Colah R. Hemoglobin Lepore Hollandia in India. Int J Lab Hematol. 2012;34:148–153. [DOI] [PubMed] [Google Scholar]
- [8].Sahu T, Sahani NC, Das S, Sahu SK. Sickle cell anemia in tribal children of Gajapati district in South Orissa. Indian J Commun Med. 2003;XXVIII:180–183. [Google Scholar]
- [9].Chandrashekar B, Soni M. Hemoglobin disorders in South India. ISRN Hematol. 2011;2011:748939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vitrano A, Calvaruso G, Lai E, et al. The era of comparable life expectancy between thalassemia major and intermedia dichotomy? Br J Haematol. 2017;176:124–130. [DOI] [PubMed] [Google Scholar]
- [12].Patel S, Meher S, Dehury S, et al. Molecular, hematological and clinical characterization of sickle cell HbE in Eastern India: the largest series in the world. Ind J Haemat Blood Transf. 2014;30:S505. [Google Scholar]
- [13].Purohit P, Mohanty PK, Panigrahi J, Patel S. Infection with Plasmodium falciparum malaria in a patient with homozygous hemoglobin E. Muller J Med Sci Res. 2019;10:36–38. [Google Scholar]
- [14].Olivieri NF, Pakbaz Z, Vichinsky E. HbE/beta-thalassaemia: a common & clinically diverse disorder. Indian J Med Res. 2011;134:522–531. [PMC free article] [PubMed] [Google Scholar]
- [15].Balgir RS. Hereditary persistence of foetal haemoglobin in a tribal family of Orissa, India. Natl Med J India. 2004;17:138–140. [PubMed] [Google Scholar]
- [16].Patel DK, Patel M, Mashon RS, Patel S, Dash PM, Das BS. Clinical and molecular characterization of βS & Gγ(Aγδβ)0-thalassemia in eastern India. Hemoglobin. 2010;34:604–609. [DOI] [PubMed] [Google Scholar]
- [17].Patel S, Dehury S, Purohit P, Meher S, Das K. Inheritance of hereditary persistence of fetal haemoglobin (HPFH) in a family of Western Odisha, India. J Clin Diagn Res. 2015;9:OD09–OD10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Patel DK, Mashon RS, Patel S, Dash PM, Das BS. β-Globin gene haplotypes linked with the Hb D-Punjab [β121(GH4)Glu→Gln, GAA>CAA] mutation in eastern India. Hemoglobin. 2010;34:530–537. [DOI] [PubMed] [Google Scholar]
- [19].Mukherjee MB, Surve RR, Gangakhedkar RR, Mohanty D, Colah RB. Hemoglobin sickle D Punjab—a case report. Indian J Hum Genet. 2005;11:154–155. [Google Scholar]
- [20].Patel DK, Purohit P, Dehury S, et al. Fetal hemoglobin and alpha thalassemia modulate the phenotypic expression of HbSD-Punjab. Int J Lab Haematol. 2013;36:444–450. [DOI] [PubMed] [Google Scholar]
- [21].Colah RB, Surve R, Sawant P, et al. HPLC Studies in hemoglobinopathies. Indian J Pediatr. 2007;74:657–662. [DOI] [PubMed] [Google Scholar]
- [22].Joutovsky A, Hadzi-Nesic J, Nardi MA. HPLC retention time as a diagnostic tool for hemoglobin variants and hemoglobinopathies: a study of 60000 samples in a clinical diagnostic laboratory. Clin Chem. 2004;50:1736–1747. [DOI] [PubMed] [Google Scholar]
- [23].Rao S, Kar R, Gupta SK, Chopra A, Saxena R. Spectrum of hemoglobinopathies diagnosed by cation exchange-HPLC & modulating effects of nutritional deficiency anemias from north India. Indian J Med Res. 2010;132:513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Khera R, Singh T, Khurana N, Gupta N, Dubey AP. HPLC in characterization of hemoglobin profile in thalassemia syndromes and hemoglobinopathies: a clinicohematological correlation. Indian J Hematol Blood Transfus. 2015;31:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Raghothama C, Rao P. Degradation of glycated haemoglobin: role of erythrocytic proteolytic enzymes and oxidant damage. Clin Chim Acta. 1997;264:13–25. [DOI] [PubMed] [Google Scholar]
- [26].Patel S, Purohit P, Jit BP, Meher S. Pregnancy outcomes in women with sickle cell disease: a retrospective study from eastern India. J Obstetr Gynaecol. 2019;39:882–884. [DOI] [PubMed] [Google Scholar]