

Abstract
Adoptive T cell therapy utilizing tumor-specific autologous T cells has shown promising results for cancer treatment. However, the limited numbers of autologous tumor-associated antigen (TAA)-specific T cells and the functional aberrancies, due to disease progression or treatment, remain factors that may significantly limit the success of the therapy. The use of allogeneic T cells, such as umbilical cord blood (CB) derived, overcomes these issues but requires gene modification to induce a robust and specific anti-tumor effect. CB T cells are readily available in CB banks and show low toxicity, high proliferation rates, and increased anti-leukemic effect upon transfer. However, the combination of anti-tumor gene modification and preservation of advantageous immunological traits of CB T cells represent major challenges for the harmonized production of T cell therapy products. In this manuscript, we optimized a protocol for expansion and lentiviral vector (LV) transduction of CB CD8+ T cells, achieving a transduction efficiency up to 83%. Timing of LV treatment, selection of culture media, and the use of different promoters were optimized in the transduction protocol. LentiBOOST was confirmed as a non-toxic transduction enhancer of CB CD8+ T cells, with minor effects on the proliferation capacity and cell viability of the T cells. Positively, the use of LentiBOOST does not affect the functionality of the cells, in the context of tumor cell recognition. Finally, CB CD8+ T cells were more amenable to LV transduction than peripheral blood (PB) CD8+ T cells and maintained a more naive phenotype. In conclusion, we show an efficient method to genetically modify CB CD8+ T cells using LV, which is especially useful for off-the-shelf adoptive cell therapy products for cancer treatment.
Keywords: cord blood, lentiviral transduction, CD8+ T cells, T cell therapy, off-the-shelf
Graphical abstract
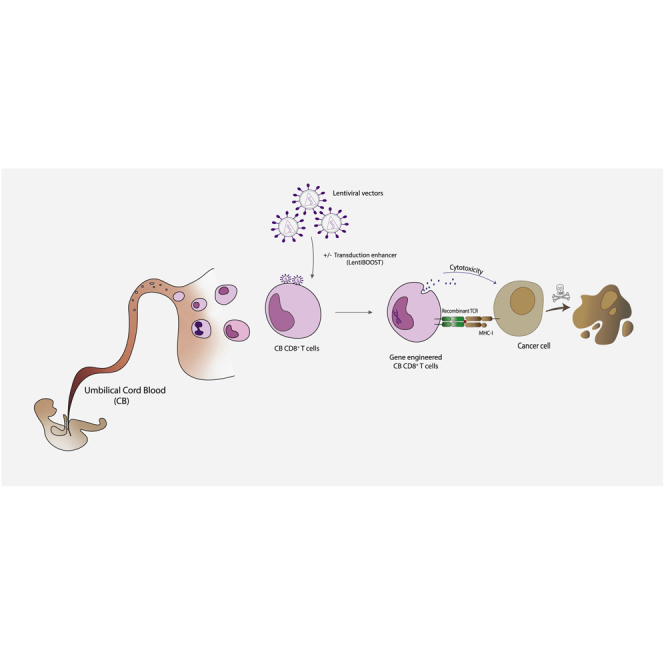
The manuscript proposes an efficient method to gene modify cord blood-derived CD8+ T cells using lentiviral vectors and a transduction enhancer (LentiBOOST). Future application of these findings can improve the generation of allogeneic T cell therapies for the treatment of cancer.
Introduction
Adoptive T cell therapy has developed into a promising treatment option for cancer patients in the last decade. The rationale toward efficient T cell therapies is to redirect the patients’ T cells against tumor antigens, either by selection and ex vivo expansion of tumor-infiltrating lymphocytes (TILs)1 or by gene-modified lymphocytes. TIL therapy,1 combined with interleukin-2 (IL-2), has shown encouraging results in the treatment of metastatic melanoma patients, with a remission rate up to 72%.2,3 Treatment with gene-modified T cell products,4 such as chimeric antigen receptor (CAR) T cells,5 has shown very promising results in several clinical trials,6 and Kymriah became the first approved cell therapy by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for B cell acute lymphoblastic leukemia (ALL) patients.7,8 An alternative gene-modification strategy for the generation of a T cell product is the transfer of a transgenic tumor-associated antigen (TAA)-specific T cell receptor (TCR) recognizing a specific peptide in the context of major histocompatibility complex (MHC). Also, this strategy has been explored in several early-phase clinical trials, predominantly in melanoma, gastrointestinal cancer, and leukemia.9 MART1-TCR-engineered T cells persisted for at least 2 months after the infusion in peripheral blood (PB) of 15 melanoma patients. Two patients with high sustained levels of circulating MART1-specific T cells 1 year post-infusion exhibited regression of metastatic melanoma lesions.10 NY-ESO-TCR-engineered T cells were well tolerated and exhibited an encouraging clinical response in multiple myeloma.11 Post-transplantation transfer of Wilm’s tumor 1 (WT1)-TCR-engineered T cells prevented relapse in all acute myeloid leukemia (AML)-treated patients in a median of 44 months post-infusion.12 These recent results indicate the potential of TCR engineering therapy, even if the tumor mutational burden is low, as, for example, in many childhood cancers.13
A commonly used method for gene-modifying T cells is the use of lentiviral vectors (LVs), which are highly efficient in transducing many cell types,14 including quiescent cells, to express a gene of interest. More specifically, the transduction efficiency in T cells is not only dependent on the proliferation status but also on the activation status. The presence of sufficient numbers of functional patient-derived cells with adequate activation potential might be hampered by disease biology, disease progression, and/or previous therapies applied (such as chemotherapy).15 It has been reported that during chemotherapy cycles, T cells drastically decrease in number;16 show signs of senescence and low proliferation capacity and activate apoptotic pathways;17 and require several months to return to physiological levels.18, 19, 20 Together, the decline in numbers of naive T (Tn) cells and stem central memory T (Tscm) cells, as well as the reduced proliferation and stimulation, potentially limits the use of autologous T cells for harmonized production of autologous T cell therapy products.
A solution to overcome this problem is to utilize a patient-unrelated cell source, such as T cells derived from healthy donors. Umbilical cord blood (CB)-derived T cells are of particular interest, as they show a naive phenotype with elevated proliferation potential and low expression of exhaustion markers compared to PB-derived T cells.21,22 Additionally, Hiwarkar et al.23 showed that CB CD8+ T cells have an increased killing effect on human leukocyte antigen (HLA)-mismatched Epstein-Barr virus (EBV)-driven tumor cells compared to other sources. The use of CB-derived cells has been mostly limited to CD34+ hematopoietic stem cell (HSC) and progenitor cells, which are amenable to high lentiviral transduction rates.24, 25, 26, 27 The feasibility of the approach to introduce CARs28, 29, 30 or recombinant TCRs (rTCRs)31 into CB-derived T cells has been shown mostly using non-viral gene transfer methods or retroviral vectors (RVs). However, as shown by Cieri et al.32, the use of LV is superior to RV, especially for the transduction of Tn and central memory T (Tcm) cells. To date and to the best of our knowledge, the use of LV to gene modify CB-derived T cells is very scarce but showed promising results.33
In the present study, we developed an efficient method, using a serum-free transduction system, to transduce CB CD8+ T cells with LV with limited effect on their naive immunophenotype. With the use of this protocol, CB CD8+ T cells are more susceptible to LV transduction compared to PB CD8+ T cells. In addition, we demonstrate how to further increase the transduction efficiency using the transduction enhancer LentiBOOST (LB) without altering CB CD8+ T cells’ functionality and maintaining the ability to expand in vitro, especially when using a low multiplicity of infection (MOI). Altogether, these results show efficient gene modification of CB CD8+ T cells using LV to potentially generate off-the-shelf T cell therapy products for cancer treatment.
Results
Method optimization for efficient lentiviral transduction of CB CD8+ T cells
Two different methods were compared for transduction efficiency: spinoculation and overnight incubation. 3 days after expansion, CB CD8+ T cells were transduced with either enhanced green fluorescent protein (EGFP) under the control of the phosphoglycerate kinase (PGK.EGFP) promoter or a synthetic promoter that contains the U3 region of a modified Moloney murine leukemic virus (MoMuLV) long terminal repeat (LTR) with myeloproliferative sarcoma virus enhancer (MND.EGFP) at a MOI of 10 using X-Vivo15 media (Figure 1). With the use of spinoculation, 6 days after transduction, 24% ± 5% of CD8+ T cells were GFP+ with the PGK.EGFP vector and 31% ± 9% with the MND.EGFP vector, whereas overnight incubation showed 48% ± 11% GFP+ cells for PGK.EGFP and 68% ± 8% for MND.EGFP (Figure 2A). Additionally, the use of two different types of media during the transduction strongly influenced the efficiency of transduction of CD8+ T cells. Overnight culture in StemMACS HSC Expansion Media XF (StemMACS) showed a significant increase in the percentage of transduced cells up to 78% ± 7.7% for PGK.EGFP and 83% ± 8.8% for MND.EGFP in contrast to X-Vivo15 (Figures 2B and 2C). Hence, overnight incubation and StemMACS medium were used for further experiments (from hereafter referred to as STND protocol). No significant differences were observed in the percentage of transduced cells if the EGFP was under the control of the MND or the PGK promoter; however, the protein expression, calculated as median fluorescence intensity (MFI), increased significantly using the MND promoter. GFP-positive CB CD8+ T cells showed a 3-fold increase in MFI compared to the CB CD8+ T cells transduced with PGK.EGFP (Figure 2D), also when normalized for vector copy number (VCN) (Figure 2E).
Figure 1.

Schematic overview of lentiviral transfer vector plasmid maps
Third-generation self-inactivating (SIN) lentiviral vectors (LVs) were constructed using the CMV promoter driving the expression of the viral transcript (pCCL). The transfer vector contains the packaging signal (ψ), the REV responsive element (RRE), central polypurine tract (cPPT), and a modified Woodchuck post-translational regulatory element (bPRE4). Expression of the reporter gene EGFP was under the control of either the PGK or MND promoter (arrows). The WT1-TCR was under the control of the MND promoter.
Figure 2.

Optimization of the lentiviral transduction method for CB CD8+ T cells
(A) Transduction efficiency, presented as percentage of GFP+ cells, comparing two methods for T cell transduction: spinoculation (black) and overnight incubation (gray) (B) Transduction efficiency, presented as percentage of GFP+ cells, comparing the use of different culture media during overnight transduction: X-Vivo15 (gray) and StemMACS media (stripes). (C) Representative flow cytometry plots of methods and media tested using both transduction with PGK.EGFP and MND.EGFP. The density plots are showing the measured CD3 and CD8 expression (on the left) and GFP expression in CD3+ CD8+ T cells (on the right) in the left (PGK.EGFP) and right (MND.EGFP) panels. (D) Protein expression, presented as MFI of EGFP signal, is statistically increased using the MND promoter. (E) qPCR analysis of the VCN-positive correlates with the MFI results. The data correspond to ≥3 independent experiments and are shown as average ± standard deviation; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
CB CD8+ T cells are more efficiently transduced than PB CD8+ T cells with LV.MND.EGFP
Autologous PB T cells are often the main cell source for adoptive cell therapy to treat cancer. Here, we developed a method to transduce allogeneic CB CD8+ T cells, and hence, we compared the two T cell sources for efficient transduction by LVs. PB CD8+ T cells were isolated from healthy donors, expanded, and transduced following the same method developed for CB CD8+ T cells. As shown in Figures 3A and 3B, this method has an advantage to transduce CB CD8+ T cells more efficiently than PB CD8+ T cells, which was observed using MND.EGFP. The immunophenotype based on markers for Tn cells (CD45RO, CD62L, CD45RA) of freshly isolated (day 0; fresh) T cells is substantially different between CB CD8+ T cells and PB CD8+ T cells. As shown in Figures 3C and 3D, fresh CB CD8+ T cells are characterized by a homogeneous population expressing classic Tn cell markers (CD45RO− CD62L+ CD45RA+), whereas fresh PB CD8+ T cells consist of a more heterogeneous population with different expression levels of both Tn and memory T (Tm) cell markers, characteristic of Tm cell subsets (CD45RO+CD62L− effector memory T [Tem] cells and CD45RO+CD62L+ Tcm cells). The expansion phase, characterized by the presence of cytokines and anti-CD3/CD28 (aCD3/CD28) beads, confers to both CB and PB CD8+ T cells an increased proportion of population expressing CD45RO and CD62L (Tcm). At the moment of transduction (day 3; expanded), expanded T cells from both sources were more similar, contrary to the initial differences shown in fresh cells (day 0) (Figures 3C−3E). Moreover, both CB and PB CD8+ T cells showed similarly high levels of the IL-2 receptor (CD25), indicating an activated state at the moment of transduction (Figure S1). Interestingly, 6 days after transduction (day 10; transduced), CB CD8+ T cells reverted to the Tn cell markers, with a specific loss of expression of CD45RO and increased expression of CD45RA, which did not occur in PB CD8+ T cells (Figure 3E). Moreover, CB and PB CD8+ T cells showed differential expression of the co-inhibitory receptor (TIM3 and LAG3) during all experimental steps (Figures 3F and S1).
Figure 3.

CB and PB CD8+ T cells show different immune phenotype and transduction efficiency
(A) Transduction efficiency, as percentage of GFP+ cells in CB CD8+ T cells (black) compared to PB CD8+ T cells (gray) using MND.EGFP. (B) Representative flow cytometry plot of CB and PB CD8+ T cells transduced with MND.EGFP. (C) Proportion of naive T (Tn) cells, effector memory T (Tem) cells, and central memory T (Tcm) cells based on the expression of CD45RO and CD62L, in both CB and PB in CD8+ fresh, expanded, and transduced T cells. (D) Representative flow cytometry plot of CD45RO-CD62L-expressing cells during the three selected time points (i.e., fresh, expanded, transduced). (E) CD45RA expression in CB (black) and PB (gray) CD8+ fresh, expanded, and transduced T cells. (F) Co-inhibitory receptor expression in CB (black) and PB (gray) CD8+ fresh, expanded, and transduced T cells. The data correspond to ≥3 independent experiments and are shown as average ± standard deviation; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
The transduction enhancer LB increases transduction efficiency and maintains proliferation capacity of CB CD8+ T cells in vitro
To further improve the transduction efficiency, we tested if the addition of the non-ionic, amphiphilic Poloxamer Synperonic F108 (LB)25,34 could further enhance the GFP expression in CB CD8+ T cells, while limiting its effect on viability and proliferation capacity of the CB CD8+ T cells and using MOIs in the range of MOI 1−20, commonly used to transduce PB T cells.35, 36, 37 As shown in Figures 4A and 4B, the use of LB increased both the percentage of transduced T cells and the MFI of the signal. With the use of an MOI 10 to transduce CB CD8+ T cells, the LB resulted in 15% more GFP+ T cells, and with a lower MOI (MOI 1), the increase in the percentage of GFP+ T cells was even more pronounced, with an average increase of 25% compared to the STND. Lentiviral VCN analysis by qPCR confirmed that in both cases (MOI 10 and MOI 1), the use of LB significantly increased the VCN (Figure 4C). The use of LB increased the transduction efficiency also when using an LV.rTCR (next paragraph) and an LV.CAR against CD19 (CAR19) (Figure S2). 6 days post-transduction, CB CD8+ T cells were stained with CellTrace Violet (CTV) and followed up over 4 days to determine the proliferation capacity. CB CD8+ T cells transduced with an MOI 1, in the presence of LB, maintained their proliferation capacity after a strong stimulation with aCD3/CD28 beads, but this was lower than untransduced cells (Figure 4E). To determine the direct influence of the LB on proliferation, untransduced cells were cultured overnight in the presence of LB. LB alone did not show any effect on the proliferation rate of untransduced CB CD8+ T cells (Figure 4E). FlowJo analysis using the proliferation tool additionally showed a trend of impaired proliferation, division, and expansion rate in CB CD8+ T cells transduced in the presence of LB but not a significant difference (Figure 4F). In addition, CB CD8+ T cells transduced with an MOI 1, in the presence of LB, did not show lower proliferation capacity (Figure S3). The use of LB only slightly and not significantly decreased viability when compared to transduced T cells in the absence of LB (Figure 4G).
Figure 4.

LentiBOOST (LB) increases transduction efficiency of CB CD8+-transduced T cells and maintains proliferation in vitro
(A) Transduction efficiency, presented as percentage of GFP+ cells, is improved by the use of LB together with LV at MOI 10 (left) and MOI 1 (right). (B) Protein expression, calculated as MFI of EGFP signal, is increased by the use of LB together with both LV at MOI 10 (left) and MOI 1 (right). (C) qPCR analysis of the VCN in CB CD8+ T cells transduced with an MOI 10 in the absence (black) or presence (black stripes) of LB or transduced with an MOI 1 in the absence (gray) or presence (gray stripes) of LB. (D) Representative flow cytometry plot of proliferation analysis using CellTrace Violet (CTV) on CB CD8+ T cells transduced with the STND method or with STND+LB. (E) Proliferation rate (as CTV-MFI of day 0/day 4) in CB CD8+ T cells untransduced and transduced in the presence or absence of LB. (F) Proliferation indexes (calculated using proliferation model of FlowJo v.10) define proliferation, division, and expansion rate of CB CD8+ T cells transduced in the presence of LB (black stripes) compared to STND protocol (black). (G) Cell viability, respectively, at day 0 and day 4 of proliferation assay in CB CD8+ T cells transduced in the presence of LB (black stripes) compared to STND protocol (black). The data correspond to ≥3 independent experiments and are shown as average ± standard deviation; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
CB CD8+ T cells transduced with a TAA-specific TCR, in the presence or absence of LB, show comparable and efficient antigen recognition, proliferation, and killing capacity
To further test the effect of LB on CB CD8+ T cell proliferation and functionality in vitro, CB CD8+ T cells transduced with a LV expressing a WT1-specific TCR in the presence of LB were tested for antigen recognition, antigen-specific proliferation, and killing capacity. To test antigen recognition, transduced CB CD8+ T cells were incubated with fluorophore-coupled tetramer molecules pulsed with the target WT1-derived peptide (pWT1126). As shown in Figures 5A and 5B, both T cells transduced, in the presence or absence of LB, showed recognition of pWT1126, with a percentage similar as the transduction efficiency, with an MOI 1 (STND: 8.9% ± 2.3%; STND + LB: 18.4% ± 2.2%) and an MOI 10 (STND: 60.5% ± 10%; STDN + LB: 80% ± 7.3%). qPCR analysis of VCN shows a higher number of integration, both when using an MOI 10 and an MOI 1, in the presence of LB (Figure 5C). After 6 days from the transduction, CB CD8+ T cells were stained with CTV and followed up over the course of 7 days in the presence of pWT1126 tetramer stimulation. WT1-TCR CB CD8+ T cells, transduced in the presence or absence of LB, showed a similar proliferation capacity (Figure 5D). The same cells were co-cultured overnight with T2 cells, loaded with pWT1126, in a 1:1 ratio between target and effector (T:E) cells. The WT1-TCR-engineered cells, transduced in the absence or presence of LB, were effectively killing, on average, 81% and 90% of target T2 cells, respectively, which was statistically significant compared to untransduced cells that only killed 2% of target cells (Figures 5E and 5G). Cytotoxic cytokine levels (interferon [IFN]-γ, tumor necrosis factor [TNF]-α, granzyme A, granzyme B, and perforin) were analyzed in the supernatant of the killing assays. WT1-TCR-transduced cells produced a higher amount of cytotoxic cytokines compared to untransduced cells, statistically significant for IFN-γ and granzyme B (p value: <0.0001), whereas there were no differences between transduced T cells in the presence or absence of LB (Figure 5F). Additional experiments to support our findings were performed with a more limiting T:E cell ratio (10:1) and showed a slightly increased killing capacity of WT1-TCR-transduced CB CD8+ T cells in the presence of LB, likely due to the presence of a larger proportion of WT1-TCR-transduced T cells (Figure S4).
Figure 5.

LB does not affect functionality of WT1-TCR-transduced CB CD8+ T cells
(A) Transduction efficiency and antigen recognition, presented as percentage of TCR Vb21.3+ T cells (left) and tetramer+ cells (right), improved by the use of LB at MOI 1 and MOI 10 compared to STND method. (B) Representative flow cytometry plot of CB CD8+ T cell transduction efficiency and pWT1126 tetramer staining. (C) qPCR analysis of the VCN in CB CD8+ T cells transduced with an MOI 10 in the absence (black) or presence (black stripes) of LB or transduced with an MOI 1 in the absence (gray) or presence (gray stripes) of LB. (D) Proliferation rate (as CTV-MFI of day 0/day 7) of CB CD8+ T cells untransduced and transduced with WT1-TCR (MOI 10) in the presence or absence of LB and stimulated with tetramer loaded with pWT1126. (E) Killing capacity of WT1-TCR CB CD8+ T cells transduced in the presence or absence of LB, depicted as percentage of alive target cells in a co-culture experiment with target cells loaded with pWT1126. (F) Levels of cytotoxic cytokines in the supernatant of co-culture experiments produced by WT1-TCR-transduced CB CD8+ T cells in the presence or absence of LB compared to untransduced cells. (G) Representative flow cytometry plot of percentage of alive target cells after overnight co-culture with WT1-TCR-transduced CB CD8+ T cells. Co-culture experiments were performed in a ratio of 1:1 (target:effector). The data correspond to ≥3 independent experiments and are shown as average ± standard deviation; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001.
Discussion
The possibility to gene modify immune cells has opened a broad range of cell therapy applications for difficult-to-treat tumor types. However, it remains challenging to obtain enough functionally proficient autologous T cells from cancer patients and to efficiently gene modify immune cells. In this manuscript, we studied the use of allogeneic CB cells as a rich source to obtain CD8+ T cells that are amenable for lentiviral transduction. The use of CB-derived cells is well established in clinical applications, such as hematopoietic cell transplantation (HCT).38, 39, 40 CB T lymphocyte cultures can result in more than 100-fold magnitude of expansion, enabling feasible upscaling processes to generate sufficient cell numbers for therapeutic purposes.41,42 Furthermore, CB lymphocytes are associated with favorable graft versus tumor effects, reaching sufficient numbers and efficiency for donor lymphocyte infusion post-HCT.43,44 Several clinical trials are testing the safety and the potential benefit of infusing CB-derived, naturally occurring lymphocytes,45,46 including T cells, natural killer (NK) cells, and cytokine-induced killer cell (CIK), to avoid relapse (ClinicalTrials.gov: NCT01630564) or viral reactivation (Clinical Trials.gov: NCT03594981) after HCT. Although the results on CB-derived T cell infusions are promising, the generation of specific, gene-modified CB T lymphocytes for therapeutic approaches is still in its infancy compared to using PB T cells. In this manuscript, we present an optimized method to obtain highly efficient lentiviral transduction of CB CD8+ T cells, which could potentially be translated for use in clinical settings. In fact, the use of serum-free StemMACS medium showed a transduction efficiency of more than 80% of CB CD8+ T cells, using a relatively low MOI, favorable for future good manufacturing practice (GMP) production and safety concerns (Figure 2). This method also confirmed the enhanced expression achieved with the use of a viral-derived promoter (MND) compared to the constitutively expressed promoters (PGK).
With the use of this protocol, the total fraction of CB CD8+ T cells was more amenable to transduction compared to the total fraction of PB CD8+ T cells. We speculate that this protocol may be beneficial by exploiting the naive characteristics of CB CD8+ T cells, in accordance to previous studies that showed an increased transduction efficiency in PB-derived Tn cells and Tscm cells.32,47 Nevertheless, we may expect an increased potency of CB CD8+ T cells due to the differences in immune phenotype between the two cell sources.48,49 Pre-clinical and clinical studies have shown the short persistence and low proliferation properties of engineered T cells derived from PB of adult patients.50 Moreover, in vivo studies have shown the superiority of Tn over Tm cells in adoptive immunotherapeutic applications.51,52 The proposed method combines high transduction efficiency with a global maintenance of the least differentiated Tscm or Tn phenotype of the CB CD8+ T cells, confirmed by the downregulation of CD45RO and the upregulation of CD45RA (Figure 3). This method increases the feasibility to use CB CD8+ T cells in clinical applications; however, its clinical significance will rely on the activation, proliferation, and long-term survival of genetically modified cells in vivo. The naive phenotype, together with the intermediate and the low expression of inhibitory receptors (TIM3, LAG3), which were maintained low in the current protocol, could be a valuable feature for improving proliferation, persistence, and efficacy in future pre-clinical and clinical studies. The difference in transduction efficiency between the cell sources may also be influenced by the expression levels of the cognate glycoprotein receptors on the target cells, defining the probability of binding between viral particle and cell membrane.53 This effect was previously shown by substituting the envelope from vesicular stomatitis virus G protein (VSV-G) to measles virus (MV)37 or baboon retroviral envelope (BaEV),53 which increased the transduction efficiency of the LV, a strategy that can be further explored.
Further improvements in the transduction efficiency was achieved using the transduction enhancer LB. The active component of LB is Poloxamer 338/Pluronic F108 and belongs to the class of A-B-A-type tri-block copolymers composed of hydrophilic ethyleneoxide and hydrophobic propyleneoxide. This structure confers a capability of interacting with hydrophobic particles and biological membranes. The advantage of using this enhancer and alternative poloxamers (Poloxamer 407) has already been proven in HSCs and mouse-derived T cells,25,34,54 and its GMP-grade formula has already been used in clinical trials. The use of LB clearly increased the transduction efficiency also in CB CD8+ T cells using different LV constructs (LV.EGFP, LV.WT1-TCR, and LV.CAR19), especially interesting for applications requiring low MOI or when vector titers are limited. As previously shown,34,55 the use of LB very minimally affects T cell viability, as shown also by using our optimized protocol (Figure 4G). With the use of a tracer molecule, we observed that LB affected the proliferation rate of CB CD8+ T cells transduced with an MOI 10, after providing both the primary and co-stimulatory signals that are required for activation and expansion of T cells via aCD3/CD28 beads. The reduced proliferation rate was visible only when using LV.EGFP and an MOI 10, whereas untransduced cells treated with the LB molecule (Figure 4E) and cells transduced with an MOI 1 (Figure S3) did not show impaired proliferation upon aCD3/CD28 stimulation (Figure S4). The effect on proliferation coincided with an increase in VCN in the transduced T cells in the presence of LB, possibly caused by genotoxic stress in the CB CD8+ T cells, supported by the observation that CB CD8+ T cells transduced at an MOI 1 did not show a decreased proliferation rate. As previously reported, self-inactivating LVs have the tendency to integrate into transcriptionally active genes in T cells,56 potentially causing genotoxic effects at higher VCNs. Interestingly, the decrease in proliferation was not visible when CB CD8+ T cells were transduced with a TAA-specific TCR in the presence of LB and stimulated with antigen-specific tetramers. Moreover, WT1-TCR CB CD8+ T cells, transduced in the presence of LB, did not show any impairment in terms of functionality, as assessed by killing capacity and cytotoxic cytokines produced (Figures 5E−5G).
In conclusion, we present an efficient expansion and transduction method for CB CD8+ T cells using LVs that is GMP compliant. Furthermore, this method shows better transduction of the total fraction of CB CD8+ T cells compared to the total fraction of PB CD8+ T cells, while preserving the CB naive phenotypic markers, increasing the chance to generate a cellular product that will persist and proliferate in vivo. Additionally, we confirmed the use of the LB as a valid transduction enhancer for CB CD8+ T cells in the field of gene and cell therapy. Overall, these results underlie the possibilities and advantages of using gene-modified CB CD8+ T cells as a source for future application in cancer cell therapy.
Materials and methods
LV production and titration
HIV-derived, self-inactivating, third-generation LVs were constructed using the cytomegalovirus (CMV) promoter driving the expression of the viral transcript (pCCL plasmid backbone). Additionally to the packaging signal (ψ), the REV responsive element (RRE), and the central polypurine tract (cPPT), the transfer vector plasmid contains a modified Woodchuck post-translational regulatory element (bPRE4).57,58 Lentiviral particles coding for EGFP were produced by transient cotransfection of the lentiviral transfer vector plasmid (LV.EGFP) and the respective packaging plasmids (pRSV-Rev, pMDLg/pRRE, and pMD2-VSV-G) to HEK293T cells (ATCC CRL-3216) using the CalPhos Mammalian Transfection Kit (Clontech Laboratories), as previously described.59 Viral supernatants were filtered through 0.45 μm low-protein-binding filters, concentrated by ultracentrifugation at 20,000 × g for 2 h, resuspended in StemMACS (Miltenyi Biotec), and stored at −80°C. We generated two LVs expressing promoter PGK.EGFP60 or a synthetic promoter MND.EGFP.61,62 Additionally, two extra LVs were generated expressing a codon-optimized human TCR alpha- and beta-chain sequence, bridged by a T2A self-cleaving, recognizing pWT1126 antigen63,64 under the control of the MND promoter (MND.WT1-TCR) (Figure 1) and a CAR19 under the control of the PGK promoter (PGK.CAR19) (Figure S2). VSV-G protein-pseudotyped lentiviral particles were titrated by serial dilution on Jurkat cells (clone E6-1; ATCC TIB-152). At 72 h post-transduction, cells were harvested and analyzed by flow cytometry for GFP expression, and viral titer was calculated.
CD8+ T cell isolation and expansion
Fresh CB or PB was collected after informed consent was obtained according to the Declaration of Helsinki. The collection protocol was approved by the Ethical Committee of the University Medical Center (UMC) Utrecht. CB or PB was processed to isolate CD8+ T cells using Ficoll (GE Healthcare Bio-Sciences AB) separation and CD8-positive magnetic bead separation according to the manufacturer’s protocol (Miltenyi Biotec). CD8+ T cells were subsequently cultured in RPMI (Fisher Scientific), supplemented with 10% human serum; 1% penicillin/streptomycin (P/S) (cytotoxic T lymphocyte media [CTL media]), with aCD3/CD28 Dynabeads (Gibco; Thermo Fisher Scientific) in a 1:3 ratio (beads:T cells); 50 U of IL-2/mL (UMC-Pharmacy); 5 ng/mL IL-7; and 5 ng/mL IL-15 (Miltenyi Biotec). CD8+ T cells were kept in culture for 3 days, resulting in an average 4.5-fold expansion rate.
T cell transduction
Expanded and stimulated CD8+ T cells were transduced at MOI 1 and 10 with two different methods: spinoculation and overnight incubation. For the spinoculation, T cells were mixed and incubated with the LV for 30 min at room temperature and centrifuged for 30 min at 800 × g at 32°C. After the centrifugation step, transduction media were replaced with complete CTL media. For the overnight transduction, T cells were mixed and incubated with the LV for 14−16 h, after which, transduction media were replaced with complete CTL media. We used two different transduction media: X-Vivo15 (Lonza) and StemMACS (Miltenyi Biotec), supplemented with 1% P/S, with or without supplementation of the transduction enhancer LB (1:100 of the total volume; SIRION Biotech).
VCN
Genomic DNA was isolated from transduced cells using a Genomic DNA Purification kit (QIAGEN). qRT-PCR was performed with SYBR Green PCR Master Mix (Thermo Fisher Scientific) and primers targeting the U3 region (FW: 5′-CTGGAAGGGCTAATTCACTC-3′) and Psi (RV: 5′-GGTTTCCCTTTCGCTTTCAA-3′). Amplification of the β-actin gene was used as a reference gene (FW primer: 5′-AGCGGGAAATCGTGCGTGAC-3′; R primer: 5′-CAATGGTGATGACCTGGCCGT-3′). Serial dilutions of a standard DNA plasmid containing one integrated copy of the LV sequence were used to plot a standard curve. Samples and standard serial dilutions were run in duplicate.
CD8+ T cell proliferation assay
Untransduced and transduced CD8+ T cells were washed with PBS to remove serum that affects staining. Then, cells were suspended in PBS at a concentration of 2.4 × 106 cells/mL and labeled with the CTV Proliferation Kit (5 mM; Thermo Fisher Scientific) at 37°C for 20 min. Subsequently, cells were washed with warm fetal calf serum and resuspended in CTL media. After 24 h, cells were analyzed with flow cytometry to confirm CTV staining. Cells were stimulated with aCD3/CD28 Dynabeads in a ratio 1:8 (beads:T cells) or with a tetramer molecule loaded with pWT1126. After 4 and 7 days, T cells were stained with Fixable Viability Dye and analyzed with flow cytometry to detect a decrease of CTV signal.
Tetramer staining
A tetramer molecule was generated according to an established protocol,65 consisting of pWT1126 stained with fluorescent-labeled streptavidin (phycoerythrin [PE] and PE-Cy7). Untransduced and transduced CD8+ T cells were treated with dasatinib (VWR International) at a final concentration of 50 nM. Cells were incubated at 37°C for 30 min to allow stabilization of the TCR on the cell surface. Cells were subsequently washed once with fluorescence-activated cell sorting (FACS) buffer, after which the tetramer staining was initiated by adding approximately 0.1 μg per peptide:MHC complex and incubated 15 min at 37°C. Without washing, an antibody mix was added to the cells: CD8-APC (clone RPA-T8, ref. 555369; BD Pharmingen); CD3-Pacific Blue (clone UCHT1, ref. 558117; Becton Dickinson [BD]); Fixable Viability Dye eFluor 780 (ref. 65-0865-14; Thermo Fisher Scientific); and TCR Vβ21.3 (lone IG125, ref. PN IM1483; Beckman Coulter). Cells were incubated with the antibodies mix for 15 min at 4°C, washed, and analyzed on a BD Fortessa using FACSDiva.
Killing assay and cytokine analysis
T2 cells (ATCC CRL-1992), were stained with CTV (following the method explained in CD8+ T cell proliferation assay above) and subsequently pulsed with 1 μg/mL of pWT1126. T2 cells were co-cultured overnight with untransduced or transduced CB-CD8+ T cells in a 1:1 or a 1:10 ratio (T:E cells). After 14−16 h, cells were stained with 7-AAD (BD Pharmingen), after which, flow cytometry analysis was performed on a BD LSRFortessa using FACSDiva, with acquisition of a fixed amount of total volume for each sample. Cytokines in supernatants were measured using the LEGENDplex assay (BioLegend). Flow cytometry analysis was performed on a BD LSRFortessa using FACSDiva, and post-acquisition analysis was performed with LEGENDplex Data Analysis Software.
Flow cytometry analysis
Transduced CD8+ T cells were stained for 15 min at 4°C with fluorescently labeled antibodies against CD3-Pacific Blue (clone UCHT1, ref. 558117; BD) and CD8-PE (clone RPA-T8, ref. 301008; BioLegend); Fixable Viability Dye eFluor 780 (ref. 65-0865-14; Thermo Fisher Scientific) was used to evaluate transduction efficiency. Fresh, expanded, and transduced cells were stained with Fixable Viability Dye eFluor 780 (ref. 65-0865-14; Thermo Fisher Scientific), CD62L-BV650 (clone DREG-56, ref. 2124160; Sony Biotechnology), CD25-PerCP Cy5.5 (clone BC96, ref. 2113130; Sony Biotechnology), TIM3-APC (clone 34482, ref. FAB2365A; R&D Systems [R&D]), CD3-AF700 (clone UCHT1, ref. 300424; Sony Biotechnology), CD45RA-BV421 (clone HI100, ref. 304118; BioLegend), CD45RO-BV711 (clone UCHL1, ref. 304236; BioLegend), LAG3-PE (ref. FAB2319P; R&D), CD8-PE-Cy7 (clone SK1, ref. 335822; BD), TCR Vβ21.3 (clone IG125, ref. PN IM1483; Beckman Coulter), and CD19 CAR detection reagent (ref. AB_2811310; Miltenyi Biotec). Flow cytometry analysis was performed on a BD LSRFortessa using FACSDiva, with acquisition of a fixed number of cells for each experiment. FACS data analysis was performed using FlowJo version (v.)10 . Data are presented as percent of cells and as MFI.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v.6.00 for Windows (GraphPad Software, La Jolla, CA, USA). Due to the small number of samples, normality was assessed using QQ-plot visualization. One-way analysis of variance and Student’s t test were used to determine differences between groups. The results are presented as average ± standard deviation (SD). p values are considered statistically significant when ∗p ≤0.05.
Acknowledgments
The authors thank the staff of the Flow Core Facility (Center for Translational Immunology, UMC Utrecht [UMCU], the Netherlands), especially Jeroen van Velzen, for the support and help in the sample analysis. The authors also thank the women for donating the CB and the nurses (Wilhelmina’s Children Hospital, UMCU, the Netherlands) for collecting it. The work done for the manuscript was supported by the Dutch Cancer Society (KWF; grant number 10474).
Author contributions
V.L.P. designed and conducted the experiments, analyzed the data, and wrote the manuscript. A.M.C. and M.P. contributed to the execution of experiments and the writing of the manuscript. E.D. helped in performing experiments. J.K. and J.J.B. scientifically contributed to the design of the project and the manuscript. N.P.v.T. and S.N. designed and supervised the experimental phase and contributed to drafting the manuscript.
Declaration of interests
N.P.v.T. is an employee of AVROBIO, Inc., Cambridge, MA, USA. J.K. is cofounder and shareholder of Gadeta; is an inventor on multiple patents dealing with γδTCR topics as well as isolation of engineered immune cells; and received research support from Miltenyi Biotec, Novartis, and Gadeta. The other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.03.015.
Supplemental information
References
- 1.Dudley M.E., Wunderlich J.R., Shelton T.E., Even J., Rosenberg S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg S.A., Yang J.C., Sherry R.M., Kammula U.S., Hughes M.S., Phan G.Q., Citrin D.E., Restifo N.P., Robbins P.F., Wunderlich J.R. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley M.E., Yang J.C., Sherry R., Hughes M.S., Royal R., Kammula U., Robbins P.F., Huang J., Citrin D.E., Leitman S.F. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim W.A., June C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017;168:724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.June C.H., Sadelain M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grigor E.J.M., Fergusson D., Kekre N., Montroy J., Atkins H., Seftel M.D., Daugaard M., Presseau J., Thavorn K., Hutton B. Risks and Benefits of Chimeric Antigen Receptor T-Cell (CAR-T) Therapy in Cancer: A Systematic Review and Meta-Analysis. Transfus. Med. Rev. 2019;33:98–110. doi: 10.1016/j.tmrv.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 7.European Medicines Agency: Committee for Medicinal Products for Human Use (CHMP) 2018. Assessment Report - Kymriah.https://www.ema.europa.eu/en/medicines/human/EPAR/kymriah [Google Scholar]
- 8.Leary M.O., Przepiorka D., George B., Theoret M. 2017. BLA Clinical Review Memorandum.https://www.fda.gov/media/107973/download [Google Scholar]
- 9.Zhang J., Wang L. The emerging world of TCR-T cell trials against cancer: A systematic review. Technol. Cancer Res. Treat. 2019;18 doi: 10.1177/1533033819831068. 1533033819831068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan R.A., Dudley M.E., Wunderlich J.R., Hughes M.S., Yang J.C., Sherry R.M., Royal R.E., Topalian S.L., Kammula U.S., Restifo N.P. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapoport A.P., Stadtmauer E.A., Binder-Scholl G.K., Goloubeva O., Vogl D.T., Lacey S.F., Badros A.Z., Garfall A., Weiss B., Finklestein J. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015;21:914–921. doi: 10.1038/nm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapuis A.G., Egan D.N., Bar M., Schmitt T.M., McAfee M.S., Paulson K.G., Voillet V., Gottardo R., Ragnarsson G.B., Bleakley M. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019;25:1064–1072. doi: 10.1038/s41591-019-0472-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gröbner S.N., Worst B.C., Weischenfeldt J., Buchhalter I., Kleinheinz K., Rudneva V.A., Johann P.D., Balasubramanian G.P., Segura-Wang M., Brabetz S., ICGC PedBrain-Seq Project. ICGC MMML-Seq Project The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–327. doi: 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- 14.Zufferey R., Dull T., Mandel R.J., Bukovsky A., Quiroz D., Naldini L., Trono D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham C., Jozwik A., Pepper A., Benjamin R. Allogeneic CAR-T Cells: More than Ease of Access? Cells. 2018;7:1–11. doi: 10.3390/cells7100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackall C.L., Fleisher T.A., Brown M.R., Magrath I.T., Shad A.T., Horowitz M.E., Wexler L.H., Adde M.A., McClure L.L., Gress R.E. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood. 1994;84:2221–2228. [PubMed] [Google Scholar]
- 17.Stahnke K., Fulda S., Friesen C., Strauss G., Debatin K.M. Activation of apoptosis pathways in peripheral blood lymphocytes by in vivo chemotherapy. Blood. 2001;98:3066–3073. doi: 10.1182/blood.v98.10.3066. [DOI] [PubMed] [Google Scholar]
- 18.Mackall C.L., Fleisher T.A., Brown M.R., Andrich M.P., Chen C.C., Feuerstein I.M., Magrath I.T., Wexler L.H., Dimitrov D.S., Gress R.E. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. 1997;89:3700–3707. [PubMed] [Google Scholar]
- 19.Fagnoni F.F., Lozza L., Zibera C., Zambelli A., Gibelli N., Ponchio L., Oliviero B., Pavesi L., Gennari R., Vescovini R. T-cell dynamics after high-dose chemotherapy in adults : elucidation of the elusive CD8 + subset reveals multiple homeostatic T-cell compartments with distinct implications for immune competence. Immunology. 2002;106:27–37. doi: 10.1046/j.1365-2567.2002.01400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onyema O.O., Decoster L., Njemini R., Forti L.N., Bautmans I., De Waele M., Mets T. Chemotherapy-induced changes and immunosenescence of CD8+ T-cells in patients with breast cancer. Anticancer Res. 2015;35:1481–1489. [PubMed] [Google Scholar]
- 21.Paloczi K. Immunophenotypic and functional characterization of human umbilical cord blood mononuclear cells. Leukemia. 1999;13(Suppl 1):S87–S89. doi: 10.1038/sj.leu.2401318. [DOI] [PubMed] [Google Scholar]
- 22.Lo Presti V., Nierkens S., Boelens J.J., van Til N.P. Use of cord blood derived T-cells in cancer immunotherapy: milestones achieved and future perspectives. Expert Rev. Hematol. 2018;11:209–218. doi: 10.1080/17474086.2018.1431119. [DOI] [PubMed] [Google Scholar]
- 23.Hiwarkar P., Qasim W., Ricciardelli I., Gilmour K., Quezada S., Saudemont A., Amrolia P., Veys P. Cord blood T cells mediate enhanced antitumor effects compared with adult peripheral blood T cells. Blood. 2015;126:2882–2891. doi: 10.1182/blood-2015-06-654780. [DOI] [PubMed] [Google Scholar]
- 24.Oki M., Ando K., Hagihara M., Miyatake H., Shimizu T., Miyoshi H., Nakamura Y., Matsuzawa H., Sato T., Ueda Y. Efficient lentiviral transduction of human cord blood CD34(+) cells followed by their expansion and differentiation into dendritic cells. Exp. Hematol. 2001;29:1210–1217. doi: 10.1016/s0301-472x(01)00695-6. [DOI] [PubMed] [Google Scholar]
- 25.Hauber I., Beschorner N., Schrödel S., Chemnitz J., Kröger N., Hauber J., Thirion C. Improving Lentiviral Transduction of CD34+ Hematopoietic Stem and Progenitor Cells. Hum. Gene Ther. Methods. 2018;29:104–113. doi: 10.1089/hgtb.2017.085. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y., Hangoc G., Campbell T.B., Goodman M., Tao W., Pollok K., Srour E.F., Broxmeyer H.E. Identification of parameters required for efficient lentiviral vector transduction and engraftment of human cord blood CD34(+) NOD/SCID-repopulating cells. Exp. Hematol. 2008;36:947–956. doi: 10.1016/j.exphem.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans J.T., Kelly P.F., O’Neill E., Garcia J.V. Human cord blood CD34+CD38- cell transduction via lentivirus-based gene transfer vectors. Hum. Gene Ther. 1999;10:1479–1489. doi: 10.1089/10430349950017815. [DOI] [PubMed] [Google Scholar]
- 28.Serrano L.M., Pfeiffer T., Olivares S., Numbenjapon T., Bennitt J., Kim D., Smith D., McNamara G., Al-Kadhimi Z., Rosenthal J. Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy. Blood. 2006;107:2643–2652. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Micklethwaite K.P., Savoldo B., Hanley P.J., Leen A.M., Demmler-Harrison G.J., Cooper L.J.N., Liu H., Gee A.P., Shpall E.J., Rooney C.M. Derivation of human T lymphocytes from cord blood and peripheral blood with antiviral and antileukemic specificity from a single culture as protection against infection and relapse after stem cell transplantation. Blood. 2010;115:2695–2703. doi: 10.1182/blood-2009-09-242263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pegram H.J., Purdon T.J., van Leeuwen D.G., Curran K.J., Giralt S.A., Barker J.N., Brentjens R.J. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia. 2015;29:415–422. doi: 10.1038/leu.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frumento G., Zheng Y., Aubert G., Raeiszadeh M., Lansdorp P.M., Moss P., Lee S.P., Chen F.E. Cord blood T cells retain early differentiation phenotype suitable for immunotherapy after TCR gene transfer to confer EBV specificity. Am. J. Transplant. 2013;13:45–55. doi: 10.1111/j.1600-6143.2012.04286.x. [DOI] [PubMed] [Google Scholar]
- 32.Cieri N., Camisa B., Cocchiarella F., Forcato M., Oliveira G., Provasi E., Bondanza A., Bordignon C., Peccatori J., Ciceri F. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–584. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 33.Tammana S., Huang X., Wong M., Milone M.C., Ma L., Levine B.L., June C.H., Wagner J.E., Blazar B.R., Zhou X. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum. Gene Ther. 2010;21:75–86. doi: 10.1089/hum.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delville M., Soheili T., Bellier F., Durand A., Denis A., Lagresle-Peyrou C., Cavazzana M., Andre-Schmutz I., Six E. A Nontoxic Transduction Enhancer Enables Highly Efficient Lentiviral Transduction of Primary Murine T Cells and Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev. 2018;10:341–347. doi: 10.1016/j.omtm.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cavalieri S., Cazzaniga S., Geuna M., Magnani Z., Bordignon C., Naldini L., Bonini C. Human T lymphocytes transduced by lentiviral vectors in the absence of TCR activation maintain an intact immune competence. Blood. 2003;102:497–505. doi: 10.1182/blood-2003-01-0297. [DOI] [PubMed] [Google Scholar]
- 36.Jones S., Peng P.D., Yang S., Hsu C., Cohen C.J., Zhao Y., Abad J., Zheng Z., Rosenberg S.A., Morgan R.A. Lentiviral vector design for optimal T cell receptor gene expression in the transduction of peripheral blood lymphocytes and tumor-infiltrating lymphocytes. Hum. Gene Ther. 2009;20:630–640. doi: 10.1089/hum.2008.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frecha C., Costa C., Nègre D., Gauthier E., Russell S.J., Cosset F.L., Verhoeyen E. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood. 2008;112:4843–4852. doi: 10.1182/blood-2008-05-155945. [DOI] [PubMed] [Google Scholar]
- 38.Ballen K.K., Gluckman E., Broxmeyer H.E. Umbilical cord blood transplantation: the first 25 years and beyond. Blood. 2013;122:491–498. doi: 10.1182/blood-2013-02-453175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boelens J.J. The power of cord blood cells. Blood. 2016;127:3302–3303. doi: 10.1182/blood-2016-04-713065. [DOI] [PubMed] [Google Scholar]
- 40.Barker J.N., Davies S.M., DeFor T., Ramsay N.K.C., Weisdorf D.J., Wagner J.E. Survival after transplantation of unrelated donor umbilical cord blood is comparable to that of human leukocyte antigen-matched unrelated donor bone marrow: results of a matched-pair analysis. Blood. 2001;97:2957–2961. doi: 10.1182/blood.v97.10.2957. [DOI] [PubMed] [Google Scholar]
- 41.Azuma H., Yamada Y., Shibuya-Fujiwara N., Yamaguchi M., Murahashi H., Fujihara M., Sato N., Fukazawa K., Ikebuchi K., Ikeda H. Functional evaluation of ex vivo expanded cord blood lymphocytes: possible use for adoptive cellular immunotherapy. Exp. Hematol. 2002;30:346–351. doi: 10.1016/s0301-472x(02)00776-2. [DOI] [PubMed] [Google Scholar]
- 42.Okas M., Gertow J., Uzunel M., Karlsson H., Westgren M., Kärre K., Ringden O., Mattsson J., Uhlin M. Clinical expansion of cord blood-derived T cells for use as donor lymphocyte infusion after cord blood transplantation. J. Immunother. 2010;33:96–105. doi: 10.1097/CJI.0b013e3181b291a4. [DOI] [PubMed] [Google Scholar]
- 43.Parmar S., Robinson S.N., Komanduri K., St John L., Decker W., Xing D., Yang H., McMannis J., Champlin R., de Lima M. Ex vivo expanded umbilical cord blood T cells maintain naive phenotype and TCR diversity. Cytotherapy. 2006;8:149–157. doi: 10.1080/14653240600620812. [DOI] [PubMed] [Google Scholar]
- 44.Kwoczek J., Riese S.B., Tischer S., Bak S., Lahrberg J., Oelke M., Maul H., Blasczyk R., Sauer M., Eiz-Vesper B. Cord blood-derived T cells allow the generation of a more naïve tumor-reactive cytotoxic T-cell phenotype. Transfusion. 2018;58:88–99. doi: 10.1111/trf.14365. [DOI] [PubMed] [Google Scholar]
- 45.Berglund S., Gertow J., Uhlin M., Mattsson J. Expanded umbilical cord blood T cells used as donor lymphocyte infusions after umbilical cord blood transplantation. Cytotherapy. 2014;16:1528–1536. doi: 10.1016/j.jcyt.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Balassa K., Rocha V. Anticancer cellular immunotherapies derived from umbilical cord blood. Expert Opin. Biol. Ther. 2018;18:121–134. doi: 10.1080/14712598.2018.1402002. [DOI] [PubMed] [Google Scholar]
- 47.Sabatino M., Hu J., Sommariva M., Gautam S., Fellowes V., Hocker J.D., Dougherty S., Qin H., Klebanoff C.A., Fry T.J. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128:519–528. doi: 10.1182/blood-2015-11-683847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gattinoni L., Klebanoff C.A., Palmer D.C., Wrzesinski C., Kerstann K., Yu Z., Finkelstein S.E., Theoret M.R., Rosenberg S.A., Restifo N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klebanoff C.A., Gattinoni L., Torabi-Parizi P., Kerstann K., Cardones A.R., Finkelstein S.E., Palmer D.C., Antony P.A., Hwang S.T., Rosenberg S.A. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robbins P.F., Dudley M.E., Wunderlich J., El-Gamil M., Li Y.F., Zhou J., Huang J., Powell D.J., Jr., Rosenberg S.A. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hinrichs C.S., Borman Z.A., Gattinoni L., Yu Z., Burns W.R., Huang J., Klebanoff C.A., Johnson L.A., Kerkar S.P., Yang S. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Costa C., Hypolite G., Bernadin O., Lévy C., Cosset F.-L., Asnafi V., Macintyre E., Verhoeyen E., Tesio M. Baboon envelope pseudotyped lentiviral vectors: a highly efficient new tool to genetically manipulate T-cell acute lymphoblastic leukaemia-initiating cells. Leukemia. 2017;31:977–980. doi: 10.1038/leu.2016.372. [DOI] [PubMed] [Google Scholar]
- 54.Uchida N., Nassehi T., Drysdale C.M., Gamer J., Yapundich M., Demirci S., Haro-Mora J.J., Leonard A., Hsieh M.M., Tisdale J.F. High-Efficiency Lentiviral Transduction of Human CD34+ Cells in High-Density Culture with Poloxamer and Prostaglandin E2. Mol. Ther. Methods Clin. Dev. 2019;13:187–196. doi: 10.1016/j.omtm.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simon B., Harrer D.C., Thirion C., Schuler-Thurner B., Schuler G., Uslu U. Enhancing lentiviral transduction to generate melanoma-specific human T cells for cancer immunotherapy. J. Immunol. Methods. 2019;472:55–64. doi: 10.1016/j.jim.2019.06.015. [DOI] [PubMed] [Google Scholar]
- 56.Field A.C., Vink C., Gabriel R., Al-Subki R., Schmidt M., Goulden N., Stauss H., Thrasher A., Morris E., Qasim W. Comparison of lentiviral and sleeping beauty mediated αβ T cell receptor gene transfer. PLoS ONE. 2013;8:e68201. doi: 10.1371/journal.pone.0068201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schambach A., Bohne J., Baum C., Hermann F.G., Egerer L., von Laer D., Giroglou T. Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 2006;13:641–645. doi: 10.1038/sj.gt.3302698. [DOI] [PubMed] [Google Scholar]
- 58.van Til N.P., Sarwari R., Visser T.P., Hauer J., Lagresle-Peyrou C., van der Velden G., Malshetty V., Cortes P., Jollet A., Danos O. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J. Allergy Clin. Immunol. 2014;133:1116–1123. doi: 10.1016/j.jaci.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 59.Tiscornia G., Singer O., Verma I.M. Production and purification of lentiviral vectors. Nat. Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 60.Singer-Sam J., Keith D.H., Tani K., Simmer R.L., Shively L., Lindsay S., Yoshida A., Riggs A.D. Sequence of the promoter region of the gene for human X-linked 3-phosphoglycerate kinase. Gene. 1984;32:409–417. doi: 10.1016/0378-1119(84)90016-7. [DOI] [PubMed] [Google Scholar]
- 61.Li H., Ginzburg Y.Z. Crosstalk between Iron Metabolism and Erythropoiesis. Adv. Hematol. 2010;2010:605435. doi: 10.1155/2010/605435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Logan A.C., Nightingale S.J., Haas D.L., Cho G.J., Pepper K.A., Kohn D.B. Factors influencing the titer and infectivity of lentiviral vectors. Hum. Gene Ther. 2004;15:976–988. doi: 10.1089/hum.2004.15.976. [DOI] [PubMed] [Google Scholar]
- 63.Wolfl M., Kuball J., Ho W.Y., Nguyen H., Manley T.J., Bleakley M., Greenberg P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110:201–210. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuball J., Dossett M.L., Wolfl M., Ho W.Y., Voss R.H., Fowler C., Greenberg P.D. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–2338. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodenko B., Toebes M., Hadrup S.R., van Esch W.J.E., Molenaar A.M., Schumacher T.N.M., Ovaa H. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat. Protoc. 2006;1:1120–1132. doi: 10.1038/nprot.2006.121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.