

Summary
Dendritic spinules are fine membranous protrusions of neuronal spines that play a role in synaptic plasticity, but their nanoscale requires resolution beyond conventional confocal microscopy, hindering live studies. Here, we describe how to track individual spinules in live dissociated cortical pyramidal neurons utilizing fluorescence labeling, optimized confocal imaging parameters, and post-acquisition iterative 3D deconvolution, employing NIS Elements software. This approach enables investigations of spinule structural dynamics and function without using super-resolution microscopy, which involves special fluorophores and/or high laser power.
For complete details on the use and execution of this protocol, please refer to Zaccard et al. (2020).
Subject areas: Cell Biology, Cell culture, Microscopy, Neuroscience
Graphical abstract
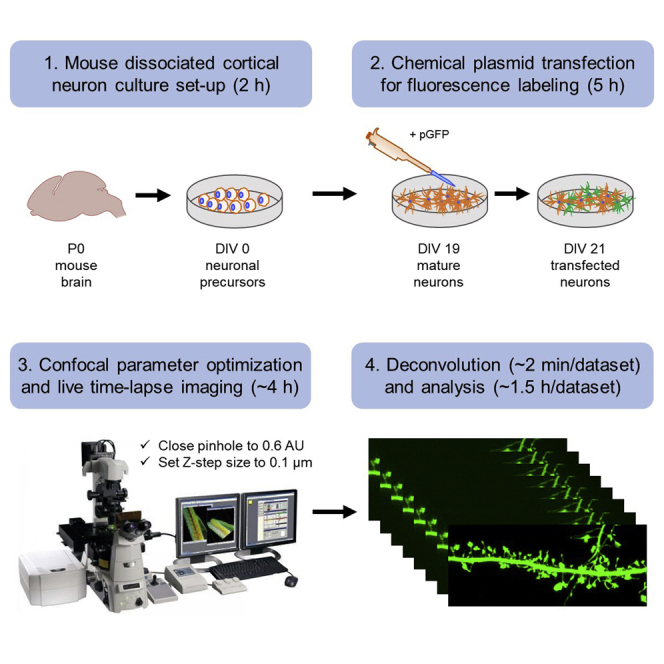
Highlights
-
•
Protocol for live time-lapse enhanced resolution confocal imaging of neurons
-
•
Utilizes fluorescence labeling, optimized confocal settings, and 3D deconvolution
-
•
Enables the visualization of thin membrane projections on spines, e.g., spinules
-
•
The structural dynamics of live individual spinules can be followed over time
Dendritic spinules are fine membranous protrusions of neuronal spines that play a role in synaptic plasticity, but their nanoscale requires resolution beyond conventional confocal microscopy, hindering live studies. Here, we describe how to track individual spinules in live dissociated cortical pyramidal neurons utilizing fluorescence labeling, optimized confocal imaging parameters, and post-acquisition iterative 3D deconvolution, employing NIS Elements software. This approach enables investigations of spinule structural dynamics and function without using super-resolution microscopy, which involves special fluorophores and/or high laser power.
Before you begin
Mouse dissociated cortical neuron culture set-up
Timing: 2 h
-
1.Before you start, prepare the high-glucose complete (c)DMEM and Neurobasal (NB) + supplements media (see “Materials and equipment” for detailed recipes).
-
a.Pre-warm high-glucose cDMEM medium, NB + supplements medium, and 0.25% trypsin in a 37°C hot water bath.
-
b.Filter high-glucose cDMEM medium using a sterile syringe and 0.22 μm filter before use.
-
a.
-
2.
Prepare 3–4 mL aliquots of cold Leibovitz's L-15 medium + Penicillin/Streptomycin (P/S) in 15 mL conical tubes, on ice (up to 8 cortices per tube). Reserve ~100 mL of L-15 medium in 50 mL conical tubes, also on ice, and fill 100 × 15 mm petri dishes half-way with cold L-15 just prior to step 3.
-
3.
In a biosafety cabinet, sacrifice P0 mouse pups by decapitation with a surgical scissors.
Note: Mice were housed inside a specific pathogen-free area on a 12:12 h light/dark plan, under steady conditions, i.e., temperatures between 20°C–26°C and 30%–60% humidity. Nourishment as well as filtered and autoclaved tap H2O were given ad libitum. Mice from 2 to 4 months in age, 1 male and 2 females per cage, were utilized for breeding pups.
-
4.
Remove the skin and extract the brains with a fine forceps, and place 1–2 brains in a petri dish containing cold L-15 medium. Place each covered petri dish on ice.
-
5.
Using a stereo microscope, remove the meninges with a fine forceps and dissect cortices, transferring each to a 15 mL conical tube containing 3–4 mL of cold L-15 medium, on ice. Add up to 8 cortices (from 4 brains) per 15 mL conical tube.
-
6.
In the biosafety cabinet, hold the 15 mL conical tube containing cortices settled to the bottom at a 90° angle. Gently remove the L-15 medium using a 1 mL pipette tip and replace with 300–400 μL of pre-warmed 0.25% trypsin. Place the tube in 37°C H2O bath for 10 min.
-
7.Mechanical dissociation of cortical neuronal precursors.
-
a.Angle the tube again and gently remove trypsin with a 1 mL pipette tip without disrupting tissue by holding the tip just above tissue and slowly rotating tube as trypsin is withdrawn. A small amount of trypsin (<50 μL) may remain.
-
b.Add 1 mL of pre-warmed and filtered high-glucose cDMEM medium.
-
c.Mechanically dissociate tissue by gently and steadily pipetting up and down 10 times with a 1 mL pipette tip that is nearly touching the bottom of the tube.
-
d.Add ~4 mL of high-glucose cDMEM medium to the tube to dilute cells for counting, swirl gently to mix.
-
e.Use a 1 mL pipette tip to slowly pass the cell suspension through a 40 μm nylon cell strainer into a 50 mL conical tube receptacle.
-
a.
CRITICAL: During mechanical dissociation, no visible clumps of cells should remain after pipetting up and down 10 times. If a few clumps remain, pipette up and down only once more. Any remaining clumps are likely a result of inadequate trypsinization and will be lost during straining, impacting cell yield. For best results, trypsinize for exactly 10 min, varying the amount of trypsin with the number of cortices, i.e., 300 μL for ≤6 cortices and 400 μL for 7–8 cortices.
-
8.Perform a live/dead stain and hemocytometer count using a diascopic and epi-fluorescence illumination microscope to determine the total number of live neuronal precursors.
-
a.To dilute cells 1:1 in Trypan blue, add 10 μL of cell suspension, mixed gently, to 10 μL of Trypan blue in a small tube. Pipette up and down gently several times to mix.
-
b.Add 10 μL of the diluted cell suspension to the hemocytometer.
-
c.Before counting, move the 15 mL tube containing cell suspension from the hood to a humidified chamber at 37°C, with 5% CO2.
-
d.Using a 10× objective, count the number of live cells, which should appear clear, small, and rounded, in 3 large, gridded hemocytometer squares and take the average.
-
e.To estimate the number of viable neuronal precursors per mL, multiply the average number of live cells per square by the dilution factor (2) by 104 (Live cells per mL=Ave number of cells per square × 2 × 104).
-
a.
Note: Each brain typically yields from 1.5–2.25 million precursors, and it is normal to see many dead cells (blue), roughly equal or exceeding the number of live cells during counting, even with good neuron preparations. The viable precursor yield depends on multiple key factors, including the age of pups when sacrificed (the closer to birth, the higher the cell yield), time between brain extraction and dissociation, and the adequacy of cortical tissue trypsinization/dissociation steps. Troubleshooting.
-
9.
Adjust the concentration of cells in high-glucose cDMEM medium to ~1.25 million cells/mL.
-
10.
Using a 1 mL pipette tip, add 1 mL of warm high-glucose cDMEM medium to each 35 mm 1.5 coverslip poly-D-lysine (PDL)-coated glass-bottom imaging dish, followed by 1 mL of well-mixed cell suspension (swirled gently to mix), for a total of 2 mL per dish. Rock each dish gently back and forth and up and down twice to evenly distribute cells.
-
11.
Transfer plated imaging dishes to a humidified chamber at 37°C with 5% CO2 and let sit undisturbed for 1 h to allow for neuronal precursor cell attachment.
-
12.
After 1 h, confirm that most precursors have attached using a diascopic and epi-fluorescence illumination microscope at 10–20× magnification. If most cells are unattached and/or floating after gently tapping the side of dish, the preparation has failed.
-
13.
In the biosafety cabinet, carefully remove high-glucose cDMEM medium from each dish using a sterile Pasteur pipette and suction, placing the tip along the inside edge. Use a 1 mL sterile pipette tip to gently add 2.5 mL total of fresh, pre-warmed NB + supplements medium along the inside edge of each dish. Keep dishes outside of the incubator no longer than a few min to maintain optimal health.
-
14.
Maintain dissociated cortical neuronal cultures in a humidified incubator at 37°C, with 5% CO2 for up to 21 days-in-vitro (DIV).
Dissociated cortical neuron culture maintenance
-
15.
On DIV 3–4, check dishes for the formation of neurites at 10× or 20× magnification using the diascopic and epi-fluorescence illumination microscope. Neurite formation is typically evident within 24–48 h of plating if cultures are healthy.
-
16.
Pre-warm and filter NB + supplements + 2× DL-2-Amino-5-phosphonopentanoic acid (APV) feeding media (see “Materials and equipment” for details) to 37°C, ~1.25 mL per dish.
-
17.
In the biosafety cabinet, use a 1 mL pipette tip against the inside dish edge to gently remove 1 mL (approximately 1/2) of media from each dish, and then discard.
-
18.
Slowly add 1.2 mL of pre-warmed NB + supplements + 2× APV media, which is added to prevent excitotoxicity, along the inside edge of the dish, taking care not to dislodge cells.
-
19.
Every 3–4 days, repeat feeding with fresh pre-warmed, filtered NB + supplements + 1× APV feeding media, as described previously.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| DMEM; With L-Glutamine; 4.5 g/L glucose and sodium pyruvate | Fisher Scientific | Cat#MT10013CV |
| Fetal Bovine Serum – Heat Inactivated | Life Technologies | Cat#MT35016CV |
| Penicillin/streptomycin 100× | Gibco | Cat#15070-063 |
| Glutamax I Supplement 100× | Life Technologies | Cat#35050-061 |
| D-(+)-glucose | Sigma-Aldrich | Cat#G7528-1KG |
| Neurobasal without phenol red | Thermo Fisher Scientific | Cat#12348017 |
| B27 | Life Technologies | Cat#17504044 |
| Leibovitz's L-15 Medium | Thermo Fisher Scientific | Cat#11415064 |
| HyClone™ Trypsin - Trypsin 0.25% | Fisher Scientific | Cat#SH3004201 |
| Lipofectamine® 2000 Transfection Reagent | Thermo Fisher Scientific | Cat#11668019 |
| Trypan blue | Gibco | Cat#15250-061 |
| DL-2-Amino-5-phosphonopentanoic acid | Abcam | Cat#ab120004 |
| Poly-D-lysine | Sigma-Aldrich | Cat#P6407-5MG |
| Opti-MEM I Reduced Serum Media | Thermo Fisher Scientific | Cat#31985070 |
| Experimental models: organisms/strains | ||
| C57BL6 mice (male and female, aged 2–4 months) | Jackson Laboratories | RRID:IMSR_JAX:000664 |
| Recombinant DNA | ||
| pGFP | Gift from Stephen Mayfield | Addgene plasmid#64904; http://n2t.net/addgene:64904; RRID:Addgene_64904 |
| p-mRuby-N1 | Gift from Michael Davidson | Addgene plasmid#54581; http://n2t.net/addgene:54581; RRID:Addgene_54581 |
| pCAG_PSD95.FingR-eGFP-CCR5TC | Gift from Don Arnold | Addgene plasmid#46295; http://n2t.net/addgene:46295; RRID:Addgene_46295 |
| pGP-CMV-GCAMP6s | Gift from Douglas Kim | Addgene plasmid#40753; http://n2t.net/addgene:40753; RRID:Addgene_40753 |
| Software and algorithms | ||
| Imaris | Oxford Instruments |
https://imaris.oxinst.com/; RRID:SCR_007370 |
| NIS Elements | Nikon Instruments | https://www.microscope.healthcare.nikon.com/en_EU/products/software/nis-elements; RRID:SCR_014329 |
| ImageJ Fiji | ImageJ Fiji |
https://fiji.sc/; RRID:SCR_002285_ |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| Other | ||
| Thermo Scientific SUN-SRi Luer-LOCK Syringe | Thermo Fisher Scientific | Cat#14-823-221 |
| Syringe filter, PES, 0.22 μm | DOT Scientific Inc. | Cat#229747 |
| Falcon® Bacteriological Petri Dishes 100 × 15 mm | Corning | Cat#351029 |
| 15 mL conical tubes | DOT Scientific Inc | Cat#416-PG |
| 50 mL conical tubes | DOT Scientific Inc | Cat#451-PG |
| Filter System, 150 mL, .22 μm, PES | Corning | Cat#431153 |
| Falcon® 40 μm Cell Strainer, Sterile | Corning | Cat#352340 |
| Zeiss SteREO Discovery.V8 stereomicroscope | Zeiss | https://www.microscopeworld.com/p-3295-zeiss-stereo-discoveryv8-manual-stereo-microscope.aspx |
| Nikon Ti Eclipse Diascopic and Epi-fluorescence illumination microscope | Nikon | https://www.nikon.com/products/microscope-solutions/lineup/inverted/ts2/ |
| 35 mm 1.5 coverslip poly-D-lysine-coated glass-bottom imaging dishes | MatTek | https://www.mattek.com/store/p35g-1-5-14-c-case/ |
| Forma II Series Water Jacketed CO2 incubator | Thermo Scientific | https://www.thermofisher.com/order/catalog/product/3110#/3110 |
| Nikon A1 R HD resonant scanning confocal microscope equipped with 2 GaAsP and 2 multi-alkali PMTs | Nikon | https://www.microscope.healthcare.nikon.com/products/confocal-microscopes/a1hd25-a1rhd25 |
| Tokai Hit Environmental imaging chamber | Tokai Hit | https://www.tokaihit.com/ |
| Microscopy Immersion Oil Type F | Nikon | Cat#MXA22168 |
| Plan Apo Lambda 100× 1.45 NA oil objective | Nikon | https://www.nikon.com/products/microscope-solutions/lineup/objectives/cfi_plan_apochromat_lambda/index.htm |
| Plan Apo VC 20× 0.75 NA DIC N2 dry objective | Nikon | https://www.nikon.com/products/microscope-solutions/lineup/objectives/cfi_plan_apochromat_vc_series/index.htm |
| X-Cite 120 Fluorescence LED Illumination System | Excelitas | https://www.excelitas.com/product-category/x-cite-fluorescence-led-illuminators |
| Nano-Drive piezo nanopositioner | Mad City Labs Inc. | http://www.madcitylabs.com/nanodrive.html |
Materials and equipment
-
•
PDL-coated dishes (Critical reagent, to be stored at 4°C)
| Reagents | Final concentration | Amount |
|---|---|---|
| PDL hydrobromide, 5 mg/mL stock | 100 μg/mL | 300 μL |
| Sterile diH2O | n/a | 15 mL |
Note: Prior to dissociated neuron culture set-up, the 35 mm 1.5 coverslip PDL-coated glass-bottom imaging dishes must be re-coated. The recipe above can be used to prepare 10 imaging dishes. Prepare in advance a stock solution of PDL at a concentration of 5 mg/mL, to be aliquoted and stored at −20°C for up to 6 months. Dilute stock PDL 50 times in sterile diH2O, mix, and add 1.5 mL of the final solution per imaging dish. Let coat for 3–4 h at 20–22°C. Rinse 3 times with sterile diH2O and let dry (with lids cracked open) in the biosafety cabinet. Parafilm and store at 4°C for up to 1 week until use.
-
•
High-glucose cDMEM medium (Critical reagent, to be stored at 4°C for up to 3 months and filtered before use)
| Reagents | Final concentration | Amount |
|---|---|---|
| DMEM; With L-Glutamine; 4.5 g/L Glucose and sodium pyruvate | n/a | 428 mL |
| Fetal Bovine Serum – Heat Inactivated | 10% | 50 mL |
| Penicillin/Streptomycin 100× | 0.7% | 3.5 mL |
| Glutamax I Supplement 100× | 0.7% | 3.5 mL |
| 20% D-(+)-glucose | 3% | 15 mL |
Note: Prepare in advance a 20% solution of D-(+)-glucose and filter sterile syringe and 0.22 μm syringe filter. Aliquots of 15 mL sterile filtered 20% D-(+)-glucose can be stored at −20°C for up to 6 months. Add the above reagents to DMEM and filter using a 150 mL, 0.22 μm Filter System. The medium can be stored at 4°C for up to 3 months.
-
•
L-15 Medium (Critical reagent, to be stored at 4°C for up to the manufacturer’s expiration date)
| Reagents | Final concentration | Amount |
|---|---|---|
| Leibovitz's L-15 Medium | n/a | 495 mL |
| Penicillin/Streptomycin 100× | 1% | 5 mL |
-
•
NB + supplements (Critical reagent, to be stored covered at 4°C for up to 3 months) and NB transfection media (to be stored covered at 4°C for up to 1 month)
| Reagents | Final concentration | Amount |
|---|---|---|
| Neurobasal (NB) without phenol red | n/a | 485 mL |
| Glutamax I Supplement 100× | 1% | 5 mL |
| B27 | 2% | 10 mL |
| Penicillin/Streptomycin 100× | 1% | 4.5 mL |
Note: Add 5 mL of Glutamax I supplement and 10 mL of B27 supplement to NB medium and mix, prior to removing and reserving 50 mL of antibiotic-free media, which can be labeled “NB transfection” medium and stored covered at 4°C for up to 1 month. Add P/S to the remaining medium to make the “NB + supplements” medium, which should be stored covered at 4°C for up to 3 months.
-
•
NB + supplements + 1×/2× APV (feeding) media (Critical reagent, to be prepared day-of-use, pre-warmed to 37°C and filtered).
| Reagents | Final concentration | Amount |
|---|---|---|
| NB + supplements medium | n/a | 20 mL |
| DL-2-Amino-5-phosphonopentanoic acid, 20 mM stock | 200 μM | 200 μL |
Note: Dilute 20 mM APV stock 100 times in NB + supplements medium to make 1× APV (above), or dilute APV stock 50 times to make 2× APV feeding medium. APV feeding media should be prepared day-of-use, pre-warmed to 37°C, and filtered using a sterile syringe and 0.22 μm filter to prevent culture contamination.
Step-by-step method details
Chemical plasmid transfection of DIV 19 cortical neurons
Transfection of neurons with a fluorescent protein-encoding plasmid to label subcellular structures, i.e., dendrites, spines, and spinules, adapted from (Srivastava et al., 2011).
-
1.Prepare in advance the following media (See “Materials and equipment” for detailed recipe), pre-warm to 37°C in an H2O bath, and then pass through a 0.22 μm filter using a sterile syringe.
-
a.NB transfection medium.
-
b.Opti-MEM 1×.
-
c.NB + supplements + 1× APV (feeding) medium.
-
a.
-
2.
In the biosafety cabinet, add 100 μL of Opti-MEM 1× to a 1.5 mL Eppendorf tube and 50 μL of Opti-MEM 1× to a second tube (set of 2 tubes per imaging dish).
-
3.Form the lipofectamine 2000 (LFA2K)-DNA complexes.
-
a.Add 7 μL of LFA2K to the first tube containing 100 μL of Opti-MEM 1×.
-
b.Add 4 μg of DNA per plasmid, i.e., p-mRuby-N1 or p-GFP, to the second tube containing 50 μL of Opti-MEM 1×.
-
c.Using a 200 μL pipette tip, transfer contents of the Opti-MEM + LFA2K tube to the plasmid DNA tube.
-
d.Pipette up and down steadily 30 times to mix LFA2K and plasmid DNA.
-
e.Transport closed tubes to 37°C with 5% CO2 incubator, open the caps, and incubate for 20–30 min to facilitate LFA2K-DNA complex formation.
-
a.
-
4.Transfect dissociated cortical neuron cultures with fluorescent protein-encoding plasmid(s).
-
a.Using a 1 mL pipette tip, remove neuronal conditioned media, reserving it in a 15 mL conical tube, to be kept at 37°C for later use.
-
b.Gently add 1.75 mL of pre-warmed, antibiotic-free NB transfection media to the inside edge of a dish, taking care not to disrupt the neuronal network.
-
c.Add contents of the Eppendorf tube, which now contains LFA2K-DNA complexes, to the dish drop-by-drop using a 1 mL pipette tip.
-
d.Gently mix by rocking the imaging dish up and down and then back and forth to evenly distribute DNA complexes. Do not swirl.
-
a.
-
5.
Incubate imaging dishes in a humidified incubator at 37°C with 5% CO2 for 3–4 h to enable sufficient uptake of complexes by neurons.
-
6.Replace transfection media containing complexes with ½ conditioned media and ½ NB + supplements + 1× APV medium.
-
a.Position a glass pipette tip along the inside edge of the dish, carefully use suction to remove all transfection media without disrupting the neuronal network.
-
b.Add 1.5 mL of reserved, pre-warmed neuron-conditioned medium and 1 mL of NB + supplements + 1× APV medium.
-
a.
-
7.
Incubate dishes in a humidified incubator at 37°C with 5% CO2 for ~48 h to allow time for adequate fluorescent protein expression.
Pause point: Allow ~48 h for adequate fluorescence protein expression in transfected neurons.
Optional: In addition to the fluorescent cell-filling plasmid, a second plasmid can be added to label a protein of interest or measure intracellular calcium transients in spinules. For example, spinules can be labeled by transfecting with p-mRuby-N1 (red) and endogenous PSD95 can be labeled with pCAG_PSD95.FingR-eGFP-CCR5TC (green). Similarly, to track spinule and local intracellular Ca2+ transients, neurons can be transfected with p-mRuby-N1 (red) and pGP-CMV-GCAMP6s (green).
Confocal parameter optimization and live time-lapse imaging
Optimized optical parameter set-up and live time-lapse confocal image acquisition
This protocol describes the optimization of confocal parameters for enhanced resolution spinule imaging using a Nikon A1R HD microscope with resonant scanning and a GaAsP multi-detector unit with a Plan Apo Lambda 100× 1.45 NA oil objective, and 488/561 nm excitation lasers. Although we used a Nikon A1R HD laser scanning system, enhanced resolution via oversampling with a smaller pinhole and pixel size coupled with deconvolution can theoretically be achieved using any research grade point scanning confocal microscope, provided that the data can be collected with appropriate S/N, sample spacing, and proper deconvolution (Biggs, 2010). The use of a piezo-electric positioner with nanometer resolution and repeatability is also critical for rapidly acquiring the high precision Z-stacks needed to track individual spinules. Our protocol is also generally applicable to imaging tissues, e.g., acute or organotypic mouse brain slice cultures, but the loss of signal associated with the imaging of deep optical tissue sections may limit the resolution benefit.
-
8.Environmental chamber set-up for live imaging.
-
a.Start microscope system according to standard protocol.
-
b.Turn on the Tokai Hit stage top incubation system, using manufacturer-recommended settings to maintain a temperature of 37°C in the environmental chamber, and open 5% CO2 tank valve.Note: If your incubation system is not equipped with a gas mixer, a tank of premixed gas (5% CO2, 20% O2, Balance N2) may be used.
-
c.Check for water in humidifier.
-
d.Switch to the Plan Apo Lambda 100× 1.45 NA oil objective and attach the objective heat collar.
-
e.Add one drop of oil to 100× 1.45 NA objective.
-
f.Switch to the Plan Apo VC 20× 0.75 NA DIC N2 dry objective.
-
g.Attach environmental chamber insert to the stage base.
-
a.
-
9.
Securely mount the sample, a 35 mm dish (No. 1.5 Coverslip, 14 mm Glass Diameter) containing healthy transfected pyramidal excitatory neurons, onto the environmental chamber stage.
-
10.
Find the sample plane of focus at 20× magnification.
-
11.
Select eye port for ocular viewing using an X-Cite 120 Fluorescence LED Illumination System with FITC filter cube. Other light sources, e.g., a mercury arc lamp, can be used as an alternative to LED illumination.
-
12.
Scan the entire glass area for healthy, brightly fluorescing neurons and save each X/Y coordinate demarking a neuron’s location by navigating to the ND Acquisition tab and XY sub-tab (leave XY box unchecked) and checking box #1, #2, etc. under Point Name. (Pause Point)
-
13.
Switch to the 100× 1.45 NA objective and adjust focus. If more oil is needed, carefully remove environmental stage with the sample still securely attached to preserve X/Y positions.
-
14.
Select Resonant scanning detector button.
-
15.
Select a scan speed of ½ frame per s.
-
16.
Adjust the pinhole to 0.6 airy units.
-
17.
Select a pixel size of 1/2 Nyquist suggested sampling size. For the 100× 1.45 NA lens, we used the following 3D pixel dimensions: X= 0.06 μm, Y= 0.06 μm, Z=0.12 μm and a scan area of 1024 pixels (width) × 512 pixels (height).
-
18.
Use Scan or Capture mode to adjust field of view, centering on the secondary branches of apical neuronal dendrites, where mushroom spines are evident.
-
19.
Adjust the laser power, photomultiplier tube (PMT) gain (named as HV by the software), and offset for each channel to achieve optimal S/N for enhanced resolution spinule imaging in the sample. Troubleshooting.
Figure 1.

Balancing laser power, gain, averaging, and noise is critical for time-lapse enhanced resolution spinule imaging
Single Z-plane images depict a secondary apical dendrite and spines of a fixed GFP-expressing DIV 21 mouse pyramidal neuron acquired with a 100× 1.45 NA objective. The use of different optical settings highlights the necessary balance between laser power, voltage gain, and line averaging to achieve optimal S/N with minimal photo-toxicity and photo-bleaching at adequate acquisition speeds. The offset was held constant in all conditions and the images had comparable levels of intensity with these settings.
(A) Upper: Imaging settings: Laser power (15%), gain (20), no averaging, and acquisition speed (65 ms/frame). Scale bar equals 10 microns. Lower: Line intensity profile (green arrow) shows an idealized S/N using high laser power, which would result in unacceptable amount of photo-toxicity and photo-bleaching for live time-lapse imaging. Scale bar equals 1 micron.
(B) Upper: Image settings: Laser power (2%), gain (45), no averaging, and acquisition speed (65 ms/frame). Lower: Line intensity profile shows poor S/N, which is suboptimal for deconvolution.
(C) Upper: Image settings: Laser power (2%), gain (45), 4× line averaging, and acquisition speed (260 ms/frame). Lower panel: Line intensity profile shows an acceptable S/N using a combination of low laser power, high gain, and averaging, which would result in negligible photo-effects at the modest expense of acquisition speed.
-
20.
Select 2× to 4× line averaging, depending on the desired Z-interval, 2× for shorter intervals and 4× if longer intervals are preferred.
-
21.
If imaging more than one channel, select Channel Series to minimize channel bleed-through, which will increase image acquisition time.
-
22.
For enhanced resolution, select a Z-step size of 0.1 μm. Set a Z-range that encompasses the entire dendrite, spines, and emerging spinules, typically from 3 to 8 μm. Add 1 or 2 additional Z-slices to the top and bottom of the range to account for possible drift during time-lapse imaging. The use of a piezoelectric Z-axis nanopositioner achieves the necessary high-speed and high-precision sampling in Z.
-
23.Set duration of imaging and Z-interval in accordance with the aims of the experiment.
-
a.For tracking long-lived spinule dynamics, which exist for ≥60 s, we recommend acquiring at speeds of 15 to 30 s per stack for a minimum duration of 15 min and a maximum duration of 2 h.
-
b.For tracking short-lived spinules, which can exist from 2 s up to 60 s, we recommend acquiring at speeds of 2 to 5 s per stack for a minimum duration of 2.5 min and a maximum duration of 30 min.
-
c.For tracking both short and long-lived spinules, we recommend acquiring at ~10 s per stack for a minimum duration of 10 min and a maximum duration of 1 h.
-
a.
-
24.
Select Run.
Three-dimensional confocal image deconvolution
The use of iterative image restoration-based 3D deconvolution software, which can afford up to a 1.5 times improvement in XY and a 1.7 times improvement in Z (Holmes and Liu, 1989, Biggs, 2010) has been possible for decades in principle. However, the application of deconvolution has been limited until recently by computer processing speed. Nikon’s enhanced resolution software offers algorithms for deconvolving point-scanning confocal datasets in minutes, depending on the processing system’s Graphics Processing Units.
-
25.
Open NIS Elements AR, i.e., Version 5.211, software.
-
26.
Navigate to File tab, select Open folder, and open a previously acquired NIS-Elements ND2 file to be deconvolved. Other file formats are not supported.
-
27.
Navigate to the Deconvolution tab and select 3D Deconvolution from the drop-down menu.
-
28.
Under the General tab, select automatic from the Deconvolution Method drop-down menu. Other parameters, e.g., Modality, Magnification, Pinhole Size, NA, Immersion Ref. Index, Calibration, Z-step, should all auto-populate.
-
29.
For the output, select create new document.
-
30.
Under Channels, check the box for each channel to be deconvolved, e.g., 488, 568, 647.
-
31.
Under the Advanced Settings tab, Preprocessing, verify that ‘do not subtract’ is already selected. Under Point Spread Function, verify that ‘Compute PSF automatically’ is already selected.
-
32.Click the Deconvolve button to begin deconvolving. Troubleshooting.
-
a.Processing frame number pop-up window will appear and show progress as processing frame number.
-
b.Performing Deconvolution pop-up window will appear showing progress in processing as progress percentage.
-
a.
-
33.
When processing is complete, navigate to the File menu and select Save As. If ‘show image window on save’ box is checked, an Image Properties pop-up window will appear. Select OK.
Note: While our protocol has been optimized for the detection of fine spinule-like protrusions in live imaging data, 3D deconvolution can be utilized for ad hoc or post hoc analysis of spinules in fixed images. Additionally, deconvolution can be used for post hoc analysis of live imaging data acquired using standard confocal parameters, although the deconvolution benefit may be weaker than expected.
Expected outcomes
This protocol was performed as described in mouse dissociated cortical neurons that were chemically transfected with p-GFP at DIV 19, allowing 2 d for fluorescent protein expression prior to imaging. DIV 21 was chosen as the optimal time-point for studying structural dynamics of cortical mushroom spines (Yoon et al., 2021), while DIV 14 would ideal for investigating spine development (Yoon et al., 2020). Time of transfection can vary from 1–4 d, depending on the plasmid used in the experiment. Live time-lapse imaging was performed using a Nikon A1 Resonant scanning confocal microscope and Tokai Hit Environmental imaging chamber at 37°C and 5% CO2. Healthy GFP-expressing neurons were first selected at 20× magnification. Secondary branches of apical dendrites on excitatory pyramidal neurons were live time-lapse imaged using a 100× 1.45 NA Plan Apo Lambda oil objective and 488 nm excitation laser.
In Figure 2A, the upper panel shows 3D reconstruction images of spines acquired using standard optical confocal parameters, before deconvolution (Methods video S1), and after deconvolution. The lower panel shows 3D reconstructions of the same spines acquired using parameters optimized for enhanced resolution, before and after deconvolution (Methods video S2). The combination of optimized confocal parameters and 3D deconvolution yields in the maximum resolution benefit, and subsequent optimal visualization of thin membrane protrusions, e.g., spinules. This protocol allows for the tracking of individual spinules over time, which often recur at the same topographical spine head locations. While the majority of spinules on cortical mushroom spines are short-lived, existing for <60 s, dynamic, and exploratory, a subset is long-lived (≥60 s) and elongated, and can be stabilized by contact with distal pre-synaptic terminals (Zaccard et al., 2020). A montage from Methods video S2 highlights a small short-lived spinule, which exists at a single time-point (Figure 2B), in contrast to an elongated long-lived spinule, which is present throughout the entire imaging duration (Figure 2C).
Figure 2.

Comparison of pre- and post-deconvolution 3D dendritic reconstructions of spines and emerging spinules acquired using standard or optimized parameters
Three-dimensional IMARIS reconstruction confocal images depicting a secondary apical dendrite, mushroom spines, and emerging spinules from a DIV 21, GFP-expressing cortical pyramidal neuron.
(A) Live time-lapse confocal Z-stacks acquired using standard settings, i.e., Z-step=0.15 μm, pinhole radius=67.69, or settings optimized for enhanced resolution, i.e., Z-step=0.1 μm, pinhole radius=30.65, are shown before and after 3D automatic iterative deconvolution. Blue arrows and insets highlight the resolution of a fine, elongated, long-lived spinule protruding from a mushroom spine in each of the images. Scale bar equals 5 microns. See also Methods videos S1 and S2.
(B) Montage from Methods video S2 of a small mushroom spine forming a short-lived spinule (red arrow) that appears in a single time-point, with an estimated lifespan of ≤12 s. Scale bar equals 1 micron.
(C) Montage from Methods video S2 highlights a very long-lived spinule (blue arrows), with a lifespan equal to or exceeding the 15 min imaging duration, emerging from a large mushroom spine. Scale bar equals 1 micron.
One-minute, time-lapse, 3D reconstruction video acquired using standard optical confocal settings with no deconvolution showing poor resolution of spinules and spine necks. Parameters: Z-step=0.15 μm, Z-range=7.7 μm, Z-interval=8 s.
Fifteen-minute, time-lapse, 3D reconstruction video of confocal imaging data acquired using confocal settings optimized for enhanced resolution followed by 3D deconvolution, showing optimal resolution of spinules and spine necks. Parameters: Z-step=0.1 μm, Z-range=7.7 μm, Z-interval=12 s.
Quantification and statistical analysis
While visualization, quantitation, and tracking of spine volumes, spinule lengths, and colocalization of protein(s) of interest in 3D over time can be performed using IMARIS 3D/4D Analysis software, this platform is expensive and therefore not accessible to all researchers. Here we describe the manual quantification of mushroom spine areas and spinule parameters over multiple time-points (Z-intervals) in maximum projection images, confirming spinule origins in 3D projections, using Fiji/ImageJ software, which is freely available to all researchers. Fiji/ImageJ commands are italicized to distinguish commands from the text. Here we estimate the time of generating spine head ROIs and spine areas to be 15 min per dataset, with each dataset containing ~12 mushroom spines, which take roughly 5 min each to quantify their spinules over a 10 min imaging duration, for a total of ~1.25 h per dataset. Each time-lapse imaging dataset follows the dynamics of spines emerging from a single dendritic segment of a transfected pyramidal neuron. Depending on transfection efficiency and cell health, one can expect to image dendritic segments from 2–4 neurons per culture dish. In general, we recommend acquiring ≥3 datasets from ≥2 independent cortical neuron preparations, for a minimum of 6 datasets (neurons) per condition to be analyzed.
-
1.Import an ND document containing deconvolved live time-lapse confocal imaging of spines into Fiji/ImageJ by dragging and dropping it into the Reading file header, which causes the Bio-formats Import Options window to pop-up.
-
a.Under Stack viewing, View stack with, select Hyperstack from the drop-down menu.
-
b.Under Dataset organization, check open files individually box.
-
c.Under Color options, check Autoscale box.
-
d.Select OK.
-
a.
-
2.Under the Image tab, select Adjust and then Brightness/Contrast from the drop-down menu.
-
a.B&C window will popup.
-
b.Select Auto button to quickly adjust brightness. B&C can also be adjusted manually using sliders.
-
a.
-
3.Optional: Image rotation for alignment of the dendrite.
-
a.Under Image tab, select Transform and Rotate from the dropdown menus.
-
b.Check preview box (crosshairs appear on image).
-
c.Use Angle slider to orient the dendrite left to right.
-
d.Select OK.
-
e.Process Stack? Query box will pop-up, select Yes button.
-
f.Save as tiff file and add “rot” to file name.
-
g.If crosshairs still appear on the image, re-open Transform and Rotate and select Cancel button.
-
a.
-
4.Optional: Image crop to include only the dendrite segment of interest.
-
a.In the toolbar, select rectangle tool.
-
b.Left click and drag to select crop area.
-
c.Under the Image tab, select Crop from the drop down.
-
d.Save as Tiff file with “crop” in filename.
-
a.
-
5.Convert to a Z maximum-projection image (over time).
-
a.Under Image tab, select Stacks and Z-project from the dropdown menu.
-
b.Z-Projection pop-up window will autofill start/stop slice, crop slices if desired.
-
c.Under Projection type in the pop-up window, check that Max Intensity is selected.
-
d.Check the All time frames box.
-
e.Select OK.
-
f.A MAX projection image will appear in which the lower Z bar disappears, while the C bar still lets you toggle between channels (if multiple) and play button scrolls through time.
-
a.
-
6.Identify a segment of the secondary apical dendrite that is of consistent length across all datasets, i.e., 20 μm (depending on field of view), with no sign of poor neuron health, i.e., plasma membrane blebbing.
-
a.In the tool bar, right click, and select straight or segmented line.
-
b.Left click on the dendrite to begin line segment, drag line along dendrite, right click to end line segment (toolbar shows line segment length in microns).
-
c.Use the shortcut, Control t to open the ROI manager and add the 20 μm segment (Alternatively, Under Analyze menu, select Tools and ROI manager to open ROI manager).
-
d.Click the string of numbers in ROI manager, select Rename to rename ROI as “dendrite segment”.
-
e.Check the Show All box.
-
f.In the ROI manager, select More and Labels, and check the boxes Show labels and Use names as labels from the Labels pop-up window, followed by OK.
-
a.
-
7.Calculate mushroom spine areas in first time point using Fiji/ImageJ (Figure 3).
-
a.If there are multiple channels in the dataset, from the Image dropdown, select Color and Split channels and select the GFP dendrite cell-fill channel, i.e., C1-Max.
-
b.From the Image drop-down menu, Select Image Adjust and Threshold.
-
c.Check drop-down threshold method is Default and B&W is selected from drop-down menus.
-
d.Check the dark background box.
-
e.Select Auto button.
-
f.Adjust upper slider as needed until most fine structures, i.e., mushroom spine necks and spinules, are just filled in. Do NOT select Apply or click anywhere on the image.
-
g.Under the Edit menu, choose Selection and Create Selection.
-
h.Use the short-cut Control t to add the dendrite and spine ROI to the ROI manager.
-
i.In the Threshold pop-up window, now select Apply (image converts to black ROI on white background, if new window pops up, leave boxes unchecked).
-
j.Select the recently added ROI in ROI manager (will be a string of numbers) and Rename, i.e., 488_dendrite.
-
k.Under Image dropdown, choose Color and then Color Picker. When the CP window pops up, check that White on Black is selected (White box over black box).
-
l.Right click on Paint brush tool, change the thickness from 10 pixels to 1 pixel, and select OK.
-
m.Moving left to right with the paint brush tool, erase a portion of the spine neck at the base of each mushroom spine to be quantified, to generate individual spine head ROIs, minus the neck.Note: For reference, a seminal publication by Harris, Jensen, and Tsao describes the generally accepted criteria for assigning categorical shape classification to dendritic spines, i.e., stubby, thin, mushroom, and branched (Harris et al., 1992). Since dendritic spinules are most frequently found on large mushroom spines (Petralia et al., 2015), we have limited our quantitation to mushroom spine parameters, e.g., spine head area and spinule number per spine, in this protocol. ROIs of interest with over-lapping objects that cannot be readily distinguished should be excluded from the analysis.
-
n.In ROI manager, select again the edited 488_dendrite ROI, then select Update to save the previous edits to spine heads.
-
o.Using the wand tool, select each spine head ROI to be quantified and use Control t to add each individual spine head ROI to the ROI manager.
-
p.In ROI manager, rename each mushroom spine head ROI, i.e., S01, S02, etc.
-
q.To numerically reorder the spine head ROIs in ROI manager (as needed), select More and then Sort.
-
r.Shift + Left click to highlight the big 488_dendrite ROI and individual numbered spine head ROIs. Select More and then Save to save ROIs for each dataset.
-
s.Select the 488_dendrite ROI in the ROI manager and select Delete, to leave only the spine head ROIs in the manager (for ease of visualization).
-
t.In the ROI manager, check Show All and Labels boxes to view labeled spine ROIs within the image. Select More and then Labels, and check the following boxes: Show labels, Use names as labels, and Draw backgrounds. Select OK.
-
u.Under the Analyze tab, select Set Measurements and check Area box, as well as additional quantitative parameter boxes if desired, e.g., Perimeter.
-
v.In ROI manager, Shift + Left click to select the spine head ROIs, select Measure, and a pop-up window of Results, i.e., spine head ROI areas, will appear.
-
w.Copy and paste measurements into an Excel file for statistical analysis.
-
a.
-
8.Individual spinule tracking and length quantification in maximum projection images (Figure 4).
-
a.Reload original (or rotated and cropped) dataset into the Reading file header.
-
b.Under Image drop-down, select Stacks and Z project to make a maximum projection image.
-
c.Zoom in along the dendrite segment being analyzed on a mushroom spine that is typical in size, using the magnifying glass tool.
-
d.Choose typical spinule classification parameters, i.e., thin protrusions of spines that are ≥0.25 μm in length and <0.25 μm in width, provided length>width (Zaccard et al., 2020).
-
e.Use the circle quadrant tool to draw a circle similar in size to the mushroom spines to be analyzed.
-
f.Use the line tool to draw lines bisecting the circle into 4 equal quadrants (QI, QII, QIII, QIV), and sub-quadrants if necessary (QIA, QIB, etc.), using Control t to add each segment to the ROI manager.
-
g.In ROI manager, highlight the circle and line segment ROIs, select More and XOR to combine all to a single ROI, and Rename the new ROI “Quad. tool”.
-
h.Use the wand tool to drag and reposition the circle quadrant tool as needed.
-
i.With the circle quadrant tool as a guide, use the play button slider to scroll through time starting at time-point 1, scanning for spinules in QI.
-
j.Right click on the line tool and select segmented line from the drop-down menu. Measure each spinule length by Left clicking and dragging the line tool from the spinule base to the tip, right clicking to end the line.
-
k.Press Control t to add the spinule length measurement to the ROI manager. Repeat for each progressive time-point with a spinule in QI, labeling each with the quadrant, followed by the individual spinule number and the time-point, e. g. QI_Spl1_T3, QI_ Spl1_T4, QI_Spl1_T5, QI_Spl2_T7.Note: An individual spinule is defined as a fine protrusion emerging from a spine head that is observed in one or more successive time-points (Z-intervals) at the same topographical spine head location. Interestingly, spinules often extend and retract repeatedly, appearing and reappearing in discontinuous time frames at the same topographical spine head location. We showed previously that the majority of spinules recur at the same locations over 10–15 min imaging durations (Zaccard et al., 2020). Our numbering system allows for individual spinule quantitation (Spl1, Spl2, etc.), as well as tracking of spinule recurrence, e.g., the number of individual spinules that recur at a particular location, e.g., QI, over time. The lifespan of an individual spinule can be estimated as the number of successive Z-intervals where the spinule is observed multiplied by the duration of the Z-interval. A spinule existing for ≥60 s duration is considered long-lived, while spinules existing for <60 s are considered short-lived, and those existing for <20 s are considered to be extremely short-lived. For example, Figure 4 shows key frames in the tracking of an individual long-lived spinule, which exists in multiple continuous time frames, and a short-lived spinule, which is detected in a single time frame.
-
l.Highlight the ROIs of each individual spinule in the ROI Manager and select Measure.
-
m.Copy and paste individual spinule length measurements in microns from the measure pop-up window to an Excel spreadsheet.
-
n.In ROI manager, highlight all the ROIs for QI, and select More and Save to save the ROI set.
-
o.Repeat the above instructions for QII, QIII, and QIV in the spine of interest.
-
a.
-
9.Confirmation of spinule origination using a 3D projection image.
-
a.Reload the original (or rotated and cropped) dataset and under Image drop-down, select Image, Stacks, and then 3D project.
-
b.3D Projection menu will auto-populate, select Y-axis from the drop-down. (alternatively, select X-axis to rotate on X-axis), and check All time points box.
-
c.Select OK.
-
d.“Projections of” image will be generated with a Z slider, which rotates the 3D image along the selected Y-axis, and the Play button (time).
-
e.Zoom in on the spine of interest using the magnifying tool and use the Z slider to rotate the image 360°.
-
f.Confirm in the 3D reconstruction that previously quantitated spinules emerge from the spine of interest rather an overlapping structure, such as a nearby filopodium that emerges directly from the dendritic shaft.
-
g.In Excel, delete any erroneous spinule length measurements.
-
a.
-
10.Optional: Record individual spinule categorical shape according to the following classifications, previously described in detail (Zaccard et al., 2020):
-
a.Tapered.
-
b.Filopodia/Thin.
-
c.Mushroom.
-
d.Transitioning (shape changes over time).
-
a.
-
11.
In Excel, calculate the number of individual spinules per spine.
-
12.
In Excel, calculate individual spinule parameters, such as mean and maximum spinule length as well as lifespan, which can be estimated by multiplying the number of successive time frames where the spinule is detected by the Z-interval duration.
-
13.
Copy and paste individual parameters, e.g., spinule number per spine (over time) from two experimental conditions (arranged into two columns) into statistical analysis software such as GraphPad Prism.
-
14.
Compare parameters such as spinule number per spine, mean and maximum spinule length, and mean and maximum spinule lifespan from two conditions using two-sided, unpaired student’s t tests or Mann-Whitney tests (depending on normality of the data). When comparing 2 conditions, we recommend normalizing spinule number per spine to spine area, considering spinule numbers increase with spine size.
Figure 3.

Steps for generating mushroom spine head regions of interest using Fiji/ImageJ
Panel shows maximum projection enhanced resolution confocal images of secondary apical cortical dendrites and spines through the progressive stages of step 7. These stages detail the process for quantitating mushroom spine head ROIs at the first imaging time-point (Z-series). Live time-lapse imaging data was acquired at Z-intervals of 20 s for a duration of 10 min. Scale bar is equal to 1 micron.
Figure 4.

Quantitation of spinules emerging from mushroom spines over time using Fiji/ImageJ
Enhanced resolution maximum projection images depict a GFP-labeled mushroom spine and the manual tracking of spinule lengths over time using Fiji/ImageJ software. Live time-lapse imaging was acquired at Z-intervals of 20 s for a duration of 10 min. The circle quadrant tool assists in tracking of individual spinules over time at the same topographical locations. Blue arrows highlight an elongated, long-lived spinule that appears continuously at location QII in the first 13 time-points, for an estimated lifespan of 260 s. The spinule disappears and another re-emerges at the same location at a later time-point (T=360 s), in a phenomenon termed spinule recurrence. The red arrow highlights a smaller, short-lived spinule that appears at a single time-point in QIII, for an estimated lifespan of ≤20 s. Scale bar equals 1 micron.
Limitations
The main limitation of this method is the requirement for a strong fluorescence signal to label spinules, as the 3D deconvolution algorithm discards “weak” pixels from the image and strengthens “strong” pixels. Hence, high S/N is paramount to success. Considering that deconvolution alters pixel values, fluorescence intensity comparisons should not be conducted on deconvolved imaging data, while distance measurements can be performed. To quantify fluorescence intensity, i.e., in a differentially labeled protein of interest, deconvolve the cell-filling channel to resolve spinules, but perform intensity quantification only on the second, non-deconvolved channel. Finally, the ability to track millisecond scale events using this method is restricted by the instrument scanning speed and the necessity for Z-stacks that encompass entire spines and their projections in 3D.
Troubleshooting
Problem 1
Low neuronal precursor yield (BEFORE YOU BEGIN, step 8)
Each brain typically yields from 1.5–2.25 million cortical neuronal precursors with this protocol, and a yield of <1.5 million precursors per brain is considered sub-optimal. The viable cell yield depends on multiple key factors, which may include the age of pups when sacrificed, time between brain extraction and dissociation, or the adequacy of trypsinization/dissociation steps.
Potential solution
Use younger pups and/or process fewer brains
In our experience, age of the pups is one influential factor in determining viable neuronal precursor yield. The closer the P0 pups sacrificed in this protocol are to birth, the higher the typical viable neuronal precursor yield. The use of P1 and older pups is not recommended as it will likely result in lower cell yields and inconsistent neuron culture health. Another way to maximize neuronal precursor yield is to minimize the time from brain extraction to mechanical dissociation. Consider processing fewer brains at once, i.e., ≤4 brains, if it takes >5 min per brain to dissect the cortices and the resulting precursor yields are low. Finally, adequate trypsinization of cortical tissue is critical for subsequent mechanical dissociation, and the presence of observable clumps after dissociation is indicative of an issue at one of these steps. For best results, trypsinize tissue in the warm H2O bath for exactly 10 min, varying the amount of trypsin with the number of cortices, i.e., 300 μL for ≤6 cortices and 400 μL for 7–8 cortices. Distribute tissue into two 15 mL tubes if processing >8 cortices (4 brains) at one time.
Problem 2
Neuronal precursors do not attach and/or fail to form neurites and healthy networks (BEFORE YOU BEGIN, step 12)
Poor attachment of neuronal precursors to the PDL substrate during plating and/or the lack of observable neurite formation by attached precursors within 24–48 h represent common early indicators of cortical dissociated neuron culture failure. These problems are often caused by issues with the PDL coating. The ability of neurons to form neurites and/or increasingly complex networks over time without signs of poor health, i.e., blebbing, can also be influenced by initial plating density.
Potential solution
Validate new lots of PDL prior to use and re-coat imaging dishes
Inadequate PDL coating represents one of the main causes of dissociated neuron culture failure, when the initial neuronal precursor yield during plating was adequate. We suggest validating new lots of PDL as needed to minimize the potential impact of inconsistent PDL quality on neuron culture health. For optimal results, we strongly recommend re-coating in-house the 35 mm 1.5 coverslip pre-PDL-coated glass-bottom imaging dishes with PDL at 0.2 mg/mL. Re-coated imaging dishes can be stored at 4°C, wrapped in parafilm for up to a week before use.
Potential solution
Increase precursor plating density
If precursors initially form healthy neurites, but only sparce networks that become increasingly degraded over time, consider increasing plating density from 1.25 million precursors per mL up to 1.5 million precursors per mL. Higher density neuron cultures are generally healthier than lower density cultures and tend to survive subsequent chemical transfection and imaging steps more readily.
Problem 3
Poor transfection efficiency (step 7)
This protocol was specifically designed for sparce labeling, typically from 0.25–1 transfected neuron per 20× ocular field of view. Sub-optimal transfection efficiency can be a result of poor neuron health prior to transfection, or it can be the result of issues related to the transfection protocol and/or selected plasmids.
Potential solution
Neuronal transfection optimization
To increase fluorescent protein expression in neurons, increase the time allowed for LFA2K-DNA complex formation from 20 min up to 30 min, and increase neuron incubation time with DNA-LFA2K complexes from 4 h up to 5 h, prior to medium change. Consider incubating transfected neurons for an additional 24–36 h prior to imaging and/or the use of alternate fluorescent protein expression constructs if fluorescent protein expression remains low.
Problem 4
Low S/N during confocal imaging (step 19)
The most common issue with the protocol is a low S/N, which can result in a suboptimal post-3D deconvolution resolution benefit (Figure 5).
Figure 5.

Deconvolution of a low S/N image results in poor resolution enhancement
IMARIS 3D reconstructions depict a secondary apical dendrite and spines from a DIV 21 mouse cortical pyramidal neuron that exhibits low GFP expression, resulting in a low S/N image and a sub-optimal deconvolution benefit. Live confocal microscopy was performed using parameters adjusted for enhanced resolution, and the resulting image is shown before and after 3D deconvolution. Arrows highlight a long spinule emerging from a mushroom spine. Scale bar equals 3 microns.
Potential solution
Adjustment of confocal parameters
Low S/N can potentially be addressed by adjusting the confocal parameters at the point of imaging. In general, all parameters that improve resolution reduce the number of photons reaching the detector. Hence, collecting enough photons to achieve sufficient S/N for enhanced resolution is a result of fine-tuning all confocal parameters against each other. For example:
Small pinhole diameters lead to improved resolution but a drastic reduction in S/N (Wilson, 2011).
Doubling the pixel time (dwell time) or doubling the number of signal acquisition (2× line averaging) improves S/N by a factor of 2. Doubling the dwell time or 2× averaging also doubles the image acquisition time. Increasing dwell time or line averaging should be used sparingly due to the dramatic increase in acquisition speed.
Larger pixels collect more photons but will result in a decrease in resolution.
Higher gain on the PMT can amplify a weak fluorescence signal, but also amplifies noise. At the point of imaging, this increase in noise is typically greater than the increase in S/N, which is not useful. Ideally, the offset should be close to zero because the deconvolution needs to identify the background noise.
Overall, the parameters chosen should represent in a “sweet spot” of the highest resolution with the lowest decrease in S/N, as illustrated previously in Figure 1. Specific confocal settings are dependent on the actual experiment as well as the microscope’s hardware and software.
Problem 5
3D deconvolution pop-up error messages (step 32)
Image under-sampled error message states “The image calibration is too high. It should be below 0.123 μm/pixel for current image optical properties. As a result, deconvolution effect can be weaker than expected”.
Potential solution
Proceed or repeat imaging with settings optimization
Proceed with deconvolution, accepting that the resolution improvement will be suboptimal, or repeat imaging using the optimized settings for pixel size described previously.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter Penzes (p-penzes@northwestern.edu).
Materials availability
This study did not generate any new unique reagents. Further information and requests for materials should be directed to and will be fulfilled by the lead contact, Peter Penzes, upon request.
Data and code availability
This study did not generate any custom code, software, or algorithms. Datasets related to the current study are available from the lead contact, Peter Penzes, upon request.
Acknowledgments
Confocal microscopy and 3D deconvolution were performed at the Northwestern’s Center for Advanced Microscopy supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. This work was supported by NIH grants, R01MH071316 and R01MH107182. Image of the Nikon A1R microscope in the graphical abstract was re-printed with permission from Nikon Instruments Inc.
Author contributions
C.R.Z.: conceptualization, methodology, investigation, visualization, writing – original draft, and writing – review and editing; DK: methodology, resources, and writing – review and editing; S.Y.: methodology and writing – review and editing; CA: conceptualization, resources, and writing – review and editing; PP: conceptualization and supervision.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100427.
Contributor Information
Colleen R. Zaccard, Email: colleen.zaccard@northwestern.edu.
Peter Penzes, Email: p-penzes@northwestern.edu.
References
- Biggs D.S. 3D deconvolution microscopy. Curr. Protoc. Cytom. 2010;12:12 19 1–20. doi: 10.1002/0471142956.cy1219s52. [DOI] [PubMed] [Google Scholar]
- Harris K.M., Jensen F.E., Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes T.J., Liu Y.H. Richardson-Lucy/maximum likelihood image restoration algorithm for fluorescence microscopy: further testing. Appl. Opt. 1989;28:4930–4938. doi: 10.1364/AO.28.004930. [DOI] [PubMed] [Google Scholar]
- Petralia R.S., Wang Y.X., Mattson M.P., Yao P.J. Structure, distribution, and function of neuronal/synaptic spinules and related invaginating projections. Neuromol. Med. 2015;17:211–240. doi: 10.1007/s12017-015-8358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava D.P., Woolfrey K.M., Penzes P. Analysis of dendritic spine morphology in cultured CNS neurons. J. Vis. Exp. 2011;53:e2794. doi: 10.3791/2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T. Resolution and optical sectioning in the confocal microscope. Journal of Microscopy. 2011;244:113–121. doi: 10.1111/j.1365-2818.2011.03549.x. [DOI] [PubMed] [Google Scholar]
- Yoon S., Parnell E., Kasherman M., Forrest M.P., Myczek K., Premarathne S., Sanchez Vega M.C., Piper M., Burne T.H.J., Jolly L.A. Usp9X controls ankyrin-repeat domain protein homeostasis during dendritic spine development. Neuron. 2020;105:506–521 e7. doi: 10.1016/j.neuron.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S., Piguel N.H., Khalatyan N., Dionisio L.E., Savas J.N., Penzes P. Homer1 promotes dendritic spine growth through ankyrin-G and its loss reshapes the synaptic proteome. Mol. Psychiatry. 2021 doi: 10.1038/s41380-020-00991-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccard C.R., Shapiro L., Martin-De-Saavedra M.D., Pratt C., Myczek K., Song A., Forrest M.P., Penzes P. Rapid 3D enhanced resolution microscopy reveals diversity in dendritic spinule dynamics, regulation, and function. Neuron. 2020;107:522–537 e6. doi: 10.1016/j.neuron.2020.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
One-minute, time-lapse, 3D reconstruction video acquired using standard optical confocal settings with no deconvolution showing poor resolution of spinules and spine necks. Parameters: Z-step=0.15 μm, Z-range=7.7 μm, Z-interval=8 s.
Fifteen-minute, time-lapse, 3D reconstruction video of confocal imaging data acquired using confocal settings optimized for enhanced resolution followed by 3D deconvolution, showing optimal resolution of spinules and spine necks. Parameters: Z-step=0.1 μm, Z-range=7.7 μm, Z-interval=12 s.
Data Availability Statement
This study did not generate any custom code, software, or algorithms. Datasets related to the current study are available from the lead contact, Peter Penzes, upon request.