

Summary
Patch-clamp and multi-electrode array electrophysiology techniques are used to measure dynamic functional properties of neurons. Whole-cell and cell-attached patch-clamp recordings in brain slices can be performed in voltage-clamp and current-clamp configuration to reveal cell-type-specific synaptic and cellular parameters governing neurotransmission. Multi-electrode array electrophysiology can provide spike activity recordings from multiple neurons, enabling larger sample sizes, and long-term recordings. We provide our guide to preparing acute rodent brain slices with example experiments and analyses intended for novice and expert electrophysiologists.
For complete details on the use and execution of this protocol, please refer to Manz et al. (2020b).
Subject areas: Cell Biology, Signal Transduction, Neuroscience
Graphical Abstract
Highlights
-
•
Viable and efficient preparation of mouse brain tissue
-
•
Comprehensive material source for acute brain slice electrophysiology
-
•
Detailed approach to whole-cell patch-clamp recording of neurons
-
•
Utility and application of multi-electrode array electrophysiology in mouse brain
Patch-clamp and multi-electrode array electrophysiology techniques are used to measure dynamic functional properties of neurons. Whole-cell and cell-attached patch-clamp recordings in brain slices can be performed in voltage-clamp and current-clamp configuration to reveal cell-type-specific synaptic and cellular parameters governing neurotransmission. Multi-electrode array electrophysiology can provide spike activity recordings from multiple neurons, enabling larger sample sizes, and long-term recordings. We provide our guide to preparing acute rodent brain slices with example experiments and analyses intended for novice and expert electrophysiologists.
Before you begin
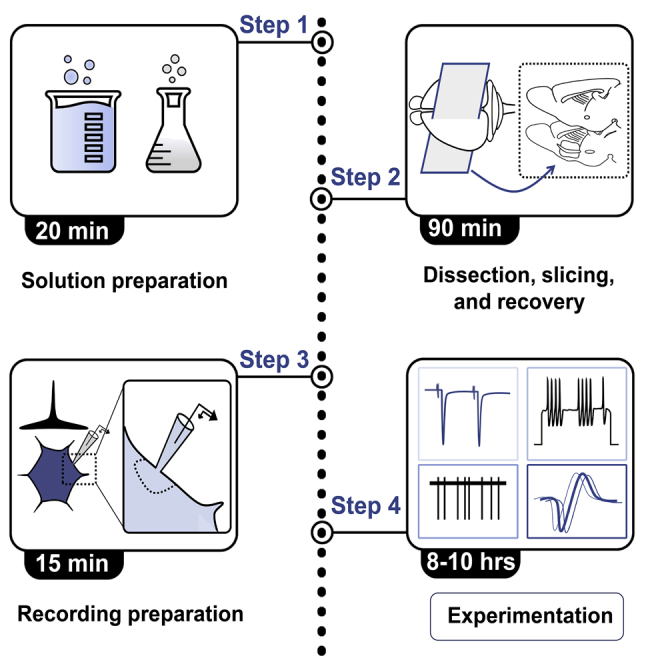
Figure 1, depicts the timing and preparation workflow for patch-clamp electrophysiology.
Figure 1.

Workflow diagram for performing patch-clamp electrophysiology
Schematic depicting the workflow and timing required to prepare each of the steps preceding the recording session. For each step outline in the black box, the key elements of each step are depicted in a smaller, colored box. In brief, 1× ACSF and NMDG-based solutions (1) are created fresh each day followed by (2) preparation of the dissection area, where utensils are gathered, vibratome assembled, and holding chambers filled. Once the dissection area is prepared and the NMDG-based solution is chilled and oxygenated, brain slices are prepared from the dissected animal (3). Slices are permitted 60-min to recover in an ACSF-containing holding chamber (4). Preparing for the recording session (5) involves setting up the ACSF perfusion system, backfilling a microfilament with a thawed aliquot of internal solution, and gathering any drugs or manipulations needed for experiments performed during the recording session (6).
Solutions, instrumentation, and dissection area preparation
Timing: 45–60 min
-
1.Solutions: Prepare the following solutions: (refer to “Materials and equipment” for detailed recipes)
-
a.artificial cerebral spinal fluid (ACSF) bathing solution
-
b.N-methyl-D-glucamine (NMDG)-based dissecting/recovery solution
-
c.Intracellular (internal) solution
-
d.Dissecting Solution for Multi-electrode array recordings
-
e.Recording Solution for Multi-electrode array recordings
-
a.
-
2.
Holding chambers:
Prepare two separate slice-holding chambers containing ACSF and NMDG-based recovery solution with chamber floors that are (a) large enough to suspend slices without overlapping and (b) sufficiently porous to permit exchange of aerated (95% O2, 5% CO2) solution (Figure 2).
Note: It is recommended that slices rest in the orientation in which they will be positioned during experimentation to ensure maximal cell health and survival. While the ACSF holding chamber for slices awaiting experimentation may be kept at room temperature (20°C–23.5°C), the recovery holding chamber should be placed in a submerged water bath at 32°C near the dissection area.
-
3.Dissection preparation (Figure 2):
-
a.Vibratome: Fill the vibratome (ie Leica VT1200S) buffer tray with 150 mL of oxygenated NMDG-based recovery solution and pack the outside perimeter of the tray with crushed ice. Continuously oxygenate the filled buffer tray.
-
b.Insert a razor blade into the blade holster, fasten securely with a screwdriver or hex-key Allen wrench, and lower the blade into the buffer tray.
-
c.Dissection area: Flatten an absorbent cotton underpad near the vibratome. Place dissecting instruments (scissors, ronguer, and forceps), a small beaker (50 mL), brain slicer matrix, specimen disk, razor blades, stainless steel microspatula, and plastic spoon in a large bucket filled with ice. A fine brush, superglue, and a plastic transfer pipette (with the narrow opening cut off 0.5” from the end) should be placed within reach in the dissection area.
-
d.Add 30 mL of oxygenated NMDG-based recovery solution to the 50 mL beaker and embed the beaker firmly in the ice. The remaining NMDG-based recovery solution should be placed in the submerged holding chamber oxygenated at 32°C and filled ≥1” above the chamber floor.
-
e.Once the dissection area and vibratome are prepared and organized, allow 10–15-min for the recovery solution in the 50 mL beaker/buffer tray and submerged holding chamber to reach ∼0°C and 32°C, respectively.
-
a.
Note: The procedures outlined above are very similar for multi-electrode array recordings, however the dissecting and recording solutions are different compared to the patch clamp electrophysiology methods and are described in the “Materials and Equipment” section.
Figure 2.

Electrophysiology and recording equipment
(A) Identification of tools and equipment needed for slicing, including holding chambers, dissecting utensils, and the assembled dissection area.
(B) Electrophysiological equipment outlining key components of the rig, perfusion system, and brain slice setup.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| CsMeSO3 | Sigma-Aldrich | 24892412 |
| CsCl | Sigma-Aldrich | 24852140 |
| NaCl | Sigma-Aldrich | 329824637 |
| HEPES | Sigma-Aldrich | 24895572 |
| EGTA | Sigma-Aldrich | 329747808 |
| TEA-Cl | Sigma-Aldrich | 57653171 |
| QX-314-Br | Tocris Bioscience | 9884487 |
| Spermine tetra-HCl | Tocris Bioscience | 9384 |
| MgCl2 | Sigma-Aldrich | 5360315 |
| K+-D-gluconate | Sigma-Aldrich | 57654209 |
| Mg2+-ATP | Sigma-Aldrich | 15126 |
| Na+-GTP | Sigma-Aldrich | 135410925 |
| NMDG | Sigma-Aldrich | 24896593 |
| Na-ascorbate | Sigma-Aldrich | 329823275 |
| Na-pyruvate | Sigma-Aldrich | 24898615 |
| NaHCO3 | Sigma-Aldrich | 24899673 |
| D-(+)-Glucose | Sigma-Aldrich | 24895335 |
| KCl | Tocris Bioscience | 4873 |
| CaCl2 (anhydrous) | Sigma-Aldrich | 329748319 |
| MgCl2⋅6H2O | Sigma-Aldrich | 643991 |
| NaH2PO4 (anhydrous) | Sigma-Aldrich | 21225609 |
| NaH2PO4-2H2O | Sigma-Aldrich | 57652311 |
| MgSO4-7H2O | Sigma-Aldrich | 329752370 |
| CaCl2-2H2O | Sigma-Aldrich | 329775159 |
| Picrotoxin | Sigma-Aldrich | 31304 |
| Apamin | Tocris Bioscience | 1652 |
| NBQX disodium salt | Tocris Bioscience | 6098006 |
| D-AP5 | Tocris Bioscience | 135342 |
| Kynurenic acid | Tocris Bioscience | 3845 |
| L-Tryptophan | Sigma-Aldrich | 24278135 |
| (R)-(-) Phenylephrine hydrochloride | Sigma-Aldrich | 57654535 |
| 8-OH-DPAT | Sigma-Aldrich | 57654276 |
| Tergazyme enzyme detergent | Sigma-Aldrich | Z273287 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J, male and female, > 3 weeks of age | The Jackson Laboratory | Stock No. 000664 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/, male and female, > 3-weeks of age | The Jackson Laboratory | Stock No: 007909 |
| Mouse: B6.Cg-Pvalb-T2A-Cre-D, male and female, > 3-weeks of age | The Jackson Laboratory | Stock No: 022863 |
| Other | ||
| MultiClamp 700B | Axon Instruments | N/A |
| Digitizer digidata | Axon Instruments | Model 1440E |
| Isolated Pulse Stimulator | A-M Systems | Model 2100 |
| Micromanipulator | Sutter Instrument | Model MP-225 |
| Fluorescent microscope | Olympus Bx51 | N/A |
| X-Y translation stage | Mike's Machine Co | N/A |
| Magnetic chamber platform | Warner | 64-1526 |
| Manual micromanipulator | Newport | MS-200M-XYZ |
| Peristaltic flow pump | Gilson | MINIPLUS3 |
| Temperature regulator | Warner | SH-27B |
| Pipette puller | Sutter Instrument | P-1000 |
| pClamp patch-clamp software | Molecular Devices | N/A |
| PatchVision software | Scientifica | PV-5000 |
| Kwik-Fil borosilicate glass capillaries | World Precision Instruments | TW150-4 |
| MicroFil syringe needle | World Precision Instruments | MF28G-5 |
| Syringe filter | Nalgene | 171-0020 |
| Tissue slicer and specimen disc | Leica | VT1200S |
| Bone rongeur | Roboz | RS-8300 |
| Microdissecting scissors | Roboz | RS-5840 |
| Blunt operating scissors | Roboz | RS-6752 |
| Stainless steel sagittal brain matrices | Roboz | SA-2275 |
| Polyethylene transfer pipette | Fisher Scientific | 13-711-5AM |
| Falcon 50 mL conical tubes | Fisher Scientific | 14-432-22 |
| Precision water bath | Thermo Fisher Scientific | TSGP2S |
| Freezing point osmometer | Advanced Instruments | Model 3250 |
| Pre-amplifier | Multi Channel Systems | N/A |
| Stage plate | Multi Channel Systems | N/A |
| Inflow | Multi Channel Systems | N/A |
| Safety | Multi Channel Systems | N/A |
| O-ring | Multi Channel Systems | N/A |
| Electrophysiology harp | Multi Channel Systems | N/A |
| Ground | Multi Channel Systems | N/A |
| Heater | Multi Channel Systems | PH01 |
| MEA recording chamber | Multi Channel Systems | 60MEA500/10iR-Ti-gr |
| Software and algorithms | ||
| MEA Spike Sorting Data Analysis Software | Plexon Inc. Offline Sorter | N/A |
Materials and equipment
-
•
To prepare 1.0 L of 10× ACSF and NMDG-based solutions, add the following reagents to a 1.0 L graduated cylinder of distilled water. Once established, 10× solutions can be stored in a 4°C refrigerator for up to 6 months.
10× ACSF stock solution
| Chemical | g/L | Concentration (mM) |
|---|---|---|
| NaCl | 69.50 | 118.93 |
| KCl | 1.86 | 2.49 |
| CaCl2 (anhydrous) | 2.77 | 2.50 |
| MgCl2⋅6H2O | 2.64 | 1.30 |
| NaH2PO4 (anhydrous) | 1.20 | 1.00 |
| distilled water | Fill up to 1 L | - |
10× NMDG-based dissecting/recovery stock solution
| Chemical | g/L | Concentration (mM) |
|---|---|---|
| KCl | 0.19 | 2.50 |
| HEPES | 5.20 | 20.00 |
| NaH2PO4 (anhydrous) | 0.17 | 1.20 |
| Glucose | 4.51 | 25.00 |
| distilled water | Fill up to 1 L | - |
1× preparations from 10× ACSF and NMDG stock solutions are made fresh each day electrophysiological recordings are performed.
-
•ACSF bathing (external) solution: ACSF, a perfusion buffer resembling CSF circulating throughout the CNS in vivo, is used to maintain acute ex vivo brain slices during and in preparation for electrophysiological analyses.
-
○To obtain 1.0 L of 1× ACSF, add 2.2 g of NaHCO3 (26.2 mM) and 1.98 g of glucose (11 mM) to a 1.0 L graduated cylinder containing 100 mL of 10× ACSF stock solution.
-
○Dilute the stirred solution to 1.0 L with filtered, deionized H2O and measure the osmotic concentration using an osmometer.
-
○
Note: The resulting osmolarity (290–300 mOsm) should be within 5 mOsm of the intracellular solution used in whole-cell patch-clamp experiments to minimize osmotic perturbations in recording quality.
1× ACSF
| Chemical | g/L | Concentration (mM) |
|---|---|---|
| NaHCO3 | 2.20 | 26.20 |
| Glucose | 1.98 | 11.00 |
| 10× ACSF stock solution | 100 mL | - |
| distilled water | Fill up to 1 L | - |
-
•
NMDG-based dissecting/recovery solution: Specialized dissecting and recovery solution is used to minimize excitotoxic cell death during slice preparation by replacing extracellular Na+ with the iso-osmolar excipient, N-methyl-D-glucamine (NMDG or meglumine).
Note: Alternative dissecting/recovery solutions are available to suit specific experimental requirements (e.g., sucrose-based dissecting solution).
-
○To obtain 250 mL of 1× N-methyl-D-glucamine (NMDG)-based recovery solution, add 0.63 g of NaHCO3 (29.9 mM), 0.08 g of Na+-pyruvate (3.0 mM), 0.25 g of Na+-ascorbate (5.0 mM), and 4.54 g of NMDG (93 mM) into a 500 mL beaker containing 25 mL of 10× NMDG stock solution. Each concentration in parentheses represents the final concentration of each reagent.
-
○Dilute up to 250 mL with filtered, deionized H2O and add 63 μL of 2.0 M stock CaCl2-2H2O (0.5 mM) and 1.25 mL of 2.0 M sock MgCl2 (10 mM) solution. The resulting pH (9.8–10.2) must be lowered carefully with concentrated HCl (10 M) until stabilized between 7.3–7.4. Each concentration in parentheses represents the final concentration of each reagent.
-
○Using an osmometer, ensure that the final osmolarity of the solution is 300–310 mOsm.
-
○
Note: The osmolarity of the NMDG-based recovery solution is slightly higher than the ACSF to reduce excitotoxic osmotic swelling during the slicing procedure. An iso-osmolar solution may also be acceptable and should be optimized by the experimenter.
1× NMDG-based dissecting/recovery
| Chemical | g/250 mL | Concentration (mM) |
|---|---|---|
| NMDG | 4.54 | 93.00 |
| Na-ascorbate | 0.25 | 5.00 |
| Na-pyruvate | 0.08 | 3.00 |
| NaHCO3 | 0.63 | 30.00 |
| 10× NMDG stock solution | 25 mL | - |
| distilled water | Fill up to 250 mL | - |
Mg2+ and Ca2+ solutions incorporated into NMDG-based solutions
| Chemical | mL/250 mL | Concentration (mM) |
|---|---|---|
| MgCl2 (2M stock) | 1.25 | 10.00 |
| CaCl2⋅2H2O (2M stock) | 0.063 | 0.500 |
-
•Intracellular (internal) solution: The appropriate internal solution used for whole-cell patch-clamp electrophysiology depends on which analyses are being performed. Given the number of different recipes reported in the literature, we provide here example recipes of internal solutions used in voltage-clamp and current-clamp configurations. Because of the importance of well-calibrated internal solutions for whole-cell patch-clamp analyses, notes and clarifying points are defined in detail below.
-
○To obtain 100 mL of Cs+-based internal solution for analyses performed in voltage-clamp configuration, add 1.152 g of Cs+ methane sulfonic acid (MeSO3, 120 mM), 0.253 g of CsCl (15 mM), 0.047 g of NaCl (8 mM), 0.238 g of HEPES (10 mM), 0.007 g of EGTA (0.2 mM), 0.166 g of TEA-Cl (10 mM), 0.171 g of QX-315 (5 mM), and 0.002 g of spermine (0.1 mM) into 100 mL of filtered, deionized H2O.
-
○Incorporate 0.2 g of Mg2+-ATP (4 mM) and 0.016 g of Na+-GTP (0.3 mM) into the stirred solution on ice to minimize autohydrolysis of GTP and ATP. Carefully titrate the pH of the solution to 7.20–7.30 using concentrated CsOH.
-
○Using a freezing-point osmometer, ensure that the final osmolarity of the solution is 290–295 mOsm and within 5 mOsm of the ACSF bathing solution.
-
○Aliquot the 100 mL of internal solution into small volumes (1.0–1.5 mL) to be used for individual experiments on recording days. Store at −20°C for up to 1 year. This is necessary to prevent the internal solution from undergoing multiple freeze-thaw cycles and decreasing the stability of each solubilized reagent.
-
○
Cs+-based internal solution
| Chemical | g/100 mL | Concentration (mM) |
|---|---|---|
| CsMeSO3 | 1.152 | 120.00 |
| CsCl | 0.253 | 15.00 |
| NaCl | 0.047 | 8.00 |
| HEPES | 0.238 | 10.00 |
| EGTA | 0.007 | 0.20 |
| TEA-Cl | 0.166 | 10.00 |
| QX-315 | 0.171 | 5.00 |
| Spermine | 0.002 | 0.10 |
| Mg2+-ATP | 0.200 | 4.00 |
| Na+-GTP | 0.016 | 0.30 |
| distilled water | Fill up to 100 mL | - |
Note: intracellular inclusion of TEA, Cs+ and QX-214 achieves broad-spectrum blockade of voltage-sensitive ion channels, thereby optimizing control over somatodendritic membrane potential. However, this may disrupt postsynaptic effector function dependent on these channels (e.g., receptor-mediated modulation of various K+ channel subtypes) and must be considered when assessing synaptic mechanisms using this internal solution.
CRITICAL: The Cs+-based internal solution described here is suitable for analyses of GABAergic and glutamatergic transmission. According to the calculated Nernst potentials (EX) of ionotropic glutamate (AMPA and NMDA) receptors and ionotropic GABAA receptors (GABAARs) using our ACSF and Cs+-based internal solution, EAMPA/NMDA is approximately 0–(+)5 mV and EGABA is −70−(−60) mV. Thus, voltage-clamp experiments performed near the resting membrane potential of most central neurons [−60−(−80 mV)] will yield minimal (low amplitude) GABAAR-mediated currents.
Alternatives: To avoid having to depolarize cells to examine GABAergic transmission, a Cl--loaded, “symmetrical Cl-” ([Cl-]external = [Cl-]internal) solution can be used instead. To obtain a Cs+-based symmetrical Cl- internal solution, MeSO3 is substituted for an additional 1.770 g of CsCl, resulting in a solution containing 130 mM total Cl- and a calculated EGABA = 0 mV.
Note: Symmetrical Cl- internal solutions used to examine GABAergic transmission at quiescent (hyperpolarized) membrane potentials will result in depolarizing “outward” currents mediated by Cl- efflux through GABAARs. While this configuration allows for longer stable recordings, the “artificial” electrochemical reversal of GABAAR function with this internal solution must be taken into consideration when assessing inhibitory synaptic function.
-
○To obtain 100 mL of K+-based internal solution for analyses performed in current-clamp configuration, add 3.126 g of K+-gluconate (135 mM), 0.029 g of NaCl (5 mM), 0.041 g of MgCl2 (2 mM), 0.238 g of HEPES (10 mM), 0.023 g of EGTA (0.6 mM) into 100 mL of filtered, deionized H2O.
-
○Incorporate 0.203 g of Mg2+-GTP (4 mM) and 0.021 g of Na+-ATP (0.4 mM) into the stirred solution on ice to minimize autohydrolysis of GTP and ATP. Carefully titrate the pH to 7.20–7.30 using concentrated KOH.
-
○Using an osmometer, ensure that the final osmolarity of the solution is 290–295 mOsm and/or within 5 mOsm of the ACSF bathing solution.
-
○Aliquot the 100 mL of internal solution into small volumes (1.0–1.5 mL) to be used for individual experiments on recording days. Store at −20°C for up to 1 year. This is necessary to prevent the internal solution from undergoing multiple freeze-thaw cycles and decreasing the stability of each solubilized reagent.
-
○
K+-based internal solution
| Chemical | g/100 mL | Concentration (mM) |
|---|---|---|
| K+-gluconate | 3.126 | 135.00 |
| NaCl | 0.029 | 5.00 |
| MgCl2 | 0.041 | 2.00 |
| HEPES | 0.238 | 10.00 |
| EGTA | 0.023 | 0.60 |
| Mg2+-GTP | 0.203 | 4.00 |
| Na+-ATP | 0.021 | 0.400 |
| distilled water | Fill up to 100 mL | - |
Note: K+-based internal solution is suitable for experiments performed in voltage-clamp configuration examining glutamatergic transmission. However, the absence of blockers for voltage-sensitive ion channels may introduce space-clamp error and should be taken into consideration when interpreting data.
Alternatives: A K+-based internal solution resembling more physiological conditions can improve cell longevity during long-term recordings. Additionally, a K+-based internal solution may be required for experiments in which the electrophysiological profile of a cell must be confirmed prior to performing any subsequent manipulations.
Dissecting solution for multi-electrode array recordings
| Chemical | g/L | Concentration (mM) |
|---|---|---|
| NaCl | 6.691 | 114.5 |
| KCl | 0.261 | 3.50 |
| NaH2PO4-2H2O | 0.156 | 1.00 |
| MgSO4-7H2O | 0.320 | 1.30 |
| CaCl2-2H2O | 0.368 | 2.50 |
| D-(+)-glucose | 1.801 | 10.0 |
| NaHCO3 | 2.999 | 35.7 |
Recording solution for multi-electrode array recordings
| Chemical | g/L | Concentration (mM) |
|---|---|---|
| NaCl | 7.247 | 124.0 |
| KCl | 0.261 | 3.50 |
| NaH2PO4-2H2O | 0.156 | 1.00 |
| MgSO4-7H2O | 0.320 | 1.30 |
| CaCl2-2H2O | 0.368 | 2.50 |
| D-(+)-glucose | 1.801 | 10.0 |
| NaHCO3 | 2.184 | 26.0 |
Solutions are made fresh the day of recording in 1.0 L of deionized water and before experimental use. Each stock solution should be stored at 4–10oC for no longer than 2-weeks.
Step-by-step method details
Dissection and slice preparation
-
1.
After the dissection area has been prepared and solutions oxygenated and chilled, anesthetize the mouse with 5% isoflurane until the pedal reflex is absent. Decapitate the mouse rostral to the cervicothoracic junction.
-
2.
Extraction and sectioning: After decapitation, extract the brain from the skull using dissecting scissors, a rongeur, forceps and microspatula and quickly submerge the brain in the 50 mL beaker containing oxygenated 0°C NMDG-based recovery solution.
-
3.
Remove the brain slicer matrix from the ice and transfer a small volume (1–2 mL) of recovery solution into the matrix. Using the plastic spoon, remove the brain from the 50 mL beaker and place it in the appropriate anatomical position on the matrix.
-
4.
Wick away residual solution from the matrix using a Kimwipe or filter paper and insert razor blades along the desired slice intervals. After obtaining the desired anatomical brain sections, apply a uniform layer of glue to the specimen disk and carefully mount each section.
-
5.
Slicing: Promptly transfer the specimen disk to the vibratome buffer tray containing oxygenated ∼0°C NMDG-based recovery solution and allow the brain sections to equilibrate for 1–2-min prior to slicing.
-
6.
Once the specimen disk is securely positioned, adjust the vibratome settings to the desired slicing speed (0.28–0.3 mm/s), vibrational amplitude (0.8–1.00 mm), and slice thickness (200-–400 μm depending on region of interest and anatomical orientation).
-
7.
Elevate the vibratome stage towards the superficial plane of the mounted brain sections and assign the appropriate start-end positions.
-
8.
As the vibratome begins to release slices of tissue containing the region of interest, use the open-ended transfer pipette to move slices to the submerged holding chamber containing oxygenated ∼32°C NMDG-based recovery solution.
Pause point:Recovery: Allow the slices to recover for 10-min in the submerged holding chamber before transferring slices to the ACSF-containing holding chamber. To improve slice health and longevity throughout recording sessions, arrange each slice on the holding chamber floor in the orientation used for experimentation. Any pharmacological manipulation requiring pre-incubation can be incorporated into the ACSF holding chamber at this point.
Note: The timing, dissection, and slice preparation methods are very similar for multi-electrode array recordings, however the solutions for dissecting and recording differ (see tables "Recording solution for multi-electrode array recordings" and "Dissecting solution for multi-electrode array recordings").
Multi-electrode array dissection
-
9.
Briefly, the brain should be removed within 30 s and 1 min, and submerged in cold dissecting solution. To specifically target the ventromedial portion of the dorsal raphe nucleus, which will be used as an example throughout this protocol based on its high proportion of serotonin cells, the experimenter will obtain 300-micron thick sections targeting −5.5 and −5.75 mm from Bregma.
-
10.
To obtain these slices, after at least 1 min of the brain being submerged in cold dissecting solution, place it on a cold piece of filter paper on an inverted Petri dish that is placed in ice. Bisect the brain in the coronal plane, halfway between the olfactory bulbs and cerebellum.
-
11.
As described above, apply a uniform layer of glue on the specimen disk and place the tissue on the disk mounting the brain such that the rostral portion is facing down on the disk and the cerebellum is facing up. Place the brain and specimen disk into the vibratome buffer tray containing oxygenated 0°C dissecting solution. Allow the tissue to rest for at least 1 min before beginning to slice the most ventral portion of the cerebellum and then removing the cerebellum.
-
12.
300-micron slices are taken until ventromedial portions of the dorsal raphe are identified by the presence of the cerebral aqueduct. These slices are then collected and placed in a Petri dish containing room temperature (20°C–23.5°C) oxygenated recording solution (Figures 7A and 7B).
-
13.
The cerebral aqueduct is cut through the middle resulting in a U, and the lateral portions are removed with a scalpel. The ventromedial portion of the DRN is directly below the cerebral aqueduct (“U”) (Figures 7A and 7B). Slices are then placed in room temperature (20°C–23.5°C) oxygenated recording solution in a plastic slice tray.
-
14.
While the slice is recovering, add 40 μM of tryptophan and 3 μM of phenylephrine to the glass media bottle containing the remaining recording solution to be used for multi-electrode array recordings. Specifically when recording from 5-HT neurons, tryptophan is provided as a substrate for 5-HT synthesis in the slice to allow for intrinsic 5-HT1A auto-inhibition, and phenylephrine is provided to stimulate noradrenergic 1b receptors and sustain spontaneous spiking of 5-HT neurons.
-
15.
Lastly, add 50 mL of the recording solution containing tryptophan and phenylephrine to a 50 mL Falcon tube, and add 1 μM of 8-OH-DPAT and label the tube. This will be used for 5-HT1A inhibition of serotonin neurons during the recording session.
Note: Multi-electrode array contacts should be cleaned with Q-tip and ethanol solution, and allowed to dry approximately 45 min before recordings begin.
Figure 7.

Setup of a multi-electrode array recording from a DRN slice
(A) A representative DRN slice after dissection, circled in red.
(B) A representative DRN slice used for recordings once the lateral tissue is removed and the cerebral aqueduct is cut resulting in a “U” shape. The ventromedial portion of the DRN, which is the area used for recordings, is located just below the cut aqueduct or “U” shape.
(C) Demonstration of the components of a multi-electrode array including the contacts and recording chamber along with the location/placement of the DRN tissue and an electrophysiology harp to keep the tissue in place.
(D) Demonstration of the baseplate of the recording stage and the O-ring, covered in vacuum grease, which is necessary to maintain a tight seal once the multi-electrode array and the components shown in (C) are firmly pushed down onto the O-ring.
(E) Once the multi-electrode array, harp, and tissue are firmly placed on the O-ring, the pre-amplifier is attached along with the heated inflow and safety lines.
(F) The overall setup for a multi-electrode array recording.
Patch-clamp electrophysiology: Pre-recording preparation
-
16.Pipettes: Prior to the recording session, pull 3–6 MΩ patch pipettes from borosilicate glass capillary tubes using a micropipette puller system. While the optimal patch pipette resistance for whole-cell recordings depends on neuronal morphology and the type of experiment being performed, pipettes with a resistance between 3–6 MΩ should be suitable for most patch-clamp experiments.
-
a.After pulling pipette tips, house them carefully in a closed, airtight container to prevent air particulate from entering the distal end of the pipette tip.
-
a.
-
17.To fill a pipette tip with internal solution, first aspirate a chilled aliquot of internal solution into a small syringe (0.5–1 mL) and place a non-sterile syringe filter (Nalgene, 4 mm diametric membrane) on the tip of the syringe.
-
a.Place a flexible, plastic MicroFil or microloader on the syringe filter tip, fill the patch pipette to 60%–75% of capacity, and rest the syringe-MicroFil assembly on ice for future use.
-
a.
-
18.
Perfusion and slice positioning: Turn on the rig, configure the acquisition software, and perfuse warmed ACSF (30±2°C) into the recording chamber at a rate of 2–3 mL/min using a gravity-driven or peristaltic pump system.
Note: ACSF entering the recording chamber should exit through an outlet into a pre-bleached Erlenmeyer collection container (2 L) connected to a vacuum system.
-
19.
When the ACSF reaches steady-state perfusion, use an open-ended transfer pipette to transfer a brain slice into the recording chamber and place a nylon thread-containing stainless-steel harp on the slice. Make sure the distance between nylon threads on the harp provides sufficient room to maneuver the patch pipette in the slice without interference.
-
20.
Once the slice is positioned correctly in the recording chamber, carefully lower the stimulating electrode (bipolar or monopolar) into the slice so that it makes direct contact with the tissue in the region of interest.
Patch-clamp electrophysiology: Patching
-
21.
Fill a patch pipette 60%–75% with internal solution and insert the pipette in the recording electrode holder. Fasten the pipette tightly with the O-ring rubber gasket and plastic washer.
-
22.
To reduce the probability of cellular debris occluding the pipette tip, deliver continuous positive pressure through the pipette using a large/medium-bore syringe connected to the recording electrode holder via plastic tubing (Figure 3B).
Note: Positive pressure will assist in identifying competent cells by moving aside neuropil and cellular processes obscuring neuronal borders.
-
23.
Using the micromanipulator in low-power magnification, maneuver the patch pipette near the stimulating electrode in the region of interest and align the microscope vertically with the patch pipette.
-
24.Initiate the membrane test on the recording software to generate a 50 ms, 10 mV voltage step looped at 20 Hz to continuously monitor pipette resistant and adjust pipette offset.
-
a.Ensure that air bubbles and particulate are completely evacuated from the pipette tip using a brief pulse of positive pressure.
-
a.
-
25.
Switch to the high-power objective and use the fine-motion micromanipulator to lower the objective slowly into the recording chamber until a column of ACSF bathing solution immerses the lens. Adjust the depth (y-axis) of the high-power objective until the patch pipette can be visualized.
-
26.
Prior to maneuvering the patch pipette through the neuropil, survey the area with the high-power objective for competent cells that (a) contain a thin, smooth cell membrane, (b) lack pyknotic or karyolitic nuclei, (c) appear turgid but easily deformable, and (d) are oriented with a membrane surface accessible to the patch pipette.
Note: To maximize the likelihood of patching a healthy cell with reliable membrane parameters, avoid cells close to the surface of the slice. While cells embedded more deeply in the slice are more challenging to visualize, these cells are less likely to exhibit gross morphological disturbances from the slicing and handling process and will survive longer recording intervals.
-
27.Once a healthy cell is selected, move the patch pipette in close apposition to the cell membrane until a small invagination of the cell membrane is visualized as a “dimple” (Figure 3B-1).
-
a.Release positive pressure by removing the syringe and apply slow, continuous negative pressure using buccal (mouth) suction (Hamill et al., 1981).
-
a.
Alternatives: Buccal suction provides greater manual dexterity of the extent and duration of negative pressure applied to the cell. An alternative approach is to use a smaller-bore (5–10 mL) syringe that is retracted to create negative pressure.
Note: Regardless of whether experiments will be performed in voltage- or current-clamp configuration, cells can be voltage-clamped near the resting membrane potential during initial break-in to reduce sudden electrochemical disturbances to the cell.
-
28.Apply brief pulses of negative pressure to rupture the patch of membrane beneath the patch pipette. A successful break-in is signaled by a precipitous reduction in membrane resistance and sharp capacitive transients time-locked with the membrane test (Figure 3B-2).
-
a.If the patch of membrane was incompletely rendered during the initial break-in, apply additional pulses of negative pressure while monitoring the membrane resistance.
-
a.
Note: If patching fails to convert to whole-cell configuration, avoid reusing the same patch pipette for subsequent attempts. The used pipette tip will contain membranous material from the unsuccessful patch that will preclude a GΩ seal from forming on subsequent cells.
-
29.
After entering whole-cell configuration, allow 3–5-min for the intracellular environment to equilibrate to the internal solution as it dialyzes the cell. This is particularly important for cells with highly arborized dendritic and axonal processes (Figure 3B-3).
Note: Important additional electrochemical parameters emerge when entering whole-cell configuration, including holding current (Iholding), membrane resistance (Rm), membrane capacitance (Cm), and access resistance (Ra). These parameters enable the experimenter to assess the patency of the patch pipette tip and stability of the seal during the recording period.
Note: Absolute values of Ih, Cm, and Rm are determined by the type of cell being studied, experimental condition, which internal solution is being used, and ultimately experimenter technique. In general, cells being examined in whole-cell voltage-clamp configuration will display a higher Rm with Cs+-based internal solutions containing Na+ and K+ ion channel blockers. In contrast, cells examined using a K+-based internal solution will display a lower, more physiological Rm that requires a greater Ih, particularly when pronounced leak conductances are present.
Figure 3.

Schematic depicting the steps to entering whole-cell configuration
(A) Schematic depicting whole-cell patch-clamp electrophysiology alongside a 40× DIC image including a “healthy” and an “unhealthy” MSNs in the NAc. Scale bar: 80 μm.
(B) Steps to entering whole-cell configuration with the associated current response to a voltage step elicited via the membrane test. (1) Using positive pressure to remove cellular debris from occluding the patch pipette, approach a healthy cell exhibiting features described in the text. (2) As the patch pipette produces a modest invagination of the membrane, remove positive pressure and apply gentle buccal suction to achieve a ~1GΩ seal (reflected by the membrane test). (3) After cell-attached configuration, apply brief pulses of negative pressure to enter whole-cell until a precipitous reduction in membrane resistance and sharp capacitive transients are observed.
(C) A 40× DIC image of a cell in the NAc patched in whole-cell configuration. Scale bar: 50 μm.
Patch-clamp electrophysiology: Recording in voltage-clamp configuration
For simplicity and broad applicability, we describe here an example of steps taken to examine the effects of receptor-specific pharmacology on glutamatergic transmission. Using the nucleus accumbens (NAc) as our model system, we will demonstrate how the metabotropic GABAB receptor (GABABR), a Gi/o-coupled G protein-coupled receptor (GPCR), alters electrically-evoked glutamatergic transmission onto medium spiny projection neurons (MSNs) (Figure 4) (Manz et al., 2019).
-
30.To record electrically-evoked excitatory postsynaptic currents (EPSCs) from MSNs in the NAc, begin by voltage-clamping the patched MSN at −70 mV using a Cs+-based internal solution according to methods outlined above (Figures 4A and 4B).
-
a.Turn on the electrical supply to the stimulating electrode and switch the discharge mode to current (I).
-
a.
-
31.Because electrical stimulation triggers neurotransmitter release from glutamatergic and GABAergic terminals, incorporate a GABAAR receptor antagonist (e.g., picrotoxin or bicuculline at 50 μM) into the ACSF perfusion medium to eliminate coincident GABAAR-mediated inhibitory postsynaptic currents (IPSCs).Pause point: Allow sufficient time for the antagonist to completely infiltrate the slice (10–15 min, depending on how long it takes for the perfusate to reach the recording chamber).
-
a.To ensure that electrically-evoked EPSCs are exclusively glutamatergic, superfuse a broad-spectrum antagonist of ionotropic glutamate receptors (kynurenic acid, KynA, 50 μM) at the end of the experiment. To assess the specific contribution of AMPA receptors (AMPARs) or NMDA receptors (NMDARs) to the electrically-evoked EPSC, superfuse instead a selective AMPAR (NBQX, 10 μM) or NMDAR antagonist (D-APV, 50 μM) and examine the corresponding change in EPSC amplitude. Each concentration represents the final concentration of the drug in the perfusing medium.
-
a.
-
32.
Using an “Episodic” protocol to discharge the stimulating electrode, stimulate with the lowest intensity and pulse duration required to evoke an EPSC in the recorded MSN. Define an interstimulus interval (ISI) of 10-s (0.1 Hz) to minimize any activity-related synaptic rundown during the recording period.
Note: The EPSC baseline reflects synaptic responses from a heterogeneous population of glutamatergic inputs, each possessing distinct pre- and postsynaptic properties. Thus, it is essential that a stable EPSC baseline is obtained prior to proceeding with subsequent experimental manipulations. Although we routinely report 10-min EPSC baselines, more time may be needed (12–15 min) to ensure that responses have completely stabilized. A stabilized recording is defined here as consistent synaptic responses, holding current, and membrane resistance (<20% change). Cells requiring considerably longer than 15 min to stabilize should be discarded from analysis, as gross electrochemical or technical disturbances to the patched cell are likely contributing to wavering EPSC responses.
-
33.
Once EPSCs have stabilized, superfuse selective GABABR agonist, (RS)-baclofen (BAC, 1 μM), into the ACSF bath for 10-mins (Figures 4C and 4D).
Note: To simplify pharmacological preparations, we recommend creating stock solutions of drug at 1000× the concentration used in the ex vivo preparation. Thus, a 1 μM solution of BAC in ACSF is obtained by incorporating 30 μL of a 1 mM BAC stock solution to a conical tube containing 30 mL of ACSF.
-
34.
Continue to examine changes in EPSC amplitude as BAC-containing ACSF begins to enter the recording chamber (Figures 4C–4E).
-
35.To begin terminating exposure to BAC, switch the perfusion line back to ACSF used to obtain the EPSC baseline. BAC will gradually exit the recording chamber during washout, returning EPSC amplitude back to baseline. Depending on the scientific question, continue to record EPSCs for 10–30-min post-washout (Figures 4C–4E).Alternatives: As the effect of BAC on EPSC amplitude stabilizes, several important experimental trajectories can be taken. To determine whether BAC-induced GABABR activity triggers a long-term change in glutamatergic synaptic strength, a selective GABABR antagonist (SCH 50911, 5 μM) can be incorporated into the ACSF bath following BAC (Atwood et al., 2014). A persistent change in EPSC amplitude in the presence of SCH 50911 indicates a long-lasting, agonist-independent change in synaptic strength. Alternatively, EPSC amplitude can be continuously monitored as BAC completely exits the ACSF bath. We recommend both trajectories be taken in different experiments so that a complete pharmacological profile of BAC can be obtained at the synapse being studied (Manz et al., 2020a, 2020b).Note: All pharmacological analyses should be performed with the appropriate control experiments. Although BAC is a well-characterized and prototypical GABABR ligand, it is essential that experiments are performed ensuring that synaptic effects elicited by BAC are mediated by GABABR. Thus, prior superfusion of SCH 50911 should completely abolish subsequent effects elicited by BAC. If SCH 50911 is used to reverse the effects of BAC, separate experiments must be conducted to determine if bath-application of SCH 50911 alone unmasks a change in EPSC amplitude.
-
a.A change in EPSC amplitude in the antagonist alone may indicate the presence of tonic receptor activity at the synapse. In the case of GABABR, an antagonist-induced increase in EPSC amplitude would suggest that endogenously-released GABA tonically inhibits glutamate release onto MSNs via GABABRs. Additional pharmacological experiments will then be needed to assess whether GABA-independent constitutive receptor activity is mediating this effect (Manz et al., 2019; Uchimura and North, 1991).
-
a.
-
36.
After the experiment is completed, flush the perfusion line and recording chamber with hot water to prevent drug contamination of subsequent experiments. If lipophilic compounds are used, flush the line with a 10% bleach solution prior to rinsing with hot water. Make sure the 10% bleach solution has completely exited the recording chamber prior to beginning future experiments.
Figure 4.

Example experiment performed in voltage-clamp configuration
(A) Schematic depicting the parasagittal plane through which NAc-containing ex vivo brain slices are prepared.
(B) Schematic of the whole-cell electrophysiological recording strategy in the NAc. Electrically-evoked EPSCs are recorded in MSNs patched in whole-cell voltage-clamp configuration with a Cs+-based internal solution.
(C) A representative experiment showing how GABABR agonist, (RS)-baclofen (BAC, 1 μM), reduces glutamatergic synaptic strength onto MSNs in the NAc. Each point reflects the amplitude of an EPSC elicited via electrical stimulation at 0.1 Hz. To eliminate GABAAR-mediated synaptic currents, GABAAR antagonist, picrotoxin (50 μM), is included in the ACSF bath throughout the recording session. After obtaining a stable 10-min EPSC baseline, BAC is superfused for 10-min, after which the line is switched to the original ACSF.
(D) Representative traces of EPSCs a\t baseline (black) and in the presence of BAC (blue).
(E) Summary graph depicting average EPSC amplitude at baseline and in the presence of BAC. Note: To compare outcomes across experiments, summary graphs of multiple experimental replicates should be depicted and analyzed following normalization.
Patch-clamp electrophysiology: Recording in current-clamp configuration
We describe here a step-by-step approach to assessing differences in intrinsic membrane parameters between neuronal subtypes in current-clamp configuration. Using the NAc as our model system, we examine action potential (AP) firing rates in MSNs and parvalbumin (PV)-expressing interneurons, which exhibit regular-spiking and fast-spiking AP profiles, respectively (Manz et al., 2020b). Importantly, both neuronal subtypes in the NAc exhibit a hyperpolarized resting membrane potential [VRMP = −70−(−90) mV] and lack intrinsic peacemaking mechanism, making them ideal current-clamp comparators (Scudder et al., 2018; Tepper et al., 2018) (Figure 5A).
-
37.
To begin measuring intrinsic membrane properties, patch an MSN in whole-cell configuration using a K+-based internal solution according to the protocol outlined above.
-
38.
After breaking-in, switch the recording mode to current-clamp and set the command current to 0 pA (IInjected, INJ = 0). This setting will result in no current being injected into the cell, resulting in a transmembrane potential equivalent to VRMP.
Note: As the intracellular solution dialyzes the cell, the membrane potential recorded at IINJ = 0 pA will gradually hyperpolarize beyond VRMP. To circumvent the contribution of this phenomenon and various junctional potentials to the recorded VRMP, compare values at defined time points after entering whole-cell configuration (Barry and Lynch, 1991).
-
39.
After recording VRMP, set the command current such that the membrane potential stabilizes at a defined value (e.g., ∼−70 mV). This will standardize the starting point at which sequential current steps will be delivered to cells with a different starting VRMP.
-
40.
To begin to assess voltage responses to intracellular (somatic) current injection, evoke sequential 400–800-ms current steps beginning at −400 pA at 0.016 Hz (1 step/min). Beginning with hyperpolarizing current steps will assist in the identification of a hyperpolarization-activated “sag” current, passive membrane conductances, and the input resistance (∼Rm) (Figure 5B).
Note: We recommend incremental current steps of 20–50 pA, with smaller increments allowing more precise measurements of rheobase, defined as the minimum current of infinite duration needed to elicit a single AP.
Alternatives: Shortening the inter-step interval may be possible in cells that are less susceptible to depolarization block. In the NAc, high-fidelity AP firing can be obtained in MSNs with an inter-step interval as short as 5-sec. However, a 1-min interval is suitable for cells in most regions and should be applied with careful consideration of the cell’s established electrophysiological properties.
Note: A voltage sag (Vsag) indicates the presence of an Ih current mediated by hyperpolarization-activated cyclic nucleotide-gated (HCN) cation channels (Oswald et al., 2009). A prominent Vsag is often present in cells that express HCN channels and exhibit autonomous peacemaking activity (He et al., 2014). MSNs and FSIs do not exhibit Ih and lack pacemaker activity (Kawaguchi et al., 1995).
-
41.
As positive current steps elicit depolarizing voltage responses, APs will begin to emerge with increasing frequency. AP frequency is defined as the number of spikes elicited during a given 400–800 pA current step. An increase in AP frequency will occur until suprathreshold current injection triggers depolarization block, a process by which excessive current injection fails to evoke APs despite a depolarizing membrane response (Figure 5B) (Dovzhenok and Kuznetsov, 2012).
-
42.
To obtain the rheobase, record the current step at which the first AP was elicited.
-
43.After obtaining these data for MSNs, the same approach can then be performed in FSIs to assess cell type-specific differences in intrinsic membrane properties.Note: Specific features of the AP waveform can be used to assess contributing factors to intrinsic excitability, such as AP threshold, amplitude, and phases of the afterhyperpolarization (AHP) (Figure 5).
-
a.To rigorously quantify AP threshold, a phase plane analysis can be performed on a single spike by plotting the first derivate of the AP (dV/dt) against the membrane potential (Vm). AP threshold is 5% of the maximum of the second derivative of this relationship (d2V/dt2), aligning closely with the inflection point of the phase plot. AP amplitude can then be assessed by measuring the peak of the AP relative to threshold (Kress et al., 2008; Sun et al., 2014).
-
b.To assess medium and slow AHP (mAHP and sAHP), the amplitude of the AHP can be quantified at cell type-specific time points following AP. A more isolated assessment of post-spike AHPs can be performed in voltage-clamp using K+-based internal solution. Briefly, set the command voltage to −50−(−60 mV) and applying a 100–400-ms voltage step to 0 mV. An outward “tail current”, indicating the mixed component AHP, will follow in close succession to the end of the voltage step. The contribution of specific K+ channels to the AHP, such as small conductance Ca2+-activated K+ (SK) channels, can be assessed using selective SK inhibitor, apamin (50 nM), and other K+ channel blockers, such as TEA, 4-aminopyridine, Ba2+, and dendrotoxin (Goldberg and Wilson, 2005; Shan et al., 2019).
-
a.
Figure 5.

Example experiment performed in current-clamp configuration
(A) Schematic of the whole-cell electrophysiological recording strategy of MSNs and PV-expressing FSIs in the NAc. Note: this experiment was performed in a PV-specific transgenic reporter line to differentiate FSIs from MSNs. Using a K+-based internal solution, MSNs and FSIs are patched in whole-cell current-clamp configuration and stabilized at ~−70 mV.
(B) Example traces of membranes responses in FSIs (black) and MSNs (blue) following hyperpolarizing (−150 pA) and depolarizing (100, 200, 350 pA) current injection (IINJ). As IINJ becomes increasingly positive, increased AP firing is observed in both cell types, with FSIs exhibiting significantly increased spike frequency relative to MSNs.
Patch-clamp electrophysiology: Recording in cell-attached and loose-patch configuration
While patch-clamp electrophysiology performed in whole-cell configuration confers direct electrical access to the cell, this process may disrupt intrinsic cellular processes requiring an intact intracellular compartment, such as autonomous AP firing (Fricker et al., 1999). To circumvent this issue, cells can be examined in cell-attached configuration by maintaining patched cells near the 1 GΩ seal without converting to whole-cell. This technique is particularly useful for long-term (> 10 min) analyses of neuronal spontaneous firing rate. Because the steps to entering cell-attached configuration are identical to those needed for whole-cell, we provide here conditions for optimizing cell-attached recordings (Figures 6A and 6B).
-
44.To perform cell-attached recordings, backfill patch pipettes with ACSF instead of an internal solution to resemble the extracellular environment. This will establish the electrochemical driving force for spontaneous AP firing in the membrane patch beneath the pipette (Figure 6A).CRITICAL: The speed with which a GΩ seal is formed can influence the duration of cell-attached recordings. To minimize premature conversion to whole-cell configuration, we recommend positive pressure be removed and only moderate, if any, negative pressure applied to the cell. This will minimize sheer stress to the patch of membrane beneath the patch pipette. As experimental technique improves, removal of positive pressure will be sufficient to obtain the GΩ seal.Alternatives: Long-term recordings in cell-attached configuration can lead to the gradual degradation of the GΩ seal. If cell-attached configuration is used to assess spontaneous AP firing, a GΩ seal may not be necessary to observe high-fidelity AP firing. In this case, loose-patch recordings may be a useful alternative.
-
a.To perform loose-patch recordings, a similar experimental approach to cell-attached configuration is taken, except patch pipette resistance is intentionally maintained below 1GΩ (50–200 MΩ). This is accomplished easiest by reusing prior patch pipettes.
-
a.
Figure 6.

Example recording performed in cell-attached configuration
(A) Schematic of the cell-attached (or loose-patch) electrophysiological recording strategy of tonically-active cholinergic interneurons (CINs).
(B) Example trace of spontaneous APs observed in CINs. Holding current (Iholding) is stabilized by voltage-clamping CINs at −70 mV (not required to observe AP firing in cell-attached).
We provide an extended representative trace of spontaneous APs observed in tonically-active cholinergic interneurons (CINs), one of few neuronal subtypes in the NAc that exhibit intrinsic pacemaker activity (Cheng et al., 2019) (Figure 6B).
Note: The representative trace depicted is from a CIN patched in cell-attached configuration with a patch pipette resistance of 4.2 MΩ backfilled with ACSF, resulting in a ∼1.4 GΩ seal that persisted for 32.5 min (Figure 6B).
Dorsal raphe nucleus (DRN) serotonin (5-HT) neuron multielectrode array preparation
-
45.
This protocol is written for use with the multi-electrode array and recording system from Multi-Channel Systems (MCS), although there are several manufacturer systems for which the instructions would be similar. We describe multi-electrode array recordings in the DRN as neurons in this region autonomously fire action potentials in an ex vivo slice. For DRN recordings 6×10 (60 electrode) perforated arrays are used mounted in the MCS recording chamber on an inverted microscope to visualize the full array and recording chamber. The diameter of each electrode is 30 microns and the space between electrodes is 100 microns resulting in the electrodes covering a total space of 1200 × 680 microns (Green et al., 2015; Siemann et al., 2019).
-
46.
Room temperature (20°C–23.5°C) oxygenated (95%O2–5%CO2) extracellular recording solution for multi-electrode array recordings (in mM: 124 NaCl, 3.5 KCl, 1 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 10 D (+) glucose, and 26 NaHCO3) containing 40 μM tryptophan and 3 μM phenylephrine is used for the recordings of DRN 5-HT neurons. Tryptophan is provided as a substrate for 5-HT synthesis in the slice to allow for intrinsic 5-HT1A auto-inhibition, and phenylephrine is provided to stimulate noradrenergic 1b receptors and sustain spontaneous spiking of 5-HT neurons.
-
47.
300-micron ventromedial DRN slices, sectioned between −5.5 and −5.75 mm from Bregma, are used for this preparation. The lateral portions are removed with a scalpel and slices rest in the recording solution in a plastic slice tray for approximately 30–45 min before recordings begin (Figures 7A and 7B).
Note: The ventromedial portion of the DRN contains approximately 70%–95% serotonin cells (Kirby et al., 2003), which gives the experimenter confidence that DRN 5-HT neurons will likely be identified and recorded from, however the DRN contains a heterogenous population of neurons (glutamatergic, GABAergic, and dopaminergic), which are more typically found in higher proportions in the lateral “wings” of the DRN (Kirby et al., 2003).
-
48.
Fill a pipette with recording solution and collect a ventromedial DRN slice from the slice tray. Carefully place the slice and recording solution onto the multielectrode array. Use Kimwipes to carefully remove excess recording solution to ensure the slice makes contact with the recording electrodes (Figure 7C).
-
49.
Under visual control through the inverted microscope carefully use a fine-bristle paintbrush dipped in recording solution to gently move and orient the slice such that the cerebral aqueduct is in line with the long axis of the electrode array rectangle as this will cover the majority of the ventromedial portion of the DRN.
-
50.
With ultra-fine forceps, under visual control, carefully place an electrophysiology harp (Multi-Channel Systems) over the slice to immobilize the slice on the electrode array.
-
51.
Gently push the multielectrode array down to seat it on the recording chamber O-ring, which has been lubricated with vacuum grease to ensure a tight seal (Figure 7D).
Note: Push gently down on the outside rim of the multielectrode array recording chamber. By pushing on the array, itself, this will result in the contacts being dirtied and could also damage or break the array.
-
52.
Place the pre-amplifier plate on top of the baseplate of the recording stage, which now contains the multielectrode array, and lock the amplifier in place, making contact with the electrode leads on along the edges of the array.
-
53.
Add a few drops of oxygenated recording solution to the slice and recording chamber to maintain slice health and cell viability.
-
54.
Place the heated (32°C) inflow and safety suction lines into the recording chamber and turn on the peristaltic pump, keeping it at a rate of 1.3 mL/min (Figure 7E).
Note: Steps 48–54 should take approximately 45 s or less to ensure cell and slice health. Adding too much recording solution to the recording chamber increases the likelihood that the electrophysiology harp and/or slice moves off of the electrodes and the desired location, however removing too much liquid will increase the likelihood of cell death and decrease slice viability.
-
55.
As the outflow line is connected to the baseplate of the recording stage, use visual verification that both the inflow and outflow lines are distributing recording solution properly, and there are no air bubbles present in the lines at the beginning or throughout the recording.
-
56.
Once it is established that the lines are working properly and oxygenated recording solution is constantly being perfused, allow the slice to settle onto the recording electrodes for approximately 45 min (Figure 7F).
Multielectrode array recording
-
57.
After waiting approximately 45 min for the slice to settle, identify the channels that are exhibiting consistent neuronal firing, and obtain a baseline recording while oxygenated recording solution is being continuously applied at a rate of 1.3 mL/min for 4–5 min.
-
58.
After obtaining a sample stable baseline activity from recorded neurons, pharmacological agents can be applied to test for synaptic, or ionic mechanisms regulating 5-HT neuron activity. The larger sample sizes available with array recording can facilitate obtaining dose-response relationships (e.g., to ADRA1b agonist, or TREK-1 K+ channel antagonists (Giannoni-Guzmán et al., 2021; Green et al., 2015), compared to single cell recording methods.
-
59.
To identify units that are putative 5-HT neurons, after experimental manipulations have been performed, briefly stop the peristaltic pump, remove the inflow line with recording solution and replace it with the 8-OH-DPAT line. Restart the pump, use a pipette to add a few drops of 8-OH-DPAT to the multielectrode array slice chamber, and continue to record for an additional 5–6 min. Place the recording solution line away from the recording area in a container (i.e., pipette tip box) to collect the solution as it drips during the recording as it is still connected to the pump.
-
60.
After 5–6 min of recording under 8-OH-DPAT conditions at a rate of 1.3 mL/min have passed, stop the pump and switch the 8-OH-DPAT and recording solution lines again, now placing the recording solution line into the recording chamber, and allowing the 8-OH-DPAT line to drip in a container (i.e., pipette tip box) outside of the recording area.
-
61.
Restart the pump again and record for 4–5 min to ensure that the slice and 5-HT neurons can recover from 5-HT1A suppression via 8-OH-DPAT. This is important as it will help differentiate if the lack of neuronal firing observed was in fact due to 5-HT1A inhibition, and not due to potential cell death.
Note: Again, make a note of when the recording solution was added to the recording chamber.
-
62.
Once 4–5 min have passed of the second application of recording solution, stop the peristaltic pump, stop the recording, remove the inflow and safety suction lines from the multielectrode recording chamber, remove the amplifier from the baseplate of the recording stage, and dispose of the excess recording and 8-OH-DPAT solutions.
-
63.
After the experiment is completed, remove the multielectrode array from the baseplate, remove the electrophysiology harp, wipe off the excess vacuum grease on the bottom edges, and place it in a container on a rocker in a cold 5% Tergazyme solution. After the array has been in 5% Tergazyme for at least 2 h and no more than 24 h, remove the array from the solution and place it in a new container filled with water on a rocker and keep it in the water container until needed for future recordings (replace the water weekly). If recording from multiple slices per day, then ideally prepare to use new/clean multi-electrode arrays for each slice. If this is not possible, wait at least 2 h or longer and confirm that the previous slice tissue has been removed and cleaned with Turgazyme in the shaker. Finally, run water through the inflow and outflow lines to clean the lines and to prevent contamination for subsequent experiments, and turn off the peristaltic pump, the safety suction line, heater, and oxygen tank.
-
64.
Briefly, for the analysis of DRN 5-HT neurons using multi-electrode arrays, multiple different software programs can be used. We utilize Offline Sorter (Plexon Inc.) and apply a Bessel filter to the raw traces that were obtained and identified as potentially serotonergic cells from the different channels during the recording. Threshold levels are determined by manually selecting the least amount of unipolar noise. A K means scan method is then applied, which will cluster the spikes based on characteristics such as amplitude, spike duration, etc. We then manually identify and verify if cells demonstrate the characteristic waveforms observed for DRN 5-HT cells, such as broad spike waveform, regular and low frequency firing rate (Figure 8B). Once a cell displays these characteristics, we then note if the cell had been responsive to 5HT1A inhibition. We calculate the firing rate during the application of recording solution and compare this to the firing rate when 8-OH-DPAT was applied to the slice. If the firing rate suppression is greater than 50% during 8-OH-DPAT application, we count the cell as serotonergic and include it in our analysis (Figure 8C), however if 50% suppression is not achieved, then the cell is not included in our calculations. In addition to single units, multiple cells can be recorded from a single electrode. To separate and track the activity of multiple cells on an electrode we use the same methods applied above, including the K means scan method. Multiple cells will be typically separated based on differing spike waveform characteristics, and for 5-HT identification utilize the same 50% suppression criteria when 8-OH-DPAT is applied to the slice. As a result, anywhere between 50 to 150 cells can reliably be recorded from in 5–7 slices (Green et al., 2015; Siemann et al., 2019). Further detail on the analysis of 5-HT DRN neurons using multi-electrode array recordings can be found in (Green et al., 2015; Siemann et al., 2019).
Figure 8.

Example of a DRN 5-HT neuronal recording using multi-electrode arrays
(A) A representative cartoon of a multi-electrode array (Multi-Channel Systems) and a bright field image of the dorsomedial portion of the DRN. Note the “U” or dissected cerebral aqueduct at the top of the image and the ventromedial DRN tissue below covering the electrodes (black dots). The scale bar (in white) is 100 μm.
(B) A representative waveform of a DRN serotonergic cell (yellow) and a non-serotonergic cell (green). This example waveform form was selected from one (Channel 14) of multiple channels on the multi-electrode array, is 53 μV, and spans a total of 3200 μsec with each grid representing 200 μs.
(C) A representative spike trace and raster plot during the initial recording along with the application of 8-OH-DPAT to induce 5HT1A inhibition. Note the firing rate of this representative cell was suppressed by more than 50% during 8-OH-DPAT application, criteria which we use to classify cells as a serotonergic. Also, in this diagram the recovery period when recording solution is added after 8-OH-DPAT is applied is not displayed, however this procedure is used in our methods and necessary to validate cell health and viability. This Figure has been previously published from Siemann et al., 2019. Note the cartoon in (A) was adapted from Multi Channel Systems, https://www.multichannelsystems.com/products/microelectrode-arrays/60mea50010ir-ti.
Expected outcomes
Outcomes from each electrophysiological recording method are depicted in each example experiment. Using whole-cell voltage-clamp configuration to assess the pharmacological effects of GABABR function on glutamatergic transmission in the NAc, our representative experiment shows that selective GABABR agonist, BAC, triggers a robust decrease in EPSC amplitude in MSNs, as described previously (Manz et al., 2019). Using current-clamp configuration to assess differences in excitability between and MSNs and FSIs, our data shows that FSIs reach a maximum AP frequency orders of magnitude greater than MSNs (Manz et al., 2020b). Finally, in cell-attached configuration, we show a representative trace of spontaneous APs in CINs, an electrophysiological feature absent in most neuronal subtypes in the NAc (Kawaguchi et al., 1995). We hope these outcomes demonstrate the broad and powerful functional utility of patch-clamp electrophysiology in assessing cellular and synaptic function.
The outcomes from multielectrode array recordings targeting the dorsal raphe nucleus is the observation, identification, and manipulation of putative DRN serotonin neurons (Figures 7 and 8). 5-HT neurons account for approximately 75%–95% of the neurons in ventromedial portion (Kirby et al., 2003), and about 84% of 5-HT neurons in this region in the mouse respond to 5-HT1A inhibition (Kiyasova et al., 2011). In addition, in this portion of the DRN, 5-HT neurons typically display a low frequency, regular firing rate (between 0.5 and 5 Hz, which averages around 1 Hz), and a broad spike waveform (Giannoni-Guzmán et al., 2021; Green et al., 2015; Siemann et al., 2019) (Figure 8). Thus, based on the known characteristics of the serotonin neurons in the ventromedial portion of the DRN, the high distribution of serotonin cells in this region, and the responses to 5-HT1A inhibition via 8-OH-DPAT (Figure 8), it is possible to identify and reliably record from multiple 5-HT neurons in the DRN. In addition, pharmacological agents can be used to create dose-response curves, and determine the response properties of multiple 5-HT neurons in the DRN with the use of multi-electrode array recordings.
Limitations
The greatest strength of whole-cell patch-clamp electrophysiology is the ability to control specific electrical parameters of a cell without grossly disturbing cellular homeostasis. However, key functional features of the cell, such as second messenger systems, molecular interactions, and ion channel activity, may be altered when the internal solution of the patch pipette dialyzes the cell. This can become problematic when examining mechanisms of plasticity that rely on highly regulated postsynaptic signaling cascades (Mathias et al., 1990; Winder and Sweatt, 2001). The highly compartmentalized ultrastructure of neuronal dendrites, including those lacking spinous subcompartments (e.g., NAc FSIs), can retain substantial biological function following intracellular dialysis (Malinow and Tsien, 1990; Malenka and Bear, 2004). In addition, the ex vivo slice preparation can sheer and dissociate long-range projections innervating a region. As a result, examining heterogenous synapses using electrical stimulation may differentially skew analysis towards inputs with more intact terminals (Ting et al., 2014). In some cases, this limitation can be overcome by shifting the slicing orientation to a different anatomical plane (e.g., sagittal to horizontal).
In line with limitations imposed by the ex vivo preparation, biophysical features of neurons may change in the absence of an intact neural network. For example, MSNs in the NAc and striatum oscillate between up- (−60 mV) and down (−80 mV) membrane states in vivo mediated by afferent glutamatergic input and voltage-sensitive cation channels (Kawaguchi et al., 1995; Mahon et al., 2006). However, this dynamic property of MSNs is absent ex vivo due to the functional decortication of top-down network activity during the slicing process. Finally, whole-cell analysis of neuromodulation and plasticity at a specific synapse may proceed through a complex polysynaptic network that goes undetected when recording from an individual cell. Although precise temporal resolution of these processes is difficult to assess using this technique alone, experiments can be conducted to address this concern, such as multi-cell recordings, pharmacological manipulations, and cell type-specific genetic, optogenetic, and chemogenetic techniques. Despite these limitations, patch-clamp techniques remain the gold standard approach to assessing cellular and synaptic function ex vivo.
One key advantage of multielectrode arrays is the ability to record from multiple neurons at once, which can inform the researcher on the cell types and response properties of neuronal populations within a specific brain region of interest. However, as with any ex vivo preparation, one main disadvantage is that these recordings can only occur in one specific brain region at a time, limiting questions surrounding circuit-level changes along with observing neural activity within an animal model in an in vivo context. 5-HT1A inhibition via 8-OH-DPAT allows us to identify 5-HT neurons pharmacologically. However, there are still a relatively small subset of DRN neurons in the ventromedial portion that can demonstrate characteristic waveforms of 5-HT cells (i.e., broad spike waveform, regular and low frequency firing rate), but do not reach our statistical threshold for identifying and counting a cell as serotonergic as the spike rate is not suppressed by 50% during the application of 8-OH-DPAT. This further indicates that while the majority of 5-HT neurons in this region demonstrate 5-HT1A inhibition (Kiyasova et al., 2011) not all 5-HT neurons will respond accordingly. In addition, the DRN can contain relatively heterogenous cell populations especially towards the lateral “wings” of the DRN, making it critical to record from a portion that is likely to contain large numbers of 5-HT cells such as the ventromedial portion (Kirby et al., 2003). Lastly, while we discuss multielectrode array recordings broadly and specifically in the context of the ventromedial portion of the DRN, it is important to note that the parameters needed to apply these methods to alternative brain regions, such as the suprachiasmatic nucleus, will have a variety of important methodological differences (Herzog et al., 1997; Kuhlman and McMahon, 2006). A key feature of these recordings is that they are stable over long periods enabling tracking of individual neuronal activity over a period of days to weeks.
Troubleshooting
Problem 1
Slice health remains poor despite proficiency in the dissection and slicing process (steps 1–8 or 9–15).
Potential solution
Several factors can contribute to poor slice health, including the ACSF and NMDG-based dissecting/recovery solutions, slicing temperature, and relative ion (Mg2+/Ca2+) balance. 10× stock ACSF and NMDG-based recovery solutions should be used within 1–2 weeks of being made. Older stock solutions (>2 weeks) may contain components that have crashed out of solution, resulting in an unpredictable electrolyte profile.
Make sure the instruments used to obtain each solution (e.g., analytical scale, osmometer, pH meter, etc.) are calibrated properly. If poor slice health persists, make sure the buffer tray and submerged holding chamber have reached the appropriate temperatures before slicing.
If these solutions fail to improve slice health, consider (a) an alternative dissection and recovery solution, such as a high sucrose-containing dissecting solution, (b) reducing the Ca2+/Mg2+ ratio to decrease intrinsic membrane excitability and excitotoxic cell death, and (c) performing transcardial perfusion of dissecting solution in deeply anesthetized mice (a preparatory procedure required for recordings in certain areas of the brain) (Fricker et al., 1999).
Problem 2
Recurring issues with Ra, GΩ seal formation, or cell longevity (steps 21–29).
Potential solution
A within-experiment change in access resistance (Ra) can be a frequent problem encountered during long-term whole-cell patch-clamp recordings. While it is typical for minor shifts (< 15%) to occur during the experiment, recurring issues with Ra often result from a steep osmotic gradient between the ACSF and internal solution. This gradient may be worsened by the incorporation of osmotically-active pharmacology into the perfusate.
A clue that this happening is if the cell size changes dramatically during the recording period and is accompanied by a change in Cm. To minimize this problem, remeasure the osmolarity of the ACSF and internal solution and make sure the difference is within 5 mOsm. If correcting the osmotic gradient is unsuccessful, the approach taken to obtain a successful patch may need to be adjusted to account for the rig’s optics, slice depth, and region of interest.
If a GΩ seal is unusually slow or difficult to obtain, the most likely culprit is an issue with one of the components of the internal solution. In general, Cs+-based and heavily Ca2+-chelated internal solutions can reach a GΩ seal more slowly than K+-based internal solutions.
If freshly prepared internal solution does not correct the problem, consider a different recipe or type of internal solution. Because the internal solution establishes the intracellular environment, avoid combining or pooling datasets using different internal solutions.
Similarly, difficulty holding cells for > 30-mins is often attributed to dialyzing the cell with a poorly calibrated (e.g., pH, ion concentration, etc.) or nucleotide-poor internal solution. Make sure that the ATP and GTP salts are incorporated into the internal solution on ice. Nucleotides may also be added to the internal solution fresh on the day of use to ensure that autohydrolysis has not occurred during storage.
Phosphocreatine, a regenerative energy source increasingly included in internal solution recipes, can be used to adjunct cellular energy demands (Baldi et al., 2016). If these solutions fail to improve cell longevity, considering switching to a K+-based internal solution that more closely resembles the physiological intracellular environment (Williams and Mitchell, 2008).
Problem 3
Electrical stimulation fails to reliably evoke synaptic responses in the recorded neuron (steps 30–35).
Potential solution
The maximum distance between the stimulating electrode and recoded neuron depends on circuit properties in the region of interest. Cells with a diffusely arborizing dendritic tree are more likely to observe synaptic currents at greater recording distances. However, to ensure that synaptic events elicited in the recorded neuron arise from a monosynaptic connection, we recommend standardizing the recording distance to 50–100 μm from the electrode. This will significantly improve synaptic efficacy and reduce the stimulation intensity required to evoke an EPSC or IPSC, allowing more sensitive electrophysiological analyses to be conducted (e.g., synaptically-evoked plasticity, train stimulation, gain operations, minimal stimulation assays, etc.).
If shortening the recording distance fails to improve synaptic responses, consider (a) trimming the existing electrode to eliminate occlusive cellular debris at the end of the electrode and/or (b) obtaining or creating a new electrode. Finally, inconsistent synaptic response (i.e., high failure rate) may occur in slices with poor or declining tissue health, as synaptic terminals, particularly dissociated long-range projections, exhibit early sensitivity to metabolic and mechanical stress (Bell and Dallas, 2018).
If poor slice health is responsible for failed synaptic responses, refer to the solution to problem 1 above.
Problem 4
Intracellular current injection fails to evoke an AP or results in premature depolarization block (steps 37–43)
Potential solution
A frequent contributor to poor AP fidelity when assessed using a K+-based internal solution in current-clamp configuration is a poor patch. A pronounced leak current at the patch pipette-cell membrane interface will significantly reduce the depolarizing response to positive intracellular current injection. To rule out this possibility, set the command current to 0 pA and measure VRMP upon entering whole-cell configuration. A VRMP value that is significantly more depolarized than established values for that cell type is indicative of a poor patch, even if a GΩ seal was obtained prior to breaking in. Alternatively, if a poor patch is present, voltage-clamping the cell at −70 mV (or a command voltage used in for voltage-clamp analyses in that region) will yield an Iholding value that is significantly more negative. Troubleshooting a poor patch is described in problem 2 above.
As the internal solution dialyzes the cell, VRMP will become increasingly hyperpolarized and AP probability will go down. Several factors contribute to this phenomenon, including a shift in voltage-sensitive ion channel kinetics and removal of intracellular effectors that support AP firing (Williams and Mitchell, 2008). Make sure the current ramp protocol is applied as soon as VRMP has stabilized.
If poor AP fidelity continues or depolarization block is present at subthreshold current steps, several modifications can be made to the step protocol. First, try changing the duration of the current step. Cells that exhibit latent firing patterns may require a longer current step duration (> 1-sec) to trigger AP firing.
Lengthen the inter-step interval to provide voltage-gated ion channels with greater time to recover from an inactivated, low conductance state (Dovzhenok and Kuznetsov, 2012). The kinetics and stoichiometric profile of ion channels mediating the AP dictate the on-off mechanics occurring between current steps.
Make sure that the ACSF perfusing the slice chamber is warmed using a closed-loop thermoregulatory system, as recordings performed at room temperature (20°C–23.5°C) may yield an artificially low AP frequency.
Problem 5
Mechanical or physical noise is disrupting multi-electrode array recordings (steps 45–56).
Potential solution
First, it is necessary that all metal and mechanical equipment has been grounded and the recording station is in a Faraday cage - the use of a floating table can help with these issues as well.
Check that either the inflow and/or the safety suction lines are attached to the rim of the multi-electrode recording chamber. If these are too far in the recording chamber and thus too close to the slice, they can cause physical noise.
Check to ensure periodically that the slice has not moved from its original position and the outflow has sufficiently pulled the slice down onto the electrodes. If this is an issue refer to problem 6 below for more detail.
If noise issues persist and the above solutions have been addressed, it is possible that the multi-electrode array contacts are dirty. It is necessary that the multi-electrode arrays are cleaned in a cold 5% Tergazyme solution for between 2 and 24 h, and then are washed with filtered water after each recording (see Multi-electrode array recording step 63 for more detail). In addition, the multi-electrode array contacts should be cleaned with a Q-tip and ethanol solution, and allowed to dry approximately 45 min before recordings begin (see the Note at the end of the Dissection section for multi-electrode array). Lastly, if these steps have been followed and there are still noise issues it is possible that the multi-electrode array itself needs to be replaced. Replacement can occur within 6 months to 1 year if recording is frequent (multiple times a week) and noise issues are becoming more prevalent.
Problem 6
Issues with the inflow and/or outflow lines creating either too much or too little solution within the multi-electrode array recording chamber (steps 47–48).
Potential solution
These issues can occur frequently, are due to multiple potential issues, and can result in noise during the recordings. First, it is important to check if there are any air bubbles in the lines. If there are bubbles in the outflow line it is important to gently push down on the rim of the multi-electrode array chamber to ensure a tighter seal occurs with the outflow line.
Also, there may be either too little or too much vacuum grease on the O-ring, which can create either air bubbles or block the outflow of the multi-electrode array. If there are still air bubbles, rub a small amount of vacuum grease under the base plate of the recording stage, as over time small holes can form. If low amounts of solution are being pulled through the outflow line, after the recording is completed, inspect and determine if too much vacuum grease was applied and as a result the outflow of the multi-electrode array and outflow line was blocked. Note, this is why it is critical to have a safety suction line as to remove any potential excess liquid that could overflow over the recording chamber and damage the amplifier. If this occurred, try to carefully remove any vacuum grease from the outflow of the multi-electrode array with Kimwipes, make sure to wash the array closer to the 24-h mark with Tergazyme, and use a compressed air duster to blow out any residual blockages from the baseplate and/or outflow line once the array is removed.
It is imperative to run water through the inflow and outflow lines before and after each recording to ensure that the lines are working properly and to not contaminate future experiments (see Multi-electrode array recording step 63). Also, use a compressed air duster to blow out any residual dirt that will accumulate over the course of multiple recordings.
Lastly, if there are still issues related to the inflow/outflow lines it is likely that the tubes have either become too dirty or are damaged after continuous use from recordings. It is important to check the lines weekly, particularly the portions connected to the peristaltic pump as these pieces tend to get damaged more frequently. If the inflow or outflow lines are replaced, it is critical to then properly calibrate the flow rates for the next recording.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Brad A. Grueter (brad.grueter@vumc.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate data or codes.
Acknowledgments
This study was supported by the National Institute on Drug Abuse (NIDA) grant R01DA040630 (to B.A.G).
Author contributions
K.M.M. and J.K.S. wrote the document. D.G.M. and B.A.G. reviewed and edited the document.
Declaration of interests
The authors declare no competing interests.
References
- Atwood B.K., Lovinger D.M., Mathur B.N. Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci. 2014;37:663–673. doi: 10.1016/j.tins.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi R., Ghosh D., Grueter B.A., Patel S. Electrophysiological measurement of cannabinoid-mediated synaptic modulation in acute mouse brain slices. Curr. Protoc. Neurosci. 2016;75:6 29 21–26 29 19. doi: 10.1002/cpns.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry P.H., Lynch J.W. Liquid junction potentials and small cell effects in patch-clamp analysis. J. Membr. Biol. 1991;121:101–117. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- Bell D.C., Dallas M.L. Using automated patch clamp electrophysiology platforms in pain-related ion channel research: insights from industry and academia. Br J Pharmacol. 2018;175:2312–2321. doi: 10.1111/bph.13916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Umschweif G., Leung J., Sagi Y., Greengard P. HCN2 channels in cholinergic interneurons of nucleus accumbens shell regulate depressive behaviors. Neuron. 2019;101:662–672.e5. doi: 10.1016/j.neuron.2018.12.018. [DOI] [PubMed] [Google Scholar]
- Dovzhenok A., Kuznetsov A.S. Exploring neuronal bistability at the depolarization block. PLoS One. 2012;7:e42811. doi: 10.1371/journal.pone.0042811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker D., Verheugen J.A., Miles R. Cell-attached measurements of the firing threshold of rat hippocampal neurones. J. Physiol. 1999;517(Pt 3):791–804. doi: 10.1111/j.1469-7793.1999.0791s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoni-Guzmán M.A., Kamitakahara A., Magalong V., Levitt P., McMahon D.G. Circadian photoperiod alters TREK-1 channel function and expression in dorsal raphe serotonergic neurons via melatonin receptor 1 signaling. J Pineal Res. 2021;70:e12705. doi: 10.1111/jpi.12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg J.A., Wilson C.J. Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J. Neurosci. 2005;25:10230–10238. doi: 10.1523/JNEUROSCI.2734-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green N.H., Jackson C.R., Iwamoto H., Tackenberg M.C., McMahon D.G. Photoperiod programs dorsal raphe serotonergic neurons and affective behaviors. Curr. Biol. 2015;25:1389–1394. doi: 10.1016/j.cub.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- He C., Chen F., Li B., Hu Z. Neurophysiology of HCN channels: from cellular functions to multiple regulations. Prog. Neurobiol. 2014;112:1–23. doi: 10.1016/j.pneurobio.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Herzog E.D., Geusz M.E., Khalsa S.B., Straume M., Block G.D. Circadian rhythms in mouse suprachiasmatic nucleus explants on multimicroelectrode plates. Brain Res. 1997;757:285–290. doi: 10.1016/s0006-8993(97)00337-5. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Wilson C.J., Augood S.J., Emson P.C. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kirby L.G., Pernar L., Valentino R.J., Beck S.G. Distinguishing characteristics of serotonin and non-serotonin-containing cells in the dorsal raphe nucleus: electrophysiological and immunohistochemical studies. Neuroscience. 2003;116:669–683. doi: 10.1016/s0306-4522(02)00584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyasova V., Fernandez S.P., Laine J., Stankovski L., Muzerelle A., Doly S., Gaspar P. A genetically defined morphologically and functionally unique subset of 5-HT neurons in the mouse raphe nuclei. J. Neurosci. 2011;31:2756–2768. doi: 10.1523/JNEUROSCI.4080-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress G.J., Dowling M.J., Meeks J.P., Mennerick S. High threshold, proximal initiation, and slow conduction velocity of action potentials in dentate granule neuron mossy fibers. J. Neurophysiol. 2008;100:281–291. doi: 10.1152/jn.90295.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman S.J., McMahon D.G. Encoding the ins and outs of circadian pacemaking. J. Biol. Rhythms. 2006;21:470–481. doi: 10.1177/0748730406294316. [DOI] [PubMed] [Google Scholar]
- Mahon S., Vautrelle N., Pezard L., Slaght S.J., Deniau J.M., Chouvet G., Charpier S. Distinct patterns of striatal medium spiny neuron activity during the natural sleep-wake cycle. J. Neurosci. 2006;26:12587–12595. doi: 10.1523/JNEUROSCI.3987-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka R.C., Bear M.F. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R., Tsien R.W. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Manz K.M., Baxley A.G., Zurawski Z., Hamm H.E., Grueter B.A. Heterosynaptic GABAB receptor function within feedforward microcircuits gates glutamatergic transmission in the nucleus accumbens core. J. Neurosci. 2019;39:9277–9293. doi: 10.1523/JNEUROSCI.1395-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz K.M., Becker J.C., Grueter C.A., Grueter B.A. Histamine H3 receptor function biases excitatory gain in the nucleus accumbens. Biol. Psychiatry. 2020;89:588–599. doi: 10.1016/j.biopsych.2020.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz K.M., Ghose D., Turner B.D., Taylor A., Becker J., Grueter C.A., Grueter B.A. Calcium-permeable AMPA receptors promote endocannabinoid signaling at parvalbumin interneuron synapses in the nucleus accumbens core. Cell Rep. 2020;32:107971. doi: 10.1016/j.celrep.2020.107971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias R.T., Cohen I.S., Oliva C. Limitations of the whole cell patch clamp technique in the control of intracellular concentrations. Biophys. J. 1990;58:759–770. doi: 10.1016/S0006-3495(90)82418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald M.J., Oorschot D.E., Schulz J.M., Lipski J., Reynolds J.N. IH current generates the afterhyperpolarisation following activation of subthreshold cortical synaptic inputs to striatal cholinergic interneurons. J. Physiol. 2009;587(Pt 24):5879–5897. doi: 10.1113/jphysiol.2009.177600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudder S.L., Baimel C., Macdonald E.E., Carter A.G. Hippocampal-evoked feedforward inhibition in the nucleus accumbens. J. Neurosci. 2018;38:9091–9104. doi: 10.1523/JNEUROSCI.1971-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L., Galaj E., Ma Y.Y. Nucleus accumbens shell small conductance potassium channels underlie adolescent ethanol exposure-induced anxiety. Neuropsychopharmacology. 2019;44:1886–1895. doi: 10.1038/s41386-019-0415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemann J.K., Green N.H., Reddy N., McMahon D.G. Sequential photoperiodic programing of serotonin neurons, signaling and behaviors during prenatal and postnatal development. Front. Neurosci. 2019;13:459. doi: 10.3389/fnins.2019.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q., Srinivas K.V., Sotayo A., Siegelbaum S.A. Dendritic Na(+) spikes enable cortical input to drive action potential output from hippocampal CA2 pyramidal neurons. Elife. 2014;3 doi: 10.7554/eLife.04551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper J.M., Koos T., Ibanez-Sandoval O., Tecuapetla F., Faust T.W., Assous M. Heterogeneity and diversity of striatal GABAergic interneurons: update 2018. Front. Neuroanat. 2018;12:91. doi: 10.3389/fnana.2018.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting J.T., Daigle T.L., Chen Q., Feng G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol. 2014;1183:221–242. doi: 10.1007/978-1-4939-1096-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura N., North R.A. Baclofen and adenosine inhibit synaptic potentials mediated by gamma-aminobutyric acid and glutamate release in rat nucleus accumbens. J Pharmacol Exp Ther. 1991;258:663–668. [PubMed] [Google Scholar]
- Williams S.R., Mitchell S.J. Direct measurement of somatic voltage clamp errors in central neurons. Nat. Neurosci. 2008;11:790–798. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]
- Winder D.G., Sweatt J.D. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat. Rev. Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate data or codes.