

Summary
This protocol provides a flow-cytometry-based procedure to classify and isolate all cells of the adult rodent subependymal zone (SEZ) neurogenic lineage, without the need for reporter mice, into different cell populations, including three neural stem cell (NSC) fractions with molecular signatures that are coherent with single-cell transcriptomics. Additionally, their cycling behavior can be assessed by means of 5-ethynyl-2′-deoxyuridine (EdU) incorporation. Our method allows the isolation of different NSC fractions and the functional assay of their cycling heterogeneity and quiescence-activation transitions.
For complete details on the use, execution, and outcomes of this protocol, please refer to Belenguer et al. (2021).
Subject areas: Cell Biology, Single Cell, Flow Cytometry/Mass Cytometry, Neuroscience, Stem Cells
Graphical Abstract
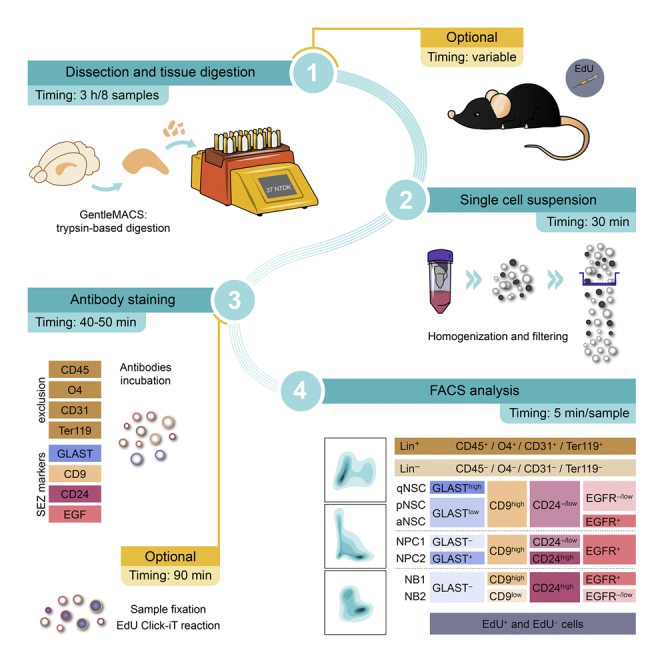
Highlights
-
•
The subependymal neurogenic lineage can be stratified with a set of surface markers
-
•
Cytometry-based classification and isolation of three distinct neural stem cell states
-
•
Nucleoside incorporation can be combined to score cycling dynamics and cell transitions
-
•
Tips for accurate dissection of the subependymal neurogenic niche
This protocol provides a flow-cytometry-based procedure to classify and isolate all cells of the adult rodent subependymal zone (SEZ) neurogenic lineage, without the need for reporter mice, into different cell populations, including three neural stem cell (NSC) fractions with molecular signatures that are coherent with single-cell transcriptomics. Additionally, their cycling behavior can be assessed by means of 5-ethynyl-2′-deoxyuridine (EdU) incorporation. Our method allows the isolation of different NSC fractions and the functional assay of their cycling heterogeneity and quiescence-activation transitions.
Before you begin
Prepare flow cytometry buffer
Timing: 15 min
| Reagent | Final concentration | Amount |
|---|---|---|
| HBSS w/o Ca and Mg (10×) | 1× | 20 mL |
| EDTA (20 mM) | 2 mM | 20 mL |
| HEPES (1 M) | 10 mM | 2 mL |
| Glucose (30%) | 0.1% | 0.67 mL |
| Bovine serum albumin (BSA) | 0.5% | 1 g |
| ddH2O | n/a | 157.33 mL |
| Total | 200 mL |
Filter, sterilize and store at 4°C for up to 1 week.
Prepare washing buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 (1:1) with L-Glutamine | - | 470 mL |
| Glucose (30%) | 0.6% | 10 mL |
| NaHCO3 (7.5%) | 0.1% | 7.5 mL |
| HEPES (1 M) | 5 mM | 2.5 mL |
| L-Glutamine (200 mM) | 2 mM | 5 mL |
| Antibiotic/Antimycotic (100×) | 1× | 5 mL |
| BSA | 4 mg/mL | 2 g |
| Total | 500 mL |
Filter, sterilize and store at 4°C for up to 1 week.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45-BUV421 (clone, rat 30-F11) | BD Biosciences | Cat#563890; RRID: AB_2651151 |
| O4-AF405 (clone, mouse O4) | R&D | Cat#FAB1326V |
| CD31-BUV421 (clone, rat 390) | BD Biosciences | Cat#563356; RRID: AB_2738154 |
| Ter119-BUV421 (clone, rat TER-119) | BD Biosciences | Cat#563998, RRID:AB_2738534 |
| GLAST-PE (clone, mouse ACSA-1) | Miltenyi | Cat#130-095-821; RRID: AB_10829184 |
| O4-Biotin (clone, mouse O4) | Miltenyi | Cat#130-095-895; RRID: AB_10828154 |
| CD24-PerCP-Cy5.5 (clone, rat M1/69) | BD Biosciences | Cat#562360; RRID: AB_11151895 |
| CD24-BB700 (clone, rat M1/69) | BD Biosciences | Cat#746122; RRID: AB_2743489 |
| CD9-Vio770 (clone, rat MZ3) | Miltenyi | Cat#130-102-384; RRID: AB_2659590 |
| CD133-Biotin (clone, rat 13A4) | eBioscience | Cat#13-1331-82; RRID: AB_466591 |
| Chemicals, peptides, and recombinant proteins | ||
| 5-Ethynyl-2′- deoxyuridine (EdU) | Life Technologies | Cat#E10187; CAS: 61135-33-9 |
| 4′, 6-diamidino-2′-phenylindole, dihydrochloride (DAPI) | Sigma | Cat#D9542; CAS: 28718-90-3 |
| BD Cytofix/CytopermTM | BD Biosciences | Cat#554722 |
| HBSS 10× | Biowest | Cat#X0507 |
| EDTA | Sigma | Cat#E6511; CAS: 10378-23-1 |
| HEPES 1M | Biowest | Cat#L0180; CAS: 7365-45-9 |
| D(+)-Glucose | PanReac | Cat#141341; CAS: 50-99-7 |
| BSA | Sigma | Cat#B4287; CAS: 9048-46-8 |
| DMEM/F12 | Gibco | Cat#11320-074 |
| Sodium bicarbonate | Sigma | Cat#S8761-500; CAS: 144-55-8 |
| L-Glutamine | Gibco | Cat#25030024; CAS: 56-85-9 |
| Antibiotic/Antimycotic (100×) | Gibco | Cat#15240062 |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Sigma | Cat#D1408 |
| Trypsin inhibitor | Sigma | Cat#T6522; CAS: 9035-81-8 |
| Epidermal growth factor (EGF)-Alexa FluorTM 488 | Molecular Probes | Cat#E13345 |
| Streptavidin-BV421 | BD Biosciences | Cat# 563259; RRID: AB_2869475 |
| Critical commercial assays | ||
| Neural Tissue Dissociation kit (T) | Miltenyi | Cat#130-093-231 |
| Click-iTTM Plus EdU Alexa FluorTM 647 Flow Cytometry Assay Kit | Thermo Fisher | Cat#C10634 |
| Dead cell removal kit (optional) | Miltenyi | Cat#130-090-101 |
| Myelin removal kit (optional) | Miltenyi | Cat#130-096-731 |
| Experimental models: organisms/strains | ||
| Mice C57BL/6J (male and female; 2–4 months old) | Jackson Labs. | Cat#000664;RRID: IMSR_JAX:000664 |
| Software and algorithms | ||
| FlowJoTM | Becton Dickinson | RRID:SCR_008520 |
| Deposited data | ||
| Data sets | Belenguer et al., 2021 | GSE138243 |
| Other | ||
| gentleMACSTM Octo Dissociator with Heaters | Miltenyi Biotec | Cat#130-042-109 |
| gentleMACS tubes | Miltenyi | Cat#130-096-334 |
| LSR-Fortessa Cytometera | Becton Dickinson | N/A |
| 15 mL centrifuge tubes | Corning | Cat#CLS430791 |
| Cell strainer (40 μm) | Labclinics | Cat# PLC93040 |
| Pasteur pipette | Labbox | Cat#PIPP-E03-1K0 |
| MS columns (optional) | Miltenyi Biotec | Cat#130-042-201 |
| LS columns (optional) | Miltenyi Biotec | Cat#130-042-401 |
| OctoMACSTM separator (optional) | Miltenyi Biotec | Cat#130-042-108 |
| MidiMACSTM separator (optional) | Thermo Fisher | Cat#130-042-302 |
| BD FACSAria IIIb sorter (optional) | Becton Dickinson | N/A |
LSR-Fortessa Cytometer: 355, 405, 488, 561 and 640 nm lasers
BD FACSAria III: 375, 405, 488, 561 and 640 nm lasers; 100 μm nozzle.
Step-by-step method details
There are two slightly different variants of this protocol. Both share the core procedure, but differ in a few critical steps, as well as in the type of analysis that they are intended for. Protocol A consists in the labeling and analysis of the SEZ lineage in non-fixated samples and can be employed for both quantitative analysis of the SEZ populations by flow cytometry or to isolate specific cell types by Fluorescent Activated Cell Sorting (FACS) for subsequent in vitro culture or molecular analysis. Protocol B includes the additional in vivo incorporation of the artificial thymidine analog EdU (5-ethynyl-2′-deoxyuridine) during the S-phase of the cell-cycle for assessment of the cycling dynamics of the identified populations. Because EdU detection requires the use of a fixative agent, this protocol is only suitable for quantitative analysis within populations. Since this protocol variant requires some exclusive extra steps, for clarification, they will be indicated as ‘only in Protocol B’ along the step-by-step method details.
Note: As indicated in the key resources table, this protocol has been set up using the gentleMACS Octo Dissociator (Miltenyi) which can homogenize up to 8 samples simultaneously. Accordingly, timing for each step of the protocol has been calculated assuming that 8 mice are processed. The procedure could be adapted to use other dissociation systems, but considerable differences in yield and population resolution might be expected.
EdU pulse and chase (only in protocol B)
Proliferation assessment in Protocol B is based on a classical pulse-chase experimental paradigm, adapted to work in combination with the cell-classifying analysis of SEZ populations. It relies on the incorporation of EdU by actively proliferating cells during a defined period of time (pulse) in which the nucleoside analog is administered intraperitoneally in a single or repeated doses that are followed by different chase regimes, depending on the experimental purpose and design. For instance, a single pulse plus 1-h chase will provide a proliferative labeling index for each cell population, whereas repeated pulses (usually 7, one every two hours, or 7×) followed by long chase times can be used to detect cells with low proliferative activity, commonly known as label-retaining cells (see, for example, Ferrón et al., 2007; Belenguer et al., 2021). The 7× pulse regime followed by a 12-h chase reveals transitions among NSC states (Belenguer et al., 2021).
-
1.
Prepare EdU at 10 mg/mL dissolved in sterile 0.9% saline solution.
-
2.
Inject EdU intraperitoneally at 50 mg/kg body weight (number and frequency of pulses can be varied). Use saline as vehicle for EdU-negative control samples.
-
3.
House injected mice in disposable or dedicated cages during the chase period and dispose of the soiled bedding as cytotoxic residues.
Dissection and tissue digestion
-
4.
Sacrifice mice according to current regulatory guidelines and institutionally approved protocols.
-
5.
Carefully extract the brains and keep them in ice-cold sterile 1× DPBS during the whole process in the appropriate plastic ware.
-
6.
Dissect the SEZs as previously described (Belenguer et al., 2016) and shown in Methods video S1. Keep them in ice-cold sterile 1× DPBS too.
CRITICAL: Carrying on excessive non-SEZ tissue during dissection will have a negative impact on the resolution of the analyses, so keep dissected pieces very thin, with minimal visible striatal fibers attached, and as homogeneous as possible between samples (Belenguer et al., 2016; Figures 1A–1D; Methods video S1). Dissection time has been calculated assuming an average-skilled experimenter.
Figure 1.

Example of the expected thickness of the dissected SEZ tissue
(A) Frontal view of the dissected piece (SEZ ventricle face).
(B) Rear view of the dissected piece (striatum face). Note that, although several striatal white fibers remain attached to the SEZ, they are scarce and it is possible to visualize areas of clean tissue between them.
(C) A partial lateral view showing the expected thickness of the tissue piece.
(D) The same frontal view as in A, showing that the dissected tissue should be thin enough to be translucent.
(E and F) Bright-field images of SEZ homogenates showing expected viable cell (orange arrowheads) to debris ratio in a suboptimal (E) vs. optimal (F) preparation.
Scale bars: A–D, 1 mm; E and F, 50 μm.
Note: In order to set the fluorescence threshold of CD9, the marker that separates non-neurogenic astrocytes from NSCs (see step 49g below), it will be necessary to include a tissue sample from a non-neurogenic region. We recommend processing a piece of cortex of similar size that can be easily obtained during the SEZ dissection (see Methods video S1).
-
7.
Mince each dissected tissue into 5–6 smaller pieces.
-
8.
Prepare C tubes (GentleMACS) and the enzymes included in the Neural Tissue Dissociation kit (T). The gentleMACS Octo Dissociator can homogenate up to 8 samples simultaneously. For each processed sample prepare:
MIX A: 1,750 μL buffer X + 200 μL enzyme T
MIX B: 20 μL buffer Y + 10 μL enzyme A.
Note: Check the provider protocol for further information:
https://www.miltenyibiotec.com/DE-en/shop/comMiltenyiDatasheet/product?productId=70
-
9.
Transfer SEZ pieces from both hemispheres to a C tube with a plastic Pasteur pipette. Remove all the DPBS with a p1000 micropipette and leave only the tissue.
Optional: In case of working with experimental conditions in which the total number of SEZ cells are expected to be low or if you are performing Protocol A to isolate high numbers of cells by FACS, it is possible to pool the SEZ from several mice and process them in the same C tube. According to the manufacturer’s instructions, as much as 1,600 mg of tissue can be digested in a single tube. We have successfully performed this protocol by pooling up the SEZs of three mice per tube at the most. As a reference to decide if your experiment requires to pool samples, take into account that we usually obtain around 100,000 live cells per mice that are further stratified in the different populations (see ‘Cytometry Analysis’ section below).
-
10.
Add to each C tube: 1,950 μL of MIX A and 30 μL of MIX B. Work as fast as possible and proceed immediately to the next steps to minimize the action of trypsin outside the automatic dissociator.
-
11.
Make sure to tighten the cap of each C tube and carefully turn them upside down. All pieces must fall onto the blades of the cap so check that none remains attached to the walls of the tube.
-
12.
Place the C tubes on the GentleMACS Octo Dissociator, place the heaters over each C tube and start preset program ‘37 NTDK’.
Single cell suspension
-
13.
Prepare one 15-mL collection tube per sample and place a 40 μm cell strainer on each one of them.
-
14.
Add 1 mL of washing buffer with a p1000 micropipette so as to leave a small drop at the center of each strainer (do not discard the flow-through).
-
15.
Prepare 3 mL/sample of washing buffer supplemented with 100 μg/mL trypsin inhibitor.
-
16.
Once the GentleMACS program is finished, remove the heaters and the C tubes. Turn the tubes upside down and make sure all the pieces fall to the bottom of the tube.
-
17.
Add 3 mL of trypsin inhibitor-supplemented washing buffer to each C tube. Close the tube and carefully turn it upside down to wash the walls and the blades.
-
18.
Centrifuge 1 min at 100 × g to pull down all the sample.
-
19.
Mechanically dissociate the remaining tissue chunks by pipetting up and down 20–30 times through a plastic Pasteur pipette (sample should appear cloudy but homogeneous). Avoid making bubbles and foam formation.
-
20.
Transfer the homogenate to the pre-wet cell strainer and collect the filtered solution in the 15-mL tube.
-
21.
Add 7 mL of washing buffer to each C tube rinsing it as in step 17 and pass it through the strainer. At this point, the volume of filtered sample should be of ≅ 11 mL.
Note: This is the best moment to split the samples in case different combinations of antibodies need to be applied to the same sample. Take into account that, as in any other FACS experiment, the necessary single-stained and fluorescence minus one (FMO) controls will be required during the analysis step. Therefore, reserve a small portion of the experimental samples to be used as controls. Usually 1/10 of a 2-SEZ homogenate is sufficient for a single-stained control, whereas we recommend 1/3 for FMO controls. Be aware that at least half of each sample must be left for experimental data acquisition. Therefore, we recommend making a pool with an aliquot of each sample, taking into account the indicated proportions, and then split it into the desired controls. Once the samples have been distributed into new 15 mL tubes, add washing buffer up to 7 mL and proceed to step 22. Alternatively, when processing less than 8 samples or when analyzing a strain/phenotype in which cell yield might be limiting, include an extra wild-type mouse to be used exclusively to generate all the necessary staining controls.
-
22.
Centrifuge collection tubes to sediment cells at 300 × g, 10 min.
Optional: During the initial setting up and optimization of this protocol, we used to include at this point an additional cleaning step with the Miltenyi’s Dead Cell Removal Kit, following the manufacturer's instructions. This procedure, that takes about 30 min, reduces the presence of dead cells and of cell and myelin debris in the sample, consequence of the enzymatic and mechanical disaggregation. The recognition and retention of dead cells and membrane fragments is performed by means of magnetic beads, so it requires a magnet (OctoMACSTM separator in key resources table) and suitable magnetic columns (MS columns in the key resources table), also provided by the kit manufacturer. As a result, initial FACS dot plots appear cleaner and the population of SEZ cells established by SSC-A vs. FSC-A gating (see step 49a below) is more clearly defined, but be aware that application of this procedure reduces the cell yield (up to 30% reduction) due to column retention and extra centrifugation steps. Therefore, we only recommend including this step in case you are not experienced at cytometry analysis or while optimizing the protocol. On the other hand, if you are performing Protocol A not just to obtain quantitative data but to isolate specific populations by FACS, it is critical to clean the sample. However, instead of using the Dead Cell Removal Kit, we recommended the use of Miltenyi’s Myelin Removal Kit, following the manufacturer’s instructions. Take into account that this procedure requires the use of a MidiMACSTM separator and LS columns (see ‘Other (optional)’ in the key resources table). Moreover, depending on the number of cells per population to be retrieved, it may be necessary to pool several mice per sample. In this case, myelin removal is absolutely necessary. Although the cell yield is similarly reduced, this step clearly improves the quality of the sorted samples.
Note: Check the provider protocol for the Dead Cell Removal and Myelin Removal Kits for further information:
https://www.miltenyibiotec.com/DE-en/shop/comMiltenyiDatasheet/product?productId=100
https://www.miltenyibiotec.com/ES-en/products/myelin-removal-beads-ii-human-mouse-rat.html#200-ul
Optional: If the experimental design requires that the total number of cells in the homogenate be estimated, remove supernatant and resuspend the pellet in washing buffer (approximately, 250 μL/SEZ). Count the number of viable cells with the method of your choice (see the expected viable cell to debris ratio in a suboptimal vs. optimal preparation in Figures 1E and 1F). As mentioned before, a homogenate of the two SEZs from a 2-month-old wild-type C57Bl6/J mouse, without any magnetic cleaning step, yields around 100,000 total live cells in the 500 μL. Before continuing with the protocol, add FACS buffer up to 6 mL and perform again step 22.
Antibody staining
-
23.
Prepare 100 μL per sample of FACS buffer with antibodies and other labeling reagents (see Table 1). The presence of BSA in the buffer increases viscosity and, hence, tip retention during pipetting, so consider adding 10–15 μL of excess per sample. Prepare all the required mixes for both samples and controls.
-
24.
Mix thoroughly but avoid foam formation.
-
25.
Once the sample is centrifuged, aspirate supernatant until 0.5–1 mL is left in the tube.
-
26.
Wait approximately 1 min to let the remaining liquid over the tube wall to slide down.
-
27.
Carefully remove all the remaining buffer without disturbing the pellet.
-
28.
Add 100 μL of antibody mix and resuspend the pellet by gently pipetting up and down. Make sure to bring back all cells to suspension, but avoid making bubbles.
-
29.
Incubate for 30 min on ice and in darkness.
-
30.
Add 2 mL of FACS buffer.
-
31.
Centrifuge at 300 × g, 10 min.
-
32.
Remove supernatant as in steps 25–27.
-
33.
If performing Protocol A, resuspend the pelleted cells in 0.5 mL of FACS buffer and proceed to analysis (steps 46–49). In the case of Protocol B, the sample requires fixation and Click-iT reaction for EdU visualization (steps 34–45) before the analysis.
Table1.
Preparation of the labeling mix
| Reagent | Label | Dilution | Protocol |
|---|---|---|---|
| anti-CD31 | BV421 | 1:100 | Both |
| anti-CD45 | BV421 | 1:100 | Both |
| anti-TER119 | BV421 | 1:200 | Both |
| anti-O4 | AF405 | 1:100 | Only Aa |
| anti-O4 | Biotinylated | 1:100 | Only Ba |
| Streptavidin | BV421 | 1:300 | Only Ba |
| EGF | AF488 | 1:300 | Both |
| anti-CD24 | PerCP- Cy5.5 | 1:300 | Only Aa |
| anti-CD24 | BB700 | 1:300 | Only Ba |
| anti-GLAST | PE | 1:100 | Both |
| anti-CD9 | APC-Vio770 | 1:20 | Both |
In protocol B, antibodies to detect O4 and CD24 have been substituted because the signal from AF405 and PerCEP-Cy5.5 fluorochromes is greatly quenched as a result of fixation. Instead, the combination of biotinylated anti-O4 with BV421-Streptavidin and BB700-conjugated anti-CD24 provides a strong fixative-resistant signal using the same laser/filter cytometer configuration.
Sample fixation and EdU click-it reaction (only in protocol B)
-
34.
Carefully vortex the pellet from step 32 to avoid aggregates during fixation.
-
35.
Add 100 μL of CytoFix/CytoPerm and resuspend cells in the fume hood.
-
36.
Incubate 20 min at 4°C in darkness.
-
37.
Add 2 mL of FACS buffer.
-
38.
Centrifuge at 300 × g, 10 min.
Pause point: At this point, supernatant can be removed and fixed cells can be resuspended in FACS buffer and stored at 4°C in darkness for several days if analysis is not performed immediately.
-
39.
Prepare the Click-iT reaction buffer following manufacturer instructions: Click-iTTM Plus EdU Alexa FluorTM 647 Flow Cytometry Assay Kit.
Note: Check the provider protocol for further information: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/mp10637.pdf
-
40.
Once centrifuged, remove supernatant as in steps 25–27. It is critical to dry all pellets similarly.
-
41.
Add 100 μL of Click-iTTM reaction buffer, resuspend cells, and incubate for 30 min at 18°C–22°C and in darkness.
-
42.
Add 2 mL of FACS buffer.
-
43.
Centrifuge at 300 × g for 10 min.
-
44.
Remove supernatant as in steps 25–27.
-
45.
Resuspend in 0.5 mL of FACS buffer and proceed to analysis.
Cytometry analysis
This procedure stratifies the cell suspension obtained from the SEZ tissue into the following populations, according to the expression of the selected markers as indicated in Figure 2A. Lin+ cells include CD45+ microglia and circulating leukocytes, CD31+ endothelial cells, Ter119+ erythrocytes, and O4+ oligodendrocytes, usually non-relevant to the analysis of the neurogenic lineage. Once Lin+ excluded, Lin‒ cells can be stratified into: (1) neuroblasts (NBs: GLAST‒/CD24high) that are further separated into: NB1 (EGFR+) and NB2 (EGFR‒/low); (2) neural progenitor cells (NPCs) comprised of two different subpopulations: NPC1 (GLAST‒/CD24‒/low) and NPC2 (GLAST+/CD24high), both EGFR+; (3) non-neurogenic astrocytes (GLAST+/CD24‒/low/CD9low), and (4) neural stem cells (NSCs: GLAST+/CD24‒/low/CD9high) which are further divided into quiescent (qNSCs: GLASThigh/EGFR‒/low), primed (pNSCs: GLASTlow/EGFR‒/low) and activated (aNSCs: GLASTlow/EGFR+).
Note: There is a small percentage of cells (around 3% of all SEZ cells) that appear negative for all the considered markers. RNA-seq profile of this population (“other”, see Belenguer et al., 2021) reveals that it most probably contains a heterogeneous mixture of different cell types: either unidentified cells that do not express the selected population-defining surface markers or low-expressing cells that fall under the positive threshold as a consequence of their antigens being affected by the enzymatic digestion.
Note: The whole set of appropriate controls for each of the fluorophores (single-stained and FMO) should be included for the setting up of this strategy.
-
46.
Mix the sample thoroughly and filter it through a 40 μm strainer.
-
47.
Add DAPI (4′, 6-diamidino-2′-phenylindole, dihydrochloride; 0.1 μg/mL) and mix briefly (only in Protocol A) for dead cell exclusion.
-
48.
Load the sample on the cytometer.
-
49.Stratify the populations as described in the following gating strategy:
-
a.Gate a region that excludes debris and most of the dead cells and erythrocytes on a SSC-A vs. FSC-A dot plot. Label the selected population as ‘SEZ cells’ (Figure 2B).
-
b.Gate on singlets using SCC-H and SCC-A (Figure 2B).
-
c.Discard dead cells by gating DAPI-negative cells (Figure 2B). This will define the ‘live cells’ population (only in Protocol A).
-
d.To set the Lin‒ population (Ter119‒, O4‒, CD31‒, and CD45‒), gate AF405 and BV421-negative cells (Figure 2B).
-
e.Divide Lin‒ cells into four quadrants according to GLAST and CD24 expression (Figure 2C).
-
f.Separate cells according to EGF binding (AF488) in the GLAST‒/CD24‒/low to obtain NPC1, in the GLAST+/CD24high quadrant to obtain NPC2, and in the GLAST‒ /CD24high quadrant to obtain EGFR+ NB1 and EGFR‒ NB2 (Figure 2D).
-
g.From the GLAST+/CD24‒/low population, gate CD9high cells to separate NSCs from non-neurogenic astrocytes (Figure 2E).
-
h.Gate on three distinct populations of NSCs on the basis of GLAST levels and EGF binding (Figures 2A and 2F).Optional (only for Protocol A): If your final purpose is to purify specific populations by FACS, perform the analysis (steps 48 and 49) in a properly equipped cell sorter (see key resource table) and adapt the sorting protocol according to your subsequent analysis (e.g., sort cells directly on the appropriate culture medium for in vitro analysis or on lysis buffer for RNA or protein extraction).Optional (only for Protocol B): To determine the EdU labeling index in each population, gate positive cells (AF647) according to an adequate FMO control from non-injected mice. This control sample must have been fixated and treated as the other samples (steps 34–45).
-
i.To obtain the percentage of each population relative to all ‘live cells’, make sure that you reverse calculate the percentage of the percentage for each of the gates that were required to define each population, until you get to the ‘live cells’ gate (Protocol A). When working with fixed cells (Protocol B), percentages can only be calculated relative to Lin- cells.
-
a.
Figure 2.

Comprehensive classification of subependymal cells
(A) Defined flow cytometry panel with the specific combination of markers used to identify the different SEZ populations.
(B) Initial steps to select Lin– cells after excluding cell aggregates and discarding CD45/O4/CD31/Ter119-positive and dead cells.
(C) Representative FACS plot showing GLAST and CD24 staining levels in the Lin– fraction and segregation of the cells into four categories.
(D) Based on the expression of the activation marker EGFR, the GLAST–CD24–/low region defines EGFR+ NPC1 (left), the GLAST+CD24high fraction corresponds to EGFR+ NPC2 (center) and the GLAST–CD24high region contains EGFR+ NB1 and EGFR– NB2 (right).
(E) Among the GLAST+CD24–/low fraction, CD9high levels distinguish between GLAST+ NSCs from non-neurogenic GLAST+ astrocytes. CD9 plot for cortex astrocytes is adapted from Belenguer et al., 2021.
(F) GLAST and EGFR levels define three NSC populations: qNSC (GLASThighEGFR–/low), pNSC (GLASTlowEGFR–/low) and aNSC (GLASTlowEGFR+).
(G) Estimated number of cells for each SEZ population considering an approximate total viable cell yield of 100,000 cells per brain.
(H and I) Representative FACS plots showing EdU staining in the Lin– fraction following two different pulse and chase regimes described in Belenguer et al., 2021: 1 shot of EdU followed by 1 h chase (H) and 7 injections of EdU (q2 h: one every 2 h) followed by a 12 h chase (I). aNSC and NB2 populations are included as examples for these two regimes.
Expected outcomes
Protocol A, for the cellular analysis of the SEZ populations of adult 2-month-old healthy wild-type C57B6/J mice, should yield more than 95% of live cells (DAPI-negative) after FSC-A vs. SSC-C gating and doublet exclusion (Figure 2B). Of all viable cells, around 25%–35% will be Lin+ cells (CD45+ microglia and circulating leukocytes, CD31+ endothelial cells, Ter119+ erythrocytes, and O4+ oligodendrocytes) that will be excluded from the analysis to leave Lin‒ cells for further stratification (Figure 2B). The classifying strategy (step 49) yields percentages of each population of interest relative to all live cells (calculated as indicated in 49i) that range approximately as follows: non-neurogenic astrocytes (2%–3%), NPC1 (2%–3%), NPC2 (2%–4%), NB1 (10%–12%), NB2 (40%–50%), and NSCs (10%–14%) distributed in qNSCs (4.5%–9%), pNSCs (1.8%–2.2%) and aNSCs (3%–4%). Note that the major source of variation is found in the population of qNSCs. After extensively performing this procedure, we have found that qNSCs are more strongly adhered to other niche components and, therefore, their efficient isolation is highly dependent on a disaggregation thorough enough to release them into suspension. Following a tissue homogenization as described in this step-by-step protocol should produce a yield of qNSCs close to 9%, whereas lower percentages might reflect suboptimal disaggregation. In any case, take these numbers as a generic baseline and note that they could vary depending on the dissection accuracy and homogenization (Figure 2G).
Protocol B, on the other hand, provides the relative number of EdU+ cells in each population. However, the percentages will be completely dependent on the number and frequency of the administered EdU doses as well as on the length of the chase period (steps 1–3). Although we cannot provide a generic single outcome for this protocol, examples of FACS plots for two different pulse-chase regimes are shown in Figures 2H and 2I. Quantitations of different EdU administration paradigms can be found in Belenguer et al., 2021.
A previously reported protocol to isolate subependymal NSCs was based on combining the use of mice expressing GFP under the control of the human GFAP promoter with the immunofluorescent detection of prominin 1 (PROM1; also known as CD133), a transmembrane glycoprotein that is present in the primary cilia of adult NSCs (Pastrana et al., 2009; Beckervordersandforth et al., 2010; Codega et al., 2014). Surprisingly, we found low percentages (around 4%) of GLAST+/GFP+ cells in GFAP::GFP reporter mice (Figure 3A). Furthermore, despite previous descriptions that most GFAP::GFP+ sorted cells are GLAST+ (Pastrana et al., 2009), we found many GFAP::GFP+ cells in the GLAST–/CD24– and GLAST–/CD24+ fractions of NPCs and NBs, respectively (Figure 3B), suggesting potential stability of the GFP reporter protein along the lineage. Accordingly, when we included in our panel the most extensively used anti-PROM1 antibody (rat anti-CD133 from eBioscience at 1:100; Codega et al., 2014), we found its signal highly associated to aNSC (whereas to a much lesser extent to pNSC and qNSC), but also present in virtually all NPCs and some EGFR+ NBs (Figure 3C). These results indicate that PROM1-based protocols may not properly distinguish between aNSCs and NPCs. Indeed, we found that, even after discarding the CD24+ cells from the GFAP::GFP+ population, GFAP::GFP+/PROM1+/EGFR+ cells, reportedly regarded as an activated NSC fraction (Codega et al., 2014; Dulken et al., 2017; Leeman et al., 2018), included around one third of GLAST– cells which have been defined as NPCs (Figure 3D; see also, Llorens-Bobadilla et al., 2015). Conversely, the GFAP::GFP–/EGFR+ fraction, previously considered to correspond to NPCs (Pastrana et al., 2009; Dulken et al., 2017), also contained GLAST+/PROM1+/EGFR+ cells, a profile compatible with an aNSC identity (Figure 3E). In line with this potential cross-contamination in the GFAP::GFP-based strategies, RNA-Seq analyses have indeed shown significant overlaps in the molecular profiles of aNSCs and NPCs (Dulken et al., 2017; Leeman et al., 2018).
Figure 3.

Comparison between GLAST and GFAP::GFP-based identification of SEZ NSCs and NPCs
(A) GFAP::GFP reporter expression and GLAST immunostaining of SEZ Lin– cells.
(B) GLAST and CD24 expression in the GFAP::GFP+ fraction.
(C) Quantification of PROM1+ cells present in the different SEZ cell types identified (n = 3).
(D) Gating strategy for aNSC and qNSC identification as in Codega et al. (Codega et al., 2014), and quantification of GLAST– GFAP::GFP+ aNSCs (n = 3).
(E) Gating strategy for NPC identification similar to Dulken et al. (Dulken et al., 2017), and distribution of GFAP::GFP– EGFR+ NPCs based on GLAST and PROM1 expression (n = 3).
(F) Representative FACS plot showing the GLAST and CD9 immunostaining of GFAP::GFP-only cells identified in panel D and distribution of GFAP::GFP+ PROM1– astrocytes based in GLAST and CD9 expression (n = 3).
Overlap between the molecular signatures of non-neurogenic astrocytes (GFAP::GFP+/PROM1–/EGFR–) and qNSCs (GFAP::GFP+/PROM1+/EGFR–) has also been reported (Dulken et al., 2017), a result that could be explained by the presence of PROM1– qNSCs in the niche astrocyte fraction, since only 52.3% of qNSCs were PROM1+ in our strategy (Figure 3C). In line with this, we found a number of GLAST+/CD9high NSCs in the population of GFAP::GFP+ cells that were negative for CD24, PROM1 and EGFR (labeled as ‘GFAP::GFP+ only’; Figures 3D and 3F). As a final note, the second PROM1 antibody used in the literature (the same rat anti-CD133 clone but supplied by BD Biosciences and used at 1:100; Beckervordersandforth et al., 2010) did not work in our hands and, therefore, we could not perform the same comparison. Although these data indicate that the molecular profiles published so far constitute very powerful tools to fine-tune sorting strategies, the use of Gfap-reporter mice and PROM1 may restrict the comprehensive analysis of cell heterogeneity in the SEZ niche.
Limitations
Our cell fractions have a significantly distinct transcriptomic profile and exhibit proliferation dynamics that are coherent with the panel marker strategy (Belenguer et al., 2021). However, in the absence of a transcriptomic post hoc validation, some sources of variability that can potentially lead to changes in the cellular landscape among different researchers must be kept in mind. We highly recommend a meticulously precise dissecting technique (see Belenguer et al., 2016 and Methods video S1). Other published dissecting procedures rely on the effect of neurosphere culture medium to select for neurosphere-forming cells (Walker and Kempermann, 2014) or on the power of single cell transcriptomics to uncover cell heterogeneity (Zywitza et al., 2018). However, our protocol aims at quantitating different SEZ populations and, therefore, only SEZ cells should go into the initial homogenate. We also recommend an automatized tissue homogenization method, even if the researchers are highly experienced in neurosphere cultures.
Our separation strategy allowed the identification of a set of NPCs (NPC2) that, despite being highly similar to previously reported progenitors, display high surface levels of GLAST and CD24 and enough differences in their transcriptome to be considered as a separate population. Determining whether these cells are a specific subtype of progenitor or reflect a transitional state along the neurogenic differentiation will require further analyses. Likewise, we found a group of cells that were negative for all the surface markers of the panel. Expression analysis of this ‘other’ population suggests a certain degree of heterogeneity (Belenguer et al., 2021), evidencing that SEZ cellular complexity might be even greater.
Aside from the potential existence of higher heterogeneity in our cell fractions, one must also keep in mind the potential effects of tissue dissociation on the cellular transcriptome (Van der Brink et al., 2017) and potential alternatives to be tested, such as cold-active proteases or transcription inhibitors.
Troubleshooting
Problem 1
Very few events appear in the initial SSC-A vs. FSC-A analysis dot plot (step 49a). This is probably due to excessive cell loss during the washing steps, especially when discarding the buffer after centrifugation.
Potential solution
Be more gentle and precise when aspirating the liquid from the centrifuged tubes: use fine-tip glass Pasteur pipettes, make sure you remove any bubbles present in the liquid surface before starting the aspiration and perform it in a single constant motion without touching the cell pellet (or the tube bottom in case the pellet is not visible).
Problem 2
When performing Protocol A, the percentage of live intact cells (DAPI-negative) in step 49c is low. Usually, after gating SEZ cells by SSC-A and FSC-A less than 5% of cells should appear stained with DAPI so >95% of live cells are expected.
Potential solution
Solution 1: Make sure you have properly placed the SSC-A vs. FSC-A gate in the initial dot plot (see Figure 2).
Solution 2: Working with live cells requires that sample handling and waiting times be reduced to the minimum. Perform the whole protocol (from mice sacrifice to analysis) in less than 5 h. Higher processing times will have a negative impact on cell viability.
Problem 3
Most cells are CD9low in step 49g in a wild-type mouse sample. This can happen with extremely poor dissection technique and excessive carrying over of striatal tissue.
Potential solution
Make sure to follow the dissection steps described in Belenguer et al. (2016) and to remove as much striatal tissue as possible during the initial SEZ dissection. Carve the reverse of the dissected piece until very few striatal white fibers are visible, without severing the SEZ surface (see Figure 1).
Problem 4
In Protocol B, CD24 staining appears very faint so the percentage of GLASTlow/CD24high NBs in step 49e is very low.
Potential solution
Make sure that you choose the proper combination of antibodies. Take into account that AF405-conjugated anti-O4 and PerCEP-Cy5-conjugated anti-CD24, recommended for Protocol A, are not compatible with the fixative agent used for detection of EdU in Protocol B. Use biotinylated anti-O4 combined with BV421-Streptavidin and BB700-anti-CD24 instead.
Problem 5
Percentage of qNSCs (GLASTlhigh/EGFR‒/low NSCs) in step 49 h is very low.
Potential solution
In our experience, qNSCs strongly adhere to other niche elements and are, therefore, more resistant to go into suspension after the tissue digestion. Make sure that you perform a thorough mechanical disaggregation of the remaining tissue chunks right after the GentleMACS enzymatic digestion, as indicated in step 19.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Isabel Fariñas (Isabel.farinas@uv.es).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The published article (Belenguer et al., 2021) includes all data sets [GEO under accession number: GSE138243] generated or analyzed during this study.
Acknowledgments
We thank the contribution of the Servicio Central de Soporte a la Investigación Experimental (SCSIE; Universidad de Valencia). This work was supported by Spanish grants from FEDER/Ministerio de Ciencia e Innovación (MICI)–Agencia Estatal de Investigación (SAF2017-86690-R), Instituto de Salud Carlos III (CIBERNED CB06/05/0086 and RETIC Tercel RD16/0011/0017), Fundación Botín-Banco Santander, and Generalitat Valenciana (Prometeo 2017/030) to I.F. G.B. and A.D.-M. have been recipients of FPU predoctoral fellowships from the Spanish Ministerio de Educación. P.D.-A. is recipient of an FPI-MICI predoctoral fellowship.
Author contributions
Conceptualization, G.B., A.D.-M., P.D.-A., J.M.M.-R., and I.F.; methodology, G.B. and J.M.M.-R.; investigation, G.B., P.D.-A., A.D.-M., and J.M.M.-R.; writing – original draft, A.D.-M., P.D.-A., and J.M.M.-R.; writing – review & editing, G.B., A.D.-M., P.D.-A., J.M.M.-R., and I.F.; visualization, A.D.-M., P.D.-A., and J.M.M.-R.; supervision, J.M.M.-R., and I.F.; project administration, I.F., J.M.M.-R.; funding acquisition: I.F.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100425.
Contributor Information
Jose Manuel Morante-Redolat, Email: jm.morante@uv.es.
Isabel Fariñas, Email: isabel.farinas@uv.es.
References
- Beckervordersandforth R., Tripathi P., Ninkovic J., Bayam E., Lepier A., Stempfhuber B., Kirchhoff F., Hirrlinger J., Haslinger A., Lie D.C. In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell. 2010;7:744–758. doi: 10.1016/j.stem.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Belenguer G., Domingo-Muelas A., Ferrón S.R., Morante-Redolat J.M., Farinas I. Isolation, culture and analysis of adult subependymal neural stem cells. Differentiation. 2016;91:28–41. doi: 10.1016/j.diff.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Belenguer G., Duart-Abadia P., Jordán-Pla A., Domingo-Muelas A., Blasco-Chamarro L., Ferrón S.R., Morante-Redolat J.M., Fariñas I. Neural stem cells are alerted by systemic inflammation through TNF-α receptor signaling. Cell Stem Cell. 2021;28:285–299.e9. doi: 10.1016/j.stem.2020.10.016. [DOI] [PubMed] [Google Scholar]
- Codega P., Silva-Vargas V., Paul A., Maldonado-Soto A.R., Deleo A.M., Pastrana E., Doetsch F. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron. 2014;82:545–559. doi: 10.1016/j.neuron.2014.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulken B.W., Leeman D.S., Boutet S.C., Hebestreit K., Brunet A. Single-cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Rep. 2017;18:777–790. doi: 10.1016/j.celrep.2016.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrón S.R., Andreu-Agulló C., Sánchez P., Mira H., Marqués-Torrejón M.A., Fariñas I. Ex vivo and in vivo assays to detect effects of exogenously added factors in the behavior of adult neural stem cells. Nat. Protoc. 2007;2:849–859. doi: 10.1038/nprot.2007.104. [DOI] [PubMed] [Google Scholar]
- Leeman D.S., Hebestreit K., Ruetz T., Webb A.E., McKay A., Pollina E.A., Dulken B.W., Zhao X., Yeo R.W., Ho T.T. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science. 2018;359:1277–1984. doi: 10.1126/science.aag3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Bobadilla E., Zhao S., Baser A., Saiz-Castro G., Zwadlo K., Martin-Villalba A. Single-cell transcriptomics reveals a population of dormant neural stem cells that become activated upon brain injury. Cell Stem Cell. 2015;17:329–340. doi: 10.1016/j.stem.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Pastrana E., Cheng L.C., Doetsch F. Simultaneous prospective purification of adult subventricular zone neural stem cells and their progeny. Proc. Natl. Acad. Sci. U S A. 2009;106:6387–6392. doi: 10.1073/pnas.0810407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink S.C., Sage F., Vértesy Á., Spanjaard B., Peterson-Maduro J., Baron C.S., Robin C., van Oudenaarden A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods. 2017;14:935–936. doi: 10.1038/nmeth.4437. [DOI] [PubMed] [Google Scholar]
- Walker T.L., Kempermann G. One mouse, two cultures: isolation and culture of adult neural stem cells from the two neurogenic zones of individual mice. J. Vis. Exp. 2014;84:e51225. doi: 10.3791/51225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zywitza V., Misios A., Bunatyan L., Willnow T.E., Rajewsky N. Single-cell transcriptomics characterizes cell types in the subventricular zone and uncovers molecular defects impairing adult neurogenesis. Cell Rep. 2018;25:2457–2469 e2458. doi: 10.1016/j.celrep.2018.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article (Belenguer et al., 2021) includes all data sets [GEO under accession number: GSE138243] generated or analyzed during this study.