

Abstract
Extracellular vesicles (EV) are lipid-bilayer enclosed vesicles in submicron size that are released from cells. A variety of molecules, including proteins, DNA fragments, RNAs, lipids, and metabolites can be selectively encapsulated into EVs and delivered to nearby and distant recipient cells. In tumors, through such intercellular communication, EVs can regulate initiation, growth, metastasis and invasion of tumors. Recent studies have found that EVs exhibit specific expression patterns which mimic the parental cell, providing a fingerprint for early cancer diagnosis and prognosis as well as monitoring responses to treatment. Accordingly, various EV isolation and detection technologies have been developed for research and diagnostic purposes. Moreover, natural and engineered EVs have also been used as drug delivery nanocarriers, cancer vaccines, cell surface modulators, therapeutic agents and therapeutic targets. Overall, EVs are under intense investigation as they hold promise for pathophysiological and translational discoveries. This comprehensive review examines the latest EV research trends over the last five years, encompassing their roles in cancer pathophysiology, diagnostics and therapeutics. This review aims to examine the full spectrum of tumor-EV studies and provide a comprehensive foundation to enhance the field. The topics which are discussed and scrutinized in this review encompass isolation techniques and how these issues need to be overcome for EV-based diagnostics, EVs and their roles in cancer biology, biomarkers for diagnosis and monitoring, EVs as vaccines, therapeutic targets, and EVs as drug delivery systems. We will also examine the challenges involved in EV research and promote a framework for catalyzing scientific discovery and innovation for tumor-EV-focused research.
Graphical abstract
Highlights
-
•
The full spectrum of small extracellular vesicles (sEV) based cancer fundamental and clinical studies is introduced.
-
•
It is one of very few comprehensive reviews that systematically introduce/summarize sEV in cancer research.
-
•
The reviewed topics will advance the frontiers of science, highlight challenges, and support higher education.
1. Introduction
Extracellular vesicles (EVs) were first observed in the 1980s as secretory vesicles released by reticulocytes and were thought to be a means of disposing cellular waste [[1], [2], [3]]. EVs have since become a focus for research and it is now well-established and accepted that EVs play crucial roles in cell-to-cell communication, contributing to various pathological conditions including heart disease, neurodegenerative diseases, mental disorders, and cancer [[4], [5], [6], [7], [8], [9], [10]].
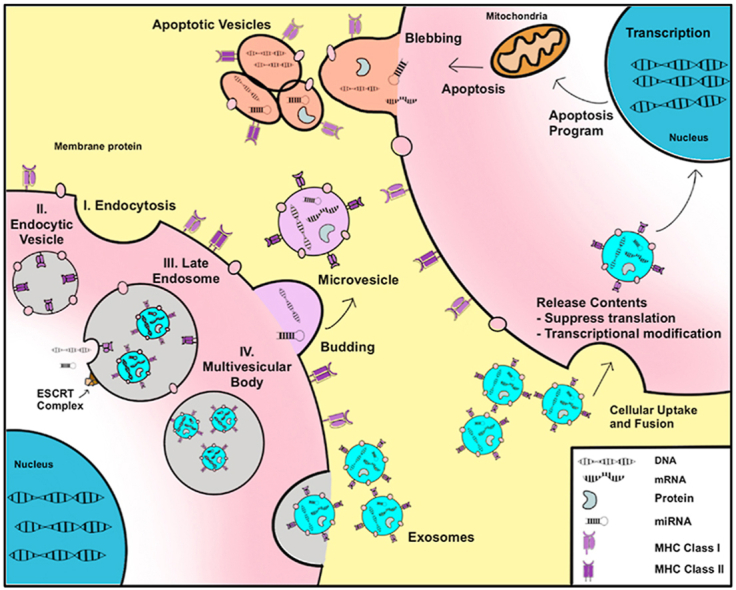
EVs are lipid-bilayer enclosed vesicles that are released by virtually every cell into extracellular space and are detectable in all somatic fluids, including blood, urine, saliva, and cerebrospinal fluid [[11], [12], [13], [14], [15]]. Based on modes of biogenesis and size distribution, EVs are classified into three subtypes: exosomes, microvesicles or micropaticles, and apoptotic bodies and form by their own specific mechanisms (Fig. 1). Among them, apoptotic bodies are the largest EVs ranging from 1 to 5 μm in diameter. They are directly generated by blebbing from apoptotic cells [16]. Microvesicles or microparticles, on the other hand, ranges from 100 to 1000 nm in size. They are formed directly from outward budding of the cell membrane [16]. The smallest EVs, termed exosomes, are 30–120 nm particles and are generated by intraluminal buds fusing with the cell membranes. Initially, the cell membrane invaginates to form the early endosomes endosomes which comprise the multivesicular body (MVB). The endosomal sorting complexes required for transport (ESCRT) complex aids in the sorting and packaging of various cargoes into these endosomes. The MVB will then fuse with either lysosomes for degradation or with the cell membrane where the endosomes are released into extracellular space, becoming exosomes [16,17]. Of note, the exact biogenesis of exosomes and microvesicles is not fully understood. To further confound classification, exosomes and microvesicles exhibit a partial overlap in size and densities and share similar markers. Because current isolation techniques are incapable of separating these two EVs with a high degree of purity, we will refer to them collectively as small EVs (sEVs) which are conventionally precipitated at high speeds (over 100,000 g fraction). sEVs contain tissue-specific signatures wherein a variety of lipids, proteins, RNAs, and DNA fragments are selectively inherited from parental cells and packaged [18]. These molecules can be transferred between local or distant cells through circulation and exhibit physiological and pathological regulatory functions [19,20]. Substantial research has examined the effects of the various EV contents and their roles in tumorigenesis and metastasis [21]. For example, miR-27a, miR130a, and miR-7641 have exhibited increased expression in sEVs isolated from colorectal cancer (CRC) patient plasma as well as CRC cells [22,23]. Likewise, sEV long non-coding RNA (lncRNA) species exhibit altered expression in many tumors. For example, metastasis associated lung adenocarcinoma transcript 1 (MALAT-1) is increased in sEVs. Growth arrest specific 5 (GAS5) exhibits a decrease in sEVs derived from non-small cell lung cancer (NSCLC) patients, and is negatively correlated with NSCLC staging [24]. sEV lncRNA HOX Transcript Antisense Intergenic RNA (HOTAIR) have been shown to instigate epithelial to mesenchymal transition (EMT) in multiple tumors including bladder cancer and gliomas [25,26]. Similarly, DNA species, including mitochondrial DNA (mtDNA), single-stranded DNA (ssDNA), and double-stranded DNA (dsDNA), have also been discovered in sEVs, and can potentially engage in horizontal transfer of the gene fragment to recipient cells, thereby contributing to the pre-metastatic niche and promoting metastasis [23,[27], [28], [29]]. In addition, sEV can induce epigenetic changes by modulating methylation of the genome in recipient cells [[30], [31], [32]]. For example, osteosarcoma derived EVs mediate LINE1 hypomethylated DNA from osteosarcoma to recipient cells causing epigenetic changes [33]. Also, typical protein markers of sEVs, such as CD9, CD81, CD63, and TSG101aid in the identification and capture of EV populations which are further characterized by tumor-specific markers such as Glypican-1 (GPC1) for pancreatic ductal adenocarcinoma [34], EpCAM for a general tumor marker [35], and HER2 for breast cancer, to name a few [36]. On the other hand, sEVs resemble their parental cells, like fingerprints, often displaying cellular constituents which derive from the parent cell [37,38]. This has contributed to their growing role as tumor biomarkers for diagnosis, prognosis, and monitoring [[39], [40], [41]]. In general, precise characterization and profiling of these internal genomic, transcriptomic, proteomic, and metabolomic molecules in sEVs would deepen understanding of intercellular communications in cancers.
Fig. 1.

Biogenesis of extracellular vesicles. Extracellular vesicles form by mechanisms specific to the type of EV. (A) Exosomes form through a complex process that utilizes the ESCRT complex. (I) Exosomes first form by endocytosis, forming the (II) early endocytic vesicle also known as the endosome. (III) Upon construction of the ESCRT complex, the endosome invaginates to form the late endosome which incorporates endocytic vesicles. (IV) This forms the multivesicular body which fuses wit h the plasma membrane and releases the exosomes into extracellular space. Exosomes can also form independent of ESCRT complexes though this mechanism is not well understood. Macrovesicles, by contrast, form as an outward budding of the plasma membrane. (B) Apoptotic bodies form as a result of blebbing during apoptosis. All EVs harbor DNA, various RNA species, proteins, and other cellular constituents which are packaged into the vesicles. These vesicles can be taken up by neighboring or distant cells. When this happens, the contents of the vesicles are released into the new cell and can enact various biological and metabolic functions in the new cell.
To promote and streamline sEV-related studies, there are minimum guidelines outlined by the International Society for Extracellular Vesicles [42]. Additionally, the following databases are available to promote further research: Vesiclepedia, ExoCarta, EVpedia, and EVmiRNA [[43], [44], [45], [46]]. Vesiclepedia (http://www.microvesicles.org/) is a web-based compendium of RNA, proteins, lipids, and metabolites derived from general EVs, which covers data from two origins, one submitted by EV researchers, the other from the data manually curated from published articles. To date, Vesiclepedia reports data from 1254 studies and has catalogued 349,988 proteins, 27,646 mRNA entries, 10,520 miRNAs, and 639 lipids [47]. This data is readily available for download and encourages other researchers to contribute their findings, ensuring annotations are up to date. A concern, however, is that the data incorporates all EV studies and does not focus on EV subtypes, despite this, the database is more comprehensive than other databases. Next, EXOcarta (http://www.exocarta.org/credits) is another web-based compendium which merely covers the exosomal genetic ingredients [48]. As of the most recent update to the database, there are 41,860 proteins, more than 7540 RNAs, and 116 lipids from 286 exosome-focused studies have been hosted in this database [49]. EVpedia (https://omictools.com/evpedia-tool) is another public database integrating vesicular molecules for sEVs research. The information is based off 2879 publications incorporating 172,080 vesicular molecules from 163 high-throughput datasets and has been visited more than 65,000 times since 2015, indicating its huge potential as a repository for EV study [45]. In addition to thesegeneral repositories which encompass all EVs and their components, some databases exist which only include specific components found in EVs. For example, the first database to focus on profiling miRNA in EVs is the EVmiRNA database (http://bioinfo.life.hust.edu.cn/EVmiRNA), whichexclusively provides detailed miRNA expression profiles in EVs and the related information of sample sources. Since 2019, 462 small RNA sequencing samples of EVs coming from 17 sources and diseases were manually curated in this database [46]. These databases provide user-friendly interfaces for easy and rapid download to facilitate EV-related research and applications, including biomarker identification for liquid biopsy and function recognition of internal genetic cargos. While many databases have been compiled to facilitate EV research, no consensus has been reached for the isolation and purification of pure EV subpopulations, thus researchers need to use these databases with caution. The difficulty in isolating pure EV samples continues to impede further developments in this field. Much of the research already mentioned utilizes different methods for isolating and purifying EVs, resulting in concern regarding reliability and validity of the data utilized to generate these EV compendiums. Low-purity isolation of sEVs with co-isolated contaminants or incomplete isolation from samples still needs to be tackled. Only the prerequisite is guaranteed: that EVs and their contents were isolated. However, without accurate characterization, it becomes difficult to accurately state that these observations were derived solely from one type of EV.
Reviews on specific topics including EV biogenesis, stem cells, tissue engineering, and a variety of diseases are available elsewhere [[50], [51], [52], [53], [54]]. Therefore, in this review we present a comprehensive and updated review with a specific focus in the cancer field. It covers the popular isolation techniques for sEVs and the state-of-the-art methods which have been reported over that last 3–5 years, especially as they pertain to cancer. It also reviews the roles of EVs in cancer pathology, diagnosis, prognosis, and drug resistance as well as their potential as drug delivery vehicles, vaccines, and therapeutic targets. Finally, we discuss the challenges and future perspectives in the sEV field, including shortcomings of current isolation and analysis approaches, the difficulties in relevant clinical translations of sEV-based diagnosis and therapeutics, and their potential roles in cancer research in the future.
2. Isolation of sEVs
For sEVs to even be considered for diagnostic use, an ideal and standardized isolation and analysis approach must address the following concerns: (1) Rapidity. sEV isolation can range from several minutes to more than 24 h. An ideal EV isolation method for clinical diagnosis should decrease the amount of time needed to isolate EVs to under an hour. (2) Isolation and retrieval efficiency. Current methods vary widely in their yield, ranging anywhere from 1.5% to nearly 100%. An optimal method should approach as close to 100% EV isolation and collection as possible. (3) Purity. Along with rapid isolation and maximum yield, a high degree of purity is also necessary. This is particularly important when considering EV isolation from complex body fluids such as blood. The current methods range from 20% to over 90%, and thus an ideal method should minimize protein and free nucleic acid contaminants. (4) Flexibility. Isolation of EVs should also integrate with downstream analyses in order to accomplish a successful diagnosis. (5) Affordability. The ideal assay or device must be capable of industry-scale production in order to decrease cost. (6) Reliability. For clinical translation, reliability of a diagnostic is imperative. An ideal method must offer consistency in the aforementioned metrics to ensure an accurate diagnosis. (7) Throughput. Toward clinical translation, it must be able to handle high volumes of sample and process multiple samples, as necessary. Lastly, (8) Ease of use and automation. The ideal assay or device must be easy to operate with minimal training. It should be one-step operation wherein the raw sample is added and the output is pure EVs for integrating into additional devices for further analysis.
While new EV isolation methods are strongly desired, issues arise when considering EV subpopulations, especially if a particular subtype is more ideal for diagnostic purposes. EVs are a diverse group which share many similarities between subtypes including density (1.1–1.2 g/ml), size (30–200 nm), solubility, charge (negative), and surface proteins (CD9, CD63, CD81, TSG101), making isolation of pure subtypes difficult to achieve. Current isolation methods of EVs fall under the following categories: density, size, solubility, charge, immunoaffinity, and lipid self-assembly. Each of these categories contains specific isolation methods discussed in this section. We will also examine the pros and cons of each method (Table 1). Overall, the greatest challenge facing clinical use of EVs is a lack of consensus regarding optimal isolation methods, assays, or devices for isolation of sEVs [55].
Table 1.
Existing techniques for sEVs isolations and their respective advantages and disadvantages.
| Technique | Advantages | Disadvantages | Volume | Time | |
|---|---|---|---|---|---|
| Density | |||||
| Ultracentrifugation [56,61,[64], [65], [66], [67], [68]] | Most widely used method; Minimal reagents and expertise required; Ability to process large volumes; Absence of chemicals which could damage EV integrity | Time and labor intensive; Comparatively low throughput; Heavy protein contamination; Requires expensive equipment with the potential for cross-contamination when the same tubes are reused; Loss and damage of EVs due to repetitive centrifugation; Sample type, rotors, and gravitational force result in varying isolation efficiencies and amount of time required to isolate EVs; Difficulty resuspending large pellets without use of mechanical force or chemicals which can degrade the EVs; Difficult to ensure sterility | Variable | ≥2 h | |
| Density Gradient Ultracentrifugation [59,61,62,[68], [69], [70], [71]] | Highest purity, considered gold standard; Sort EVs by density, allowing for isolation of specific subpopulations; Iodixanol and sucrose most common for generating gradients—few chemicals therefore required; | Time and labor intensive; Requires expensive equipment; Extremely low recovery efficiency; Low throughput; Recovery dependent on type of fluid from which exosomes are being harvested, rotors, and gravitational force as well as percentage of sucrose and iodixanol concentrations; Viral particle contamination possible if sucrose gradient is used; Larger particles with similar density cannot be isolated with purity; Iodixanol is toxic | 200 μl | 16–22 h | |
| Acoustofluidics [68,72] | Rapid; Easily integrated into an automatic system; Requires minimal sample; Maintains EV integrity; Scalable | High protein contamination | 20 μl | 7–30 min | |
| Solubility | |||||
| Precipitation [61,68,71,[73], [74], [75]] | Low labor required; Commercially available kits; Specific for EV solubility; Minimal pre-treatment required; High yield; Preserves EV integrity | Commercial kits can be costly; Co-precipitates guaranteed; Aggregation which can interfere with downstream analyses; Lowest purity, often containing protein and nucleic acid contaminants, especially when processing plasma or serum samples; Additional steps required to remove PEG | Variable | 2–24 h | |
| Size | |||||
| Ultrafiltration [61,[76], [77], [78]] | Highly adaptable and versatile: Easily integrated into microfluidic devices or with other EV isolation methods; Enables isolation of specific EV sizes; Can be multi-step to enhance purity; Rapid isolation; Minimal chemical and training requirements; Potential for clinical setting; | Risk of pore clogging which can damage EVs as well as result in filter burst, introducing contaminants; Potential for deformation of EV structure if wrong filter sizes are used; May not be as effective at generating pure samples as UC; Low throughput/cannot process large volumes | 10 μl-150 ml | <1 h | |
| Size Exclusion Chromatography [61,[79], [80], [81], [82]] | Rapid; Integrity of EVs maintained; Simple with minimal equipment; Commercial kits available; Can integrate with other methods; Scalable; Repeatable; High purity; Efficient | Co-isolation of similarly sized proteins; Fairly low yield; Fraction is diluted and may require concentration for downstream applications | 200 μl-20 ml | <1 h | |
| Immunoaffinity | |||||
| Antibody [61,[83], [84], [85]] | Specific to EV subpopulations; Rapid; Can be combined with other methods to enhance purity; Provides very pure samples | Comparatively low yield; Issues arise when downstream analyses require intact vesicles as it is difficult to harvest EVs from antibodies; Capture efficiency is altered by sample media; Can be costly | 10 μl–10 ml | ~1 h | |
| Aptamer [[86], [87], [88], [89]] | Very specific for target; Minimal nonspecific binding; Easily separate from target; Scalable; Inexpensive; High purity | Growing field with comparatively fewer publications; Variable yield dependent on aptamer used | 5 μl–2 ml | ~2 h | |
| Lipid Self Assembly | |||||
| Lipid Nanoprobes [[90], [91], [92]] | Rapid isolation; Easily modified to enhance downstream analysis; High yield; Easy to use; Does not require bulky or expensive equipment | Relatively new; Low purity; Expensive reagents; Strict storage methods to maintain integrity of reagents | <1 ml | ~15 min | |
| Charge | |||||
| Dielectrophoresis [[93], [94], [95]] | Rapid; Requires minimal sample and reagents; Cost-effective; Can separate specific EV subpopulations; Good purity | Can potentially damage EV membranes; Low yield | 25–200 μl | 20–90 min | |
| Ion Exchange Chromatography/Electrostatic interaction [[96], [97], [98], [99]] | High purity; Able to isolate EVs from complex body fluids; Adaptable method | Cannot isolate specific sub-populations of EVs; Non-specific binding | Variable | 40 min–3 h | |
2.1. Density-based sEV isolation
2.1.1. Differential ultracentrifugation
Differential UC (UC) is the most prevalent method utilizing size and density for isolation of sEVs. UCt has been widely used in both fundamental and clinical studies over the last decades. Various UC protocols exist forseparatig sEVs from cell culture supernatant and body fluids [56]. Initial low-speed centrifugation (400×g) is used to deplete intact cells, followed by 1000–20,000×g to remove cellular debris and medium/large vesicles. Then, high speed (100,000–150,000×g) at 4 °C for at least 2 h is used to precipitate sEVs [57,58]. Subsequently, the pellet of sEVs are resuspended with PBS for direct characterization or other molecular analyses. Alternatively, resuspended sEVs can be stored at −80 °C or in liquid nitrogen indefinitely before further actions [59]. UC can process multiple samples with minimal expertise required while providing a moderate yield without the usage of damaging chemicals [60,61]. However, UC still has disadvantages such as lengthy procedures which offer inconsistent recovery efficiency, protein contamination, low throughput, and the necessity for expensive equipment. Of note, the inconsistent recovery efficiency results in an isolation efficiency of 10–70% and is influenced by external parameters including g force, rotor type (e.g., fixed angle or swinging bucket), sample viscosity, and sedimentation distance However, one study found that incorporating a single 60% iodixanol cushion to generate a modest gradient increased sEV yield from 30% to 70% and protein contamination from 0.3% to 6% [62]. Thus, sEV yield can be improved with slight modifications though at risk for increased protein contaminants [58,63]. Additionally, complete resuspension of aggregated or agglomerated EVs often requires shear force or trypsin, which potentially damages the isolated sEVs [59]. Finally, the large ultracentrifuges required for such high g forces may not be available in the clinical settings of underserved areas [56,60]. In brief, these inherent limitations hamper the relevant clinical translation.
2.1.2. Density gradient ultracentrifugation
Density gradient ultracentrifugation (dgUC) is the gold standard for isolation of sEVs from other particles by utilizing their respective densities [100]. It follows UC and combines with linear sucrose or iodixanol density gradient/cushions to float sEVs. Exosomes, the smallest subtypes of EVs, exhibit typical floatation densities of 1.08–1.22 g/mL. This density makes them distinct from other molecules, thus providing an advantage of dgUC for enriching exosomes with high purity [101]. For example, one study examined patient serum and used several methods for isolating sEVs and compared their yield and purity [102]. The serum contained an average of 1.44 × 1011 sEVs. For UC and dgUC, an average of 6.27 × 109 and 4.93 × 109 particles, respectively, were recovered from 200 μl of serum. When comparing protein co-sediments, dgUC contained about 30 μg of protein while UC captured 40–50 fold more. Similarly, other vesicles with different flotation density can be specifically enriched in respective fractions. In brief, samples are pretreated with UC as previously described to obtain the crude sEV pellets followed by resuspension in PBS. Then, sEV suspension is carefully added onto the sucrose or iodixanol gradient solution without disturbing the interphase. Next, samples are ultracentrifuged at 160,000×g for 16–22 h at 4 °C to reach equilibrium. Afterward, vesicles with respective densities can be sequentially harvested [58,69]. Many studies report a preference for iodixanol gradients over sucrose gradients for sEV isolation as iodixanol more efficiently isolates particles of specific densities without co-capturing most protein and viral contaminants [59,61,62]. Despite the advantage of harvesting very pure sEVs, the application of dgUC is impeded by the lengthy and labor intensive process, which may span two days, and result in low recovery efficiency of total EVs (10–50%) due to repetitive centrifugation [61,79,103]. Moreover, it is difficult to isolate sEVs from other particles, such as lipoproteins, which have similar sedimentation rates [102,104]. Given that UC and dgUC are the most common methods, any new methods for sEV isolation should be compared to these two standards and should provide comparable yields and purity.
2.1.3. Acoustic wave isolation
Acoustic wave isolation (AWI) is a label-free method for direct isolation of EVs from body fluids and is often integrated into microfluidic chips [68,72]. Acoustic waves (AW) operate by creating a gradient via acoustic radiation forces and acoustic streaming drag forces within the liquid medium [68]. When these forces interact with any particles within the medium, certain particles will be scattered while other particles will aggregate. This effectively results in the isolation of the particles of interest which associate with specific frequencies. Acoustic separation is also scalable and capable of interacting with a range of bioparticles which span tens of nanometers to hundreds of micrometers. For example, AW have been used to isolate and lyse sEVs from small amounts of patient plasma (20 μl) [105]. This method detected 13x greater levels of liver cancer derived sEV-miR-21 compared to control samples in under 30 min. Additionally, AWI has been used to separate sEVs from whole blood with 99.99% efficiency and other EVs with 98.4% purity [106]. However, there are still issues with effectively, and efficiently, isolating nanoparticles which are less than 100 nm, thereby limiting its usage towards certain subpopulations of sEVs [68]. Once this issue is addressed, acoustic wave separation can potentially isolate sEVs with high purity, yield, and integrity, which would make it an ideal method for point-of-care (POC) applications.
2.2. Solubility-based EV precipitation
Polymer-based precipitation (PBP) of sEVs frequently uses polyethylene glycol (PEG). PEG interacts with water molecules to decrease sEV solubility resulting in sEV precipitation. Precipitation of EVs involves the incubation of the sample with the polymer solution for a few hours or overnight followed by low speed centrifugation to harvest sEVs [73,74]. Several commercial kits are available on the market, including ExoQuick (System Biosciences, USA) and Total Exosome Isolation Reagent (Invitrogen, USA) as well as ExoPrep (HansaBioMed, Estonia), Exosome Purification Kit (Norgen Biotek, Canada), and miRCURY Exosome Isolation Kit (Exiqon, Denmark) [61]. PBP of sEVs is quickly becoming the preferred method for sEV isolation as it is easily scaled up, requires much less labor, and offers high yield while maintaining sEV integrity compared to UC. However, the pellet of PEG/sEV mixture will not only harbor sEVs, but also free proteins, such as lipoproteins, immunoglubulins, and viral particles [61,74,75]. In fact, heavy protein contamination is the main concern for PBP, limiting its relevant applications. The protein contaminants, adsorbed free RNAs, and residual PEG could interfere with EV biofunction and cell interaction assays.
To alleviate protein co-precipitation, studies have optimized the PEG concentration and molecular weight. One study reported a solution of media and 8% PEG6000 yielded the least amount of protein contamination while enhancing purity with a wash step incorporating UC [71]. The authors reported a level of purity comparable to both differential UC and dgUC while requiring less time and labor. Another study compared PEG6000, 8000, and 20,000 and found the greatest particle yield using PEG6000 at 10–12% concentration, while PEG8000 and PEG20000 yielded 8–10% [107]. This study also used a wash step with UC to remove excess PEG for harvesting purer EV samples. Studies have also confirmed that PEG is much more cost effective, even reporting costs of less than $0.01 per mL of sample, which is in stark contrast to the high costs of commercial kits [71,108]. Thus, PBP of EVs can be modified to fit the time and cost requirements of the study. However, despite some protocols requiring an overnight incubation, the overall labor and cost of EV isolation can be significantly decreased if using PEG. The other concern is the aggregation of PEG/EVs. High molecular weight of PEG, high centrifugal force, and long processing times could cause difficulties in EV resuspension. While this method is simple and easy, residual PEG, free proteins, and free nucleic acid contaminants may make precipitation-based isolation unsuitable for EV functional studies.
2.3. Size-based EV isolation
2.3.1. Ultrafiltration
Ultrafiltration (UF) utilizes decreasing filter pore sizes in order to isolate particles of a specific size and may incorporate ultracentrifugation or gel filtration chromatography for increased purity [61,80,[109], [110], [111]]. The filters used for progressive filtration have pore sizes with diameters of 0.8, 0.45, 0.22, and 0.1 μm. Larger particle sizes are filtered out while the EVs are concentrated into specific size fractions by the 0.22 and 0.1 μm filters. In theory, this should ensure purified EV fractions, however, non-EV proteins and other particles of a similar size are likely to be concentrated by respective pore sizes. Another disadvantage to UF is clogging of the filter pores. This can result in sample loss and filter burst, which can interfere with purity and yield. Sample loss by clogging the filter can be mitigated by additional wash steps, though this carries a risk of introducing proteins into the EV concentrate. However, it has been reported that UF can isolate EVs with purity and numbers similar to, and more efficiently than, UC [61,109,112]. For example, one study compared five different centrifugal filters (Amicon Ultra-2 10k Regenerated cellulose (RC), Amicon Ultra-2 100k RC, Vivaspin 2 PES 10k, Vivaspin 2 CTA 10k, and Vivaspin 2 Hydrosart 10k) to establish which was most effective at isolating and purifying EVs [113]. Using 1010 EV/mL spiked into PBS, it was determined that the Amicon 10k RC exhibited the highest recovery of EVs and the lowest recovery of protein.
UF has been incorporated into many microfluidic devices to facilitate effective point-of-care diagnostics. In 2017, a device was created that utilized four layers of Poly (methyl methacrylate) (PMMA) and double-sided adhesives and incorporated a 200 nm pore filter and a 30 nm pore filter [76]. The flow, containing the EVs, is forced through the 200 nm pore filter, through the isolation chamber, and then up through the 30 nm pore filter into the waste chamber. The isolation chamber contains the flow depleted of particles greater than 200 nm and less than 30 nm. The device captured significantly greater levels of sEVs from bladder cancer patients compared to controls and this diagnostic exhibited a sensitivity of about 80% and a specificity of 90%. Additionally, the device recovered 74% of sEVs, which is superior to UC. Another device, called Exodisc, was able to purify sEVs from whole blood using 600 nm and 20 nm pore filters under relatively low centrifugal force (<500 g) Exodisc was also used to enrich sEVs for prostate cancer detection, and found a greater yield in prostate cancer patients compared to controls.77 78. Similarly, another device, the Exosomal Total Isolation Chip (ExoTIC) uses a series of smaller filter pores (200-100-80-50) and a syringe pump to isolate and purify sEVs and is reported to yield approximately 4-1000-fold greater sEV numbers compared to precipitation and UC [114]. ExoTIC sports several other advantages in its design including the ability to swap out filter sizes, can withstand typical pressures which result in filter damage, utilizes small volumes of fluid ranging from 10 μL to 150 mL, and can incorporate multiple filtration steps with minimal cost and time.
While UF can isolate sEVs in under an hour with greater purity compared to UC, it has disadvantages. UF suffers from effectively isolating sEVs from blood, plasma, and serum due to their high protein contents. By contrast, UF may be more suitable for less complex fluids such as cell culture supernatant, cerebrospinal fluid, and urine as these fluids contain less proteins. Another potential issue with UF, is that while it can process fairly small volumes of sample, the accuracy and specificity of a diagnostic may be impaired as the contents derived from such small volumes of specimens may not support multiplex analyses.
2.3.2. Size exclusion chromatography
Size exclusion chromatography (SEC) is a column separation approach based on size differences of EVs to isolate EVs from samples such as cell culture supernatant, blood, and urine [81,115,116]. It is constructed by heterogeneous porous beads packed in a column to form a “maze-like” internal structure. The beads consist of numerous openings and shafts of various sizes. When the sample is loaded, EVs and other sample contents move through the pores in the beads. Smaller molecules will take much longer to elute than larger particles due to a more hindered path as the smaller ones get trapped in the beads [58]. Thus, the separation of EVs occurs via differential SEC as the sample contents pass through the beaded columns. To enhance efficiency of SEC, many commercial kits have also been developed by Sigma-Aldrich (USA), Izon Science (UK), and GE Healthcare (Sweden) [61]. SEC can also be scaled to match customized requirements. For example, sizes of the pores and beads can be altered to isolate specific molecules and EV types, and when coupled with longer columns, can also enhance resolution, while wider columns can maximize processing of larger volumes Additionally, because these samples are subjected to minimal pressure, the structural integrity and biological properties of exosomes is guaranteed [58,61]. However, this process make take longer due to the limited force from gravity flow pressure [117].
Bind Elute SEC (BE-SEC) and SEC are also comparable to UF and UC in that the sEVs isolated from optimal fractions may be similar in size. BE-SEC relies on trapping particles smaller than sEVs, thereby allowing the sEVs to elute with the flow through, rather than the beads as is the case with SEC. A major advantage of SEC isolation of EVs is that it results in almost no sample loss compared to UF and UC. Despite this, UC may still yield purer samples compared to SEC and UC is still necessary to concentrate samples eluted from SEC [70,118]. Moreover, many papers have published SEC coupled with methods, such as UF, UC, and dUC to further enhance the efficiency and purity of EVs [62,79,80,109,111,[119], [120], [121]]. These combination methods do improve purity compared to either method alone, with menial loss to yield. However, these coupled isolation methods also make the process more complicated, which hinders clinical translation. These types of optimization experiments are imperative if the field of EV research is to reach a consensus for standard isolation procedures.
2.4. Immunoisolation of EVs
Immunoaffinity for EV isolation is based on antigen-antibody or antigen-aptamer reactions [122]. While there are many proteins which have been characterized on the surface of sEVs, the most commonly used are CD9, CD63, CD81, Alix, and heat shock proteins [61,123]. Other surface markers can be used as well for capture of specific sub-populations of EVs. For example, one study chose chondroitin sulfate proteoglycan 4 (CSPG4) to capture a subpopulation of melanoma-specific sEVs [84]. This study utilized mini-SEC to remove plasma proteins from melanoma patient plasma before isolating sEVs with immunoisolation beads. Streptavidin-coated beads with biotinylated anti-CSPG4 were used to isolate a fraction of melanoma-derived sEVs. They reported a mean capture efficiency of 98% for these melanoma-specific sEVs from purified patient plasma. They also reported that attempts to capture sEVs from plasma without mini-SEC resulted in blockages of the antibodies, and thus prevented capture. Another commonly used EV marker is EpCAM, which is often used to identify and capture tumor specific EVs [124]. Because of this potential for isolation of disease-specific EVs, specific antibodies and aptamers targeting disease-derived EVs has gained immense attention [[125], [126], [127]].
2.4.1. Antibody-conjugated methods
One publication used antibody cocktail-conjugated magnetic nanowires to isolate sEVs from the plasma of breast and lung cancer patients [128]. This nanowire was functionalized with CD9, CD63, and CD81 in order to pulldown as many sEVs as possible. The number of sEVs isolated from the supernatant of four different cell culture models (MDA-MB-231, HeLa, MCF7, and HCT116) was compared between magnetic DynaBeads™ functionalized with anti-CD9 or anti-CD81; the magnetic nanowires (MNW) functionalized with either anti-CD9 (CD9_MNW), CD81 (CD81_MNW), or all three (CD9, CD63, and CD81, Abs_MNW); and UC. CD9 DynaBeads™ captured ≤20 × 108 total sEVs/ml across all cell lines; CD81 DynaBeads™ captured ≤20 × 108 sEVs from MDA-MB-231 and HeLa, while capturing about 50 and 40 × 108 sEVs for MCF7 and HCT116, respectively; across all cell lines, UC isolated ≤20 × 108 sEVs. The MNWs, by contrast, captured greater numbers of sEVs, regardless of antibody used. For CD9_MNWs, ≤ 40 × 108 sEVs, ~80 × 108, ~60 × 108, and ~30 × 108 sEVs from MDA-MB-231, HeLa, MCF7, and HCT116, respectively. The CD81_MNWs captured similar numbers of sEVs as the CD9_MNWs, excepting HCT116, in which about 110 × 108 sEVs were isolated. The Abs_MNWs captured around 1.5–2x more sEVs from HeLa and MCF7 supernatant (~120 and ~150 × 108, respectively) compared to the CD9 and CD81_MNWs, while capturing about 70 × 108 from HCT116. In summary, the Abs_MNWs were superior to UC for isolating sEVs. Furthermore, when they used the MNWs to capture sEVs from the plasma of breast and lung cancer patients, they found that the cancer patients exhibited a threefold increase in circulating sEVs compared to the healthy controls. They also compared the MNW system to exosome isolation kits Exoquick and Invitrogen's total exosome isolation (TEI) kit. Nanoparticle tracking analysis determined that the MNW system isolated about 6.3 × 109 particles/ml compared to ExoQuick and Invitrogen which isolated about 2.4 × 109 and 1.73 × 109 particles/ml, respectively. The MNW system therefore exhibits much higher yields than both UC and precipitation methods, and reportedly takes 1 h to capture, isolate, and elute sEVs for about $11 per 1 ml sample. Furthermore, this method requires a minimum of 250 μl of plasma, which is a smaller volume than other methods. This method is therefore more efficient than UC, while providing greater yield, with less sample, and is presumably a purer preparation, though it is unclear in the paper what level of purity this method provides.
This superior efficiency of immunoisolation over commercial kits has been reported in other research which examined EV capture from prostate cancer patient plasma [83]. In this publication, the research team utilized Prostate Specific Membrane Antigen to capture prostate-specific EVs from 100 μl of patient plasma. They utilized atomic force microscopy to establish the height and volume of sEVs present in the immunocapture set up compared to the ExoQuick, ExoSpin, and TEI commercial kits. The sEVs isolated from plasma, under AFM, exhibited only events of sEV size, with no other events being observed, thereby suggesting a very pure sample of sEVs. By comparison, TEI and Exospin revealed few objects of sEV size, while exhibiting an abundance of proteins. ExoQuick did not show any topography >100 nm in height, but a monolayer of protein was reported. This data shows that immunoaffinity methods are superior to precipitation methods, and supports that immunoaffinity produces much higher yields, and highly pure samples, when compared to commercial kits.
sEVs are often damaged when separated from their antibody. A recent study aimed to solve this issue by using their previously designed microfluidic device, called the OncoBean [85,129]. Their previous device utilized bean-shaped microposts (118 μm l x 50 μm w x 100 μm h) functionalized with biotin-conjugated EpCAM to capture circulating tumor cells. They modified the device by replacing the biotin-conjugated EpCAM antibodies with desthiobiotin-conjugated sEV markers CD9, CD63, and CD81. The advantage of using desthiobiotin is that it has lower binding affinity to avidin, and when eluted with a biotin solution, facilitates the release of the captured molecule with no damage [85,90]. This method facilitated release of nearly all sEVs from the microposts upon biotin elution without causing structural damage. This work suggests a vast improvement over the current immunoisolation methods in terms of maintaining the integrity of isolated sEVs for downstream analysis.
Another study examined the effectiveness of an EV capture device with pillars in the shape of intestinal microvilli, called NanoVilli, to enhance capture efficiency by offering a much larger surface area for EVs to bind to Ref. [124]. They designed this device using a silicon wafer and silicon nanowires to attach the EpCAM antibodies. The device was used to isolate EVs from 13 NSCLC patients. For this, they used 200 μl of plasma from each patient. The isolated EVs were then analyzed for the presence of CD74-ROS1 and EGFR T790 M mutations. Furthermore, the NanoVilli was used to isolate EVs and monitor the presence of these mutations in response to treatment over 279 days. One patient, for example, exhibited an EV-EGFR T790 M mutation copy number decrease from 225 to 9 with visible tumor shrinkage by day 146, thereby proving the feasibility of NanoVilli to isolate EVs for monitoring patient response to therapy. With NanoVilli able to capture a majority of EVs from the sample in 30 min, and can effectively monitor patients undergoing chemotherapy, NanoVilli exhibits obvious advantages over current EV isolation methods. However, it is unclear to the purity of these EV isolations as the paper does not discuss protein contamination.
2.4.2. Aptamer-conjugated methods
Aptamers are strands of nucleic acids which fold into 3D configurations that bind specifically to their target ligands with high affinity and specificity [86]. Aptamers offer benefits over antibodies by exhibiting very low immunogenicity and therefore a low chance of nonspecific binding, low variation between batches, and high specificity and sensitivity. While aptamers have been around for several decades, their use in EV capture and isolation is new, with PubMed reporting about 100 papers published between 2017 and 2020, with the most common reported use of aptamers being for detection rather than isolation. Aptamers for EV isolation is a growing field which requires further study and offers strong potential for the future of EV isolation.
A common method is to conjugate magnetic beads with CD63-binding aptamers [87,88]. This has been done for capturing liver cancer-derived sEVs [87], breast cancer sEVs [88], and prostate cancer (PCa) sEVs [130]. Liver cancer sEVs were captured by these CD63 aptamer-conjugated beads and amplified via cascade reactions between the capture probes, fluorescent probes affixed to AuNPs and a reaction between the AuNPs and β-mercaptoethanol to emit a fluorescence signal [87]. They tested the system on the serum from two healthy controls and two liver cancer patients and found that the healthy controls exhibited < 2 × 107 particles/μl. By contrast, the liver cancer patients exhibited 2 to just over 2.5 × 107 particles/μl, which were also consistent with NTA. Similarly, the breast cancer study utilized CD63 initially to isolate sEVs, but found little difference in sEV numbers in tumor vs control samples [88]. However, upon using MUC1, a common tumor marker, for sEV capture, they found a significant increase from <10% of MUC1-sEVs in control patients to 25–35% sEV capture in breast cancer patients. Another platform was developed for the isolation and detection of prostate cancer (PCa) specific sEVs using prostate specific membrane antigen (PSMA), instead of CD63, for sEV capture from urine [130]. This platform is composed of superparamagnetic conjunctions, instead of AuNPs, and molecular beacons (SMC-MB). The SMC contains Fe3O4 cores and aptamers specific for capture, while incorporating fluorescent-hairpin complexes for amplification. Ten ml of urine was collected from 20 PCa patients and 50 healthy donors for analysis using this device. Fluorescent intensity was greater for the PCa compared to the controls. Furthermore, SMC-MB shows promise for isolating specific EVs with superior purity and comparable yield to UC. Similarly, MUC1 was used for capture of tumor-specific sEVs from breast cancer patients. In general, the specificity, purity, yield, and POC potential of these aptamer capture systems compared to UC make them a superior method for sEV isolation towards specific tumor diagnosis.
Immunoisolation yields highly pure samples of EVs in a comparatively short time compared to other methods, and, with immunoaffinity methods, it is possible to isolate specific subpopulations of EVs. However, preparing immunoisolation beads is costly. For example, ThermoFisher's Dynabeads™ can cost anywhere from $548 for 2 ml up to nearly $11,000 for 100 ml, for one example of commercially available beads. Antibodies and aptamers to functionalize the beads can cost $269 for 2 μl for antibodies, while aptamers can offer a less expensive alternative of $100 for 1 mg, and these prices can vary widely, depending on the manufacturer and product. Furthermore, while immunoisolation exhibits greater purity, it does offer variable yield, depending on volume used, the antibodies used for capture (which vary due to tumor heterogeneity), and sample type (plasma, cell culture, blood, etc) [61]. Taken together, these factors have the potential to negatively affect isolation efficiency. Additionally, even if the correct antibodies are used, thereby fixing the issue raised by tumor heterogeneity, using the antibody to isolate EV from plasma/serum is still very challenging due to the high-abundance of serum proteins. It is also difficult to remove EVs bound to their affinity molecules without damaging them and causing issues for downstream functional analysis [61,131]. And while many papers have been discussed here in which these issues have been addressed, immunoisolation methods still require consensus and standardization before they can be viable for POC.
On the other hand, the selection of sEV membrane antigens for isolation of sEV is also pivotal. The most common of these markers include surface tetraspanins CD9, CD63, and CD81 [132]. While the majority of previous papers suggest CD63 as the optimal choice for sEVs, a recent publication suggests CD9 or CD81 may be more precise markers [133]. Extensive analysis was conducted using pancreatic cancer and lung cancer cell culture, tissue samples (tumor versus adjacent normal), and plasma/serum samples from pancreatic or lung cancer patients. Comparisons of expression were conducted from plasma and serum between PDAC or lung cancer patients and controls without cancer. The analysis compared total protein expression to determine which proteins were the most highly expressed in tumor derived sEVs. With regards to the traditional EV markers, CD81 and CD9 were observed in over 77% of the sEVs. By contrast, CD63, which has been considered the most common sEV marker, appeared to be more highly expressed in murine cell culture sEVs and rarely found in sEVs collected from body fluids. Moreover, the analysis found that beta-actin (ACTB), moesin (MSN), and ras-related protein 1b (RAP1B) were markers that were found across sEVs ranging from 60 to 80 nm and 90–120 nm as well as exomeres which are non-vesicular particles < 50 nm. However, they found that stomatin (STOM) is present only on sEVs (60–120 nm). Taken together, this paper highlights the need for specific determination of sEV membrane antigens in order to maximize sEV isolation.
2.5. Lipid nanoprobe
Lipid nanoprobes (LNP) are an emerging and novel tool for EV isolation [90]. The lipid nanoprobe is comprised of a diacyl lipid, DSPE-PEG-biotin, labeling probe and a NeutrAvidin (NA)-coated magnetic sub-micrometer particle capture probe. The labeling probe tags EVs in solution and then interacts with the capture probe in order to isolate the EVs. The NA-biotin reaction-based isolation takes 15 min and exhibited an isolation efficiency of approximately 80% when maximized using 10 nmol of labeling probe. Additionally, when DSPE-PEG-desthiobiotin is used for the labeling probe, the desthiobiotin can be displaced with biotin, effectively freeing the captured EVs. The release efficiency was about 84 ± 3% within 30 min. The captured EVs remained intact upon release and were effectively analyzed for their DNA, RNA and protein cargoes and found that these EVs were able to carry out their biological functions in wound healing assays with MCF-7 cells. Additionally, they found that the DNA, RNA, and protein profiles of EVs captured using their LNP were consistent with the profiles of EVs isolated via UC. To verify the clinical relevance of this method, the LNP system was used to isolate EVs from 100 μl of blood plasma from stage IV NSCLC patients. The contents of the captured EVs were successfully analyzed for the presence of common NSCLC mutations such as EGFRL858R and KRASG12D via PCR and NGS. LNP allows for shorter isolation time of 15 min when compared to UC, which can require more than 22 h. Isolation efficiency and cargo composition obtained from LNP and UC showed great similarity, indicating good reliability of LNP for isolation of EVs. Moreover, the LNP system did not require expensive equipment or require extensive time and intensive lab work, which makes LNP superior to existing methods such as UC and dgUC. However, the clinical translation of these LNP has limitations with the processing capability of the system. Therefore, to overcome this concern the LNP system was further improved by grafting the LNPs on a fabricated silica nanostructure surface [91]. The LNP were modified to include cholesterol and PEG1000 to improve capture. The isolation efficiency for this modified system was 28.8 ± 5.3% at a flow rate of 10 μl min−1 when isolating EVs from up to 2 ml of PANC1 and MDA-MB-231 cell culture media. Compared to UC, this method also provides higher purity, removing 96.5% of plasma protein, while UC removes about 71% of protein from plasma. This fabrication design allowed for an increase in the EV binding surface area, as well as an increase in sample volume compared to their previous device, when the sample is passed through the LNP system. Hence, concerns with isolation of EVs from tumor derived plasma with low EV levels were eliminated. Also, this redesigned system in combination with digital droplet polymerase chain reaction (ddPCR) facilitated more precise downstream molecular analyses of mutation allele frequency from tumor derived EVs. This would enable development of a patient specific treatment as mutation allele frequency is wide ranging. In addition, this system could potentially aid in early diagnosis of cancer as the combination of LNP and ddPCR provide high sensitivity and specificity and can detect allele frequency lower than 0.01%.
Another paper aimed to improve capture efficiency by affixing DSPE-PEG-biotin labels to the wing of a butterfly, Morpho Menelaus, which was integrated into a microfluidic chip and exhibited a 70% isolation efficiency of EVs in under 30 min for cell culture, while the isolation efficiency for plasma was significantly less, at 51% [92]. The structure of the butterfly wing incorporates natural 3D micro-groove structures which create a vortex when liquid is poured onto it, thereby increasing the potential for EVs to be caught on the structures. The ridges themselves run parallel on the wing, with distances ranging from 0.5 to 5 μm. The wings also possess a natural photonic crystal, which enhances fluorescent intensity, thereby making the wings attractive for use in biological detection. Initial analyses were conducted using EVs isolated from the cell culture media of MDA-MB-231 and MCF10a for the control. Fluorescent intensity of captured EVs suggested that 88.1% of the EVs from the cell culture media were captured, by contrast, when the back of the wings were used (the non-photonic crystal side) to isolate the EVs, the fluorescence suggested only 30% of the EVs were captured. Washing with triethylamine successfully eluted all EVs off the wings, as well, as evidenced by no visible EVs on the wings under SEM imaging. The isolation efficiency of the butterfly wing was compared to that of UC, whereas the modified LNP wings were able to capture nearly 76% of EVs, UC isolated about 14.3%. EVs captured from the cell culture media were analyzed for the presence of GPC1 and found that the MDA-MB-231 exhibited lower Ct values for GPC1, then for MCF10a, showing that the EVs from the tumor cell line expressed greater levels of GPC1 compared to the control. Finally, they examined the wing chip compared to a flat chip and found that the wing chip exhibited a capture efficiency of 74.3% compared to 39.6% on the flat chip for breast cancer patient plasma. The paper does not specify the volumes of cell culture media or plasma used, however, despite this, the data supports the validity of the usage of a wing-modified microfluidic chip and it's incorporation of LNPs to efficiently and rapidly isolate and release intact EVs for diagnostics and downstream analyses.
Taken together, these studies show increasing promise for LNP capture of EVs. This type of efficiency and high-throughput offers strong potential for the future of EVs use in cancer diagnosis and treatment. There are disadvantages however, including the high cost of reagents and strict storage conditions. Improper storage can result in formation of micelles or liposomes. It is also not possible to isolate specific subpopulations of EVs with lipid nanoprobes alone, and would require additional steps like SEC or other immunoaffinity capture methods. Another disadvantage is that the lipid probe/lipophilic protein interaction cannot be eliminated. However, the technology is still young and therefore remains under intense investigation.
2.6. Charge-based sEV isolation
2.6.1. Ion exchange chromatography/electrostatic adsorption
Ion exchange chromatography (IEC) takes advantage of EVs' negative charges to separate and purify them from other particles [96]. EVs with negative charges bind to the positively charged chromatographic column and detach from it by enhancing the ionic strength of the mobile phase. The advantage to IEC is that it enables easy isolation of EVs, however, this method does not allow for isolation of specific subpopulations, and binding is non-specific. Furthermore, when processing serum and plasma samples, the abundance of negatively charged proteins can be adsorbed, leading to heavy protein contamination. Regardless, this method has shown promise for isolating EVs from certain types of body fluids like amniotic fluid (AF) [97]. In one study, 16 ml of AF was collected from a single woman. A reported 83.8% capture efficiency and isolation of EVs in the sample was observed with a purity of 80%. Another study reported a rapid, single-step, scalable anion exchange column-based chromatography (AIEC) to isolate EVs and compared it with “gold-standard” UC and tangential flow filtration (TFF) on size distribution, surface marker, and morphology [98]. Results displayed that EVs isolated by AIEC had relatively higher yield and more expression of EV markers compared to the UC isolated EVs. Additionally, AIEC exhibited superior purity compared to TFF with reduced contamination from debris and proteins in isolated EV samples. A further study created a device utilizing 2 μm ZnO nanowires coated in 10 nm thick Al2O3 embedded in a PDMS microfluidic chip [99]. The slightly positive charge of the nanowire was able to capture EVs, and other negatively charged free-floating elements from 1 ml of urine, with a reported 99% collection efficiency. This high efficiency is largely due to the solution's pH of 6–8, resulting in the positive charge of the ZnO due to isoelectric point. Any objects captured on the chip were subjected to 1 ml of lysis buffer in order to analyze the EV-miRNA contents to facilitate the development of a tumor diagnostic. Using this method to collect and isolate EV-miRNA before microarray analysis, they report identifying about 700–1000 different miRNA species, which was nearly fivefold higher compared to. While IEC appears to offer high capture efficiency from complex samples, it suffers in isolating pure samples due to the negative charges of proteins and nucleic acid species, thus IEC may be better used for preliminary isolation steps of EVs. Additionally, previous studies have reported co-isolation of viruses with EV fractions, also due to the negative charges of viral particles, therefore contamination of viruses, nucleic acids, and proteins continues to be an issue for IEC [134]. Regardless, IEC offers a potentially novel method for fairly rapid EV isolation, and, when integrated with other methods for analysis, shows promise for cancer diagnostics.
2.6.2. Dielectrophoresis
Dielectrophoresis (DEP) uses electric fields to separate particles by charge [93]. More polarized particles move faster towards the electrode while lesser polarized particles move slower. This method has been utilized to separate, trap, and sort different types of cells. DEP advantages include label-free isolation which helps in cost effectiveness and the method is reported to take minimal time compared to conventional methods. A disadvantage to the method, however, is that the electric current has been reported to damage cell membranes, and by extension, EV membranes are likely to be damaged as well by the currents, which could potentially interfere with downstream applications. Another major disadvantage is the requirement of isotonic sucrose solution for sample preparation prior to DEP. The sucrose raises the molarity for the solution to physiological levels in order to minimize the chances of the EVs being damaged by the change in currents. However, this also results in ions forming a barrier around the electrodes. This weakens the overall charge, thereby only EVs which hover long enough around the electrodes can be captured. There are several devices that incorporate DEP to isolate EVs [94,95,[135], [136], [137]]. Three papers report using alternating current electrokinetic (ACE) devices [94,136]. The first, utilizes ACE for isolation and recovery of glioblastoma EVs from plasma samples [94]. EVs were captured on the chip by AC and then fluorescently labeled to validate their presence. Additionally, they were able to identify glioblastoma-specific EGFRvIII using 30–50 μl of patient plasma. The entire analysis including isolation, labeling, and EV-RNA analysis took 30 min. Purity and yield was not reported. This same chip was used for the detection of PDAC from 25 μl of whole blood [136]. The samples were incubated with CD63 and GPC1 fluorescent antibodies to validate the capture of PDAC-specific sEVs. PDAC patients exhibited a larger fluorescence range (5–20 fluorescence units) compared to the benign pancreatic diseases (<5–10 units) and healthy controls (≤5). Furthermore, this diagnostic method is reported to have a 99% sensitivity and 82% specificity and could detect PDAC sEVs in 90 min. Similarly, the third work which uses the same device also utilized 30–50 μl of patient sample to detect the presence of EV-Tau-5 and EV-GFAP (Glial fibrillary acidic protein), two proteins which are commonly deregulated in patients with glioblastomas [95]. ACE isolation and on-chip IF for identification of EV-Tau and (GFAP) took about 90 min. The device reportedly captured 60–70% of the EVs. The relative fluorescence of the concentration of GFAP in glioblastoma, meningioma, and patients with metastasis was about 1.7–2.2, 1.7–2.2, and 1.2 to almost 4 units while the healthy controls did not exhibit any values over 1.7. The concentration for EV-tau for the same was 5–~7.5, <2.5 to ~6, and >2.5 to over 7.5. The healthy patients did not exhibit and values over 2.6. Furthermore, EV-GFAP exhibited a sensitivity of 93% and a specificity of 38% with a positive predictive value (PPV) of 74% and a negative predictive value (NPV) of 75%. For EV-Tau, the sensitivity, specificity, PPV, and NPV are 67%, 75%, 83%, and 55%. Thus, the two markers may provide confirmation of a glioblastoma diagnosis. However, they do report that all the patients exhibited sizeable brain tumors and as such, they are unsure if this method would work for early detection. This ACE device offers rapid and sensitive POC diagnostic potential. Two other studies utilized insulated dielectrophoresis (iDEP), which enables trapping of the EVs [135,137]. The first study used MCF-7-derived EVs. With their system, they were able to trap the EVs between the insulated post [135]. The process took 20 s to trap and separate the EVs. This method used only 100 μl of MCF7 cell culture media. The second study used an insulator-based dielectrophoretic device interfaced with an array of borosilicate micropipettes [137]. They analyzed cell culture media, plasma, serum, and saliva. The process used 200 μl and took 20 min under low voltage (10 V cm−1). The device captured as many as 1.1 × 1012 particles/ml compared to dgUC, which isolated 6.95 × 1010 particles/ml from serum. Across all sample types, the iDEP method reported two orders of magnitude greater EV recovery than differential ultracentrifugation.
Taken together, all dielectrophoresis methods appear to require minimal special reagents for the isolation and recovery of EVs. In general, these methods are rapid and cost-effective, and at lower voltages, are less likely to damage the EV membrane if it is required for downstream analyses. Another advantage is that all these devices require minimal sample volume compared to conventional means and these methods appear to provide high yield. Furthermore, dielectrophoresis is also able to separate EV particles by size, which is another benefit over other methods [135].
2.7. Lateral flow assays
Other methods exist as well which aim to isolate EVs with high purity and yield with greater efficiency. Lateral flow assays, while commonly used for detection, are also able to sort and isolate sEVs. A preliminary study reported enrichment of medium-sized exosomes using lateral flow [138]. To do this, they used a nano-deterministic lateral displacement (nano-DLD) pillar array. In this system, they report the ability of this nano-DLD to separate particles ranging from 20 to 110 nm. The nano-DLD separates these particles based on their movement through the device, with larger particles bumping between pillars while the smaller particles have a zig-zagging and partial bumping path, with both sizes collecting in respective parts of the device. Through this method, they were able to separate 50 nm beads from 110 nm beads with a separation resolution of 1.5. They used 200 μl of human urine-derived exosomes to test the ability of the device to separate different-sized sEVs and found the nano-DLD enriched sEVs within the size range of 60–70 nm in about 2 h. The paper does not examine the isolation efficiency or retrieval efficiency of this method, nor is it clear how effective this method is at deriving pure samples. In a follow-up design, however, around 1000 of these nano-DLDs were arrayed on a chip for the analysis of prostate cancer-sEVs [139]. With this arrangement, they were able to process samples to a maximum of 900 μl/hr. The study examined the ability of the device to concentrate and isolate EVs of different sizes (bump vs zigzag particles, as previously described) compared to the volume of serum or urine samples used. Approximately 2.0 E+09 particles/ml of urine EVs and about 1.0 E+12 particles/ml of serum EVs were introduced onto the device. As the EVs flowed through the device, they concentrated into either the zigzag or bump collection chambers. Additionally, the nano-DLD array was able to concentrate these sEVs into the bump fraction at an approximate 3-fold increase compared to the input volume for both urine and serum samples in an hour's time. Furthermore, the device was ~2–4x more effective at concentrating serum EVs compared to UC, dgUC, qEV SEC, and ExoEasy, but not so with urine EVs. The device also exhibited a significantly greater yield of about 30–70% of serum EVs compared to the other methods while this was true only in comparison to dgUC for urine samples. When they modified this device further, by using 3840 parallel nano-DLD arrays, they were able to increase concentration from 2.6x to about 60x for urine samples and with 50x greater purity compared to the initial device. They then tested the modified array using serum EVs from 9 prostate cancer patients. They compared the number of RNA species collected from EVs isolated by UC (2 ml) and the device (0.5 ml). Greater miRNA percentage and less rRNA was observed in EVs isolated by the device compared to EVs isolated by UC. The authors attribute this to the device's ability to enrich for sEVs compared to UC. Furthermore, they report that most of the prostate cancer-specific markers were expressed in both isolation methods; but while UC takes several hours, the nano-DLD took only an hour to process the samples. Taken together, the nano-DLD chip exhibits comparable, and in many ways superior, performance to the gold standard. However, the paper is unclear if the nano-DLD chip can isolate sEVs with a purity that is comparable to dgUC.
2.8. Summary
There are other methods for isolating EVs as well that are not discussed in this work. These other methods include precipitation with sodium acetate, protamine. protein with organic solvents or affinity using phosphatidylserine, heparin, binding of heat shock proteins, and lectins [61]. Each with their own advantages and disadvantages, which were not introduced in detail. Overall, isolation methods should ensure vesicle integrity. Intact vesicles enable a more thorough and reliable analysis of internal contents such as protein and nucleic acids, which can provide greater insight into the mechanisms of particular tumors and provide a more specific diagnostic. An ideal EV isolation method for clinical implementation should offer consistently high purity and yield while maintaining intact vesicles via a rapid, cost effective, and easy-to-use method which is both sensitive and specific [140]. Thus, current EV isolation techniques remain unviable for clinical settings and new methods which address the aforementioned requirements are strongly desired [[141], [142], [143], [144], [145], [146], [147], [148], [149]].
3. Direct on-chip detection of sEVs after isolation
While isolation of pure EVs is most important for downstream analyses, it is also necessary to develop more efficient and cost-effective methods for EV detection. Direct detection of EVs can be labor intensive and costly, however, with the development of various on-chip detection methods, less time and effort are required for accurate identification of EVs for clinical application. Both are amenable to clinical application, though ELISA offers a more direct observation via detectable color changes while bio and nanosensors may require specialized equipment to measure fluorescence or record electrical signals.
3.1. Highly sensitive ELISA and lateral flow immunoassays
ELISA is often used in conjunction with lateral flow immunoassays (LFIA). A LFIA platform was developed using the common sEV markers CD9, CD63, and CD81 [150]. Their LFIA system utilizes Au-conjugated anti-CD9 for the capture line and Au-conjugated anti-CD63 for the detection/control line. The limit of detection (LOD) is 8.54 × 105 EV/μl and the assay takes 15 min to complete. A follow-up study incorporated a tumor marker, MHC class I chain-related protein A (MICA), and found that it was detectable with the same LFIA platform with the ability to detect 5 × 107 EV/μl in 15 min [151].
In a recent publication, researchers engineered porous superparamagnetic gold-loaded ferric oxide nanocubes (Au–NPFe2O3NC) with the intention of isolating exosomes from other EVs [152]. Au–NPFe2O3NC are able to functionalize with multiple probes, thereby increasing capture efficiency and that are able to isolate, mix, separate, and purify samples due to the magnetic properties of the nanocubes. Additionally, the nanocubes also exhibit peroxidase-like activity, and thus act as nanoenzymes for direct detection of captured and isolated sEVs. For sEV capture, the Au–NPFe2O3NC were functionalized with anti-CD63, of which 5 μl were mixed with 100 μl of placental choriocarcinoma (BeWo cell line) cell culture media. The sEV-laden Au–NPFe2O3NC were then transferred to electrodes modified with placental alkaline phosphatase (PLAP) to specifically capture placenta-derived sEVs. To detect the presence of these specific exosomes, Au–NPFe2O3NC possesses intrinsic peroxidase-like activity that catalyzes the oxidation of TMB in the presence of H2O2, resulting in the colorimetric detection of placenta-derived exosomes.
ELISA has also been used to develop a droplet-based assay for counting single exosome-sized sEVs, which they call ExoELISA [153]. This paper reports the use of magnetic beads functionalized with anti-CD63 to capture sEVs and anti-GPC-1 for secondary antibodies for tumor-sEVs. This was further conjugated with enzymatic reporter β-Galactosidase. The beads with immunocomplex are then encapsulated into microdroplets using a microfluidic chip, such that only one bead with a captured sEV are encased in single microdroplets. Once the fluorescein-di-β-d-galactopyranoside substrate is catalyzed by the enzyme, fluorescein is emitted, and the exosome concentration can be determined. The reported LOD of this assay is about 10 sEVs/μl. ExoELISA was used to distinguish between breast cancer patients, benign breast disease, and breast cancer patients post-surgery. About a 5–7 greater fold change of exosomes/μl was observed in breast cancer patients compared to the other groups, and a significant drop in exosome numbers was observed in the two breast cancer patients post-surgery. Thus, ExoELISA exhibits strong potential for use in early detection as well as monitoring patients after treatment.
3.2. Biosensors and nanosensors
Several technologies are emerging from the biotechnology sector to shorten the time needed to isolate sEVs without sacrificing yield or purity. Regardless, even these methods still exhibit shortcomings, such as complex fabrication methods or issues with scaling up, which must be addressed and overcome for sEVs to be viable for tumor detection. To address these issues, many types of biosensors have been examined including plasmon resonance [154,155], quantum dots [156,157], and others [158,159]. Bio and nanosensors offer rapid and sensitive detection and readouts with strong potential for compatibility with clinical settings. They are commonly used commercially to detect the presence of bacteria in food supply or to monitor fermentation. Glucometers are the most common usage of biosensors in medicine, with the majority being used for at-home glucose monitoring [160]. Recently, they have become of great interest to the medical field for monitoring metabolic diseases, viruses and bacterial infections, and cancer [161]. Biosensors utilize electrochemical, piezoelectric, optical, or thermal means to transduce a biological signal into a quantifiable measurement. Because biosensors and nanosensors can encompass so many different types of devices, this section is broken into specific biosensing subsections.
3.2.1. Surface plasmon resonance
Surface plasmon resonance (SPR) relies on light to excite electrons, resulting in resonant oscillation currents. These oscillations are highly sensitive to any disturbance, such as the addition of molecules, which makes SPR attractive for detecting various analytes [[162], [163], [164]]. When an analyte binds to the nanoparticles on the conducting surface, changes in the oscillation and refractive index are observed and visualization can be enhanced with fluorescent labels. This makes SPR highly amenable for incorporation into microfluidic detection of nanoparticles such as sEVs or even submicroscopic particles like miRNAs [164].
A nanoplasmon-enhanced scattering (nPES) assay was developed for the detection of PDAC-derived sEVs with as little as 1 μl of plasma [154]. The silica sensor chip is functionalized with anti-CD81 to capture sEVs. Samples are added to each well of the sensor chip. Then, the gold nanospheres (AuS) and gold nanorods (AuR), which are functionalized with anti-CD63 (AuS-anti-CD63) and anti-CD9 (AuR-anti-CD9), respectively, are added to each well for sEVs labeling, forming AuS-EV-AuR complexes. The two different Au nanoparticle (GNP) labels are used due to their different optical properties which, when the distance is < 200 nm apart, results in a color shift and resultant scattering of yellow light for greater signal intensity. Additionally, the nPES exhibited a LOD of 0.23 ng/μl compared to ELISA which could not detect sEV concentrations lower than 10 ng/μl. Furthermore, each well of the nPES utilizes 1 μl of diluted plasma compared to 50 μl of undiluted plasma for ELISA detection and is more cost effective. To assess the clinical application of nPES, the AuS was modified with anti-EphA2 (anti-EphA2-AuS) for specific detection of PDAC sEVs. EphA2 (ephrin type-A receptor 2) was chosen due to its significant overexpression in pancreatic cancer tissues compared to chronic pancreatitis and normal pancreatic tissue as well as for its association with tumor progression and metastasis. The nPES functionalized with CD81−CD9-EphA2 was able to discriminate pancreatic cancer patients by stage (I-III) and could distinguish PDAC patients from chronic pancreatitis and normal controls (N = 48–49 per group) with greater sensitivity and specificity compared to CA19-9, the current standard. Furthermore, EphA2 levels decreased in patients (N = 23) who underwent successful neoadjuvant chemotherapy/radiation, but not in patients with poor responses to treatment, while CA19-9 levels did not exhibit significant changes regardless of successful or poor response to treatment.
A small, compact SPR system was previously developed for the detection of sEVs from lung cancer [155]. This biosensor was designed using a glass slide with a 2 nm titanium layer followed by 49 nm of Au film. PDMS with a 6 mm hole was bound to the glass to serve as a sample well. The chip was then functionalized with NeutrAvidin and biotinylated anti-EGFR and anti-PD-L1 for tumor detection, and anti-IgG for the control. sEVs isolated from 50 μl of serum from NSCLC patients (N = 5) and normal controls (N = 5) was resuspended in 50 μl of PBS and used to test the device and compare it to ELISA. No difference was observed in EV-EGFR between normal and cancer patients, however, there was a difference in sEV-PD-L1 levels, suggesting the usefulness of PD-L1 as a diagnostic for NSCLC. Additionally, the LOD for this device was 2 × 1010 sEVs/mL which was only slightly more sensitive than ELISA, which was reported to have a LOD of 4 × 1010 EV/mL. Thus, SPR may offer a viable alternative to tumor detection than current methods.
3.2.2. Quantum dots
Quantum dots (QDs) are inorganic colloid tracers used in signal transduction labeling and offer a direct and sensitive means for detecting sEVs [156]. A QD system was developed wherein sEVs are first captured from samples by magnetic beads functionalized with CD9 or CD63 antibodies. These captured sEVs are then examined for breast and colon cancer specificity by CdSeQDs functionalized with biotinylated HER2 and FAM143B antibodies. When the CdSeQDs are dissolved by nitric acid, Cd2+ ions are released and detected by square-wave anodic stripping voltammetry (SWASV), thereby providing exact quantification of sEVs with HER2 (breast cancer) or FAM143B (colon cancer). Using 10 μl of cell culture media from SW-48 (colon cancer) and BT-474 (breast cancer) a LOD of about 100 sEVs was reached, which is 10x higher than that of NTA or qNano (103 EV/μl) and outperforms ELISA. The serum from nine colon cancer patients of varying stages and one healthy control were obtained to test the clinical feasibility of this method. The SWASV readout for the healthy control was 5 μA cm−2 while the SWASV of cancer patients ranged from 38 μA cm−2 to 95 μA cm−2, with the current density increasing with advanced staging. The capture and analysis take about 2 h to complete and requires special equipment, which may hinder its use in the clinic.
Another microfluidic chip utilizes micropillars to evenly disperse EV-captured-beads and QD probes for clearer fluorescence observation [157]. The initial capture beads were functionalized with anti-CD9 and then mixed with QD probes for tumor sEV detection. The tumor-labeling QD probes utilized carcinoembryonic antigen (CEA, lung adenocarcinoma marker), fragments of cytokeratin 19 (Cyfra21-1, squamous cancer marker), and pro-gastrin-releasing peptide (ProGRP, small cell lung carcinoma marker). The chip and micropillar array were made of PDMS. Patient plasma samples were analyzed for the expression of these specific lung cancer markers. Ten lung cancer patients and 10 healthy controls were used. Plasma samples were optimized to 10 μl with 8 nM of QD probes. In the patient samples, Higher fluorescent intensities were observed for lung cancer patients compared to controls. Furthermore, the differences between CEA concentrations determined by the device and clinical testing was minimal. Thus, such a device may one day be useful as a minimally invasive, rapid, and sensitive alternative for detection of specific types of lung cancer.
3.2.3. Others
One ovarian cancer biosensor utilizes 3D-nanostructured herringbone (nano-HB) microelements to promote increased surface area for probes to enhance sEV binding efficiency, promote mass transfer of the bioparticles, and allow for drainage of the boundary fluids, thereby aiding in maximizing surface binding of the particles [158]. The nano-HB device utilizes 2 μl of plasma sample and reports a LOD of 10 sEVs/μl. The chip can directly sEVs captured on the chip were analyzed for the presence of common ovarian cancer markers HER2, EGFR, FRα, CA125, EpCAM and CD24 derived from SKOV2 and OVCAR3 cell lines. The expressions of these nano-HB-captured sEV-mRNA were analyzed via digital droplet PCR (ddPCR) and exhibited similar expression levels as the sEVs isolated by UC from the same cell lines. In addition, the fluorescence of the six aforementioned markers of sEVs captured by the nano-HB was comparable to the fluorescence of the same markers analyzed via commercial ELISA kits. Because FRα is present in low levels, they used it to test the sensitivity of the nano-HB. The nano-HB was able to detect FRα+ sEVs from 2 μl of ovarian cancer patient plasma with a LOD of 103 total sEVs/μl. Finally, to test the validity of the nano-HB for clinical use, 20 ovarian cancer patients and 10 healthy controls were examined. FRα, CD24, and EpCAM was utilized for the analysis, and when calibrated for protein expression, the nano-HB is reported to have a LOD for FRα, CD24, and EpCAM of 100 fg/ml, 10 fg/ml, and 10 fg/ml, respectively. Furthermore, these proteins alone, and in combination exhibits AUCs of 1.00 (CD24), 1.00 (EpCAM), 0.995 (FRα), and 1.00 (all three markers). Thus, this platform exhibits high sensitivity and may be applicable for clinical settings with additional testing and validation using a larger sample size.
Another device was developed using silicon and graphene coating [159]. This label free device uses a reduced graphene oxide field effect transistor. The device was functionalized with CD63 for specific capture of exosomes/sEVs. sEV concentration was determined by level of impedance with a decrease in current corresponding to increased sEV concentrations. With just CD63 sEVs, the voltage value was -ΔCNPV 48 mV. The device exhibits a LOD of 33 sEVs/μl, suggesting high sensitivity. The device was able to detect sEVs in 10 μl of serum from 6 prostate cancer patients compared to 8 healthy controls. The device detected greater numbers of sEVs from prostate cancer patients (-ΔCNPV of ~40–80 mV) compared to the healthy controls (-ΔCNPV of ~30–40 mV). Furthermore, the captured sEVs could dissociates from the probes and recapture with minimal loss of quality. The device provides rapid detection, requiring only 30 min incubation time with the sEVs, and minimal sample for detection. However, the device suffers from a lack of tumor-specific markers, thus the device may be able to detect the presence of a tumor, but not the type.
3.3. Summary
Direct on-chip detection of sEVs is optimal for clinical settings. On-chip detection should be rapid, sensitive, specific, and require little to no additional equipment. Devices should also be easy to operate to minimize user error. ELISA-based on-chip detection methods offer visible color changes to indicate the presence of the target, and generally take between 2 and 4 h to complete and exhibits high sensitivity, reporting a LOD ranging from 10 EV/μl to 1000 EV/μl [152,153,165]. Similarly, LFIA also takes about 2 h but is not as sensitive as ELISA [166]. SPR, on the other hand, has the potential to be label-free, though labels enhance detection by offering visible color shifts and may be more sensitive than traditional ELISAs [154,155]. Quantum dots provide colorimetric changes as well in the presence of bound EV targets, and can process small sample sizes, 10 μl, in about 20 min, with a high sensitivity of 100 EV/μl [156,157]. Lastly, other devices such as bio and nanosensors can produce results with minimal quantities of sample as well, 2–10 μl, within 30 min, and can be designed for specificity while maintaining sensitivity, with a LOD of femtograms [158,159]. These newer methods may offer alternatives to current diagnostics while utilizing sEVs as the detection medium. Thus, if sEVs are ever to become alternatives to current detection methods, much work is needed to streamline the process, including requirements for minimum sample volume, rapidity, and high sensitivity and specificity.
4. sEVs in cancer biology
Decades of sEV research has provided evidence showing sEV-mediated intercellular communication and crosstalk between healthy cells, tumor cells, and the tumor microenvironment. Tumor development and progression is marked by constant inflammation, giving rise to satellite instability and acquisition of mutations that alter the tumor at all stages, from commencement to progression, invasion, metastasis and even recurrence. Tumor-derived sEVs, and their contents, therefore, reflect the evolving tumor cells at all its stages. Because sEVs are so integral in intercellular communication, they are able to modify the tumor microenvironment, thereby promoting tumor progression as shown in Fig. 2. Angiogenesis, vascular leakage, formation of premetastatic niches, modulation of immune response and drug resistance are a few areas in which sEVs play a significant role in promoting tumor progression [19,36]. In this section, we therefore discuss, in detail, the role of sEVs in tumorigenesis and tumor progression over the last 5–8 years. Additional work done by peers beyond the timeline of this review can be referred [[167], [168], [169], [170], [171]].
Fig. 2.

Role of sEVs Cargo in Cancer Biology. Transfer of tumor derived sEVs cargo molecules alter recipient cell phenotype and thus cancer biology.
4.1. Role of sEVs in angiogenesis
Tumors rely on angiogenesis for nutrient acquisition and metastasis [172]. The tumor microenvironment, as well as the sEVs secreted by the tumor cells, promote vascular growth via excitation of endothelial cells [173]. For example, pancreatic cancer-derived sEVs have promoted angiogenesis via >1.5-fold change upregulation of Human Umbilical Vein Endothelial Cells (HUVECs) [174]. The increased expression of phospho-Akt and phospho-ERK1/2 in HUVECs, along with tubule formation caused by the dynamin dependent endocytosis behavior, demonstrates the angiogenic phenomenon of the pancreatic cancer derived exosomes. Angiogenesis is primarily caused by Vascular Endothelial Growth Factor (VEGF), a soluble proangiogenic factor secreted by endothelial cells, tumor cells, and tumor derived sEVs [175]. It has even been proposed that sEVs carrying VEGF may be major contributors to early tumor angiogenesis [176]. Researchers have demonstrated the activation of endothelial cells by tumor derived sEVs occurs via VEGF [177]. Activation of endothelial cells by sEVs enhanced proliferation, migration, and growth of endothelial cells. The primary aortic ring (AoR) supported this claim as exosome mediated activation of endothelial cells showed approximately 2-fold change in AoR on sEVs treatment. In addition, Dio-Ac-LDL results showed that the recovery of endothelial cells was increased after coculture with AS-Tspan8 exosomes. This suggests a crucial role for sEVs in tumorigenic angiogenesis. Additionally, sEVs can cause tumor angiogenesis of MDA-MB-231 cells by the activation of a specialized type of VEGF receptors known as 90 kDa VEGF (VEGF90K) [178]. VEGF90K was formed by crosslinking VEGF165K and catalyzed by enzyme tissue transglutaminase, which interacts with sEVs via Hsp90 chaperone protein. The team further observed Hsp90 localized to sEVs VEGF from breast cancer cells and interfered with the effectiveness of breast cancer monoclonal antibody, Bevacizumab. This suggests that VEGF-activated Hsp90 promotes tumor viability. However, inhibition of Hsp90 by 17AAG (17-N-allylamino-17-demothoxygeldanamycin) in the presence of EV-VEGF90K rendered the tumor cells sensitive to treatment with Bevacizumab. Similar results were observed using MDA-MB-231 tumor xenografts (p-value 0.001). Blocking Hsp90 in conjunction with anti-cancer drugs may therefore provide therapeutic benefit. Other sEV proteins which are known to promote angiogenesis, and could therefore be targets of anti-angiogenic drugs, include carbonic anhydrase 9, annexin II, myoferlin, and WNT4 [[179], [180], [181], [182]]. sEV carbonic anhydrase 9 in Renal Cell Carcinoma aids angiogenesis by promoting migration (p < 0.01) and tube formation (p < 0.05) associated with upregulation of MMP2 in-vitro, however requires validation in-vivo [179]. Likewise, overexpression of sEV annexin II in malignant and premetastatic breast cancer cells promotes tPA-dependent angiogenesis (pre-treatment with tPA antibody decreased the endothelial tube formation by approximately 3 folds with compared to control) both in-vitro and in-vivo. The underlying mechanism involved sEV Annexin II causing macrophage-mediated activation of the p38MAPK, NF-κB, and STAT3 pathways and increased secretion of IL6 and TNFα. Furthermore, in-vivo analysis revealed metastatic exosomes can create a microenvironment favorable for metastasis. Priming with Annexin II depleted sEVs reduced brain (~4-fold) and lung (~2-fold) metastasis [180]. Whereas depletion of myoferlin, commonly overexpressed in cancers and found in sEVs from different breast and pancreatic cancer cell lines leads to a significantly modulated sEVs protein load. Such sEVs have shown reduced functionality in transferring nucleic acids to HUVEC and they also have reduced capability to cause migration and proliferation in HUVEC (Breast Cancer: migration p < 0.02, proliferation p < 0.001 Pancreatic Cancer: migration p < 0.001, proliferation p < 0.001) [181]. In colorectal cancer, sEVs mediate Wnt/β-catenin signaling in endothelial cells under hypoxia promoting tumor growth and angiogenesis as sEV Wnt4 increases β-catenin nuclear translocation in endothelial cells. β-catenin is a critical player in signaling of proliferation and migration of endothelial cells inhibited by ICG001 [182]. In addition to the proteins, sEV RNA also plays a significant role in angiogenesis. Currently, sEV miRNAs are being studied extensively to understand their role in tumor angiogenesis. A study showed that delivery of EV-miR-130a from gastric cancer cells to HUVECs promoted angiogenesis and tumor growth by targeting c-MYB in both in vitro and in vivo models [183]. C-MYB functions as a transcription factor and plays an important role in angiogenesis and various biological processes like cell proliferation and migration [[184], [185], [186]]. Therefore, based on the role of c-MYB in angiogenesis, C-MYB expression is positively correlated with prognosis of gastric cancer. However, the overexpression of EV-miR-130a resulted in significantly lower expression of its downstream target c-MYB promoting poor prognosis of gastric cancer. Likewise, leukemia derived extravesicular miR-92a, when transfected with Cy3, enhanced the migration and vessel formation in endothelial cells [187]. The downstream target was identified as integrin α5 however, the underlying signaling pathway was not fully identified. On the other hand, miR-92a has been reported to inhibit angiogenesis under some conditions [188,189]. Therefore, tumor sEVs are known to affect the angiogenic process by engaging in intercellular communication via the various cargos which exosomes shuttle. In addition, conditions such as hypoxia [190,191] and loose cell to cell [192] contact have also been documented for facilitating the angiogenic properties of tumor cells. However, earlier studies which report sEV involvement in these processes may need to be revisited in light of newer sEV isolation techniques which yield purer sEV samples such that accuracy of these earlier observations is maintained.
4.2. sEVs mediate vascular leakage in cancer
It is estimated that 90% of all cancer related deaths occur due to metastasis [193]. Vascular leakage is considered one of the major factors promoting metastasis [194]. During metastatic progression tumor soluble factors like VEGF, Angptl4, CCL2, SDF-1, etc. activate different signaling pathways that weaken the inter endothelial junctions increasing the vascular permeability [193]. This defective vascular anatomy allows cancer to metastasize and colonize at distant locations [195]. Therefore, vascular leakage leading to tumor angiogenesis and progression is of concern [196]. sEVs have also shown to play an important role in mediating vascular leakage. Currently, tumor derived sEVs are being extensively studied as they can alter cellular signaling that potentially triggers vascular leakage [197] and recruitment of bone marrow progenitor cells [198]. Some research groups have successfully described the underlying process and/or mechanism by which sEVs cause vascular leakage. For example, miR-105 from breast cancer derived EVs targets tight junction protein ZO-1, destroying the natural barrier of tight cell to cell contact against metastasis [192]. Over expression miR-105 in non-metastatic cancer cells can cause metastasis and vascular leakage to distant organs. Similar results were obtained while studying lung cancer derived small extravesicular miR-23a [199]. Delivery of small extravesicular miR-23a to endothelial cells inhibited the expression of tight junction protein ZO-1, favoring migration and prognosis. Whereas in colorectal cancer, small extravesicular miR-200c, miR-141 and miR-429 have been reported to alter the vascular barrier by regulating ZEB proteins [200]. ZEB proteins are known to induce EMT of epithelial cells into migratory mesenchymal cells. E-cadherin is considered the downstream target of ZEB and downregulation of E-cadherin is also considered as a hallmark of EMT. In epithelial cells, E-cadherin serves as cell adhesion molecule therefore its downregulation in tumor metastasis indicates reduction in cell adhesion of tumor cells facilitating migration and invasion [201]. Collectively, activity of sEV miRNAs from tumor cells in these studies have shown to impact tumor prognosis due to loss of tight junctions. Vascular leakage is also considered as a characteristic behavior leading to formation of pre metastatic niches in metastatic cancers [202]. In brain metastasis, the Blood Brain Barrier (BBB) is considered as the most important vascular barrier. It functions in restricting the penetration of various molecules into the brain and thus aids in preventing potential metastatic progression of the brain to a certain extend. Under such strict conditions, permeability of cancer cells or cancer derived sEVs can likely be associated with metastasis phenomena [203]. For example, transport of Semaphorin 3A (in Sema3A/NRP1-dependent manner p < 0.05) and VEGF-A factor (targeting brain endothelial cells p < 0.05) from glioblastoma derived sEVs promoted vascular permeability, leading to metastasis of distant organs [204,205]. However, modulating the permeability of these cancer cells has allowed researchers to use sEVs as a therapeutic tool in case of brain metastasis [206]. Researchers believe that having a better understanding of exosomal functions would help in understanding the mechanism underlying brain metastasis. Various research reports claim that small extravesicular miRNAs (let7 family and miR-34a) affect the homeostasis of the tight junctions of blood brain barrier [[207], [208], [209]]. MiR-34a is reported to mediate BBB through a mitochondrial mechanism. Cytochrome c was identified as the downstream target of miR-34a. In vitro studies reveal that miR-34a targets the breakdown of BBB in the cerebrovascular endothelial cell monolayer. This occurs because of reduced mitochondrial oxidative phosphorylation, adenosine triphosphate production, and decreased cytochrome c levels. Similarly, sEV miR-181c from brain metastatic breast cancer cells can destroy the blood brain barrier and promote brain metastasis [210]. This finding was supported by analyzing the expression of miR-181c in sera from breast cancer patients. MiR-181c in sEVs collected from brain metastasis patients (serum was significantly higher p < 0.05) compared with non-brain metastasis patients. The mechanism leading to destruction of the BBB was demonstrated to be the degradation of PDPK1 gene (responsible for actin dynamics) by miR-181c. Together, these findings suggest that sEV miR-181c derived from metastatic brain tumor cells trigger abnormal PDPK1-regulated actin localization as miR-181c interacts with endothelial cells, thereby resulting in a weakening of the cell-to-cell contact. Vascular leakage is marked by increased permeability and directly contributes to metastasis. These scientific findings have shown that sEVs play a crucial role in promoting leaky vessels. Thus, the mechanisms contributing to vascular leakage are currently under study to determine optimal therapies. Combination tumor therapies should therefore include drugs that would target and mitigate vascular leakage. Further research is necessary in order to fully understand the mechanisms by which sEVs mediate vascular permeability and how they interfere with tight cell-to-cell junctions.
4.3. Communication of sEVs with neighboring non-tumor cells
The tumor microenvironment is a highly potent and complex region surrounding the tumor cells. It aids in their growth, invasion, and metastasis [211]. The tumor microenvironment is comprised of the tumor cells as well as the cells which support tumor growth. These include fibroblasts, immune cells, adipose tissue, blood and lymphatic vessel networks, and the extracellular matrix [212]. Researchers even claim that non-tumor cells can potentially account for more than 50% of the net tumor mass [213]. In addition, the tumor microenvironment also constitutes complex communication pathways between the tumor cells and the cells which support the tumor. EVs are known to play a key role in these communication pathways. A group of researchers showed that sEVs derived from several types of cancers (mesothelioma, prostate cancer, bladder cancer, colorectal cancer, and breast cancer) were able to communicate with normal stromal fibroblasts [214]. They observed that TGF-β1 from these tumor cells sEVs caused the fibroblasts to differentiate into myofibroblasts, which are important in degrading the extracellular matrix and contributing to metastases. Several other research groups reported that transforming growth factor β (TGF- β) from tumor derived sEVs are also capable of converting normal fibroblasts to Cancer Associated Fibroblasts (CAFs) [214,215]. Transformation of normal fibroblasts to CAFs was also seen in adipose tissue derived mesenchymal stem cells on treatment with breast cancer derived sEVs. Breast cancer derived exosomes were capable of causing this change by increasing the expression of TGF- β (average fold change ~3.5 folds), vascular endothelial growth factor (VEGF) (average fold change ~5 folds), stromal cell-derived factor 1 (SDF-1) (average fold change >20 folds), and C–C motif chemokine ligand 5 (CCL5)) (average fold change >10 folds) [216].
The effects of cancer derived sEVs are seen on both tumor cells as well as non-tumor cells such as endothelial cells, fibroblasts, and other healthy tissue cells. sEVs released from glioblastoma promote primary tumor growth and endothelial cell proliferation (p < 0.002) by delivering the EGFRvIII mRNA, miR-21 and angiogenic protein (angiogenin, IL-6 and IL-8) contents within them [217]. They stimulate an angiogenic phenotype in normal brain endothelial cells which initiates the proliferation of other glioma cells. The angiogenic proteins contained within them aids this process (angiogenin, FGF., IL-6, IL-8, TIMP-1, VEGF and TIMP-2). These angiogenic proteins potentially interact with cognate receptors on the surface of endothelial cells to promote angiogenesis. Under certain conditions sEVs undergo lysis to release the proteins within them. This process is supported by the acidic environment caused by tumor [218]. Whereas angiogenic proteins like angiogenin require transportation across the membrane to cause a biological effect, which could be facilitated by the microvesicles [219]. From the mRNAs found by the research team in glioblastoma sEVs, EGFR mRNA in particular interested them as EGFRvIII mutant splice variant is found in many glioblastomas and could serve as a biomarker [220]. This suggests the use of tumor sEVs as a multicomponent delivery vehicle for mRNA, miRNA and proteins to communicate genetic information as well as signaling proteins to surrounding cells. Similarly, transfer of RNA molecules (Hsa-miR-574–3p, Hsa-miR-501–3p, Hsa-miR-1290, Hsa-miR-2110, Hsa-miR-107, Hsa-miR-331–3p, Hsa-miR-375, Hsa-miR-21, Hsa-miR-625, Hsa-miR-301a, Hsa-miR-143–3p, Hsa-miR-196a-5p, Hsa-miR-196a-3p) from pancreatic cancer cell derived sEVs to recipient osteoblasts which facilitated communication between them [221]. This sEV mediated communication altered the functioning of osteoblasts such that it supports pancreatic cancer cell proliferation. Pancreatic cell derived sEVs improved the osteoblast viability along with a favorable environment for growth of pancreatic cancer cells when co-cultured with osteoblasts that were pretreated with sEVs. In addition, characterization of the RNA cargo of sEVs produced by the bone metastatic pancreatic cancer cell line was significantly enriched in genes relating to cell surface signaling, cell–cell interaction, and protein translation.
RNA molecules are most abundantly found in extravesicular cargo and thus are of interest amongst researchers [57,222,223]. Transfer of such exosomal mRNA and miRNA from mast cells to recipient cells is capable of regulating gene expression [31]. Likewise, researchers have observed the phenomenon of sEVs from miR-1227 enriched ameboid tumor cells of RWPE-2 prostate cancer cells (isolated from 200 μl of plasma) to enhance cancer associated fibroblast migration (fold change >1.5 when compared to control) [224]. Distinct classes of miRNAs were also expressed at higher levels in sEVs derived from the tumorigenic cells when compared to their non-tumorigenic counterpart. In addition, highly expressed miRNAs like miR-100–5p, miR-21–5p, and miR-139–5p from prostate cancer stem cells and exosomes from the bulk tumor on transfection into prostate fibroblasts, significantly increased the expression of MMP 2, MMP 9, and MMP 13, as well as receptor activator nuclear factor κB ligand (RANKL) expression leading to increased fibroblast migration [225]. Whereas in-vitro analysis of liver cancer derived sEVs containing miR-1247–3p have been reported to transform normal fibroblast to cancer associated fibroblasts (CAFs) promoting primary tumor growth as well as proliferation in lung tissue cells [222]. Since most studies involving miRNA mediated communication are done in vitro, it limits our knowledge of their functions in living systems. However, the role of miRNA in mediating cellular communication has been strongly supported by various research reports studying tumor growth, progression, and metastasis.
4.4. Role of cancer derived sEVs in immune regulation
The phenomenon of immune regulation is very crucial in inflammatory diseases like cancer [226]. Intercellular communication is considered key to immune regulation and thus sEVs play an important role. In addition, based on their origin from cancer cells, sEVs exhibit heterogeneities in their membranes as well as their cargo. These cargoes include growth factors, proteins, genes, cytokines, chemokines, and even miRNAs [227]. It is known that VEGF, IL10, and other soluble factors released by tumor cells and the tumor microenvironment can alter normal immune response [228]. Hodgkin lymphoma derived sEVs promote the activation of normal fibroblasts into cancer associated fibroblasts, which leads to the secretion of pro-inflammatory cytokines (IL-1α, IL-6, and TNF-α), growth factors like G-CSF and GM-CSF and even pro-angiogenic factors VEGF that support tumor growth [229]. Collectively, these factors promote Hodgkin lymphoma via TNF-α/NF-κB signaling thereby directing sEV dependent phenotype changes in fibroblasts. This sEV dependent transformation of normal fibroblast to CAFs was confirmed with a significant elevation in α-SMA expression within xenograft model treated with Hodgkin lymphoma sEVs compared to control (~3-fold change). Another unique study showcased the ability of Burkitt's lymphoma derived sEVs to activate CD8+ T cells by significantly improving the dendritic cell cross processing ability [230]. Recent studies suggest that tumor derived sEVs are also capable of modulating the dendritic cell property of eliciting an immune response [231,232]. Dendritic cells are important antigen presenting cells as they can coordinate both innate and acquired immunity. Therefore, alterations in processing of dendritic cells antigens and intercellular signaling can be done to promote a long-lasting anti-tumor response. Currently, the immunomodulatory influence of sEVs with MUC1 tumor glycoantigen has been studied on clinical grade DCs [230]. Phagocytosis plays an important role in antigen loading of dendritic cells used as cancer vaccines. In the phagosomal region, results indicate a reduction in phagocytosis (p < 0.01) along with increase in pH (from 7.01 to 7.35 pH) (p < 0.05) of dendritic cells upon interaction with tumor derived sEVs. The increase in pH suggests a reduction in antigen cross processing efficiency of the dendritic cells confirming an altered immune response in clinical grade dendritic cells. These functional changes in dendritic cells were compared to their normal functioning. Therefore, this study points towards the significant role sEVs play in dendritic cell immune modulation which can be used to develop immunotherapy vaccines. The transfer of non-coding RNAs is another direction that EVs follow to cause immune modulation. In colorectal cancer the transfer of miRNAs (miR210, miR193a, miR19a), long coding RNAs (MAGEA3, CRNDE-h) and circular RNAs (circ-KLDHC10, circRTN4) leads to their interaction with respective mRNA transcripts causing deregulation of the mRNA [[233], [234], [235]]. This causes their transformation into oncogenes that promote progression and metastasis of cancer. These deregulated mRNAs also modulate cells (epithelial, endothelial and fibroblast) responsible for causing an immune response and drug resistance within the tumor microenvironment by promoting their proliferation.
For sEVs derived from cancer cells, immune regulation works both ways i.e. in the form of suppression or activation of immune response against cancer. This claim is supported by study involving sEVs derived from metastatic melanomas carry programmed death-ligand 1 (PD-L1) on their surface [236]. This programmed death-ligand 1 interacts with the corresponding programmed death – protein 1 causing inactivation of CD8 T cells. Even stimulation with interferon-γ increases the amount of programmed death-ligand 1 on the surface of exosomes thereby suppressing the function of CD8 T cells and facilitating tumor growth. While some other studies claim that exosomes cause a cytotoxic T cell reaction which generates an antitumor immune response with the help of tumor specific antigens like CEA, HER2, mesothelin, CD24 and EpCAM present within them [[237], [238], [239]]. Likewise, sEVs from irradiated tumor cells appear to exhibit irradiated effects on neighboring nontumor cells [240,241]. Thus, it can be concluded that cancer derived sEVs have a variable function in immune regulation.
Toll-like receptors on the othr hand are a type of structural pattern recognition receptors which upon activation can promote cancer growth. These receptors are known to activate the NF-ϰB complex which leads to secretion of pro-inflammatory cytokines and miRNAs from sEVs in order to enhance the inflammation and cancer growth [175]. Similar results were observed while studying lung metastasis caused from liver cancer [222]. Here the activation of toll like receptors resulted due to the transformation of fibroblasts in myofibroblasts. However, production of multiple cytokines can potentially activate an anti-cancer response as they are chemical messengers regulating innate and adaptive immunity [242,243]. Cytokines are also known to enhance the detection of tumor cells with the help of cytotoxic effector cells. Stimulating immune effector cells and stromal cells helps in this process. In recent years animal studies involving cancer therapy have shown that cytokines can be translated into several cancer therapies. These findings are supported with cytokines like GM-CSF, IL-7, IL-12, IL-15, IL-18 and IL-21 that are being studied in clinical trials for patient with advanced cancer [243,244].
4.5. Cancer derived sEVs causing extracellular matrix (ECM) remodeling
The extracellular matrix is a macromolecule network which serves as a physical and biochemical support for neighboring cells thereby regulating their functioning [245,246]. Thus, extracellular matrix is considered as an important part of the tumor microenvironment. In cancer patients, it is subjected to dynamic changes resulting in remodeling of the ECM. Intercellular communicators are responsible for causing these dynamic changes to the ECM [202]. Researchers believe that remodeling of extracellular matrix causes quantitative as well as qualitative changes in the ECM which promotes growth and metastasis of cancer [247,248]. Fibrosarcoma derived sEVs promote cell adhesion and motility in tumor cells by matrix remodeling caused by small extravesicular fibronectin [249]. Remodeling of ECM also favors tumor-stromal interaction. In addition, the functional as well as molecular difference in normal cells demonstrated in response to sEVs from normal cells and cancer cells explains a strong molecular mechanism responsible for modulating the ECM related genes resulting in its remodeling [250]. BBOX1 and EFEMP1 are examples of such genes observed by researchers. Their overexpression when compared to control is reported to be responsible for cancer growth and invasion [251,252].
Small extravesicular cargo like Extracellular Matrix Metalloproteinase Inducer (EMMPRIN) has been reported to trigger the ECM remodeling by production ofMatrix Metalloproteinase (MMPs) in fibroblasts [253]. Moreover, tumor derived sEVs transform the fibroblasts into myofibroblasts which secrete MMPs. These MMPs then remodel the ECM by breaking it down and releasing the growth factor within them, promoting cancer proliferation and metastasis [202]. Another group of researchers reported similar results where introduction of Rab27b in metastatic breast cancer cells resulted in an exocytic secretion of sEVs, triggering MMP2 expression. This in turn facilitated cancer migration and invasion as a result of matrix remodeling [254]. Growth and progression of breast cancer cells can also be promoted by cancer derived sEVs [216]. Conversion of adipose derived mesenchymal stem cells to cancer associated fibroblasts by tumor derived sEVs resulted in ECM remodeling. Elevated expression of myofibroblast associated functional factor (α- SMA) in a dose dependent manner, along with tumor promoting cytokines (SDF-1, VEGF, CCL5, and TGF β), in the Adipose tissue derived Mesenchymal Stem Cells (ADSCs) on exosome treatment supported their findings.
These scientific studies suggest that tumor derived sEVs influences the biological behavior of cells and thus can remodel the ECM. However, its underlying mechanism is complex and still requires study.
4.6. sEVs initiate the formation of the pre-metastatic niche
While studying distribution of secondary growths in breast cancer, concept of “pre-metastatic niche” was introduced when tumor microenvironment was observed to be responsible for metastasis leading to invasion of different tumors to distant organs [255]. The ability of primary tumor to metastasize and spread to distant regions within the human body is supported by the formation of pre-metastatic niche. This occurs as a result of intercellular communication between tumor and stromal cells. In cellular communication, tumor derived extracellular vesicles are reported to contribute significantly to the formation of pre-metastatic niche by transferring the cargo content within them. However, the underlying mechanism remains unclear. It is still believed that sEVs while transporting their cargo, stimulate vascular leakage which allows them to selectively interact with cells from distant organs [256]. Melanoma derived sEVs promote vascular leakage in the lymph nodes leading to elevated levels of vascular growth factors (Tnfa, Tnfaip2, Vegfb, Hif1a, Thbs1) [257]. This ability of sEVs to modulate pathophysiological processes like vascular leakage, support pre-metastatic niche formation and subsequent metastasis. In addition, previous studies have also reported that hypoxic tumor derived exosomes promote angiogenesis as well as vascular leakage resulting in pre-metastatic niches causing tumor progression [191,258]. Exposure of breast cancer and glioblastoma cells to hypoxic conditions in these studies led to increased expression of tumor derived hypoxic sEVs to facilitate their growth and invasion. These hypoxic sEVs secrete growth factors, cytokines, miRNA (miR-210) that are known to promote angiogenesis and vascular leakage facilitating pre-metastatic niche formation.
sEVs are also able to promote the formation of pre-metastatic niches by upregulating the inflammatory molecules and suppressing immune response [259]. As inflammation favors tumor growth and metastasis, the inflammatory microenvironment is an important factor in formation of pre-metastatic niches. Production of VEGF, TNF-α, TGF-β IL-6, and IL-10 is stimulated by inflammatory microenvironment which results in migration of myeloid cells and ultimately forming pre-metastatic sites [260]. Tumor derived small extravesicular integrins are able to target distant organs by regulating pro inflammatory factors [261]. Tumor derived small extravesicular integrins are responsible for elevated levels of pro inflammatory factor S100, which promote cellular, as well as molecular, changes in distant organ cells. In addition, upregulation of pro inflammatory factors also results in recruitment of inhibitory immune cells (tumor associated macrophages, tumor associated neutrophils, regulatory T cells and myeloid derived suppressor cells) to distant organs and may potentially inhibit the antitumor immune response [259,262,263].
Collectively, these studies highlight the contribution of sEVs in metastatic disease progression by formation of pre-metastatic niches in distant organs. However, a thorough understanding of the mechanisms sEVs use to transfer specific cargo contents to alter biological processes and conditions favoring pre-metastatic niche formation is needed.
4.7. Organotropic metastasis by sEVs
Organotropism can be defined as biological changes in an organ due to an external stimulus. Moreover, it can be said that organotropic metastasis is an extension to pre-metastatic niches. Previously, various cellular components have been reported to cause oraganotropic metastasis. For example, high levels of chemokine receptors CXCR4 and CCR7 along with high expression chemokine receptive ligands CXCL12 and CCL21 in distant organs promote organotropic metastasis of lung due to breast cancer metastasis [264]. Similarly, periostin and tenascin are some other cellular molecules promoting organotropic metastasis of breast cancer and melanoma respectively [265,266]. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. MSI1-dependent NOTCH signaling was determined to be the underlying pathway supporting metastasis. Likewise, melanoma cells produce periostin as a key niche component for wound metastasis of melanoma. sEVs also play a vital role in promoting organotropic metastasis of various cancers. Breast cancer derived exosomal content like miR-181c, ave been reported to potentially cause organotropic metastasis in lung, brain, bone, and skin [210,267,268]. These breast-cancer-derived sEVs are also capable of redirecting tumor metastases from bone to lung by contributing to the pre-metastatic niche [269]. Small extravesicular integrins also explain organotropic metastasis. Pancreatic cancer derived exosomal Integrin αvβ5 stimulates metastasis of the liver whereas exosomal integrins α6β4 and α6β1 (confine to laminin rich regions of the lung) from 4175-LuT breast cancer cells stimulated lung metastasis [261,270]. Targeting the integrins helped researchers in decreasing the sEVs uptake and thus metastasis. Similar results were demonstrated where β1-integrin from hepatic cancer derived sEVss triggered lung metastasis [222]. NF-ϰB was identified as the signaling pathway. In addition to the studies discussed above and current scientific findings suggest small extravesicular integrins and proteins which cause organotropic metastasis can potentially be translated into clinical use as cancer biomarkers in the future.
4.8. sEVs promote coagulation/microemboli
In cancer patients suffering metastasis, the risk of developing thrombotic problems is extremely high. These thrombotic conditions are also reported to be a major factor contributing to deaths in metastatic cancer patients [271]. Thrombotic conditions result in biological changes caused by tumor cells that lead to coagulation as well as platelet accumulation. Cancer cells promote this biological condition as it helps prevent their recognition by immune cells [272]. Numerous reports claim that sEVs are involved in promoting the coagulation leading to cancer progression. For instance, microvesicles derived from either cancer cells, platelets or inflammatory cells are capable of facilitating coagulation [273]. Recently, researchers have observed podoplanin expressing sEVs in plasma of pancreatic and colorectal cancer patients [274]. Podoplanin is a membrane glycoprotein belonging to mucin type proteins that are expressed on the surface of cancer cells [275,276]. These proteins exhibit in-vitro platelet aggregation based on podoplanin expression [277]. Platelet aggregation from podoplanin expression usually results in coagulation and even venous thromboembolism (VTE) [278,279]. Hence, it can be said that tumor derived sEVs are potentially responsible for coagulation or microemboli. However, activated platelets have also been reported to cause accumulation of sEVs at thrombotic sites. This occurs as a result of P-selectin glycoprotein ligand 1 (PSGL-1) and integrins present on the surface of sEVs membranes [280]. Treatment with P-selectin blocking antibody, RGD peptide, and clopidogrel in animal models significantly inhibits the accumulation of sEVs [280,281]. Some other groups of researchers have also reported that in cancer patients with high thrombotic risk, an increased amount of microvesicular tissue factors which promote coagulation is seen [273,282,283]. A study demonstrated formation of arterial thrombus promoted the binding of activated platelets [284]. The platelets were transformed to an activated state by P-selectin and P-selectin glycoprotein ligand 1 (PSGL-1) dependent tissue factor enriched sEVs derived from monocytes. In general, metastatic cancer cells express approximately 1000 folds higher levels of tissue factor than non-metastatic cancers [285]. Overexpression of tissue factor also has a correlation with cancer prognosis and procoagulant activity of tumor cells in certain types of cancers [[286], [287], [288], [289]]. High levels of K-ras-dependent tumorigenic and angiogenic tissue factor in colorectal carcinoma derived sEVs were observed by researchers contributing to its poor prognosis [290]. Activation of K-ras oncogene and inactivation of the p53 tumor suppressor, in MEK/mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3′-kinase (PI3K) dependent manner were identified as the underlying mechanism. Based on scientific evidence it can thus be concluded that cancer derived sEVs significantly contribute to cancer invasion and metastatic progression by promoting coagulation. They are capable of triggering coagulation by expressing tissue factor, P-selectin ligands, etc. Tumor cells circulate in the blood stream and localize at metastatic sites. This causes platelet aggregation and is dependent on coagulation factors carried by sEVs, as mentioned earlier [289]. Therefore, alterations in sEVs to reverse coagulation and thus metastatic progression serves as a direction for development of therapies against cancer.
4.9. sEVs in epithelial to mesenchymal transition (EMT)
In invasive cancer, EMT is a characteristic phenomenon in which static epithelial cells transform into dynamic mesenchymal cells [291]. It is also believed that a relationship exists between sEVs derived from mesenchymal cells and the triggering of EMT in epithelial cells [292]. However, it is yet pending scientific validation. Overexpression of HRAS in Madin Darby canine kidney epithelial cells contributed to EMT and the selective packaging of mesenchymal markers, such as vimentin, into sEVs [292]. This selective packaging was noted to occur in response to reduction in thrombospondin-1 and epithelial markers E-cadherin, and EpCAM. Similar effects caused by linoleic acid, stimulated MDA-MB-231 derived sEVs in MCF10A epithelial cells [293]. In Normal epithelial prostate cell lines RWPE-1 and PNT-2 treatment with sEVs from biopsies of prostate primary tumors or plasma from prostate cancer patients, stimulated alteration in prostate cancer related gene expression, thereby promoting EMT as well as cancer progression in the cell lines [294]. A study demonstrated that sEVs shed from invasive bladder cancer cell lines (T24 and UMUC3) could induce the expression of mesenchymal markers (α-smooth muscle actin, S100A4 and snail) in primary urothelial cells [295]. Increase in mesenchymal markers was seen along with varied gene expression between the two cell lines. Also, down regulation of epithelial markers E-cadherin and β-catenin is closely related to bladder cancer progression [296,297]. Of these, E-cadherin is generally present in very low amount or completely absent in more than 80% of muscle-invasive bladder cancer [298]. As downregulation of E-cadherin causes release and translocation into the nucleus. This activates WNT signaling leading to EMT and cancer metastasis. Also, full length tissue factor (flTF) III and CD142 from tumor derived sEVs have promoted EMT and aggressive metastases in tumors [299]. Previous studies have reported that TGF-β from sEVs of tumors are important regulators in EMT [300]. The role of itinerant exosomes in EMT as the transport of morphogens and RNA is explored. This transport influences cell polarity causing EMT in-vivo, however it requires further investigation [301]. Extravesicular miR-200a, miR-200b, miR-200c, miR-429 and miR-141 from the miR-200 family are also well known regulators of EMT which modulate Zeb1 and Zeb2 expression [302]. sEVs obtained from melanoma cells induce EMT in primary melanocytes [303]. Importantly, EMT-derived sEVs mediated this through Let-7i. This finding was supported by evaluation of a Let-7i mimic in invasion assays and immunofluorescence evaluation. Moreover, MAPK pathway activation was observed following introduction of melanoma sEVs which induced EMT. EMT-associated miRNAs (miR-191 and Let-7a) were found to be enriched in serum sEVs obtained from melanoma patients in comparison with non-melanoma subjects. Similar findings were reported in which human lung adenocarcinoma A549 cells derived sEVs containing TGF-β1 induced EMT [304]. On EMT elevated miR-23a levels were seen that promoted malignant progression by enhancing the transcriptional activity and abundance of β-catenin. Taken together, small extravesicular protein and miRNA content can induce phenotypic as well as physiological changes via autocrine signaling. Therefore, developing strategies targeting sEVs released during disease condition or pathological processes might serve as a breakthrough in inhibiting various characteristics of sEVs. This in turn could provide an in-depth understanding of sEVs released during the EMT with key insights into the EMT process and therapeutic opportunities aimed at halting and limiting metastatic spread.
4.10. sEVs regulate therapeutic resistance
Evolution of cancer by sEVs mediated multi drug resistance is most commonly reported by scientists [305,306]. sEVs can facilitate drug resistance in several ways. The role of mesenchymal stem cell derived sEVs in promoting immune suppression as well as chemo resistance of gastric cancer is studied [307]. The molecular mechanism underlying this resistance involved sEVs mediated activation of calcium/calmodulin-dependent protein kinases (CaM-Ks) and Raf/MEK/ERK kinase cascade in gastric cancer cells. Whereas, multidrug resistance proteins like LRP and MRP are reported to be regulated by mesenchymal stem cell exosomes in gastric cancer cells fighting against 5 fluorouracil and cisplatin [308]. Small extravesicular RNAs have also been reported to enhance therapeutic resistance by educating the cancer cells to alter their chemo resistance to chemotherapeutic drugs. sEVs miR-1247–3p from hepatocellular carcinoma cells (HCC) demonstrated high resistance to chemotherapeutic drug sorafenib when compared to control [222]. These findings were further confirmed by blocking IL-6 or IL-8 with neutralizing antibody which partially reversed the resistance ability to sorafenib of HCC cells when incubated with media from fibroblasts pre-treated by miR1247–3p. In addition, the ability of sEVs to transfer the multi drug resistance ability from docetaxel-resistant variant of MCF-7 to drug-sensitive variant of MCF-7 is showcased by monitoring P-glycoprotein expression of MCF-7 variants [309]. P-glycoprotein overexpression is considered one of the mechanisms facilitating drug resistance by carrying a variety of anti-cancer and cardiovascular drugs along other immunosuppressants [16,310,311]. A significant increase in P-glycoprotein levels of drug sensitive MCF-7 cells on incubation with sEVs from the drug resistant variant of MCF-7. Together, sEVs from the drug resistant variant of MCF-7 were capable of educating drug sensitive variant of MCF-7 breast cancer cells with the help of intercellular communication facilitated by sEVs [309]. sEVs from tumor microenvironment also mediate communication with cancer cells improving their resistance capability against chemotherapy and radiation therapy [305]. Coculturing stromal cell derived sEVs loaded with 5′triphosphate enhanced transposable RNAs in breast cancer cells [312]. The interaction of stromal cells with breast cancer cells resulted in paracrine and juxtacrine signaling events to drive stroma mediated resistance. Such sEVs cargos can also promote antiviral signaling by activating pattern recognition receptor RIG – I. Upon activation, the expression of interferon related DNA damage resistance signatures like ISG15, IFIT1, MX1, OAS1, and STAT1 influencing radiation therapy and chemo resistance are enhanced in cancer cells sensitive to chemo and radiation therapy. Further it was also reported that paracrine antiviral and juxtacrine Notch3 pathways combine into STAT1 which enhances transcriptional responses to Notch3. This results in proliferative expansion educating sensitive cancer cells to develop resistance against chemo and radiation therapy [305,312]. However, the transfer of small extravesicular miR21 from malignant adipocytes and fibroblasts to ovarian cancer cells inhibits apoptosis and promotes chemoresistance by interaction with Apoptotic protease activating factor 1 (APAF1), known to facilitate tumor apoptosis [313]. In addition, cancer cells can not only transmit therapeutic resistance through horizontal transfer of sEVs cargo but also through drug efflux pumps. Small extravesicular P- glycoprotein, Multidrug resistance-1 (MDR-1), and ATP Binding Cassette Subfamily B Member 1 (ABCB1) are a few, well studied drug efflux pumps whose transfer exhibit MDR in prostate cancer, ovarian cancers, acute T lymphoblastic leukemia and osteosarcoma [[314], [315], [316], [317]]. sEVs have emerged as important modulators of drug resistance through a variety of mechanisms described above. Drug resistance is still one of the biggest hurdles in developing cancer therapies. Therefore, further development of an approach to determine the accurate biological composition of sEVs will not only explain their role in cancer with more details but also would aid in the fight against cancer.
4.11. sEVs in hypoxia
In inflammatory diseases like cancer, blood supply plays an important role in its growth and maintenance [318]. Lack of oxygen levels in cancer cells triggers the clinical condition of hypoxia [319]. Hypoxia stimulates vascular leakage, angiogenesis and immune suppression in cancer cells. Generally, in a hypoxic environment cancer cells tend to release extracellular vesicles stimulating angiogenesis of cancer [320]. However, the underlying mechanism of sEVs promoting angiogenesis is complex. In some tumors there is a periodic cycling of oxygen resulting in acute hypoxia [321]. Under such low hypoxic condition many cancer cell types secrete sEVs due to selective transport of miRNAs and proteins which promote angiogenesis and thus proliferation of cancer cells [319]. Small extravesicular miR-210 highly promoted angiogenesis of breast cancer cells under hypoxia by suppressing the target genes and activating endothelial cells by miR-210 uptake by HIF-1alpha/VHL transcriptional system but not HIF-2alpha [322]. Similarly, hypoxic melanoma derived sEVs transfer exosomal miR-9 to endothelial cells triggering angiogenesis via JAK-STAT signaling [323]. Hypoxic cancer cells have been observed to trigger pro-angiogenic pathways due to the release of cancer derived sEVs leading to triggering of TF/VIIa–dependent activation in a paracrine manner [324]. Activation of VEGF by sEVs derived from hypoxic glial cancer cells due to changes in pro-angiogenic pathway is one such example [325]. Additionally, glial cells stimulated with hypoxic sEVs showed a significant upregulation of small nucleolar RNA, C/D box 116–21 (SNORD116-21) transcript among others while significantly downregulated the potassium voltage-gated channel subfamily J member 3 (KCNJ3) message. Similarly, sEVs which transferred EGFR to endothelial cells also promoted angiogenesis by activating VEGF/VEGFR2 pathways [326]. The transcription factor hypoxia inducing factor (HIF), is also considered to be a major contributor to tumor progression. It is responsible for managing angiogenesis, apoptosis and therapeutic resistance. Recently, it was demonstrated that small extravesicular HIF1α supports the invasive potential of nasopharyngeal carcinoma. A type of oncoprotein known as latent membrane protein 1 (LMP1) is responsible for facilitating conditions which increase the HIF1α in exosomes derived from glial cancer cells [327]. HIF also conditions tumor cells for the stress of a hypoxic environment [320]. In conclusion, hypoxic sEVs from cancer cells promote cancer invasion and progression by stimulation of various physiological conditions like angiogenesis. Therefore, investigating methodologies to activate downstream hypoxia tolerance mechanisms in target cell sEVs will serves as a future direction for prevention on cancer.
5. sEVs DNA contents
Interest in exploring the genetic material present within sEVs cargo has grown with the knowledge that sEVs can facilitate horizontal gene transfer [328,329]. Research reports have demonstrated the presence of DNA in sEVs [28,29,330]. This small extravesicular DNA is classified as genomic (gDNA) or mitochondrial (mtDNA) based on their origin [[331], [332], [333], [334], [335]]. Whereas, the cell line and surrounding factors can be further used to classify them as single stranded (ssDNA) or double stranded DNA (dsDNA) [27,336]. DNA from sEVs have also been reported with possible origin from cell free DNA (cfDNA) found in apoptotic bodies [337,338], vesicles released by apoptotic cells [339], or cfDNA attached to the surface of sEVs [340,341]. Various studies report, sEVs DNA can be enclosed with the sEVs or may be present on the surface of sEVs [28,29,[342], [343], [344]]. DNA ranging from 100 base pairs (bp) to 2.5 kilobase pairs (kB) can be enclosed within sEVs [29]. Researchers have demonstrated that pancreatic cancer derived sEVs can carry >10 kb fragments of dsDNA [343]. These dsDNA fragments aid in the detection of mutations in KRAS and p53. In addition, serum sEVs from pancreatic ductal adenocarcinoma containing dsDNA is known to span all chromosomes. This enables the serum sEVs to be used in analyzing genomic DNA mutations. On the contrary, studies also demonstrate that DNA is mainly attached to the outer surface of sEVs [329,345]. Treatment of sEVs with nucleases like DNase I and Exonuclease III, result in reduction of the overall DNA from HCT116 derived sEVs (p < 0.05 and p < 0.01 respectively) [341]. Whereas, comparing DNA from untreated sEVs group with DNase pretreated sEVs group revealed a decrease in dsDNA longer than 2.5 kB as a result of enzymatic cleavage [29]. Suggesting that long dsDNA fragments are present in sEVs but may not be enclosed with the sEVs membrane protecting them from DNase activity. In addition, extravesicular DNA on the surface of sEVs functions to alter the potential of sEVs to attach to fibronectin [332]. This would potentially help in determining the interaction between sEVs and tumor microenvironment. On the other hand, dsDNA from prostate cancer cells stimulate mutations [334]. Analysis of genomic DNA fragments of MLH1, PTEN, and TP53 genes from the prostate cancer derived sEVs supported their discovery. TP53 and PTEN genes in sEVs are significantly mutated in both prostate cancers (localized and castration-resistant). Moreover, they also play a pivotal role in cancer initiation, progression and treatment resistance [346]. Similar observations were made while studying cell culture supernatants [29,334,343], plasma from tumor bearing mice [29], as well as serum and plasma from cancer patients [343,[347], [348], [349], [350], [351]]. Interestingly, knockdown of sEVs release in human fibroblasts and different malignant cell lines results in collection of damaged DNAs in the cytoplasm [352]. Such cytoplasmic DNA can be perceived by DNA detecting proteins, the actuation of which results in genomic DNA damage, cell senescence, or apoptosis (via stimulation of the cGAS/STING inflammatory pathway). This demonstrates that sEVs function to maintain cellular homeostasis by removal of damaged DNAs from the cytoplasm.
Currently, DNA packing in sEVs is being studied extensively. It is observed that extravesicular DNA from sources such as ectosomes, sEVs and apoptotic bodies have distinctive DNA sequences that are shared [334]. This sheds light on EVs having independent packaging mechanisms for their subtypes but remain to be fully determined. Therefore, while studying mechanisms of nuclear content loading to exosomes in ovarian cancer, researchers explored the relation of cancer cell micronuclei (markers of genomic instability) with nuclear contents in sEVs [353]. It was observed that 10% sEVs (from cancer cells) and <1% sEVs (from blood and ascites) carried nuclear contents. Genomic instability induced by topotecan and olaparib facilitated micronuclei and nuclear content production. In addition, a direct interaction between micronuclei and sEVs markers such as Tetraspanins existed. Tetraspanins are largely found in sEVs and are thought to be responsible for trafficking cargo into these organelles but have been reported to contain a more limited collection of the diverse molecules present in the extracellular environment than assumed [[354], [355], [356]]. On the other hand, small EVs including exosomes do not vehicle DNA release even though dsDNA can be obtained as a byproduct from ultracentrifugation and sucrose density gradient purification [352]. An autophagy and multivesicular endosome-dependent, but exosome-independent mechanism has been reported to be responsible for such extracellular DNAs [356]. It is due to the presence of dsDNA and histones intracellularly in CD63 positive compartments that are true to the size of multivesicular endosomes and that intracellular CD63-and LC3B-decorated compartments may vehicle their dsDNA cargo to the plasma membrane. Additionally, tumor derived sEVs have been reported to carry dsDNA using dsDNA-specific shrimp DNase and atomic force microscopy (AFM) [29]. Interestingly sEVs DNA is known to represent the entire genome and can also dictate of potential cancer progression. Taken together, the ability of sEVs to carry DNA allows for them to be harvested and analyzed via liquid biopsies, offering a less invasive alternative to the more traditional biopsies used to monitor the genetic changes in patients during disease progression [334,357].
6. sEVs in cancer liquid biopsy
In 2020, The American Cancer Society estimates a diagnosis of about 1.8 million new cancer cases and 606,520 cancer deaths in the United States. The increasing morbidity and mortality rates inspire scientists to further develop more effective and less invasive diagnostics. Accurate detection of malignancies at an early stage will not only ameliorate the survival rate but also contribute significantly to the quality of the patient's life. Currently, Biomedical Imaging techniques such as Computed Tomography (CT), Magnetic Resonance Imaging (MRI) and pathological fine-needle aspiration biopsy are being used for detection of cancer [358,359]. However, aspiration tissue biopsy is invasive and can cause bleeding, squamous metaplasia, fibrosis, and necrosis during collection [360]. Additionally, medical imaging results are not always reliable as they show some benign lesions which appear to have similar traits as malignant ones, making the medical imaging-based differential diagnosis challenging [361]. These shortcomings impel scientists to develop new strategies for diagnosis of cancers. In recent years, liquid biopsy has emerged as a viable and newer method for early cancer detection. Like traditionally used needle biopsy, this technique allows for diagnosis and treatment of cancer [362]. Liquid biopsies allow for the analysis of blood, plasma, and serum for tumor detection [363]. Through liquid biopsies, it is possible to identify circulating tumor cells, sEVs, and tumor-derived cell-free DNA. In addition, it allows for real time monitoring of patients undergoing cancer treatment by analyzing the effectiveness of the treatment and identifying therapeutic targets. Because sEVs are mediators of cellular communication and carry various tumor signatures, they exhibit an advantage over the traditional methods of cancer diagnosis. For example, liquid biopsies of small volume (1–2 mL of sample) can be examined for tumor derived sEVs [364]. Therefore, sEVs and sEVs-derived molecules have been adopted as additional markers for diagnosis in cancers, such as prostate cancer, breast cancer, lung cancer, and ovarian cancer [[365], [366], [367], [368]]. In this section, we will further discuss, in detail, the various small extravesicular biomarkers in cancer detection.
6.1. Identified sEVs DNA biomarkers
Over last 60 years, there has been no new RNA and protein marker translation into clinical use as DNA mutation is considered unequivocal for cancer diagnosis and precision therapy when compared to protein or RNA biomarkers. While exploring sEVs cargos, several research groups have demonstrated the presence of sEVs-DNA, including mitochondria DNA (mtDNA), single-stranded DNA (ssDNA), double-stranded DNA (dsDNA) [[27], [28], [29]]. sEVs DNA is highly involved in the tumor-favorable effects, like formation of pre-metastatic niche, modulation of extracellular space for cancer progression, etc. Additionally, detection of oncogenetic mutations in sEVs DNA has made researchers hypothesize their role as a biomarker for cancer. Initially, studies reported traditional cell-free DNA (cfDNA) isolated from blood useful for identifying genetic mutations [369]. As cfDNA is isolated from dying cells, it results in a reduced sensitivity that can lead to difficulties in identification of specific mutations [370]. Thus, sensitivity of cfDNA is reserved. However, the emerging sEVs-DNA, might provide a reliable and consistent alternative to replace cfDNA [29,371,372]. Additionally, latest study indicates vast majority of cfDNA mutations in cancer patients are highly correlated to leukocytes suggesting that current cfDNA based liquid biopsies are less reliable [373]. Of note, a study compared the mutations within sEVs DNA and fragmented circulating cfDNA collected from patients with NSCLC by using amplification-refractory-mutation-system-based PCR assay [374]. Their results showed no correlation between the level of sEV DNA and cfDNA. The detection sensitivity and specificity of using sEVs DNA as biomarkers for early diagnosis of NSCLC were 25.7% and 96.6%, respectively. This is superior to the cfDNA executing the same function for early diagnosis of NSCLC, which exhibited a sensitivity of 14.2% and specificity of 91.7%. In addition, sEVs derived from serum under various conditions including 4 °C, room temperature, and repeated freeze-thaw cycle were tested for the stability of sEVs and sEVs DNA. Results showed that serum sEVs and their DNA remained stable at 4 °C (24–168h), room temperature (12–48h), and repeated freeze-thaw circle (up to five times) [371]. Collectively, these studies demonstrate the use of sEVs DNA to be a promising diagnostic tool for cancer due to their better sensitivity, specificity, and stability. However, one of the biggest challenges for future cfDNA applications will be to elucidate the underlying biology of DNA release into the circulation. Table 2 compares forms of liquid biopsies (sEV, ctDNA, and CTC) for cancer diagnosis.
Table 2.
Comparison of three forms of liquid biopsies, including sEVs, ctDNA, and CTC.
| sEVs | ctDNA | CTC | |
|---|---|---|---|
| Origination | Liver tumor cells | Dying tumor cells | Live solid tumor |
| Required sample volume | ≤1 ml plasma | 5–10 ml blood | 5–60 ml blood |
| Sample storage | Plasma: −80 °C for years | Whole blood: ≤96 h | Whole blood: ≤96 h, a few hours for viable cells |
| Counting & DNA amount | 108-1011/ml plasma; 5–200 ng/ml plasma | 10–400 ng/ml plasma | 1-100 cells/7.5 ml blood; ~10 pg/cell |
| Integrity of DNA | Medium: >10k bp [343] | Low: 100–300 bp [375,376] | High: intact DNA |
| DNA analysis | Yes | Yes | Yes |
| RNA analysis | Yes | No | Yes |
| Proteomics analysis | Yes | No | Yes, but not at single cell level |
| Lipidomic analysis | Yes | No | Yes, but not at single cell level |
| FDA approved assays | Not available | Available | Available |
Over the years use of sEVs-DNA to diagnose cancers throughout copy-number variation detection, mutation detection has been studied extensively. Researchers have the copy number of mitochondrial DNA (mtDNA) within whole blood, circulating cell-free plasma, and sEVs collected from patients with late-stage ovarian cancer. The highest copy number was observed in sEVs followed by cell-free plasma and then whole blood (sEVs > cell-free plasma > whole blood), indicating the reliability of sEVs DNA-copy number in cancer diagnosis [377]. Additionally, levels of sEVs mtDNA are correlated with late stage cancer, indicating that mtDNA copy number variation exhibits potential for predicting cancer progression and metastasis [378]. Subsequently, a study determined that chromosomal DNA was abundant in large sEVs isolated from PCa patient's plasma. The copy number variation of mutated genes such as MYC, AKT1, PTK2, KLE10, and PTEN that was found in the PCa-derived sEVs was reflective of those mutations within the cells themselves. This demonstrates the vast potential for sEVs to be used in diagnosis as well as enabling a deeper understanding of the mutations driving a particular tumor [379]. Circular extrachromosomal DNA (ecDNA), a type of long stranded DNA has been reported to promote cancer heterogeneity and chemoresistance [380]. These ecDNAs have oncogenes encoded on them that are highly expressed genes in the transcription of tumors, linking increased copy number with high transcription levels. However, little is known about their ability to be wrapped into sEVs. In recent years, sEVs DNA has also been reported to facilitate diagnosis of ground-glass opacity (GGO), an early-stage lung lesion hard to be diagnosed in CT [381]. A protein-based molecular-affinity method (heparin/polymer coated microspheres) was developed to isolate sEVs with 81% isolation efficiency from patient's plasma using targeted sequencing researchers have further analyzed mutations in sEVs-DNA collected from patients with GGO and in tissue-DNA. Results show 35.4% of sEVs DNA mutations found in tissue DNA whereas 29% of tissue DNA is found in sEV DNA. The concordance between the two types of DNAs was 39.8%, and EGFR, TP53, and NF1 were identified to have higher expression than other oncogenes as well as antioncogenes associated with lung adenocarcinoma. This suggests the combinational protein based molecular affinity microspheres and targeted sequencing can potentially aid diagnosis of malignant GGO. However, sEVs DNA mutations are not exactly same as tissue DNA mutations. Biased tissue biopsies potentially cause these inconsistences, therefore use of liquid biopsies can serve as an asset by reflecting the overall status of tumors.
6.2. Identified sEVs RNA biomarkers
Since discovery of extracellular mRNAs and miRNAs, researchers have confirmed the function of RNAs in sEVs and have explored the various physiological and pathological phenomena mediated by them. Interestingly, sEVs RNAs are resistant to RNases from the body fluids [35]. Also, their biological cargo that is specific to the origin of the cells they emerged from can be detected at different locations within the body [35,382]. Together these factors allow for the use of small extravesicular RNA in cancer diagnosis and monitoring.
MicroRNAs (miRNA) are small, non-coding, and single-stranded RNA sequences with 18–22 nucleotides, crucial in regulation of protein coding genes [383]. In the past decade, researchers have determined a correlation between the expression level of miRNA and cancers [[384], [385], [386]]. In lung cancer derived EVs the reduced expression of let-7 from metastatic tissue samples is correlated with poor prognosis [385]. Likewise, let-7 and miR-18a from serum samples of multiple melanoma sEVs showed a significant impact on the overall and progression free survival [387]. This suggests that circulating small extravesicular miRNAs aid in distinguishing patients with poor prognosis. Interestingly, sEVs isolated from serum are identified to have the highest amount of miRNA compared to those in cell-free plasma, revealing that sEVs could be a reliable source of miRNA for biomarker determination in cancer diagnosis [388,389]. Similar findings were reported while studying Esophageal Squamous Cell Cancer (ESCC) where high level of exosomal miR-1246 from serum was observed when compared to biopsy samples of ESCC [390]. Elevated miR-1246 level in serum has been reported to serve as a diagnostic tool for ESCC with a sensitivity of 71.3% and specificity of 73.9%. Upregulation of exosomal miR-21 in serum from patients with ESCC compared to its expression in patents with benign disease without systematic inflammation is yet another example [391]. The expression level of small extravesicular miR-21 was also correlated with the advanced cancer stage, lymph node status, and tumor progression (TNM classification). Table 3 further summarizes various small extravesicular miRNAs as biomarkers influencing cancer growth and progression with changes in their expression levels.
Table 3.
miRNAs derived from sEVs as a biomarker for cancer diagnosis.
| miRNA species | Expression condition | Source | Cancer | Reference |
|---|---|---|---|---|
| miR-27a & miR130a | Increase | plasma | CRC | [22] |
| miR-7641 | Upregulated | SW620-cell lysates | CRC | [23] |
| miR-17–5p | Upregulated | serum | NSCLC | [392] |
| miR-451a | High upregulation in NSCLC patients with recurrence | Plasma | NSCLC | [393] |
| let-7d-3p & miR-30d-5p | Decreased expression level in CINI II + group when compared with the CINI I+ group | plasma | Cervical cancer | [394] |
| miR-1246 | Downregulated | Serum | Aggressive prostate cancer | [395] |
| miR-21.miR-222 & miR-124–3p | Higher in high grade gliomas | Serum | Glioma | [396] |
| miR-210 | Increase | Serum | Glioma | [397] |
| miR-223–3p | Higher in invasive ductal carcinoma in situ (DCIS) breast cancer | Plasma | DCIS breast cancer | [398] |
| 268miR-23b | Lower expression in patients than healthy people | Plasma | Gastric cancer | [399] |
| miR-6807–5p & miR-6856–5P | Higher expression | Urinary | Gastric cancer | [400] |
| miR-191, miR-21, & miR-451a | Elevated expression | Serum | Pancreatic neoplasm | [401] |
| 266miR-122 & miR-21 | Decreased | Serum | Hepatocellular carcinoma | [402] |
| miR-204–5p | Elevated | Urinary of transgenic mice overexpressing human PRCC-TFE3 fusion gene | Xp 11.2 Translocation renal cell carcinoma | [403] |
| miR-145 & miR-200c | Higher expression | Serum | Ovarian Cancer | [404] |
Functionally small extravesicular mRNAs not only translate into proteins but also dictate the transcriptional aspect of tumors to aid cancer detection extent [35,364]. One such example is the study involving urinary sEVs mRNA that aids in discriminating malignant patients with prostate cancer from benign patients [405]. Similarly, hnRNPH1 mRNA within serum sEVs derived from hepatocellular carcinoma (HCC) could distinguish HCC from chronic hepatitis B by the AUC value of 0.865 with 85.2% sensitivity and 76.5% specificity [204]. Additionally, the combination of hnRNPH1 mRNA with α-fetoprotein (AFP) to diagnose HCC, yielded an increase in AUC value to 0.891 with a sensitivity of 87.5% and a specificity of 84.8%, which is higher than hnRNPH1 mRNA alone. Researchers have also found small extravesicular mRNA to mediate metastasis in ovarian cancer [406]. MMP1 mRNA from ovarian cancer sEVs is reported to incite apoptosis in mesothelial cells resulting in peritoneal metastasis of ovarian cancer. This suggests the use of MMP1 carrying sEVs as biomarkers for early stage detection of peritoneal metastasis. Another example of liquid biopsy is for the accurate diagnosis of colorectal cancer. The combination of VEGF and CD133 mRNA from sEVs have been reported to demonstrate 100% sensitivity, 80% specificity and 93% accuracy [407]. Additionally, quantification of mRNA from urinary sEVs of prostate cancer patients has revealed androgen receptor splice variant 7 mRNA as a potential sEVs based biomarker [408].
Accumulating evidence suggests that RNA transcripts of more than 200 bp found in sEVs interact with DNA, RNA and protein molecules [409,410]. During their interaction changes in the expression profiles of such Long non-coding RNAs (lncRNAs) results in tumor growth and metastasis [411]. Furthermore, long noncoding RNAs have been investigated in circulation sEVs and are highly correlated with tumor stage and overall survival rate of patients [[412], [413], [414], [415], [416], [417]]. A study involving lncRNA growth arrest-specific transcript 5 (GAS5) collected from serum sEVs of patients with NSCLC (n = 64) and healthy control (n = 40) demonstrated that sEVslncRNA GAS5 was downregulated in NSCLC patients and inversely correlated with tumor size and advanced TNM classification [418]. ROC analysis further showed that the lncRNA GAS5 could be utilized to distinguish patients with stage I NSCLC with an AUC of 0.822. Also, exosomal lncRNA HOTTIP, another important lncRNA in cancer progression, derived from sEVs in serum of gastric cancer (GC) patients was analyzed using RT-PCR [417]. The results showed that HOTTIP was upregulated in GC patients (n = 160) compared to the healthy controls (n = 120), indicating the potential for HOTTIP as a biomarker for GC diagnosis. Availability of nucleic acid-based detection technologies like qPCR and RNA sequencing have proven to be an asset in cancer detection. Advances in these technologies would allow for having a more thorough understanding of sEVs RNAs and their role in early cancer detection. In recent years use of combinational technology integrating machine learning with nanofluidic technology to diagnose pancreatic cancer has allowed for better diagnosis efficiency [419]. However, with sEVs RNA based cancer diagnoses majority of the studies are limited to cell lines. In addition, clinical studies have sample size limited to 30 making clinical validation by FDA challenging. Thus, FDA approved sEVs RNA based technologies for cancer diagnosis are very limited (miRNA or other RNA).
6.3. DNA methylation in sEVs
DNA methylation is an epigenetic process. It involves covalent transfer of a methyl group (CH3) to the CpG dinucleotides in C-5 position of the DNA cytosine ring by DNA methyltransferases (DNMTs) [420]. This results in functional modification of genes (which do not alter the nucleotide sequence) and gene expression depending on how the methylation drivers are read and interpreted. Therefore, DNA methylation acts as a switch for transcriptional activation or repression of genes that are essential for development and proper cellular functioning. Till date, DNMT1, DNMT3A, DNMT3B, DNMT2 and DNMT3L are known mammalian DNMTs [[421], [422], [423], [424]]. Whereas activation induced cytidine deaminase (AICDA) and thymine DNA glycosylase (TDG) are examples of enzymes that demethylate DNA and protect unmethylated regions of mammalian genomes from de novo methylation [425,426].
Currently cancer is considered a genetic disorder, involving nuclear mutations in oncogenes that develop into tumors [[427], [428], [429]]. Typically, a tumor contains several driver genes that regulate tumorigenic characteristics. Functional genetic changes due to the nuclear mutations are noticed in almost all types of cancer cells. These genetic changes are also considered to be the cause of uncontrolled growth of cells, angiogenesis, enhanced vascular leakage, immune resistance, invasion, and metastasis typically observed in cancer [430,431]. Such functional changes in cancer related genes caused by epigenetic mechanisms like DNA methylation, histone modification and microRNA (miRNA), or long noncoding RNA (lncRNA) regulation are hot topics in cancer research [30]. Even though DNA methylation plays a pivotal role in biological processes, prominent and untypical forms of methylation are observed in cancers [432]. These changes affect the hallmarks of cancer and also suggest for DNA methylation as biomarker for early detection and treatment of cancer since they occur during early tumorigenesis [433]. In fact, DNA methylation is considered an alternative pathway to cancer [434]. Similarly, sEVs due to their complex bioactive cargo, can cause malignant transformation of normal cells. The significant contribution of sEVs is cancer initiation, growth, diagnosis, and treatment are well supported with numerous research studies discussed earlier. Additionally, research reports suggest DNA, RNA, and protein contents in sEVs can induce epigenetic changes by modulating methylation of the genome in recipient cells [33]. For example, osteosarcoma derived sEVs mediate global LINE1 hypomethylation in mesenchymal stem cells of osteogenic lineage, but not pre-osteoblasts [33]. Also, tumor cell DNA exhibits methylation at CpG region in the form of clusters and global hypomethylation at intergenic region of the genome. These methylation processes have also been reported to regulate physiochemical properties like hydrophobicity, adsorption towards metal surfaces and flexibility [[435], [436], [437], [438], [439]]. sEVs from leukemia cells have shown globally elevated DNA methylation levels in recipient cells [440]. Promoter regions of tumor suppressor P53 and RIZ1 genes were hypermethylated resulting in the elevated DNA methylation and to the increased level of DNMT3a and DNMT3b mRNA and protein. Interestingly, protein and mRNA levels of AICDA was also increased in recipient cells. Together these results suggest that leukemic progression can be promoted due to genomic instability in the recipient cells. Further, treatment of sEVs with RNase resulted in decrease of DNMT3a, DNMT3b, and AICDA levels confirming leukemia derived sEVs modulate methylation in recipient cell via microvesicular RNA transmission. In K562 leukemia cell line breakpoint cluster region-Abelson leukemia gene human homolog 1 (BCR-ABL1) onco mRNA was dominant. Collectively, sEVs can transmit enzymes responsible for both methylation and demethylation of recipient cells resulting in changes facilitating tumor initiation, growth and metastasis. Likewise, DNA methylation mediated decrease in small extravesicular miR-652–5p from esophageal squamous cell carcinoma (OSCC) is reported to be correlated with TNM stages, lymph node metastasis, and short overall survival [441]. Hypermethylation at the promoter sites has resulted in this change. Poly (ADP-ribose) glycohydrolase (PARG) and vascular endothelial growth factor A (VEGFA) were identified as the direct targets of miR-652–5p. Whereas miR-652–5p agomir delivery significantly repressed the tumor growth and metastasis and PARG, VEGFA protein expression in nude mice were also inhibited. This suggests the use of serum miR-652–5p as a tumor marker to predict the overall survival and as a therapeutic target in OSCC. In endometrial cancer, DNMT1 has shown to be capable of enhancing cancer cell metastasis by inducing EMT [442]. However, transfer of sEVs miR-148b from CAFs to endometrial cancer cells suppressed tumor metastasis by directly binding to DNMT1. These outcomes suggest the loss of miR‐148b in the sEVs of CAFs with a corresponding increase in the exchange of stromal cell derived miR‐148b may serve as a potential treatment to prevent endometrial cancer progression. There is a wealth of research on sEVs RNA and proteins, however only a few works have been done on sEVs DNA. EV DNAs are known to have similar solution and surface based properties to cellular gDNA, which indicates that they might carry similar methylation levels and patterns like their parent cell gDNA [443]. The physicochemical properties of sEVs DNA can also be utilized to develop a simple and multiplex liquid biopsy test for cancer. Researchers have even developed liposome-based model to improve the isolation of sEVs DNA that is free from cfDNA. This system evaluates the methylation dependent physiochemical properties of sEVs DNA that can be used to develop tests for detecting cancer sEVs DNA. In gastric cancers, the use of gastric juice DNA for molecular diagnostics is considered impracticable because the DNA is easily degraded by gastric acidity [444]. Hence, researchers have suggested the use of gastric washes to address this concern. Using gastric washes, they have further developed a method for early detection of gastric cancer by analyzing DNA methylation of MINT25 and sex determining region Y-Box 17 (SOX17) [[444], [445], [446]]. Additionally, sEVs from gastric juices of gastric cancer patients were looked at to determine if they contain significant amounts of tumor related methylated DNA [447]. Bisulfite pyrosequencing analyses for methylation levels of LINE1 and tumor related SOX17 gene revealed that methylated DNA is efficiently packed in sEVs. This was supported by comparing nuclear DNA and sEVs DNA from gastric cancer cells where LINE1 methylation was reduced and methylation levels of SOX17 were the same. sEVs DNA from gastric juices further enabled researchers to detect SOX17 DNA methylation that dictates the nuclear DNA methylation level of the corresponding tumor. Collectively, these findings shed light on the functional molecular content of tumor derived sEVs to carry tumor methylated DNA and their potential use for methylation analysis. However, the underlying mechanism for packaging of tumor methylated DNA is yet to be explored.
6.4. Identified sEVs protein biomarkers
Proteins constitute a large amount of the sEVs cargo and can be used in cancer diagnosis and treatment [448,449]. They possess good tissue permeability making them available throughout the human body for sampling [450]. Small samples volumes have been reported to be efficient for use in clinical diagnosis. Compared to serological markers small extravesicular proteins are identified to have high sensitivity and specificity [449]. Additionally, they also demonstrate greater stability due to protection from lipid bilayer membrane morphology [451]. Small extravesicular protein markers are therefore attractive targets for cancer detection and warrant great attention.
A major step towards treating disease like cancer is early detection. Phosphoproteins from plasma sEVs are used in early detection of various cancer types. Their characteristic potent nature and active phosphates in blood are the reason for their use as diagnostic markers [449]. Hundreds of phosphoproteins have been reported as diagnostic markers. Researchers have used techniques like Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) and Parallel Reaction Monitoring (PRM) to detect protein biomarkers. For example, X-box-binding protein 1 (NFX1), cGMP-dependent protein kinase 1 (PKG1), epithelial cell adhesion molecule (EpCAM), tight junction protein 2 (TJP2), nuclear transcription factor, and Ral GTPase-activating protein subunit alpha-2 (RALGAPA2), which showed significant upregulation in breast cancer patients [366]. Similarly, overexpression of Glypican-1 (GPC-1), a cell surface proteoglycan found in sEVs derived from tumor cells, helps in early detection of breast and pancreatic cancer [449]. In pancreatic cancer patients, GPC-1 positive sEVs have helped differentiate between early and terminal stages [34]. GPC-1 positive sEVs in blood of pancreatic cancer patients, increased significantly and were better prognostic markers compared to CA19-9. After resection of pancreatic lesions, GPC-1 positive sEVs have been reported to correlate with the patient's clinical outcome. On the contrary, GPC-1 has also demonstrated inability to distinguish between pancreatic cancers and non-cancerous controls [452]. Further the researchers found macrophage migration inhibitory factor (MIF) facilitating hepatic metastasis. Suggesting its use as an early stage diagnostic marker for hepatic metastasis of pancreatic cancer [34]. Additionally, MIF also serves as a prognostic maker and initiates pre metastatic niche formation for pancreatic cancer metastasizing to the liver [453]. Researchers have developed ultrasensitive and inexpensive nanoplasmon enhanced scattering (nPES) assay to quantify tumor derived sEVs from as little as 1 μl of plasma [154]. Antibody conjugated gold nanospheres and nanorods interact with sEVs that are captured by sEVs specific antibodies on a sensor chip to produce a local plasmon effect. Using this technology, ephrin type-A receptor 2 (EphA2), a pancreatic cancer sEVs biomarker was identified to distinguish healthy patient from pancreatic cancer patients. Recently, a microfluidic chip was designed to detect low levels of ovarian cancer derived exosome in plasma that is reported to be undetectable by standard microfluidic systems for biosensing [158]. The three-dimensional herringbone nanopatterns of this design allowed for increase in surface area, microscale mass transfer, drainage of boundary fluid, and increased efficiency as well as speed of exosome binding. Application of the device in differentiating ovarian cancer patients (2ul plasma sample) and controls expressing CD24, epithelial cell adhesion molecule and folate receptor alpha proteins suggested sEVs folate receptor to be a potential biomarker for early detection of ovarian cancer and that the nanolithography-free nanopatterned device should facilitate the use of liquid biopsies for cancer diagnosis. In addition to these advances, liquid biopsy samples in the form of saliva and urine exhibit an advantage over the traditional methods of cancer diagnosis. Research studies reported significantly higher expression of sEVs EGFR and leucine-rich alpha-2-glycoprotein (LRG1) from urinary sEVs in lung cancer patients [454]. Additionally, Mimecan, Cystatin-SA, transforming protein RhoA, Thrombospondin-1, Protein lifeguard 3, Azurocidin, Tetraspanin, CD81 antigen, Antileukoproteinase, Dipeptidyl peptidase 4, saliva sEVs proteins (Fold change greater than 2) are potential biomarkers for lung cancer patients [455]. Recently, researchers showcased sEVs proteins: alpha-2-HS-glycoprotein (AHSG) and extracellular matrix protein 1 (ECM1) as biomarkers for non-small cell lung cancer (NSCLC) [451]. These sEVs proteins were highly expressed and the diagnostic capacity with AUC values of 0.795 and 0.739 in NSCLC and early stage patients, respectively was reported, Similarly, various small extravesicular proteins have been identified as diagnostic and prognostic markers. sEVs proteins like CD24 and claudin-4, can fill in as promising biomarkers of ovarian malignancy [456,457]. Whereas, EGFR, EGFRvIII, and CD63 were identified in serum exosomes of glioblastoma patients [458]. Numerous melanoma-specific exosome proteins, for example, caveolin-1, were also distinguished in clinical examples [459]. Table 4 further summarizes various sEVs protein markers in cancer. Thus, it can be said that extravesicular proteins are not only aid in diagnosis and treatment of cancer but also help in real time monitoring and evaluating reoccurrence of cancer.
Table 4.
Exosomal Protein markers in Cancer.
| Cancer Types | sEVs Protein | Application | Reference |
|---|---|---|---|
| Glioma | IL13QD | Detection and Relapse | [460] |
| Cholangiocarcinoma | VNN1, CRP, FIBG, IGHA1, and A1AG1 | Diagnosis | [461] |
| Colorectal cancer | CD147 | Detection and diagnosis | [462] |
| Colorectal cancer | GPC1 | Diagnosis and treatment | [463] |
| Pancreatic cancer | ZIP4 | Diagnosis | [464] |
| Gastric cancer | EGFR | Detection and diagnosis | [465] |
| Pancreatic Cancer | MIF | Prognosis and treatment | [453] |
| Pancreatic Cancer | Vimentin | Diagnosis and treatment | [466] |
| Lung Cancer | BPIFA1, CRNN, MUC5B, and IQGAP | Detection | [467] |
| Lung Cancer | Vasorin | Treatment | [468] |
| Lung Cancer | LG3BP and PIGR | Diagnosis | [461] |
| Lung Cancer | NY-ESO-1 | Diagnosis | [469] |
| Breast Cancer | Her2 | Diagnosis | [470] |
| Breast Cancer | PKG1, RALGAPA2, NFX1, TJP2 | Diagnosis | [366] |
| Colorectal Cancer | CEA | Diagnosis | [471] |
| Prostate Cancer | PSA | Diagnosis | [472] |
| Prostate Cancer | GGT1 | Diagnosis | [473] |
6.5. Identified small extracellular lipid biomarkers
To identify diagnostic and prognostic biomarkers, protein and nucleic acid content of sEVs has been thoroughly studied [474]. Likewise, various research groups have broken down the sEVs lipid profile to better understand the lipid signature related to diseases [475]. Lipids are known to play a role in sEVs release and their protection, additionally they also serve as biomarkers [476]. During sEVs release the fatty acyl composition of lipids dictates their structure to be conical (head group at cone end) or that like lysolipids [477,478]. Whereas the lipid from the sEVs provide better stability in varied extracellular environments and facilitates their uptake by recipient/target cells [476].
Over the years lipidomic studies have reported several small extravesicular lipids as promising biomarkers [[479], [480], [481], [482]]. Recently, a high-throughput mass spectrometry quantitative lipidomic analysis was performed to profile lipids within urinary exosomes from patients with prostate cancer and healthy individuals [480]. 107 lipid species were quantified in urinary exosomes. The 107 lipid species were enriched for 36 of the most abundant lipids. These 36 were then analyzed in prostate cancer patients (n = 15) and healthy individuals (n = 13). Results exhibited significant differences in 9 of these lipid species between the cancer and healthy patients, with phosphatidylserine (PS) and LacCer exhibiting the highest incidences. When PS and LacCer were combined, they were able to discriminate between the two groups with 93% sensitivity and 100% specificity. A study also revealed 277 lipid species that were profiled from the cell and sEVs of malignant prostate cancer cell [483]. Among them, a great enrichment of glycosphingolipids, such as Hex and lactosylceramide (LacCer), were identified, showing high potential for markers in diagnosis of prostate cancer and potentially other cancers, as well. In addition, comparison of lipid composition from serum sEVs of pancreatic cancer patients has revealed around 270 lipids that were significantly dysregulated between the serum exosome of PC patients and healthy controls [482]. Importantly, LysoPC 22:0, PC (P-14:0/22:2) and PE (16:0/18:1) were associated with tumor stage, CA19-9, CA242 and tumor diameter. Also, PE 16:0/18:1 was found to be significantly correlated with patient overall survival. However, the evaluation of the data presented by researchers in the form of relative signal intensities of m/z signals is challenging. This is because it can contain a combination of fatty acyl groups which were not thought of being present in cellular membranes. Small extravesicular lipids from plasma of multiple sclerosis (MS) patients and healthy volunteers has also been analyzed by scientists [484]. Results show sulfatides, belonging to glycosphingolipids class, that are highly expressed in brain were found in these sEVs. Also, sEVs that were isolated from MS patients showed higher levels of sulfatide C16:0 compared to healthy controls. Likewise, SM 34:1 was reported to be more abundantly found in sEVs from secondhand smoke exposure asthmatics compared to healthy controls [481].
7. sEVs in cancer therapeutics
Extracellular vesicles are known to engage in crosstalk between both nearby cells and distant tissues [[485], [486], [487], [488], [489], [490]]. The cargo held within EVs includes mRNA, miRNA DNA, lipids, proteins, and tRNA. These components, when taken up by a distant cell, can enact a cellular response [491]. Similarly, EVs can stimulate or dampen the immune response in the same way by transferring their contents from an antigen-presenting cell (APC) to a T-cell or vice versa. In fact, a study examined the effects of exosomal miR-335 derived from T-cells When the exosomal miR-335 from the T-cells was transferred to dendritic cells, a decrease in the expression of SOX4—a gene necessary for B-cell maturation—was observed [492]. This is an example of immune dampening. Likewise, EVs can stimulate the immune response. It is well-established that dendritic cells can take up exosomes loaded with MHC class II molecules and use that exosome-derived MHC class II to activate Helper T cells, thereby stimulating an immune response [[493], [494], [495], [496], [497]]. Recently, it was observed that DC-derived exosomes could directly stimulate CD8 T-cell response [498]; CD8 T-cells give rise to cytotoxic T-lymphocytes which are necessary for tumor cell destruction [499]. Inadvertently, tumor-derived exosomes can result in the suppression of CD8 T-cells and contribute to tumor progression [500]. These interactions between EVs, immune cells, and target cells suggest promise for use in cancer therapeutics, and thus, this section aims to discuss how sEVs may be used as novel cancer treatments (Fig. 3).
Fig. 3.

sEVs in cancer therapy. Naïve T-cells can be activated by sEVs from tumor and tumor-educated immune cells, thereby priming them against tumors. sEVs loaded with chemotherapeutics and toxic proteins can be used to deliver therapy directly to tumor cells, thereby avoiding the systemic toxicity that is common when patients undergo therapy. Drugs which interfere with sEV formation can also be delivered to tumor cells. This prevents tumor cells from creating sEVs and may result in inhibiting tumor progression.
7.1. Vaccines
Using EVs for vaccines against cancer has existed since the 1990s, though the early studies were met with limited success [[501], [502], [503], [504], [505], [506]]. The basic premise regardless, remains the same. To prime the immune response against a tumor, a specific subset of T-lymphocytes, the CD8+T-cells, need to be activated. Priming of naïve CD8+ T-cells requires stimulation of the T-cell receptor (TCR) with the antigen-loaded MHCI of the APC and co-stimulation of APC CD80 and CD28 of the naïve CD8+T-cell [507]. This results in proliferation of the cytotoxic lymphocyte (CTL). Further release of specific cytokines such as Interleukin-12 (IL-12) and interferon alpha (IFNα) by the APC actuate CTL effector functions. CTL effectors kill tumor cells by releasing tumor necrosis factor α (TNFα), IFNγ, and cytolytic granzyme B. Additional release of interleukin-15 (IL-15) results in the development of memory CTLs. Vaccines are useful in developing these memory lymphocytes.
Extracellular vesicles released by tumor-activated DCs are often loaded with these immune-activation receptors and cytokines and thus may be useful in priming the innate and adaptive immune responses against tumors [508]. One study utilized such a method to prime CD4+ helper T-cells (TH) against human epidermal growth factor receptor 2 (HER2)-positive, trastuzumab-resistant metastatic breast cancer [509,510]. These DC-derived sEVs were introduced to immature TH cells. These sEVs expressed MHCI with tumor antigen and CTL activation receptors CD80 and CD40L as well as cytokine, IL-2, which is necessary for differentiating T-cells into effector and memory cells. TH recycled the exosomal MHCI + tumor antigen complex as well as CD80 and CD40L and fused those receptors to their own membranes. Furthermore, the TH were able to express IL-2, thereby promoting the activation of new CTLs and the rescuing of exhausted CTLs. The reactivation of these CTLs led to a complete inhibition of tumor growth and inhibited tumor formation upon later challenge in athymic mice, thereby suggesting the efficacy of this type of vaccination. Similarly, EVs isolated from the ascites of T-cell lymphoma (TCL)-bearing mice were used to vaccinate naïve mice. This elicited an anti-TCL immune response in the vaccinated mice, but did not invoke an anti-mammary tumor response, thus suggesting specificity of this anti-tumor vaccine [511].
Another type of vaccine used exosomes derived from non-metastatic melanoma cell lines (ExoNM) [512]. These ExoNM were shown to inhibit melanoma metastasis to the lungs in mice by nearly 10-fold compared to mice receiving exosomes from metastatic melanoma cell lines (ExoM). It is reported that in this study, these ExoNM educate patrolling monocytes in the bone marrow, which leads to activation of the innate immune response and abrogation of tumor progression at the premetastatic niche. These data demonstrate that exosomes have the capacity to inhibit lung metastasis. Another study utilized sEVs derived from activated M1 macrophages (anti-tumor macrophages), in conjunction with the melanoma-associated tyrosinase related protein 2 (Trp2) peptide vaccine to actuate the immune response [513]. They found that M1-derived sEVs contributed to CTL activation and efficacy of Trp2 vaccine. Additionally, when they challenged the activated CTLs with the Trp2 peptide, the CTLs responded by secreting large amounts of IFNγ. While this does not suggest the exosomes alone are useful as vaccines, it does support their potential usage as a vaccine adjuvant.
Exosomes have also proven useful as vaccines for HPV-associated neoplasms [514]. A mutant variation of a protein found in HIV, called Nef, was used to act as a vector incorporating HPV16-E7 tumor-associated antigen. When this DNA vector incorporating mutant Nef and HPV16-E7 (Nefmut/E7) was injected intramuscularly, the cells released greater amounts of endogenous exosomes containing HPV16-E7. When they examined the mice that had been injected with the plasmid vector containing Nefmut/E7, they found the mice had developed CTL responses to both Nef and E7 antigens through the endogenous release of exosomes from these mice inoculated with these DNA vectors. The majority of mice which had been inoculated with Nefmut/E7 exhibited a constant tumor burden of <150 mm3, with some mice showing a complete reduction, compared to control mice and mice which had been inoculated with only Nefmut, which showed a steadily increasing tumor burden over the course of the 30 day monitoring period. Additionally, they replicated this method using HER2 instead of E7 and found that mouse models predisposed to developing HER2-positive mammary tumors exhibited a delay in tumor formation from 17 weeks, for mice which did not receive the vaccine, to 26 weeks of age in inoculated mice [515]. These studies indicate that intramuscular inoculation of Nefmut in conjunction with tumor-specific DNA can potentially generate endogenously engineered exosomes which are capable of educating CD8+ CTLs against tumor-specific antigens.
Vaccines are useful tools for actuating the immune response against viruses as well as tumors. Exosomes and exosome-like particles provide a novel vaccination method against tumors. They can activate immune responses in CTLs as well as support the immune response as adjuvants for other types of vaccines. Additionally, vectors introduced into the body can aid in the production of endogenous exosomes possessing antigen, thereby promoting a native adaptive immune response in the host and effectively boosting the immune response against specific tumor antigens.
7.2. As drug delivery vesicles
EVs may prove beneficial as drug delivery vehicles due to their stability in body fluids, ability to transport various cargos, and their immunogenicity. The main issue regarding EVs for use in this manner, however, involves the issue of targeting, which will be discussed in this section. Despite this obstacle, there has been several studies and reviews that have examined and discussed exosomes as drug carriers [117,[516], [517], [518], [519], [520], [521], [522], [523]]. This section aims to discuss some of the studies that have focused on sEVs as drug delivery systems, the types of drugs that exosomes can deliver, how sEVs are prepared from specific cell types and primed to be drug carriers, and how they may be engineered for targeting specific tumor cell types. Compared with commercial liposomes and polymeric nanoparticles, sEVs as a natural delivery system can evade phagocytosis, have extended blood half-life, and exhibit optimal biocompatibility without potential long-term safety issues [524,525]. sEVs can fuse with the cellular plasma membrane and deliver drugs directly into cytoplasm. By evading the engulfment by lysosomes, EVs remarkably enhance delivery efficiency of vulnerable molecules. In addition, the small size of EVs facilitates their extravasation, translocation through physical barriers, and passage through extracellular matrix. sEVs as drug delivery nanocarriers have two advantages: (1) sEVs derived from certain cells express surface antigen CD47 (i.e., “Don't eat me” signal) and, as a result, have extended blood circulation times because they are not quickly cleared by monocytes. sEVs generated by certain cells can also achieve tumor- or tissue-specific delivery. Currently the major challenge that sEVs drug delivery faces is the low yield of sEVs, as a single cell secretes only ~50 sEVs/hour. To harvest a sufficient amount of sEVs, abundant cells and a long period of time are required, which makes large-scale applications unrealistic.
7.2.1. Drug type: siRNA, peptide, toxic protein, chemical drugs
One of the main issues regarding treating tumors is systemic toxicity due to nonspecific targeting of cells by current therapies. sEVs loaded with siRNA, toxic proteins, or chemotherapy can potentially enhance specific targeting of cancer cells resulting in increased response to therapy and a decrease in negative side effects. To load these cargos into sEVs, cargo can be co-incubated with the donor cells to increase cargo uptake and exosome secretion [526]. Another commonly used method is electroporation, which administers electrical current to aqueous nanosized pores that results in increased cell permeability and allows cargo to cross the membrane [527]. Other methods include sonication, thawing, freeze and thaw cycles, and extrusion [528]. This section will explain how these certain siRNA, proteins and drugs are applied within cancer therapy.
Small interfering RNA (siRNA) can target and silence messenger RNA (mRNA) that are responsible for cell proliferation [529]. In fact, a study has examined the effects of priming sEVs with siRNA specifically targeting KRASG1D2, a gene that is associated with cell proliferation exclusively in pancreatic ductal adenocarcinoma (PDAC) [530]. Mice were injected with PANC1 cells and then treated with exosomes loaded with the siRNA. Results showed that EVs loaded with the siRNA were able to successfully target KRASG12D, resulting in a nearly fourfold decrease of KRASG12D transcripts and an overall decrease in cell proliferation and increased apoptosis in the pancreatic tumor cells treated with this siRNA. Additionally, mice which received treatment showed a fourfold reduction in pancreatic tumor mass and all mice which received the treatment were still alive after 87 days, whereas the mice which did not receive siKRASG12D had all succumbed to the tumors. However, mice which received the treatment did eventually succumb after 120 days, thus their survival had been substantially increased compared to control mice. Finally, these benefits were not observed in mice treated with liposome-delivery of the siRNA, thereby suggesting sEVs-delivery of siRNA is more effective.
sEVs can deliver different types of drug molecules such as anti-inflammatory drugs, anti-cancer drugs or anti-fungal drugs [531]. An early study examined sEVs loaded with curcumin, a hydrophobic bioactive anti-inflammatory compound found in turmeric [532]. Curcumin was mixed with sEVs derived from mouse lymphoma cell culture model, EL-4. When equal amounts of curcumin were mixed into PBS or PBS and sEVs, the concentration of curcumin in PBS and exosomes increased fivefold compared to PBS alone, suggesting that the exosomes participated in increasing the solubility of curcumin. Furthermore, this study found that mice treated with a LD50 dose of curcumin without sEVs exhibited poor survival, whereas mice which received sEVs-encapsulated curcumin exhibited a greater overall survival as well as a nearly fourfold decrease in inflammatory cytokines. Another study analyzed the effect of sEVs-curcumin on pancreatic cancer cell viability [533]. Treatment with sEVs-curcumin showed a nearly 50% decrease in cell viability after 48 h in both PANC1 and MIA PaCa-2 pancreatic cancer cell culture models. A third study utilized murine exosomes, functionalized with specific integrins (iRGD), from immature dendritic cells to encapsulate doxorubicin, a common chemotherapy, to treat MDA-MB-231 breast cancer xenografts [534]. The iRGD-exosomes were administered intravenously to mice bearing tumors of approximately 0.2 mm3 in size. These iRGD-exosomes were observed localizing to the tumor site whereas exosomes without iRGD dispersed throughout the body and collected in liver and spleen and did not collect in the tumor. When iRGD-exosomes containing doxorubicin was administered to mice, tumor growth was drastically inhibited, showing only a fourfold increase in volume compared to untreated mice which exhibited a 15-fold increase in tumor burden. Additionally, mice treated with the iRGD-exosome-doxorubicin did not exhibit morbidity or mortality during the treatment, suggesting little to no toxicity from the delivery method. Furthermore, the mice treated with the iRGD-exosome-doxorubicin exhibited similar serum levels as control mice of cardiac damage markers, creatine kinase MB isoenzyme and aspartate aminotransferase, compared to mice treated with free doxorubicin, which exhibited nearly double the amount of the cardiotoxic serum markers as the control mice. Taken together, these studies show that sEVs and exosomes are not only more efficient at localizing to tumor sites, but also superior at mitigating systemic toxicity compared to freely-circulating chemotherapies. The use of toxic proteins has been well studied over several decades. Toxic proteins are specific proteins which can interrupt cell function, alter cell composition, and promote cell death. To target these cancer cells, specific ligands, antibodies or receptors are identified within the cancer cell and this allows these immunotoxins to attach and target the cancer cell to deliver and release the specific toxic cargo into the cell cytosol to induce cell death [535,536]. Most immunotoxins target elongation factor-2 (EF-2) which results in the suppression of protein synthesis, eventually leading to apoptosis in healthy and tumor cells [535,537]. It is therefore imperative that targeted therapy incorporating immunotoxins is highly discriminatory to minimize uptake of the immunotoxins in healthy cells. Toxic proteins have clearly been used with success to treat various types of tumors, even without exosomes. However, exosome-delivery may prove more beneficial at mitigating toxicity and increasing efficacy.
Toxic proteins tend to lack specificity on their own, oftentimes not discriminating between healthy and cancer cells [537,538]. One study functionalized sEVs with the K4-peptide, an antimicrobial peptide, to better cluster the exosomes to E3-EGFR expressing MDA-MB-231 cell [539]. These functionalized sEVs were loaded with saporin, a plant-derived ribosome-inhibiting protein. They observed that the K4-peptide on the sEVs was more effective at clustering, and therefore activating, the EGFR. This resulted in increased uptake of the saporin-loaded sEVs and subsequent apoptosis of the tumor cells, resulting in 30% decline in cell viability compared to the control. The donor cells from which the sEVs arise may also contain native immunotoxins. For example, a study found that sEVs derived from NK cells contain cytotoxic perforin, granulysin, and granzymes A and B which are the tumor-fighting arsenal of the NK cells [540]. When cell lines were incubated with these activated NK cell-derived sEVs, an overall decrease in tumor cell survival was observed. These studies thus show promise for targeted delivery of immunotoxins by sEVs.
Toxic proteins have been widely researched over the decades for their treatment of tumors. Unfortunately, many of these immunotoxins lack specificity and therefore systemic toxicity is an issue. However, sEVs may be useful in mitigating this concern. Utilizing sEVs to deliver toxic proteins is a fairly untapped field of research, and thus information regarding sEVs delivery of toxic proteins is limited. As more information of EV biogenesis and functionalizing sEVs for effective targeting is gathered, the use of toxic proteins in cancer treatment may exhibit a resurgence as more research is conducted on sEVs-mediated delivery of these old types of therapies.
7.2.2. Donor cell types for preparation of sEVs
A variety of donor cells have been used as the source for exosome-based delivery systems such as dendritic cells (DC), T-Cells, B-cells, mesenchymal stem cells (MSC), specific cancer derived cells, immune cells, and stem cells. This section aims to focus on discussing some of the more commonly used cell-types for exosome harvest. Since exosomes often display characteristics like the cells from which they are derived, various cell types are considered when determining which attributes will most successfully contribute to the desired drug-delivery method.
Exosomes derived from immune cells have been widely studied and have exhibited a crucial role in antigen-specific immunity and tolerance [541]. DC derived exosomes display membrane proteins such as MHC-I and MHC-II, CD86 molecules, and a milk fat globule protein MFG-E8 that are useful for targeting, docking and cell fusion [541,542]. However, immature DCs can show immunosuppressive functions due to certain expressions of co-stimulatory molecules, therefore, DCs are primed with stimulatory agents to produce their therapeutic functions [543]. For instance, when these DC derived exosomes are primed with antigens, this activates both helper and killer T-Cells as well as B-cells [544]. One study has concluded that exosomes derived from α-fetoprotein (AFP)-expressing DCs not only showed a potent antigen-immunogenetic response, but also an ability to significantly suppress tumor growth in hepatocellular carcinoma tumors in mice models [545]. This study therefore supported DC-derived exosomes, specifically primed with tumor-specific antigens, as a promising cancer therapy.
Mesenchymal stem cells (MSC) are known to aid in tissue repair by a paracrine manner and thus may prove a novel use for cancer treatments [546]. In fact, it is the only known cell donor to mass produce sEVs, making MSCs a suitable candidate for exosome and sEVs harvest and drug therapy [547]. MSC-sEVs have exhibited an ability to decrease inflammation as well as display strong immunosuppressive properties [548]. However, some studies have shown that MSC-cells can contribute to angiogenesis and carcinogenesis [549,550]. On the other hand, an early study found that MSC-sEVs suppressed the expression of vascular endothelial growth factor (VEGF) in cancer cells via expression of miR-16, which contributes to the inhibition of angiogenesis in vitro and in vivo [551]. Regardless, MSC-sEVs can be loaded with therapeutic RNA, chemotherapeutics, and proteins for tumor destruction [552]. For example, another older study examined the effects of Paclitaxel loaded into human bone marrow-derived mesenchymal stem cells (hMSCs) and its effect on breast cancer xenografts [553]. To assess this, the PTX-loaded hMSC sEVs were loaded into xenograft MDA-MB-231 breast cancer mouse model. The results showed that the PTX-loaded hMSC sEVs inhibited tumor growth and decreased in size by nearly 50% compared to controls. Additionally, sEVs derived from irradiated MSCs may increase the efficacy of radiotherapy and decrease the rate of metastasis in melanoma mouse models [554]. Overall, early studies suggest that MSC-sEVs can exert strong anti-cancer therapeutic effects.
Specific Immune cell-derived sEVs have shown remarkable abilities to suppress immune responses in response to cancer therapy. Depending on the type of sEVs and which antigens they express, sEVs can enhance metastasis and cell proliferation, or they can promote cancer immunosurveillance [555]. For example, immune cells associated with innate immune responses, such as Natural killer cells (NK), play a substantial role in immunosurveillance and host defense against cancer due to their expression of cytotoxic proteins and pro-apoptotic activities on cancer cells [556]. Immune cell derived sEVs are also associated in acquired immune responses as indicated by their ability to activate Dendritic cells and T-cells [557]. Dendritic cells contain antigens including MHC-I and MHC-II surface molecules that stimulate CD8+ and CD4+ T cell. As a result, both the CD8+ and CD4+ T cells promote the eradication of tumor cells [558].
Tumor-derived sEVs mimic the biological roles of the tumor cell. Their roles in suppressing host anti-tumor activity, response to immunotherapy and mediating drug therapy are important to consider for its uses in cancer therapy. One big advantage of these types of sEVs is that they contain specific antigen surface molecules that can prime immune cells to induce immunogenic responses, therefore increasing therapeutic efficiency [528]. These sEVs contain MHC-1 molecules, HSP70 and antigens that are used to gear the immune response against cancer [559]. One study has demonstrated that hepatocellular carcinoma (HCC)-derived exosomes carried an array of HCC antigens that elicited a stronger immune response than cell lysates in vitro and in vivo [560]. DCs, DC lysates (DClys), and DCs educated by tumor-derived exosomes (DCTEX) were compared for their abilities to activate cytolytic T-cell activities. The DCs alone resulted in <20% cytolysis by CTLs while DClys and DCTEX lysates caused <40% and 40–50% T-cell mediated cytolysis, respectively. An overall decreased tumor burden over 25 days was also observed in mice treated with DCTEX (<500 mm3) compared to mice treated with DClys (<600 mm3) and DCs alone (~1000 mm3). Additionally, the tumor immune microenvironment was significantly improved in HCC mice, with a decrease in pro-tumorigenic regulatory T-cells (<40% of activated T-cells compared to controls) and an increase in CD8+ T-cells (<30% of activated T-cells compared to controls). Furthermore, 100% of the mice treated with DCTEX exhibited significantly prolonged survival and decreased tumor burden 58 days after tumor challenge, while none of the mice in the control groups had survived passed day 58. Significant levels of anti-tumor cytokines were also observed in DCTEX-treated mice, showing 2–3 fold higher levels of IL2 and IFNᵧ compared to controls. Thus, sEVs donated by tumors appear to exhibit a strong anti-tumor response when used to educate DCs.
The use of donor cell sEVs has been encouraging in response to cancer therapy. Undeniably, there are situations wherein these sEVs are either promoting angiogenesis and tumorigenesis or suppressing tumor responses and promoting immunosurveillance. By understanding the biological properties and roles of these sEVs derived from specific donor cells, it may be possible to develop new and improved therapeutic approaches.
7.2.3. Non-specific targeting of tumors using sEVs
Drug delivery is conducted via specific targeting or non-specific targeting. Specific targeting involves usage of specific receptors or ligands that allow the drug to home to the cell or tissue of interest. There are several methods for achieving targeting using exosomes, which will be covered in the next section. This section will focus on non-specific targeting. Non-specific targeting is a more passive process wherein the drug is administered intravenously or orally. In this scenario, the drug circulates throughout the body and may be taken up by any type of cell. This often leads to a clearing of the drug before enough builds in the tissue of interest, thereby resulting in low response to treatment. To overcome this, a larger dose of the drug is needed to enact a therapeutic response. This can potentially lead to accumulation of the medication in the heart, liver, spleen, and other organs, resulting in negative side effects. This is often observed in cancer patients treated with chemotherapy [561].
sEVs can be used for non-specific targeting of tumors as well and with fewer side effects compared to chemotherapy. For example, it is known that the common chemotherapeutic, doxorubicin, causes cardiotoxicity [534,[562], [563], [564], [565]]. Though one study found that the IC50 of exosome-loaded doxorubicin (Exo-Dox) in cell culture models (HEK293, BT-20, SK-BR-3, HUVEC, PASMC, and hiPS cardiomyocytes) exhibited a potency of 20x more than free dox, 5x more than liposomal dox (Myocet), and 50x more potent than liposomal dox (Doxil) and there was little difference in uptake between cardiomyocyte cultures and tumor cell cultures [563]. Studies which utilized Exo-Dox found minimal accumulation of doxorubicin in cardiomyocytes. In fact, Exo-Dox appeared to reduce cardiotoxicity by 40% compared to free doxorubicin in mouse xenografts [564]. Additionally, mice treated with Exo-Dox exhibited normal behaviors and displayed only a 10% weight reduction compared to mice treated with free dox, which exhibited 25% weight reduction and an observable decrease in normal behaviors. Furthermore, Exo-Dox was just as effective as free dox, with both conditions exhibiting a 30% decrease in tumor volume compared to untreated mice. A follow up study found that even when concentrations of Exo-Dox, Dox, and Doxil were increased to 5 mg/kg, mice treated with Exo-Dox and Doxil did not exhibit any decline while mice treated with Dox began to lose weight [565]. Tumors treated with Exo-Dox were observed to have twice as much doxorubicin as tumors treated with free-dox, suggesting more efficient uptake of Exo-Dox.
Another study examined the potential toxicity of wildtype sEVs compared to engineered exosomes [566]. In this study, they used 8.5 μg of sEVs from HEK293T and engineered cells loaded with a modified protein containing pre-miR-199a-3p for monitoring uptake. These sEVs were injected into the mice intravenously three times a week for three weeks. They found no significant histopathological changes in the organs of the mice and no immune responses were observed. They did find accumulation of the sEVs in the pancreas though did not observe any in the liver or spleen. Overall, the study suggests minimal cytotoxicity across all major organs and tissues. However, the reporters do suggest further studies are needed to determine if a higher dosage would result in cytotoxicity.
It has been observed that non-specific targeting of tumors using sEVs and liposomes resulted in a 10-fold higher accumulation of sEVs in the tumor mass compared to liposomes [567,568]. The reason for increased uptake of sEVs by tumor cells is likely due to higher vascularization as well as the abnormal vascularization associated with tumors [568,569]. Additionally, another study observed an increased uptake of tumor-derived exosomes by the tumors themselves by taking advantage of this increased vascularity [569]. However, this group did PEGylate the sEVs to ensure longer circulation times. Regardless, PEGylated sEVs derived from tumors were found to accumulate with greater numbers in tumors and tumor-associated immune cells compared to normal cells. This study thus suggests that specific targeting can be obtained using tumor-derived sEVs with minimal alterations.
Non-specific targeting of tumors using sEVs holds a great deal of promise. Several studies have been conducted that confirm minimal, if any, systemic toxicity when exosomes/sEVs or exosome-mimetics are used to deliver chemotherapeutics [523,534,570]. sEVs delivery of chemotherapeutics has also been shown to overcome drug resistance in cancer stem cells [528,571]. Additionally, sEVs provide a more targeted delivery to the tumor cells, even without modifications to enhance targeting. However, sEVs-mediated drug delivery must overcome rapid clearance by the liver and spleen in order to sustain long enough in circulation to reach the tumor site.
7.2.4. Approaches for targeting
In most cases, the use of normal sEVs remains a challenge due its low transfection efficiency. Altering the surface level proteins of an sEVs has been commonly used to fix this dilemma. This can be achieved through cell engineering of donor cells, recipient cells, or through the addition of targeting ligands, in which these donor cells are genetically engineered to induce the expression of fusion proteins or by using click chemistry to add the targeting ligands [528]. In this section, we will go more into depth on how these processes achieve targeting.
.An earlier study modified donor cells to express the transmembrane domain of platelet derived growth factor receptor fused to the GE11 peptide in order to achieve better targeting of exosomes to EGFR + breast cancer cells [572]. In this study, they found that the sEVs functionalized with GE11 were less likely to enact EGFR-associated responses in the cells compared to sEVs functionalized with EGF. These GE11 expressing sEVs were used to deliver let-7a miR to these tumor cells in nude mouse models. Via fluorescent imaging assays, it was observed that GE11-sEVs were 3 times as likely to collect in EGFR + tumors then were control sEVs. Additionally, they did not accumulate in major organs, supporting a strong tumor-specific delivery method. Tumors were injected with luciferin, and in those mice which were treated with let-7a delivered by GE11-sEVs, minimal signal was observed. A more recent study transfected HEK293T cells with tLyp-1 peptide to generate tLyp-1 labeled sEVs [573]. The tLyp-1 peptide is known to interact with specific receptors which exhibit high expression in neoplastic vessels and tumor cells. These tLyp-1 sEVs were loaded with siRNA for the SOX2 gene. Encapsulation efficiency of the siRNA into targeting sEVs was nearly 15% greater than the encapsulation of the siRNA into natural sEVs. When administered to A549 cells, the sEVs functionalized for targeting tLyp-1 were about 1.5x more effective at delivering the siRNA and knocking down SOX2 than natural sEVs or liposome-based delivery system, Lipo3000. Even though these studies were proven successful towards targeting, the use of using the donor's own cells for sEV harvest remains a challenge due to time-consuming production and acquiring the specific sEVss from the patient's own body fluids [574].
A common ligand for targeting tumor cells is Transferrin, a transmembrane glycoprotein, which is abundant on cancer cells compared to healthy cells [575,576]. In fact, a few studies have shown that the overexpression of transferrin on the surface of lung cancer cells aids better delivery of anti-cancer drugs to the cancer cells [[577], [578], [579]]. Therefore, the addition of a transferrin conjugated protein to sEVs can increase targeting and cellular uptake of anti-cancer drugs. However, other ligands can also be used. For example, a previously mentioned study engineered immature dendritic cells from mice to express membranal protein, Lysosome-associated membrane protein 2 (LAMP2B) [534]. While we previously discussed this study under our drug delivery subsection, it is important to note that the sEVs were generated by modifying the immature dendritic cells to over express the LAMP2B protein fused with αv integrin-specific iRGD peptide to generate sEVs which could target αv integrin-positive breast cancer cells. As was previously discussed, the sEVs targeted the tumor cells more effectively, exhibited greater therapeutic potential then free dox, and exhibited minimal systemic toxicity.
Click chemistry has been increasingly popular since it does not alter the structure or the biocompatibility of sEVs. Click chemistry is a copper-catalyzed azide alkyne cycloaddition, which is suitable for the attachment of small biomolecules to sEVs [580]. For example, the cycloaddition of Arg-Gly-Asp-D-Try-Lys peptide was added to the surface of mesenchymal stem cell (MSC) derived sEVs [581]. This modification enabled targeting and treatment of ischemic reactive cerebral vascular endothelial cells. This type of sEVs-functionalization can therefore be helpful in the treatment of cancers. For example, a recent study utilized click chemistry to functionalize sEVs with biocompatible Cy5 fluorescent labeling in order to track the sEVs [582]. These Cy5-labeled sEVs accumulated rapidly, and 1.53 fold greater than sEVs labeled with carbocyanine lipophilic dye, in the tumors of mouse model and exhibited a surprisingly low level of accumulation in organs usually affected by systemic dosages. While more work is required to determine how well this type of functionalization would work for treatment, it does suggest potential for monitoring patients.
Triple negative breast cancer (TNBC) is difficult to treat due to the lack of estrogen receptors, progesterone receptors, and human epidermal growth factor receptor 2. A new study, however, has shown that it is possible to use HER2+ EVs to functionalize TNBC cells with the HER2 receptor, thereby rendering TNBC cells susceptible to chemotherapy [583]. HER2+ BT-474 cells were used to generate these HER2+ EVs. Introducing these HER2+ EVs into MDA-MB-231 triple negative cells showed HER2 expression increase of 2.47 and 8.75-fold higher at 6- and 12-h time periods when compared to negative controls. To ascertain the effectiveness of conferring HER2 receptors to TNBC cells, liposomes loaded with paclitaxel (LP-PTX) or tagged with HER2 antibody (Ab-LP-PTX) were introduced to these HER2+ MDA-MB-231 cells. They found that the Ab-LP-PTX had released 45% of its load compared to the 85% released by LP-PTX, suggesting a more controlled and targeted release by the Ab-tagged liposomes. This was verified when no fluorescent signal was observed when the Ab-LP-PTX was introduced to MDA-MB-231 control cells which had not been educated by the HER2+ EVs. To test this in mice, educated MDA-MB-231 cells were injected into mice and then treated with Ab-LP-PTX and free PTX. Mice treated with the Ab-LP-PTX showed a 34.6% decrease in tumor weight and a 33.3% decrease in tumor volume over the course of 18 days. Thus, taken together, it is possible to confer HER2 onto HER2-cells using HER2+ EVs and then specifically target HER2+ cells for drug delivery by labeling drug-loaded liposomes with HER2 antibody.
The incorporation of either genetically engineering donor cells or targeting ligands have shown favorable responses for targeting specific cell types or organs with sEVs. However, manipulation of the donor cells and sEVs can affect the biocompatibility and structure. Therefore, consideration should be taken when determining which methods should be used and how that may influence the morphology of the donor cells or exosomes [584].
7.2.5. Preparation of engineered sEVs
7.2.5.1. Mechanical extrusion
sEVs can also be engineered via mechanical extrusion of cells to form nanovesicles that mimic naturally-produced sEVs. For example, studies have shown that these cell-engineered nanovesicles (CNVs) can mediate cellular communication, much like standard sEVs and have been found to improve wound healing [585,586]. Extrusion is also more efficient for producing greater quantities of vesicles compared to isolating sEVs [585,587]. Mechanical extrusion involves forcing cells through a microporous filter, resulting in the shredding of membranes into fragments that spontaneously reform into small extracellular vesicle-like structures which contain fragments of DNA, RNA, as well as proteins and other cellular components [587]. When engineering sEVs in this manner, if the cells being extruded are mixed with the drug for loading, the drug will be encapsulated when the membranes reform [520]. Thus, it is possible to utilize these structures in place of sEVs. Two studies from the same lab have shown the viability of these CNVs in cell-to-cell communication, similarly to natural sEVs. In the first study, these nanovesicles were derived from embryonic stem cells (ESCs), mechanically extruded, and the appropriate sizes isolated via dgUC [585]. They then treated fibroblasts with these CNVs to determine if they were readily taken up by the fibroblasts. To verify the presence of the ESC CNVs, they analyzed the RNA of treated versus untreated groups for the presence of pluripotent markers Oct3/4 and Nanog and found that only the fibroblasts which were treated with the CNVs expressed these pluripotent markers. They then conducted a wound healing assay on the fibroblast groups treated with CNVs compared to untreated groups and found increased proliferation in the treated groups (188, 151, and 84%) compared to the untreated groups (168, 103, and 62%). Furthermore, there was also a 2- and 4-times greater increase in VEGFα and TGFβ expressions, respectively, in the treated groups compared to the untreated. The second study utilized mesenchymal stem cells (MSCs) to generate the CNVs (MSCNVs) and compared the efficiency of MSCNVs to EVs derived from MSCs (MSCEVs) in wound healing [586]. They found that the fibroblasts treated with MSCNVs were better at stimulating cell proliferation compared to fibroblasts treated with MSCEVs and no treatment. Then, they conducted a wound assay in nude mice, where one group was treated with MSCNVs and the other did not receive treatment. Both wounds were covered with tegaderm during the healing process. After 7 days, they analyzed the tissues of the two mice and found that the wound treated with MSCNVs had increased to a thickness of 543 μm while the untreated wound had a skin thickness of 423 μm After 13 days, the wound that was treated with MSCNVs had almost completely filled the dermis and epidermis with blood vessels, whereas the wound that remained untreated did not exhibit significant vascular penetration. Both studies utilized sEVs mimetics via mechanical extrusion of cells and found that extruded sEVs mimetics behave similarly to naturally produced sEVs, and may even be more effective than normal sEVs in wound healing.
While sEVs-like particles can be generated via extrusion, sEVs derived from cancer can also be extruded with other particles to increase their uptake or drug delivery effectiveness. In one study, mechanical extrusion through 100 nm filter was used to functionalize 70 nm positively charged gold nanoparticles with branched polyethylene imine (AuNP-BPEI) to maximize interaction with the negatively charged sEVs membranes [588]. The study examined the effects of membrane proteins on sEVs and their uptake in cancer cells and macrophages. They did this by using proteinase K to dissolve the proteins on the sEVs membranes (PK-EV) and found that macrophage uptake of PK-EVs were 67% more than sEVs with intact membrane proteins. Meanwhile, 4T1 cells took up 27% fewer PK-EVs compared to sEVs with proteins, suggesting specific targeting of tumor cells by tumor-derived sEVs. Then they examined whether the surface proteins remained intact through the extrusion process by analyzing protein composition before extrusion and after. There was no significant difference in the membrane proteins before and after extrusion with AuNP-BPEI. Finally, they analyzed the uptake of the sEVs-coated AuNP-BPEI compared to AuNP-BPEI without sEVs coating and observed a slight, but significant 15% decrease in macrophage uptake of sEVs-coated AuNP-BPEI in relation to AuNP-BPEI. This study suggest the potential for using sEVs membranes, not the sEVs themselves, for cloaking drugs and improving targeted delivery of these treatments directly to cancer cells.
7.2.5.2. Ultrasonication
Sonication is another means by which sEVs can be generated or encase drugs for therapeutic targeting. Sonication utilizes sound waves to weaken the membranes of sEVs and cells, resulting in more efficient dismantling of the cells and reforming into nanovesicles [587,589] or more efficient drug loading of the sEVs [520,590,591]. For the former, ultrasonication was utilized on human umbilical cord mesenchymal stem cells (huMSCs) to engineer sEVs (eEVs) for skin treatment [589]. The cells were sonicated for 1 min to shear the cells. The lipid bilayer membranes reformed, encapsulating biomolecules and mimicking naturally secreted EVs (nsEVs). They reported that nsEVs and the eEVs did not exhibit any significant differences in structure or size. Sonication generated ~20-fold greater sEVs and is reported to be 100-fold faster than traditional EVs harvest. To verify if the eEVs could educate cells similarly to nsEVs, human dermal fibroblasts (HDFs) were inoculated with either eEVs or nsEVs in a 96-well plate. After 24 h, the control, nsEVs, and eEVs groups were examined. The cell number increased from 44.8 ± 3.2 cells to 56.7 ± 2.9 (control), 45.8 ± 3.6 to 117.2 ± 4.1 (nsEVs), and 42 ± 3.6 to 137.2 ± 9.4 (eEVs), thereby showing that nsEVs and eEVs could significantly affect the proliferation of HDFs. Similarly, nsEVs and eEVs increased proliferation in a wound healing assay. After 48 h, the wound width was observed to be 27.1 ± 10.6%, 5.3 ± 3.1%, and 2.7 ± 1.3% for the control, nsEVs, and eEVs groups respectively. When a wound assay was conducted on mice, the wounds which had been treated with nsEVs and eEVs exhibited superior healing after 14 days with the wounds decreasing in size to 4.5 ± 2.7 and 4.1 ± 0.7%, respectively, compared to the control (23.2 ± 8.9%). Furthermore, in the mice treated with nsEVs or eEVs, there were greater numbers of fibroblasts compared to the control, thereby confirming the wound-healing affects of both nsEVs and eEVs. Additionally, this study suggests that there is not a significant difference between the effectiveness of nsEVs and EVs generated by ultrasonication in wound healing, which further supports the potential for eEVs to be used in place of nsEVs for various purposes.
As has already been discussed in this review, sEVs innately target tumor cells, whether it is from an increased uptake by the tumors or direct homing. Thus, sEVs offer a strong alternative for drug delivery. One group compared different methods for loading PTX into sEVs, specifically exosomes, and assessed the ability of PTX-loaded exosomes (exoPTX) to enhance therapy in multiple drug resistant (MDR) tumors [591]. The exosomes were first harvested from macrophage-like RAW 264.7 cells. The loading capacities (LC) of PTX into these exosomes for incubation, electroporation, and sonication were compared. Sonication exhibited the greatest LC (28.29 ± 1.38%), meanwhile, incubation exhibited the lowest (1.44 ± 0.38) and electroporation also displayed a low LC, 5.3 ± 0.43%. Additionally, the LC of sonication is also reportedly higher than commercial formulations such as Taxol (~1%) or Abraxane (~10% LC). The delivery efficiency of exoPTX was compared to other drug delivery methods such as liposomes and polystyrene nanoparticles of the same size. Using fluorescent labels, it was observed that a drug resistant Lewis lung carcinoma cell line (3LL-M27) cells took up nearly 30x more exosomes than liposomes or nanoparticles. The IC50 of exoPTX, PTX, and taxol were also compared in MDCK (Madin-Darby Canine Kidney) cells expressing the drug efflux transporter Pgp (MDCKMDR1), drug sensitive MDCKWT, and 3LL-M27. In all cell lines, exoPTX exhibited an IC50 of far less than taxol and PTX. For 3LL-M27, MDCKWT, and MDCKMDR1, the IC50s (ng/ml) are as follows: 13.57 ± 1.33, 23.33 ± 3.77, and 187.5 ± 38.65 compared to that for taxol (23.16 ± 1.88, 69.54 ± 11.5, and 1708.67 ± 299.93) and PTX (126.41 ± 31.31, 428.77 ± 63.37, and >10,000). Finally, the antineoplastic effect of exoPTX was examined in C57BL/6 murine models injected with 3LL-M27 cells. 48 h after tumor cell injection, exoPTX, Taxol, or saline were intranasally administered every other day and tumor metastasis and growth were monitored for 22 days. The data presented in the paper shows a significant inhibition of metastasis by exoPTX compared to controls and taxol. This work therefore shows the effectiveness of using sonication to load drugs into EVs and overcoming drug resistance in resistant tumors.
7.2.5.3. Exosome/liposome hybrid
Fusing sEVs and liposomes may increase stability as well as promote uptake by target cells [592]. In this paper, exosomes from RAW 267.4 cells were fused with liposomes via freeze-thaw cycles. The efficiency of this method was tested using fluorescently labeled liposomes in a liposome-liposome system and an exosome-liposome system. The efficiency for the double liposome system was 2.1 ± 0.1 compared to the exosome-liposome system, 3.3 ± 0.2, after 10 freeze-thaw cycles. Furthermore, exosomes fused with liposome membranes did not exhibit any obvious morphological changes. CMS7 lines, which are normally HER2-negative, were then altered to express HER2 so they could collect HER2-expressing exosomes to test fusion with liposomes. They found that CMS7-HER2 derived exosomes fused with liposomes at a higher efficiency (3.5 ± 0.2 to 9 ± 0.8, depending on the liposome used) compared to the exosomes harvested from RAW 267.4 cells (1.7–7.6 ± 0.5). To determine the cellular uptake efficiency, HeLa cells were incubated with fluorescently labeled exosome-liposome hybrids for 4 h and found that neutral and anionic lipids did not affect uptake whereas cationic profiles did, while the hybrids incorporating PEG-lipids significantly increased cellular uptake by almost two fold compared to unmodified exosomes, as determined by mean fluorescence (400 for unmodified exosomes and 800 for PEG-lipid-exosome hybrids). This study therefore shows the potential effectiveness of using an exosome-liposome hybrid for drug delivery by increasing circulation in the bloodstream, due to PEGylation of the hybrid, while simultaneously increasing its cellular uptake. Exosome/liposome hybrids have also exhibited potential for delivering CRISPR/Cas9 gene editing to target cells, thereby eliminating the need for potentially harmful viral vectors for delivery [593]. Exosomes from HEK293FT cells were harvested via PEG6000 precipitation before hybridization. The exosomes were incubated with liposomes and CRISPR/Cas9 interference system targeting mRunx2 gene or a CRISPR/Cas9 cleavage system targeting hCTNNB1 gene for 12 h at 37 °C before incubating with murine MSCs. They found that the CRISPR system successfully decreased Runx2 expression by about half in these MSCs compared to the untreated groups. By contrast, exosomes derived from transfected cells exhibited little effect on Runx2 expression in the MSCs. Similarly, in the cleavage system, only the experimental group that was treated with exosome hybrid exhibited successful gene editing as evidenced by the presence of the mismatch, compared to the lipofectamine and exosome only groups. This data therefore suggests that exosome/liposome hybrids can successfully deliver functional CRISPR/Cas9 systems while only liposome or exosome could not.
Another paper describes the hybridization of exosomes and liposomes via PEG-mediated fusion to develop a drug delivery method [594]. EVs were harvested from human umbilical vein endothelial cells (HUVEC) and mixed with rhodamine-labeled liposomes generated by extrusion. The liposomes and exosomes were incubated in a 30% PEG 8000 concentration for 2 h before imaging. EVs possessing phosphatidylserine (PS) were tagged with Annexin V-FITC, whereas the liposomes contained rhodamine. These hybrids contained most of the rhodamine lipid fraction (69%) whereas the liposomes and exosomes that were incubated in the absence of PEG exhibited less rhodamine integration, suggesting less successful fusion. To determine the uptake efficiency of the hybrid compared to liposomes or sEVs alone, sEVs harvested from MSC cells were used and mixed with the liposomes in a 1/1 ratio with PEG. Two groups were used, one wherein the liposomes were PEGylated and the other group in which the liposomes were not PEGylated. The PEGylated or non-PEGylated hybrids were then introduced to THP-1 derived macrophages. PEGylated hybrids were about 8-fold less internalized compared to non-PEGylated ones, showing that PEGylated hybrids were less likely to be opsonized by the macrophages, and thus more likely to make it to their target. To test drug loading efficiency and delivery, sEVs were fused with a mTHPC-encapsulated liposomes, called Foslip, in the presence of PEG solution. Clinically, mTHPC is an antitumor photosensitizer, which, when activated by light, results in apoptosis of targeted cells. The mTHPC-loaded hybrids were compared to the encapsulation performance of sEVs generated by cells which preloaded with the drug. The hybrid exhibited 90% encapsulation efficiency compared to 3% for the sEVs harvested from parent cells preloaded with mTHPC. Cytotoxicity was measured in CT26 colon cancer cells. Hybrid sEVs, Foslip, and free mTHPC were administered to culture groups at three different doses (2.5, 0.5, and 0.1 μM) and incubated for 4 h. Nearly 60–70% of the hybrid EVs were internalized compared to 10–20% for Foslip or free mTHPC. Upon irradiation with 650 nm laser at 10 J/cm2, every condition, except for the lowest dose of 0.1 μM of free mTHPC, exhibited nearly 100% toxicity. Taken together, sEVs can be hybridized with liposomes to improve drug loading efficiency and better performance than sEVs or liposome-delivery methods alone.
Once sEVs are engineered, several methods exist for further preparing them for use as drug delivery systems. While it is possible to generate sEVs from parent cells pre-loaded with drugs, this method is inefficient because sEVs isolation methods are laborious or inconsistent, and seldom produce enough sEVs for effective therapy. Thus, it is possible to load drugs into sEVs via extrusion, ultrasonication, or hybridizing them with lipids. Mechanical extrusion can risk damaging the sEVs membranes. However, sEVs can also be prepared via extrusion, and with greater efficiency than traditional isolation methods. These fabricated sEVs successfully mimic natural sEVs, enacting similar, or even better, wound healing abilities compared to natural sEVs. Similarly, ultrasonication can also produce sEVs-like particles that exhibit similar characteristics and functions as standard sEVs, including the ability to affect cellular metabolism. Drug loading via ultrasonication is also efficient compared to passive loading methods and appears to be more efficient at delivering drugs. Hybridization of sEVs with liposomes exhibits great versatility for drug delivery and design, with these hybrids able to deliver different types of therapies while also being more malleable to modifications which increase their circulation time. Furthermore, hybridization also offers a benefit over liposomes, which suffer from lower cellular uptake compared to sEVs or sEVs hybrids.
7.3. As the treatment target: eliminate cancer-derived extracellular vesicle
It is widely known and accepted that exosomes modulate immune response, prime distant sites for metastases, and contribute to chemoresistance [[595], [596], [597], [598], [599], [600], [601], [602]]. Disrupting these actions may prove a beneficial strategy for treating cancer patients. Previous studies have shown that macrophages will phagocytose exogenous exosomes [[603], [604], [605]]. For example, one study injected CD9 and CD63 antibodies dissolved in PBS into mice [605]. They observed a decrease in breast cancer metastasis to the lungs, especially in mice injected with anti-CD63 as determined by the number of metastatic foci. For both anti-CD9 and anti-CD63 treated groups, the foci numbered around 25 with very little variation, compared to the control which showed above 25 foci and extensive variation. However, they did not observe a decrease in primary tumor size or neovascularization. This study suggests the feasibility of introducing methods for targeting specific tumor sEVs subpopulations to minimize metastases.
Exosome populations can also be targeted using drugs that disrupt their formation. These methods include the ESCRT-independent and ESCRT-dependent mechanisms. It has previously been established that syndecan-syntenin-ALIX signaling is involved in exosomal biogenesis and could potentially be a therapeutic target for minimizing exosomes release [41,606]. Additionally, ceramides can be targeted via sphingomyelinases [607,608]. Rab27a and Rab27b have also been implicated in sEVs development and release, with inhibition of Rab27a resulting in a 50–75% decrease in sEVs population from tumor cells, and a 25–50% decrease in tumor size and metastases. Other studies have also reported that inhibition of Rab27a results in decreased tumor cell proliferation and metastasis [41,608,609]. Interfering with the uptake of sEVs may also be effective at impeding the functions of tumor-derived exosomes. For example, chlorpromazine is known to inhibit clathrin-mediated endocytosis and has been shown to decrease sEVs uptake by 41% [41,610]. Blood filtration has also been proposed as a means of removing tumor-derived sEVs and mitigating the immunosuppressive effects of sEVs [41,611,612]. Still, more studies have been conducted identifying proton pump inhibitors as potential targets for decreasing exosome formation and release [41,320]. Thus, there are several ways in which exosomes and sEVs can be targeted that could potentially mitigate tumor progression.
EVs are crucial in cell-to-cell communication and contribute a great deal to tumor progression and metastasis. However, with such a diverse function, any broad methods for targeting sEVs could prove disastrous as such methods could interfere with the functions of healthy cells. Targeting sEVs has potential if specific tumor subpopulations of sEVs can be eliminated. Regardless, targeting tumor-derived sEVs is not enough of a treatment on its own, but it can certainly support traditional therapies and mitigate drug resistance, metastases, and immunosuppressive effects of tumors.
7.4. Summary
sEVs offer a valuable alternative to the current therapeutic options. sEVs are unique molecules with a wide array of application. They have been explored for use as novel vaccines against tumors due to their inherent characteristics, including ability to activate the immune response. Furthermore, sEVs can evade degradation by immune cells and are taken up in greater numbers by tumors, which makes them useful as drug delivery vehicles. However, it is necessary to choose the right cell type from which to harvest sEVs for use as a vaccine or drug delivery vehicle. For example, tumor derived and dendritic cell derived sEVs may provide more efficient delivery vehicles for chemotherapy and vaccines due to their inherent roles in priming the immune responses towards cancer. However, it is difficult to harvest enough sEVs from these cell types in order to develop efficient delivery methods for medical use. Thus, mechanical extrusion, ultrasonication, and hybridizing exosomes with liposomes provide an alternative for mass production of viable sEVs-like structures and maximizing drug and vaccine loading into these sEVs. Finally, it is possible to target sEVs as a means of anti-tumor therapy by interrupting their formation. Much research is still needed to validate sEVs for therapy, but the current research suggests great promise as the field continues to grow and its methods improve.
8. Conclusion
Extracellular vesicles are important modulators of inter- and intracellular communication and are capable of enacting cellular responses when their cargoes are internalized. The lipid bilayer membrane of EVs confer stability in body fluids, allowing them to enact responses in distant cells via their cargoes. These cargoes include DNA, mRNA, miRNA, lncRNA, and proteins as well as other cellular constituents. The internal and external contents of EVs therefore offer strong potential for non-invasive tumor biomarkers as well as assessing patient prognosis and monitoring response to therapy. However, EVs-based diagnostics offer major challenges. One challenge is that EVs are a diverse group with three major subtypes: exosomes, microvesicles, and apoptotic bodies. Each subtype exhibits specific characteristics, compositions, and functions which confound standardized isolation protocols. Thus, current EVs isolation methods suffer from protein contamination and co-isolation of other size-similar molecules, thereby making the isolation of specific EVs sub-populations difficult. Additionally, other methods which may yield high purity, may also yield high losses of sEVs. This is concerning, especially when determining a pure sEVs-based tumor diagnostic. Regardless, technology and methods continue to be improved upon, with combinations of methods often offering higher purity and yield.
Numerous studies have examined the relationship between tumor derived sEVs and the tumor microenvironment. These studies have determined that sEVs support the tumor microenvironment through several key processes which involve the transfer of their cargoes to neighboring and distant cells. These pro-tumorigenic-cargo-laden EVs contribute to angiogenesis and the pre-metastatic niche, tumor growth, increased vascular permeability, subversion of the immune response, facilitation of EMT programs, and the reprogramming of stromal cells [175]. Additionally, studies have identified a significant association between hypoxia [613,614] and acidity [[615], [616], [617]] with tumor aggressiveness, and, evidently, both appear to contribute to increased EV release [618,619]. Interestingly, studies have implicated hypoxia in inducing the release of pro-angiogenic EVs [620,621] and EVs derived from hypoxic environments have contributed to hypoxia tolerance in other cells [620,622]. While more research is required to establish the precise role of each EVs subpopulation in tumor progression, one recent study has suggested that exosomes are the most bioactive and effective at promoting hypoxic cell survival in the tumor microenvironment [619].
Liquid biopsies complement and resolve the limitations of traditional biopsies by facilitating assessment and analysis of tumor tissue when acquiring tissue samples are impossible or if specimens were inadequate [623]. Additionally, liquid biopsy enables a better understanding of the wide array of mutations present within the heterogenous tumor. Traditionally, liquid biopsy refers to the collection of circulating tumor cells (CTCs) and cell-free nucleic acids from peripheral blood through venipuncture. This offers considerably fewer risks, resources, and expertise than serial biopsy which requires an experienced physician to perform. Furthermore, liquid biopsies offer a noninvasive method for determining specific tumor mutations which inform therapy options and provide a means to monitor patient response to therapy as well as tumor recurrence. Traditional liquid biopsies, however, suffer from low levels of detection as CTCs are rare and cell-free nucleic acids are often degraded in circulation. Current FDA-approved methods for liquid biopsy therefore exhibit low sensitivity and specificity. A recent clinical study, however, utilized liquid biopsy to analyze methylation signatures in cell-free DNA and was able to detect 50 different types of tumors, with increasing sensitivity and specificity as staging progressed [624]. However, liquid biopsies utilizing EVs may offer a more robust tumor detection as EVs resist degradation in the bloodstream, unlike cell-free nucleic acids, and are considerably more abundant than CTCs [[625], [626], [627]]. EVs have also shown potential for early tumor detection even before the onset of clinical symptoms as well as ability to distinguish between benign lesions and tumors [40,618,626,628,629]. However, EVs carry a plethora of markers that offer diagnostic capability; thus, optimization of biomarker selection is necessary to justify the significant financial and time demands of assay development for clinical application.
Current chemo- and radiotherapies often result in systemic toxicity which can manifest in any organ or multiple organs, depending on the therapy and dosage administered and its duration [630]. Immunotherapy decreases these negative side effects by targeting specific tumors. However, some types of immunotherapy, such as monoclonal antibodies, still induce negative side effects such as fever, muscle pain, nausea and vomiting, as well as other flu-like symptoms [631]. sEVs, on the other hand, have exhibited promise in tumor targeting with minimal to no observed toxicity [566,632,633]. sEVs have been engineered to deliver chemotherapy, toxic proteins, and tumor suppressing miRs/siRNA [39,531]. Even non-targeted delivery of sEVs have shown minimal toxicity and an increased uptake by tumor cells compared to healthy cells [528,634]. sEVs-mediated drug delivery also enables treatment to effectively pass the blood-brain barrier, which is an advantage over traditional chemotherapy [39,635]. Efficacy of sEVs-based delivery systems, however, relies on identifying the optimal cell population for sEVs harvest. sEVs for drug delivery have come from immune cell populations as well as tumor cell populations, thereby enhancing anti-tumor immune response in the host [636]. However, there are issues that must be overcome for sEVs-based drug delivery systems to be viable in the clinic. These systems still rely heavily on identifying the most viable subpopulations of sEVs and isolating those purified samples. Despite apparent tolerance of sEVs, and an observed lack of toxicity, dosage must be optimized to minimize clearance and ensure enough drug-carrying sEVs are sequestered by the tumor. Fortunately, sEVs structure can be modified to enhance circulation time and ensure arrival to and uptake at the tumor site [524,[637], [638]].
Despite the hurdles facing clinical use of EVs, they remain incredibly versatile. As the field of EVs research progresses and technology improves, standardized isolation methods which enhance purity will be determined and agreed upon for clinical applications. Once a standardized isolation protocol is determined, the next barrier to clinical EVs application will involve enrichment for tumor-specific markers that can diagnose, inform treatment, and facilitate patient monitoring. Deeper understanding of the mechanisms governing EVs communication between cells will inform novel treatment options which include not only utilizing EVs targeted drug delivery, but also potentially targeting EVs to enhance current and future antitumor therapies. It is this versatility which secures the future of EVs research and ensures their role in the future of medicine.
Author contributions
Writing of original draft: K.A. A.M. Y.W. and N.R.; Review & editing: K.A. A.M. W.M. W.A. and Y.W. All authors have read and agreed to the published version of the manuscript.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
This work was partially supported by Binghamton University Faculty Start-up Fund (grant No. 910252–35), Binghamton University S3IP Award (No. ADLG195), and National Cancer Institute 1R01CA230339-01 subaward.
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Contributor Information
Wenjun Mao, Email: maowenjun1@njmu.edu.cn.
Waseem Asghar, Email: wasghar@fau.edu.
Yuan Wan, Email: ywan@binghamton.edu.
References
- 1.Harding C., Heuser J., Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983;97:329–339. doi: 10.1083/jcb.97.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan B.T., Johnstone R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: selective externalization of the receptor. Cell. 1983;33:967–978. doi: 10.1016/0092-8674(83)90040-5. [DOI] [PubMed] [Google Scholar]
- 3.Pan B.T., Teng K., Wu C., Adam M., Johnstone R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985;101:942–948. doi: 10.1083/jcb.101.3.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maia J., Caja S., Strano Moraes M.C., Couto N., Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Frontiers in Cell and Developmental Biology. 2018;6 18 doi: 10.3389/fcell.2018.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathieu M., Martin-Jaular L., Lavieu G., Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019:21 9–2117. doi: 10.1038/s41556-018-0250-9. [DOI] [PubMed] [Google Scholar]
- 6.Meldolesi J. Current biology review exosomes and ectosomes in intercellular communication. Curr. Biol. 2018;28:R435–R444. doi: 10.1016/j.cub.2018.01.059. [DOI] [PubMed] [Google Scholar]
- 7.Bellin G. Exosome in cardiovascular diseases: a complex world full of hope. Cells. 2019;8:166. doi: 10.3390/cells8020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saeedi S., Israel S., Nagy C., Turecki G. The emerging role of exosomes in mental disorders. Transl. Psychiatry. 2019:9 1–11. doi: 10.1038/s41398-019-0459-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu W. Role of exosomes in central nervous system diseases. Front. Mol. Neurosci. 2019;12 240 doi: 10.3389/fnmol.2019.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jan A.T. Perspective insights of exosomes in neurodegenerative diseases: a critical appraisal. Front. Aging Neurosci. 2017;9 doi: 10.3389/fnagi.2017.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arraud N. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J. Thromb. Haemostasis. 2014;12:614–627. doi: 10.1111/jth.12554. [DOI] [PubMed] [Google Scholar]
- 12.Salih M., Zietse R., Hoorn E.J. Urinary extracellular vesicles and the kidney: biomarkers and beyond. Am. J. Physiol. Physiol. 2014;306:F1251–F1259. doi: 10.1152/ajprenal.00128.2014. [DOI] [PubMed] [Google Scholar]
- 13.Winck F.V. Insights into immune responses in oral cancer through proteomic analysis of saliva and salivary extracellular vesicles. Sci. Rep. 2015;5:16305. doi: 10.1038/srep16305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiasserini D. Proteomic analysis of cerebrospinal fluid extracellular vesicles: a comprehensive dataset. J. Proteomics. 2014;106:191–204. doi: 10.1016/j.jprot.2014.04.028. [DOI] [PubMed] [Google Scholar]
- 15.Maas S.L.N., Breakefield X.O., Weaver A.M. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. 2017;27:172–188. doi: 10.1016/j.tcb.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Pol E., Boing A.N., Harrison P., Sturk A., Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012;64:676–705. doi: 10.1124/pr.112.005983. [DOI] [PubMed] [Google Scholar]
- 17.Raposo G., Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalluri R. The biology and function of exosomes in cancer. J. Clin. Invest. 2016;126:1208–1215. doi: 10.1172/JCI81135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yanez-Mo M. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles. 2015;4:27066. doi: 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kahlert C., Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. 2013;91:431–437. doi: 10.1007/s00109-013-1020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee Y., El Andaloussi S., Wood M.J.A. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012;21:R125–R134. doi: 10.1093/hmg/dds317. [DOI] [PubMed] [Google Scholar]
- 22.Liu X. Circulating exosomal miR-27a and miR-130a act as novel diagnostic and prognostic biomarkers of colorectal cancer. Cancer Epidemiol. Prev. Biomarkers. 2018;27:746–754. doi: 10.1158/1055-9965.EPI-18-0067. [DOI] [PubMed] [Google Scholar]
- 23.Chen M. Distinct shed microvesicle and exosome microRNA signatures reveal diagnostic markers for colorectal cancer. journals.plos.org. 2019;14 doi: 10.1371/journal.pone.0210003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cruz De los Santos M., Dragomir M.P., Calin G.A. The role of exosomal long non-coding RNAs in cancer drug resistance. Cancer Drug Resist. 2019;2:1178–1192. doi: 10.20517/cdr.2019.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Y. The role of exosomal noncoding RNAs in cancer. Mol. Canc. 2019;18 37 doi: 10.1186/s12943-019-0984-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng J., Meng J., Zhu L., Peng Y. Exosomal noncoding RNAs in Glioma: biological functions and potential clinical applications. Mol. Canc. 2020:19 1–1914. doi: 10.1186/s12943-020-01189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guescini M., Genedani S., Stocchi V., Agnati L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural. Transm. 2010;117:1–4. doi: 10.1007/s00702-009-0288-8. [DOI] [PubMed] [Google Scholar]
- 28.Balaj L. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thakur B.K. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24:766–769. doi: 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian Z., Shen Q., Yang X., Qiu Y., Zhang W. The role of extracellular vesicles: an epigenetic view of the cancer microenvironment. BioMed Res. Int. 2015:2015. doi: 10.1155/2015/649161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valadi H. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 32.Vallabhaneni K.C. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget. 2015;6:4953–4967. doi: 10.18632/oncotarget.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mannerström B. Epigenetic alterations in mesenchymal stem cells by osteosarcoma-derived extracellular vesicles. Epigenetics. 2019;14:352–364. doi: 10.1080/15592294.2019.1585177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melo S.A. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kosaka N. Exploiting the message from cancer: the diagnostic value of extracellular vesicles for clinical applications. Exp. Mol. Med. 2019;51:1–9. doi: 10.1038/s12276-019-0219-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto T., Kosaka N., Ochiya T. Latest advances in extracellular vesicles: from bench to bedside. Sci. Technol. Adv. Mater. 2019;20:746–757. doi: 10.1080/14686996.2019.1629835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Liu Y., Liu H., Tang W.H. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9 doi: 10.1186/s13578-019-0282-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Q. Clinical applications of exosome membrane proteins. Precis. Clin. Med. 2020;3:54–66. doi: 10.1093/pcmedi/pbaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tai Y.L. Basics and applications of tumor-derived extracellular vesicles. J. Biomed. Sci. 2019;26:1–17. doi: 10.1186/s12929-019-0533-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makler A., Asghar W. Exosomal biomarkers for cancer diagnosis and patient monitoring. Expert Rev. Mol. Diagn. 2020:20 387–400. doi: 10.1080/14737159.2020.1731308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tai Y.L., Chen K.C., Hsieh J.T., Shen T.L. Exosomes in cancer development and clinical applications. Canc. Sci. 2018:109 2364–2374. doi: 10.1111/cas.13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Théry C. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018;7 doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalra H. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012;10 doi: 10.1371/journal.pbio.1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathivanan S., Fahner C.J., Reid G.E., Simpson R.J. ExoCarta 2012: database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2011;40:D1241–D1244. doi: 10.1093/nar/gkr828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim, D. et al. EVpedia: a Community Web Portal for Extracellular Vesicles Research. academic.oup.com. [DOI] [PMC free article] [PubMed]
- 46.Liu T. EVmiRNA: a database of miRNA profiling in extracellular vesicles. Nucleic Acids Res. 2018;47:89–93. doi: 10.1093/nar/gky985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pathan M. Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019;47:D516–D519. doi: 10.1093/nar/gky1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simpson R.J., Kalra H., Mathivanan S. Exocarta as a resource for exosomal research. J. Extracell. Vesicles. 2012;1 doi: 10.3402/jev.v1i0.18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keerthikumar, S., Chisanga, D., … D. A.-J. Of Molecular & 2016, Undefined. ExoCarta: a Web-Based Compendium of Exosomal Cargo. Elsevier. [DOI] [PMC free article] [PubMed]
- 50.Latifkar A., Hur Y.H., Sanchez J.C., Cerione R.A., Antonyak M.A. New insights into extracellular vesicle biogenesis and function. J. Cell Sci. 2019;132 doi: 10.1242/jcs.222406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hur Y.H., Cerione R.A., Antonyak M.A. Extracellular vesicles and their roles in stem cell biology. Stem Cell. 2020;38:469–476. doi: 10.1002/stem.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alqurashi H., Ortega Asencio I., Lambert D.W. The emerging potential of extracellular vesicles in cell-free tissue engineering and regenerative medicine. Tissue Eng. Part B Rev. ten.teb.2020. 2020:222. doi: 10.1089/ten.teb.2020.0222. [DOI] [PubMed] [Google Scholar]
- 53.Malloci M. Extracellular vesicles: mechanisms in human health and disease. Antioxidants Redox Signal. 2019;30:813–856. doi: 10.1089/ars.2017.7265. [DOI] [PubMed] [Google Scholar]
- 54.Akbari A. Free and hydrogel encapsulated exosome-based therapies in regenerative medicine. Life Sci. 2020;249 doi: 10.1016/j.lfs.2020.117447. [DOI] [PubMed] [Google Scholar]
- 55.Ayers L., Pink R., Carter D.R.F., Nieuwland R. Clinical requirements for extracellular vesicle assays. J. Extracell. Vesicles. 2019;8 doi: 10.1080/20013078.2019.1593755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Livshits M.A. Isolation of exosomes by differential centrifugation: theoretical analysis of a commonly used protocol. Sci. Rep. 2015;5:17319. doi: 10.1038/srep17319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abels E.R., Breakefield X.O. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell. Mol. Neurobiol. 2016;36:301–312. doi: 10.1007/s10571-016-0366-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taylor D.D., Shah S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods. 2015;87:3–10. doi: 10.1016/j.ymeth.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 59.Momen-Heravi F. Isolation of extracellular vesicles by ultracentrifugation. Methods Mol. Biol. 2017;1660:25–32. doi: 10.1007/978-1-4939-7253-1_3. [DOI] [PubMed] [Google Scholar]
- 60.Lucchetti D., Fattorossi A., Sgambato A. Extracellular vesicles in oncology: progress and pitfalls in the methods of isolation and analysis. Biotechnol. J. 2019;14:1700716. doi: 10.1002/biot.201700716. [DOI] [PubMed] [Google Scholar]
- 61.Konoshenko M.Y., Lekchnov E.A., Vlassov A.V., Laktionov P.P. Isolation of extracellular vesicles: general methodologies and latest trends. BioMed Res. Int. 2018:1–27. doi: 10.1155/2018/8545347. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duong P., Chung A., Bouchareychas L., Raffai R.L. Cushioned-Density Gradient Ultracentrifugation (C-DGUC) improves the isolation efficiency of extracellular vesicles. PloS One. 2019;14 doi: 10.1371/journal.pone.0215324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Witwer K.W. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles. 2013;2 doi: 10.3402/jev.v2i0.20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li P., Kaslan M., Lee S.H., Yao J., Gao Z. Progress in exosome isolation techniques. Theranostics. 2017;7:789. doi: 10.7150/thno.18133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Deun J. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles. 2014;3:24858. doi: 10.3402/jev.v3.24858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Greening D.W., Xu R., Ji H., Tauro B.J., Simpson R.J. 1295 vols. 179–209. Humana Press Inc.; 2015. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 67.Carnino J.M., Lee H., Jin Y. Isolation and characterization of extracellular vesicles from Broncho-alveolar lavage fluid: a review and comparison of different methods. Respir. Res. 2019;20:240. doi: 10.1186/s12931-019-1210-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu M. Acoustofluidic separation of cells and particles. Microsystems and Nanoengineering. 2019:5 1–18. doi: 10.1038/s41378-019-0064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iwai K., Minamisawa T., Suga K., Yajima Y., Shiba K. Isolation of human salivary extracellular vesicles by iodixanol density gradient ultracentrifugation and their characterizations. J. Extracell. Vesicles. 2016;5:30829. doi: 10.3402/jev.v5.30829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Monguio-Tortajada M., Galvez-Monton C., Bayes-Genis A., Roura S., Borras F.E. Extracellular vesicle isolation methods: rising impact of size-exclusion chromatography. Cell. Mol. Life Sci. 2019;76:2369–2382. doi: 10.1007/s00018-019-03071-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rider M.A., Hurwitz S.N., Meckes D.G. ExtraPEG: a polyethylene glycol-based method for enrichment of extracellular vesicles. Sci. Rep. 2016;6:1–14. doi: 10.1038/srep23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo S.-C., Tao S.-C., Dawn H. Microfluidics-based on-a-chip systems for isolating and analysing extracellular vesicles. J. Extracell. Vesicles. 2018;7:1508271. doi: 10.1080/20013078.2018.1508271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szatanek R., Baran J., Siedlar M., Baj-Krzyworzeka M. Isolation of extracellular vesicles: determining the correct approach. Int. J. Mol. Med. 2015;36:11–17. doi: 10.3892/ijmm.2015.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Niu Z. Polymer-based precipitation preserves biological activities of extracellular vesicles from an endometrial cell line. PloS One. 2017;12 doi: 10.1371/journal.pone.0186534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abramowicz A., Widlak P., Pietrowska M. Proteomic analysis of exosomal cargo: the challenge of high purity vesicle isolation. Mol. Biosyst. 2016;12:1407–1419. doi: 10.1039/c6mb00082g. [DOI] [PubMed] [Google Scholar]
- 76.Liang L.-G. An integrated double-filtration microfluidic device for isolation, enrichment and quantification of urinary extracellular vesicles for detection of bladder cancer. Sci. Rep. 2017;7:46224. doi: 10.1038/srep46224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Woo H.K. Exodisc for rapid, size-selective, and efficient isolation and analysis of nanoscale extracellular vesicles from biological samples. ACS Nano. 2017;11:1360–1370. doi: 10.1021/acsnano.6b06131. [DOI] [PubMed] [Google Scholar]
- 78.Sunkara V. Fully automated, label-free isolation of extracellular vesicles from whole blood for cancer diagnosis and monitoring. Theranostics. 2019;9:1851–1863. doi: 10.7150/thno.32438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Onódi Z. Isolation of high-purity extracellular vesicles by the combination of iodixanol density gradient ultracentrifugation and bind-elute chromatography from blood plasma. Front. Physiol. 2018;9:1479. doi: 10.3389/fphys.2018.01479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oeyen E. Ultrafiltration and size exclusion chromatography combined with asymmetrical-flow field-flow fractionation for the isolation and characterisation of extracellular vesicles from urine. J. Extracell. Vesicles. 2018;7:1490143. doi: 10.1080/20013078.2018.1490143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lozano-Ramos I. Size-exclusion chromatography-based enrichment of extracellular vesicles from urine samples. J. Extracell. Vesicles. 2015;4:27369. doi: 10.3402/jev.v4.27369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lane R.E., Korbie D., Trau M., Hill M.M. Optimizing size exclusion chromatography for extracellular vesicle enrichment and proteomic analysis from clinically relevant samples. Proteomics. 2019;19 doi: 10.1002/pmic.201800156. [DOI] [PubMed] [Google Scholar]
- 83.Brett S.I. Immunoaffinity based methods are superior to kits for purification of prostate derived extracellular vesicles from plasma samples. Prostate. 2017;77:1335–1343. doi: 10.1002/pros.23393. [DOI] [PubMed] [Google Scholar]
- 84.Sharma P. Immunoaffinity-based isolation of melanoma cell-derived exosomes from plasma of patients with melanoma. J. Extracell. Vesicles. 2018;7:1435138. doi: 10.1080/20013078.2018.1435138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lo T.W. Microfluidic device for high-throughput affinity-based isolation of extracellular vesicles. Lab Chip. 2020;20:1762–1770. doi: 10.1039/c9lc01190k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tran P.H.L. Aptamer-guided extracellular vesicle theranostics in oncology. Theranostics. 2020:10 3849–3866. doi: 10.7150/thno.39706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gao M.L., He F., Yin B.C., Ye B.C. A dual signal amplification method for exosome detection based on DNA dendrimer self-assembly. Analyst. 2019;144:1995–2002. doi: 10.1039/c8an02383b. [DOI] [PubMed] [Google Scholar]
- 88.Zhang K. Rapid capture and nondestructive release of extracellular vesicles using aptamer-based magnetic isolation. ACS Sens. 2019;4:1245–1251. doi: 10.1021/acssensors.9b00060. [DOI] [PubMed] [Google Scholar]
- 89.Yu Q. Development of a lateral flow aptamer assay strip for facile identification of theranostic exosomes isolated from human lung carcinoma cells. Anal. Biochem. 2020;594:113591. doi: 10.1016/j.ab.2020.113591. [DOI] [PubMed] [Google Scholar]
- 90.Wan Y. Rapid magnetic isolation of extracellular vesicles via lipid-based nanoprobes. Nat. Biomed. Eng. 2017;1:58. doi: 10.1038/s41551-017-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wan Y. Enrichment of extracellular vesicles with lipid nanoprobe functionalized nanostructured silica. Lab Chip. 2019;19:2346–2355. doi: 10.1039/c8lc01359d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han S. Isolation and analysis of extracellular vesicles in a Morpho butterfly wing-integrated microvortex biochip. Biosens. Bioelectron. 2020;154:112073. doi: 10.1016/j.bios.2020.112073. [DOI] [PubMed] [Google Scholar]
- 93.Rahman N.A., Ibrahim F., Yafouz B. Dielectrophoresis for biomedical sciences applications: a review. Sensors. 2017;17 doi: 10.3390/s17030449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ibsen S.D. Rapid isolation and detection of exosomes and associated biomarkers from plasma. ACS Nano. 2017;11:6641–6651. doi: 10.1021/acsnano.7b00549. [DOI] [PubMed] [Google Scholar]
- 95.Lewis J. A pilot proof-of-principle analysis demonstrating dielectrophoresis (DEP) as a glioblastoma biomarker platform. Sci. Rep. 2019;9:1–10. doi: 10.1038/s41598-019-46311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tengattini S. Chromatographic approaches for purification and analytical characterization of extracellular vesicles: recent advancements. Chromatographia. 2019:82 415–424. [Google Scholar]
- 97.Kosanovic M., Milutinovic B., Goc S., Mitic N., Jankovic M. Ion-exchange chromatography purification of extracellular vesicles. Biotechniques. 2017;63:65–71. doi: 10.2144/000114575. [DOI] [PubMed] [Google Scholar]
- 98.Heath N. Rapid isolation and enrichment of extracellular vesicle preparations using anion exchange chromatography. Sci. Rep. 2018;8:5730. doi: 10.1038/s41598-018-24163-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yasui T. Unveiling massive numbers of cancer-related urinary-microRNA candidates via nanowires. Sci. Adv. 2017;3 doi: 10.1126/sciadv.1701133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thery C., Amigorena S., Raposo G., Clayton A. Curr Protoc Cell Biol Chapter 3. Unit. 2006;3:22. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- 101.Chulpanova D.S., Kitaeva K.V., James V., Rizvanov A.A., Solovyeva V.V. Therapeutic prospects of extracellular vesicles in cancer treatment. Front. Immunol. 2018;9:1534. doi: 10.3389/fimmu.2018.01534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brennan K. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020;10:1–13. doi: 10.1038/s41598-020-57497-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Coumans F.A.W. Methodological guidelines to study extracellular vesicles. Circ. Res. 2017;120:1632–1648. doi: 10.1161/CIRCRESAHA.117.309417. [DOI] [PubMed] [Google Scholar]
- 104.Karimi N. Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cell. Mol. Life Sci. 2018;75:2873–2886. doi: 10.1007/s00018-018-2773-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ramshani Z. Extracellular vesicle microRNA quantification from plasma using an integrated microfluidic device. Commun. Biol. 2019;2:1–9. doi: 10.1038/s42003-019-0435-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu M. Isolation of exosomes from whole blood by integrating acoustics and microfluidics. Proc. Natl. Acad. Sci. U.S.A. 2017;114:10584–10589. doi: 10.1073/pnas.1709210114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ludwig A.K. Precipitation with polyethylene glycol followed by washing and pelleting by ultracentrifugation enriches extracellular vesicles from tissue culture supernatants in small and large scales. J. Extracell. Vesicles. 2018;7:1528109. doi: 10.1080/20013078.2018.1528109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.García-Romero N. Polyethylene glycol improves current methods for circulating extracellular vesicle-derived DNA isolation. J. Transl. Med. 2019;17:75. doi: 10.1186/s12967-019-1825-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Benedikter B.J. Ultrafiltration combined with size exclusion chromatography efficiently isolates extracellular vesicles from cell culture media for compositional and functional studies. Sci. Rep. 2017;7:1–13. doi: 10.1038/s41598-017-15717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu R., Simpson R.J., Greening D.W. 1545 vols. 91–1545116. Humana Press Inc.; 2017. A protocol for isolation and proteomic characterization of distinct extracellular vesicle subtypes by sequential centrifugal ultrafiltration. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 111.Guerreiro E.M. Efficient extracellular vesicle isolation by combining cell media modifications, ultrafiltration, and size-exclusion chromatography. PloS One. 2018;13 doi: 10.1371/journal.pone.0204276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kornilov R. Efficient ultrafiltration-based protocol to deplete extracellular vesicles from fetal bovine serum. J. Extracell. Vesicles. 2018;7:1422674. doi: 10.1080/20013078.2017.1422674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vergauwen G. Confounding factors of ultrafiltration and protein analysis in extracellular vesicle research. Sci. Rep. 2017;7:1–12. doi: 10.1038/s41598-017-02599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu F. The exosome total isolation chip. ACS Nano. 2017;11:10712–10723. doi: 10.1021/acsnano.7b04878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lobb R.J. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles. 2015;4:27031. doi: 10.3402/jev.v4.27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baranyai T. Isolation of exosomes from blood plasma: qualitative and quantitative comparison of ultracentrifugation and size exclusion chromatography methods. PloS One. 2015;10 doi: 10.1371/journal.pone.0145686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Batrakova E.V., Kim M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Contr. Release. 2015;219:396–405. doi: 10.1016/j.jconrel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Takov K., Yellon D.M., Davidson S.M. Comparison of small extracellular vesicles isolated from plasma by ultracentrifugation or size-exclusion chromatography: yield, purity and functional potential. J. Extracell. Vesicles. 2019;8:1560809. doi: 10.1080/20013078.2018.1560809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Koh Y.Q., Almughlliq F.B., Vaswani K., Peiris H.N., Mitchell M.D. Exosome enrichment by ultracentrifugation and size exclusion chromatography. Front. Biosci. (Landmark Ed. 2018;23:865–874. doi: 10.2741/4621. [DOI] [PubMed] [Google Scholar]
- 120.Brenner G. Efficient isolation of extracellular vesicles from blood plasma based on iodixanol density gradient ultracentrifugation combined with bind-elute chromatography. J. Extracell. Vesicles. 2018;7:152–153. [Google Scholar]
- 121.Gheinani A.H. Improved isolation strategies to increase the yield and purity of human urinary exosomes for biomarker discovery. Sci. Rep. 2018;8:3945. doi: 10.1038/s41598-018-22142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boriachek K. Biological functions and current advances in isolation and detection strategies for exosome nanovesicles. Small. 2018;14:1702153. doi: 10.1002/smll.201702153. [DOI] [PubMed] [Google Scholar]
- 123.Tucher C. Extracellular vesicle subtypes released from activated or apoptotic T-lymphocytes carry a specific and stimulus-dependent protein cargo. Front. Immunol. 2018;9 doi: 10.3389/fimmu.2018.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dong J. Bio-inspired NanoVilli chips for enhanced capture of tumor-derived extracellular vesicles: toward non-invasive detection of gene alterations in non-small cell lung cancer. ACS Appl. Mater. Interfaces. 2019;11:13973–13983. doi: 10.1021/acsami.9b01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kanwar S.S., Dunlay C.J., Simeone D.M., Nagrath S. Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes. Lab Chip. 2014;14:1891–1900. doi: 10.1039/c4lc00136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.He M., Crow J., Roth M., Zeng Y., Godwin A.K. Integrated immunoisolation and protein analysis of circulating exosomes using microfluidic technology. Lab Chip. 2014;14:3773–3780. doi: 10.1039/c4lc00662c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Oksvold M.P., Neurauter A., Pedersen K.W., Clifton N.J. 1218 vols. 465–481. 2015. Magnetic bead-based isolation of exosomes. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 128.Lim J. Direct isolation and characterization of circulating exosomes from biological samples using magnetic nanowires. J. Nanobiotechnol. 2019;17:1. doi: 10.1186/s12951-018-0433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murlidhar V. A radial flow microfluidic device for ultra-high-throughput affinity-based isolation of circulating tumor cells. Small. 2014;10:4895–4904. doi: 10.1002/smll.201400719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li P. Ultrasensitive and reversible nanoplatform of urinary exosomes for prostate cancer diagnosis. ACS Sens. 2019;4:1433–1441. doi: 10.1021/acssensors.9b00621. [DOI] [PubMed] [Google Scholar]
- 131.Nakai W. A novel affinity-based method for the isolation of highly purified extracellular vesicles. Sci. Rep. 2016;6:33935. doi: 10.1038/srep33935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Campos-Silva C. High sensitivity detection of extracellular vesicles immune-captured from urine by conventional flow cytometry. Sci. Rep. 2019;9:1–12. doi: 10.1038/s41598-019-38516-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hoshino A. Extracellular vesicle and particle biomarkers define multiple human cancers. Cell. 2020;182:1044–1061. doi: 10.1016/j.cell.2020.07.009. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Popović M., de Marco A. Canonical and selective approaches in exosome purification and their implications for diagnostic accuracy. Transl. Cancer Res. 2017;7:S209–S225. [Google Scholar]
- 135.Ayala-Mar S., Perez-Gonzalez V.H., Mata-Gómez M.A., Gallo-Villanueva R.C., González-Valdez J. Electrokinetically driven exosome separation and concentration using dielectrophoretic-enhanced PDMS-based microfluidics. Anal. Chem. 2019;91:14975–14982. doi: 10.1021/acs.analchem.9b03448. [DOI] [PubMed] [Google Scholar]
- 136.Lewis J.M. Integrated analysis of exosomal protein biomarkers on alternating current electrokinetic chips enables rapid detection of pancreatic cancer in patient blood. ACS Nano. 2018;12:3311–3320. doi: 10.1021/acsnano.7b08199. [DOI] [PubMed] [Google Scholar]
- 137.Shi L. Rapid and label-free isolation of small extracellular vesicles from biofluids utilizing a novel insulator based dielectrophoretic device. Lab Chip. 2019;19:3726–3734. doi: 10.1039/c9lc00902g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wunsch B.H. Nanoscale lateral displacement arrays for the separation of exosomes and colloids down to 20nm. Nat. Nanotechnol. 2016;11:936–940. doi: 10.1038/nnano.2016.134. [DOI] [PubMed] [Google Scholar]
- 139.Smith J.T. Integrated nanoscale deterministic lateral displacement arrays for separation of extracellular vesicles from clinically-relevant volumes of biological samples. Lab Chip. 2018;18:3913–3925. doi: 10.1039/c8lc01017j. [DOI] [PubMed] [Google Scholar]
- 140.Wan Y., Zheng S.Y. Enhanced detection of tumour-secreted vesicles. Nat. Biomed. Eng. 2019;3:421–422. doi: 10.1038/s41551-019-0415-2. [DOI] [PubMed] [Google Scholar]
- 141.Jeppesen D.K. Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J. Extracell. Vesicles. 2014;3 doi: 10.3402/jev.v3.25011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen C.Y., Hogan M.C., Ward C.J. Purification of exosome-like vesicles from urine. Methods Enzymol. 2013;524:225–241. doi: 10.1016/B978-0-12-397945-2.00013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Böing A.N. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles. 2014;3 doi: 10.3402/jev.v3.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lv L.L. Isolation and quantification of MicroRNAs from urinary exosomes/microvesicles for biomarker discovery. Int. J. Biol. Sci. 2013;9:1021–1031. doi: 10.7150/ijbs.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rekker K. Comparison of serum exosome isolation methods for microRNA profiling. Clin. Biochem. 2014;47:135–138. doi: 10.1016/j.clinbiochem.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 146.Caradec J. Reproducibility and efficiency of serum-derived exosome extraction methods. Clin. Biochem. 2014;47:1286–1292. doi: 10.1016/j.clinbiochem.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 147.Yuana Y., Levels J., Grootemaat A., Sturk A., Nieuwland R. Co-isolation of extracellular vesicles and high-density lipoproteins using density gradient ultracentrifugation. J. Extracell. Vesicles. 2014;3 doi: 10.3402/jev.v3.23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hutcheson J.D. Enrichment of calcifying extracellular vesicles using density-based ultracentrifugation protocol. J. Extracell. Vesicles. 2014;3:25129. doi: 10.3402/jev.v3.25129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Musante L. A simplified method to recover urinary vesicles for clinical applications, and sample banking. Sci. Rep. 2014;4 doi: 10.1038/srep07532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Oliveira-Rodríguez M. Development of a rapid lateral flow immunoassay test for detection of exosomes previously enriched from cell culture medium and body fluids. J. Extracell. Vesicles. 2016;5 doi: 10.3402/jev.v5.31803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.López-Cobo S. Immunoassays for scarce tumour-antigens in exosomes: detection of the human NKG2D-Ligand, MICA, in tetraspanin-containing nanovesicles from melanoma. J. Nanobiotechnol. 2018;16:47. doi: 10.1186/s12951-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Boriachek K. Avoiding pre-isolation step in exosome analysis: direct isolation and sensitive detection of exosomes using gold-loaded nanoporous ferric oxide nanozymes. Anal. Chem. 2019;91:3827–3834. doi: 10.1021/acs.analchem.8b03619. [DOI] [PubMed] [Google Scholar]
- 153.Liu C. Single-exosome-counting immunoassays for cancer diagnostics. Nano Lett. 2018;18:4226–4232. doi: 10.1021/acs.nanolett.8b01184. [DOI] [PubMed] [Google Scholar]
- 154.Liang K. Nanoplasmonic quantification of tumour-derived extracellular vesicles in plasma microsamples for diagnosis and treatment monitoring. Nat. Biomed. Eng. 2017;1:21. doi: 10.1038/s41551-016-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu C. Sensitive detection of exosomal proteins via a compact surface plasmon resonance biosensor for cancer diagnosis. ACS Sens. 2018;3:1471–1479. doi: 10.1021/acssensors.8b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Boriachek K. Quantum dot-based sensitive detection of disease specific exosome in serum. Analyst. 2017;142:2211–2219. doi: 10.1039/c7an00672a. [DOI] [PubMed] [Google Scholar]
- 157.Bai Y. Rapid isolation and multiplexed detection of exosome tumor markers via queued beads combined with quantum dots in a microarray. Nano-Micro Lett. 2019;11:1–11. doi: 10.1007/s40820-019-0285-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang P. Ultrasensitive detection of circulating exosomes with a 3D-nanopatterned microfluidic chip. Nat. Biomed. Eng. 2019;3:438–451. doi: 10.1038/s41551-019-0356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yu Y. Electrical and label-free quantification of exosomes with a reduced graphene oxide field effect transistor biosensor. Anal. Chem. 2019;91:10679–10686. doi: 10.1021/acs.analchem.9b01950. [DOI] [PubMed] [Google Scholar]
- 160.Mehrotra P. Biosensors and their applications - a review. Journal of Oral Biology and Craniofacial Research. 2016:6 153–159. doi: 10.1016/j.jobcr.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Noah N.M., Ndangili P.M. Current trends of nanobiosensors for point-of-care diagnostics. J. Anal. Methods Chem. 2019;2019 doi: 10.1155/2019/2179718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Unser S., Bruzas I., He J., Sagle L. Localized surface plasmon resonance biosensing: current challenges and approaches. Sensors. 2015;15:15684–15716. doi: 10.3390/s150715684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gorodkiewicz E., Lukaszewski Z. Recent progress in surface plasmon resonance biosensors (2016 to mid-2018) Biosensors. 2018;8 doi: 10.3390/bios8040132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bellassai N., D'Agata R., Jungbluth V., Spoto G. Surface plasmon resonance for biomarker detection: advances in non-invasive cancer diagnosis. Frontiers in Chemistry. 2019;7:570. doi: 10.3389/fchem.2019.00570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lee J. Enhanced paper-based ELISA for simultaneous EVs/exosome isolation and detection using streptavidin agarose-based immobilization. Analyst. 2020;145:157–164. doi: 10.1039/c9an01140d. [DOI] [PubMed] [Google Scholar]
- 166.Oliveira-Rodríguez M. Point-of-care detection of extracellular vesicles: sensitivity optimization and multiple-target detection. Biosens. Bioelectron. 2017;87:38–45. doi: 10.1016/j.bios.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 167.Kanada M., Bachmann M.H., Contag C.H. Signaling by extracellular vesicles advances cancer hallmarks. Trends in Cancer. 2016;2 84–94 doi: 10.1016/j.trecan.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 168.Anderson H.C., Mulhall D., Garimella R. 90 vols. 1549–1557. Laboratory Investigation; 2010. (Role of Extracellular Membrane Vesicles in the Pathogenesis of Various Diseases, Including Cancer, Renal Diseases, Atherosclerosis, and Arthritis). [DOI] [PubMed] [Google Scholar]
- 169.Valenti R. Tumor-released microvesicles as vehicles of immunosuppression. Canc. Res. 2007:67 2912–2915. doi: 10.1158/0008-5472.CAN-07-0520. [DOI] [PubMed] [Google Scholar]
- 170.Pilzer D., Gasser O., Moskovich O., Schifferli J.A., Fishelson Z. Springer Seminars in Immunopathology; 2005. Emission of Membrane Vesicles: Roles in Complement Resistance, Immunity and Cancer; pp. 27 375–387. [DOI] [PubMed] [Google Scholar]
- 171.Chaput N., Taïeb J., André F., Zitvogel L. The potential of exosomes in immunotherapy. Expet Opin. Biol. Ther. 2005:5 737–747. doi: 10.1517/14712598.5.6.737. [DOI] [PubMed] [Google Scholar]
- 172.Sato S., Weaver A.M. Extracellular vesicles: important collaborators in cancer progression. Essays Biochem. 2018;62:149–163. doi: 10.1042/EBC20170080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Song W. Tumor-derived extracellular vesicles in angiogenesis. Biomed. Pharmacother. 2018;102:1203–1208. doi: 10.1016/j.biopha.2018.03.148. [DOI] [PubMed] [Google Scholar]
- 174.Chiba M., Kubota S., Sato K., Monzen S. Exosomes released from pancreatic cancer cells enhance angiogenic activities via dynamin-dependent endocytosis in endothelial cells in vitro. Sci. Rep. 2018;8:11972. doi: 10.1038/s41598-018-30446-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Han L., Lam E.W.F., Sun Y. Extracellular vesicles in the tumor microenvironment: old stories, but new tales. Mol. Canc. 2019;18:59. doi: 10.1186/s12943-019-0980-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ahmadi M., Rezaie J. Tumor cells derived-exosomes as angiogenenic agents: possible therapeutic implications. J. Transl. Med. 2020;18:249. doi: 10.1186/s12967-020-02426-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nazarenko I. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Can. Res. 2010;70:1668–1678. doi: 10.1158/0008-5472.CAN-09-2470. [DOI] [PubMed] [Google Scholar]
- 178.Feng Q. A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumour angiogenesis. Nat. Commun. 2017;8:14450. doi: 10.1038/ncomms14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Horie K. Exosomes expressing carbonic anhydrase 9 promote angiogenesis. Biochem. Biophys. Res. Commun. 2017;492:356–361. doi: 10.1016/j.bbrc.2017.08.107. [DOI] [PubMed] [Google Scholar]
- 180.Maji S. Exosomal annexin II promotes angiogenesis and breast cancer metastasis. Mol. Canc. Res. 2017;15:93–105. doi: 10.1158/1541-7786.MCR-16-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Blomme A. Myoferlin is a novel exosomal protein and functional regulator of cancer-derived exosomes. Oncotarget. 2016;7:83669–83683. doi: 10.18632/oncotarget.13276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Huang Z., Feng Y. Exosomes derived from hypoxic colorectal cancer cells promote angiogenesis through wnt4-induced beta-catenin signaling in endothelial cells. Oncol. Res. 2017;25:651–661. doi: 10.3727/096504016X14752792816791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yang H. Exosome-derived miR-130a activates angiogenesis in gastric cancer by targeting C-MYB in vascular endothelial cells. Mol. Ther. 2018;26:2466–2475. doi: 10.1016/j.ymthe.2018.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 184.Ramsay R.G., Barton A.L., Gonda T.J. Targeting c-Myb expression in human disease. Expert Opin. Ther. Targets. 2003;7:235–248. doi: 10.1517/14728222.7.2.235. [DOI] [PubMed] [Google Scholar]
- 185.Knopfová L. C-Myb regulates matrix metalloproteinases 1/9, and cathepsin D: implications for matrix-dependent breast cancer cell invasion and metastasis. Mol. Canc. 2012;11:15. doi: 10.1186/1476-4598-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liang J. MicroRNA-103a inhibits gastric cancer cell proliferation, migration and invasion by targeting c-Myb. Cell Prolif. 2015;48:78–85. doi: 10.1111/cpr.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Umezu T., Ohyashiki K., Kuroda M., Ohyashiki J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene. 2013;32:2747–2755. doi: 10.1038/onc.2012.295. [DOI] [PubMed] [Google Scholar]
- 188.Daniel J.M. Inhibition of miR-92a improves re-endothelialization and prevents neointima formation following vascular injury. Cardiovasc. Res. 2014;103:564–572. doi: 10.1093/cvr/cvu162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Murata K. Inhibition of miR-92a enhances fracture healing via promoting angiogenesis in a model of stabilized fracture in young mice. J. Bone Miner. Res. 2014;29:316–326. doi: 10.1002/jbmr.2040. [DOI] [PubMed] [Google Scholar]
- 190.Umezu T. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014;124:3748–3757. doi: 10.1182/blood-2014-05-576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kucharzewska P. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. U. S. A. 2013;110:7312–7317. doi: 10.1073/pnas.1220998110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhou W. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Canc. Cell. 2014;25:501–515. doi: 10.1016/j.ccr.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Garcia-Roman J., Zentella-Dehesa A. Vascular permeability changes involved in tumor metastasis. Canc. Lett. 2013;335:259–269. doi: 10.1016/j.canlet.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 194.Aghajanian A., Wittchen E.S., Allingham M.J., Garrett T.A., Burridge K. Endothelial cell junctions and the regulation of vascular permeability and leukocyte transmigration. J. Thromb. Haemostasis. 2008;6:1453–1460. doi: 10.1111/j.1538-7836.2008.03087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Maeda H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012;88:53–71. doi: 10.2183/pjab.88.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Azzi S., Hebda J.K., Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol. 2013;3:211. doi: 10.3389/fonc.2013.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Peng J., Wang W., Hua S., Liu L. Roles of extracellular vesicles in metastatic breast cancer. Breast Cancer. 2018;12 doi: 10.1177/1178223418767666. 1178223418767666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Peinado H. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hsu Y.L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene. 2017;36:4929–4942. doi: 10.1038/onc.2017.105. [DOI] [PubMed] [Google Scholar]
- 200.Holzner S. Colorectal cancer cell-derived microRNA200 modulates the resistance of adjacent blood endothelial barriers in vitro. Oncol. Rep. 2016;36:3065–3071. doi: 10.3892/or.2016.5114. [DOI] [PubMed] [Google Scholar]
- 201.Vandewalle C., Van Roy F., Berx G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009;66:773–787. doi: 10.1007/s00018-008-8465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Becker A. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Canc. Cell. 2016;30:836–848. doi: 10.1016/j.ccell.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lockman P.R. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Canc. Res. 2010;16:5664–5678. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Treps L. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene. 2016;35:2615–2623. doi: 10.1038/onc.2015.317. [DOI] [PubMed] [Google Scholar]
- 205.Treps L., Perret R., Edmond S., Ricard D., Gavard J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles. 2017;6:1359479. doi: 10.1080/20013078.2017.1359479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kikuchi S., Yoshioka Y., Prieto-Vila M., Ochiya T. Involvement of extracellular vesicles in vascular-related functions in cancer progression and metastasis. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20102584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lopez-Ramirez M.A., Reijerkerk A., de Vries H.E., Romero I.A. Regulation of brain endothelial barrier function by microRNAs in health and neuroinflammation. Faseb. J. 2016;30:2662–2672. doi: 10.1096/fj.201600435RR. [DOI] [PubMed] [Google Scholar]
- 208.Song J., Yoon S.R., Kim O.Y. miR-Let7A controls the cell death and tight junction density of brain endothelial cells under high glucose condition. Oxid Med Cell Longev. 2017;2017:6051874. doi: 10.1155/2017/6051874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Bukeirat M. MiR-34a regulates blood-brain barrier permeability and mitochondrial function by targeting cytochrome c. J. Cerebr. Blood Flow Metabol. 2016;36:387–392. doi: 10.1177/0271678X15606147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tominaga N. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015;6:6716. doi: 10.1038/ncomms7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Quail D.F., Joyce J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chen F. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015;13:45. doi: 10.1186/s12916-015-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Balkwill F.R., Capasso M., Hagemann T. The tumor microenvironment at a glance. J. Cell Sci. 2012;125:5591–5596. doi: 10.1242/jcs.116392. [DOI] [PubMed] [Google Scholar]
- 214.Webber J., Steadman R., Mason M.D., Tabi Z., Clayton A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Can. Res. 2010;70:9621–9630. doi: 10.1158/0008-5472.CAN-10-1722. [DOI] [PubMed] [Google Scholar]
- 215.De Wever O., Demetter P., Mareel M., Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Canc. 2008;123:2229–2238. doi: 10.1002/ijc.23925. [DOI] [PubMed] [Google Scholar]
- 216.Cho J.A., Park H., Lim E.H., Lee K.W. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2012;40:130–138. doi: 10.3892/ijo.2011.1193. [DOI] [PubMed] [Google Scholar]
- 217.Skog J. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Taraboletti G. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH 1. Neoplasia. 2006;8:96–103. doi: 10.1593/neo.05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Xu Z. ping, Tsuji T., Riordan J.F., Hu G. fu. Identification and characterization of an angiogenin-binding DNA sequence that stimulates luciferase reporter gene expression. Biochemistry. 2003;42:121–128. doi: 10.1021/bi020465x. [DOI] [PubMed] [Google Scholar]
- 220.Nishikawa R. Immunohistochemical analysis of the mutant epidermal growth factor, ΔEGFR, in glioblastoma. Brain Tumor Pathol. 2004;21:53–56. doi: 10.1007/BF02484510. [DOI] [PubMed] [Google Scholar]
- 221.Probert C. Communication of prostate cancer cells with bone cells via extracellular vesicle RNA; a potential mechanism of metastasis. Oncogene. 2019;38:1751–1763. doi: 10.1038/s41388-018-0540-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Fang T. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018;9:191. doi: 10.1038/s41467-017-02583-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sun Z. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Canc. 2018;17:147. doi: 10.1186/s12943-018-0897-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Morello M. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle. 2013;12:3526–3536. doi: 10.4161/cc.26539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sanchez C.A. Exosomes from bulk and stem cells from human prostate cancer have a differential microRNA content that contributes cooperatively over local and pre-metastatic niche. Oncotarget. 2016;7:3993–4008. doi: 10.18632/oncotarget.6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Disis M.L. Immune regulation of cancer. J. Clin. Oncol. 2010;28:4531–4538. doi: 10.1200/JCO.2009.27.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Meldolesi J. Extracellular vesicles, news about their role in immune cells: physiology, pathology and diseases. Clin. Exp. Immunol. 2019;196:318–327. doi: 10.1111/cei.13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Landskron G., De la Fuente M., Thuwajit P., Thuwajit C., Hermoso M.A. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. doi: 10.1155/2014/149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Dorsam B. Hodgkin lymphoma-derived extracellular vesicles change the secretome of fibroblasts toward a CAF phenotype. Front. Immunol. 2018;9:1358. doi: 10.3389/fimmu.2018.01358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Dionisi M. Tumor-derived microvesicles enhance cross-processing ability of clinical grade dendritic cells. Front. Immunol. 2018;9:2481. doi: 10.3389/fimmu.2018.02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pulendran B. The varieties of immunological experience: of pathogens, stress, and dendritic cells. Annu. Rev. Immunol. 2015;33:563–606. doi: 10.1146/annurev-immunol-020711-075049. [DOI] [PubMed] [Google Scholar]
- 232.Qian C., Cao X. Dendritic cells in the regulation of immunity and inflammation. Semin. Immunol. 2018;35:3–11. doi: 10.1016/j.smim.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 233.To K.K., Tong C.W., Wu M., Cho W.C. MicroRNAs in the prognosis and therapy of colorectal cancer: from bench to bedside. World J. Gastroenterol. 2018;24:2949–2973. doi: 10.3748/wjg.v24.i27.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Colangelo T. The miR-27a-calreticulin axis affects drug-induced immunogenic cell death in human colorectal cancer cells. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Siveen K.S. The role of extracellular vesicles as modulators of the tumor microenvironment, metastasis and drug resistance in colorectal cancer. Cancers. 2019;11 doi: 10.3390/cancers11060746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Chen G. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–386. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zech D., Rana S., Buchler M.W., Zoller M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell Commun. Signal. 2012;10:37. doi: 10.1186/1478-811X-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wu C.J. Graft-versus-leukemia target antigens in chronic myelogenous leukemia are expressed on myeloid progenitor cells. Clin. Canc. Res. 2005;11:4504–4511. doi: 10.1158/1078-0432.CCR-05-0036. [DOI] [PubMed] [Google Scholar]
- 239.Berchem G. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-beta and miR23a transfer. OncoImmunology. 2016;5 doi: 10.1080/2162402X.2015.1062968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Jabbari N., Nawaz M., Rezaie J. Ionizing radiation increases the activity of exosomal secretory pathway in MCF-7 human breast cancer cells: a possible way to communicate resistance against radiotherapy. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20153649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Braunstein M.J., Kucharczyk J., Adams S. Targeting toll-like receptors for cancer therapy. Targeted Oncol. 2018;13:583–598. doi: 10.1007/s11523-018-0589-7. [DOI] [PubMed] [Google Scholar]
- 242.Fitzgerald W. A system of cytokines encapsulated in ExtraCellular vesicles. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-27190-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Waldmann T.A. Cytokines in cancer immunotherapy. Cold Spring Harb Perspect Biol. 2018;10 doi: 10.1101/cshperspect.a028472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Teti A. Regulation of cellular functions by extracellular matrix. J. Am. Soc. Nephrol. 1992;2:S83. doi: 10.1681/ASN.V210s83. [DOI] [PubMed] [Google Scholar]
- 245.Frantz C., Stewart K.M., Weaver V.M. The extracellular matrix at a glance. J. Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Matusiewicz M. Extracellular matrix remodeling. In: Schwab M., editor. Encyclopedia of Cancer. Springer Berlin Heidelberg; 2011. 1362–1365. [DOI] [Google Scholar]
- 247.Bonnans C., Chou J., Werb Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sung B.H., Ketova T., Hoshino D., Zijlstra A., Weaver A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015;6:7164. doi: 10.1038/ncomms8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Qadir F. Transcriptome reprogramming by cancer exosomes: identification of novel molecular targets in matrix and immune modulation. Mol. Canc. 2018;17:97. doi: 10.1186/s12943-018-0846-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Dawany N.B., Dampier W.N., Tozeren A. Large-scale integration of microarray data reveals genes and pathways common to multiple cancer types. Int. J. Canc. 2011;128:2881–2891. doi: 10.1002/ijc.25854. [DOI] [PubMed] [Google Scholar]
- 251.Han A.L. Fibulin-3 promotes muscle-invasive bladder cancer. Oncogene. 2017;36:5243–5251. doi: 10.1038/onc.2017.149. [DOI] [PubMed] [Google Scholar]
- 252.Sidhu S.S., Mengistab A.T., Tauscher A.N., LaVail J., Basbaum C. The microvesicle as a vehicle for EMMPRIN in tumor-stromal interactions. Oncogene. 2004;23:956–963. doi: 10.1038/sj.onc.1207070. [DOI] [PubMed] [Google Scholar]
- 253.Hendrix A. Effect of the secretory small GTPase Rab27B on breast cancer growth, invasion, and metastasis. J. Natl. Cancer Inst. 2010;102:866–880. doi: 10.1093/jnci/djq153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Paget S. The distribution of secondary growths in cancer of the breast. Canc. Metastasis Rev. 1889;8:98–101. 1989. [PubMed] [Google Scholar]
- 255.Martins V.R., Dias M.S., Hainaut P. Tumor-cell-derived microvesicles as carriers of molecular information in cancer. Curr. Opin. Oncol. 2013;25:66–75. doi: 10.1097/CCO.0b013e32835b7c81. [DOI] [PubMed] [Google Scholar]
- 256.Hood J.L., San R.S., Wickline S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Can. Res. 2011;71:3792–3801. doi: 10.1158/0008-5472.CAN-10-4455. [DOI] [PubMed] [Google Scholar]
- 257.King H.W., Michael M.Z., Gleadle J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Canc. 2012;12:421. doi: 10.1186/1471-2407-12-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Guo Y. Effects of exosomes on pre-metastatic niche formation in tumors. Mol. Canc. 2019;18:39. doi: 10.1186/s12943-019-0995-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Deng J. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Canc. Cell. 2012;21:642–654. doi: 10.1016/j.ccr.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hoshino A. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.McAllister S.S., Weinberg R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014;16:717–727. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Liu Y., Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 2016;94:509–522. doi: 10.1007/s00109-015-1376-x. [DOI] [PubMed] [Google Scholar]
- 263.Muller A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 264.Oskarsson T. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fukuda K. Periostin is a key niche component for wound metastasis of melanoma. PloS One. 2015;10 doi: 10.1371/journal.pone.0129704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Jin L. Breast cancer lung metastasis: molecular biology and therapeutic implications. Canc. Biol. Ther. 2018;19:858–868. doi: 10.1080/15384047.2018.1456599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Coleman R.E., Rubens R.D. The clinical course of bone metastases from breast cancer. Br. J. Canc. 1987;55:61–66. doi: 10.1038/bjc.1987.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Weidle U.H., Birzele F., Kollmorgen G., Ruger R. The multiple roles of exosomes in metastasis. CANCER GENOMICS PROTEOMICS. 2017;14:1–15. doi: 10.21873/cgp.20015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Alipoor S.D. The potential biomarkers and immunological effects of tumor-derived exosomes in lung cancer. Front. Immunol. 2018;9:819. doi: 10.3389/fimmu.2018.00819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Stein P.D. Incidence of venous thromboembolism in patients hospitalized with cancer. Am. J. Med. 2006;119:60–68. doi: 10.1016/j.amjmed.2005.06.058. [DOI] [PubMed] [Google Scholar]
- 271.Sierko E., Wojtukiewicz M.Z. Inhibition of platelet function: does it offer a chance of better cancer progression control? Semin. Thromb. Hemost. 2007;33:712–721. doi: 10.1055/s-2007-991540. [DOI] [PubMed] [Google Scholar]
- 272.Rak J. Microparticles in cancer. Semin. Thromb. Hemost. 2010;36:888–906. doi: 10.1055/s-0030-1267043. [DOI] [PubMed] [Google Scholar]
- 273.Mege D. The origin and concentration of circulating microparticles differ according to cancer type and evolution: a prospective single-center study. Int. J. Canc. 2016;138:939–948. doi: 10.1002/ijc.29837. [DOI] [PubMed] [Google Scholar]
- 274.Kufe D.W. Mucins in cancer: function, prognosis and therapy. Nat. Rev. Canc. 2009;9:874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Quintanilla M., Montero-Montero L., Renart J., Martin-Villar E. Podoplanin in inflammation and cancer. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20030707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Suzuki-Inoue K. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J. Biol. Chem. 2007;282:25993–26001. doi: 10.1074/jbc.M702327200. [DOI] [PubMed] [Google Scholar]
- 277.Riedl J. Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood. 2017;129:1831–1839. doi: 10.1182/blood-2016-06-720714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Mir Seyed Nazari P., Riedl J., Pabinger I., Ay C. The role of podoplanin in cancer-associated thrombosis. Thromb. Res. 2018;164(Suppl):S34–S39. doi: 10.1016/j.thromres.2018.01.020. [DOI] [PubMed] [Google Scholar]
- 279.Thomas G.M. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J. Exp. Med. 2009;206:1913–1927. doi: 10.1084/jem.20082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Mezouar S., Darbousset R., Dignat-George F., Panicot-Dubois L., Dubois C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int. J. Canc. 2015;136:462–475. doi: 10.1002/ijc.28997. [DOI] [PubMed] [Google Scholar]
- 281.Tilley R.E., Holscher T., Belani R., Nieva J., Mackman N. Tissue factor activity is increased in a combined platelet and microparticle sample from cancer patients. Thromb. Res. 2008;122:604–609. doi: 10.1016/j.thromres.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Hron G. Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb. Haemostasis. 2007;97:119–123. [PubMed] [Google Scholar]
- 283.Falati S. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J. Exp. Med. 2003;197:1585–1598. doi: 10.1084/jem.20021868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Mueller B.M., Reisfeld R.A., Edgington T.S., Ruf W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc. Natl. Acad. Sci. U. S. A. 1992;89:11832–11836. doi: 10.1073/pnas.89.24.11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Milsom C., Rak J. Tissue factor and cancer. Pathophysiol. Haemostasis Thrombosis. 2008;36:160–176. doi: 10.1159/000175154. [DOI] [PubMed] [Google Scholar]
- 286.Kasthuri R.S., Taubman M.B., Mackman N. Role of tissue factor in cancer. J. Clin. Oncol. 2009;27:4834–4838. doi: 10.1200/JCO.2009.22.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.van den Berg Y.W., Osanto S., Reitsma P.H., Versteeg H.H. The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood. 2012;119:924–932. doi: 10.1182/blood-2011-06-317685. [DOI] [PubMed] [Google Scholar]
- 288.Gil-Bernabe A.M., Lucotti S., Muschel R.J. Coagulation and metastasis: what does the experimental literature tell us? Br. J. Haematol. 2013;162:433–441. doi: 10.1111/bjh.12381. [DOI] [PubMed] [Google Scholar]
- 289.Yu J.L. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood. 2005;105:1734–1741. doi: 10.1182/blood-2004-05-2042. [DOI] [PubMed] [Google Scholar]
- 290.Salnikov A.V. Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PloS One. 2012;7 doi: 10.1371/journal.pone.0046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Tauro B.J. Oncogenic H-ras reprograms Madin-Darby canine kidney (MDCK) cell-derived exosomal proteins following epithelial-mesenchymal transition. Mol. Cell. Proteomics. 2013;12:2148–2159. doi: 10.1074/mcp.M112.027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Galindo-Hernandez O., Serna-Marquez N., Castillo-Sanchez R., Salazar E.P. Extracellular vesicles from MDA-MB-231 breast cancer cells stimulated with linoleic acid promote an EMT-like process in MCF10A cells. Prostagl. Leukot. Essent. Fat. Acids. 2014;91:299–310. doi: 10.1016/j.plefa.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 293.Souza A.G. Extracellular vesicles as drivers of epithelial-mesenchymal transition and carcinogenic characteristics in normal prostate cells. Mol. Carcinog. 2018;57:503–511. doi: 10.1002/mc.22775. [DOI] [PubMed] [Google Scholar]
- 294.Franzen C.A. Urothelial cells undergo epithelial-to-mesenchymal transition after exposure to muscle invasive bladder cancer exosomes. Oncogenesis. 2015;4:e163. doi: 10.1038/oncsis.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Baumgart E. Identification and prognostic significance of an epithelial-mesenchymal transition expression profile in human bladder tumors. Clin. Canc. Res. 2007;13:1685–1694. doi: 10.1158/1078-0432.CCR-06-2330. [DOI] [PubMed] [Google Scholar]
- 296.Paliwal P., Arora D., Mishra A.K. Epithelial mesenchymal transition in urothelial carcinoma: twist in the tale. Indian J. Pathol. Microbiol. 2012;55:443–449. doi: 10.4103/0377-4929.107777. [DOI] [PubMed] [Google Scholar]
- 297.Sylvester R.J. Natural history, recurrence, and progression in superficial bladder cancer. ScientificWorldJournal. 2006;6:2617–2625. doi: 10.1100/tsw.2006.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Han X., Guo B., Li Y., Zhu B. Tissue factor in tumor microenvironment: a systematic review. J. Hematol. Oncol. 2014;7:1–8. doi: 10.1186/s13045-014-0054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Miyazono K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009;85:314–323. doi: 10.2183/pjab.85.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Lakkaraju A., Rodriguez-Boulan E. Itinerant exosomes: emerging roles in cell and tissue polarity. Trends Cell Biol. 2008;18:199–209. doi: 10.1016/j.tcb.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Gopal S.K. Extracellular vesicles: their role in cancer biology and epithelial-mesenchymal transition. Biochem. J. 2017;474:21–45. doi: 10.1042/BCJ20160006. [DOI] [PubMed] [Google Scholar]
- 302.Xiao D. Melanoma cell-derived exosomes promote epithelial-mesenchymal transition in primary melanocytes through paracrine/autocrine signaling in the tumor microenvironment. Canc. Lett. 2016;376:318–327. doi: 10.1016/j.canlet.2016.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Kim J. Exosome cargo reflects TGF-beta1-mediated epithelial-to-mesenchymal transition (EMT) status in A549 human lung adenocarcinoma cells. Biochem. Biophys. Res. Commun. 2016;478:643–648. doi: 10.1016/j.bbrc.2016.07.124. [DOI] [PubMed] [Google Scholar]
- 304.Han L. Extracellular vesicles in the tumor microenvironment: therapeutic resistance, clinical biomarkers, and targeting strategies. Med. Res. Rev. 2017;37:1318–1349. doi: 10.1002/med.21453. [DOI] [PubMed] [Google Scholar]
- 305.Soekmadji C., Nelson C.C. The emerging role of extracellular vesicle-mediated drug resistance in cancers: implications in advanced prostate cancer. BioMed Res. Int. 2015;2015:454837. doi: 10.1155/2015/454837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ji R. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric cancer. Cell Cycle. 2015;14:2473–2483. doi: 10.1080/15384101.2015.1005530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Corcoran C. Docetaxel-resistance in prostate cancer: evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PloS One. 2012;7 doi: 10.1371/journal.pone.0050999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Lv M.M. Exosomes mediate drug resistance transfer in MCF-7 breast cancer cells and a probable mechanism is delivery of P-glycoprotein. Tumour Biol. 2014;35:10773–10779. doi: 10.1007/s13277-014-2377-z. [DOI] [PubMed] [Google Scholar]
- 309.Yang X., Weng Z., Mendrick D.L., Shi Q. Circulating extracellular vesicles as a potential source of new biomarkers of drug-induced liver injury. Toxicol. Lett. 2014;225:401–406. doi: 10.1016/j.toxlet.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 310.Zhou S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica. 2008;38:802–832. doi: 10.1080/00498250701867889. [DOI] [PubMed] [Google Scholar]
- 311.Boelens M.C. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159:499–513. doi: 10.1016/j.cell.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Au Yeung C.L. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016;7:11150. doi: 10.1038/ncomms11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Maacha S. Extracellular vesicles-mediated intercellular communication: roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Canc. 2019;18:55. doi: 10.1186/s12943-019-0965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Zhang F.F. Microvesicles mediate transfer of P-glycoprotein to paclitaxel-sensitive A2780 human ovarian cancer cells, conferring paclitaxel-resistance. Eur. J. Pharmacol. 2014;738:83–90. doi: 10.1016/j.ejphar.2014.05.026. [DOI] [PubMed] [Google Scholar]
- 315.Bebawy M. Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia. 2009;23:1643–1649. doi: 10.1038/leu.2009.76. [DOI] [PubMed] [Google Scholar]
- 316.Torreggiani E., Roncuzzi L., Perut F., Zini N., Baldini N. Multimodal transfer of MDR by exosomes in human osteosarcoma. Int. J. Oncol. 2016;49:189–196. doi: 10.3892/ijo.2016.3509. [DOI] [PubMed] [Google Scholar]
- 317.Munn L.L. vol. 9. Wiley Interdiscip Rev Syst Biol Med; 2017. (Cancer and Inflammation). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Rajabi M., Mousa S.A. The role of angiogenesis in cancer treatment. Biomedicines. 2017;5 doi: 10.3390/biomedicines5020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Liu S.-S.S.-L., Sun P., Li Y., Liu S.-S.S.-L., Lu Y. Exosomes as critical mediators of cell-to-cell communication in cancer pathogenesis and their potential clinical application. Transl. Cancer Res. 2019;8:298–311. doi: 10.21037/tcr.2019.01.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Magagnin M.G., Koritzinsky M., Wouters B.G. Patterns of tumor oxygenation and their influence on the cellular hypoxic response and hypoxia-directed therapies. Drug Resist. Updates. 2006;9:185–197. doi: 10.1016/j.drup.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 321.Camps C. hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin. Canc. Res. 2008;14:1340–1348. doi: 10.1158/1078-0432.CCR-07-1755. [DOI] [PubMed] [Google Scholar]
- 322.Zhuang G. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012;31:3513–3523. doi: 10.1038/emboj.2012.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Svensson K.J. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 2011;108:13147–13152. doi: 10.1073/pnas.1104261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Kore R.A. Hypoxia-derived exosomes induce putative altered pathways in biosynthesis and ion regulatory channels in glioblastoma cells. Biochem Biophys Rep. 2018;14:104–113. doi: 10.1016/j.bbrep.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Al-Nedawi K., Meehan B., Kerbel R.S., Allison A.C., Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. U. S. A. 2009;106:3794–3799. doi: 10.1073/pnas.0804543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Aga M. Exosomal HIF1alpha supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene. 2014;33:4613–4622. doi: 10.1038/onc.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Grull M.P., Mulligan M.E., Lang A.S. Small extracellular particles with big potential for horizontal gene transfer: membrane vesicles and gene transfer agents. FEMS Microbiol. Lett. 2018;365 doi: 10.1093/femsle/fny192. [DOI] [PubMed] [Google Scholar]
- 328.Fischer S. Indication of horizontal DNA gene transfer by extracellular vesicles. PloS One. 2016;11 doi: 10.1371/journal.pone.0163665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Gezsi A., Kovacs A., Visnovitz T., Buzas E.I. Systems biology approaches to investigating the roles of extracellular vesicles in human diseases. Exp. Mol. Med. 2019;51:33. doi: 10.1038/s12276-019-0226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Guescini M. C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp. Cell Res. 2010;316:1977–1984. doi: 10.1016/j.yexcr.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 331.Németh A. Antibiotic-induced release of small extracellular vesicles (exosomes) with surface-associated DNA. Sci. Rep. 2017;7:1–16. doi: 10.1038/s41598-017-08392-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Sansone P. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. U.S.A. 2017;114:E9066–E9075. doi: 10.1073/pnas.1704862114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Lazaro-Ibanez E. Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: apoptotic bodies, microvesicles, and exosomes. Prostate. 2014;74:1379–1390. doi: 10.1002/pros.22853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.García-Romero N. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget. 2017;8:1416–1428. doi: 10.18632/oncotarget.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Lee T.H. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 2014;451:295–301. doi: 10.1016/j.bbrc.2014.07.109. [DOI] [PubMed] [Google Scholar]
- 336.Halicka H.D., Bedner E., Darzynkiewicz Z. Segregation of RNA and separate packaging of DNA and RNA in apoptotic bodies during apoptosis. Exp. Cell Res. 2000;260:248–256. doi: 10.1006/excr.2000.5027. [DOI] [PubMed] [Google Scholar]
- 337.Holmgren L., Bergsmedh A., Spetz A.L. Horizontal transfer of DNA by the uptake of apoptotic bodies. Vox Sang. 2002;83(Suppl 1):305–306. doi: 10.1111/j.1423-0410.2002.tb05323.x. [DOI] [PubMed] [Google Scholar]
- 338.Orozco A.F. Membrane protected apoptotic trophoblast microparticles contain nucleic acids: relevance to preeclampsia. Am. J. Pathol. 2008;173:1595–1608. doi: 10.2353/ajpath.2008.080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Miranda K.C. Potential biomarkers for renal disease. 2015;78:191–199. doi: 10.1038/ki.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Kawamura Y., Yamamoto Y., Sato T.A., Ochiya T. Extracellular vesicles as trans-genomic agents: emerging roles in disease and evolution. Canc. Sci. 2017;108:824–830. doi: 10.1111/cas.13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Cai J. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013;5:227–238. doi: 10.1093/jmcb/mjt011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Kahlert C. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014;289:3869–3875. doi: 10.1074/jbc.C113.532267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Lee T.H. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 2014;451:295–301. doi: 10.1016/j.bbrc.2014.07.109. [DOI] [PubMed] [Google Scholar]
- 344.Shelke G., Jang S.C., Yin Y., Lässer C., Lötvall J. Human mast cells release extracellular vesicle-associated DNA. Matters (Zürich) 2016;2 [Google Scholar]
- 345.Taylor B.S. Integrative genomic profiling of human prostate cancer. Canc. Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.San Lucas F.A. Minimally invasive genomic and transcriptomic profiling of visceral cancers by next-generation sequencing of circulating exosomes. Ann. Oncol. 2016;27:635–641. doi: 10.1093/annonc/mdv604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Allenson K. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol. 2017;28:741–747. doi: 10.1093/annonc/mdx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Möhrmann L. Liquid biopsies using plasma exosomal nucleic acids and plasma cell-free DNA compared with clinical outcomes of patients with advanced cancers. Clin. Canc. Res. 2018;24:181–188. doi: 10.1158/1078-0432.CCR-17-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Yang S. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Canc. Biol. Ther. 2017;18:158–165. doi: 10.1080/15384047.2017.1281499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Castellanos-Rizaldos E. Exosome-based detection of EGFR T790M in plasma from non–small cell lung cancer patients. Clin. Canc. Res. 2018;24:2944–2950. doi: 10.1158/1078-0432.CCR-17-3369. [DOI] [PubMed] [Google Scholar]
- 351.Takahashi A. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Yokoi A. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019;5:1–17. doi: 10.1126/sciadv.aax8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Andreu Z., Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014;5:1–12. doi: 10.3389/fimmu.2014.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Willms E., Cabañas C., Mäger I., Wood M.J.A., Vader P. Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in cancer progression. Front. Immunol. 2018;9 doi: 10.3389/fimmu.2018.00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Jeppesen D.K. Reassessment of exosome composition. Cell. 2019;177:428–445. doi: 10.1016/j.cell.2019.02.029. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Sadovska L., Eglitis J., Line A. Extracellular vesicles as biomarkers and therapeutic targets in breast cancer. Anticancer Res. 2015;35:6379–6390. [PubMed] [Google Scholar]
- 357.Keshavarzi M. Molecular imaging and oral cancer diagnosis and therapy. J. Cell. Biochem. 2017;118:3055–3060. doi: 10.1002/jcb.26042. [DOI] [PubMed] [Google Scholar]
- 358.Kasivisvanathan V. MRI-targeted or standard biopsy for prostate-cancer diagnosis. N. Engl. J. Med. 2018;378:1767–1777. doi: 10.1056/NEJMoa1801993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Edizer D.T., Server E.A., Yiğit Ö., Yıldız M. Role of fine-needle aspiration biopsy in the management of salivary gland masses. Turkish Arch. Otorhinolaryngol. 2016;54:105. doi: 10.5152/tao.2016.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Lee C.-T. What do we know about ground-glass opacity nodules in the lung? Transl. lung cancer Res. 2015;4:656. doi: 10.3978/j.issn.2218-6751.2015.04.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Zhao Z. Extracellular vesicles as cancer liquid biopsies: from discovery, validation, to clinical application. Lab Chip. 2019;19:1114–1140. doi: 10.1039/c8lc01123k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Crowley E., Di Nicolantonio F., Loupakis F., Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013;10:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 363.Torrano V. Vesicle-MaNiA: extracellular vesicles in liquid biopsy and cancer. Curr. Opin. Pharmacol. 2016;29:47–53. doi: 10.1016/j.coph.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Biggs C.N. Prostate extracellular vesicles in patient plasma as a liquid biopsy platform for prostate cancer using nanoscale flow cytometry. Oncotarget. 2016;7:8839. doi: 10.18632/oncotarget.6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Chen I.-H. Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. Proc. Natl. Acad. Sci. Unit. States Am. 2017;114:3175–3180. doi: 10.1073/pnas.1618088114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Cui S., Cheng Z., Qin W., Jiang L. Exosomes as a liquid biopsy for lung cancer. Lung Canc. 2018;116:46–54. doi: 10.1016/j.lungcan.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 367.Nawaz M. Extracellular vesicles in ovarian cancer: applications to tumor biology, immunotherapy and biomarker discovery. Expert Rev. Proteomics. 2016;13:395–409. doi: 10.1586/14789450.2016.1165613. [DOI] [PubMed] [Google Scholar]
- 368.Olmedillas López S. KRAS G12V mutation detection by droplet digital PCR in circulating cell-free DNA of colorectal cancer patients. Int. J. Mol. Sci. 2016;17:484. doi: 10.3390/ijms17040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Stroun M., Lyautey J., Lederrey C., Olson-Sand A., Anker P. About the possible origin and mechanism of circulating DNA: apoptosis and active DNA release. Clin. Chim. Acta. 2001;313:139–142. doi: 10.1016/s0009-8981(01)00665-9. [DOI] [PubMed] [Google Scholar]
- 370.Jin Y. DNA in serum extracellular vesicles is stable under different storage conditions. BMC Canc. 2016;16:753. doi: 10.1186/s12885-016-2783-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Kalluri R., LeBleu V.S. 81 vols. 275–280. Cold Spring Harbor Laboratory Press; 2016. Discovery of double-stranded genomic DNA in circulating exosomes. (Cold Spring Harbor Symposia on Quantitative Biology). [DOI] [PubMed] [Google Scholar]
- 372.Razavi P. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019;25:1928–1937. doi: 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Wan Y. Nanoscale extracellular vesicle-derived DNA is superior to circulating cell-free DNA for mutation detection in early-stage non-small-cell lung cancer. Ann. Oncol. 2018;29:2379–2383. doi: 10.1093/annonc/mdy458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Bettegowda C. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Newman A.M. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Keserű J.S. Detection of cell-free, exosomal and whole blood mitochondrial DNA copy number in plasma or whole blood of patients with serous epithelial ovarian cancer. J. Biotechnol. 2019;298:76–81. doi: 10.1016/j.jbiotec.2019.04.015. [DOI] [PubMed] [Google Scholar]
- 377.Ruivo C.F., Adem B., Silva M., Melo S.A. The biology of cancer exosomes: insights and new perspectives. Can. Res. 2017;77:6480–6488. doi: 10.1158/0008-5472.CAN-17-0994. [DOI] [PubMed] [Google Scholar]
- 378.Vagner T. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles. 2018;7:1505403. doi: 10.1080/20013078.2018.1505403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Wu S. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. 2019;575:699–703. doi: 10.1038/s41586-019-1763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Mao W. Isolation and retrieval of extracellular vesicles for liquid biopsy of malignant ground-glass opacity. Anal. Chem. 2019;91:13729–13736. doi: 10.1021/acs.analchem.9b03064. [DOI] [PubMed] [Google Scholar]
- 381.Pavlou M.P., Diamandis E.P. The cancer cell secretome: a good source for discovering biomarkers? Journal of Proteomics. 2010;73:1896–1906. doi: 10.1016/j.jprot.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 382.Ono S., Lam S., Nagahara M., Hoon D. Circulating microRNA biomarkers as liquid biopsy for cancer patients: pros and cons of current assays. J. Clin. Med. 2015;4:1890–1907. doi: 10.3390/jcm4101890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Calin G.A. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. Unit. States Am. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Takamizawa J. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Can. Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 385.Wang G.-K. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010;31:659–666. doi: 10.1093/eurheartj/ehq013. [DOI] [PubMed] [Google Scholar]
- 386.Manier S. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood. 2017;129:2429–2436. doi: 10.1182/blood-2016-09-742296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Cheng L., Sharples R.A., Scicluna B.J., Hill A.F. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracell. Vesicles. 2014;3:23743. doi: 10.3402/jev.v3.23743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Mitchell P.S. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. Unit. States Am. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Takeshita N. Serum microRNA expression profile: MiR-1246 as a novel diagnostic and prognostic biomarker for oesophageal squamous cell carcinoma. Br. J. Canc. 2013;108:644–652. doi: 10.1038/bjc.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Tanaka Y. Clinical impact of serum exosomal microRNA‐21 as a clinical biomarker in human esophageal squamous cell carcinoma. Cancer. 2013;119:1159–1167. doi: 10.1002/cncr.27895. [DOI] [PubMed] [Google Scholar]
- 391.Zhang Y., Zhang Y., Yin Y., Li S. Detection of circulating exosomal miR-17-5p serves as a novel non-invasive diagnostic marker for non-small cell lung cancer patients. Pathol. Pract. 2019;152466 doi: 10.1016/j.prp.2019.152466. [DOI] [PubMed] [Google Scholar]
- 392.Kanaoka R. Usefulness of plasma exosomal MicroRNA-451a as a noninvasive biomarker for early prediction of recurrence and prognosis of non-small cell lung cancer. Oncol. 2018;94:311–323. doi: 10.1159/000487006. [DOI] [PubMed] [Google Scholar]
- 393.Zheng M. Exosomal let-7d-3p and miR-30d-5p as diagnostic biomarkers for non-invasive screening of cervical cancer and its precursors. Mol. Canc. 2019;18:76. doi: 10.1186/s12943-019-0999-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Bhagirath D. microRNA-1246 is an exosomal biomarker for aggressive prostate cancer. Can. Res. 2018;78:1833–1844. doi: 10.1158/0008-5472.CAN-17-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Santangelo A. A microRNA signature from serum exosomes of patients with glioma as complementary diagnostic biomarker. J. Neuro Oncol. 2018;136:51–62. doi: 10.1007/s11060-017-2639-x. [DOI] [PubMed] [Google Scholar]
- 396.Fengming L.A.N., Xiao Y.U.E., Tingyi X.I.A. Exosomal microRNA–210 is a potentially non–invasive biomarker for the diagnosis and prognosis of glioma. Oncol. Lett. 2020;19:1967–1974. doi: 10.3892/ol.2020.11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Yoshikawa M. Exosome-encapsulated microRNA-223-3p as a minimally invasive biomarker for the early detection of invasive breast cancer. Oncol. Lett. 2018;15:9584–9592. doi: 10.3892/ol.2018.8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Kumata Y. Exosome-encapsulated microRNA-23b as a minimally invasive liquid biomarker for the prediction of recurrence and prognosis of gastric cancer patients in each tumor stage. Oncol. Rep. 2018;40:319–330. doi: 10.3892/or.2018.6418. [DOI] [PubMed] [Google Scholar]
- 399.Iwasaki H. A novel urinary microRNA biomarker panel for detecting gastric cancer. J. Gastroenterol. 2019;54:1061–1069. doi: 10.1007/s00535-019-01601-w. [DOI] [PubMed] [Google Scholar]
- 400.Goto T. An elevated expression of serum exosomal microRNA-191, − 21, −451a of pancreatic neoplasm is considered to be efficient diagnostic marker. BMC Canc. 2018;18:116. doi: 10.1186/s12885-018-4006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Suehiro T. Serum exosomal microRNA-122 and microRNA-21 as predictive biomarkers in transarterial chemoembolization-treatedhepatocellular carcinoma patients. Oncol. Lett. 2018;16:3267–3273. doi: 10.3892/ol.2018.8991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Kurahashi R. MicroRNA-204-5p: a novel candidate urinary biomarker of Xp11.2 translocation renal cell carcinoma. Canc. Sci. 2019;110:1897–1908. doi: 10.1111/cas.14026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Kim S. Serum exosomal miRNA-145 and miRNA-200c as promising biomarkers for preoperative diagnosis of ovarian carcinomas. J. Canc. 2019;10:1958–1967. doi: 10.7150/jca.30231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Royo F. Different EV enrichment methods suitable for clinical settings yield different subpopulations of urinary extracellular vesicles from human samples. J. Extracell. Vesicles. 2016;5 doi: 10.3402/jev.v5.29497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Yokoi A. Malignant extracellular vesicles carrying MMP1 mRNA facilitate peritoneal dissemination in ovarian cancer. Nat. Commun. 2017;8 doi: 10.1038/ncomms14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Cha B.S., Park K.S., Park J.S. Signature mRNA markers in extracellular vesicles for the accurate diagnosis of colorectal cancer. J. Biol. Eng. 2020;14:1–9. doi: 10.1186/s13036-020-0225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Woo H.K. Urine-based liquid biopsy: non-invasive and sensitive AR-V7 detection in urinary EVs from patients with prostate cancer. Lab Chip. 2019;19:87–97. doi: 10.1039/c8lc01185k. [DOI] [PubMed] [Google Scholar]
- 408.Guttman M. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Huarte M. The emerging role of lncRNAs in cancer. Nat. Med. 2015;21:1253. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 410.Yang G., Lu X., Yuan L. LncRNA: a link between RNA and cancer. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2014;1839:1097–1109. doi: 10.1016/j.bbagrm.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 411.Qu L. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Canc. Cell. 2016;29:653–668. doi: 10.1016/j.ccell.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 412.Li Z. Tumor-derived exosomal lnc-Sox2ot promotes EMT and stemness by acting as a ceRNA in pancreatic ductal adenocarcinoma. Oncogene. 2018;37:3822–3838. doi: 10.1038/s41388-018-0237-9. [DOI] [PubMed] [Google Scholar]
- 413.Conigliaro A. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Canc. 2015;14:155. doi: 10.1186/s12943-015-0426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Tan S.K. Serum long noncoding RNA HOTAIR as a novel diagnostic and prognostic biomarker in glioblastoma multiforme. Mol. Canc. 2018;17:74. doi: 10.1186/s12943-018-0822-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Lin L.-Y. Tumor-originated exosomal lncUEGC1 as a circulating biomarker for early-stage gastric cancer. Mol. Canc. 2018;17:84. doi: 10.1186/s12943-018-0834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Zhao R. Exosomal long noncoding RNA HOTTIP as potential novel diagnostic and prognostic biomarker test for gastric cancer. Mol. Canc. 2018;17:68. doi: 10.1186/s12943-018-0817-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Li C. Tumor‐derived exosomal lncRNA GAS5 as a biomarker for early‐stage non‐small‐cell lung cancer diagnosis. J. Cell. Physiol. 2019 doi: 10.1002/jcp.28678. [DOI] [PubMed] [Google Scholar]
- 418.Ko J. Combining machine learning and nanofluidic technology to diagnose pancreatic cancer using exosomes. ACS Nano. 2017;11:11182–11193. doi: 10.1021/acsnano.7b05503. [DOI] [PubMed] [Google Scholar]
- 419.Lister R. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Robertson K.D. DNA methylation and chromatin - unraveling the tangled web. Oncogene. 2002;21:5361–5379. doi: 10.1038/sj.onc.1205609. [DOI] [PubMed] [Google Scholar]
- 421.Ashapkin V.V., Kutueva L.I., Vanyushin B.F. Dnmt2 is the most evolutionary conserved and enigmatic cytosine DNA methyltransferase in eukaryotes. Russ. J. Genet. 2016;52:237–248. [PubMed] [Google Scholar]
- 422.Bourc’his D. Dnmt3L and the establishment of maternal genomic imprints. Science (80-. ) 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 423.Dakhlallah D.A. Circulating extracellular vesicle content reveals de novo DNA methyltransferase expression as a molecular method to predict septic shock. J. Extracell. Vesicles. 2019;8 doi: 10.1080/20013078.2019.1669881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Popp C. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Shen L. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011:144 646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 427.Vogelstein B. Cancer genome landscapes. Science. 2013:340 1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Hou J.P., Ma J. DawnRank. Discovering personalized driver genes in cancer. Genome Med. 2014;6:56. doi: 10.1186/s13073-014-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Fang J.H. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology. 2018;68:1459–1475. doi: 10.1002/hep.29920. [DOI] [PubMed] [Google Scholar]
- 430.Zeng Z. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-07810-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Mahmood N., Rabbani S.A. DNA methylation readers and cancer: mechanistic and therapeutic applications. Frontiers in Oncology. 2019;9:489. doi: 10.3389/fonc.2019.00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Locke W.J. DNA methylation cancer biomarkers: translation to the clinic. Front. Genet. 2019;10:1150. doi: 10.3389/fgene.2019.01150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Wajed S.A., Laird P.W., DeMeester T.R. DNA methylation: an alternative pathway to cancer. Ann. Surg. 2001:234 10–20. doi: 10.1097/00000658-200107000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Bird A. DNA methylation patterns and epigenetic memory. Gene Dev. 2002:16 6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 435.Sina A.A.I. Epigenetically reprogrammed methylation landscape drives the DNA self-assembly and serves as a universal cancer biomarker. Nat. Commun. 2018;9:1–13. doi: 10.1038/s41467-018-07214-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Lee J.Y., Lee T.H. Effects of DNA methylation on the structure of nucleosomes. J. Am. Chem. Soc. 2012;134:173–175. doi: 10.1021/ja210273w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Geahigan K.B., Meints G.A., Hatcher M.E., Orban J., Drobny G.P. The dynamic impact of CpG methylation in DNA. Biochemistry. 2000;39:4939–4946. doi: 10.1021/bi9917636. [DOI] [PubMed] [Google Scholar]
- 438.Kaur P. Hydrophobicity of methylated DNA as a possible mechanism for gene silencing. Phys. Biol. 2012;9 doi: 10.1088/1478-3975/9/6/065001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Zhu X. BCR-ABL1-positive microvesicles transform normal hematopoietic transplants through genomic instability: implications for donor cell leukemia. Leukemia. 2014;28:1666–1675. doi: 10.1038/leu.2014.51. [DOI] [PubMed] [Google Scholar]
- 440.Gao P. DNA methylation-mediated repression of exosomal miR-652-5p expression promotes oesophageal squamous cell carcinoma aggressiveness by targeting PARG and VEGF pathways. PLoS Genet. 2020;16 doi: 10.1371/journal.pgen.1008592. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 441.Li B. Loss of exosomal miR‐148b from cancer‐associated fibroblasts promotes endometrial cancer cell invasion and cancer metastasis. J. Cell. Physiol. 2019;234:2943–2953. doi: 10.1002/jcp.27111. [DOI] [PubMed] [Google Scholar]
- 442.Sina A.A.I. Methylation dependent gold adsorption behaviour identifies cancer derived extracellular vesicular DNA. Nanoscale horizons. 2020;5:1317–1323. doi: 10.1039/d0nh00258e. [DOI] [PubMed] [Google Scholar]
- 443.Watanabe Y. Sensitive and specific detection of early gastric cancer with DNA methylation analysis of gastric washes. Gastroenterology. 2009;136:2149–2158. doi: 10.1053/j.gastro.2009.02.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Oishi Y. Hypermethylation of Sox17 gene is useful as a molecular diagnostic application in early gastric cancer. Tumor Biol. 2012;33:383–393. doi: 10.1007/s13277-011-0278-y. [DOI] [PubMed] [Google Scholar]
- 445.Baba S. Gastric wash-based molecular testing for antibiotic resistance in helicobacter pylori. Digestion. 2011;84:299–305. doi: 10.1159/000332570. [DOI] [PubMed] [Google Scholar]
- 446.Yamamoto H. Detection of DNA methylation of gastric juice-derived exosomes in gastric cancer. Integr. Mol. Med. 2014;1 [Google Scholar]
- 447.Li W. Role of exosomal proteins in cancer diagnosis. Mol. Canc. 2017:16 1–1612. doi: 10.1186/s12943-017-0706-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Li A., Zhang T., Zheng M., Liu Y., Chen Z. Exosomal proteins as potential markers of tumor diagnosis. J. Hematol. Oncol. 2017;10 doi: 10.1186/s13045-017-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Boukouris S., Mathivanan S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteonomics Clin. Appl. 2015:9 358–367. doi: 10.1002/prca.201400114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Niu L. Tumor‐derived exosomal proteins as diagnostic biomarkers in non‐small cell lung cancer. Canc. Sci. 2019;110:433. doi: 10.1111/cas.13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Lai X. A microRNA signature in circulating exosomes is superior to exosomal glypican-1 levels for diagnosing pancreatic cancer. Canc. Lett. 2017;393:86–93. doi: 10.1016/j.canlet.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Costa-Silva B. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015;17:816–826. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Li Y., Zhang Y., Qiu F., Qiu Z. Proteomic identification of exosomal LRG1: a potential urinary biomarker for detecting NSCLC. Electrophoresis. 2011;32:1976–1983. doi: 10.1002/elps.201000598. [DOI] [PubMed] [Google Scholar]
- 454.Sun Y. Systematic comparison of exosomal proteomes from human saliva and serum for the detection of lung cancer. Anal. Chim. Acta. 2017;982:84–95. doi: 10.1016/j.aca.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 455.Zhao Z., Yang Y., Zeng Y., He M. A microfluidic ExoSearch chip for multiplexed exosome detection towards blood-based ovarian cancer diagnosis. Lab Chip. 2016;16:489–496. doi: 10.1039/c5lc01117e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Li J. Claudin-containing exosomes in the peripheral circulation of women with ovarian cancer. BMC Canc. 2009;9:244. doi: 10.1186/1471-2407-9-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Shao H. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med. 2012;18:1835–1840. doi: 10.1038/nm.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Logozzi M. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PloS One. 2009;4 doi: 10.1371/journal.pone.0005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Madhankumar A.B. Interleukin-13 conjugated quantum dots for identification of glioma initiating cells and their extracellular vesicles. Acta Biomater. 2017;58:205–213. doi: 10.1016/j.actbio.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 460.Arbelaiz A. Serum extracellular vesicles contain protein biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2017;66:1125–1143. doi: 10.1002/hep.29291. [DOI] [PubMed] [Google Scholar]
- 461.Tian Y. Protein profiling and sizing of extracellular vesicles from colorectal cancer patients via flow cytometry. ACS Nano. 2018;12:671–680. doi: 10.1021/acsnano.7b07782. [DOI] [PubMed] [Google Scholar]
- 462.Li J. GPC1 exosome and its regulatory miRNAs are specific markers for the detection and target therapy of colorectal cancer. J. Cell Mol. Med. 2017;21:838–847. doi: 10.1111/jcmm.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Jin H. Exosomal zinc transporter ZIP4 promotes cancer growth and is a novel diagnostic biomarker for pancreatic cancer. Canc. Sci. 2018;109:2946. doi: 10.1111/cas.13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Zhang H. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017;8:1–11. doi: 10.1038/ncomms15016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.An M. Quantitative proteomic analysis of serum exosomes from patients with locally advanced pancreatic cancer undergoing chemoradiotherapy. J. Proteome Res. 2017;16:1763–1772. doi: 10.1021/acs.jproteome.7b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Sun Y. Comparative proteomic analysis of exosomes and microvesicles in human saliva for lung cancer. J. Proteome Res. 2018;17:1101–1107. doi: 10.1021/acs.jproteome.7b00770. [DOI] [PubMed] [Google Scholar]
- 467.Huang A. Exosomal transfer of vasorin expressed in hepatocellular carcinoma cells promotes migration of human umbilical vein endothelial cells. Int. J. Biol. Sci. 2015;11:961–969. doi: 10.7150/ijbs.11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Sandfeld-Paulsen B. Exosomal proteins as prognostic biomarkers in non-small cell lung cancer. Mol. Oncol. 2016;10:1595. doi: 10.1016/j.molonc.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Fang S. Clinical application of a microfluidic chip for immunocapture and quantification of circulating exosomes to assist breast cancer diagnosis and molecular classification. PloS One. 2017;12 doi: 10.1371/journal.pone.0175050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Yokoyama S. Clinical implications of carcinoembryonic antigen distribution in serum exosomal fraction—measurement by ELISA. PloS One. 2017;12 doi: 10.1371/journal.pone.0183337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Logozzi M. Increased PSA expression on prostate cancer exosomes in in vitro condition and in cancer patients. Canc. Lett. 2017;403:318–329. doi: 10.1016/j.canlet.2017.06.036. [DOI] [PubMed] [Google Scholar]
- 472.Kawakami K. Gamma-glutamyltransferase activity in exosomes as a potential marker for prostate cancer. BMC Canc. 2017;17 doi: 10.1186/s12885-017-3301-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Fais S. Evidence-based clinical use of nanoscale extracellular vesicles in nanomedicine. ACS Nano. 2016;10:3886–3899. doi: 10.1021/acsnano.5b08015. [DOI] [PubMed] [Google Scholar]
- 474.Chen S. Lipidomic characterization of extracellular vesicles in human serum. J. Circ. Biomarkers. 2019;8 doi: 10.1177/1849454419879848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Skotland T., Sagini K., Sandvig K., Llorente A. An emerging focus on lipids in extracellular vesicles. Adv. Drug Deliv. Rev. 2020 doi: 10.1016/j.addr.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 476.Harayama T., Riezman H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018;19:281–296. doi: 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- 477.Simons K., Sampaio J.L. Membrane organization and lipid rafts. Cold Spring Harbor Perspectives in Biology. 2011:3 1–17. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Del Boccio P. A hyphenated microLC-Q-TOF-MS platform for exosomal lipidomics investigations: application to RCC urinary exosomes. Electrophoresis. 2012;33:689–696. doi: 10.1002/elps.201100375. [DOI] [PubMed] [Google Scholar]
- 479.Skotland T. Molecular lipid species in urinary exosomes as potential prostate cancer biomarkers. Eur. J. Canc. 2017;70:122–132. doi: 10.1016/j.ejca.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 480.Hough K.P. Unique lipid signatures of extracellular vesicles from the airways of asthmatics. Sci. Rep. 2018;8:1–16. doi: 10.1038/s41598-018-28655-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Tao L. Metabolomics identifies serum and exosomes metabolite markers of pancreatic cancer. Metabolomics. 2019;15:86. doi: 10.1007/s11306-019-1550-1. [DOI] [PubMed] [Google Scholar]
- 482.Llorente A. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2013;1831:1302–1309. doi: 10.1016/j.bbalip.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 483.Moyano A.L. Sulfatides in extracellular vesicles isolated from plasma of multiple sclerosis patients. J. Neurosci. Res. 2016;94:1579–1587. doi: 10.1002/jnr.23899. [DOI] [PubMed] [Google Scholar]
- 484.Cheng M. Circulating myocardial microRNAs from infarcted hearts are carried in exosomes and mobilise bone marrow progenitor cells. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-08895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Jiang N. Exosomes mediate epithelium-mesenchyme crosstalk in organ development. ACS Nano. 2017;11:7736–7746. doi: 10.1021/acsnano.7b01087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Lan J. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Can. Res. 2019;79:146–158. doi: 10.1158/0008-5472.CAN-18-0014. [DOI] [PubMed] [Google Scholar]
- 487.Li Q., Wang H., Peng H., Huyan T., Cacalano N.A. Exosomes: versatile nano mediators of immune regulation. Cancers. 2019;11 doi: 10.3390/cancers11101557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Ren J. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8:3932–3948. doi: 10.7150/thno.25541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Momen-Heravi F., Bala S. vol. 9. 2018. pp. 34838–34854. (Extracellular Vesicles in Oral Squamous Carcinoma Carry Oncogenic miRNA Profile and Reprogram Monocytes via NF-Κb Pathway. Oncotarget). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Jella K.K. vol. 6. Vaccines; 2018. (Exosomes, Their Biogenesis and Role in Inter-cellular Communication, Tumor Microenvironment and Cancer Immunotherapy). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Mittelbrunn M. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011;2 doi: 10.1038/ncomms1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Segura E. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood. 2005;106:216–223. doi: 10.1182/blood-2005-01-0220. [DOI] [PubMed] [Google Scholar]
- 493.Hao S. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology. 2007;120:90–102. doi: 10.1111/j.1365-2567.2006.02483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Tkach M. Qualitative differences in T‐cell activation by dendritic cell‐derived extracellular vesicle subtypes. EMBO J. 2017;36:3012–3028. doi: 10.15252/embj.201696003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Wahlund C.J.E. Exosomes from antigen-pulsed dendritic cells induce stronger antigen-specific immune responses than microvesicles in vivo. Sci. Rep. 2017;7:1–9. doi: 10.1038/s41598-017-16609-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Benites B.D., Alvarez M.C., Saad S.T.O. Small particles, big effects: the interplay between exosomes and dendritic cells in antitumor immunity and immunotherapy. Cells. 2019;8:1648. doi: 10.3390/cells8121648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Lindenbergh M.F.S., Stoorvogel W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu. Rev. Immunol. 2018;36 doi: 10.1146/annurev-immunol-041015-055700. [DOI] [PubMed] [Google Scholar]
- 498.Admyre C., Johansson S.M., Paulie S., Gabrielsson S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur. J. Immunol. 2006;36:1772–1781. doi: 10.1002/eji.200535615. [DOI] [PubMed] [Google Scholar]
- 499.Maybruck B.T., Pfannenstiel L.W., Diaz-Montero M., Gastman B.R. Tumor-derived exosomes induce CD8+ T cell suppressors. J. Immunother. Cancer. 2017;5 doi: 10.1186/s40425-017-0269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Zitvogel L. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med. 1998;4:594–600. doi: 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]
- 501.André F. vol. 20. 2002. Tumor-derived exosomes: a new source of tumor rejection antigens. (Vaccine). [DOI] [PubMed] [Google Scholar]
- 502.Hsu D.H. Exosomes as a tumor vaccine: enhancing potency through direct loading of antigenic peptides. J. Immunother. 2003;26:440–450. doi: 10.1097/00002371-200309000-00007. [DOI] [PubMed] [Google Scholar]
- 503.Chaput N. Exosome-based immunotherapy. Canc. Immunol. Immunother. 2004:53 234–239. doi: 10.1007/s00262-003-0472-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Andre F., Escudier B., Angevin E., Tursz T., Zitvogel L. Exosomes for cancer immunotherapy. Ann. Oncol. 2004;15 doi: 10.1093/annonc/mdh918. [DOI] [PubMed] [Google Scholar]
- 505.Altieri S.L., Khan A.N.H., Tomasi T.B. Exosomes from plasmacytoma cells as a tumor vaccine. J. Immunother. 2004;27:282–288. doi: 10.1097/00002371-200407000-00004. [DOI] [PubMed] [Google Scholar]
- 506.Ramanathan S. Cytokine synergy in antigen-independent activation and priming of naive CD8+ T lymphocytes. Crit. Rev. Immunol. 2009:29 219–239. doi: 10.1615/critrevimmunol.v29.i3.30. [DOI] [PubMed] [Google Scholar]
- 507.Nikfarjam S., Rezaie J., Kashanchi F., Jafari R. Dexosomes as a cell-free vaccine for cancer immunotherapy. J. Exp. Clin. Canc. Res. 2020:39 1–3920. doi: 10.1186/s13046-020-01781-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Li R., Chibbar R., Xiang J. Novel EXO-T vaccine using polyclonal CD4+ T cells armed with HER2-specific exosomes for HER2-positive breast cancer. OncoTargets Ther. 2018:11 7089–7093. doi: 10.2147/OTT.S184898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Wang L. Exosomal pMHC-I complex targets T cell-based vaccine to directly stimulate CTL responses leading to antitumor immunity in transgenic FVBneuN and HLA-A2/HER2 mice and eradicating trastuzumab-resistant tumor in athymic nude mice. Breast Canc. Res. Treat. 2013;140:273–284. doi: 10.1007/s10549-013-2626-7. [DOI] [PubMed] [Google Scholar]
- 510.Menay F. Exosomes isolated from ascites of T-cell lymphoma-bearing mice expressing surface CD24 and HSP-90 induce a tumor-specific immune response. Front. Immunol. 2017;8 doi: 10.3389/fimmu.2017.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Plebanek M.P. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat. Commun. 2017;8:1–12. doi: 10.1038/s41467-017-01433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Cheng L., Wang Y., Huang L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node. Mol. Ther. 2017;25:1665–1675. doi: 10.1016/j.ymthe.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Di Bonito P. Antitumor HPV E7-specific CTL activity elicited by in vivo engineered exosomes produced through DNA inoculation. Int. J. Nanomed. 2017;12:4579–4591. doi: 10.2147/IJN.S131309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Anticoli S. Engineered exosomes emerging from muscle cells break immune tolerance to HER2 in transgenic mice and induce antigen-specific CTLs upon challenge by human dendritic cells. J. Mol. Med. 2018;96:211–221. doi: 10.1007/s00109-017-1617-2. [DOI] [PubMed] [Google Scholar]
- 515.Antimisiaris S.G., Mourtas S., Marazioti A. Exosomes and exosome-inspired vesicles for targeted drug delivery. Pharmaceutics. 2018;10 doi: 10.3390/pharmaceutics10040218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Arrighetti N. Exosome-like nanovectors for drug delivery in cancer. Curr. Med. Chem. 2018;25 doi: 10.2174/0929867325666180831150259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Bunggulawa E.J. Recent advancements in the use of exosomes as drug delivery systems. J. Nanobiotechnol. 2018;16:81. doi: 10.1186/s12951-018-0403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Kibria G., Ramos E.K., Wan Y., Gius D.R., Liu H. Exosomes as a drug delivery system in cancer therapy: potential and challenges. Mol. Pharm. 2018;15:3625–3633. doi: 10.1021/acs.molpharmaceut.8b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Luan X. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017;38:754–763. doi: 10.1038/aps.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Saari H. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Contr. Release. 2015;220:727–737. doi: 10.1016/j.jconrel.2015.09.031. [DOI] [PubMed] [Google Scholar]
- 521.Vader P., Mol E.A., Pasterkamp G., Schiffelers R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016:106 148–156. doi: 10.1016/j.addr.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 522.Vázquez-Ríos A.J. Exosome-mimetic nanoplatforms for targeted cancer drug delivery. J. Nanobiotechnol. 2019;17:85. doi: 10.1186/s12951-019-0517-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Murphy D.E. Extracellular vesicle-based therapeutics: natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019;51 doi: 10.1038/s12276-019-0223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Jabbari N., Akbariazar E., Feqhhi M., Rahbarghazi R., Rezaie J. Breast cancer‐derived exosomes: tumor progression and therapeutic agents. J. Cell. Physiol. 2020;235:6345–6356. doi: 10.1002/jcp.29668. [DOI] [PubMed] [Google Scholar]
- 525.Emam S.E. Liposome co-incubation with cancer cells secreted exosomes (extracellular vesicles) with different proteins expressions and different uptake pathways. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-32861-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Bolhassani A., Khavari A., Oraf Z. Application of Nanotechnology in Drug Delivery. InTech; 2014. Electroporation – advantages and drawbacks for delivery of drug, gene and vaccine. [DOI] [Google Scholar]
- 527.Walker S. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics. 2019;9:8001–8017. doi: 10.7150/thno.37097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Mahmoodi Chalbatani G. Small interfering RNAs (siRNAs) in cancer therapy: a nano-based approach. Int. J. Nanomed. 2019;14:3111–3128. doi: 10.2147/IJN.S200253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Kamerkar S. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Ha D., Yang N., Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm. Sin. B. 2016;6:287–296. doi: 10.1016/j.apsb.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Sun D. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010;18:1606–1614. doi: 10.1038/mt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Osterman C.J.D. Curcumin modulates pancreatic adenocarcinoma cell-derived exosomal function. PloS One. 2015;10 doi: 10.1371/journal.pone.0132845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Tian Y. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 534.Aruna G. Immunotoxins. A review of their use in cancer treatment. J. Stem Cells Regen. Med. 2006;1:31–36. doi: 10.46582/jsrm.0101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Gill D.M., Pappenheimer A.M., Brown R., Kurnick J.T. Studies on the mode of action of diphtheria toxin. VII. Toxin-stimulated hydrolysis of nicotinamide adenine dinucleotide in mammalian cell extracts. J. Exp. Med. 1969;129:1–21. doi: 10.1084/jem.129.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Hassan R., Alewine C., Pastan I. New life for immunotoxin cancer therapy. Clin. Canc. Res. 2016:22 1055–1058. doi: 10.1158/1078-0432.CCR-15-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Akbari B. Immunotoxins in cancer therapy: review and update. Int. Rev. Immunol. 2017:36 207–219. doi: 10.1080/08830185.2017.1284211. [DOI] [PubMed] [Google Scholar]
- 538.Nakase I. Receptor clustering and activation by multivalent interaction through recognition peptides presented on exosomes. Chem. Commun. 2017;53:317–320. doi: 10.1039/c6cc06719k. [DOI] [PubMed] [Google Scholar]
- 539.Jong A.Y. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles. 2017;6 doi: 10.1080/20013078.2017.1294368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Steinman R.M. Decisions about dendritic cells: past, present, and future. Annu. Rev. Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 541.Mignot G., Roux S., Thery C., Ségura E., Zitvogel L. Prospects for exosomes in immunotherapy of cancer • Introduction • Exosome Characterization-Exosome genesis-Exosome composition-Exosome purification • Exosomes and the immune system-Immunostimulatory properties of DEX-The use of tumor-derived exosomes as. J. Cell Mol. Med. 2006;10 doi: 10.1111/j.1582-4934.2006.tb00406.x. www.jcmm.ro [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Robbins P.D., Morelli A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014:14 195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Tian H., Li W. Dendritic cell-derived exosomes for cancer immunotherapy: hope and challenges. Ann. Transl. Med. 2017;5 doi: 10.21037/atm.2017.02.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Yang X., Chen J., Wang N., Liu Z., Li Y. Clinical use of dendritic cell-derived exosomes for hepatocellular carcinoma immunotherapy: how far we are? J. Hepatol. 2018;69:984–986. doi: 10.1016/j.jhep.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 545.Wang X. Fetal dermal mesenchymal stem cell-derived exosomes accelerate cutaneous wound healing by activating Notch signaling. Stem Cell. Int. 2019 doi: 10.1155/2019/2402916. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Yeo R.W.Y. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013:65 336–341. doi: 10.1016/j.addr.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 547.Park J.S., Suryaprakash S., Lao Y.H., Leong K.W. Engineering mesenchymal stem cells for regenerative medicine and drug delivery. Methods. 2015;84:3–16. doi: 10.1016/j.ymeth.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Gong M. Mesenchymal stem cells release exosomes that transfer miRNAs to endothelial cells and promote angiogenesis. Oncotarget. 2017;8:45200–45212. doi: 10.18632/oncotarget.16778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Karaoz E., Sun E., Demir C.S. Mesenchymal stem cell-derived exosomes do not promote the proliferation of cancer cells in vitro. Int. J. Physiol. Pathophysiol. Pharmacol. 2019;11:177–189. [PMC free article] [PubMed] [Google Scholar]
- 550.Lee J.K. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PloS One. 2013;8 doi: 10.1371/journal.pone.0084256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Vakhshiteh F., Atyabi F., Ostad S.N. Mesenchymal stem cell exosomes: a two-edged sword in cancer therapy. Int. J. Nanomed. 2019;14:2847–2859. doi: 10.2147/IJN.S200036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Pessina A. Mesenchymal stromal cells primed with paclitaxel provide a new approach for cancer therapy. PloS One. 2011;6 doi: 10.1371/journal.pone.0028321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.de Araujo Farias V. Exosomes derived from mesenchymal stem cells enhance radiotherapy-induced cell death in tumor and metastatic tumor foci. Mol. Canc. 2018;17:122. doi: 10.1186/s12943-018-0867-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Yan W., Jiang S. Immune cell-derived exosomes in the cancer-immunity cycle. Trends in Cancer. 2020 doi: 10.1016/j.trecan.2020.02.013. [DOI] [PubMed] [Google Scholar]
- 555.Watzl C., Urlaub D. Molecular mechanisms of natural killer cell regulation. Front. Biosci. 2012;17:1418–1432. doi: 10.2741/3995. [DOI] [PubMed] [Google Scholar]
- 556.Groot Kormelink T., Mol S., de Jong E.C., Wauben M.H.M. The role of extracellular vesicles when innate meets adaptive. Semin. Immunopathol. 2018:40 439–452. doi: 10.1007/s00281-018-0681-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Andre F. Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002;360:295–305. doi: 10.1016/S0140-6736(02)09552-1. [DOI] [PubMed] [Google Scholar]
- 558.Gao D., Jiang L. Exosomes in cancer therapy: a novel experimental strategy. Am. J. Cancer Res. 2018;8:2165–2175. [PMC free article] [PubMed] [Google Scholar]
- 559.Rao Q. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology. 2016;64:456–472. doi: 10.1002/hep.28549. [DOI] [PubMed] [Google Scholar]
- 560.Senapati S., Mahanta A.K., Kumar S., Maiti P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduction and Targeted Therapy. 2018;3 doi: 10.1038/s41392-017-0004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.De Angelis A. Doxorubicin cardiotoxicity and target cells: a broader perspective. Cardio-Oncology. 2016;2:2. doi: 10.1186/s40959-016-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Schindler C. Exosomal delivery of doxorubicin enables rapid cell entry and enhanced in vitro potency. PloS One. 2019;14 doi: 10.1371/journal.pone.0214545. e0214545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Toffoli G. Exosomal doxorubicin reduces the cardiac toxicity of doxorubicin. Nanomedicine. 2015;10:2963–2971. doi: 10.2217/nnm.15.118. [DOI] [PubMed] [Google Scholar]
- 564.Hadla M. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine. 2016;11:2431–2441. doi: 10.2217/nnm-2016-0154. [DOI] [PubMed] [Google Scholar]
- 565.Zhu X. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles. 2017;6 doi: 10.1080/20013078.2017.1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Smyth T.J., Redzic J.S., Graner M.W., Anchordoquy T.J. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim. Biophys. Acta Biomembr. 2014;1838:2954–2965. doi: 10.1016/j.bbamem.2014.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Wang J., Zheng Y., Zhao M. Exosome-based cancer therapy: implication for targeting cancer stem cells. Front. Pharmacol. 2017;7 doi: 10.3389/fphar.2016.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Emam S.E. Cancer cell-type tropism is one of crucial determinants for the efficient systemic delivery of cancer cell-derived exosomes to tumor tissues. Eur. J. Pharm. Biopharm. 2019;145:27–34. doi: 10.1016/j.ejpb.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 569.Yong T. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-11718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Ma J. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016;26:713–727. doi: 10.1038/cr.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Ohno S.I. Systemically injected exosomes targeted to EGFR deliver antitumor microrna to breast cancer cells. Mol. Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Bai J. Engineered targeting tLyp-1 exosomes as gene therapy vectors for efficient delivery of siRNA into lung cancer cells. Asian J. Pharm. Sci. 2019 doi: 10.1016/j.ajps.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Kooijmans S.A.A., Schiffelers R.M., Zarovni N., Vago R. Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: new nanotools for cancer treatment. Pharmacol. Res. 2016;111:487–500. doi: 10.1016/j.phrs.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 574.Singh M. Transferrin as A targeting ligand for liposomes and anticancer drugs. Curr. Pharmaceut. Des. 1999;5:443–451. [PubMed] [Google Scholar]
- 575.Shen Y. Transferrin receptor 1 in cancer: a new sight for cancer therapy. Am. J. Cancer Res. 2018;8:916–931. [PMC free article] [PubMed] [Google Scholar]
- 576.Tan S., Wang G. Lung cancer targeted therapy: folate and transferrin dual targeted, glutathione responsive nanocarriers for the delivery of cisplatin. Biomed. Pharmacother. 2018;102:55–63. doi: 10.1016/j.biopha.2018.03.046. [DOI] [PubMed] [Google Scholar]
- 577.Qi H. Blood exosomes endowed with magnetic and targeting properties for cancer therapy. ACS Nano. 2016;10:3323–3333. doi: 10.1021/acsnano.5b06939. [DOI] [PubMed] [Google Scholar]
- 578.Guo Y., Wang L., Lv P., Zhang P. Transferrin-conjugated doxorubicin-loaded lipid-coated nanoparticles for the targeting and therapy of lung cancer. Oncol. Lett. 2015;9:1065–1072. doi: 10.3892/ol.2014.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Smyth T. Surface functionalization of exosomes using click chemistry. Bioconjugate Chem. 2014;25:1777–1784. doi: 10.1021/bc500291r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Tian T. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials. 2018;150:137–149. doi: 10.1016/j.biomaterials.2017.10.012. [DOI] [PubMed] [Google Scholar]
- 581.Song S. In situ one-step fluorescence labeling strategy of exosomes via bioorthogonal click chemistry for real-time exosome tracking in vitro and in vivo. Bioconjugate Chem. 2020;31:1562–1574. doi: 10.1021/acs.bioconjchem.0c00216. [DOI] [PubMed] [Google Scholar]
- 582.Quinn Z., Mao W., Xia Y., John R., Wan Y. Conferring receptors on recipient cells with extracellular vesicles for targeted drug delivery. Bioact. Mater. 2021;6:749–756. doi: 10.1016/j.bioactmat.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Liu C., Su C. Design strategies and application progress of therapeutic exosomes. Theranostics. 2019:9 1015–1028. doi: 10.7150/thno.30853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Jeong D. Nanovesicles engineered from ES cells for enhanced cell proliferation. Biomaterials. 2014;35:9302–9310. doi: 10.1016/j.biomaterials.2014.07.047. [DOI] [PubMed] [Google Scholar]
- 585.Han C. Mesenchymal stem cell engineered nanovesicles for accelerated skin wound closure. ACS Biomater. Sci. Eng. 2019;5:1534–1543. doi: 10.1021/acsbiomaterials.8b01646. [DOI] [PubMed] [Google Scholar]
- 586.Zhu Q., Heon M., Zhao Z., He M. Microfluidic engineering of exosomes: editing cellular messages for precision therapeutics. Lab Chip. 2018;18:1690–1703. doi: 10.1039/c8lc00246k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Van Deun J. Feasibility of mechanical extrusion to coat nanoparticles with extracellular vesicle membranes. Cells. 2020;9:1797. doi: 10.3390/cells9081797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Wang L. Preparation of engineered extracellular vesicles derived from human umbilical cord mesenchymal stem cells with ultrasonication for skin rejuvenation. ACS Omega. 2019;4:22638–22645. doi: 10.1021/acsomega.9b03561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.García-Manrique P., Matos M., Gutiérrez G., Pazos C., Blanco-López M.C. Therapeutic biomaterials based on extracellular vesicles: classification of bio-engineering and mimetic preparation routes. J. Extracell. Vesicles. 2018;7 doi: 10.1080/20013078.2017.1422676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Kim M.S. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2016;12:655–664. doi: 10.1016/j.nano.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Sato Y.T. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016;6:1–11. doi: 10.1038/srep21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Lin Y. Exosome–liposome hybrid nanoparticles deliver CRISPR/Cas9 system in MSCs. Adv. Sci. 2018;5 doi: 10.1002/advs.201700611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Piffoux M., Silva A.K.A., Wilhelm C., Gazeau F., Tareste D. Modification of extracellular vesicles by fusion with liposomes for the design of personalized biogenic drug delivery systems. ACS Nano. 2018;12:6830–6842. doi: 10.1021/acsnano.8b02053. [DOI] [PubMed] [Google Scholar]
- 594.Biswas S. Exosomes produced by mesenchymal stem cells drive differentiation of myeloid cells into immunosuppressive M2-polarized macrophages in breast cancer. J. Immunol. 2019:ji1900692. doi: 10.4049/jimmunol.1900692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Federici C. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PloS One. 2014;9 doi: 10.1371/journal.pone.0088193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Gamperl H. Extracellular vesicles from malignant effusions induce tumor cell migration: inhibitory effect of LMWH tinzaparin. Cell Biol. Int. 2016;40:1050–1061. doi: 10.1002/cbin.10645. [DOI] [PubMed] [Google Scholar]
- 597.Huang Z., Yang M., Li Y., Yang F., Feng Y. Exosomes derived from hypoxic colorectal cancer cells transfer wnt4 to normoxic cells to elicit a prometastatic phenotype. Int. J. Biol. Sci. 2018;14:2094–2102. doi: 10.7150/ijbs.28288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Li X.Q. Exosomes derived from gefitinib-treated EGFR-mutant lung cancer cells alter cisplatin sensitivity via up-regulating autophagy. Oncotarget. 2016;7:24585–24595. doi: 10.18632/oncotarget.8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Wada J. Surface-bound TGF-beta1 on effusion-derived exosomes participates in maintenance of number and suppressive function of regulatory T-cells in malignant effusions. Anticancer Res. 2010;30:3747–3757. [PubMed] [Google Scholar]
- 600.Yoshida K. Exosomes containing ErbB2/CRK induce vascular growth in premetastatic niches and promote metastasis of bladder cancer. Canc. Sci. 2019;110:2119–2132. doi: 10.1111/cas.14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Zhang Z. LMP1-positive extracellular vesicles promote radioresistance in nasopharyngeal carcinoma cells through P38 MAPK signaling. Cancer Med. 2019;8:6082–6094. doi: 10.1002/cam4.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Feng D. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11:675–687. doi: 10.1111/j.1600-0854.2010.01041.x. [DOI] [PubMed] [Google Scholar]
- 603.Imai T. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J. Extracell. Vesicles. 2015;4:26238. doi: 10.3402/jev.v4.26238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Nishida-Aoki N. Disruption of circulating extracellular vesicles as a novel therapeutic strategy against cancer metastasis. Mol. Ther. 2017;25:181–191. doi: 10.1016/j.ymthe.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Baietti M.F. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012;14:677–685. doi: 10.1038/ncb2502. [DOI] [PubMed] [Google Scholar]
- 606.Trajkovic K. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science (80-. ) 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 607.Bobrie A. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Can. Res. 2012;72:4920–4930. doi: 10.1158/0008-5472.CAN-12-0925. [DOI] [PubMed] [Google Scholar]
- 608.Ostrowski M. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010;12:19–30. doi: 10.1038/ncb2000. [DOI] [PubMed] [Google Scholar]
- 609.Tian T. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014;289:22258–22267. doi: 10.1074/jbc.M114.588046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Ichim T.E. Exosomes as a tumor immune escape mechanism: possible therapeutic implications. J. Transl. Med. 2008;6 doi: 10.1186/1479-5876-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Zheng H. The roles of tumor-derived exosomes in non-small cell lung cancer and their clinical implications 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. J. Exp. Clin. Canc. Res. 2018;37 doi: 10.1186/s13046-018-0901-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Petrova V., Annicchiarico-Petruzzelli M., Melino G., Amelio I. The hypoxic tumour microenvironment. Oncogenesis. 2018:7 1–13. doi: 10.1038/s41389-017-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Al Tameemi W., Dale T.P., Al-Jumaily R.M.K., Forsyth N.R. Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol. 2019;7:4. doi: 10.3389/fcell.2019.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.El-Kenawi A. Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br. J. Canc. 2019;121:556–566. doi: 10.1038/s41416-019-0542-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Rohani N. Acidification of tumor at stromal boundaries drives transcriptome alterations associated with aggressive phenotypes. Can. Res. 2019;79:1952–1966. doi: 10.1158/0008-5472.CAN-18-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Li W., Zhou Y., Shang C., Sang H., Zhu H. 2020. Effects of Environmental pH on the Growth of Gastric Cancer Cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Logozzi M., Spugnini E., Mizzoni D., Di Raimo R., Fais S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Canc. Metastasis Rev. 2019:38 93–101. doi: 10.1007/s10555-019-09783-8. [DOI] [PubMed] [Google Scholar]
- 618.Patton M.C., Zubair H., Khan M.A., Singh S., Singh A.P. Hypoxia alters the release and size distribution of extracellular vesicles in pancreatic cancer cells to support their adaptive survival. J. Cell. Biochem. 2020;121:828–839. doi: 10.1002/jcb.29328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Zonneveld M.I., Keulers T.G.H., Rouschop K.M.A. Extracellular vesicles as transmitters of hypoxia tolerance in solid cancers. Cancers. 2019;11 doi: 10.3390/cancers11020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Almeria C. Hypoxia conditioned mesenchymal stem cell-derived extracellular vesicles induce increased vascular tube formation in vitro. Front. Bioeng. Biotechnol. 2019;7:292. doi: 10.3389/fbioe.2019.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Endzeliņs E. Extracellular vesicles derived from hypoxic colorectal cancer cells confer metastatic phenotype to non-metastatic cancer cells. Anticancer Res. 2018;38:5139–5147. doi: 10.21873/anticanres.12836. [DOI] [PubMed] [Google Scholar]
- 622.Underwood J.J. Liquid biopsy for cancer: review and implications for the radiologist. Radiology. 2020:294 5–29417. doi: 10.1148/radiol.2019182584. [DOI] [PubMed] [Google Scholar]
- 623.Liu M.C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020;31:745–759. doi: 10.1016/j.annonc.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Pang B. Extracellular vesicles: the next generation of biomarkers for liquid biopsy-based prostate cancer diagnosis. Theranostics. 2020;10:2309–2326. doi: 10.7150/thno.39486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Osti D. Clinical significance of extracellular vesicles in plasma from glioblastoma patients. Clin. Canc. Res. 2019;25:266–276. doi: 10.1158/1078-0432.CCR-18-1941. [DOI] [PubMed] [Google Scholar]
- 626.Johnsen K.B., Gudbergsson J.M., Andresen T.L., Simonsen J.B. What is the blood concentration of extracellular vesicles? Implications for the use of extracellular vesicles as blood-borne biomarkers of cancer. Biochim. Biophys. Acta Rev. Canc. 2019:1871 109–116. doi: 10.1016/j.bbcan.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 627.Sharma R., Huang X., Brekken R.A., Schroit A.J. Detection of phosphatidylserine-positive exosomes for the diagnosis of early-stage malignancies. Br. J. Canc. 2017;117:545–552. doi: 10.1038/bjc.2017.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Lea J. Detection of phosphatidylserine-positive exosomes as a diagnostic marker for ovarian malignancies: a proof of concept study. Oncotarget. 2017;8:14395–14407. doi: 10.18632/oncotarget.14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Livshits Z., Rao R.B., Smith S.W. An approach to chemotherapy-associated toxicity. Emerg. Med. Clin. 2014:32 167–203. doi: 10.1016/j.emc.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 630.Kimiz-Gebologlu I., Gulce-Iz S., Biray-Avci C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018:45 2935–2940. doi: 10.1007/s11033-018-4427-x. [DOI] [PubMed] [Google Scholar]
- 631.Saleh A.F. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale. 2019;11:6990–7001. doi: 10.1039/c8nr08720b. [DOI] [PubMed] [Google Scholar]
- 632.Rayamajhi S., Nguyen T.D.T., Marasini R., Aryal S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019;94:482–494. doi: 10.1016/j.actbio.2019.05.054. [DOI] [PubMed] [Google Scholar]
- 633.Chin A.R., Wang S.E. Cancer-derived extracellular vesicles: the ‘soil conditioner’ in breast cancer metastasis? Canc. Metastasis Rev. 2016;35:669–676. doi: 10.1007/s10555-016-9639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Elsharkasy O.M. Extracellular vesicles as drug delivery systems: why and how? Adv. Drug Deliv. Rev. 2020 doi: 10.1016/j.addr.2020.04.004. [DOI] [PubMed] [Google Scholar]
- 635.Balachandran B., Yuana Y. Extracellular vesicles-based drug delivery system for cancer treatment. Cogent Med. 2019;6 [Google Scholar]
- 636.Watson D.C. Efficient production and enhanced tumor delivery of engineered extracellular vesicles. Biomaterials. 2016;105:195–205. doi: 10.1016/j.biomaterials.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Kooijmans S.A.A. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Contr. Release. 2016;224:77–85. doi: 10.1016/j.jconrel.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 638.Mentkowski K.I., Snitzer J.D., Rusnak S., Lang J.K. Therapeutic potential of engineered extracellular vesicles. AAPS J. 2018;20 doi: 10.1208/s12248-018-0211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]