

Abstract

Anthracyclines are effective drugs in the treatment of various cancers, but their use comes with severe side effects. The archetypal anthracycline drug, doxorubicin, displays two molecular modes of action: DNA double-strand break formation (through topoisomerase IIα poisoning) and chromatin damage (via eviction of histones). These biological activities can be modulated and toxic side effects can be reduced by separating these two modes of action through alteration of the aminoglycoside moiety of doxorubicin. We herein report on the design, synthesis, and evaluation of a coherent set of configurational doxorubicin analogues featuring all possible stereoisomers of the 1,2-amino-alcohol characteristic for the doxorubicin 3-amino-2,3-dideoxyfucoside, each in nonsubstituted and N,N-dimethylated forms. The set of doxorubicin analogues was synthesized using appropriately protected 2,3,6-dideoxy-3-amino glycosyl donors, equipped with an alkynylbenzoate anomeric leaving group, and the doxorubicin aglycon acceptor. The majority of these glycosylations proceeded in a highly stereoselective manner to provide the desired axial α-linkage. We show that both stereochemistry of the 3-amine carbon and N-substitution state are critical for anthracycline cytotoxicity and generally improve cellular uptake. N,N-Dimethylepirubicin is identified as the most potent anthracycline that does not induce DNA damage while remaining cytotoxic.
Introduction
The anthracycline drug doxorubicin (adriamycin, Figure 1A, 1a) is one of the most used anticancer drugs in history, and is annually prescribed to over one million cancer patients.1 While doxorubicin is effective against a wide variety of cancers, including leukemia, non-Hodgkin’s lymphoma, and breast cancer, its use is limited by severe side effects. Cardiotoxicity, the main treatment-limiting side effect, emerges in a cumulative manner, and for this reason treatment with doxorubicin is restricted to a maximum of six to eight treatment cycles.2 Thousands of analogues of doxorubicin produced by (mutant) enzymes or prepared by organic (semi-)synthesis, have been tested for their biological activities in the past decades, with the aim to identify more effective anthracyclines with limited side effects.3−5 However, only a few of these anthracycline analogues have been approved for use in clinical practices. The 4′-epimer of doxorubicin, epirubicin (Figure 1A, 2a), is one of these clinically approved doxorubicin variants, and has a higher cardiotoxic risk threshold regarding its cumulative dose (for epirubicin, >720 mg/m2; for doxorubicin, >500 mg/m2).6 Currently, epirubicin is used in the treatment of breast, ovarian, gastric, and lung cancers, as well as several types of lymphomas.7 This illustrates that new effective and less toxic anthracyclines can be developed, which may allow more intense, longer, and more effective treatment of cancer patients, with limited long-term side effects for cancer survivors.
Figure 1.
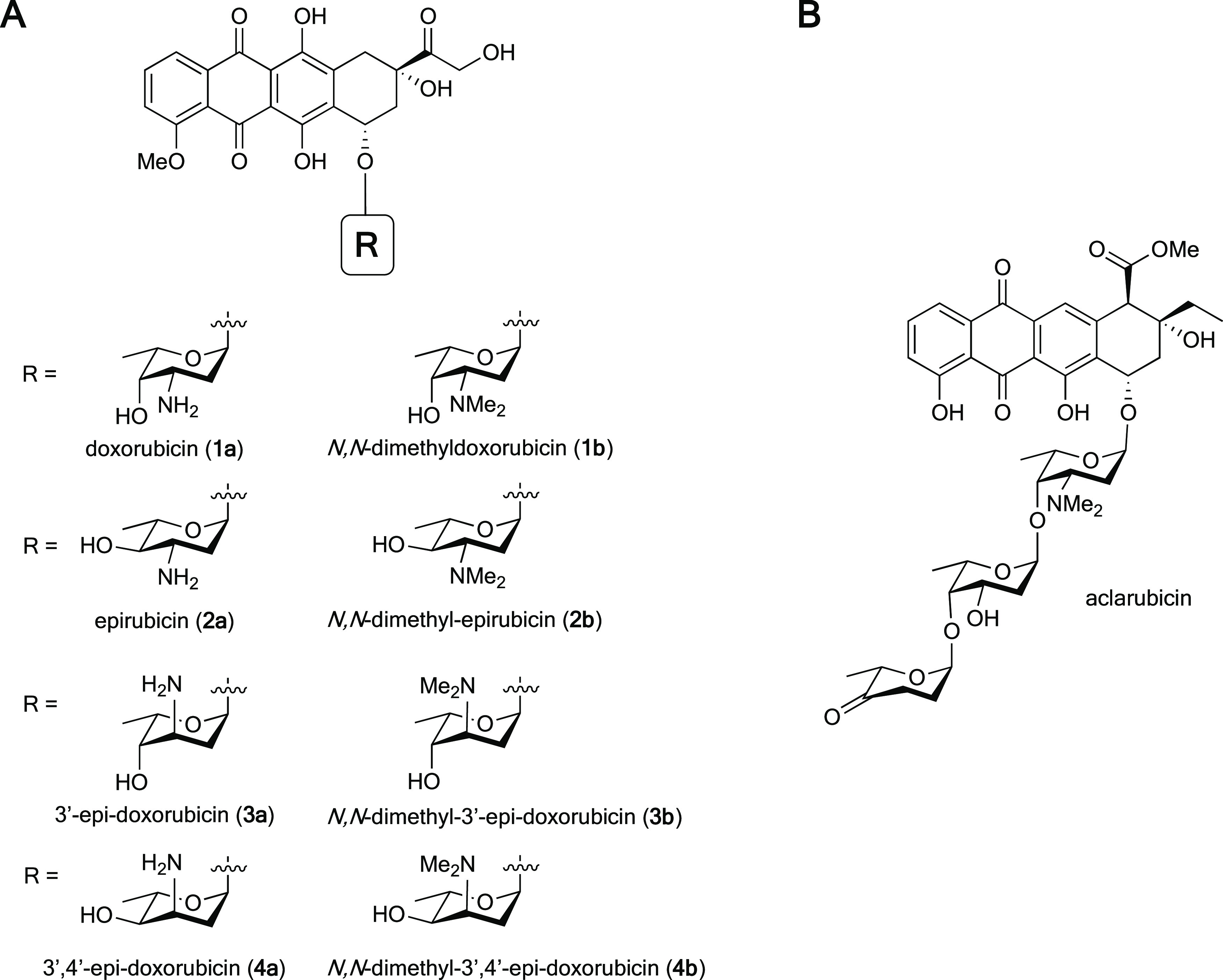
(A) Chemical structures of doxorubicin (1a) and derivatives (1b, 2b, 3b, 4b), differing in stereochemistry and N,N-dimethylation on the sugar moiety. (B) Chemical structure of aclarubicin.
Deeper understanding of the molecular mode of action of anthracycline drugs is key for the development of new and improved (in terms of efficacy) analogues. One key feature of doxorubicin is the formation of DNA double-strand breaks due to topoisomerase IIα poisoning.8,9 Doxorubicin inhibits topoisomerase IIα after the generation of DNA double-strand breaks and before re-ligation of DNA, thus resulting in DNA damage. For decades, this mode of action was thought to be the main mechanism for the remarkable anticancer activity of doxorubicin and its structural analogues. However, aclarubicin (Figure 1B), another anthracycline analogue used in Japan and China, is at least equally effective in the treatment of acute myeloid leukemia (AML), but does not produce such DNA double-strand breaks.9 Moreover, aclarubicin is much less cardiotoxic.10 Aclarubicin functions by a recently discovered second activity of anthracyclines: eviction of histones from the chromatin.9,11 These evicted histones are then replaced by new nascent ones, resulting in epigenetic alterations.12 Furthermore, previous work from our labs showed that the combination of histone eviction and DNA double-strand break formation, as exerted by doxorubicin, is responsible for the major side effects of this compound: cardiotoxicity and secondary tumor formation.10N,N-Dimethyldoxorubicin (Figure 1A, 1b), a close doxorubicin analogue featuring the dimethylamine characteristic for aclarubicin, mirrors the biological activity of aclarubicin, by only inducing histone eviction (and not DNA damage).10 Like aclarubicin, N,N-dimethyldoxorubicin is an effective anticancer agent that lacks the severe side effects displayed by doxorubicin in various mouse models.10 These data suggest that separating DNA from chromatin damage activities, as found in the anthracyclines currently used in the clinic, results in drugs lacking the major long-term side effects. In addition, chromatin damage appeared to be the major cause for the anticancer activity of these compounds. Our follow-up studies on a series of doxorubicin/aclarubicin hybrid structures, varying in the tetracyclic aglycon, the sugar moiety (from the doxorubicin monosaccharide up to the aclarubicin trisaccharide), and the N-alkylation pattern revealed that dimethylated structures fail to induce DNA double-strand breaks while remaining cytotoxic.13 These findings raise the question whether there is a structure–activity relationship for stereoisomeric analogues of doxorubicin, and whether this may lead to potential new effective anticancer drugs. To this end, we herein report the synthesis and evaluation of a coherent, focused library of diastereomers of doxorubicin (1a) in the 1,2-amino alcohol arrangement of the 3-amino-2,3-dideoxy-l-fucoside (daunosamine) characteristic for this compound. Compounds 1a/b, 2a/b, 3a/b, 4a/b featuring both the free amine and the N,N-dimethylated, some of which have been reported previously10,14,15 have been generated. Our general synthesis route for the preparation of these compounds is based on the use of gold-catalyzed glycosylation reactions of alkynylbenzoate (ABz) donors. Most of these proceed with excellent stereoselectivity, which we propose to be rooted in the oxocarbenium ion-like intermediates formed in these reactions. Subsequently, we evaluated the biological activities of the set of anthracyclines by testing their abilities to induce DNA damage and histone eviction, their cellular uptake, and their cytotoxicity. Compounds featuring an equatorially oriented, dimethylated sugar amine proved to be the most cytotoxic and in general are also taken up best by cells. From these, N,N-dimethylepirubicin (2b) was identified (next to the previously described N,N-dimethyldoxorubicin (1b)) as the most potent anthracycline in this coherent set of epimeric doxorubicin analogues, with an excellent cytotoxicity profile and no DNA double-strand break activity.
Results and Discussion
Synthesis of the Set of (N,N-Dimethyl)doxorubicin Stereoisomers
We started with the development of a synthetic methodology to prepare the focused library depicted in Figure 1 (compounds 2b, 3a/b, 4a/b). Recently, we reported the synthesis of N,N-dimethyldoxorubicin (1b),10 whereas doxorubicin (1a) and epirubicin (2a) are both commercially available. Our synthetic method is based on gold(I)-mediated glycosylation chemistry, developed by Yu and co-workers,16 that in our hands has proven effective in the creation of the anthracycline α-fucosidic linkages.13,17 In the assembly of doxorubicin/aclarubicin hybrids, we found that the use of an allyloxycarbamate (Alloc) to mask the amino group of the 2,3-dideoxy-3-aminofucose in combination with relatively labile silyl ethers to protect the hydroxyl groups is very effective for the assembly of the anthracycline targets.13 Thus, alkynylbenzoate donors 9, 12, and 16 were designed and assembled as depicted in Scheme 1. p-Methoxyphenolates 6 and 10 were prepared from precursor 5(18) (a mixture of 33:67 R/S at C3) by treatment with p-methoxyphenol in the presence of catalytic trimethylsilyl trifluoromethanesulfonate (TMSOTf) to give equatorial azide 6 in 50% yield and axial azide 10 in 7%. Deacylation under Zemplén conditions was followed by triethylsilylation of the resultant alcohol. Then, the azide was reduced under Staudinger conditions and converted to the allyloxycarbamate to give 7 and 11. The p-methoxyphenolates were then subjected to oxidative hydrolysis of the anomeric p-methoxyphenolate using Ag(DPAH)2,19 delivering the lactols, which were transformed into the required alkynylbenzoates (ABz) by a Steglich esterification with o-cyclopropylethynylbenzoic acid (8),20 providing donor glycosides 9 and 12.
Scheme 1. Synthesis of Aminosugar Alkynylbenzoate Donors 9, 12, and 16.
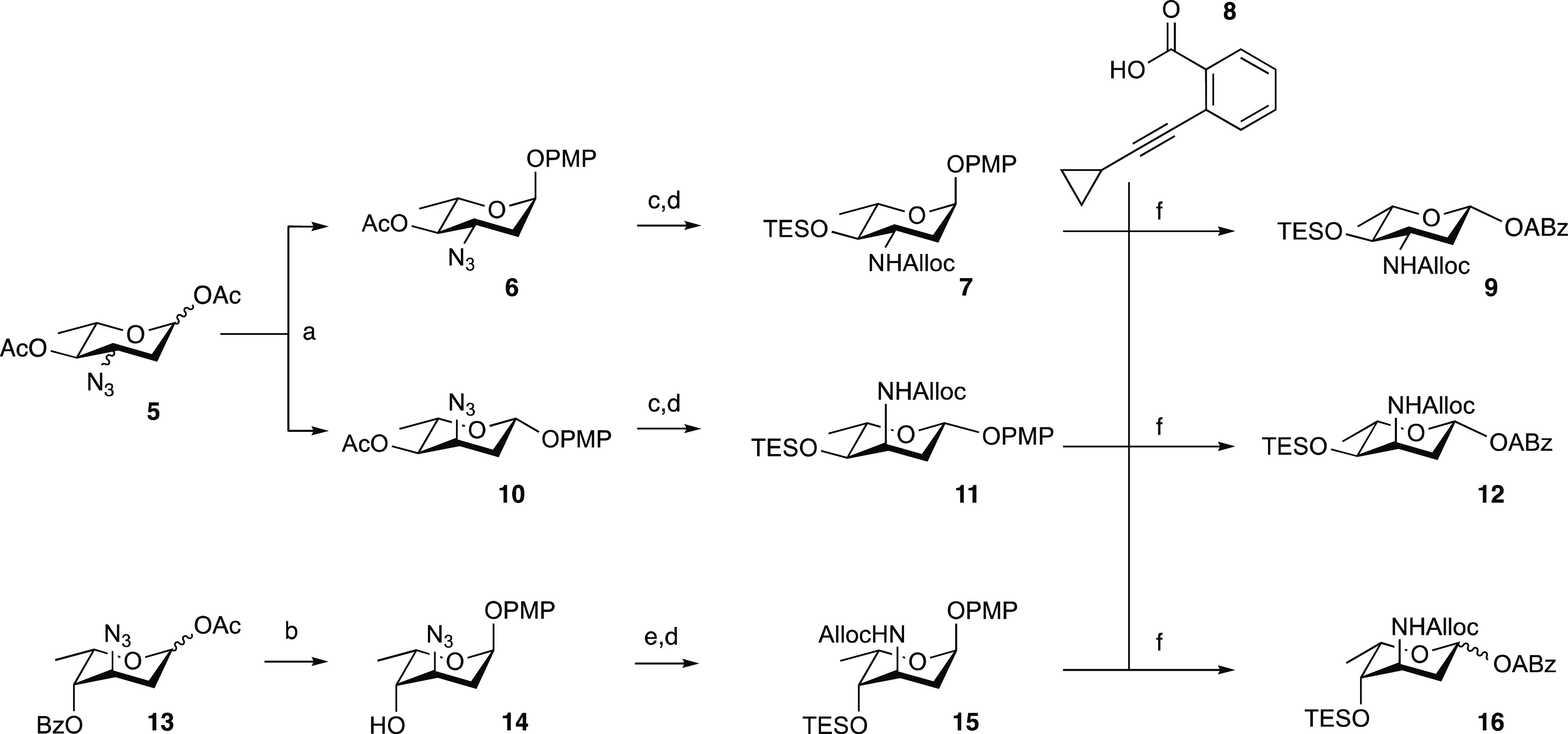
Reagents and conditions: (a) p-methoxyphenol, TMSOTf, dichloromethane (DCM), 0 °C, 50% for 6, 7% for 10; (b) (i) p-methoxyphenol, BF3·OEt2, DCM, −60 to −40 °C; (ii) NaOMe, MeOH, 70% over two steps; (c) (i) NaOMe, MeOH; (ii) TESOTf, pyr., DCM, 95% over two steps from 6, quant. over two steps from 10; (d) (i) polymer-bound PPh3, tetrahydrofuran (THF), H2O; (ii) allyl chloroformate, pyr., DCM, quant. over two steps for 7; quant. over two steps for 11; quant. over two steps for 15; (e) TESOTf, pyr., quant.; (f) (i) silver(II) di(hydrogen dipicolinate) hydrate, NaOAc, MeCN, H2O, 0 °C; (ii) ethylcarbodiimide hydrochloride (EDCI·HCl), 4-dimethylaminopyridine (DMAP), N,N-diisopropylethylamine (DIPEA), DCM, 49% over two steps for 9 (β-anomer only), 57% over two steps for 12 (β-anomer only), 79% over two steps for 16 (1:3 α/β).
In preparing for the synthesis of the 3-epi-daunosamine donor 16, acetate 13(21) was converted to the p-methoxyphenolate by the action of BF3·OEt2 giving, after deacylation, compound 14. Triethylsilylation of the 4-hydroxyl and conversion of the azide into the allyloxycarbamate yielded 15. Removal of the anomeric p-methoxyphenol group and installation of the alkynylbenzoate was then achieved as described for donors 9 and 12 to give donor 16. The three alkynylbenzoate donors 9, 12, and 16 were used, alongside daunosamine donor 17 that we previously assembled,13 in glycosylation reactions toward doxorubicin analogues 2b–3a/b–4a/b (Scheme 2). Treatment of a mixture of donor 9 and protected doxorubicinone acceptor 18(22) with a catalytic amount of PPh3AuNTf2 in DCM at room temperature led to the formation of anthracycline 19 in 80% yield as an 8:1 α/β-mixture. The desired α-anomer could be readily separated to provide the desired axially linked 19. The analogous glycosylation of 12 and 18 proceeded with poor stereoselectivity and provided 20 as a 1.5:1 α/β mixture in 50% yield. The condensation of donor 16, having two axial substituents at C-3 and C-4, led to the formation of the protected doxorubicin analogue 21 with excellent stereoselectivity and the desired product was obtained as a single anomer in 56% yield. The glycosylation of daunosamine donor 17 and acceptor 18 also delivered the desired α-anomer with excellent stereoselectivity, forming 22 in 56% yield.
Scheme 2. Synthesis of (N,N-Dimethyl)doxorubicins 2b–3a/b–4a/b.

(a) 10 mol% PPh3AuNTf2, DCM, 80% for 19 (8:1 α/β); 50% for 20 (1.5:1 α/β); 56% for 21 (>20:1 α/β), 56% for 22 (>20:1 α/β); (b) Pd(PPh3)4, NDMBA, DCM; (c) NaBH(OAc)3, aqueous (aq.) CH2O, EtOH, 61% over two steps from 19; (d) HF·pyr., pyr., 0 °C, quant. for 2b; 35% over three steps for 4b; 61% over two steps for 3a, 48% over three steps for 3b; (e) lyophilization from aq. HCl, 54% over three steps.
We propose that the observed stereoselectivity—or lack thereof—in these glycosylations can be understood upon perusal of the intermediate oxocarbenium ions (or oxocarbenium ion-like species), their conformational behavior, and the direction nucleophiles may take toward forming a glycosidic linkage. The dideoxy nature of the used donors makes them relatively reactive (“armed”), and the anomeric cation thus readily forms upon activation of the alkynylbenzoate anomeric leaving group.23 First-order nucleophilic substitution (SN1)-like pathways are further promoted by the use of the non-nucleophilic counterion triflimide (NTf2).24 The intermediate oxocarbenium ion can adopt different conformations (often close to half-chair structures) and preferred conformations are the result of stereoelectronic effects as exerted by the substituents on the ring.25 Electronegative atoms (such as oxygen and nitrogen) prefer to adopt an axial orientation when mounted at C-3 or C-4. Alkyl groups will preferentially adopt a pseudo-equatorial orientation for steric reasons.25−27 In addition, an incoming nucleophile, which will preferentially attack this oxocarbenium ion from the β-face (to deliver the product through a favorable chairlike transition state), will experience 1,3-diaxial interactions with the substituent at C-3 as well as the C-6 methyl group. Therefore, the incoming doxorubicinone nucleophile will preferentially attack the all-equatorial 3H4 conformer of 9, leading to the formation of the α-product in good selectivity (Scheme 3A). The 3H4 and 4H3 half-chair oxocarbenium ions emerge from activating donor 12 each place one of the electronegative substituents in an axial position and will therefore be of comparable stability (Scheme 3B). The trajectories of incoming nucleophiles on these ions will experience similar steric interactions, explaining the poor selectivity observed in the glycosylation of donor 12 and acceptor 18. Zeng et al. have previously reported that glycosylations of similar ristosaminyl alkynylbenzoate donors to various glycosyl acceptors proceeded with comparably poor selectivity.28 The excellent stereoselectivity of donor 16 can be traced back to the 3H4 half-chair oxocarbenium ion, which places both the C-3 and C-4 electronegative groups in an axial position, while having the C-6-methyl oriented equatorially (Scheme 3C). Finally, the 3H4 half-chair oxocarbenium ion generated from donor 17 puts just the C-3 substituent in a favorable axial position (Scheme 3D). However, the incoming nucleophile would suffer 1,3-diaxial interactions with the C-3 and C-6 substituents in the 4H3 oxocarbenium ion conformation. This together accounts for the high stereoselectivity found for the glycosylation found between donor 17 and acceptor 18.
Scheme 3. Mechanistic Rationale for the Stereoselectivity Found in the Glycosylations of Donors 9, 12, 16, and 17 with Acceptor 18.
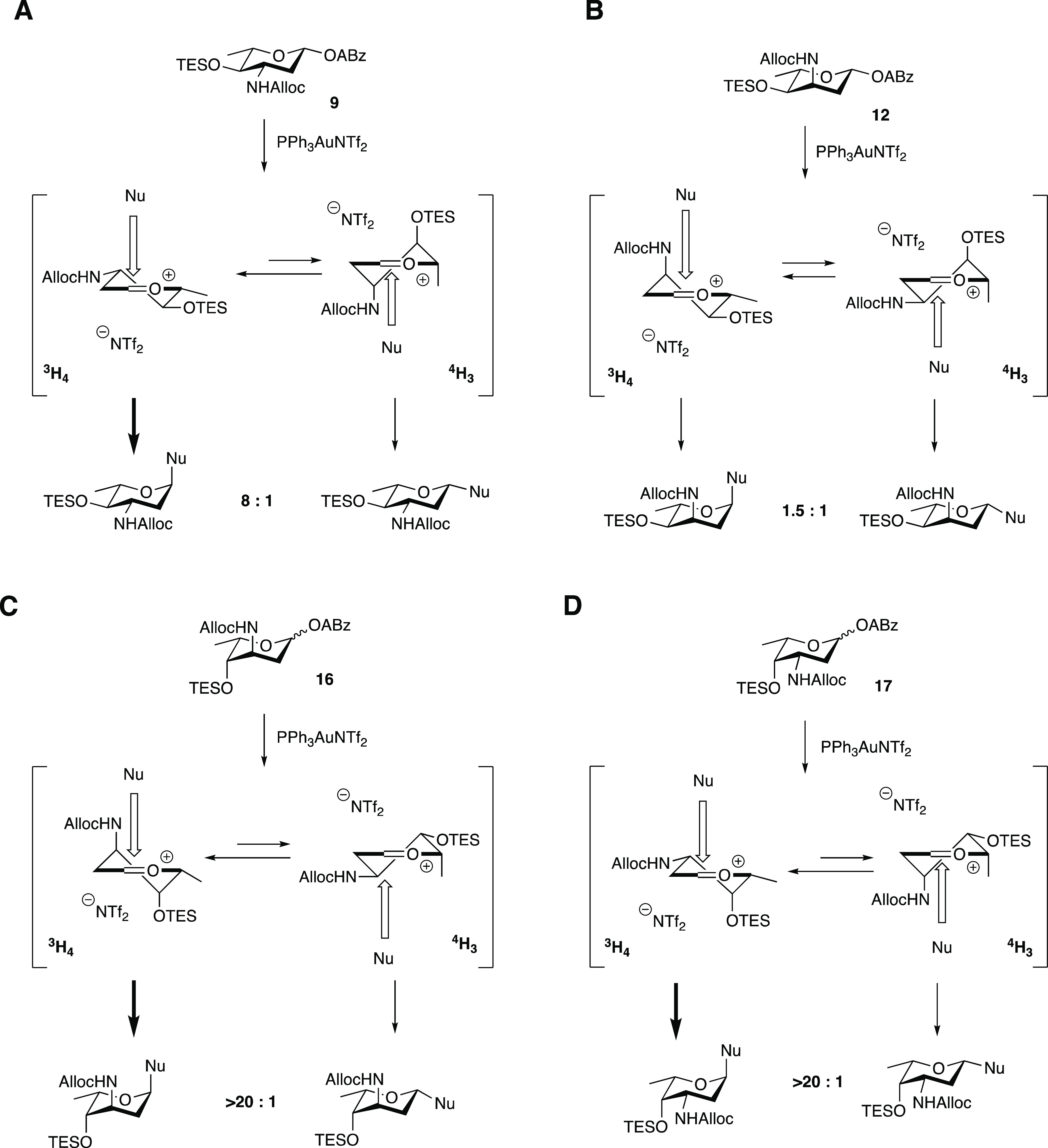
The equilibria between the 3H4 and 4H3 conformers of the oxocarbenium ions are indicated. The bold arrows indicate the most favorable product forming pathways. Nu = acceptor 18.
Alloc removal of 20 and subsequent desilylation delivered 4a, which was turned into its HCl salt for solubility. Reductive amination of the amine formed upon Alloc removal from 20 and desilylation delivered 4b. Using a similar sequence of reactions, 21 was transformed into 3a and 3b. Of note, NMR analysis of compound 4b indicated that the sugar ring adopts a 4C1 conformation, rather than the 1C4 conformation, taken up by its nonmethylated counterpart 4a (see Figure S4 for annotated NMR spectra). The observed conformation of the l-megosamine sugar moiety in 4b is consistent with that found in the macrolide megalomycin.29 As a result, the tertiary amine in 4b points away from the aglycone, and the overall shape of anthracycline 4b differs significantly from the other generated compounds.
DNA Damage Capacity and Histone Evicting Property of the (N,N-Dimethyl)doxorubicin Stereoisomers
Doxorubicin and its analogues used in the clinic have two main activities: DNA damage and chromatin damage.9,10 Dimethylation of the amine can separate these activities, but sugar epimers of doxorubicin have not been evaluated for this. Therefore, we evaluated our panel of (N,N-dimethyl)doxorubicin isomers for these biological activities. DNA double-strand break formation by the various compounds was determined indirectly by visualization of γH2AX (a post-translational modification on histone H2A that occurs as part of the DNA damage response) by Western blot analysis (Figure 2A,B).30 In addition, the degree of DNA breaks was assessed more directly using constant-field gel electrophoresis (CFGE, Figure 2C,D).
Figure 2.

DNA damage formation by the (N,N-dimethyl)doxorubicin stereoisomers (1a/b, 2a/b, 3a/b, 4a/b). K562 cells were treated for 2 h with 10 μM of the indicated compounds, and etoposide (10 μM) was used as a positive control. (A) DNA double-strand breaks were measured indirectly by visualization of the γH2AX levels by Western blot. Actin was used as a loading control, and molecular weight markers are indicated. (B) Quantification of the γH2AX signal normalized to actin, and relative to etoposide. Results are presented as mean ± standard deviation (SD) of three independent experiments. Ordinary one-way analysis of variance (ANOVA); *P < 0.05, **P < 0.01, ***P < 0.001. (C) DNA break formation by the various compounds was directly analyzed by CFGE. The position of intact and broken DNA is indicated. (D) Quantification of the fraction of broken DNA relative to etoposide. Results are presented as mean ± SD of four independent experiments. Ordinary one-way ANOVA; ****P < 0.0001.
The four anthracyclines bearing a free amine in their sugar, being doxorubicin (1a), epirubicin (2a), 3′-epi-doxorubicin (3a), and 3′,4′-epi-doxorubicin (4a), induced DNA breaks. For the analogues featuring a tertiary amine, DNA double-strand break formation was absent or reduced, compared to their primary amine counterparts. Dimethyldoxorubicin (1b) and dimethyl-epi-doxorubicin (2b) induced (almost) no DNA breaks, yet N,N-dimethyl-3′-epi-doxorubicin (3b) and N,N-dimethyl-3′,4′-epi-doxorubicin (4b) did produce DNA breaks, although significantly less so than their nonmethylated counterparts (Figure 2B,D). Overall, the orientation of the 4-OH function (1a vs 2a, 1b vs 2b, 3a vs 4a, 3b vs 4b) had little effect on their DNA damaging activity.
Since our previous findings indicate that chromatin damage, rather than DNA damage, is the most dominant cytotoxic mechanism of tumor killing by anthracycline drugs,10,12 we investigated the ability of our panel of compounds (1a/b, 2a/b, 3a/b, 4a/b) to induce histone eviction. To do so, part of the nucleus of MelJuSo cells stably expressing PAGFP-H2A was photoactivated, and release of these fluorescent histones was followed over time upon treatment with the different compounds (Figures 3A and S1). In all cases, the N,N-dimethylated variants (1b, 2b, 3b, and 4b) were more potent in evicting histones than their free amine counterparts (Figure 3A). Of the dimethylated compounds, N,N-dimethyl-3′-epidoxorubicin (3b) showed the lowest histone evicting activity, with 3′-epidoxorubicin (3a) being the only compound that failed to evict histones.
Figure 3.

Chromatin damage capacity and cytotoxicity of epimeric (N,N-dimethyl)doxorubicin analogues. (A) Quantification of histone eviction measured as PAGFP-H2A release from photoactivated nuclear regions after administration of 10 μM of the indicated doxorubicin isomers (in colors on right). Ordinary two-way ANOVA, Turkey’s multiple comparison test; ****P < 0.0001. (B) Cytotoxicity of 1a/b, 2a/b, 3a/b, 4a/b in A549 and FM3 cells. Cells were treated for 2 h with different concentrations of the indicated isomers followed by drug removal. Cell survival was determined 72 h post drug removal using CellTiter Blue. Colors correspond to the drugs shown in (A). Data are shown as mean ± SD from four independent experiments. (C) Color code table depicting the IC50 for compounds 1a/b, 2a/b, 3a/b, 4a/b determined for the 14 tumor cell lines tested. Red (high IC50 = low cytotoxicity) to yellow (medium IC50) to green (low IC50 = high cytotoxicity). IC50 for 4a vs 4b: Ordinary two-way ANOVA with Sidak’s multiple comparison test; ****P < 0.0001.
Subsequently, the cytotoxicity of the compounds was determined in a panel of 14 different tumor cell lines in vitro (Figures 3B,C and S2). With the exception of compound 3b, all compounds with tertiary amines have a lower IC50 value in the tested tumor cell lines than their nonmethylated counterparts. Furthermore, the compounds with the amine in an equatorial position (1a/b, 2a/b, and 4b) are effective at killing most of the cell lines, down to nanomolar concentrations, while the compounds with the amine in axial configuration (3a/b, 4a) show poor cytotoxicity. The difference in cytotoxicity between 3a versus 4a, and 3b versus 4b is remarkable. 3′-epi-Doxorubicin 3a and 3′,4′-epi-doxorubicin 4a both show poor cytotoxicity, and the dimethylated variant of 3a, compound 3b, is not more effective, while the dimethylated variant of 4a, being 4b, is significantly more cytotoxic (Figure 3C). Possibly, this is due to the fact that the sugar in N,N-dimethylated 4b exists in a different conformation than in 4a, placing the C-3 dimethylamino group in an equatorial orientation (similarly to 1a/b and 2a/b), pointing away from the aglycone rather than toward it (as for compounds 3a/b and 4a, Figure S4).
Another critical factor for the effectiveness of drugs is their ability to enter the cell. Since all variants in our library are fluorescent, this could be determined by flow cytometry. K562 and MeIJuSo cells were treated with compounds 1a/b, 2a/b, 3a/b, 4a/b and intracellular fluorescence was measured 2 h post treatment (Figure 4A,B). Compounds 1b, 2b, and 4b all featuring the N,N-dimethyl moiety are taken up much more efficiently than the corresponding primary amines, 1a, 2a, and 4a, respectively. This was not the case for 3a, which was taken up more efficiently than the other primary amine epimers. The difference in uptake with its dimethylated variant 3b is small. Overall, it can be concluded that the cytotoxicity of the here studied anthracyclines is mainly determined by their histone eviction effectivity (Figures 4C and S3A), which strongly correlates to the rate of uptake of the compounds (Figure 4A,B). Compounds featuring an N,N-dimethyl moiety are more effective histone evictors (Figure 4D) and are therefore more cytotoxic than the corresponding compounds having a primary amine. The orientation of the OH group at the 4′ position has very little effect on cytotoxicity (Figure S3A,B). Additionally, the stereochemistry of the fucose-carbon (C-3) bearing the amine functionality has a major influence on the IC50 values of the compounds in vitro. Compounds featuring an equatorial (or outward facing, in the case of 4b) amine are the most effective (1a/b, 2a/b, and 4b; Figure 4E).
Figure 4.

Structure–function relationship of our library of (N,N-dimethyl)doxorubicin isomers. (A, B) Uptake of the different isomers 2 h post treatment with 1 μM of the indicated compound for K562 (A) and MelJuSo (B) cells. Relative fluorescence to the parental compound doxorubicin is plotted for K562 and MelJuSo cells. Data are shown as mean ± SD. (C) Histone eviction speed (the time at which 25% of the initial signal is reduced) is correlated with IC50 of the various doxorubicin isomers. (D) N,N-Dimethylation of the sugar of the analogues enhances the histone eviction speed. (E) Equatorial positioning of the amine improves the cytotoxicity of the doxorubicin analogues. Two-tailed Pearson r correlation *p < 0.05, **p < 0.01.
Conclusions
Despite the widespread use of doxorubicin for the treatment of various cancers for nearly 5 decades, its structure–activity relationship is still not fully understood. Although doxorubicin is a very effective anticancer drug, its use is limited by cumulative cardiotoxicity and treatment-related secondary tumors. Chromatin damage by eviction of histones is a new mode of action of anthracyclines,9 which brings renewed interest to develop new doxorubicin analogues. We showed that the anthracyclines, N,N-dimethyldoxorubicin (2b) and aclarubicin, are unable to generate DNA breaks, yet induce chromatin damage via eviction of histones.10 These analogues remain equally potent to doxorubicin, but without the induction of cardiotoxicity and secondary tumor formation. Here, we synthesized a focused library of stereoisomers with respect to the 1,2-amino-alcohol characteristic for the daunosamine sugar within doxorubicin and the four possible stereoisomers both as primary and tertiary (dimethylated) amines. The set of doxorubicin analogues were obtained by glycosylating the doxorubicin aglycon with suitably protected alkynylbenzoate donors. The focused library was evaluated for histone eviction activity and DNA double-strand cleavage capacity. The doxorubicin isomers with the amine positioned axially show poor histone eviction activity and display limited cytotoxicity compared to their equatorial amine counterparts. The exception to this is 4b, in which the sugar moiety has shown to undergo a ring-flip in solution. Possibly, this configuration, having an outward-facing amine as a result of this conformational change, causes the observed activities. Remarkably, the N,N-dimethylated variants showed strongly improved cellular uptake, some up to 10-fold, compared to their nonmethylated counterparts. How anthracyclines are taken up by cells is unclear, but the N,N-dimethylation likely increases the basicity of the amine, as well as the log P, both of which could influence diffusion through the cell membrane. The rate of histone eviction correlates strongly with the cellular uptake, which influences their cytotoxicity. Further chemical modifications of anthracyclines aimed at improving cellular uptake will help in achieving cytotoxicity at lower concentrations.
We have previously shown that N,N-dimethyldoxorubicin (1b) completely abolishes DNA double-strand break formation while not affecting the ability to kill tumor cells, compared to doxorubicin (1a), which does induce DNA damage.10 This appears to be a general theme, as N,N-dimethylepirubicin (2b) also lacks DNA damage capacity and is more cytotoxic than epirubicin (2a). Because N,N-dimethylepirubicin (2b) displays potent antitumor activity in vitro, it makes us believe this compound could be an attractive lead for further development toward new, possibly more effective anthracyclines. More generally, we feel our results, based on the synthesis and evaluation of this focused library of close structural and stereochemical analogues, warrant the assessment of more such compound collections. These would feature, for instance, selected variations in the aglycon, in the sugar part (instead of stereoisomers as presented here also regio-isomers and/or glycosylated derivatives) and in the nature of the amine (next to methylation also other alkyl substituents). Thus, evaluating the chemical space around old anticancer drugs can detect new activities and improve these drugs, as illustrated by this study.
Experimental Section
Materials and Methods for Biological Evaluation
Chemicals
Doxorubicin and epirubicin were purchased from Accord Healthcare Limited, U.K., and etoposide was purchased from Pharmachemie, NL.
Cell Culture
K562 cells (B. Pang, Stanford University), HCT116 cells (T. van Hall, LUMC, The Netherlands), BXPC-3 cells (ATCC CRL-1687), PC3, and DU145 cells (C. Robson, Newcastle University, U.K.) were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 8% fetal calf serum (FCS). A549 cells (R. Bernards, NKI, The Netherlands), FM3 cells (D. Peeper, NKI, The Netherlands), U87 MG (ATCC HTB-14), U118 MG (ATCC HTB-15), U2Os cells (ATCC HTB-96), Hela cells (ATCC CCL-2), and SKBR3 (R. Beijersbergen, NKI, The Netherlands) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 8% FCS. BT474 cells (R. Beijersbergen, NKI, The Netherlands) were maintained in DMEM/F12 medium supplemented with 8% FCS. MelJuSo cells were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 8% FCS. MelJuSo cells stably expressing PAGFP-H2A were maintained in IMDM supplemented with 8% FCS and G-418, as described. Cell lines were maintained in a humidified atmosphere of 5% CO2 at 37 °C and regularly tested for the absence of mycoplasma.
Western Blot and Constant-Field Gel Electrophoresis (CFGE)
Cells were treated with drugs at indicated dose for 2 h. Subsequently, the drugs were removed by extensive washing and the cells were collected and processed immediately for the assays. The cells were lysed directly in sodium dodecyl sulfate (SDS) sample buffer (2% SDS, 10% glycerol, 5% β-mercaptoethanol, 60 mM Tris–HCl pH 6.8, and 0.01% bromophenol blue). Lysates were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) followed by Western blotting. Primary antibodies used for blotting: γH2AX (1:1000, 05-036, Millipore), β-actin (1:10 000, A5441, Sigma). DNA double-strand breaks were quantified by constant-field gel electrophoresis as described.31 Images were quantified using ImageJ software.
Microscopy
PAGFP-H2A photoactivation and time-lapse confocal imaging were performed as described9 on a Leica SP8 confocal microscope system, 63× lens, equipped with a climate chamber. Loss of fluorescence after different treatments was quantified using ImageJ software.
Cell Viability Assay
Cells were seeded into 96-well plates. Twenty-four hours after seeding, the cells were treated with indicated drugs for 2 h. Subsequently, the drugs were removed by extensive washing and cultured for an additional 72 h. Cell viability was measured using a CellTiter Blue viability assay (Promega). Relative survival was normalized to the untreated control and corrected for background signal.
Flow Cytometry for Measuring Drug Uptake in Cells
Cells were treated with 1 μM of the indicated compounds for 2 h. Samples were washed, collected, and fixed with paraformaldehyde. The samples were analyzed by flow cytometry using BD FACS Aria II, with a 561 nm laser and a 610/20 nm detector. Data were analyzed using FlowJo software.
Quantification and Statistical Analysis
Each experiment was assayed in triplicate, unless stated otherwise. All error bars denote SD. Statistical analyses were performed using Prism 8 software (GraphPad, Inc.). Two-tailed Pearson analysis was used to determine correlations, ns, not significant, *p < 0.05, **p < 0.01.
Experimental Details on the Synthesis of 2b–3a/b–4a/b
All reactions were carried out in oven-dried glassware under a nitrogen atmosphere, unless indicated otherwise. Chemicals were obtained from commercial sources and were used as received. Reactions involving HF·pyridine were carried out in high-density polyethylene (HDPE) tubes. Solvents used in reactions were dried on molecular sieves 4 or 3 Å. Reaction progress was monitored by thin-layer chromatography (TLC) on Merck F254 silica TLC plates visualized by 254 or 365 nm UV light and/or spraying with Hanessian’s stain (prepared by dissolving (NH4)6Mo7O24·4H2O (25 g/L) and (NH4)Ce(SO4)4·2H2O (10 g/L) in 10% aq. H2SO4). Column chromatography was carried out on silica gel (60 Å, 40–63 μm), obtained from screening devices BV. Neutralized silica gel was prepared by portion-wise suspending of silica gel (500 g) in H2O (1.7 L) containing NH4OH (25% solution, 100 mL), stirring for 30 min, filtering off, and drying the residue at 150 °C. Toluene, MeOH, and DCM were distilled prior to use in small-scale column chromatography (<0.5 mmol) and size-exclusion chromatography. Size-exclusion chromatography was carried out on a Sephadex LH-20 using a 1:1 MeOH/DCM mixture for elution. NMR spectra were recorded on a Bruker AV-400, AV-500, or AV-600 NMR spectrometer. Chemical shifts (d) are reported in parts per million (ppm), relative to tetramethylsilane (TMS) or residual solvent signals. Peaks were assigned using correlation spectroscopy (COSY) and heteronuclear single quantum coherence spectroscopy (HSQC). High-resolution mass spectrometry (HRMS) spectra were measured on a Waters Synapt G2-Si time-of-flight (TOF) mass spectrometer using an electrospray ion source (ESI) in positive mode (source voltage, 3.5 kV) and an internal lock mass LeuEnk [M + H]+ = 556.2771. Final compounds were lyophilized from 10% aqueous pyridine (v/v). Hydrochloride salts of final compounds were obtained by lyophilization of the free bases from aq. HCl (10–3 M, 1 mL/μmol, 2 equiv).
General Procedure A: Oxidative Hydrolysis of p-Methoxyphenyl Glycosides
To a solution of the glycoside in MeCN/H2O (0.03 M, 1:1 v/v) were added NaOAc (10 equiv) and Ag(DPAH)2·H2O (2.5 equiv). The reaction mixture was stirred for 1 h, diluted with sat. aq. NaHCO3, and extracted thrice with DCM. Combined organics were dried over MgSO4, and solvent was removed in vacuo. Column chromatography gave the crude hemiacetals.
General Procedure B: Steglich Esterification with o-Cyclopropylbenzoic Acid
To a solution of the hemiacetal in DCM (0.1 M) were added DIPEA (9 equiv), DMAP (1 equiv), EDCI·HCl (3.5 equiv), and freshly prepared o-cyclopropylethynylbenzoic acid (8) (3 equiv). After consumption of the starting hemiacetal, the mixture was diluted with DCM and washed with sat. aq. NaHCO3 and brine, dried over MgSO4, and concentrated in vacuo. Column chromatography gave the corresponding anomeric alkynylbenzoates.
General Procedure C: Glycosylation of Alkynylbenzoate Donors
To a solution of the alkynylbenzoate donor and 14-O-tert-butyldimethylsilyl-doxorubicinone (18) (1.5 equiv) in DCM (0.05 M) were added activated molecular sieves (4 Å), and the mixture was stirred for 30 min. Subsequently, a freshly prepared 0.1 M DCM solution of PPh3AuNTf2 (prepared by stirring 1:1 PPh3AuCl and AgNTf2 in DCM for 30 min) (0.1 equiv) in DCM was added dropwise. After stirring 30 min, the mixture was filtered and concentrated in vacuo. Column chromatography gave the desired anthracyclines.
General Procedure D: Global Desilylation
A solution of the amine in pyridine (0.01–0.05 M) was cooled to 0 °C. HF·pyridine (70 wt % HF, ± 3.8 mL/mmol starting material, 146 equiv) was added, and the reaction mixture was stirred for 1–5 h at this temperature. Solid NaHCO3 was added to quench, and the mixture was stirred until cessation of effervescence. Salts were then filtered off, and the filtrate was diluted with DCM (10 volumes), washed with H2O, dried over Na2SO4, and concentrated in vacuo. Column chromatography on neutralized silica gel gave the deprotected anthracyclines.
p-Methoxyphenyl-4-O-acetyl-3-azido-2,3-dideoxy-α-l-rhamnopyranoside (6) and p-Methoxyphenyl-3-azido-4-O-acetyl-2,3,6-trideoxy-β-l-ribohexapyranoside (10)
: A mixture of acetates 5 (24.8 g, 96.4 mmol) and p-methoxyphenol (12.6 g, 101 mmol, 1.05 equiv) was coevaporated thrice with toluene and subsequently dissolved in DCM (480 mL). Activated 4 Å molecular sieves were added, and the mixture was allowed to stir for 30 min. Thereafter, TMSOTf (1.70 mL, 9.6 mmol, 0.1 equiv) was added at 0 °C and the mixture was stirred for a further 3 h at that temperature. It was then filtered into sat. aq. NaHCO3, after which the organic layer was separated, washed with brine, dried over MgSO4, and concentrated in vacuo. Column chromatography (7:93 EtOAc/pentane) gave compound 6 as a white solid (11.8 g, 36.6 mmol, 50%) in addition to 10 as a clear oil (2.26 g, 7.03 mmol, 7%). Analytical data for 6: 1H NMR (400 MHz, chloroform-d) δ 7.03–6.92 (m, 2H), 6.92–6.78 (m, 2H), 5.47 (d, J = 2.7 Hz, 1H), 4.75 (t, J = 9.8 Hz, 1H), 4.07 (ddd, J = 12.3, 9.9, 5.0 Hz, 1H), 3.93 (dq, J = 9.8, 6.3 Hz, 1H), 3.77 (s, 3H), 2.36 (ddd, J = 13.3, 4.9, 1.1 Hz, 1H), 2.14 (s, 3H), 1.86 (td, J = 12.9, 3.5 Hz, 1H), 1.13 (d, J = 6.3 Hz, 3H). 13C{1H} NMR (101 MHz, chloroform-d) δ 170.1, 155.0, 150.4, 117.6, 114.7, 95.5, 76.8, 75.5, 66.7, 57.6, 55.7, 35.5, 20.9, 17.6. HRMS (ESI) m/z: [M + Na]+: calcd for C15H19N3O5Na: 344.1217; found 344.1233. Analytical data for 10: 1H NMR (400 MHz, chloroform-d) δ 6.98–6.93 (m, 2H), 6.84–6.79 (m, 2H), 5.29 (dd, J = 8.6, 2.3 Hz, 1H), 4.75 (dd, J = 8.8, 3.3 Hz, 1H), 4.27 (app q, J = 3.6 Hz, 1H), 4.09 (dq, J = 8.8, 6.3 Hz, 1H), 3.77 (s, 3H), 2.23 (ddd, J = 13.8, 4.4, 2.4 Hz, 1H), 2.15 (s, 3H), 2.15–2.05 (app m, 1H), 1.27 (d, J = 6.4 Hz, 3H). 13C{1H} NMR (101 MHz, chloroform-d) δ 170.2, 155.0, 150.9, 117.8, 114.5, 96.4, 74.0, 68.6, 57.4, 55.7, 35.1, 20.7, 18.1. HRMS (ESI) m/z: [M + H]+ calcd for C15H19N3O5: 344.12169, found 344.1223.
p-Methoxyphenyl-3-allyl-N-carbamate-2,3-dideoxy-4-O-triethylsilyl-α-
l-rhamnopyranoside (7): Acetate 11 (11.8 g, 37.2 mmol) was dissolved in MeOH (200 mL) and DCM (50 mL), NaOMe (400 mg, 7.40 mmol, 0.2 equiv) was added, and the reaction was stirred for 2 days. Dry ice was added to quench, and the reaction mixture was concentrated in vacuo. Column chromatography (25:75 EtOAc/pentane) afforded the title compound as a white solid (10.38 g, 3.72 mmol, quant.). 1H NMR (400 MHz, chloroform-d) δ 7.07–6.97 (m, 2H), 6.91–6.82 (m, 2H), 5.50 (d, J = 2.2 Hz, 1H), 3.99 (ddd, J = 12.1, 9.4, 4.9 Hz, 1H), 3.86 (dq, J = 9.4, 5.9 Hz, 1H), 3.81 (s, 3H), 3.25 (t, J = 9.4 Hz, 1H), 2.40 (ddd, J = 13.1, 4.9, 1.4 Hz, 1H), 2.24 (s, 1H), 1.89 (ddd, J = 13.2, 12.2, 3.5 Hz, 1H), 1.29 (d, J = 6.2 Hz, 3H). 13C{1H} NMR (101 MHz, chloroform-d) δ 154.8, 150.4, 117.6, 114.6, 95.6, 76.0, 68.3, 60.3, 55.7, 35.2, 17.8. HRMS (ESI) m/z: [M + H]+ calcd for C13H18N3O4: 280.1297; found 280.1292.
The alcohol (370 mg, 1.32 mmol) was dissolved in dimethylformamide (DMF) (2.23 mL) and pyridine (320 μL, 3.96 mmol, 3 equiv) and cooled to 0 °C. TESOTf (0.51 mL, 2.38 mmol, 1.8 equiv) was added, and the reaction mixture was stirred for 1.5 h at the same temperature. Et2O (80 mL) was added, and the reaction mixture was washed with H2O five times, dried over MgSO4, and concentrated in vacuo. Column chromatography (5:95 EtOAc/pentane) afforded the azide as a colorless oil (494 mg, 1.26 mmol, 95%). 1H NMR (400 MHz, chloroform-d) δ 7.04–6.98 (m, 2H), 6.89–6.82 (m, 2H), 5.47 (d, J = 2.3 Hz, 1H), 3.89 (ddd, J = 12.2, 9.2, 4.9 Hz, 1H), 3.85–3.81 (m, 1H), 3.80 (s, 3H), 3.19 (t, J = 9.2 Hz, 1H), 2.41 (ddd, J = 13.3, 4.9, 1.4 Hz, 1H), 1.88 (ddd, J = 13.2, 12.2, 3.5 Hz, 1H), 1.24 (d, J = 6.3 Hz, 3H), 1.03 (t, J = 7.9 Hz, 9H), 0.73 (qd, J = 7.9, 3.5 Hz, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 154.8, 150.5, 117.6, 114.6, 95.5, 76.4, 69.2, 61.4, 55.6, 35.7, 18.2, 6.9, 5.2. HRMS (ESI) m/z: [M + Na]+ calcd for C19H31N3O4SiNa: 416.1976; found 416.2671.
To a solution of the azide (1.57 g, 4.00 mmol) in THF/H2O (40 mL, 10:1 v/v) was added PPh3 (3.15 g, 20.0 mmol, 5 equiv), and the reaction mixture was stirred for 2 days at 50 °C. Solvent was removed in vacuo, and the residue was coevaporated with toluene twice. The crude amine was dissolved in DCM (30 mL) and pyridine (1.0 mL, 12.9 mmol, 3.2 equiv). Allyl chloroformate (0.64 mL, 24.0 mmol, 6 equiv) was added at −20 °C, and the resulting mixture was stirred for 2 h at that temperature. It was then poured into H2O and extracted with DCM thrice, combined organics were dried over MgSO4, and the solvent was removed in vacuo. Purification by column chromatography (0:100–5:95 EtOAc/pentane) afforded the title compound as a colorless oil (1.85 g, 4.00 mmol, quant.). 1H NMR (400 MHz, chloroform-d) δ 7.08–6.93 (m, 2H), 6.89–6.76 (m, 2H), 5.94 (ddt, J = 16.5, 11.0, 5.7 Hz, 1H), 5.41 (d, J = 2.3 Hz, 1H), 5.33 (d, J = 16.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.4 Hz, 1H), 4.59 (d, J = 5.7 Hz, 3H), 4.08 (dtd, J = 12.1, 9.2, 4.6 Hz, 1H), 3.83 (dq, J = 9.0, 6.3 Hz, 1H), 3.77 (s, 3H), 3.28 (t, J = 9.1 Hz, 1H), 2.32 (ddd, J = 13.3, 4.7, 1.5 Hz, 1H), 1.88 (t, J = 12.8 Hz, 1H), 1.21 (d, J = 6.3 Hz, 3H), 0.96 (t, J = 7.9 Hz, 9H), 0.62 (q, J = 7.7 Hz, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 169.7, 154.7, 150.9, 133.0, 118.0, 117.7, 114.6, 95.8, 76.6, 69.7, 65.7, 55.8, 51.0, 36.7, 18.6, 7.1, 5.5. HRMS (ESI) m/z: [M + Na]+ calcd for C22H24NO5Na: 474.2282; found 474.2289.
o-Cyclopropylethynylbenzoyl-3-N-allyloxycarbonyl-2,3-dideoxy-4-O-triethylsilyl-β-
l-rhamnopyranoside (9): Glycoside 7 (903 mg, 2.00 mmol) was hydrolyzed according to general procedure A. Column chromatography (30:70–50:50 EtOAc/pentane) afforded hemiacetal. The hemiacetal was esterified according to general procedure B. Column chromatography (0:100–15:85 Et2O/pentane) afforded the title compound as an off-white solid (498 mg, 0.97 mmol, 49%). 1H NMR (400 MHz, chloroform-d) δ 7.91 (dd, J = 8.0, 1.4 Hz, 1H), 7.53–7.43 (m, 1H), 7.40 (dd, J = 7.7, 1.4 Hz, 1H), 7.33–7.23 (m, 1H), 6.00 (dd, J = 9.6, 2.3 Hz, 1H), 5.93 (ddt, J = 16.5, 10.9, 5.7 Hz, 1H), 5.39–5.26 (m, 1H), 5.23 (d, J = 10.4 Hz, 1H), 4.73 (d, J = 8.8 Hz, 1H), 4.58 (d, J = 5.8 Hz, 2H), 3.76 (dtd, J = 12.1, 9.1, 4.7 Hz, 1H), 3.55 (dq, J = 8.6, 6.2 Hz, 1H), 3.33–3.22 (m, 1H), 2.40 (ddd, J = 12.7, 4.8, 2.4 Hz, 1H), 1.85 (q, J = 11.6 Hz, 1H), 1.51 (tt, J = 8.2, 6.1 Hz, 1H), 1.34 (d, J = 6.2 Hz, 3H), 0.96 (t, J = 7.9 Hz, 9H), 0.93–0.84 (m, 4H), 0.63 (q, J = 8.0 Hz, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 164.2, 155.5, 134.3, 134.1, 132.8, 132.1, 131.5, 131.1, 130.9, 127.4, 127.0, 125.2, 118.0, 99.8, 92.6, 75.7, 75.0, 74.5, 65.8, 53.3, 36.6, 18.6, 9.1, 8.9, 7.0, 5.4, 0.8. HRMS (ESI) m/z: [M + Na]+ calcd for C28H39NO6SiNa: 536.2444; found 536.2437.
7-[3-N-Allyloxycarbonyl-2,3-dideoxy-4-O-triethylsilyl-α-l-rhamnopyranoside]-14-O-tert-butyldimethylsilyldoxorubicinone (19)
: According to general procedure C, glycosyl donor 17 (411 mg, 0.800 mmol) was coupled to 14-O-tert-butyldimethylsilyl-doxorubicinone 18 (555 mg, 1.05 mmol, 1.3 equiv). Column chromatography (3:97–10:90 EtOAc/toluene) afforded the title compound as a red solid (545 mg, 0.64 mmol, 80%, 8:1 α/β). Spectral data for the α-anomer: 1H NMR (400 MHz, chloroform-d) δ 13.96 (d, J = 2.6 Hz, 1H), 13.26 (d, J = 2.8 Hz, 1H), 8.03 (d, J = 6.4 Hz, 1H), 7.78 (td, J = 8.2, 2.2 Hz, 1H), 7.39 (d, J = 7.7 Hz, 1H), 5.99–5.78 (m, 1H), 5.47–5.40 (m, 1H), 5.27 (d, J = 15.3 Hz, 2H), 5.18 (d, J = 10.6 Hz, 1H), 5.03–4.86 (m, 2H), 4.57 (s, 1H), 4.08 (d, J = 2.1 Hz, 3H), 3.82 (d, J = 7.5 Hz, 1H), 3.63 (d, J = 9.6 Hz, 1H), 3.29 (s, 1H), 3.21 (d, J = 18.7 Hz, 1H), 2.98 (dd, J = 19.3, 2.8 Hz, 1H), 2.34 (d, J = 14.8 Hz, 1H), 2.16 (d, J = 15.1 Hz, 1H), 2.10 (d, J = 13.4 Hz, 1H), 1.81 (s, 1H), 1.30 (dd, J = 6.3, 2.1 Hz, 3H), 1.04–0.91 (m, 19H), 0.62 (qd, J = 7.9, 2.1 Hz, 6H), 0.14 (d, J = 2.1 Hz, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 211.9, 187.3, 186.9, 161.2, 156.6, 156.1, 135.8, 134.5, 134.0, 121.1, 111.6, 111.5, 90.0, 77.6, 77.3, 77.2, 76.9, 66.9, 36.0, 34.2, 18.8, 5.5. HRMS (ESI) m/z: [M + Na]+ calcd for C43H61NO13Si2Na: 878.3574; found 878.3599.
N,N-Dimethyl-4′-epi-doxorubicin (2b)
: To a solution of 19 (135 mg, 0.158 mmol) and 1,3-dimethylbarbituric acid (74 mg, 0.47 mmol, 3 equiv) in DCM (15 mL) was portion-wise added Pd(PPh3)4 (18.5 mg, 0.016 mmol, 0.1 equiv). The reaction mixture was stirred overnight, and the solvent was removed in vacuo. The residue was submitted to column chromatography (3:97–15:85 acetone/toluene) to afford the free amine as a red solid. 1H NMR (400 MHz, chloroform-d) δ 13.93 (s, 1H), 8.00 (dd, J = 7.8, 1.1 Hz, 1H), 7.76 (dd, J = 8.4, 7.8 Hz, 1H), 7.39 (dd, J = 8.6, 1.1 Hz, 1H), 5.43 (d, J = 3.8 Hz, 1H), 5.25 (dd, J = 4.1, 2.1 Hz, 1H), 5.03–4.82 (m, 2H), 4.73 (s, 1H), 4.08 (s, 3H), 3.73 (dq, J = 8.9, 6.2 Hz, 1H), 3.16 (dd, J = 19.0, 2.0 Hz, 1H), 3.06 (t, J = 8.9 Hz, 1H), 2.92 (d, J = 18.9 Hz, 1H), 2.87 (dt, J = 8.7, 4.1 Hz, 1H), 2.34 (dt, J = 14.7, 2.2 Hz, 1H), 2.18–2.05 (m, 1H), 1.95 (dd, J = 13.4, 4.4 Hz, 1H), 1.62–1.54 (m, 1H), 1.29 (d, J = 6.3 Hz, 3H), 1.05–0.93 (m, 18H), 0.69 (q, J = 8.1 Hz, 6H), 0.14 (s, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 211.7, 187.1, 186.7, 161.1, 156.5, 155.9, 135.8, 135.6, 134.4, 134.1, 120.9, 119.9, 118.5, 111.5, 111.3, 100.7, 80.4, 70.0, 69.6, 66.8, 56.8, 50.3, 37.9, 35.8, 34.0, 26.0, 18.7, 18.4, 7.1, 5.7, −5.2. HRMS (ESI) m/z: [M + H]+ calcd for C39H59NO11Si2: 772.3543; found 772.3568.
To a solution of the amine in EtOH (10 mL) were added formaldehyde solution (aqueous 37% w/v, 0.31 mL, 3.9 mmol, 30 equiv) and sodium trisacetoxyborohydride (52 mg, 0.24 mmol, 1.95 equiv). After stirring for 1.5 h, the reaction mixture was partitioned between DCM and sat. aq. NaHCO3 and extracted thrice with DCM. Combined organics were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (1:200–10:90 acetone/toluene) afforded the title compound as a red solid (77 mg, 96 μmol, 61% over two steps). 1H NMR (400 MHz, chloroform-d) δ 13.97 (s, 1H), 13.26 (s, 1H), 8.02 (d, J = 7.6 Hz, 1H), 7.78 (t, J = 7.9 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 5.50 (d, J = 3.9 Hz, 1H), 5.25 (d, J = 3.1 Hz, 1H), 5.06–4.85 (m, 2H), 4.79 (s, 1H), 4.09 (s, 3H), 3.82–3.68 (m, 1H), 3.32–3.12 (m, 2H), 3.00 (d, J = 18.8 Hz, 1H), 2.51 (d, J = 13.4 Hz, 1H), 2.37 (d, J = 14.7 Hz, 1H), 2.14 (d, J = 4.6 Hz, 6H), 1.89–1.79 (m, 1H), 1.60 (td, J = 12.9, 4.2 Hz, 1H), 1.31 (d, J = 6.1 Hz, 3H), 1.02–0.91 (m, 18H), 0.60 (qd, J = 7.6, 3.4 Hz, 6H), 0.14 (s, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 211.8, 187.3, 186.8, 161.1, 156.6, 156.1, 135.8, 135.7, 134.6, 134.3, 121.1, 119.9, 118.5, 111.5, 111.4, 101.5, 73.6, 71.2, 69.8, 66.8, 61.8, 56.8, 40.7, 35.8, 34.0, 26.2, 26.0, 18.8, 18.7, 7.2, 5.6, −5.3. HRMS (ESI) m/z: [M + H]+ calcd for C41H62NO11Si2: 800.3856; found 800.3888.
According to general procedure D, the above dimethylamine (33 mg, 41 μmol) was desilylated. Column chromatography on neutral silica (0:100–20:80 MeOH/DCM) afforded the title compound as a red solid (24 mg, 41 μmol, quant.). 1H NMR (500 MHz, chloroform-d) δ 13.95 (s, 1H), 13.16 (s, 1H), 7.99 (dd, J = 7.7, 1.1 Hz, 1H), 7.77 (dd, J = 8.5, 7.6 Hz, 1H), 7.39 (dd, J = 8.6, 1.1 Hz, 1H), 5.56 (d, J = 3.9 Hz, 1H), 5.29 (s, 1H), 5.26 (dd, J = 4.2, 2.1 Hz, 1H), 4.88–4.71 (m, 2H), 4.08 (s, 3H), 3.80 (dq, J = 9.0, 6.2 Hz, 1H), 3.23–3.12 (m, 2H), 2.90 (d, J = 18.7 Hz, 1H), 2.63 (ddd, J = 13.3, 9.9, 3.8 Hz, 1H), 2.38 (dt, J = 14.8, 2.2 Hz, 1H), 2.20 (s, 6H), 2.17 (dd, J = 14.7, 4.1 Hz, 1H), 1.84 (ddd, J = 13.2, 4.0, 1.3 Hz, 1H), 1.63 (td, J = 13.0, 4.2 Hz, 1H), 1.35 (d, J = 6.2 Hz, 3H). 13C{1H} NMR (126 MHz, chloroform-d) δ 214.2, 187.2, 186.7, 161.1, 156.4, 155.8, 135.9, 135.5, 133.9, 133.9, 120.9, 120.0, 118.6, 111.6, 111.4, 101.5, 76.8, 71.5, 70.8, 69.8, 65.7, 61.9, 56.8, 40.0, 35.7, 34.0, 25.9, 18.4. HRMS (ESI) m/z: [M + H]+ calcd for C29H35NO11: 572.2126; found 572.2134.
p-Methoxyphenyl-3-epi-azido-α-l-daunosamine (14)
: Anomeric acetate 13 (7.92 g, 24.8 mmol) and p-methoxyphenol (4.62 g, 37.2 mmol, 1.5 equiv) were coevaporated with toluene. DCM (880 mL) and activated molecular sieves (4 Å) were added, and the reaction was stirred for 30 min at room temperature. BF3·OEt2 (7.65 mL, 62 mmol, 2.5 equiv) was added at −60 °C, and the reaction mixture was allowed to gradually warm up from −60 to −40 °C over 2.5 h. The reaction mixture was poured into a saturated aqueous solution of NaHCO3, extracted with DCM, washed twice with NaOH, dried over MgSO4, and concentrated in vacuo. The residue was dissolved in MeOH (42 mL), to which NaOMe (284 mg, 5.0 mmol, 0.2 equiv) was added after which the reaction mixture was stirred for 4 days. Dry ice was added, and the reaction mixture was concentrated in vacuo. Column chromatography (8:92–40:60 EtOAc/pentane) afforded the title compound as a white solid (3.98 g, 15.7 mmol, 70%). 1H NMR (400 MHz, chloroform-d) δ 7.13–6.94 (m, 2H), 6.94–6.73 (m, 2H), 5.46 (dd, J = 4.3, 1.4 Hz, 1H), 4.43 (qd, J = 6.7, 1.4 Hz, 1H), 4.00 (dt, J = 4.2, 3.1 Hz, 1H), 3.80 (s, 3H), 3.43 (s, 1H), 2.35 (dt, J = 15.3, 4.4 Hz, 1H), 2.26–2.05 (m, 2H), 1.22 (d, J = 6.7 Hz, 3H). 13C{1H} NMR (101 MHz, chloroform-d) δ 154.8, 151.0, 117.8, 114.6, 95.3, 69.0, 62.7, 57.1, 55.7, 29.7, 27.0, 16.3. HRMS (ESI) m/z: [M + H]+ calcd for C13H18N3O4: 280.1297; found 280.1292.
p-Methoxyphenyl-3-epi-N-allyloxycarbonyl-4-O-triethylsilyl-α-l-daunosamine (15)
: To a solution of 14 (1.22 g, 4.38 mmol) in DMF (7.4 mL) and pyridine (1.06 mL, 13.1 mmol, 3 equiv) was added TESOTf (1.8 mL, 7.88 mmol, 1.8 equiv) at 0 °C, and the reaction mixture was stirred for 1 h. It was subsequently poured into EtOAc, washed with H2O, dried over MgSO4, and concentrated in vacuo. The residue was dissolved in THF/H2O (165 mL, 10:1 v/v), PPh3 (2.30 g, 8.76 mmol, 2 equiv) was added, and the reaction mixture was stirred for 3 days after which it was concentrated in vacuo. The amine thus obtained was coevaporated with toluene thrice and dissolved in DCM (31.4 mL) and pyridine (1.1 mL, 14 mmol, 3.2 equiv). Allyl chloroformate (0.70 mL, 6.6 mmol, 1.5 equiv) was added at −25 °C, and the reaction mixture was stirred at that temperature for 1 h. The mixture was left to warm to room temperature and diluted with DCM, washed with H2O, dried over MgSO4, and concentrated in vacuo. Purification by column chromatography (2:98:1–40:60:1 Et2O/pentane/Et3N) afforded the title compound as a colorless oil (1.98 g, 4.38 mmol, quant. over three steps). 1H NMR (400 MHz, chloroform-d) δ 7.07–6.98 (m, 2H), 6.91–6.77 (m, 2H), 6.29 (d, J = 7.8 Hz, 1H), 5.96 (ddt, J = 17.2, 10.3, 5.6 Hz, 1H), 5.48 (d, J = 3.7 Hz, 1H), 5.41–5.16 (m, 2H), 4.72–4.50 (m, 2H), 4.26–4.13 (m, 1H), 3.90 (ddt, J = 7.9, 5.4, 2.7 Hz, 1H), 3.78 (s, 3H), 3.53 (d, J = 3.4 Hz, 1H), 2.52–2.32 (m, 1H), 1.79 (ddt, J = 14.4, 2.4, 1.2 Hz, 1H), 1.16 (d, J = 6.5 Hz, 3H), 1.06–0.95 (m, 9H), 0.80–0.64 (m, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 155.7, 155.1, 150.7, 133.1, 118.0, 117.9, 114.7, 97.6, 69.4, 65.7, 63.4, 55.8, 49.1, 27.9, 17.2, 7.0, 4.9. HRMS (ESI) m/z: [M + Na]+ calcd for C19H31N3O4SiNa: 474.2282; found 474.2289.
o-Cyclopropylethynylbenzoyl-3-epi-N-allyloxycarbonyl-4-O-triethylsilyl-l-daunosamine (16)
: Glycoside 15 (990 mg, 2.19 mmol) was hydrolyzed according to general procedure A. The hemiacetal thus obtained was esterified according to general procedure B. Column chromatography (0:100–15:85 Et2O/pentane) afforded the title compound as a pale-yellow oil (879 mg, 1.71 mmol, 79%, 3:1 α/β). Spectral data for the α-anomer: 1H NMR (400 MHz, chloroform-d): δ 7.96 (dd, J = 8.0, 1.4 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H, NH), 7.47 (dt, J = 7.5, 1.9 Hz, 1H), 7.44–7.38 (m, 1H), 7.34–7.27 (m, 1H), 6.09 (dd, J = 9.2, 2.5 Hz, 1H), 5.93 (ddd, J = 16.2, 10.8, 5.4 Hz, 1H), 5.37–5.22 (m, 2H), 4.59 (s, 2H), 4.00 (dt, J = 6.5, 2.9 Hz, 2H), 3.57 (s, 1H), 2.42–2.34 (m, 1H), 1.88–1.78 (m, 1H), 1.31–1.27 (m, 3H), 1.27–1.23 (m, 1H), 0.98 (t, J = 7.9 Hz, 9H), 0.91–0.88 (m, 4H), 0.73–0.65 (m, 8H). 13C{1H} NMR (100 MHz, chloroform-d): δ 164.7, 155.6, 134.9, 134.3, 132.1, 130.8, 127.2, 99.9, 91.8, 74.6, 71.5, 69.3, 65.9, 50.7, 30.6, 17.0, 9.0, 7.1, 4.85, 0.8. HRMS (ESI) m/z: [M + Na]+ calcd for C28H39NO6SiNa: 536.24389; found 536.24362.
7-[3-epi-N-Allyloxycarbonyl-4-O-triethylsilyl-α-l-daunosamino]-14-O-tert-butyldimethylsilyldoxorubicinone (21)
: According to general procedure C, glycosyl donor 16 (384 mg, 0.769 mmol) was coupled to 14-O-tert-butyldimethylsilyl-doxorubicinone 18 (593 mg, 1.12 mmol, 1.5 equiv). Column chromatography (2:98–10:90 acetone/toluene) and size-exclusion chromatography (Sephadex LH-20, 1:1 DCM/MeOH v/v) afforded the title compound as a red solid (368 mg, 0.43 mmol, 56%). 1H NMR (400 MHz, chloroform-d) δ 13.95 (s, 1H), 13.24 (s, 1H), 7.99 (dd, J = 7.7, 1.0 Hz, 1H), 7.76 (t, J = 8.1 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 6.71 (d, J = 7.7 Hz, 1H), 6.00–5.80 (m, 1H), 5.50 (d, J = 3.7 Hz, 1H), 5.40–5.01 (m, 3H), 4.74 (s, 2H), 4.51 (dt, J = 5.5, 1.5 Hz, 2H), 4.20–4.13 (m, 1H), 4.11 (s, 1H), 4.08 (s, 3H), 3.81–3.71 (m, 1H), 3.49 (d, J = 3.6 Hz, 1H), 3.19 (d, J = 18.8 Hz, 1H), 3.02 (d, J = 18.8 Hz, 1H), 2.39–2.09 (m, 3H), 1.21 (d, J = 6.5 Hz, 3H), 0.96 (d, J = 24.0 Hz, 19H), 0.70 (q, J = 7.7 Hz, 6H), 0.15 (d, J = 1.3 Hz, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 210.9, 187.1, 186.7, 161.1, 156.8, 155.8, 135.8, 135.6, 135.1, 133.7, 133.3, 121.0, 119.8, 118.5, 117.3, 111.5, 111.2, 99.4, 76.1, 69.5, 67.0, 66.9, 65.3, 63.6, 56.8, 48.7, 35.2, 33.5, 27.4, 25.9, 18.5, 17.2, 7.1, 4.9, −5.3. HRMS (ESI) m/z: [M + Na]+ calcd for C43H61NO13Si2Na: 878.35740; found 878.35817.
3′-epi-Doxorubicin (3a)
: To a solution of 21 (46 mg, 54 μmol) in DCM (5 mL) were added 1,3-dimethylbarbituric acid (25 mg, 0.16 mmol, 3 equiv) and Pd(PPh3)4 (27 mg, 5.4 μmol, 0.1 equiv). The reaction mixture was stirred for 1.5 h, after which solvent was removed in vacuo. The residue was submitted to column chromatography (0:100–10:90 acetone/toluene) to afford the crude amine. The crude amine was desilylated according to general procedure D. Column chromatography on neutral silica (0:100–50:50 MeOH/DCM) afforded the title compound as a red solid (18 mg, 33 μmol, 61% over two steps). 1H NMR (500 MHz, chloroform-d) δ 13.93 (s, 1H), 8.04 (dd, J = 7.7, 1.1 Hz, 1H), 7.79 (dd, J = 8.5, 7.7 Hz, 1H), 7.39 (dd, J = 8.6, 1.1 Hz, 1H), 5.46–5.37 (m, 1H), 5.25 (dd, J = 3.7, 2.4 Hz, 1H), 4.79 (s, 2H), 4.59–4.42 (m, 1H), 4.09 (s, 3H), 3.79–3.66 (m, 1H), 3.08 (d, J = 18.9 Hz, 1H), 2.39 (dt, J = 14.7, 2.2 Hz, 1H), 2.21–2.04 (m, 2H), 1.94–1.80 (m, 2H), 1.46 (d, J = 12.1 Hz, 2H), 1.29 (d, J = 6.7 Hz, 3H). 13C{1H} NMR (126 MHz, chloroform-d) δ 214.8, 187.2, 186.8, 161.2, 156.3, 156.0, 135.8, 135.7, 134.6, 134.0, 121.1, 119.9, 118.5, 111.6, 111.3, 100.0, 76.4, 72.0, 68.3, 65.7, 62.9, 48.7, 35.5, 34.1, 31.2, 16.4. HRMS (ESI) m/z: [M + H+] calcd for C27H30NO11: 544.1813; found 544.1812.
N,N-Dimethyl-3′-epidoxorubicin (3b)
: To a solution of 21 (34 mg, 40 μmol) in DCM (4.0 mL) was added 1,3-dimethylbarbituric acid (19 mg, 119 μmol, 3 equiv). Pd(PPh3)4 (5 mg, 4 μmol, 0.1 equiv) was added, and the reaction mixture was stirred for 1 h, after which it was concentrated in vacuo. The residue was submitted to column chromatography (0:100–15:85 acetone/toluene) to give the crude amine. To a solution of this amine in EtOH (2.5 mL) were added formaldehyde (aqueous 37% w/v solution, 0.1 mL, mmol, 30 equiv) and sodium trisacetoxyborohydride (14 mg, 66 μmol, 1.65 equiv). The reaction mixture was stirred for 2 h and partitioned between DCM and sat. aq. NaHCO3. The aqueous layer was extracted with DCM, combined organics were dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (0:100–10:90 acetone in toluene) afforded the dimethylamine as a red solid (23 mg, 29 μmol, 72%). 1H NMR (400 MHz, chloroform-d) δ 13.85 (s, 1H), 13.31 (s, 1H), 8.02 (dd, J = 7.7, 1.1 Hz, 1H), 7.76 (dd, J = 8.5, 7.7 Hz, 1H), 7.38 (dd, J = 8.6, 1.2 Hz, 1H), 5.34 (d, J = 3.7 Hz, 1H), 5.18 (dd, J = 3.6, 2.3 Hz, 1H), 5.00–4.90 (m, 2H), 4.24 (h, J = 5.7 Hz, 1H), 4.08 (s, 3H), 3.77–3.71 (m, 1H), 3.44–3.26 (m, 1H), 3.22–2.98 (m, 2H), 2.39–2.26 (m, 1H), 2.15 (s, 6H), 2.12–1.98 (m, 3H), 1.82–1.70 (m, 1H), 1.21 (d, J = 6.5 Hz, 3H), 1.03–0.93 (m, 19H), 0.71–0.56 (m, 6H), 0.14 (d, J = 3.7 Hz, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 213.0, 187.2, 186.8, 161.1, 156.5, 156.3, 135.8, 135.7, 135.4, 134.7, 121.2, 119.8, 118.4, 111.3, 111.1, 99.9, 76.8, 68.9, 67.7, 66.8, 64.7, 64.0, 56.8, 44.0, 35.7, 34.0, 26.0, 25.6, 18.8, 17.5, 7.1, 5.1, −5.1, −5.3. HRMS (ESI) m/z: [M + H]+ calcd for C41H62NO11Si2: 800.3856; found 800.3880.
According to general procedure D, the above dimethylamine (19 mg, 24 μmol) was desilylated. Column chromatography on neutral silica (0:100–20:80 MeOH:DCM) afforded the title compound as a red solid (9.0 mg, 16 μmol, 66%). 1H NMR (500 MHz, chloroform-d) δ 13.95 (s, 1H), 13.27 (s, 1H), 8.04 (dd, J = 7.7, 1.1 Hz, 1H), 7.80 (dd, J = 8.5, 7.7 Hz, 1H), 7.41 (dd, J = 8.6, 1.1 Hz, 1H), 5.36 (dd, J = 5.5, 3.9 Hz, 1H), 5.28 (dd, J = 3.7, 2.3 Hz, 1H), 4.80 (s, 2H), 4.38 (qd, J = 6.8, 3.4 Hz, 1H), 4.10 (s, 3H), 3.74 (dd, J = 6.6, 3.4 Hz, 1H), 3.23 (dd, J = 18.9, 2.0 Hz, 1H), 3.04 (d, J = 18.8 Hz, 1H), 2.52–2.41 (m, 2H), 2.23 (s, 6H), 2.11 (dd, J = 14.7, 3.8 Hz, 1H), 1.98 (dt, J = 14.0, 3.8 Hz, 1H), 1.60 (ddd, J = 13.7, 8.0, 5.5 Hz, 1H), 1.32 (d, J = 6.8 Hz, 3H). 13C{1H} NMR (126 MHz, chloroform-d) δ 215.0, 187.2, 186.8, 161.1, 156.3, 156.0, 135.7, 134.3, 134.3, 121.0, 119.9, 111.5, 111.3, 99.7, 77.4, 77.1, 76.9, 76.4, 76.4, 68.5, 68.2, 66.9, 65.7, 62.6, 56.8, 35.6, 34.0, 26.4, 26.4, 15.1. HRMS (ESI) m/z: [M + H]+ calcd for C29H35NO11: for 572.2126; found 572.2131.
p-Methoxyphenyl-3-N-allyloxycarbonyl-4-O-triethylsilyl-2,3,6-trideoxy-3-azido-β-l-ribohexapyranoside (11)
: To a solution of 10 (2.26 g, 7.03 mmol) in MeOH (120 mL) was added sodium methoxide (76 mg, 1.41 mmol, 0.2 equiv). After stirring for 2 days, the reaction mixture was quenched by addition of dry ice. Solvent was removed in vacuo and coevaporated thrice with toluene. The residue was purified by column chromatography (10:90–50:50 Et2O:pentane) to afford the alcohol as a pale-yellow oil (1.96 g, 7.03 mmol, quant.). 1H NMR (400 MHz, chloroform-d) δ 7.03–6.89 (m, 2H), 6.90–6.76 (m, 2H), 5.27 (dd, J = 8.9, 2.3 Hz, 1H), 4.11 (q, J = 3.7 Hz, 1H), 3.80 (dt, J = 8.7, 6.3 Hz, 1H), 3.76 (s, 3H), 3.48 (td, J = 8.3, 3.6 Hz, 1H), 2.28 (ddd, J = 13.8, 4.2, 2.3 Hz, 1H), 2.13–1.99 (m, 1H), 1.32 (d, J = 6.3 Hz, 3H). 13C{1H} NMR (101 MHz, chloroform-d) δ 155.0, 151.1, 117.7, 114.6, 96.3, 72.4, 70.8, 60.7, 55.7, 35.1, 18.2. HRMS (ESI) m/z: [M + Na]+ calcd for C13H17N3O4Na: 302.1111; found 302.1118.
To a solution of the above alcohol (1.12 g, 4.00 mmol) in pyridine (20 mL) was added TESOTf (1.29 mL, 1.59 g, 6.00 mmol, 1.5 equiv). The reaction mixture was stirred for 30 min and then diluted with DCM, washed twice with 1 M HCl, sat. aq. NaHCO3 and brine, dried over MgSO4, and concentrated in vacuo. Purification by column chromatography (0:100–7:93 Et2O:pentane) afforded the title compound as a colorless oil (1.57 g, 4.0 mmol, quant.). 1H NMR (400 MHz, chloroform-d) δ 6.97–6.90 (m, 2H), 6.86–6.75 (m, 2H), 5.25 (dd, J = 9.0, 2.3 Hz, 1H), 3.98 (q, J = 3.5 Hz, 1H), 3.96–3.87 (m, 1H), 3.77 (s, 3H), 3.62 (dd, J = 8.7, 3.3 Hz, 1H), 2.17 (ddd, J = 13.6, 4.0, 2.3 Hz, 1H), 1.96 (ddd, J = 13.6, 9.0, 3.4 Hz, 1H), 1.28 (d, J = 6.3 Hz, 3H), 1.00 (t, J = 7.9 Hz, 9H), 0.68 (qd, J = 8.3, 7.9, 1.7 Hz, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 155.0, 151.2, 117.8, 114.6, 96.5, 74.8, 70.8, 60.8, 55.8, 35.8, 18.5, 7.0, 5.0. HRMS (ESI) m/z: [M + Na]+ calcd for C19H31N3O4SiNa: 416.1976; found 416.1983.
To a solution of the above azide (1.57 g, 4.00 mmol) in THF/H2O (40 mL, 10:1 v/v) was added PPh3 (2.10 g, 8.00 mmol, 2 equiv), and the reaction mixture was stirred overnight at 50 °C. Solvent was removed in vacuo, and the residue was coevaporated with toluene twice. The crude amine was dissolved in DCM (30 mL), and pyridine (1 mL) to which allyl chloroformate (0.64 mL, 24 mmol, 6 equiv) was added at −20 °C. This mixture was stirred for 1 h at this temperature. It was then poured into H2O and extracted with DCM thrice, organic layers dried over MgSO4, and concentrated in vacuo. Purification by column chromatography (1:200 Et3N:pentane) afforded the title compound as a colorless oil (1.81 g, 4.00 mmol, quant.). 1H NMR (400 MHz, chloroform-d) δ 7.03–6.91 (m, 2H), 6.87–6.70 (m, 2H), 5.93 (ddt, J = 17.2, 10.4, 5.7 Hz, 1H), 5.43–5.25 (m, 2H), 5.22 (dq, J = 10.4, 1.4 Hz, 1H), 5.04 (s, 1H), 4.65–4.45 (m, 2H), 4.11 (q, J = 4.9, 4.2 Hz, 1H), 3.77 (s, 4H), 3.63 (dd, J = 6.7, 4.0 Hz, 1H), 2.51 (dd, J = 14.0, 6.3 Hz, 1H), 1.90 (ddd, J = 13.5, 7.0, 3.7 Hz, 1H), 1.28 (d, J = 6.5 Hz, 3H), 0.96 (t, J = 7.9 Hz, 9H), 0.63 (q, J = 7.7 Hz, 6H). 13C{1H} NMR (101 MHz, chloroform-d) δ 156.3, 154.8, 151.4, 132.9, 117.9, 117.6, 114.6, 97.0, 72.6, 71.9, 65.7, 55.8, 48.4, 19.2, 6.9, 5.0. HRMS (ESI) m/z: [M + Na]+ calcd for C23H37NO6SiNa: 474.2282; found 474.2290.
o-Cyclopropylethynylbenzoyl-3-N-allyloxycarbonyl-4-O-triethylsilyl-2,3,6-trideoxy-l-ribohexapyranoside (12)
: Glycoside 11 (1.71 g, 3.80 mmol) was hydrolyzed according to general procedure A. Column chromatography (90:10 EtOAc/pentane) afforded the hemiacetal. The resulting hemiacetal was esterified according to general procedure B. Column chromatography (0:100–10:90 Et2O:pentane) afforded the title compound as a pale-yellow oil (1.11 g, 2.20 mmol, 57% over two steps, 1:3 α/β). Spectral data for the β-anomer: 1H NMR (500 MHz, chloroform-d) δ 7.91 (dd, J = 8.0, 1.3 Hz, 1H), 7.47 (dd, J = 7.9, 1.3 Hz, 1H), 7.41 (tt, J = 7.7, 1.2 Hz, 1H), 7.33–7.27 (m, 1H), 6.27 (dd, J = 6.6, 2.8 Hz, 1H), 5.93 (dddd, J = 18.0, 10.5, 6.1, 5.2 Hz, 1H), 5.32 (dq, J = 17.2, 1.3 Hz, 1H), 5.23 (dq, J = 10.4, 1.3 Hz, 1H), 5.06 (s, 1H), 4.59 (q, J = 6.8, 6.2 Hz, 2H), 4.15 (tq, J = 6.6, 3.7 Hz, 1H), 3.91 (p, J = 6.5 Hz, 1H), 3.67 (dd, J = 6.1, 3.9 Hz, 1H), 2.55–2.39 (m, 1H), 1.93 (ddd, J = 13.5, 6.6, 3.8 Hz, 1H), 1.53 (tt, J = 8.1, 5.6 Hz, 1H), 1.32 (d, J = 6.6 Hz, 3H), 0.97 (dd, J = 8.3, 7.5 Hz, 9H), 0.92–0.83 (m, 4H), 0.63 (q, J = 7.8 Hz, 6H). 13C{1H} NMR (126 MHz, chloroform-d) δ 164.6, 156.2, 134.4, 132.9, 131.9, 131.2, 130.6, 127.1, 125.2, 117.9, 99.8, 92.1, 74.6, 73.6, 71.5, 65.8, 47.6, 32.2, 19.3, 9.0, 6.9, 5.0, 0.8. HRMS (ESI) m/z: [M + Na]+ calcd for C28H39NO6SiNa: 536.2434; found 536.2450.
7-[3-Allyl-N-allyloxycarbonyl-4-O-triethylsilyl-2,3,6-trideoxy-l-ribohexapyranoside]-14-O-tert-butyldimethylsilyldoxorubicinone (20)
: According to general procedure C, glycosyl donor 12 (128 mg, 0.240 mmol) was coupled to 14-O-tert-butyldimethylsilyldoxorubicinone 18 (198 mg, 0.37 mmol, 1.5 equiv). Column chromatography (0:100–5:95 EtOAc/pentane to 1:99–3:97 acetone/toluene) gave the title compound as a red solid (103 mg, 0.120 mmol, 50%, 1.5:1 α/β). Spectral data for the α-anomer: 1H NMR (500 MHz, chloroform-d) δ 13.92 (s, 1H), 13.22 (s, 1H), 7.98 (dd, J = 7.7, 1.1 Hz, 1H), 7.74 (t, J = 8.1 Hz, 1H), 7.37 (d, J = 8.5 Hz, 1H), 5.82 (ddt, J = 17.3, 10.6, 5.4 Hz, 1H), 5.71 (d, J = 6.7 Hz, 1H), 5.40 (dd, J = 4.4, 2.3 Hz, 1H), 5.26–5.17 (m, 2H), 5.07 (dq, J = 10.5, 1.5 Hz, 1H), 4.95–4.78 (m, 2H), 4.54–4.37 (m, 3H), 4.07 (s, 3H), 3.93 (dq, J = 12.5, 6.5 Hz, 2H), 3.49 (dd, J = 8.6, 4.1 Hz, 1H), 3.17 (dd, J = 18.9, 1.8 Hz, 1H), 3.00 (d, J = 18.8 Hz, 1H), 2.34 (dt, J = 14.7, 2.1 Hz, 1H), 2.17 (dd, J = 14.7, 4.2 Hz, 1H), 2.09 (ddd, J = 14.5, 4.5, 2.3 Hz, 1H), 1.86 (dt, J = 14.4, 4.3 Hz, 1H), 1.29 (d, J = 6.3 Hz, 3H), 0.99–0.93 (m, 18H), 0.71–0.59 (m, 6H), 0.15 (s, 6H). 13C{1H} NMR (126 MHz, chloroform-d) δ 211.3, 186.9, 186.6, 161.0, 156.4, 156.3, 155.7, 135.6, 135.5, 134.5, 134.0, 133.1, 120.9, 119.7, 118.4, 116.9, 111.4, 111.2, 98.9, 72.5, 66.8, 66.4, 65.1, 56.7, 48.4, 35.6, 33.7, 33.4, 25.8, 18.5, 18.1, 6.8, 4.8, −5.5. HRMS (ESI) m/z: [M + Na+] calcd for C43H61NO13Si2Na: 878.3574; found 878.3602.
3′-4′-Di-epi-doxorubicin Hydrochloride (4a)
: To a solution of 20 (45 mg, 53 μmol) in DCM (5 mL) were added 1,3-dimethylbarbituric acid (25 mg, 0.16 mmol, 3 equiv) and Pd(PPh3)4 (6.0 mg, 5.3 μmol, 0.1 equiv). The reaction mixture was stirred overnight, and solvent was removed in vacuo. The residue was submitted to column chromatography (1:99–15:85 acetone/toluene) to give the crude amine. The crude amine thus obtained was desilylated according to general procedure D. Column chromatography on neutral silica (0:100–50:50 MeOH/DCM), and lyophilization from aqueous 10 μM HCl (1 mL/μmol, 2 equiv) afforded the title compound as a dark red solid (17 mg, 29 μmol, 54% over two steps). 1H NMR (500 MHz, DMSO-d6) δ 13.98 (s, 1H), 13.22 (s, 1H), 7.92 (q, J = 4.4, 3.8 Hz, 2H), 7.80–7.55 (m, 4H), 6.29 (s, 1H), 6.18 (d, J = 4.8 Hz, 1H), 5.30 (d, J = 3.3 Hz, 1H), 5.04–4.89 (m, 2H), 4.73–4.43 (m, 2H), 3.99 (s, 3H), 3.98–3.91 (m, 1H), 3.46 (dt, J = 9.3, 4.5 Hz, 3H), 3.17 (d, J = 18.8 Hz, 1H), 3.00 (d, J = 18.8 Hz, 1H), 2.24 (d, J = 14.8 Hz, 1H), 2.11–1.81 (m, 3H), 1.28 (d, J = 6.1 Hz, 3H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 214.0, 187.2, 187.1, 161.4, 156.1, 155.1, 136.9, 135.3, 135.1, 134.6, 120.5, 120.4, 119.6, 111.4, 111.3, 98.7, 75.7, 68.9, 68.6, 65.0, 64.4, 57.2, 49.7, 40.4, 35.6, 32.1, 31.7, 18.0. HRMS (ESI) m/z: [M + H+] calcd for C27H31NO11: 544.1813; found 544.1814.
3′-4′-Di-epi-dimethyldoxorubicin (4b)
: To a solution of 20 (73 mg, 85 μmol) in DCM (6 mL) was added 1,3-dimethylbarbituric acid (40 mg, 0.26 mmol, 3 equiv). Pd(PPh3)4 (9.0 mg, 8.5 μmol, 0.1 equiv) was added, and the reaction mixture was stirred overnight. After concentrating in vacuo, the residue was submitted to silica gel column chromatography (0:100–15:85 acetone/toluene) to give the crude amine. To a solution of the crude amine in EtOH (4 mL) was added formaldehyde (aqueous 37% w/v solution, 0.13 mL) and sodium trisacetoxyborohydride (22.5 mg, 106 μmol, 1.25 equiv). The reaction mixture was stirred for 2 h, partitioned between DCM and sat. aq. NaHCO3, and the aqueous layer was extracted thrice with DCM. Combined organics were dried over Na2SO4 and concentrated in vacuo, followed by purification through column chromatography (0:100–20:80 acetone/toluene) to give the crude dimethylamine. This was then desilylated according to general procedure D. Column chromatography on neutral silica (0:100–7:93 MeOH:DCM) afforded the title compound as a red solid (17 mg, 30 μmol, 35% over three steps). 1H NMR (500 MHz, chloroform-d) δ 13.99 (s, 1H), 13.22 (s, 1H), 8.03 (dd, J = 7.7, 1.1 Hz, 1H), 7.79 (dd, J = 8.5, 7.7 Hz, 1H), 7.40 (dd, J = 8.6, 1.1 Hz, 1H), 5.38 (dd, J = 8.8, 4.8 Hz, 1H), 5.30 (dd, J = 4.1, 2.1 Hz, 1H), 4.85–4.71 (m, 2H), 4.09 (s, 3H), 4.02 (dd, J = 6.8, 5.3 Hz, 1H), 3.64–3.53 (m, 1H), 3.24 (dd, J = 18.8, 2.1 Hz, 1H), 2.95 (d, J = 18.7 Hz, 1H), 2.55 (dt, J = 14.6, 2.2 Hz, 1H), 2.37–2.31 (m, 6H), 2.31 (s, 6H), 2.15 (dd, J = 14.7, 4.1 Hz, 1H), 2.00–1.91 (m, 1H), 1.53 (td, J = 13.0, 8.8 Hz, 1H), 1.38 (d, J = 6.7 Hz, 3H). 13C{1H} NMR (126 MHz, chloroform-d) δ 214.9, 187.3, 186.7, 161.2, 156.5, 156.0, 135.9, 135.7, 134.4, 133.9, 121.0, 120.0, 118.5, 111.6, 111.4, 100.3, 76.6, 71.4, 69.0, 68.3, 65.8, 60.1, 56.8, 43.0, 36.0, 33.9, 29.0, 18.7. HRMS (ESI) m/z: [M + H+] calcd for C29H35NO11: 572.2126; found 572.2131. 13C{1H}-GATED NMR (chloroform-d, 126 MHz) δ 100.31 (JC1′, H1′ = 169 Hz, C-1).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c00220.
Biological evaluation of the panel of (N,N-dimethyl)doxorubicin glycosyl diastereomers as well as 1H, 13C, COSY, and HSQC NMR data for compounds 2a/b–4a/b and intermediates in their synthesis (PDF)
Author Contributions
∥ D.P.A.W. and S.Y.v.d.Z. contributed equally to this work.
This work was supported by grants from the Dutch Cancer Society KWF (JN) and by the Institute for Chemical Immunology, an NWO Gravitation project funded by the Ministry of Education, Culture and Science of the Netherlands to HO and JN.
The authors declare the following competing financial interest(s): J. Neefjes is a shareholder in NIHM, that aims to produce aclarubicin for clinical use.
Supplementary Material
References
- Grand View Research. Doxorubicin Market by Application (Ovarian, Multiple Myeloma, Kaposi Sarcoma, Leukemia, Bone Sarcoma, Breast, Endometrial, Gastric, Liver, Kidney, Other Cancers) and Segment Forecasts, 2018–2024, 2016.
- Lefrak E. A.; Pit’ha J.; Rosenheim S.; Gottlieb J. A. A Clinicopathologic Analysis of Adriamycin Cardiotoxicity. Cancer 1973, 32, 302–314. 10.1002/1097-0142(197308)32:2<302::AID-CNCR2820320205>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Krohn K. In Anthracycline Chemistry and Biology I Biological Occurence and Biosynthesis, Synthesis and Chemistry; Balzani V.; de Meijere A.; Houk K. N.; Kessler H.; Lehn J.-M.; Ley S. V.; Schreiber S. L.; Thiem J.; Trost B. M.; Vögtle F.; Yamamoto H., Eds.; Springer Verlag, 2008. [Google Scholar]
- Weiss R. B. The Anthracyclines: Will We Ever Find a Better Doxorubicin?. Semin. Oncol. 1992, 19, 670–686. [PubMed] [Google Scholar]
- Booser D. J.; Hortobagyi G. N. Anthracycline Antibiotics in Cancer Therapy. Drugs 1994, 47, 223–258. 10.2165/00003495-199447020-00002. [DOI] [PubMed] [Google Scholar]
- Mele D.; Nardozza M.; Spallarossa P.; Frassoldati A.; Tocchetti C. G.; Cadeddu C.; Madonna R.; Malagù M.; Ferrari R.; Mercuro G. Current Views on Anthracycline Cardiotoxicity. Heart Fail. Rev. 2016, 21, 621–634. 10.1007/s10741-016-9564-5. [DOI] [PubMed] [Google Scholar]
- Launchbury A. P.; Habboubi N. Epirubicin and Doxorubicin: A Comparison of Their Characteristics, Therapeutic Activity and Toxicity. Cancer Treat. Rev. 1993, 19, 197–228. 10.1016/0305-7372(93)90036-Q. [DOI] [PubMed] [Google Scholar]
- Nitiss J. L. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang B.; Qiao X.; Janssen L.; Velds A.; Groothuis T.; Kerkhoven R.; Nieuwland M.; Ovaa H.; Rottenberg S.; van Tellingen O.; Janssen J.; Huijgens P.; Zwart W.; Neefjes J. Drug-Induced Histone Eviction from Open Chromatin Contributes to the Chemotherapeutic Effects of Doxorubicin. Nat. Commun. 2013, 4, 1908 10.1038/ncomms2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao X.; van der Zanden S. Y.; Wander D. P. A.; Borràs D. M.; Song J.-Y.; Li X.; van Duikeren S.; van Gils N.; Rutten A.; van Herwaarden T.; van Tellingen O.; Giacomelli E.; Bellin M.; Orlova V.; Tertoolen L. G. J.; Gerhardt S.; Akkermans J. J.; Bakker J. M.; Zuur C. L.; Pang B.; Smits A. M.; Mummery C. L.; Smit L.; Arens R.; Li J.; Overkleeft H. S.; Neefjes J. Uncoupling DNA Damage from Chromatin Damage to Detoxify Doxorubicin. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 15182–15192. 10.1073/pnas.1922072117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F.; Kemp C. J.; Henikoff S. Doxorubicin Enhances Nucleosome Turnover around Promoters. Curr. Biol. 2013, 23, 782–787. 10.1016/j.cub.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang B.; de Jong J.; Qiao X.; Wessels L. F. A.; Neefjes J. Chemical Profiling of the Genome with Anti-Cancer Drugs Defines Target Specificities. Nat. Chem. Biol. 2015, 11, 472–480. 10.1038/nchembio.1811. [DOI] [PubMed] [Google Scholar]
- Wander D. P. A.; van der Zanden S. Y.; van der Marel G. A.; Overkleeft H. S.; Neefjes J.; Codée J. D. C. Doxorubicin and Aclarubicin: Shuffling Anthracycline Glycans for Improved Anticancer Agents. J. Med. Chem. 2020, 63, 12814–12829. 10.1021/acs.jmedchem.0c01191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikowski T.; Bretner M.; Najda A.; Cova L.. New Derivatives of Epirubicin, Their Medicinal Application and Pharmaceuticaly Acceptable Forms of Drugs. WO2007/075094A1, 2006.
- Arlandini E.; Vigevani A.; Arcamone F. Interaction of New Derivatives of Daunorubicin and Doxorubicin with DNA. Part II. Farm. Ed. Sci. 1980, 35, 65–78. [PubMed] [Google Scholar]
- Yu B. Gold(I)-Catalyzed Glycosylation with Glycosyl o-Alkynylbenzoates as Donors. Acc. Chem. Res. 2018, 51, 507–516. 10.1021/acs.accounts.7b00573. [DOI] [PubMed] [Google Scholar]
- Wander D. P. A.Understanding Anthracyclines: Synthesis of a Focused Library of Doxorubicin/Aclarubicin—Inspired Structures; Leiden University, 2019. [Google Scholar]
- Fan E.; Shi W.; Lowary T. L. Synthesis of Daunorubicin Analogues Containing Truncated Aromatic Cores and Unnatural Monosaccharide Residues. J. Org. Chem. 2007, 72, 2917–2928. 10.1021/jo062542q. [DOI] [PubMed] [Google Scholar]
- Noshita T.; Sugiyama T.; Kitazumi Y.; Oritani T. Reinvestigation of Phenolic Ferrier Reaction: Selective Synthesis of Aryl O-Δ 2 -Glycosides. Biosci., Biotechnol., Biochem. 1995, 59, 2052–2055. 10.1271/bbb.59.2052. [DOI] [Google Scholar]
- Ma Y.; Li Z.; Shi H.; Zhang J.; Yu B. Assembly of Digitoxin by Gold(I)-Catalyzed Glycosidation of Glycosyl o-Alkynylbenzoates. J. Org. Chem. 2011, 76, 9748–9756. 10.1021/jo201850z. [DOI] [PubMed] [Google Scholar]
- Renneberg B.; Li Y.-M.; Laatsch H.; Fiebig H.-H. A Short and Efficient Transformation of Rhamnose into Activated Daunosamine, Acosamine, Ristosamine and Epi-Daunosamine Derivatives, and Synthesis of an Anthracycline Antibiotic Acosaminyl-ε-Iso-Rhodomycinone. Carbohydr. Res. 2000, 329, 861–872. 10.1016/S0008-6215(00)00257-3. [DOI] [PubMed] [Google Scholar]
- Horton D.; Weckerle W. A Preparative Synthesis of 3-Amino-2,3,6-Trideoxy-l-Lyxo-Hexose (Daunosamine) Hydrochloride from d-Mannose. Carbohydr. Res. 1975, 44, 227–240. 10.1016/S0008-6215(00)84166-X. [DOI] [PubMed] [Google Scholar]
- The stereochemical outcome of the glycosylations indicates that long-range participation of the N-Alloc group does not play a significant role in the studied glycosylations; a Hansen T.; Elferink H.; van Hengst J. M. A.; Houthuijs K. J.; Remmerswaal W. A.; Kromm A.; Berden G.; van der Vorm S.; Rijs A. M.; Overkleeft H. S.; Filippov D. V.; Rutjes F. P. J. T.; van der Marel G. A.; Martens J.; Oomens J.; Codée J. D. C.; Boltje T. J. Characterization of Glycosyl Dioxolenium Ions and Their Role in Glycosylation Reactions. Nat. Commun. 2020, 11, 2664 10.1038/s41467-020-16362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Komarova B. S.; Tsvetkov Y. E.; Nifantiev N. E. Design of α-Selective Glycopyranosyl Donors Relying on Remote Anchimeric Assistance. Chem. Rec. 2016, 16, 488–506. 10.1002/tcr.201500245. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Li J.; Zhu Y.; Li Y.; Yu B. Mechanistic Insights into the Gold(I)-Catalyzed Activation of Glycosyl Ortho-Alkynylbenzoates for Glycosidation. J. Am. Chem. Soc. 2013, 135, 18396–18405. 10.1021/ja4064316. [DOI] [PubMed] [Google Scholar]
- Hansen T.; Lebedel L.; Remmerswaal W. A.; van der Vorm S.; Wander D. P. A.; Somers M.; Overkleeft H. S.; Filippov D. V.; Désiré J.; Mingot A.; Bleriot Y.; van der Marel G. A.; Thibaudeau S.; Codée J. D. C. Defining the SN1 Side of Glycosylation Reactions: Stereoselectivity of Glycopyranosyl Cations. ACS Cent. Sci. 2019, 5, 781–788. 10.1021/acscentsci.9b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero J. A. C.; Tabacco S. A.; Woerpel K. A. Stereochemical Reversal of Nucleophilic Substitution Reactions Depending upon Substituent: Reactions of Heteroatom-Substituted Six-Membered-Ring Oxocarbenium Ions through Pseudoaxial Conformers. J. Am. Chem. Soc. 2000, 122, 168–169. 10.1021/ja993366o. [DOI] [PubMed] [Google Scholar]
- Ayala L.; Lucero C. G.; Romero J. A. C.; Tabacco S. A.; Woerpel K. A. Stereochemistry of Nucleophilic Substitution Reactions Depending upon Substituent: Evidence for Electrostatic Stabilization of Pseudoaxial Conformers of Oxocarbenium Ions by Heteroatom Substituents. J. Am. Chem. Soc. 2003, 125, 15521–15528. 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]
- Zeng J.; Sun G.; Yao W.; Zhu Y.; Wang R.; Cai L.; Liu K.; Zhang Q.; Liu X.-W.; Wan Q. 3-Aminodeoxypyranoses in Glycosylation: Diversity-Oriented Synthesis and Assembly in Oligosaccharides. Angew. Chem., Int. Ed. 2017, 56, 5227–5231. 10.1002/anie.201700178. [DOI] [PubMed] [Google Scholar]
- Bartner P.; Boxler D. L.; Brambilla R.; Mallams A. K.; Morton J. B.; Reichert P.; Sancilio F. D.; Surprenant H.; Tomalesky G.; Lukacs G.; Olesker A.; Thang T. T.; Valente L.; Omura S. The Megalomicins. Part 7. A Structural Revision by Carbon-13 Nuclear Magnetic Resonance and X-Ray Crystallography. Synthesis and Conformational Analysis of 3-Dimethylamino- and 3-Azido-D- and -L-Hexopyranosides, and the Crystal Structure of 4″-O-(4-Iodobe. J. Chem. Soc., Perkin Trans. 1 1979, 1600–1624. 10.1039/P19790001600. [DOI] [Google Scholar]
- Kuo L. J.; Yang L. X. γ-H2AX-A Novel Biomaker for DNA Double-Strand Breaks. In Vivo 2008, 305–309. [PubMed] [Google Scholar]
- Olive P. L.; Wlodek D.; Banáth J. P. DNA Double-Strand Breaks Measured in Individual Cells Subjected to Gel Electrophoresis. Cancer Res. 1991, 51, 4671–4676. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.