

Abstract
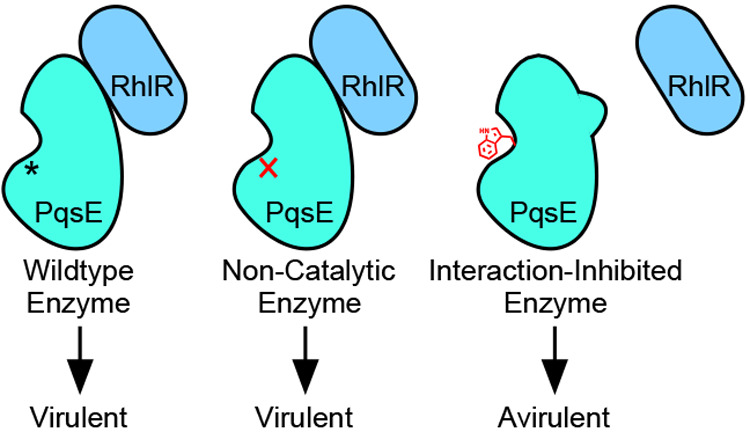
Pseudomonas aeruginosa is an opportunistic human pathogen that causes fatal infections. There exists an urgent need for new antimicrobial agents to combat P. aeruginosa. We conducted a screen for molecules that bind the virulence-controlling protein PqsE and characterized hit compounds for inhibition of PqsE enzymatic activity. The binding conformations of two inhibitory molecules, BB391 and BB393, were identified by crystallography, and inhibitor binding was mimicked by the substitution of PqsE residues E182 and S285 with tryptophan. Comparison of the inhibitor-mimetic mutations to the catalytically inactive PqsE D73A protein demonstrated that catalysis is not responsible for the role PqsE plays in driving virulence factor production. Rather, the PqsE E182W protein fails to interact with the quorum-sensing receptor, RhlR, and our results suggest that it is this interaction that is responsible for promoting virulence factor production in P. aeruginosa. These findings provide a new route for drug discovery efforts targeting PqsE.
Introduction
Pseudomonas aeruginosa is a human pathogen that causes life-threatening hospital-acquired infections in immunocompromised patients.1P. aeruginosa lung infections also occur in cystic fibrosis sufferers,2 and infections are frequent in severe burn wounds.3 Traits that drive P. aeruginosa clinical pathogenicity include the ability to form biofilms,4 the production of virulence factors,5 and antibiotic resistance,6 which combine to render P. aeruginosa infections increasingly unresponsive to treatment.7,8 Thus, there is an urgent need for new antimicrobial agents targeting this major pathogen.
P. aeruginosa virulence factor production and biofilm formation are both under the control of quorum sensing. Quorum sensing is a process of bacterial cell–cell communication that involves the synthesis, release, and detection of small molecule signals, called autoinducers.9 Multiple quorum-sensing systems exist in P. aeruginosa. The most well-studied are the Las and Rhl quorum-sensing systems, which rely on the LasR and RhlR receptors binding the acyl homoserine lactone autoinducers 3-oxo-C12-homoserine lactone (3-oxo-C12-HSL) and C4-homoserine lactone (C4-HSL), respectively.10−12 Somewhat less understood is the Pqs quorum-sensing system, which employs a class of alkyl quinolone signal molecules, with 2-heptyl-3-hydroxy-4-quinolone (also known as the Pseudomonas Quinolone Signal, PQS) as the most comprehensively studied autoinducer of the quinolone class.13 These three quorum-sensing circuits share overlapping regulons. For instance, Las signaling activates both the Rhl and Pqs systems, and in turn, the Rhl and Pqs systems both control the production of a redox-sensitive toxin called pyocyanin. Pyocyanin is present in the sputum of cystic fibrosis patients with advanced P. aeruginosa infections14,15 and is largely responsible for killing Caenorhabditis elegans in a fast-kill model of infection.5 Pyocyanin also endows P. aeruginosa cultures with their characteristic blue-green color. Therefore, the production of pyocyanin is often measured as a convenient readout of P. aeruginosa virulence and is considered an output of both Rhl and Pqs quorum sensing.
One particularly intriguing member of the P. aeruginosa Pqs quorum-sensing network is pqsE, encoding the metallo-β-hydrolase enzyme, PqsE. pqsE is a member of the pqsABCDE operon that encodes all of the necessary components for alkyl quinolone biosynthesis. PqsE has been characterized as an esterase that catalyzes the hydrolysis of 2-amino-benzoyl acetyl CoA to 2-amino-benzoyl acetate (2-ABA), a step in the synthesis of PQS.16 Curiously, pqsE is the only member of the pqsABCDE operon that is not absolutely required for PQS production, as other esterases can substitute for this function.16 Nonetheless, deletion of pqsE leads to the complete loss of both pyocyanin production in laboratory-grown cultures and infectivity in a mouse model,17 suggesting a crucial role for PqsE in P. aeruginosa pathogenicity. These observations raise the question of whether there exist additional physiologically relevant reactions catalyzed by PqsE and/or whether PqsE carries out functions that do not depend on catalysis. PqsE has been linked to activation of RhlR,18,19 and indeed, the virulence and biofilm defects of the P. aeruginosa ΔpqsE mutant closely mirror those of the ΔrhlR mutant.17,20 These findings hint that the relevant function of PqsE in P. aeruginosa virulence is associated with the Rhl quorum-sensing system, not the Pqs quorum-sensing system.
The above findings have made PqsE a promising target for the development of P. aeruginosa quorum-sensing inhibitors. Unlike RhlR, PqsE can be easily purified, and high-resolution X-ray crystal structures of the protein exist with and without small molecules present in the binding pocket.21−24 The structures reveal the putative, iron-binding catalytic site, which has been probed by alanine-scanning mutagenesis.23,25,26 PqsE has measurable in vitro enzyme activity with synthetic substrates, making it amenable to high-throughput small molecule screening. Indeed, previous screening efforts have identified molecules that bind PqsE and inhibit its ability to hydrolyze synthetic thioester substrates.23,27 However, to date, no small molecule PqsE catalytic inhibitor has been discovered that suppresses pyocyanin production in P. aeruginosa. Zender et al. did report that their identified PqsE enzymatic inhibitors were internalized by P. aeruginosa and inhibited PqsE in vivo. However, no reduction in pyocyanin production occurred.23 This finding, combined with data demonstrating that PqsE can sensitize RhlR to C4-HSL in a heterologous E. coli assay,18 again suggest that enzymatic activity might not be the pertinent activity of PqsE with respect to its role in driving virulence factor production.
In this study, we present small molecules discovered in a high-throughput differential scanning fluorimetry screen targeting PqsE, and we demonstrate their use in probing PqsE activities, both enzymatic and nonenzymatic. We solve the X-ray crystal structures of PqsE bound to two enzyme inhibitors obtained through this screen, BB391 and BB393, and we investigate the interactions these inhibitors form in the active site of PqsE. The structural insight afforded by these PqsE–ligand complexes allowed us to develop a structure-guided mutagenesis strategy to determine whether targeting the PqsE active site is an effective means for suppressing P. aeruginosa virulence. We uncover a surprising mechanism underpinning PqsE-driven virulence phenotypes involving a direct protein–protein interaction with the RhlR quorum-sensing receptor. Both the chemical tools and the basic biological insight revealed by this work can promote the discovery of new antimicrobial agents targeting PqsE.
Results
High-Throughput Differential Scanning Fluorimetry Screen Identifies Small Molecule PqsE Binders
To discover molecules capable of binding PqsE, without limiting the search to those targeting the putative active site, we conducted a differential scanning fluorimetry (DSF) screen of a 120 000-compound library of drug-like small molecules. This strategy allowed us to identify molecules capable of binding and causing melting temperature (Tm) shifts of purified, wildtype (WT) 6xHis-PqsE. For information about the high-throughput screening strategy, refer to the extended methods and see Figures S1 and S2.
Our initial DSF screen was conducted with each compound administered at a concentration of 37 μM. The screen yielded 933 molecules with |ΔTm| values of at least 0.75 °C (Figure 1). Retesting of these 933 compounds (n = 4) reduced the hit number to 510 compounds with |ΔTm| values of at least 0.5 °C, yielding an initial, confirmed hit rate of 0.4%. To eliminate nonspecific binders, this set of molecules was counter-screened against the purified carbonic anhydrase protein. The remaining compounds (425) were subsequently tested in four-point dose–response DSF assays with PqsE to yield 174 candidate molecules with specific, dose-dependent binding (final hit rate of 0.1%). A total of 121 molecules with good solubility, MW < 500 Da, and low polar surface area were selected for further analysis.
Figure 1.

Differential scanning fluorimetry screen of a small molecule library containing 120 000 compounds. See text for details.
Our first goal was to assess whether the molecules that bound PqsE were also capable of inhibiting its enzymatic activity. To do this, we measured dose-dependent enzyme inhibition by each hit compound using a synthetic ester as the substrate for PqsE. Of the 121 lead molecules, 27 displayed activity as PqsE esterase inhibitors, having IC50 values <100 μM. All of the molecules that possessed enzyme inhibitory activity had positive ΔTm values. The molecules that bound PqsE but showed no enzyme inhibition up to a test concentration of 100 μM could be divided into two subgroups: those with positive, stabilizing ΔTm values and those with negative, destabilizing ΔTm values (Figure 1). The molecules possessing destabilizing activity were not investigated further. To date, PqsE enzymatic activity has not been conclusively linked to the role PqsE plays in promoting P. aeruginosa virulence phenotypes. Our goal in this work is to characterize the mechanism(s) of action of the PqsE enzyme inhibitor compounds and use what we learn to test whether targeting the active site of this important enzyme can suppress P. aeruginosa virulence.
X-ray Crystallography of Compounds BB391 and BB393 Bound to PqsE Identifies the Residues Involved in Protein–Ligand Interactions in the PqsE Active Site
We focused on two potent PqsE enzyme inhibitors, BB391 and BB393 (Figure 2a and b, respectively). Both are highly soluble in the buffer used previously to crystallize PqsE, making them suitable for use in structural analyses. PqsE crystals were grown and subsequently soaked with saturating amounts of each compound to obtain crystals of the PqsE-BB391 and PqsE-BB393 complexes. The PqsE active site contains two iron atoms coordinated by a total of five histidine and two aspartate side chains, with the irons bridged by D178 and a water molecule.25 Both irons are hexacoordinate with a water molecule or ligand atom occupying an axial position. The active site is accessible to solvent at one end and essentially blocked by the E182 side chain at the other. The binding pocket is relatively narrow in one direction, orienting planar ligands edge-on to the iron atoms, and the central portion is lined primarily with hydrophobic residues (L193, F195, F276, L277, L281, H282, M286). The cocrystal structures demonstrate that both BB391 and BB393, as expected, bind in the reported PqsE active site (Figure 2), with no evidence of the protein undergoing any conformational change upon ligand binding. In the case of BB391, N2 of the indazole ring coordinates directly with the Fe2 ion (Fe–N bond 2.11 Å), and N1 participates in hydrogen bonding with the axial water ligand of Fe1 at the solvent-exposed end of the pocket (Figure 2a,c). Regarding BB393, no direct coordination of the Fe ions is observed, and the nitrogen atom of the thiazole group forms a short hydrogen bond (2.27 Å) with the axial water ligand of Fe2 (Figure 2b,d). Also, in the PqsE-BB393 structure, a water molecule occupies the location of the N2 atom of BB391 in the PqsE-BB391 structure, as the amidothiazole of BB393 is too small to make direct interactions with Fe2. Both BB391 and BB393 possess amide bonds, and both crystal structures reveal that the carbonyl oxygens of the amides form a hydrogen bond with the side-chain hydroxyl group of S285 (2.36 Å with BB391 and 2.35 Å with BB393). This hydrogen bond is formed by both BB391 and BB393 despite the direction of the amide bond being reversed in their respective binding poses. BB393 makes one additional polar contact through the O2 of the urea moiety with the backbone amide of the tyrosine at position 72 (Y72), an interaction that BB391 lacks. Thus, although BB391 and BB393 possess different core scaffolds, they both display similar interactions with PqsE in the interior iron-containing region of the ligand binding pocket, suggesting conformational restrictions in this portion of the binding site as a consequence of hydrogen bonding from S285 to the ligand amide moieties and the overall flattened shape of the binding pocket.
Figure 2.

Crystal structures of PqsE-BB391 and PqsE-BB393 complexes. Surface and cartoon representations of full-length PqsE shown with iron atoms (orange spheres). In a and b, key interactions for each ligand are depicted with hydrogen bonds shown as red dashed lines and direct bonds are shown as solid lines. Water molecules are depicted as black spheres. In c and d, side chains of select amino acids, including the 69HXHXDH74 motif, that form the PqsE active site are shown in gray with nitrogen atoms in blue and oxygen atoms in red. BB391 and BB393 nitrogen and oxygen atoms use the same coloring scheme. Carbon atoms are shown in pink for BB391 and light blue for BB393, and the sulfur atom in BB393 is in yellow. Hydrogen bonds, water molecules, and iron atoms appear as in a and b.
Compared to previously reported PqsE structures, the structure of the PqsE-BB393 complex highlights the ability of a small molecule to make weak secondary interactions in a shallow, hydrophobic region at the solvent-exposed periphery of the ligand binding pocket. The BB393 C10 chiral center is situated in a hydrophobic groove formed between the face of the α-helix consisting of PqsE residues S104–L116 and the backbone of the 69HXHXDH74 motif (Figure 2c,d) characteristic of this class of hydrolase enzymes. The orientation of C10 in this groove additionally forces the morpholine ring of BB393 out of the pocket and into the solvent. This structure suggests that additional, nonpolar moieties could be accommodated on compounds that are situated at the solvent-facing region peripheral to the binding pocket, encouraging further lead optimization to introduce substituents that bind in this hydrophobic groove. Moreover, the PqsE-BB393 structure shows that the (R)-enantiomer of BB393 is present, even though racemic BB393 was used for soaking. The hydrogen bond between the main-chain of Y72 and the urea moiety of BB393 (2.81 Å) likely influences the stereoselectivity at this site by reducing the conformational flexibility of this region of BB393. This result suggests a stereoselective interaction between BB393 and PqsE that could be exploited to potentially improve potency.
Fluorescent Probe Design and Competitive Binding Assays
The PqsE–BB391 structure showed that there is space at the solvent-facing portion of the binding pocket that could accommodate derivatization at the para position on the BB391 phenyl group. Thus, we generated a fluorescent probe based on the BB391 core scaffold to enable competitive binding assays for mechanistic studies of PqsE inhibitors. The fluorescent probe, BB562 (Figure 3a), did indeed bind to purified WT PqsE as shown using fluorescence polarization, with an apparent binding constant (Kapp) in the midnanomolar range (Figure 3b). This result demonstrates that BB562 can be used as a tool to assess whether small molecules have sufficient affinity to compete for binding in the active site.
Figure 3.

Compounds that bind in the PqsE active site compete with a fluorescent BB391-based probe. (a) Structure of a 5-FAM-containing fluorescent probe called BB562. The BB391 portion is in black, the linker is in blue, and the 5-FAM moiety is in green. (b) Binding curve for WT PqsE to BB562. Kapp was determined from two independent experiments performed in triplicate. (c) Competition of select screening hit compounds from the DSF screen, including BB391 and BB393, against BB562 for binding to PqsE. The fluorescence polarization value for PqsE-BB562 in the absence of a competitor is defined as 100% probe bound. The dotted line at 0 represents the level of background fluorescence polarization which was subtracted from the results depicted here. (d) Calculated EC50 values for compounds in c, determined from two independent experiments performed in triplicate. Error bars are present in all cases and represent standard deviations. Some error bars are smaller than the symbols.
The fluorescent BB562 probe was used in competitive binding assays to determine whether hit molecules identified from the primary DSF screen were capable of competing for binding in the PqsE active site. Consistent with their binding poses in the crystal structures, both BB391 and BB393 competed with BB562 for binding WT PqsE with EC50 values of 255 nM and 183 nM, respectively (Figure 3c,d Bin 1). Additionally, we tested molecules from each of the other categories of screening hits: the noninhibiting binders (Bin 2) and the destabilizers (Bin 3). Notably, these screening hits were only weakly competitive at best (compound 2b) or were incapable of competing with the probe (compounds 2a, 3a, and 3b). Curiously, the destabilizing compounds (3a and 3b) do not appear to affect the ability of PqsE to bind the BB562 probe. In the original screen, the negative Tm shifts induced by these compounds reduced the observed Tm from ∼68 °C to roughly 50–60 °C. One possible explanation for the results in Figure 3 is that because the competitive binding assay is performed at RT, under this condition, PqsE is not sufficiently destabilized by compounds 3a and 3b to affect its ability to bind BB562 in the active site.
PqsE Residues E182 and S285 Are Required for Inhibition by BB391 but Not BB393
Guided by the crystal structures of PqsE bound to BB391 and BB393, we next investigated the amino acids responsible for forming the crucial protein–ligand interactions. We focused on two residues: S285 and E182 (Figure 2c,d). Our rationale is as follows: the hydroxyl group of S285 participates in hydrogen bonding with the amide groups of both BB391 and BB393, suggesting that S285 can mediate an important interaction between the protein and each ligand. E182 has previously been suggested to act as a gating residue for the most interior region of the PqsE binding pocket. Specifically, PqsE E182A possesses increased hydrolytic activity with a synthetic phosphodiester substrate, bis(p-nitrophenyl) phosphate, due to elimination of a steric clash when the glutamic acid residue is replaced with the shorter, uncharged alanine residue (ref (25) and verified in Figure S3).
To determine whether the presence of the PqsE E182 and S285 residues is necessary for binding and/or inhibition by BB391 and BB393, we constructed PqsE E182A and PqsE S285A and purified the two mutant proteins. As a control, we also generated and purified PqsE S160A, as the crystal structures show that residue S160 is not involved in binding either molecule (Figure 2c,d). All proteins purified in this study exhibited >95% purity as judged by SDS-PAGE analysis (Figure S4). We also assayed a previously reported PqsE-inhibiting fragment molecule, 2-(pyridin-3′-yl) benzoic acid (called C1).23 The X-ray crystal structure of C1 bound to PqsE indicates that unlike BB391 and BB393, C1 does not interact with S285, nor does it abut E182. Consistent with this observation, the ΔTm values for C1 binding to PqsE E182A, PqsE S285A, and PqsE S160A were similar to those for C1 binding to WT PqsE (WT ΔTm = 8.7 °C and ΔTm = 10 °C, 8.5 °C, and 12.1 °C for PqsE E182A, PqsE S285A, and PqsE S160A, respectively, Figure 4a–d). We note that the PqsE S160A control protein has a considerably lower intrinsic Tm than WT PqsE in the absence of a small molecule (61.6 °C vs 67.2 °C, respectively) possibly explaining the increased ΔTm observed for C1 binding to PqsE S160A. The PqsE S160A protein behaved like WT PqsE with respect to stabilization by our molecules, as both BB391 and BB393 induced shifts in the PqsE S160A Tm similar to that for WT PqsE (ΔTm WT PqsE = 3.4 and 7.2 °C, and ΔTm PqsE S160A = 5.0 and 8.5 °C for BB391 and BB393, respectively; Figure 4a,b). BB391, by contrast, lost the ability to shift the Tm of PqsE E182A and PqsE S285A, suggesting that BB391 has reduced affinity for these PqsE variants (Figure 4c,d). Surprisingly, neither the PqsE E182A nor the S285A mutations altered the stabilizing effect of BB393 (Figure 4c,d). We reason that, unlike C1 and BB391, BB393 makes more extensive interactions in the binding pocket of PqsE, and therefore its affinity for PqsE is not perturbed by the loss of any single interaction with any single amino acid.
Figure 4.

PqsE E182 and PqsE S285 are required for binding to BB391 but not BB393. First-derivative plots (dF/dT is defined as the change in SYPRO Orange fluorescence divided by the change in temperature) of melting curves for (a) WT PqsE, (b) PqsE S160A, (c) PqsE E182A, and (d) PqsE S285A in the presence of 5% DMSO (black solid lines), 100 μM C1 (black dashed lines), 100 μM BB391 (pink lines), and 100 μM BB393 (blue lines). In all cases, PqsE protein was at 5 μM. The peak of each curve is defined as the Tm. (e) BB391 and (f) BB393 esterase inhibition dose–response curves for the designated PqsE proteins. PqsE proteins and MU-butyrate were used at concentrations of 200 nM and 2 μM, respectively. Two independent experiments were performed in triplicate. Error bars represent standard deviations.
We investigated whether the amino acid substitutions we introduced into PqsE affect its esterase activity and whether the changes impinge on the abilities of BB391 and BB393 to function as PqsE inhibitors. In the absence of inhibitors, all three mutant proteins (PqsE E182A, PqsE S285A, and the control protein PqsE S160A) displayed 69–109% of WT PqsE esterase activity against a synthetic substrate 4-methylumbelliferyl butyrate (MU-butyrate;28,29Figure S5). Thus, none of these three residues is required for catalysis. However, BB391 exhibited significantly decreased potency against PqsE E182A compared to WT PqsE and PqsE S160A. An accurate assessment of the IC50 of BB391 for PqsE S285A could not be determined due to its incomplete inhibition profile, but the projected IC50 was at least a log unit higher than that observed for WT PqsE (Figure 4e). Thus, PqsE residues E182 and S285 are important for BB391 to function as an inhibitor. Strikingly, the PqsE E182A and S285A substitutions had almost no effect on the inhibitory potency of BB393 (Figure 4f). The interpretation for PqsE S285 and BB391 is straightforward from the crystal structure: S285 hydrogen bonds with BB391, and substitution with an alanine eliminates this interaction. Regarding the ability of BB393 to inhibit PqsE S285A, we hypothesize that due to the larger size of BB393 relative to BB391, and its more extensive interactions with PqsE residues, individual amino acid substitutions in PqsE are not capable of perturbing BB393 binding. The results with PqsE E182A are less obvious because of the lack of hydrogen bonding with either of the two inhibitors and in light of a previous study showing an increased capacity for PqsE E182A to interact with particular artificial substrates.25 We hypothesize that PqsE residue E182 plays a role in stabilizing the interior region of the binding pocket, and mutation to alanine changes the context of inhibitor binding in this region. The BB391 indazole ring appears to bind more deeply in the PqsE active site pocket than does the thiazole ring of BB393, possibly explaining why the E182A mutation affects inhibition by BB391 more severely than it affects inhibition by BB393.
Structure-Guided Introduction of Tryptophan Residues to Produce PqsE Proteins That Mimic the Inhibited State
To gain deeper mechanistic insight into whether PqsE enzyme inhibitors have the potential to suppress virulence in P. aeruginosa, we used a structure-guided approach to design inhibitor-binding mimetic mutations in the PqsE enzyme active site. On the basis of the crystal structures of BB391 and BB393 bound to PqsE and the above results of our analyses with the purified proteins containing alanine substitutions, we engineered and purified PqsE E182W, PqsE S285W, and PqsE E182W/S285W rationalizing that these alterations could simulate the binding of a space-filling inhibitory ligand. As a reminder, neither the E182 nor the S285 residue is required for catalysis (Figure S5), making them appropriate residues for substitution with the bulky tryptophan residue without issues arising from loss of catalytic activity that would confound our studies. Because PqsE is a metallo-enzyme and contains a conserved HXHXDH motif, we reasoned that the aspartic acid, D73, in this motif is the key residue involved in the PqsE catalytic mechanism, consistent with validated mechanisms for other metallo-hydrolases.30 Thus, we also constructed and purified the PqsE D73A variant (Figure S4) to compare the behavior of a catalytically inactive PqsE protein to the potential inhibitor mimic variants, as we do not necessarily expect those different PqsE states to exhibit identical activities. In agreement with our predictions regarding the identities of catalytic residues in the active site, PqsE D73A had undetectable activity in the in vitro enzyme assay for hydrolysis of MU-butyrate (Figure 5a). PqsE E182W and PqsE E182W/S285W each displayed less than 10% of the catalytic activity of WT PqsE (Figure 5a). On the other hand, the PqsE S285W mutant displayed modestly increased hydrolytic activity (124%) compared to WT PqsE. Thus, PqsE D73A is catalytically inactive, PqsE E182W and PqsE E182W/S285W behave as if they have an inhibitor bound, and PqsE S285W behaves like WT PqsE with respect to enzyme activity.
Figure 5.

PqsE E182W and PqsE E182W/S285W mimic the effect of PqsE inhibition in vitro. (a) Hydrolysis of 4-methyl-umbelliferyl butyrate by the designated PqsE proteins. Values are represented as % of the WT PqsE activity. Two independent experiments were performed in triplicate. ****P < 0.0001 in one-way ANOVA compared to WT. (b) First derivative plots (dF/dT is defined as the change in SYPRO Orange fluorescence divided by the change in temperature) of melting curves for the designated PqsE proteins. The peak of each curve is defined as the Tm. (c) Binding of the designated PqsE proteins to BB562. Binding curves were generated from two independent experiments performed in triplicate. Error bars are present in all cases and represent standard deviations. Some error bars are smaller than the symbols.
To further compare the characteristics of the catalytically inactive mutant to the putative inhibitor-bound PqsE mimetics, we determined their melting temperatures using DSF. PqsE variants containing the E182W substitution had higher intrinsic Tm values than WT PqsE, showing that insertion of a tryptophan residue at this location stabilizes the protein (Figure 5b). Specifically, the Tm shifts for PqsE E182W and PqsE E182W/S285W were similar to the ΔTm observed following the addition of high concentrations of BB391 or BB393 to WT PqsE (ΔTm ≈ 10 °C), suggesting that introduction of a tryptophan residue at position 182 mimics the effect of a bound inhibitor. The PqsE E182W substitution clearly provides the stabilizing interactions observed in the PqsE E182W/S285W protein since PqsE S285W had the same Tm as WT PqsE (Figure 5b). Moreover, this result shows that the single S285W substitution in PqsE does not produce a phenotype mimicking the inhibitor-bound state. Each of the PqsE single tryptophan variants exhibited a ∼5–10-fold reduction in affinity for the BB562 fluorescent probe (Kapp = 5.1 μM and 2.2 μM for PqsE E182W and PqsE S285W, respectively, vs 0.4 μM for WT PqsE), and the PqsE E182W/S285W double mutant protein failed to bind BB562 (Figure 5c). Collectively, these in vitro results show that introduction of a tryptophan in place of the E182 residue in the PqsE active site produces effects similar to those that occur following treatment with the BB391 or BB393 inhibitors, and the PqsE E182W alteration can be combined with the S285W alteration to nearly completely block small molecule binding in the active site. By contrast, the PqsE D73A protein exhibited a similar Tm to that of WT PqsE (Figure 5b) and bound the BB562 probe with a similar affinity as WT PqsE (Kapp = 0.9 μM for PqsE D73A; Figure 5c), suggesting that the effect of tryptophan insertion at position 182 is related to blocking substrate access to the active site and not to affecting the catalytic mechanism. We note that although the Kapp for PqsE D73A is relatively unchanged compared to WT PqsE, the maximum change in fluorescence polarization observed for this mutant was lower than that for WT PqsE. We interpret this result to indicate a difference in the conformation of the BB562-bound mutant protein relative to WT PqsE bound to BB562, rather than altered affinity for the probe. It is unlikely that mutation of PqsE E182 or S285 to tryptophan perfectly recapitulates the effects of BB391 or BB393 binding in the active site. Nonetheless, these mutants provide powerful tools to explore the consequences of filling the PqsE active site. For simplicity, in the remainder of this work, we refer to these PqsE E182W and PqsE S285W mutants as “inhibitor mimetics.”
Mimicking Inhibitor Binding to PqsE Suppresses P. aeruginosa Virulence Traits in Vivo
Our results show that BB391 and BB393 represent promising starting points for in vivo inhibitors of PqsE enzymatic activity. However, when administered to cultures of P. aeruginosa PA14, neither molecule caused any decrease in pyocyanin production (Figure S6a). Likewise, neither molecule drove altered transcription of the virulence associated hcnA (hydrogen cyanide production) and phzH (phenazine biosynthesis) genes, both of which are known to be regulated by PqsE (Figure S6b,c, respectively).31 Two possibilities could explain this result: first, the molecules are inactive in vivo due to low cell permeability, metabolism, and/or efflux, or second, targeting the active site of PqsE with small molecule inhibitors does not affect the P. aeruginosa pyocyanin virulence trait. The latter explanation has been proposed to underpin the finding that, when administered to P. aeruginosa PAO1, the inhibitor C1 did not affect pyocyanin production.23 To distinguish between these possibilities for our inhibitors, we introduced WT PqsE and the inhibitor bound mimetic PqsE proteins into ΔpqsE P. aeruginosa PA14 and assessed whether PqsE-driven virulence phenotypes were altered. We also examined the effects of the catalytically inactive PqsE D73A variant. In all cases, the PqsE alleles were fused to C-terminal His tags. Western blot analysis showed that the different PqsE proteins were soluble and produced at similar levels in ΔpqsE P. aeruginosa PA14 (Figure S7).
Unlike WT P. aeruginosa PA14, the ΔpqsE strain carrying the empty pUCP18 vector produced almost no pyocyanin (2% of WT; Figure 6a). The strain carrying the catalytically inactive PqsE D73A variant produced pyocyanin levels similar to the WT strain. While this result supports the interpretation of Zender et al.,23 that PqsE plays a nonenzymatic regulatory role in virulence, it is surprising in light of other mutagenic analyses showing that amino acid substitutions predicted to affect catalytic activity also affected pyocyanin production in vivo.26 The ΔpqsE P. aeruginosa PA14 strains carrying PqsE alleles with the E182W substitution displayed striking impairments in pyocyanin production (compared to WT PqsE, PqsE E182W = 14% and PqsE E182W/S285W = 27% activity; Figure 6a). Surprisingly, the P. aeruginosa strain carrying the PqsE E182W/S285W mutant protein produced more pyocyanin than did the strain carrying PqsE E182W. We return to this point below. Consistent with our in vitro results, the strain harboring the PqsE S285W variant showed no change in pyocyanin phenotype compared to the strain harboring WT PqsE. Together, the results with the catalytically inactive PqsE D73A and the PqsE E182W and E182W/S285W variants that mimic the inhibitor bound state suggest there could be a nonenzymatic activity of PqsE that is primarily responsible for the virulence phenotypes in P. aeruginosa. Underscoring the relevance of the pyocyanin production profiles, analogous results were obtained for transcription of the virulence genes hcnA and phzH in P. aeruginosa carrying the different PqsE variants (Figure 6b,c, respectively).
Figure 6.

Mimicking PqsE enzyme inhibition in P. aeruginosa decreases pyocyanin production. (a) Shown is pyocyanin production by the ΔpqsE P. aeruginosa strain carrying the designated pqsE alleles on the pUCP18 plasmid. The “-” symbol designates the strain carrying the empty pUCP18 vector. The OD695 values of cell-free culture fluids were normalized to the OD600 of the cultures. The % pyocyanin production reported is relative to that of the strain carrying WT PqsE. The results shown are the average of two biological replicates. Error bars represent standard deviations. (b) Relative abundance of the hcnA transcript in the designated strains compared to WT, determined by qRT-PCR. (c) As in b for phzH. Results shown are the average of three biological replicates performed in quadruplicate. ns = not significant, ****P < 0.0001, ***P < 0.0005, **P < 0.005, *P < 0.05 in one-way ANOVA compared to WT.
PqsE Sensitizes RhlR to C4-HSL through a Protein–Protein Interaction and Inhibitor Mimetic Mutations Disrupt This Activity
We considered the possibility that the putative nonenzymatic role for PqsE in promoting P. aeruginosa PA14 virulence could concern the connection of PqsE to the Rhl quorum-sensing system. To explore this hypothesis, we took advantage of the previous finding that coexpression of pqsE together with rhlR in recombinant Escherichia coli sensitizes RhlR to exogenous addition of its native autoinducer, C4-HSL, by 2–20-fold.18,23 Because this effect occurs in a heterologous system, it enables study of the synergistic PqsE–RhlR activity in isolation. We hypothesize that it is this coupled PqsE–RhlR activity, not PqsE catalytic activity, that is key for PqsE to promote virulence. If so, our PqsE inhibited mimetics could display decreased abilities to sensitize RhlR to C4-HSL in this assay. To test this possibility, we constructed an E. coli strain harboring three plasmids: a plasmid with rhlR expressed under an inducible PBAD promoter, a plasmid with a PrhlA-luxCDABE bioluminescent reporter of RhlR transcriptional activity, and a pACYC184 plasmid that drives constitutive expression of WT pqsE, pqsE D73A, pqsE E182W, pqsE S285W, or pqsE E182W/S285W. Compared to the empty pACYC184 vector control, introduction of WT PqsE enhanced the RhlR response to 200 nM C4-HSL by ∼20-fold (Figure 7a). Introduction of PqsE D73A drove an equivalent augmentation of the RhlR response (Figure 7a), showing that the PqsE-driven enhancement of RhlR activity in this system does not require PqsE catalytic activity. Likewise, PqsE S285W was fully capable of sensitizing RhlR to C4-HSL. However, both PqsE E182W and PqsE E182W/S285W were severely defective in sensitizing RhlR to C4-HSL (23% and 30% of WT activity, respectively). Thus, the ability of PqsE to stimulate RhlR activity in this assay tracks with the ability of PqsE to drive pyocyanin production in vivo.
Figure 7.

PqsE interacts with RhlR and increases RhlR sensitivity to C4-HSL. (a) Bioluminescence output from E. coli carrying rhlR, rhlA-luxCDABE, and the designated pqsE alleles on the pACYC184 plasmid in response to 200 nM C4-HSL, normalized to the OD600 of the cultures. The “-” symbol designates the strain carrying the empty pACYC184 vector. The results shown are the average of two biological replicates performed in duplicate. Error bars represent standard deviations. ns = not significant, ****P < 0.0001, ***P < 0.0005 in one-way ANOVA compared to WT. (b) SDS-PAGE analysis of input (I) and elution (E) samples from PqsE–RhlR pull-down assays. 6xHis-PqsE was immobilized on Ni resin and exposed to lysate containing RhlR. On the gel, 6xHis-PqsE appears as a ∼34 kDa band and RhlR-mBTL as a ∼28 kDa band.
To test whether PqsE and RhlR form a complex, we purified each of our PqsE proteins containing N-terminal 6xHis tags and immobilized them on Ni resin. We exposed the immobilized PqsE proteins to lysate containing RhlR. As previously reported, addition of the artificial RhlR ligand called meta-bromo-thiolactone (mBTL) to E. coli cultures is required to isolate soluble, folded RhlR.32 Hence, we refer to the RhlR purified in this study as RhlR-mBTL. We next eluted the proteins and used SDS-PAGE to assess whether RhlR-mBTL had bound to WT PqsE and any of the variants. Strikingly, WT PqsE, PqsE D73A, and PqsE S285W formed complexes with RhlR-mBTL. However, almost no RhlR-mBTL copurified with PqsE E182W or PqsE E182W/S285W (Figure 7b). We interpret this result to mean that PqsE and RhlR physically interact, and this interaction is related to the activation of pyocyanin production in vivo. Our evidence also suggests that PqsE catalytic activity is dispensable for this function. It is curious that the PqsE E182W mutation strongly impairs the PqsE–RhlR-mBTL interaction given that the E182 residue is buried in the core of the protein (Figure 2). This finding suggests that this key residue does not itself interact with RhlR. Rather, we propose that there is an allosteric connection between the active site of PqsE and the RhlR-interaction site.
Although the PqsE E182W and S285W mutations were designed to mimic the binding of small molecule inhibitors in the active site, neither BB391 nor BB393 suppressed the interaction between PqsE and RhlR (Figure S8). This result implies that the inhibitor mimic mutations do not fully replicate the effects of BB391 and BB393 binding. Perhaps this finding should not be surprising given that BB391 and BB393 binding to PqsE did not drive large-scale conformational changes (Figure 2). Our current efforts are focused on structural characterization of PqsE E182W and linking conformational changes in the mutant protein to the RhlR interaction interface.
As mentioned in the preceding section, the P. aeruginosa PqsE E182W/S285W strain produced more pyocyanin than the P. aeruginosa PqsE E182W strain (Figure 6). The pulldown interaction assay in Figure 7 mirrors this finding, showing that the PqsE E182W/S285W mutant exhibited more interaction with RhlR-mBTL than did the PqsE E182W mutant. We do not yet understand the molecular basis for these parallel results, but it appears that alteration of PqsE S285 to tryptophan, when in the context of the PqsE E182W mutation, somewhat abrogates the defect caused by the change of E182 to tryptophan.
Discussion
P. aeruginosa remains a significant human health burden with tragic outcomes for immunocompromised individuals who acquire infections. P aeruginosa is a member of the ESKAPE class of highly resistant pathogens, making development of antimicrobials that are effective in treating P. aeruginosa infections a high-priority pursuit.7,24,33 In this study, we contribute to this effort by identifying small molecules targeting the P. aeruginosa virulence-controlling protein, PqsE. Successful modulation of PqsE could deliver a new class of antimicrobial agents that function by disrupting quorum-sensing-mediated cell–cell communication. Our approach exploited differential scanning fluorimetry to identify high-affinity PqsE binders, a strategy that was unbiased as to binding site or mechanism of action. We classified hit molecules as inhibitory or noninhibitory in a secondary assay quantifying PqsE enzymatic activity. Our follow-up characterization was focused on inhibitory compounds with chemical properties most amenable to further synthetic diversification. However, we note that the screen yielded an additional ∼80 confirmed binders that did not affect PqsE enzyme activity, as well as molecules that destabilized PqsE. All of these uncharacterized screening hits present opportunities for future analyses, mechanistic investigation, and potential therapeutic development. We highlight the compounds that destabilized PqsE as destabilization of PqsE could represent an especially attractive mechanism for impairing PqsE function to control virulence in vivo.
Based on their high affinity binding and desirable chemical features, hit molecules BB391 and BB393 were prioritized as amenable to structural analysis to identify their binding conformations in the active site of PqsE. The crystal structures presented here of the PqsE-BB391 and PqsE-BB393 complexes represent the first inhibitor-bound crystal structures of PqsE in which the bound inhibitor is not a small nonoptimized fragment but rather, a larger, drug-like ligand. These structures provide valuable information that can be used to inform future medicinal chemistry campaigns against PqsE. Moreover, the structures enabled analyses of the roles key residues play in the PqsE active site. Indeed, the structures guided the design of amino acid substitutions to generate PqsE variants that mimic the enzyme in an inhibitor-bound state. Tryptophan scanning mutagenesis has historically been used to enable structural characterization of transmembrane channels and other metastable proteins.34−37 However, the strategy of introducing tryptophan substitutions to mimic ligand binding as used here is underexplored and presents a potentially exciting route to pursue for validation of promising drug targets and investigation of inhibitor mechanisms of action for soluble cytoplasmic proteins.
In the realm of inhibitor target validation, P. aeruginosa presents a particularly daunting problem, as this pathogen is remarkably impermeable to small molecule reagents, possesses multiple drug efflux pumps, and voraciously metabolizes compounds of varied structures.38,39 Thus, it has often been impossible to accurately assess whether compounds with interesting in vitro activities are nonfunctional in vivo in P. aeruginosa or, alternatively, whether such compounds simply do not reach the cytoplasmic target for the reasons mentioned above. Our strategy of generating PqsE inhibitor mimetics enabled us to garner evidence suggesting there is indeed merit to the approach of inhibiting PqsE to suppress P. aeruginosa virulence. Moreover, our data confirm that, although the mechanism may not involve inhibition of catalytic activity, small molecule binding in the PqsE active site may suppress in vivo virulence traits. The work here also provides tools for new screens to identify molecules that affect the PqsE–RhlR interaction. For instance, the heterologous reporter system used in this study could be easily customized to enable high-throughput small molecule screening, potentially enabling identification of inhibitors of the PqsE–RhlR coupled activity that can subsequently be tested in P. aeruginosa.
Our data suggest that while PqsE catalytic activity (as judged by the catalytically inactive PqsE D73A variant) is not involved in regulating virulence factor production, mimicking the PqsE inhibited state (as judged by the PqsE E182W and PqsE E182W/S285W variants) does indeed confer a profound defect in a P. aeruginosa virulence phenotype: pyocyanin production. The question of whether targeting the PqsE active site has the potential to decrease P. aeruginosa virulence has been posed previously.23 The conundrum was that small molecules that inhibited PqsE in vitro were unable to suppress pyocyanin production in vivo. These results were interpreted to mean that PqsE has a nonenzymatic regulatory role in controlling P. aeruginosa virulence factor production. Our findings using our series of PqsE inhibitor mimetic mutants support this supposition. Furthermore, the in vitro physical interaction of each of our PqsE mutants with RhlR correlates with the corresponding in vivo pyocyanin production level that occurs when each mutant protein is present in P. aeruginosa PA14. We propose that complex formation enables PqsE to sensitize RhlR to its cognate autoinducer, C4-HSL, which upregulates RhlR-controlled virulence phenotypes. We caution that this protein–protein interaction has yet to be confirmed in P. aeruginosa, a focus of our ongoing work. Nonetheless, our data show that the PqsE variants that are incapable of interacting with RhlR in vitro closely mirror their inabilities to promote pyocyanin production and to drive transcription of hcnA and phzH in P. aeruginosa. These results support our interpretation that it is this PqsE–RhlR physical interaction, and not PqsE enzymatic activity, that is relevant to virulence. Moreover, we posit that targeting the PqsE–RhlR interaction should be prioritized in the pursuit of antimicrobial compounds that disrupt PqsE and, in turn, quorum-sensing-directed virulence.
Finally, it is curious that the tryptophan substitution at PqsE residue 182 (PqsE E182W) that disrupts the ability of PqsE to interact with RhlR is buried deep within the PqsE active site (Figure 2c,d). This location makes it unlikely that E182 participates directly in the PqsE–RhlR protein–protein interaction interface. Rather, the finding suggests that the PqsE enzyme active site may be allosterically connected to the RhlR-interaction site. The physiological importance of this allostery remains unknown, but our discovery of this interaction opens up the possibility for follow-up investigations into the identities of substrates and products of PqsE-catalyzed hydrolysis. Specifically, the only annotated substrate for PqsE is 2-ABA-CoA. As mentioned in the Introduction, PqsE is not absolutely required for converting 2-ABA-CoA to 2-ABA in the PQS biosynthetic pathway. In the new context of PqsE residing in close physical proximity to RhlR, it will be particularly interesting to identify additional in vivo PqsE substrates. Are there small molecule products of PqsE catalysis that subsequently bind to and modulate RhlR? Alternatively, does PqsE prevent small molecules from interacting with RhlR by catalyzing their hydrolysis? Does ligand binding to either protein affect this protein–protein interaction? If so, does the identity of the bound ligand determine the strength of the PqsE–RhlR interaction? We speculate that our unveiling of the PqsE–RhlR protein–protein interaction may accelerate progress in the discovery of the physiologically relevant in vivo reaction catalyzed by PqsE. Identification of that biochemical pathway could answer long-standing questions about the crucial, yet currently mysterious, function that PqsE plays in P. aeruginosa quorum sensing.
Materials and Methods
Strains, Media, and Molecular Procedures
P. aeruginosa UCBPP-PA14 was used for all experiments and strain constructions (referred to here as PA14). PqsE variants were constructed using a previously reported site-directed mutagenesis protocol with pqsE cloned on a plasmid.40 Primers were designed by following the Agilent Quikchange mutagenesis guidelines. Electrocompetent P. aeruginosa strains were transformed as described previously.41 All strains generated in this study are listed in Table S1. Unless otherwise stated, strains were grown in Luria–Bertani broth (Fisher). Antibiotics were used as follows: ampicillin (200 μg/mL), kanamycin (100 μg/mL), tetracycline (10 μg/mL), and carbenicillin (400 μg/mL). qRT-PCR analyses of select transcripts in PA14 strains were performed as described.17
Protein Expression and Purification
6xHis-PqsE was expressed and purified from recombinant E. coli as described previously27 with modifications. Briefly, protein was purified from E. coli BL21(DE3) harboring a pET28b vector containing the DNA encoding 6xHis-PqsE and variants. Cultures were grown with agitation at 37 °C to OD600 = 0.5. Protein production was induced by the addition of 1 mM IPTG followed by incubation with shaking at 37 °C for 4 h. The cells were harvested by centrifugation at 4000 rpm for 20 min and stored frozen at −80 °C. The pellet was thawed in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 20 mM imidazole, pH 8.0) supplemented with EDTA-free protease inhibitor tablets (Roche), and subsequently, the cells were lysed by sonication. The lysate was cleared by centrifugation at 12 000 rpm for 30 min, followed by incubation with Ni-NTA resin (Qiagen) for 1.5 h, before being packed into a gravity flow column. The column was washed with 10 column volumes of lysis buffer, and 6xHis-PqsE protein was eluted in elution buffer (50 mM Tris-HCl, 150 mM NaCl, 500 mM imidazole, pH 8.0). The eluate was subjected to overnight dialysis into gel filtration buffer (50 mM Tris-HCl, 150 mM NaCl, pH 8.0), prior to concentration to ∼1 mL and injection onto a Superdex 200 size-exclusion column for FPLC-assisted purification (Akta GE). Fractions containing a pure 34 kDa protein (determined by SDS-PAGE) were collected, pooled, and concentrated to ∼2 mg mL–1 followed by storage at −80 °C. Purification of PqsE protein for use in crystallography analyses had the following modifications: during overnight dialysis, the 6xHis tag was removed by the addition of biotinylated thrombin (Millipore 69022). Prior to size-exclusion chromatography, the biotinylated thrombin was removed on streptavidin-coated resin, and a second Ni purification was performed to remove any remaining PqsE containing the 6xHis tag. Following size-exclusion chromatography, the pooled PqsE-containing fractions were concentrated to ∼10 mg mL–1 and stored at 4 °C until use.
High-Throughput Differential Scanning Fluorimetry Screen
Compounds were screened for the ability to bind and shift the melting temperature (Tm) of purified 6xHis-PqsE using differential scanning fluorimetry (DSF). Compound solutions were prepared in DMSO and subsequently diluted in assay buffer (50 mM Tris, 150 mM NaCl, 2 mM MnCl2, pH 8.0) to achieve a final screening concentration of 37 μM compound in 1.85% DMSO. Compound solutions were transferred to the wells of a 384-well PCR plate (Matrix) using a PlateMate Plus liquid handler (Matrix) and control wells were prepared with DMSO (negative control) or 100 μM C1 (positive control). Plates were subjected to centrifugation for 10 s at 1000 rpm prior to the addition of a PqsE and dye master mix in assay buffer, so that the final concentrations of PqsE and SYPRO Orange were 4 μM and 20×, respectively. Plates were sealed with transparent seals (Excel Scientific) and subjected to centrifugation for 2 min at 2000 rpm. SYPRO Orange fluorescence (ex, 470 nm; em, 623 nm) was measured in a ViiA 7 Real-Time PCR system over an increasing temperature gradient from 50 to 90 °C (temperature increased at a rate of 0.1 °C/s). For the initial high-throughput screen, Tm was determined from first derivative plots of the raw fluorescence curves using custom scripts. In all subsequent experiments, Tm was determined using the Prism 8.0 software. Follow-up DSF assays were performed under similar conditions, with some modifications. Briefly, PqsE proteins were tested at 5 μM, compounds were assayed at 100 μM in a final concentration of 5% DMSO, and SYPRO Orange was used at 10×. The assay buffer in follow-up assays was 50 mM Tris, 150 mM NaCl, 200 μM MnCl2, and 10% glycerol, at pH 8.0, and measurements were made in a Quant Studio 6 Flex System (ThermoFisher).
Esterase Assay Measuring MU-Butyrate Hydrolysis
To measure PqsE enzyme activity, purified 6xHis-PqsE in assay buffer (50 mM Tricine, 0.01% Triton X-100, pH 8.5) was added to the wells of an opaque 384-well plate (Corning 3571) at 125 nM. MU-butyrate (Sigma) in assay buffer was added to the wells at a final concentration of 2 μM in a total volume of 20 μL per well. The plate was immediately incubated at RT in a Synergy plate-reader (BioTek), and fluorescence was monitored every 30 s for 30 min (ex, 360 nm; em, 450 nm). The fluorescence intensity measured after 2.5 min was plotted and normalized to the activity of WT PqsE. To assess the potencies of putative PqsE inhibitors, purified PqsE proteins were added at 200 nM to wells along with different concentrations of inhibitors supplied in DMSO, with a final DMSO concentration of 5%. Subsequently, MU-butyrate substrate was added at a final concentration of 2 μM, and the plate was incubated at RT for 20 min, prior to fluorescence quantitation. Control wells containing the inhibitor dilution series with MU-butyrate but lacking PqsE were included to establish the baseline fluorescence values, as well as to account for any inhibitor-derived fluorescence. IC50 curves were generated using the Prism 8.0 software.
Protein Crystallography
Crystals were grown at 22 °C using the hanging drop vapor diffusion method with a ∼10 mg mL–1 protein solution mixed at a 1:1 ratio with crystallization buffer (0.1 M HEPES pH 7.5, 0.2 M MgCl2, 32% (w/v) PEG 400). Crystals typically formed within 48 h at 22 °C. PqsE-ligand cocrystals were formed by soaking existing PqsE crystals with compound at 70–150 μM concentration in the crystallization drop for up to 48 h. No additional cryoprotectant was required prior to flash-cooling in liquid nitrogen. Crystals grew in space group P3221 with a = b = 60.7 Å, c = 146.2 Å, α = β = 90°, γ = 120 °C in a crystal form previously reported for PqsE with a single protomer in the asymmetric unit.25 Data were collected from flash-cooled crystals maintained at 100 K at the AMX (17-ID-1) beamline of the National Synchrotron Light Source II, processed with XDS42 and merged with AIMLESS.43 The starting model for the structure was from PDB entry 2Q0I.25 The structure was iteratively rebuilt with Coot44 and refined with PHENIX45 with ligands built into unambiguous differences in density in the active site. Ligands were refined with partial occupancy (0.81 for BB391 and 0.82 for BB393) with a TLS B-factor model for the protein atoms. Final refinement statistics are shown in the Supporting Information. The final structures were deposited in the Protein Data Bank with identifiers 7KGW (PqsE-BB391) and 7KGX (PqsE-BB393).
Fluorescence Polarization Compound Binding and Competition Assays
Dilutions of purified 6xHis-PqsE proteins were prepared in assay buffer (50 mM Tricine, 2 μM MnCl2, 0.01% Triton X-100, pH 8.5) and added to the wells of an opaque 384-well plate (Corning 3571) at a final volume of 20 μL per well. BB562 was diluted in assay buffer and added to the wells at a final concentration of 250 nM, and the plate was incubated at RT for 30 min. Fluorescence polarization was measured in a Synergy Neo2 plate-reader (Biotek) with excitation and emission wavelengths of 485 and 530 nm, respectively. Kapp values were determined using the Prism 8.0 software. For competition assays, 6xHis-PqsE was diluted to a final concentration of 250 nM in assay buffer and added to wells. The specified concentrations of competitor molecules in DMSO were added to the wells such that the final DMSO concentration was 5%. The protein–inhibitor complexes were allowed to form at RT for 10 min, followed by the addition of BB562 at a final concentration of 250 nM. The plate was incubated at RT for 30 min prior to the measurement of fluorescence polarization. EC50 values were determined using a variable slope inhibition curve fit to the data in the Prism 8.0 software.
Pyocyanin Assay
Overnight cultures of the ΔpqsE P. aeruginosa strain carrying the pUCP18 vector or the vector harboring WT or mutant pqsE genes were grown from single colonies in LB medium supplemented with carbenicillin at 37 °C. The cultures were diluted 1:1000 in 2 mL of fresh LB liquid medium with antibiotics and grown with shaking at 37 °C for 17 h. One milliliter of each culture was subjected to centrifugation at 13 000 rpm for 3 min. The clarified supernatants were collected, and the OD695 measured in a Beckman Coulter DU730 Spectrophotometer. The pellets were resuspended in PBS, and OD600 was measured to determine the cell density of each sample. Pyocyanin production was determined by normalizing OD695 of the clarified supernatant to the OD600 of the resuspended pellet. When added, test compounds were supplied at 100 μM in DMSO (final DMSO concentration = 1%) prior to the 17 h shaking incubation.
PqsE–RhlR Coupled PrhlA-lux Assay
Overnight cultures of E. coli strains harboring plasmids with rhlR driven by the PBAD promoter, luxCDABE under the PrhlA promoter, and either the pACYC184 vector or pACYC184 harboring WT pqsE or mutant pqsE alleles were grown from single colonies at 37 °C in LB medium supplemented with ampicillin, kanamycin, and tetracycline. The overnight cultures were diluted 1:100 into fresh LB medium containing antibiotics and 0.1% arabinose and added to the wells of a black, clear-bottomed 96-well plate at 150 μL per well. C4-HSL was added to the wells at 200 nM (1% DMSO). Plates were incubated for 8 h at 37 °C with shaking. Subsequently, bioluminescence and OD600 were measured in an Envision 2104 plate reader. Bioluminescence values are reported normalized to cell density (OD600).
PqsE–RhlR Interaction Pull-Down Assay
RhlR was produced using a previously reported method.32 Briefly, 25 mL cultures of E. coli BL21 (DE3) carrying rhlR on pET23b were grown to OD600 = 0.5, and expression of rhlR was induced by the addition of 1 mM IPTG, simultaneous with the addition of 100 μM mBTL. After 4 h at 25 °C, cells were collected by centrifugation, and pellets were frozen. For use, frozen pellets were resuspended in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 20 mM imidazole, pH 8.0) and lysed by sonication. Lysates were cleared by centrifugation at 13 000 rpm for 30 min, and the soluble fraction was collected. Lysates (100 μL) were combined with purified 6xHis-PqsE proteins at a final concentration of 9 μM 6xHis-PqsE in 200 μL total volume. The mixtures were allowed to incubate at 4 °C for 30 min prior to the addition of a 10 μL settle volume of MagneHis Ni Resin (Promega). Incubation was continued for an additional 1 h at 4 °C with turning. After incubation, aliquots of the lysate/PqsE/bead mixture were collected as “input” samples and prepared for SDS-PAGE analysis. The remaining mixtures were pelleted using a magnetic stand, and the resin was washed three times with lysis buffer. Ni-bound 6xHis-PqsE complexes were eluted in 200 μL elution buffer (50 mM Tris-HCl, 150 mM NaCl, 500 mM imidazole, pH 8.0), and prepared for SDS-PAGE analysis. Input and elution samples were subjected to SDS-PAGE on stain-free gels (Bio-Rad) and subsequently imaged after UV activation of the in-gel protein stain.
Acknowledgments
We thank members of the Bassler laboratory for helpful advice and discussions, especially G. Whitney for performing the qRT-PCR experiments. The high-throughput DSF screen and initial enzyme assays with potential hit molecules were performed at Bienta. Molecules used in this study were synthesized at WuXi AppTec. Protein crystallography was performed in the Macromolecular Crystallography Core Facility at Princeton University. The AMX beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory was also used under Contract DE-SC0012704. This work was supported by the Howard Hughes Medical Institute, NIH grant 2R37GM065859, and National Science Foundation grant MCB-1713731 to B. Bassler, NIH grant F32GM134583 to I. Taylor, and New York Community Trust Foundation grant P19-000454 to J. Paczkowski. The content herein is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00049.
One table, eight figures, extended screening methods, crystallography and refinement statistics, and synthetic methods (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Driscoll J. A.; Brody S. L.; Kollef M. H. (2007) The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67, 351–368. 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- Smith E. E.; Buckley D. G.; Wu Z.; Saenphimmachak C.; Hoffman L. R.; D’Argenio D. A.; Miller S. I.; Ramsey B. W.; Speert D. P.; Moskowitz S. M.; Burns J. L.; Kaul R.; Olson M. V. (2006) Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U. S. A. 103, 8487–8492. 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder I. A. (1993) P. aeruginosa Burn Infections: Pathogenesis and Treatment, in Pseudomonas aeruginosa as an Opportunistic Pathogen, pp 275–295, Springer, Boston, MA. [Google Scholar]
- Maurice N. M.; Bedi B.; Sadikot R. T. (2018) Pseudomonas aeruginosa Biofilms: Host Response and Clinical Implications in Lung Infections. Am. J. Respir. Cell Mol. Biol. 58, 428–439. 10.1165/rcmb.2017-0321TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M.-W.; Rahme L. G.; Sternberg J. A.; Tompkins R. G.; Ausubel F. M. (1999) Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc. Natl. Acad. Sci. U. S. A. 96, 2408. 10.1073/pnas.96.5.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z.; Raudonis R.; Glick B. R.; Lin T.-J.; Cheng Z. (2019) Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 37, 177–192. 10.1016/j.biotechadv.2018.11.013. [DOI] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Osmon S.; Ward S.; Fraser V. J.; Kollef M. H. (2004) Hospital mortality for patients with bacteremia due to Staphylococcus aureus or Pseudomonas aeruginosa. Chest 125, 607–616. 10.1378/chest.125.2.607. [DOI] [PubMed] [Google Scholar]
- Miller M. B.; Bassler B. L. (2001) Quorum sensing in bacteria. Annu. Rev. Microbiol. 55, 165–199. 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- Pearson J. P.; Passador L.; Iglewski B. H.; Greenberg E. P. (1995) A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 92, 1490–1494. 10.1073/pnas.92.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley M.; Lee K. M.; Greenberg E. P. (1999) Identification of genes controlled by quorum sensing in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 96, 13904–13909. 10.1073/pnas.96.24.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesci E. C.; Pearson J. P.; Seed P. C.; Iglewski B. H. (1997) Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 179, 3127–3132. 10.1128/JB.179.10.3127-3132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeb S.; Fletcher M. P.; Chhabra S. R.; Diggle S. P.; Williams P.; Cámara M. (2011) Quinolones: from antibiotics to autoinducers. FEMS Microbiology Reviews 35, 247–274. 10.1111/j.1574-6976.2010.00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau G. W.; Hassett D. J.; Ran H.; Kong F. (2004) The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol. Med. 10, 599–606. 10.1016/j.molmed.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Choi W.; Choe S.; Lin J.; Borchers M. T.; Kosmider B.; Vassallo R.; Limper A. H.; Lau G. W. (2020) Exendin-4 restores airway mucus homeostasis through the GLP1R-PKA-PPARγ-FOXA2-phosphatase signaling. Mucosal Immunol. 13, 637–651. 10.1038/s41385-020-0262-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees S. L.; Fetzner S. (2015) PqsE of Pseudomonas aeruginosa Acts as Pathway-Specific Thioesterase in the Biosynthesis of Alkylquinolone Signaling Molecules. Chem. Biol. 22, 611–618. 10.1016/j.chembiol.2015.04.012. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Moustafa D. A.; Stergioula V.; Smith C. D.; Goldberg J. B.; Bassler B. L. (2018) The PqsE and RhlR proteins are an autoinducer synthase-receptor pair that control virulence and biofilm development in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 115, E9411–E9418. 10.1073/pnas.1814023115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow J. M.; Sund Z. M.; Ellison M. L.; Wade D. S.; Coleman J. P.; Pesci E. C. (2008) PqsE functions independently of PqsR-Pseudomonas quinolone signal and enhances the rhl quorum-sensing system. J. Bacteriol. 190, 7043–7051. 10.1128/JB.00753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groleau M.-C.; de Oliveira Pereira T.; Dekimpe V.; Déziel E. (2020) PqsE Is Essential for RhlR-Dependent Quorum Sensing Regulation in Pseudomonas aeruginosa. mSystems 5, e00194-20. 10.1128/mSystems.00194-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Moustafa D.; Smith C. D.; Goldberg J. B.; Bassler B. L. (2017) The RhlR quorum-sensing receptor controls Pseudomonas aeruginosa pathogenesis and biofilm development independently of its canonical homoserine lactone autoinducer. PLoS Pathog. 13, e1006504 10.1371/journal.ppat.1006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Loughlin C. T.; Miller L. C.; Siryaporn A.; Drescher K.; Semmelhack M. F.; Bassler B. L. (2013) A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 110, 17981–17986. 10.1073/pnas.1316981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursier M. E.; Moore J. D.; Heitman K. M.; Shepardson-Fungairino S. P.; Combs J. B.; Koenig L. C.; Shin D.; Brown E. C.; Nagarajan R.; Blackwell H. E. (2018) Structure-Function Analyses of the N-Butanoyl l-Homoserine Lactone Quorum-Sensing Signal Define Features Critical to Activity in RhlR. ACS Chem. Biol. 13, 2655–2662. 10.1021/acschembio.8b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender M.; Witzgall F.; Drees S. L.; Weidel E.; Maurer C. K.; Fetzner S.; Blankenfeldt W.; Empting M.; Hartmann R. W. (2016) Dissecting the Multiple Roles of PqsE in Pseudomonas aeruginosa Virulence by Discovery of Small Tool Compounds. ACS Chem. Biol. 11, 1755–1763. 10.1021/acschembio.6b00156. [DOI] [PubMed] [Google Scholar]
- Baldelli V.; D’Angelo F.; Pavoncello V.; Fiscarelli E. V.; Visca P.; Rampioni G.; Leoni L. (2020) Identification of FDA-approved antivirulence drugs targeting the Pseudomonas aeruginosa quorum sensing effector protein PqsE. Virulence 11, 652–668. 10.1080/21505594.2020.1770508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.; Jensen V.; Seeliger J.; Feldmann I.; Weber S.; Schleicher E.; Häussler S.; Blankenfeldt W. (2009) Structure elucidation and preliminary assessment of hydrolase activity of PqsE, the Pseudomonas quinolone signal (PQS) response protein. Biochemistry 48, 10298–10307. 10.1021/bi900123j. [DOI] [PubMed] [Google Scholar]
- Folch B.; Déziel E.; Doucet N. (2013) Systematic Mutational Analysis of the Putative Hydrolase PqsE: Toward a Deeper Molecular Understanding of Virulence Acquisition in Pseudomonas aeruginosa. PLoS One 8, e73727 10.1371/journal.pone.0073727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan J. S.; Tota M. R.; Taylor I. R.; Stergioula V.; Hone G. A. B.; Smith C. D.; Henke B. R.; Carson K. G.; Bassler B. L. (2020) Discovery of PqsE Thioesterase Inhibitors for Pseudomonas aeruginosa Using DNA-Encoded Small Molecule Library Screening. ACS Chem. Biol. 15, 446–456. 10.1021/acschembio.9b00905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaneechoutte M.; Verschraegen G.; Claeys G.; Flamen P. (1988) Rapid identification of Branhamella catarrhalis with 4-methylumbelliferyl butyrate. J. Clin. Microbiol. 26, 1227–1228. 10.1128/JCM.26.6.1227-1228.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kübler D.; Ingenbosch K. N.; Bergmann A.; Weidmann M.; Hoffmann-Jacobsen K. (2015) Fluorescence spectroscopic analysis of the structure and dynamics of Bacillus subtilis lipase A governing its activity profile under alkaline conditions. Eur. Biophys. J. 44, 655–665. 10.1007/s00249-015-1061-6. [DOI] [PubMed] [Google Scholar]
- Page M. I.; Badarau A. (2008) The mechanisms of catalysis by metallo beta-lactamases. Bioinorg. Chem. Appl. 2008, 576297. 10.1155/2008/576297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampioni G.; Falcone M.; Heeb S.; Frangipani E.; Fletcher M. P.; Dubern J.-F.; Visca P.; Leoni L.; Cámara M.; Williams P. (2016) Unravelling the Genome-Wide Contributions of Specific 2-Alkyl-4-Quinolones and PqsE to Quorum Sensing in Pseudomonas aeruginosa. PLoS Pathog. 12, e1006029 10.1371/journal.ppat.1006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCready A. R.; Paczkowski J. E.; Cong J.-P.; Bassler B. L. (2019) An autoinducer-independent RhlR quorum-sensing receptor enables analysis of RhlR regulation. PLoS Pathog. 15, e1007820 10.1371/journal.ppat.1007820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister P. D.; Wolter D. J.; Hanson N. D. (2009) Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp L. L.; Zhou J.; Blair D. F. (1995) Features of MotA proton channel structure revealed by tryptophan-scanning mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 92, 7946–7950. 10.1073/pnas.92.17.7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen T.; Rasmussen A.; Singh S.; Galbiati H.; Edwards M. D.; Miller S.; Booth I. R. (2015) Properties of the Mechanosensitive Channel MscS Pore Revealed by Tryptophan Scanning Mutagenesis. Biochemistry 54, 4519–4530. 10.1021/acs.biochem.5b00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallée-Bélisle A.; Michnick S. W. (2012) Visualizing transient protein-folding intermediates by tryptophan-scanning mutagenesis. Nat. Struct. Mol. Biol. 19, 731–736. 10.1038/nsmb.2322. [DOI] [PubMed] [Google Scholar]
- Tang Q.; Villar M. T.; Artigues A.; Thyfault J. P.; Apte U.; Zhu H.; Peterson K. R.; Fenton A. W. (2019) Mutational mimics of allosteric effectors: a genome editing design to validate allosteric drug targets. Sci. Rep. 9, 9031. 10.1038/s41598-019-45202-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. E. (1998) Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria. Clin. Infect. Dis. 27, S93–S99. 10.1086/514909. [DOI] [PubMed] [Google Scholar]
- Breidenstein E. B. M.; de la Fuente-Núñez C.; Hancock R. E. W. (2011) Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 19, 419–426. 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- McCready A. R.; Paczkowski J. E.; Henke B. R.; Bassler B. L. (2019) Structural determinants driving homoserine lactone ligand selection in the Pseudomonas aeruginosa LasR quorum-sensing receptor. Proc. Natl. Acad. Sci. U. S. A. 116, 245. 10.1073/pnas.1817239116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.-H.; Kumar A.; Schweizer H. P. (2006) A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: Application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397. 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. (2013) How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Baker M. L.; Bunkóczi G.; Chen V. B.; Croll T. I.; Hintze B.; Hung L. W.; Jain S.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Poon B. K.; Prisant M. G.; Read R. J.; Richardson J. S.; Richardson D. C.; Sammito M. D.; Sobolev O. V.; Stockwell D. H.; Terwilliger T. C.; Urzhumtsev A. G.; Videau L. L.; Williams C. J.; Adams P. D. (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct Biol. 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.