

Abstract
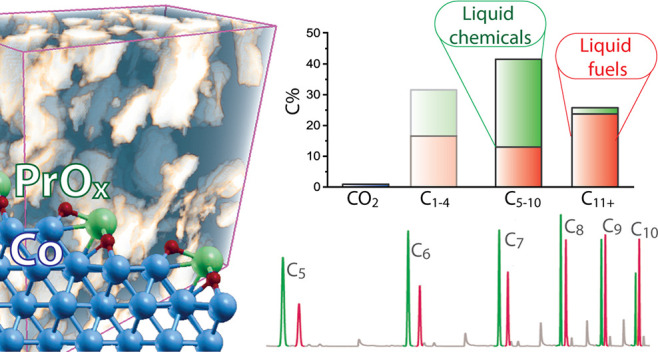
Adjusting hydrocarbon product distributions in the Fischer–Tropsch (FT) synthesis is of notable significance in the context of so-called X-to-liquids (XTL) technologies. While cobalt catalysts are selective to long-chain paraffin precursors for synthetic jet- and diesel-fuels, lighter (C10–) alkane condensates are less valuable for fuel production. Alternatively, iron carbide-based catalysts are suitable for the coproduction of paraffinic waxes alongside liquid (and gaseous) olefin chemicals; however, their activity for the water–gas-shift reaction (WGSR) is notoriously detrimental when hydrogen-rich syngas feeds, for example, derived from (unconventional) natural gas, are to be converted. Herein the roles of pore architecture and oxide promoters of Lewis basic character on CoRu/Al2O3 FT catalysts are systematically addressed, targeting the development of catalysts with unusually high selectivity to liquid olefins. Both alkali and lanthanide oxides lead to a decrease in turnover frequency. The latter, particularly PrOx, prove effective to boost the selectivity to liquid (C5–10) olefins without undesired WGSR activity. In situ CO-FTIR spectroscopy suggests a dual promotion via both electronic modification of surface Co sites and the inhibition of Lewis acidity on the support, which has direct implications for double-bond isomerization reactivity and thus the regioisomery of liquid olefin products. Density functional theory calculations ascribe oxide promotion to an enhanced competitive adsorption of molecular CO versus hydrogen and olefins on oxide-decorated cobalt surfaces, dampening (secondary) olefin hydrogenation, and suggest an exacerbated metal surface carbophilicity to underlie the undesired induction of WGSR activity by strongly electron-donating alkali oxide promoters. Enhanced pore molecular transport within a multimodal meso-macroporous architecture in combination with PrOx as promoter, at an optimal surface loading of 1 Prat nm–2, results in an unconventional product distribution, reconciling benefits intrinsic to Co- and Fe-based FT catalysts, respectively. A chain-growth probability of 0.75, and thus >70 C% selectivity to C5+ products, is achieved alongside lighter hydrocarbon (C5–10) condensates that are significantly enriched in added-value chemicals (67 C%), predominantly α-olefins but also linear alcohols, remarkably with essentially no CO2 side-production (<1%). Such unusual product distributions, integrating precursors for synthetic fuels and liquid platform chemicals, might be desired to diversify the scope and improve the economics of small-scale gas- and biomass-to-liquid processes.
Keywords: hierarchical porosity, promotion effects, linear olefins, (bio) syngas, DFT calculations
Introduction
The widespread availability of (unconventional) natural gas resources make gas-to-liquid (GTL) technologies an attractive alternative to current refining, which relies on centralized and dwindling crude oil supplies, for the production of fuels and chemicals.1,2 The Fischer–Tropsch (FT) synthesis forms the core of GTL processes, enabling the valorization of natural gas into synthetic hydrocarbon fuels and specialty lubricants via syngas (CO+H2) as a versatile intermediate.3 In a future scenario where personal and short-distance transport becomes largely electrified, synthetic GTL fuels are expected to play a central role in the heavy-duty ground transport and aviation sectors, where volumetric energy density considerations make liquid fuels a nearly irreplaceable choice.
Long-chain FT n-paraffin products (C11+) are excellent precursors for sulfur-free jetfuels–via hydroisomerization of the C11–16 fraction4,5—and high-cetane diesel fuels—via hydrocracking of heavier waxes.6 However, the broad statistical (Anderson–Schulz–Flory)7 hydrocarbon product distribution inherent to this polymerization reaction inevitably results in the coproduction of hydrocarbon fractions of lower value as fuel precursor. This includes tail-gases (C1–4) but also a significant fraction (typically >35 wt %) of condensable C5–10 alkanes, which are not suitable precursors for the aforementioned synthetic fuels because of their low boiling (<450 K) and flash (<320 K) points.
The FT reaction mechanism involves the surface polymerization of unbranched hydrocarbons from monocarbonated species, followed by chain-termination via either β-H-elimination or α-H-addition hydrogenation, leading to the desorption of α-olefin or n-paraffin products, respectively. Studies on model 2D catalysts have shown the former termination pathway to be favored and thus α-olefins to be the major primary reaction products.8 However, in technical porous catalysts, the final FT product mixture often does not reflect this intrinsic surface kinetics, and the condensable products are largely enriched in n-paraffins. After primary desorption, α-olefin products might readsorb on the catalyst surface and undergo secondary reactions, primarily hydrogenation to the corresponding n-paraffins but also chain-reinsertion and double-bond migration into internal isomers.9,10 This secondary processing is known to be pore-transport-enhanced, and thus, its extent depends not only on the reactivity of the catalyst surface but also on the pore residence time of olefin products as they egress by diffusion to the continuous phase.11 Favoring primary α-olefins over secondary n-paraffins products would add value to the FT lighter condensate (C5–10) slate.
Liquid linear α-olefins (LAO) are valuable commodity chemicals. LAO in the C5–10 range find applications as polymer comonomers, alkylating agents for the production of alkylbenzenes, precursors of specialty lubricants (via oligomerization) and raw materials for the synthesis of low-MW fatty acids, organosilanes and thiols for functional coatings,12−14 amines,15 as well as aldehyde and alcohol solvents and plasticizers via hydroformylation with syngas.16 Industrially, C5–10 LAO are largely produced by ethylene oligomerization, which yields mostly even-numbered hydrocarbon chains.17,18 Alternative production routes involve n-paraffin dehydrogenation, wax cracking, alcohol dehydration, or fatty acid decarboxylation/ethenolysis but are less widespread industrially.19,20 The coproduction of synthetic C5–10 α-olefins concomitantly to C11+ paraffinic diesel and jetfuel precursors in GTL processes could provide a future-proof source of two high-demand commodities from syngas. Moreover, downstream conversion of the former to C6–11n-alcohols via hydroformylation enables the formulation of high-cetane, drop-in syngas-derived fuels which exploit the remarkable soot inhibition properties of middle-chain n-alcohols.21,22
However, there are currently no specific catalysts leading to such unconventional product distribution from hydrogen-rich syngas (H2/CO ∼ 2). FeCx-based FT catalysts provide high olefin selectivities, particularly in the C10– fraction. However, their prominent activity for the water–gas-shift reaction (WGSR) leads to the rejection of a significant share of the oxygen in the syngas feed in the form of CO2.23,24 While this feature is valuable to convert syngas feeds with substoichiometric H2/CO ratios (0.7–1.2), for example, derived from coal gasification,25 it encumbers their use to process syngas derived from natural gas reforming or biomass steam gasification,26,27 with higher H2 contents (H2/CO ∼ 2). In the latter cases, the coproduction of CO2 would entail lower conversion efficiencies per-pass and significant tail-gas recycle compression costs.28 As a result, oxide-supported cobalt nanoparticles are the catalysts of choice in high-yield GTL technologies because of their high specific reaction rates at mild temperature (∼473 K), high selectivity to waxes, and intrinsic inactivity for the WGSR.29 However, their remarkable (secondary) hydrogenation reactivity efficiently depletes primary α-olefins, resulting in essentially paraffinic products. The design of CoMnOx30−32 catalysts has recently unlocked high selectivities to light (C2–4) olefins, remarkably, after dual promotion with Na and S, with very low WGSR activity.32 However, chain growth probabilities ≤0.5 limit their selectivity to liquid products.
Designing the pore architecture of Co-based FT catalysts to achieve fast pore transport rates has proven effective to minimize secondary hydrogenation and boost the share of liquid olefins in the products. This strategy proved useful to curb undesired overcracking in tandem FT/hydrocracking processes for the direct conversion of syngas into wax-free liquid hydrocarbons.33,34 The combination of pore transport enhancement with surface promotion effects could provide a blueprint toward unconventional FT product distributions uniting high selectivities to C5–10 α-olefin chemicals and heavier paraffinic fuel precursors while retaining the essentially null CO2 side-production inherent to cobalt-based FT catalysts. Here we address the combined effect of porosity and Lewis basic oxide promoters to develop γ-Al2O3-supported cobalt FT catalysts with high selectivity to liquid C5–10 olefins from H2-rich syngas.
Experimental Section
Synthesis of CoRu/γ-Al2O3 Catalysts
Meso-macroporous γ-Al2O3 was synthesized via soft-templating from an aqueous gel incorporating pseudoboehmite (75% Al2O3, Disperal P2, Sasol) and a polyethylene glycolether nonionic surfactant (Tergitol 15-S-7) as porogen (Al:EO:H2O = 1:8.1:49, EO: ethylene oxide subunits in the surfactant), followed by drying the mixture at 343 K for 72 h, and 393 K for 3 h, and subsequent calcination at 823 K (0.5 K min–1) in stagnant air. Mesoporous γ-Al2O3 supports were obtained by dehydration of high-purity pseudoboehmite precursors (Sasol Materials). CoRu/γ-Al2O3 catalysts were then synthesized on sieve fractions (0.2–0.4 mm) of the γ-Al2O3 supports via incipient wetness impregnation with a solution of Co(NO3)2·6H2O (1.5 M) and ruthenium(III) nitrosyl nitrate (Ru/Co = 0.007) in dilute nitric acid. After they were dried at 343 K under Ar flow (200 cm3 gcat–1) for 10 h, the precursors were decomposed by heating to 623 K for 4 h (1 K min–1) in a vertical downward flow reactor. Promoted CoRu/γ-Al2O3 catalysts were synthesized by wet impregnation with nitrate precursors of the respective promoter element dissolved in 0.5 M HNO3, followed by removal of H2O in a rotary evaporator (323 K) and calcination in air flow (200 cm3 gcat–1) for 4 h at 623 K. Catalysts were denoted xM-CoRu/AOm(p), where M indicates the identity of the promoter in the case of promoted catalysts and x specifies its surface specific content (Mat nm–2). AOm or alternatively AOmM designate either monomodal mesoporous or multimodal meso-macroporous γ-Al2O3 supports, respectively. For the former, p additionally denotes the average mesopore diameter.
Characterization Methods
N2 physisorption isotherms were recorded at 77 K using a Micromeritics 3Flex V4.04 device after degassing at 523 K under vacuum for 12 h. Hg intrusion porosimetry was performed in a Micromeritics AutoPore IV 951 apparatus after the sample (0.2–0.4 mm particles) was dried at 383 K for 72 h. H2 chemisorption isotherms were recorded at 373 K using an ASAP 2010C (Micromeritics) after in situ reduction in flowing H2 at 673 K for 5 h (2 K min–1) and degassing the sample at 1.3 Pa. High-angle annular dark-field scanning-transmission electron microscopy was performed with a spherical aberration-corrected beam (Cs) Hitachi HD-2700 microscope equipped with a cold field-emission gun and two EDAX Octane T Ultra W EDS detectors, operated at 200 kV. Samples were embedded in a low-viscosity resin, sectioned to ∼150 nm thick slices using a Reichert Ultracut ultramicrotome, and collected on Cu TEM grids (300 mesh) supporting a lacey carbon film. X-ray photoelectron spectra were recorded on a SPECS spectrometer with a Phoibos 150 MCD-9 detector and a nonmonochromatic (AlKα = 1486.6 eV) X-ray source after in situ reduction of pelletized samples under H2 flow at 673 K, and in certain instances exposure to FT reaction conditions in situ (T = 473 K, P = 10 bar, H2/CO = 2), followed by in vacuo transfer to the photoelectron spectroscopy chamber. FTIR spectroscopy experiments were performed in reactor cells featuring KRS-5 windows and mounted on a Bruker Vertex70 spectrometer. CO was applied as surface probe molecule at 298 and 110 K in order investigate metallic and oxide surface Lewis sites, respectively, on the in situ reduced catalysts. Prior to XPS and CO-FTIR experiments, catalysts had been reduced ex situ in H2 (70 cm3 min–1) at 673 K for 5 h (heating rate 1 K min–1), followed by a metal passivation treatment in flow of 1% O2/N2 for 1 h at room temperature to limit in situ reduction in the cells to the passivation overlay. CO2-TPD-MS was carried out on a Micromeritics TPD/2900 connected to a quadrupole mass spectrometer (Pfeiffer) after in situ reduction under flow of 10% H2/Ar (50 cm3 min–1) at 673 K for 2 h (heating rate 10 K min–1).
Catalysis
Catalytic experiments were performed in a fixed-bed high-Cr 316 stainless steel reactor loaded with 0.2–0.4 mm catalyst particles diluted with SiC granules (46 grit). Prior to reaction, the catalyst was reduced in situ under a flow of H2 at (200 cm3 min–1) at 673 K (2 K min–1 to 423 K, followed by 0.83 K min–1 to 673 K) for 5 h at atmospheric pressure. The reaction was carried out at 473 K and 20 bar using a feed with molar composition 30% CO/60% H2/10% Ar (Ar as internal GC standard). The stream leaving the reactor was depressurized and periodically analyzed online with a GC (Agilent 7890B) equipped with two TCD detectors and one FID detector, while liquid and solid hydrocarbons were collected in high-pressure traps, and analyzed offline. Product selectivities are reported on a carbon basis in the pseudosteady state, that is, after at least 16 h on-stream at the corresponding feed space velocity (WHSV).
First-Principles Density Functional Theory (DFT) Calculations
Periodic DFT calculations were performed using the Vienna Ab-initio Simulation Package (VASP).35,36 The exchange-correlation energy was calculated with the PBE37 form of the generalized gradient approximation (GGA) functional. The electron–ion interaction was modeled by the projector-augmented wave (PAW)38 method. Spin-polarized calculations were performed to account for the magnetic properties of cobalt with a plane wave cutoff energy of 600 eV.
Additional experimental and computational details are provided in the Supporting Information.
Results and Discussion
Catalyst Design
The porosity of the carrier material is known to play a central role for pore mass transport phenomena and selectivity in the cobalt-catalyzed Fischer–Tropsch synthesis.9,39 Table S1 in the Supporting Information summarizes the textural properties determined for the series of γ-Al2O3 support materials. Their porous structure was assessed by Hg intrusion porosimetry in order to probe the meso- as well as the macropore regimes. Figure 1a shows the corresponding pore size distributions. Monomodal and narrow size distributions peaking at 6.8 and 11.2 nm were observed for AOm(7) and AOm(11), respectively. In the case of the wider-pore AOm(24), the dominant pore population centered at 24.3 nm appeared complemented by a shoulder at 49.6 nm, suggesting the presence of a second, minor population of wider openings. Pore volume contributions in the macropore regime (>50 nm) were essentially negligible in all cases (≤0.05 cm3 g–1), indicating that this set of materials represents essentially mesoporous supports covering a wide range of pore diameters. A multimodal pore size distribution was ascertained in the case of AOmM, with mesopore modes peaking at 8.8 and 37 nm, respectively, alongside a significant contribution from macropores (0.46 cm3 g–1) with diameters extending in a wider range centered at around 1 μm. In contrast to the even outer surface observed by SEM for the monomodal mesoporous carriers (Figure 1b,c), macropores were found to protrude to the outer surface of the microparticles in AOmM (Figure 1d,e). Focused-ion-beam scanning-electron-tomography (FIB-SEM) was applied to image the internal macropore architecture of this hierarchically porous support. As observed in the cross-sectional SEM micrograph (Figure 1f) and the corresponding reconstructed tomogram (Figure 1g), irregularly shaped macropores were found to be evenly distributed within the inner volume of the material.
Figure 1.

Porosity assessment for γ-Al2O3 support materials. (a) Pore size distributions as derived by Hg intrusion porosimetry for the series of γ-Al2O3 support materials. (b,c) Representative SEM micrographs for microparticles of AOm(11) and AOm(24), respectively, showing no signs of macropores on their outer surface; (d,e) Representative scanning electron micrographs for microparticles of AOmM showing the percolation of macropores to the outer surface. (f) Cross-sectional SEM micrograph after focus-ion-beam (FIB) milling of the resin-embedded AOmM support. Lighter gray regions correspond to mesoporous Al2O3 regions, while dark gray patches correspond to macropores cross sections. (g) 3D-rendered view of a reconstructed FIB-SEM tomogram for AOmM. Purple regions correspond to mesoporous Al2O3 and white regions to intraparticle macropores.
Quantitative image analysis of the FIB-SEM tomogram revealed a high degree of connectivity for the macropore network, with an average coordination at the intersecting nodes of the macropore network model of 3.1 ± 1.3. More importantly, the network of interconnected macropores results in a significant partitioning of the mesoporous γ-Al2O3 domains, reducing the average transport distances within mesoporous regions down below 5 μm (Figure 2). Compared with mesoporous domains extending over the entire microparticle diameter (200–400 μm) in the case of the series of strictly mesoporous γ-Al2O3 supports, these shorter mesopore transport distances are expected to significantly enhance pore molecular transport.40
Figure 2.
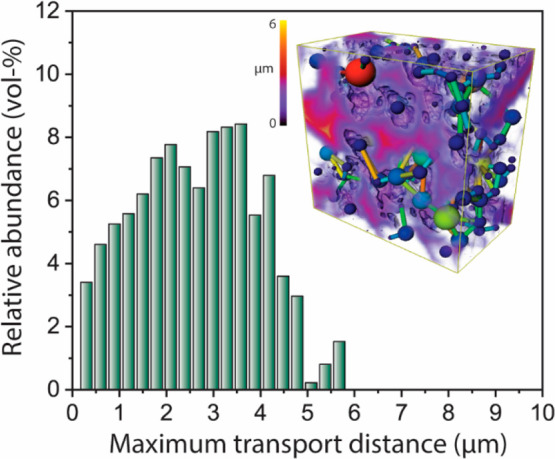
Histogram for the maximum Euclidean distance from mesopore regions to the nearest boundary with the macropore network as derived from 3D image analysis of the reconstructed FIB-SEM tomogram for the bimodally meso-macroporous γ-Al2O3 support material (AOmM). The inset shows the 3D contour plot for the Euclidean distance to nearest macropore as well as the computed Pore Network Model, with macropores shown as balls and throats connecting them as bars. See details for the 3D tomographic image quantification in the Supporting Information.
Ruthenium-promoted41 cobalt FT catalysts were synthesized on the set of Al2O3 support materials. In all cases, the nominal cobalt surface-specific loading was set constant to 9.0 ± 1.0 Coat nm–2 in order to achieve a uniform surface density of metal sites in the series of catalysts. This translated into Co loadings in the range of 9–22 wt % Co on the set of γ-Al2O3 carriers which showed BET specific surface areas spanning 96–300 m2 g–1 (Figure S1, Table S1). As oxide promoters, a series of alkali (Na, K, Cs) and lanthanide (La, Pr, Sm) oxides were additionally deposited on the surface of the cobalt FT catalysts at preset surface-specific loadings in the range of 0.1–4.0 Mat nm–2. The two series of promoters share a common Lewis basic character, which in the case of the alkali oxides stems from the strong e-donor character of their O2– groups, while in the case of lanthanide oxides it is related to the electrodonating capacity of Lnx+ cations. Moreover, while those lanthanide elements studied as promoters share very similar ionic radii in the range of 96–103 pm, the ionic radii increases notably with the atomic number, from 102 pm for Na to 167 pm for Cs, for the series of alkali promoter elements.42 Bulk energy dispersive X-ray spectroscopy (EDS) analysis showed experimental promoter loadings in good agreement with the nominal contents, with relative deviations ≤20% (Table S2).
Co3O4 spinel was the only cobalt phase detected in all catalysts in their as-calcined state (Figure S2). Cobalt reducibility was studied with H2-temperature-programmed reduction (Figures S3 and S4). Two well-defined H2 consumption events were registered in the temperature range of 400–700 K, which are characteristic for the stepwise reduction of Co3O4 to metallic cobalt via CoO as intermediate.43 While these two reduction bands peaked at 450 and 655 K, respectively, for an unpromoted CoRu/AOmM catalyst, both reduction events shifted progressively to higher temperatures, by up to 77 and 33 K, respectively, on incorporation of a lanthanide oxide such as PrOx at increasing contents up to 1.0 Prat nm–2, suggesting that interaction of the oxide promoter and the cobalt (oxide) species retards the reduction of the latter. Higher promoter loadings up to 3.0 Prat nm–2, however, led to a slight increase in reducibility as exemplified by a progressive down-shift of the CoO-to-Co0 reduction temperature (Figure S3), possibly as a result of a reduced interaction of cobalt species with the strongest acid sites on the Al2O3 carrier. A remarkably similar trend was observed when an alkali oxide such as NaOx was applied as promoter (Figure S4). Interestingly, catalysts incorporating a reference surface content of 1.0 Mat nm–2 of different promoter oxides representative of the entire series studied herein (i.e., PrOx, NaOx, and CsOx) showed essentially identical H2-TPR traces, indicating that the surface atomic loading rather than the identity of the promoter determined cobalt reducibility, likely as a result of similar degrees of promoter-cobalt interaction in all cases. On the basis of the H2-TPR profiles, a reduction temperature of 673 K was selected to achieve essentially full cobalt reduction prior to catalysis.
For reasons which are going to be detailed later, further characterization studies focused on the series of catalysts incorporating PrOx as promoter. Cobalt and promoter speciation were studied on selected catalysts after reduction activation by means of X-ray photoelectron spectroscopy (XPS). Figure 3a shows the XP spectra in the Co 2p and Pr 3d5/2 regions for 1.0Pr-CoRu/AOmM. Similar spectra were obtained for 3.0Pr-CoRu/AOmM, with a higher PrOx loading (Figure S5). As expected from the H2-TPR profiles, metallic Co0 (BE Co 2p3/2 = 777.2–777.5 eV) is the major near-surface cobalt species after reduction (≥90%). The minor Co2+ contributions (BE Co 2p3/2 = 780.4–780.9 eV) are likely overrepresented after the in situ catalyst reduction preceding XPS experiments because of the poorer gas–solid hydrodynamics around pelletized samples and thus less efficient water convective evacuation, compared with packed-bed configurations applied for activation prior to catalysis. Further analysis of metal core–electron BEs for selected catalysts after in situ reduction and Fischer–Tropsch catalysis suggested that Ru, while intimately alloyed with Co after reductive activation, tends to segregate into Ru-rich aggregates under reaction conditions (see Table S3 and accompanying discussion). Moreover, the addition of praseodymium leads to a slight down-shift (by 0.2–0.3 eV), of the Co 2p BE for metallic cobalt, suggestive for a slight depletion in electronic charge on the near-surface Co0 atoms and therefore the electron-withdrawing character of the PrOx species. The latter persists in oxidic form, predominantly as Pr4+, with ≤30% atomic contributions from Pr3+, both after catalyst reduction and following FT reaction conditions (Table S3).
Figure 3.

Surface characterization of PrOx-promoted Co-based FT catalysts. (a) X-ray photoelectron spectrum in the Co 2p and Pr 3d5/2 spectral regions for 1.0Pr-CoRu/AOmM after H2 reduction. (b) Cobalt-specific H2 chemisorption uptake for the series of Pr-CoRu/AOmM catalysts supported on multimodal meso-macroporous γ-Al2O3 as a function of the surface-specific praseodymium content. Error bars correspond to the standard error as determined from three independent chemisorption experiments on selected catalysts. The line is meant as a guide to the eye and it applies to the series of Pr-CoRu/AOmM catalysts.
The dispersion and spatial distribution of cobalt and PrOx on selected catalysts after H2 reduction was assessed by means of Cs aberration-corrected scanning-transmission electron microscopy (Cs-STEM) and EDS nanospectroscopy on ultramicrotomed catalyst cross sections. Remarkably uniform spatial distributions for cobalt nanoparticles were ascertained at both the meso- and nanoscopic scales for an unpromoted CoRu/AOmM catalyst (Figure S6). The thermal decomposition of the nitrate catalyst precursors applied herein and developed previously by de Jong and co-workers44 effectively prevents the clustering of cobalt (oxide) nanocrystals, which therefore occupy the available support surface area with essentially maximum interparticle spacing. Figures 4 and S7 show representative micrographs and EDS elemental maps for the corresponding PrOx-promoted 1.0Pr-CoRu/AOmM. The excellent cobalt dispersion is preserved following the incorporation of the oxide promoter. Moreover, PrOx appears as a nonparticulate phase, also evenly distributed on the catalyst surface at all analysis length scales. No preferential association of PrOx species with cobalt was observed. Instead, praseodymium oxide appears highly dispersed and closely associated with the γ-Al2O3 carrier, likely due to a stronger interaction facilitated by the complementary mild basic and acidic surface character of these two oxides, respectively. Similarly, excellent dispersions and uniform spatial distributions for Co and PrOx were also observed at higher Pr loadings (Figure S8).
Figure 4.

Bright field Cs-STEM and EDS microanalysis on ultramicrotomed catalyst cross sections of 1.0Pr-CoRu/AOmM. (a) Representative BF-STEM mesoscale micrograph and; (b) the corresponding ED spectrum. Spectral contributions from the embedding carbonaecous resin and the copper grid are marked with asterisks. (c,d,e) EDS compositional maps for the region imaged in (a) for Al, Co, and Pr, respectively, obtained from the corresponding EDS K-lines. (f) Representative nanoscale EDS compositional map for Co. (g) Surface-averaged cobalt nanoparticle size distribution.
Geometrical surface-averaged mean cobalt nanoparticle sizes, as determined from analysis of the STEM-EDX results, were similar (ds = 10.2 ± 0.9 nm) for all catalysts studied, discarding any significant effect of PrOx incorporation on cobalt dispersion. The exposed metal surface area was quantified with H2-chemisorption. Figure 3b shows its evolution with the Pr content for the series of Pr-CoRu/AOmM catalysts. A metal surface area of 69 m2 gCo–1 was determined for the unpromoted CoRu/AOmM, in fair agreement with the value of 61 m2 gCo–1 derived on geometrical considerations from STEM-EDS results. A ca. 12% drop in the exposed metal surface area is experienced upon incorporation of even minute amounts of the lanthanide oxide (0.4 Prat nm–2), suggesting the blockage of a fraction of the surface metal centers on the Co nanoparticles. Further increasing Pr loading up to 2.0 Prat nm–2 resulted in no statistically significant modifications of the metal surface area. Only upon further increasing the Pr content up to ≥3.0 Prat nm–2 was the available metal surface area further decreased, down to about 71% of that in the unpromoted catalyst. These results suggest that only a comparatively minor fraction of the metal centers become blocked by PrOx species, already from comparatively low promoter loadings. The surplus praseodymium oxide interacts preferentially with the γ-Al2O3 surface and does not lead to further metal blockage for Pr loadings up to 2 Prat nm–2, beyond which point the promoter coverage on the metal increases more markedly with loading, likely as a result of decreased availability of binding centers on γ-Al2O3 for further PrOx uptake. At an identical reference oxide promoter surface loading of 1.0 Mat nm–2, NaOx led to a drop in the exposed Co surface of 12%, similar to that observed for PrOx, while for CsOx this decrease reached 26%, suggesting that the capacity of the oxide promoters to block the metal surface scales with the cation size.
Fischer–Tropsch Synthesis
The Fischer–Tropsch synthesis performance was evaluated in a fixed-bed reactor setup at industrially relevant conditions. The reaction temperature was set to 473 K, at the lower end of the technically significant temperature range for cobalt-based FT catalysis, in order to minimize secondary hydrogenation activity. Unpromoted CoRu/Al2O3 catalysts showed cobalt-specific reaction rates (cobalt-time-yield, CTY) in the range of 89–242 mmol CO gCo–1 h–1, which translated into similar turnover frequencies (TOF) per unit surface metal atom of 2.5 ± 1.0 × 10–2 s−1. At similar CO conversion levels of 20 ± 3%, CH4 selectivity spanned the range of 7.9–11.6%, while the selectivity to CO2 remained ≤0.6% in all cases, illustrating the inactivity of cobalt FT catalysts to the WGSR (Table 1, Table S4). ASF chain-growth probabilities (α) were comparatively similar in the range of 0.79–0.83, resulting in selectivities to condensable C5+ hydrocarbons of 74.3–80.8%. In spite of the comparable chain-length product distributions, the share of olefins in the C5–10 fraction depended conspicuously on the support pore diameter, increasing from 33.4% to 54.7% as the support pore size increased from 7 nm (monomodally mesoporous) to the more open pore architecture of the multimodal meso-macroporous AOmM. These results illustrate the direct dependence of the olefinicity for liquid product fractions on the effective (mesopore) transport distances and thus α-olefin pore residence times within the catalyst microparticles.
Table 1. Cobalt-Time-Yield (CTY), Product Selectivities, and Hydrocarbon Chain-Growth Probability (α) for Co-Based FT-Catalysts Supported on a Multimodal Meso-Macroporous γ-Al2O3 Support, and Optionally Modified with Various Alkali and Lanthanide Oxide Promotersa.
| catalyst | CTYb [mmol CO gCo−1 h−1] | S(CO2) [C%] | S(CH4) [C%] | S(C5+) [C%] | S(C5–10 Olef.) [C%] | αc [-] | C2–4 Olef.d [%] | C5–10 Olef.e [%] |
|---|---|---|---|---|---|---|---|---|
| CoRu/AOmM | 88.9 | 0.6 | 7.9 | 79.2 | 13.3 | 0.80 | 56.5 | 54.7 |
| 1.0Na-CoRu/AOmM | 32.9 | 1.0 | 8.0 | 76.0 | 14.4 | 0.77 | 54.3 | 56.2 |
| 3.0Na-CoRu/AOmM | 8.5 | 8.1 | 10.8 | 53.2 | 10.1 | 0.66 | 36.1 | 53.1 |
| 1.0K-CoRu/AOmM | 27.5 | 1.3 | 8.6 | 74.4 | 13.9 | 0.78 | 54.1 | 57.2 |
| 1.0Cs-CoRu/AOmM | 10.9 | 2.9 | 11.4 | 65.0 | 12.1 | 0.71 | 56.7 | 54.6 |
| 1.0La-CoRu/AOmM | 43.1 | 0.8 | 8.2 | 73.2 | 16.2 | 0.75 | 54.7 | 58.1 |
| 1.0Pr-CoRu/AOmM | 36.7 | 0.9 | 8.4 | 70.2 | 19.9 | 0.75 | 57.5 | 61.5 |
| 3.0Pr-CoRu/AOmM | 12.4 | 1.3 | 12.4 | 54.6 | 19.5 | 0.66 | 53.0 | 61.7 |
| 1.0Sm-CoRu/AOmM | 47.3 | 1.1 | 9.2 | 73.6 | 14.6 | 0.76 | 54.8 | 53.2 |
Reaction conditions: T = 473 K, P = 20 bar, H2/CO = 2, WHSV = 5.1–11.0 h–1, CO conversion = 20 ± 3%.
Cobalt-time-yield.
Chain-growth probability.
Molar olefin abundance within the C2–4 hydrocarbon product fraction.
Molar olefin abundance within the C5–10 hydrocarbon product fraction.
The addition of any of the Lewis basic promoters studied, whether alkali or lanthanide oxide, resulted in a decrease in CTY with respect to the unpromoted counterpart. This is illustrated in Table 1 for a set of CoRu/AOmM catalysts incorporating different promoter oxides at selected loadings of 1.0 and 3.0 Mat nm–2. These results tie in well with previous reports of a decreasing FT activity by incorporation of basic oxides of alkali and alkaline earth metals45−47 or lanthanum48,49 on Co-based FT catalysts. At a comparable loading of 1.0 Mat nm–2, the decrease in CTY was more pronounced (63–88%) and increased with increasing the cation size and decreasing electronegativity for the series of alkali promoters (Na < K < Cs), while it was less prominent and rather similar (47–59%) among the different lanthanide oxides studied.
Product selectivity was evaluated at an equivalent CO conversion level of 20%. All promoters led to a decrease in the chain-growth probability determined from the linearized ASF plots (Figure S9) and thus a decrease in selectivity to C5+ hydrocarbons. Both reductions47 and increments50,51 in C5+ selectivity have been reported previously for alkali-modified Co/γ-Al2O3 catalysts. Our results systematically point to a lower effective chain propagation upon modification with Lewis basic promoters, regardless of their identity. With regard to product olefinicity, while the effect was found to be limited for alkali oxides, lanthanide oxides such as La and most particularly Pr led to a significant increase in the olefin abundance within the C5–10 product fraction up to ca. 62%. Moreover, this enhancement in olefinicity for the lighter condensate products was achieved while retaining the essentially null WGSR activity which is intrinsic to metallic cobalt, with selectivities to CO2 ≤ 1.3%. In contrast, the addition of alkali oxides led to enhanced WGSR activities (SCO2 up to 8%) at comparable surface contents, in line with previous observations.46,47,51 Therefore, while alkali and alkaline earth (hydroxides) have been previously proposed as promoters to synthesize cobalt-based FT catalysts with enhanced selectivities to lower (C2–4) olefins at higher operation temperatures,30,31,52 it emerges from our results that lanthanide oxides, particularly PrOx, are preferred as promoters to target high selectivity to liquid (C5+) olefins at milder operation conditions.
Figure 5 summarizes the impact of the promoter surface loading on activity and selectivity for the series of xPr-CoRu/AOmM catalysts. TOF decreased progressively with the Pr content, mirroring the behavior observed for CTY. This decrease in site-specific activity was pronounced already from low Pr contents and up to a Pr loading of 1.0 Prat nm–2, indicating that comparably low promoter surface coverages on cobalt are sufficient to notably reduce the intrinsic CO hydrogenation activity. Regarding product selectivity, praseodymium contents up to 1.0 Prat nm–2 had only marginal effects on the selectivity to CH4 and CO2, which remained within 8.0–8.4% and 0.6–0.9%, respectively. The overall selectivity to C5+ condensates did show a measurable and progressive decrease from 79.2% to 70.2% with increasing Pr content in the same range, as a result of a decrease in α from 0.80 to 0.75. More remarkably, the olefin abundance within the C5–10 fraction increased systematically to exceed 61% at a promoter loading of 1.0 Prat nm–2. Further increments in Pr loading up to 3 Prat nm–2 led to a more pronounced decrease in SC5+, with CH4 selectivity reaching up to 12.4%, while the selectivity to olefins within the C5–10 slate essentially plateaued off. Hence, a Pr loading of 1.0 Prat nm–2 was found to maximize the selectivity to liquid olefins while preserving a low methanation activity and CO2 side-production.
Figure 5.

Impact of PrOx promoter loading on catalyst performance. Evolution of (a) the cobalt-time-yield (metal-specific CO conversion rate) and the turnover frequency (TOF), per unit surface metal site as quantified with H2 chemisorption; and (b) the C5–C10 olefin abundance and hydrocarbon selectivities obtained with Pr-CoRu/AOmM catalysts supported on a multimodal meso-macroporous γ-Al2O3, as a function of the surface-specific praseodymium loading. Reaction conditions: T = 473 K, P = 20 bar, H2/CO = 2, WHSV = 8.7–18.0 h–1, CO conversion = 20 ± 3%.
Moreover, open porosity and PrOx promotion both enhanced the selectivity to linear alcohols, which improved from 5.0 C% for the mesoporous CoRu/AOm(7) to 8.3 C% for the multimodally porous CoRu/AOmM and reached 11.7 C% for 1.0Pr-CoRu/AOmM, for which an alcohol chain growth probability of αOH = 0.73 was determined (Figure S10). For the latter catalyst, this added up to an overall 67% carbon abundance of chemicals (1-alcohols and olefins) in the C5–10 product fraction. The formation of n-alcohols on cobalt-based catalysts under syngas conversion conditions has been previously associated with the presence of Co2C/Co0 interfaces on the working catalysts.53 After prolonged exposure to FT reaction conditions, no signs for Co2C crystallites could be detected by XRD for neither the unpromoted CoRu/AOmM nor for 1.0Pr-CoRu/AOmM (Figure S11). However, contact with the PrOx promoter might create small CoCx surface domains which, undetectable by XRD, promote alcohol synthesis. Jointly, these results illustrate that, PrOx-promotion, albeit at the expense of the overall CO conversion rate, led to a FT product distribution which is desirable for a combined production of paraffinic middle-distillates and waxes (C11+) and higher value C5–10 chemicals, essentially without CO2 side-production, from H2-rich syngas.
The systematic set of catalysts addressed in this study enabled disentangling the contributions of pore architecture and oxide promotion to this unusual product distribution. Neither the multimodal meso-macroporous structure nor the addition of PrOx as surface promoter modified stability (∼200 h on-stream) under Fischer–Tropsch reaction conditions (Figure S12). Figure 6a shows the evolution of the olefin-to-paraffin (O/P) molar ratio in the range of C3–10 as a function of the hydrocarbon chain length for both unpromoted and PrOx-modified (1.0 Prat nm–2) catalysts supported on γ-Al2O3 carriers of varying pore diameter. For every catalyst, the O/P-ratio decreases with the hydrocarbon chain length, an effect which is ascribed to the chain-length dependent average pore residence time of α-olefin primary products, thus the extent to which they undergo secondary hydrogenation, under reaction conditions.9 In all cases, the addition of PrOx as promoter led to a substantial increase in olefinicity. As observed in Figure 6b, the magnitude of this effect scales systematically with the support porosity, being more marked for narrow-pore catalysts and decaying progressively in magnitude as the catalyst pore architecture becomes more open. Therefore, while the role of PrOx is likely the same in all cases, that is, reducing the hydrogenation capability of surface metal sites responsible for secondary olefin hydrogenation, the magnitude of the promotion depends as well on the effective (meso)pore transport lengths for α-olefin primary products. In the study space addressed, the combined effect of porosity design and PrOx promotion leads to a ca. 2-fold raise in olefin selectivity within the liquid C5–10 products.
Figure 6.

Combined effects of support porosity and chemical promotion on the selectivity to liquid olefins. Evolution of (a) the olefin-to-paraffin molar ratio with the carbon chain-length for C3–10 hydrocarbon products and; (b) the olefin molar abundance within the C5–10 liquid products with the average support pore diameter, obtained with various cobalt-based FT catalysts supported on unimodal mesoporous or multimodal meso-macroporous γ-Al2O3 carriers of varying pore diameters either in their unpromoted form (open symbols and dotted lines) or promoted with PrOx (1.0 Prat nm2) (closed symbols and solid lines). In panel (b), catalysts to the right of the x-axis interrupt show a multimodal pore architecture with both meso- and macropores and thus no specific average pore diameter has been assigned. Reaction conditions: T = 473 K, P = 20 bar, H2/CO = 2, WHSV = 5.5–33.0 h–1, CO conversion = 20 ± 3%. The gray-shaded area on panel (b) illustrates the increase in olefinicity for the C5–10 liquid FT products due to the incorporation of PrOx as promoter.
The hydrogenation of olefins on metal nanoparticles is considered to be a structure insensitive reaction under H2 atmospheres, and hence, activity scales linearly with the available metal surface area.54,55 Compared with standard hydrogenation conditions, under Fischer–Tropsch reaction settings, molecular CO is known to be the dominant adsorbate on metallic cobalt (and ruthenium) FT catalysts, leaving fewer available vacant sites for olefin adsorption and H2 dissociation.56 For the series of catalysts studied here, no correlation was found between the cobalt surface area and the O/P (Figure S13), suggesting that simply a reduction in the exposed metal surface does not explain, on its own, the lower extent of olefin hydrogenation. It thus stands to reason to infer that PrOx brings about a preferential dampening of the cobalt hydrogenation reactivity.
In addition to hydrogenation, α-olefin primary FT products might undergo other secondary reactions, which include not only reinsertion into growing hydrocarbon chains but also double-bond isomerization to linear 2-alkenes, which may be catalyzed by metallic sites,8 as well as acidic or basic centers of the oxide catalyst support.57,58 A number of interesting downstream upgrading routes for liquid FT olefins (e.g., hydrosylilation, hydroformylation, etc.) benefit from high selectivity to end-of-chain functionalization.59−62 Therefore, the terminal-to-internal (T/I) ratio in the C5–10 olefin products is of interest as a figure-of-merit for FT catalysts designed to achieve high selectivity to liquid olefins. Under FT reaction conditions, the T/I olefin ratio decreased progressively with increasing the hydrocarbon chain length in all cases (Figure S14), reflecting that olefin isomerization secondary reactions are also transport-enhanced. As a result, in the absence of oxide promoters, the α-olefin abundance in the C5–10 olefin products increased with increasing the support pore size, from 79.5% for CoRu/AOm(7) to 92.2% in the case of the hierarchically porous CoRu/AOmM. On top of these porosity effects, the addition of PrOx as promoter on the surface of the meso-macroporous CoRu/AOmM catalysts led to a further and significant enhancement in the terminal olefin abundance, already from very low Pr loadings of 0.1 Prat nm–2. For Pr contents ≥1.0 Prat nm–2, abundances of α-olefins in excess of 95% in the C5–10 fraction were achieved. Essentially all olefin products in this product fraction were found to be linear, as no branched olefin isomers could be detected (Figure EM2b in the Supporting Information). These high structural and regio-isomery selectivities are desired for downstream olefin functionalization into linear specialty chemicals (e.g., alcohols via olefin (reductive) hydroformylation with syngas).
Additional insights into the promotional effects on secondary olefin double-bond shift reactions were gathered from the analysis of the steroisomery of 2-butene products. Similarly to the T/I ratio, the impact of the PrOx promoter on the 2-butene stereoselectivity was apparent already from very low Pr loadings (Figure S15). The cis/trans 2-butene ratio in the products increased steeply from ca. 1.4 for the unpromoted CoRu/AOmM to >1.9 for 0.1Pr-CoRu/AOmM. Further, less pronounced increments were observed on progressively increasing Pr content, leveling off at ca. 2.2 for 3.0 Prat nm–2. Experimental cis/trans 2-butene ratios are in all cases higher than the value of 0.53 predicted under equilibrium conditions at the reaction temperature of 473 K63 and are therefore the result of kinetic control. The increment observed in both the abundance of terminal olefins as well as in the share of cis isomers within 2-olefins on promotion with PrOx suggests the inhibition of acid-catalyzed secondary olefin double-bond isomerization, which proceeds via carbocation intermediates and is thus expected to favor trans products. On the contrary, Lewis basic sites are less efficient isomerization centers and are expected to promote carbanion routes via H-abstraction, which favor cis isomers.57
Also with regard to the preservation of terminal isomers within the C5–10 olefin products, PrOx was the most effective promoter among those investigated. Albeit less marked, increments in the C5–10 olefin T/I ratio were also observed on modification of CoRu/AOmM with alternative promoters, including oxides of alkali metals and alternative lanthanides such as Sm (Figure S16). With LaOx, in contrast, even though its addition enhanced the olefinicity of this hydrocarbon product fraction, it decreased the T/I olefin ratio in the entire chain-length range compared with the unpromoted benchmark. This effect is tentatively ascribed to the previously reported and rather exclusive ability of lanthanum—within the early lanthanide elements—to facilitate the development of stronger acid sites, that is, being more reactive for olefin isomerization via carbocation routes, when deposited on the surface of γ-Al2O3.64
In Situ FTIR Spectroscopy
PrOx Effects on Metal Surface Sites
Following in situ catalyst reduction, CO-FTIR spectroscopy was applied at room temperature to investigate the metal surface topology in selected catalysts, that is, unpromoted CoRu/AOmM, and the corresponding catalysts additionally incorporating 1.0 and 3.0 Prat nm–2, respectively. As shown in Figure 7, bands in the spectral region 1800–2100 cm–1, ascribed to υ(CO) in surface cobalt carbonyls, developed on increasing the CO dose. Additionally, bands in the region 1500–1800 cm–1 indicated the presence of carboxyl and carbonyl compounds on the oxide support after H2-reduction activation. Spectra deconvolution was applied to discriminate different contributions. Figure 7 shows the corresponding deconvolutions at PCO ∼ 25 mbar, that is, prior to the detection of significant coverage-driven band shifts (see Figure S17 for further deconvolution details at various CO dosages). Similar contributions were observed in all cases, suggesting that the addition of PrOx did not modify the metal surface topology to a great extent. Comparatively broader bands peaking at ca. 1870 ± 6 cm–1 and 1947 ± 5 cm–1 can be ascribed to CO adsorbed in 3-fold and bridged configurations, respectively, on cobalt.65,66 Moreover, already from the lowest CO dosages, a weaker and narrower band emerged at ca. 1992–2000 cm–1. This is the spectral region where atop cobalt carbonyls have been detected on low-coordination surface metal centers on stepped and defect-rich sputtered cobalt monocrystals,66 and hence, the band might be associated with CO adsorbed at surface defects (e.g., step-edges on the cobalt nanoparticles). This band progressively decreased in relative intensity and clearly shifted to lower wavenumbers on incorporating PrOx, suggesting that oxide promoter species might interact with lower coordination surface cobalt atoms, blocking a fraction of these sites for CO binding, while enhancing the electron backdonation of the remaining centers to CO*. At higher CO doses, the most prominent band at ca. 2030 cm–1, corresponding to atop CO adsorption on cobalt facets,66,67 dominated this spectral region. Finally, an additional evident implication of the incorporation of PrOx as promoter was observed at higher wavenumbers. For the unpromoted CoRu/AOmM, a band centered at 2059 cm–1 became evident at PCO > 10–15 mbar, clearly as a new contribution rather than as a result of blueshifts of bands developed at lower CO doses. This υ(CO) frequency is typically assigned to CO linearly adsorbed on cobalt terraces reconstructed by C*, originated from CO dissociation, which might already occur at room temperature.56,66,68 While this contribution was also observed for 1.0Pr-CoRu/AOmM, a higher Pr content of 3 Prat nm2 evidently led to its disappearance (see also Figure S18a for a CO dosage-resolved analysis), suggesting that at high oxide surface loadings, PrOx species have also modified the reactivity of extended terraces on the cobalt nanoparticles. At yet higher CO doses (PCO > 25 mbar), atop metal carbonyl bands underwent a progressive blueshift, as a result of the establishment of dipole–dipole interactions among vicinal CO* adsorbates at high surface coverages. Moreover, several comparatively narrow absorption bands emerged in the region of 1850–2000 cm–1, indicating the generation of molecular cobalt (sub)carbonyl compounds and their readsorption on the catalyst surface (Figure S19).69 Interestingly, this phenomenon became progressively exacerbated on incorporation of increasingly higher PrOx contents. This observation suggests a progressively higher fraction of surface metal atoms to be depleted in electronic density and therefore more prone to react to the electron donor CO into molecular adducts, at the Co/PrOx contact points.
Figure 7.

In situ CO-FTIR investigation on promoter effects. FTIR spectra recorded at 298 K in the υ(CO) region after increasing CO doses in the range PCO = 0–25 mbar (left); deconvolution of the FTIR spectrum recorded at 298 K after having dosed ca. 25 mbar CO (center); and deconvolution of the FTIR spectrum recorded at 110 K after having dosed 2 mbar CO (right, see Figure S20 in the Supporting Information for the full set of spectra at increasing pCO = 0–2 mbar), on the in situ reduced (a) CoRu/AOmM, (b) 1.0Pr-CoRu/AOmM, and (c) 3.0Pr-CoRu/AOmM catalysts.
PrOx Effects on Surface Acid Sites
Next to modifications on the metallic function, the impact of the PrOx species on the catalyst support surface was also assessed by CO-FTIR at cryogenic temperatures (110 K). At this temperature, CO probes not only metallic sites, but additionally, it serves as a sensitive reporter for surface Lewis centers. Upon admission of CO in the IR cell, following catalyst reduction, surface metal carbonyl bands (1900–2100 cm–1) developed already from the lowest CO doses, as a result of the stronger CO binding to metallic sites, while signals in the range of 2100–2200 cm–1, associated with CO adsorbed on the oxide supports, emerged on further increasing PCO up to 2 mbar (Figure S20). Figure 7 shows deconvolution results for spectra recorded at the highest CO surface coverage for the unpromoted CoRu/AOmM as well as those counterparts incorporating 1.0 and 3.0 Prat nm–2, respectively (see Figure S21 for deconvolution details at various CO dosages). The corresponding cobalt-free Al2O3 and 3.0Pr/AOmM materials were also studied separately to support band assignment (Figure S22). For all catalysts studied, a contribution peaking at 2119 cm–1 can be ascribed to atop CO adsorption on partially oxidized Coδ+ sites.70 Given the high overall degree of cobalt reduction ascertained by XPS, these partially oxidized centers are likely located at the metal–support interface. A prominent band centered at ca. 2150 ± 4 cm–1 is associated with CO adsorbed via H-bond interactions with surface OH groups on the alumina surface.71−73 Contributions peaking at 2174 and 2187 cm–1 for the unpromoted CoRu/AOmM catalyst can be ascribed to CO bonded to coordinatively unsaturated (cus) Al3+ Lewis centers of medium and higher acid strength, respectively.71,72 On incorporation of PrOx at increasingly higher surface loadings, the band peaking at ca. 2187–2190 cm–1, associated with the strongest Al2O3 Lewis sites, vanished progressively (see also Figure S18b for a CO dosage-resolved analysis), while a band developed at 2168 cm–1. The latter, also observed on the metal-free PrOx/AOmM, can thus be assigned to a new type of surface Lewis acid sites associated with cus praseodymium cations. The comparatively lower υ(CO) indicates that this new type of sites bears lower acid strength (electron-withdrawing character) compared with those on the pristine Al2O3. While such acidity inhibition was evident already at promoter loadings of 1.0 Mat nm–2, CO2-TPD studies showed that the development of strong surface basic centers was only noticeable at higher promoter contents of 3.0 Mat nm–2 (Figure S23). These results furnish direct evidence that PrOx species are also responsible for the depletion of the strongest Lewis acid sites on the alumina oxide support, in favor of weaker acid centers and basic sites associated with the lanthanide oxide, explaining its decisive role in inhibiting the acid-catalyzed olefin double bond isomerization and thus enhancing the terminal regioselectivity as well as cis stereoselectivity of liquid olefin products.
DFT Insights into Promotional Effects
Preferential Promoter Location on Cobalt
Density functional theory calculations were performed to further rationalize those metal-oxide promotional effects observed experimentally. Na2O and PrO2 oxides were selected as representative for the experimentally investigated alkali and lanthanide oxide promoters, respectively. First, the structure and adsorption energetics of the promoters on cobalt were assessed. Face-centered-cubic (fcc) Co0 was the predominant polymorph in all catalysts, both unpromoted and oxide-promoted, after activation via reduction in hydrogen flow (Figure S24). Hence, few-atom-layer thick slabs for Co(111) and Co(211) metal facets were considered as representative models for flat and stepped cobalt surfaces, respectively, in order to contemplate both extended terraces as well as step-edge topological motifs present on the surface of fcc cobalt nanoparticles (Figure CM1 and CM2).
Figure 8a summarizes the results for the adsorption energetics of the oxide promoter species on the different cobalt surfaces at various surface coverages. Further details on the optimized structural models as well as the atop, bridge and hollow (fcc and hcp) surface adsorption sites considered are provided in the Supporting Information (section 2, Computational Methods). Higher Eads indicate that PrO2 monomers bind stronger to cobalt surfaces, regardless of surface topology, compared with Na2O counterparts. At a comparable 0.11 ML promoter surface coverage on Co(111) terraces, the most stable adsorption configuration for Na2O was found with sodium occupying cobalt fcc hollow and bridge sites and oxygen anions on fcc sites, while for PrO2, oxygen ions occupied hcp and fcc hollow sites, and Pr a hcp hollow metal site. On the stepped Co(211) surfaces, the preferred adsorption structure for Na2O is with Na atoms located at the cobalt hcp sites on the lower terrace and only the oxygen ion siting at the step-edge, while PrO2 adopts an adsorption structure with oxygen anions on hcp hollow sites on the step and lower terrace, respectively. Details on the predicted structures at higher promoter coverages are provided in the Supporting Information. As shown in Figure 8a, the adsorption of both oxide monomer species is found to be energetically more favored on Co(211). In line with the experimental CO-FTIR results, these predictions bolster the preferential population of step-edge sites on the metal nanoparticles in the FT catalysts with promoter species at low oxide coverages.
Figure 8.

DFT calculations on promoter effects. (a) Adsorption energies for PrO2 and Na2O on flat Co(111) and stepped Co(211) cobalt surfaces as a function of the oxide coverage (fractional monolayer (ML)). The top views on the models show the corresponding optimized structures for a 0.11 ML oxide coverage in each case. Color codes: Pr: yellow, Na: orange, O: red, Co: gray and blue. For clarity cobalt step-edges on Co(211) are shown in blue. (b) Relative adsorption energies for H* and CO* on Co(111) and Co(211) cobalt surfaces either as neat or precovered with 0.11 ML PrO2. (c) Relative adsorption energies for ethylene (C2H4*) and CO* on Co(111) and Co(211) cobalt surfaces either as neat or precovered with 0.11 ML PrO2.
Promoter Effects on Cobalt
Next, the impact of promoters on the adsorption energetics for H* and CO* species was investigated. Calculations focused on a promoter coverage of 0.11 ML, given that higher oxide surface contents, that is, of 0.25 ML on Co(111) and 0.17 ML on Co(211), resulted in either CO desorption to the gas phase or decomposition of the promoter structures, suggesting that comparatively lower oxide coverages are attainable experimentally on cobalt. Figure 8b summarizes the results for PrO2. As observed, the presence of PrO2 species on cobalt leads to a decrease in the ratio Eads[H*]/Eads[CO*] compared with the bare metal surface, primarily as a result of a notable increment in the binding energy for molecular CO on the oxide-decorated cobalt surface (Tables S5 and S6). This effect is slightly more pronounced on Co(111) terraces, whereon several energetically feasible adsorption sites could be identified for hydrogen and CO, with different spatial proximity to oxygen anions or lanthanide cations (section 2 in the Supporting Information), with H* and CO* adsorption energies spanning 276–262 kJ mol–1 and 240–226 kJ mol–1, respectively. Qualitatively similar results were obtained for the case of Na2O (Tables S5 and S6).
A Bader charge analysis indicates that, on bare cobalt surfaces, both CO* and H* adsorbates withdraw electron density from the surface cobalt atoms, in line with previous experimental findings,74,75 and regardless of the metal surface topology considered (Table S7). Addition of either Na2O or PrO2 oxide promoters results in additional electronic charge accumulation on H* and CO*. On the one hand, this suggests an enhanced electron backdonation from cobalt atoms in the vicinity of the oxide promoter species to CO*, which is in line with those red shifts observed using FTIR for υ(CO) in atop cobalt carbonyls at low-coordination metal sites in PrOx-promoted catalysts (vide supra). On the other hand, it contributes to a slight reduction of Eads[H*]32 alongside a marked increase of Eads[CO*]. Even though the actual surface coverage remains a matter of debate,56 it is known that under relevant FT reaction conditions, the surface of cobalt catalysts is by and large occupied by molecular CO. Surface CO* coverages in the range from ca. 0.6 ML to essentially a monolayer have been proposed under operation conditions.57−59 Therefore, the increase in Eads[CO*] relative to Eads[H*] is unlikely to result in major further increments in the CO* coverage on the metal. However, comparatively minor CO* coverage increments have been proposed to be sufficient to hinder the dissociative H2 adsorption by decreasing the availability of those surface vacant sites involved in hydrogen activation.57 This effect can explain the lower hydrogenation activity observed on the promoted cobalt catalysts, which results in both lower overall CO hydrogenation reaction rates as well as higher selectivity to olefins.
Next to the primary hydrogenation ability, further calculations were performed to gain insight into secondary effects induced by the oxide promoters. The extent to which α-olefin primary reaction products undergo secondary hydrogenation is a function of their bed and pore residence times—adjustable via catalyst porosity—but also the driving force for olefin adsorption on the metal. The latter was explored using ethylene as a model olefin. As shown in Figure 8c, a 0.11 ML coverage of PrO2 on cobalt is predicted to lead to a remarkable decrease in Eads[C2H4*]/Eads[CO*] compared with neat cobalt, by 71% and 36% for flat Co(111) and stepped Co(211) surfaces, respectively. This lower driving force for olefin readsorption might additionally contribute to the dampening of secondary olefin hydrogenation observed experimentally on PrOx-promoted catalysts.
A second important secondary effect of oxide promoters relates to their capacity to prompt the WGSR on the essentially shift-inactive cobalt FT catalysts. This effect is generally ascribed to the development of (surface) cobalt carbide species under reaction conditions.76 In this regard, Bader charge analyses for the surface metal atoms revealed significant differences in the electronic effects induced by Na2O and PrO2 oxides on cobalt (Table S7). While Na2O acts as strongly electron-donating species, PrO2 units behave overall as electron-withdrawing groups, a behavior which dovetails with the different origin of the basic character of the two oxides. This is particularly apparent on the stepped Co(211) surface, which is the energetically most favorable location of both oxide promoters at low surface coverages. Even in the presence of electron acceptor CO* adsorbate molecules, that is, a situation more representative of the working catalyst, surface decoration with Na2O leads to a decrease in the net Bader charge on cobalt, from 0.63 to 0.50 with respect to the unpromoted metal surface (Table S7). In contrast, the presence of PrO2 results in notably more electropositive cobalt atoms, with a net Bader charge of 1.50. These markedly different behaviors have implications for the carbophilicity, which might serve as an indication for the propensity of cobalt surfaces to undergo carbidization. Adsorption energies for C* were evaluated (see section 2 in the Supporting Information). The adsorption energy for C* on clean Co(111) and Co(211) surfaces is determined to be 691 and 705 kJ mol–1, respectively. While Eads[C*] increases, to 713 and 714 kJ mol–1, respectively, on decoration with PrO2, a higher increment, to 714 and 721 kJ mol–1, was found after deposition of Na2O, making the cobalt surface more carbophilic. Reinforcing these computational predictions and in line with previous reports of a facilitated carbide formation upon incorporation of alkali promoters,77,78 XRD analysis of catalysts recovered after the FT tests revealed the development of Co2C crystalline domains in 3.0Na-CoRu/AOmM, that is, the catalyst showing the most marked increase in CO2 selectivity (Figure S11). These results allow thus to surmise that the enhanced carbon affinity and thus a facilitated development of surface carbide species during catalysis underlie the undesired boosting of the WGSR activity observed experimentally for NaOx and other alkali oxides.
Conclusion
A battery of cobalt-based Fischer–Tropsch catalysts synthesized on either monomodal mesoporous or multimodal meso-macroporous alumina support materials enables untwining and assessing pore transport and oxide surface promotion effects on the selectivity to liquid (C5+) olefin chemicals. Under relevant reaction conditions, reducing mesopore transport distances for primary products down to the (sub)micrometer regime, in hierarchically porous meso-macroporous catalysts, contributes to a higher preservation of olefins in the condensable hydrocarbon products. The incorporation of promoters of Lewis basic character (alkali and lanthanide oxides) leads in all cases to a decrease in the metal surface specific CO hydrogenation rate (TOF) and hydrocarbon chain-growth probability. Lanthanide oxides, particularly praseodymium oxide, proved efficient as promoters to further boost the share of α-olefins within the light condensate (C5–10) products, notably, while preserving key features of cobalt-based catalysts such as low activity to the WGSR which was, on the contrary, exacerbated by alkali oxide promoters at similar surface contents. DFT calculations predict oxide promoters on cobalt surfaces to locate preferentially at surface metal step-edges and associate their promotion effect to an enhanced adsorption competition of CO for surface metal sites compared to hydrogen and olefins, thereby dampening (secondary) hydrogenation. Moreover, our results suggest an enhanced metal surface carbophility because of the strong electron donor character of alkali oxides compared to lanthanide oxides as the likely reason for the undesired triggering of WGSR activity by the former. Next to surface promotion on the metal nanoparticles, in situ CO-FTIR spectroscopy furnishes evidence for the blockage of the strongest Al3+cus Lewis acid centers on the γ-Al2O3 support upon PrOx incorporation, explaining a second promotional effect which enhances the terminal regioselectivity in the liquid olefin products via inhibition of acid-catalyzed double-bond shift secondary reactions. Integrating a multimodal meso-macroporous architecture with PrOx as promoter, at an optimal loading of 1 Prat nm–2, leads to an unconventional product distribution which reconciles features of cobalt and iron carbide-based FT catalysts, respectively. Selectivities to C5+ products >70 C% are achieved in combination to light condensate products (C5–10) enriched in added-value chemicals (67 C%), predominantly α-olefins, remarkably with essentially no CO2 side-production (<1%). These results illustrate how porosity design and surface promotion effects are valuable and complementary tools to adjust the FT product distribution toward the combined production of paraffinic precursors for synthetic fuels and liquid commodity chemicals from hydrogen-rich (H2/CO = 2) syngas.
Acknowledgments
Sasol Materials is very gratefully acknowledged for the supply of high-purity pseudoboehmite alumina precursors. M. G. Farpón (ITQ) is acknowledged for 2-butene olefin isomer equilibrium composition determinations with Aspen Plus (AspenTech). J.M. Salas (ITQ) is acknowledged for his contributions to the CO-FTIR studies. A. Muñoz and M.D. Soriano (ITQ) are acknowledged for the XPS experiments, and M. Arribas (ITQ) for H2-chemisorption measurements. M. Meyer and V. Dietl (MPI-KOFO) and V. Recio (ITQ) are acknowledged for technical assistance with lab work and off-line GC product analyses. Prof. A. Lorke (UDE) is gratefully acknowledged for access to dual-beam facilities. We thank Prof. F. Schüth (MPI-KOFO) for the allocation of lab space and supportive access to research instrumentation. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under Grant Agreement no. 817612 (REDIFUEL). Additionally, parts of this research received funding by the Max Planck Society, the Bundesministerium für Bildung und Forschung (grant number 01DG17019), the Spanish Ministry of Science and Innovation (‘Severo Ochoa Excellence Program’ (SEV-2016-0683)) and the European Research Council (grant ERC-2019-CoG-864195). DFT calculations were performed at the EFES computing cluster located in the Izmir Institute of Technology (IZTECH). A.C.K. thanks IZTECH for the allocation of computational time.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c05027.
The authors declare no competing financial interest.
Supplementary Material
References
- Wood D. A.; Nwaoha C.; Towler B. F. Gas-to-liquids (GTL): A review of an industry offering several routes for monetizing natural gas. J. Nat. Gas Sci. Eng. 2012, 9, 196–208. 10.1016/j.jngse.2012.07.001. [DOI] [Google Scholar]
- Al-Douri A.; Sengupta D.; El-Halwagi M. M. Shale gas monetization – A review of downstream processing to chemicals and fuels. J. Nat. Gas Sci. Eng. 2017, 45, 436–455. 10.1016/j.jngse.2017.05.016. [DOI] [Google Scholar]
- Dry M. E. The Fischer–Tropsch process - commercial aspects. Catal. Today 1990, 6 (3), 183–206. 10.1016/0920-5861(90)85002-6. [DOI] [Google Scholar]
- Rye L.; Blakey S.; Wilson C. W. Sustainability of supply or the planet: a review of potential drop-in alternative aviation fuels. Energy Environ. Sci. 2010, 3 (1), 17–27. 10.1039/B918197K. [DOI] [Google Scholar]
- Gutiérrez-Antonio C.; Gómez-Castro F. I.; de Lira-Flores J. A.; Hernández S. A review on the production processes of renewable jet fuel. Renewable Sustainable Energy Rev. 2017, 79, 709–729. 10.1016/j.rser.2017.05.108. [DOI] [Google Scholar]
- Dry M. E. High quality diesel via the Fischer–Tropsch process – a review. J. Chem. Technol. Biotechnol. 2002, 77 (1), 43–50. 10.1002/jctb.527. [DOI] [Google Scholar]
- Anderson R. B.; Friedel R. A.; Storch H. H. Fischer–Tropsch reaction mechanism involving stepwise growth of carbon chain. J. Chem. Phys. 1951, 19 (3), 313–319. 10.1063/1.1748201. [DOI] [Google Scholar]
- Schulz H. Major and minor reactions in Fischer–Tropsch synthesis on cobalt catalysts. Top. Catal. 2003, 26 (1), 73–85. 10.1023/B:TOCA.0000012988.86378.21. [DOI] [Google Scholar]
- Iglesia E. Design, synthesis, and use of cobalt-based Fischer–Tropsch synthesis catalysts. Appl. Catal., A 1997, 161 (1), 59–78. 10.1016/S0926-860X(97)00186-5. [DOI] [Google Scholar]
- Schulz H.; Claeys M. Reactions of α-olefins of different chain length added during Fischer–Tropsch synthesis on a cobalt catalyst in a slurry reactor. Appl. Catal., A 1999, 186 (1), 71–90. 10.1016/S0926-860X(99)00165-9. [DOI] [Google Scholar]
- Iglesia E.; Reyes S. C.; Madon R. J. Transport-enhanced α-olefin readsorption pathways in Ru-catalyzed hydrocarbon synthesis. J. Catal. 1991, 129 (1), 238–256. 10.1016/0021-9517(91)90027-2. [DOI] [Google Scholar]
- Labat Y. Some industrial sulfur compounds; Novelties in their manufacture, main uses and potential developments. Phosphorus, Sulfur Silicon Relat. Elem. 1993, 74 (1–4), 173–194. 10.1080/10426509308038107. [DOI] [Google Scholar]
- Nakajima Y.; Shimada S. Hydrosilylation reaction of olefins: recent advances and perspectives. RSC Adv. 2015, 5 (26), 20603–20616. 10.1039/C4RA17281G. [DOI] [Google Scholar]
- Sarma B. B.; Kim J.; Amsler J.; Agostini G.; Weidenthaler C.; Pfänder N.; Arenal R.; Concepción P.; Plessow P.; Studt F.; Prieto G. One-pot cooperation of single-atom Rh and Ru solid catalysts for a selective tandem olefin isomerization-hydrosilylation process. Angew. Chem., Int. Ed. 2020, 59 (14), 5806–5815. 10.1002/anie.201915255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T. E.; Hultzsch K. C.; Yus M.; Foubelo F.; Tada M. Hydroamination: Direct addition of amines to alkenes and alkynes. Chem. Rev. 2008, 108 (9), 3795–3892. 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- Franke R.; Selent D.; Börner A. Applied hydroformylation. Chem. Rev. 2012, 112 (11), 5675–5732. 10.1021/cr3001803. [DOI] [PubMed] [Google Scholar]
- Agapie T. Selective ethylene oligomerization: Recent advances in chromium catalysis and mechanistic investigations. Coord. Chem. Rev. 2011, 255 (7), 861–880. 10.1016/j.ccr.2010.11.035. [DOI] [Google Scholar]
- Al-Jarallah A. M.; Anabtawi J. A.; Siddiqui M. A. B.; Aitani A. M.; Al-Sa’doun A. W. Ethylene dimerization and oligomerization to butene-1 and linear α-olefins: A review of catalytic systems and processes. Catal. Today 1992, 14 (1), 1–121. 10.1016/0920-5861(92)80128-A. [DOI] [Google Scholar]
- van der Klis F.; Le Nôtre J.; Blaauw R.; van Haveren J.; van Es D. S. Renewable linear alpha olefins by selective ethenolysis of decarboxylated unsaturated fatty acids. Eur. J. Lipid Sci. Technol. 2012, 114 (8), 911–918. 10.1002/ejlt.201200024. [DOI] [Google Scholar]
- Amghizar I.; Vandewalle L. A.; van Geem K. M.; Marin G. B. New trends in olefin production. Engineering (Beijing) 2017, 3 (2), 171–178. 10.1016/J.ENG.2017.02.006. [DOI] [Google Scholar]
- Zubel M.; Heuser B.; Pischinger S. 1-Octanol tailor-made fuel for lower soot emissions. MTZ. Worldw. 2017, 78 (3), 58–61. 10.1007/s38313-016-0178-z. [DOI] [Google Scholar]
- Heuser B.; Vorholt A.; Prieto G.; Graziano B.; Schönfeld S.; Messagie M.; Cardellini G.; Tuomi S.; Sittinger N.; Hermanns R.; Ramawamy S.; Kanth Kosuru C.; Hoffmann H.; Schulz L.; Yadav J.; Weide M.; Schnorbus T.. REDIFUEL: Robust and efficient processes and technologies for Drop-in renewable FUELs for road transport. Transport Research Arena 2020, Helinski, Finland, April 2020.
- de Smit E.; Weckhuysen B. M. The renaissance of iron-based Fischer–Tropsch synthesis: on the multifaceted catalyst deactivation behaviour. Chem. Soc. Rev. 2008, 37 (12), 2758–2781. 10.1039/b805427d. [DOI] [PubMed] [Google Scholar]
- Botes F. G.; Niemantsverdriet J. W.; van de Loosdrecht J. A comparison of cobalt and iron based slurry phase Fischer–Tropsch synthesis. Catal. Today 2013, 215, 112–120. 10.1016/j.cattod.2013.01.013. [DOI] [Google Scholar]
- Rath L. K.; Longanbach J. R. A perspective on syngas from coal. Energy Sources 1991, 13 (4), 443–459. 10.1080/00908319108909001. [DOI] [Google Scholar]
- Pala L. P. R.; Wang Q.; Kolb G.; Hessel V. Steam gasification of biomass with subsequent syngas adjustment using shift reaction for syngas production: An Aspen Plus model. Renewable Energy 2017, 101, 484–492. 10.1016/j.renene.2016.08.069. [DOI] [Google Scholar]
- van Steen E.; Claeys M. Fischer–Tropsch catalysts for the biomass-to-liquid (BTL)-process. Chem. Eng. Technol. 2008, 31 (5), 655–666. 10.1002/ceat.200800067. [DOI] [Google Scholar]
- Wang P.; Chen W.; Chiang F.-K.; Dugulan A. I.; Song Y.; Pestman R.; Zhang K.; Yao J.; Feng B.; Miao P.; Xu W.; Hensen E. J. M. Synthesis of stable and low-CO2 selective ε-iron carbide Fischer–Tropsch catalysts. Sci. Adv. 2018, 4 (10), eaau2947. 10.1126/sciadv.aau2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodakov A. Y.; Chu W.; Fongarland P. Advances in the development of novel cobalt Fischer–Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels. Chem. Rev. 2007, 107 (5), 1692–1744. 10.1021/cr050972v. [DOI] [PubMed] [Google Scholar]
- Liang Q.; Chen K.; Hou W.; Yan Q. CO hydrogenation over nanometer spinel-type Co/Mn complex oxides prepared by sol-gel method. Appl. Catal., A 1998, 166 (1), 191–199. 10.1016/S0926-860X(97)00255-X. [DOI] [Google Scholar]
- Zhou W.-G.; Liu J.-Y.; Wu X.; Chen J.-F.; Zhang Y. An effective Co/MnOx catalyst for forming light olefins via Fischer–Tropsch synthesis. Catal. Commun. 2015, 60, 76–81. 10.1016/j.catcom.2014.10.027. [DOI] [Google Scholar]
- Xie J.; Paalanen P. P.; van Deelen T. W.; Weckhuysen B. M.; Louwerse M. J.; de Jong K. P. Promoted cobalt metal catalysts suitable for the production of lower olefins from natural gas. Nat. Commun. 2019, 10 (1), 167. 10.1038/s41467-018-08019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts N.; Trotuş I.-T.; Swertz A.-C.; Schüth F.; Prieto G. In situ hydrocracking of Fischer–Tropsch hydrocarbons: CO-prompted diverging reaction pathways for paraffin and α-olefin primary products. ACS Catal. 2016, 6 (7), 4229–4238. 10.1021/acscatal.6b00904. [DOI] [Google Scholar]
- Duyckaerts N.; Bartsch M.; Trotuş I.-T.; Pfänder N.; Lorke A.; Schüth F.; Prieto G. Intermediate product regulation in tandem solid catalysts with multimodal porosity for high-yield synthetic fuel production. Angew. Chem., Int. Ed. 2017, 56 (38), 11480–11484. 10.1002/anie.201705714. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47 (1), 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54 (16), 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Blöchl P. E. Projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50 (24), 17953–17979. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Iglesia E.; Reyes S. C.; Madon R. J.; Soled S. L.. Selectivity control and catalyst design in the Fischer–Tropsch synthesis: Sites, pellets, and reactors. In Advances in Catalysis, Eley D. D., Pines H., Weisz P. B., Eds. Academic Press: 1993; Vol. 39, pp 221–302. [Google Scholar]
- Parlett C. M. A.; Wilson K.; Lee A. F. Hierarchical porous materials: Catalytic applications. Chem. Soc. Rev. 2013, 42 (9), 3876–3893. 10.1039/C2CS35378D. [DOI] [PubMed] [Google Scholar]
- Jacobs G.; Das T. K.; Zhang Y. Q.; Li J. L.; Racoillet G.; Davis B. H. Fischer–Tropsch synthesis: Support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl. Catal., A 2002, 233 (1–2), 263–281. 10.1016/S0926-860X(02)00195-3. [DOI] [Google Scholar]
- Shannon R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, 32 (5), 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Øyvind B.; Magnus R.; Sølvi S.; Wouter v. B.; Anders H.. Identification of cobalt species during temperature programmed reduction of Fischer–Tropsch catalysts. In Stud. Surf. Sci. Catal.; Davis B. H., Occelli M. L., Eds. Elsevier: 2007; Vol. 163, pp 255–272. [Google Scholar]
- Munnik P.; de Jongh P. E.; de Jong K. P. Control and impact of the nanoscale distribution of supported cobalt particles used in Fischer–Tropsch catalysis. J. Am. Chem. Soc. 2014, 136 (20), 7333–7340. 10.1021/ja500436y. [DOI] [PubMed] [Google Scholar]
- Borg Ø.; Eri S.; Blekkan E. A.; Storsæter S.; Wigum H.; Rytter E.; Holmen A. Fischer–Tropsch synthesis over γ-alumina-supported cobalt catalysts: Effect of support variables. J. Catal. 2007, 248 (1), 89–100. 10.1016/j.jcat.2007.03.008. [DOI] [Google Scholar]
- Lillebø A. H.; Patanou E.; Yang J.; Blekkan E. A.; Holmen A. The effect of alkali and alkaline earth elements on cobalt based Fischer–Tropsch catalysts. Catal. Today 2013, 215, 60–66. 10.1016/j.cattod.2013.03.030. [DOI] [Google Scholar]
- Gaube J.; Klein H. F. The promoter effect of alkali in Fischer–Tropsch iron and cobalt catalysts. Appl. Catal., A 2008, 350 (1), 126–132. 10.1016/j.apcata.2008.08.007. [DOI] [Google Scholar]
- Vada S.; Kazi A. M.; Bedu-Addo F. K.; Chen B.; Goodwin J. G.. La Promotion of Co Fischer–Tropsch Catalysts. In Studies in Surface Science and Catalysis; Curry-Hyde H. E., Howe R. F., Eds.; Elsevier: 1994; Vol. 81, pp 443–448. [Google Scholar]
- Ribeiro M. C.; Gnanamani M. K.; Garcia R.; Jacobs G.; Rabelo-Neto R. C.; Noronha F. B.; Gomes I. F.; Davis B. H. Tailoring the product selectivity of Co/SiO2 Fischer–Tropsch synthesis catalysts by lanthanide doping. Catal. Today 2020, 343, 80–90. 10.1016/j.cattod.2018.10.064. [DOI] [Google Scholar]
- Eri S.; Goodwin J. G. Jr.; Marcelin G.; Riis T.. Catalyst for production of hydrocarbons. Patent No. US4880763A, Nov. 14, 1989.
- Gavrilović; Save; Blekkan The effect of potassium on cobalt-based Fischer–Tropsch catalysts with different cobalt particle sizes. Catalysts 2019, 9 (4), 351. 10.3390/catal9040351. [DOI] [Google Scholar]
- Verhaak M. J. F. M.Process for preparing olefins from synthesis gas using a cobalt and manganese containing catalyst. Patent No. EP2439185A1, Apr. 11, 2012.
- Nebel J.; Schmidt S.; Pan Q.; Lotz K.; Kaluza S.; Muhler M. On the role of cobalt carbidization in higher alcohol synthesis over hydrotalcite-based Co-Cu catalysts. Chin. J. Catal. 2019, 40 (11), 1731–1740. 10.1016/S1872-2067(19)63344-9. [DOI] [Google Scholar]
- Dorling T. A.; Moss R. L. The structure and activity of supported metal catalysts: I. Crystallite size and specific activity for benzene hydrogenation of platinum/silica catalysts. J. Catal. 1966, 5 (1), 111–115. 10.1016/S0021-9517(66)80130-6. [DOI] [Google Scholar]
- Somorjai G. A.; Carrazza J. Structure sensitivity of catalytic reactions. Ind. Eng. Chem. Fundam. 1986, 25 (1), 63–69. 10.1021/i100021a009. [DOI] [Google Scholar]
- Zijlstra B.; Broos R. J. P.; Chen W.; Oosterbeek H.; Filot I. A. W.; Hensen E. J. M. Coverage effects in CO dissociation on metallic cobalt nanoparticles. ACS Catal. 2019, 9 (8), 7365–7372. 10.1021/acscatal.9b01967. [DOI] [Google Scholar]
- Brouwer D. M. The mechanism of double-bond isomerization of olefins on solid acids. J. Catal. 1962, 1 (1), 22–31. 10.1016/0021-9517(62)90005-2. [DOI] [Google Scholar]
- Hattori H. Heterogeneous basic catalysis. Chem. Rev. 1995, 95 (3), 537–558. 10.1021/cr00035a005. [DOI] [Google Scholar]
- Falbe J.; Bahrmann H.; Lipps W.; Mayer D.. Aliphatic alcohols. In Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Elvers B., Ed.; Wiley-VCH: Weinheim, 2009 [Google Scholar]
- Torres G. M.; Frauenlob R.; Franke R.; Börner A. Production of alcohols via hydroformylation. Catal. Sci. Technol. 2015, 5 (1), 34–54. 10.1039/C4CY01131G. [DOI] [Google Scholar]
- Jia X.; Huang Z. Conversion of alkanes to linear alkylsilanes using an iridium–iron-catalysed tandem dehydrogenation–isomerization–hydrosilylation. Nat. Chem. 2016, 8 (2), 157–161. 10.1038/nchem.2417. [DOI] [PubMed] [Google Scholar]
- Edwards D.; Crudden C.; Yam K. One-Pot Carbon Monoxide-Free Hydroformylation of Internal Olefins to Terminal Aldehydes. Adv. Synth. Catal. 2005, 347, 50–54. 10.1002/adsc.200404228. [DOI] [Google Scholar]
- Equilibrium cis/trans 2-butene composition as simulated with Aspen HYSYS using an RGibbs reactor at T = 473 K and P = 20 bar and the Soave–Redlich–Kwong (PSRK) group-contribution equation of state.
- Yamamoto T.; Hatsui T.; Matsuyama T.; Tanaka T.; Funabiki T. Structures and acid–base properties of La/Al2O3 role of La addition to enhance thermal stability of γ-Al2O3. Chem. Mater. 2003, 15 (25), 4830–4840. 10.1021/cm034732c. [DOI] [Google Scholar]
- Toomes R. L.; King D. A. The adsorption of CO on Co{1010}. Surf. Sci. 1996, 349 (1), 1–18. 10.1016/0039-6028(95)01049-1. [DOI] [Google Scholar]
- Weststrate C. J.; van de Loosdrecht J.; Niemantsverdriet J. W. Spectroscopic insights into cobalt-catalyzed Fischer–Tropsch synthesis: A review of the carbon monoxide interaction with single crystalline surfaces of cobalt. J. Catal. 2016, 342, 1–16. 10.1016/j.jcat.2016.07.010. [DOI] [Google Scholar]
- Song D.; Li J.; Cai Q. In Situ Diffuse Reflectance FTIR Study of CO Adsorbed on a Cobalt Catalyst Supported by Silica with Different Pore Sizes. J. Phys. Chem. C 2007, 111 (51), 18970–18979. 10.1021/jp0751357. [DOI] [Google Scholar]
- Couble J.; Bianchi D. Experimental Microkinetic Approach of the Surface Reconstruction of Cobalt Particles in Relationship with the CO/H2 Reaction on a Reduced 10% Co/Al2O3 Catalyst. J. Phys. Chem. C 2013, 117 (28), 14544–14557. 10.1021/jp401762u. [DOI] [Google Scholar]
- Friedel R. A.; Wender I.; Shufler S. L.; Sternberg H. W. Spectra and structures of cobalt carbonyls1. J. Am. Chem. Soc. 1955, 77 (15), 3951–3958. 10.1021/ja01620a004. [DOI] [Google Scholar]
- Prieto G.; Concepción P.; Murciano R.; Martínez A. The impact of pre-reduction thermal history on the metal surface topology and site-catalytic activity of Co/SiO2 Fischer–Tropsch catalysts. J. Catal. 2013, 302, 37–48. 10.1016/j.jcat.2013.02.022. [DOI] [Google Scholar]
- Hadjiivanov K.; Tsyntsarski B.; Venkov T.; Klissurski D.; Daturi M.; Saussey J.; Lavalley J. C. FTIR spectroscopic study of CO adsorption on Co–ZSM-5: Evidence of formation of Co+(CO)4 species. Phys. Chem. Chem. Phys. 2003, 5 (8), 1695–1702. 10.1039/b300844d. [DOI] [Google Scholar]
- El-Nadjar W.; Bonne M.; Trela E.; Rouleau L.; Mino A.; Hocine S.; Payen E.; Lancelot C.; Lamonier C.; Blanchard P.; Courtois X.; Can F.; Duprez D.; Royer S. Infrared investigation on surface properties of alumina obtained using recent templating routes. Microporous Mesoporous Mater. 2012, 158, 88–98. 10.1016/j.micromeso.2012.03.006. [DOI] [Google Scholar]
- Hadjiivanov K.Chapter two - Identification and characterization of surface hydroxyl groups by infrared spectroscopy. In Advances in Catalysis; Jentoft F. C., Ed.; Academic Press: 2014; Vol. 57, pp 99–318. [Google Scholar]
- Habermehl-Cwirzen K. M. E.; Kauraala K.; Lahtinen J. Hydrogen on cobalt: The effects of carbon monoxide and sulphur additives on the D2/Co(0001) System. Phys. Scr. 2004, 77. 10.1238/Physica.Topical.108a00077. [DOI] [Google Scholar]
- Kizilkaya A. C.; Niemantsverdriet J. W.; Weststrate C. J. Ammonia adsorption and decomposition on Co(0001) in relation to Fischer–Tropsch synthesis. J. Phys. Chem. C 2016, 120 (7), 3834–3845. 10.1021/acs.jpcc.5b11609. [DOI] [Google Scholar]
- Gnanamani M. K.; Jacobs G.; Shafer W. D.; Sparks D. E.; Hopps S.; Thomas G. A.; Davis B. H. Low temperature water–gas shift reaction over alkali metal promoted cobalt carbide catalysts. Top. Catal. 2014, 57 (6), 612–618. 10.1007/s11244-013-0219-7. [DOI] [Google Scholar]
- Dai Y.; Yu F.; Li Z.; An Y.; Lin T.; Yang Y.; Zhong L.; Wang H.; Sun Y. Effect of Sodium on the Structure-Performance Relationship of Co/SiO2 for Fischer–Tropsch Synthesis. Chin. J. Chem. 2017, 35 (6), 918–926. 10.1002/cjoc.201600748. [DOI] [Google Scholar]
- Li Z.; Zhong L.; Yu F.; An Y.; Dai Y.; Yang Y.; Lin T.; Li S.; Wang H.; Gao P.; Sun Y.; He M. Effects of Sodium on the Catalytic Performance of CoMn Catalysts for Fischer–Tropsch to Olefin Reactions. ACS Catal. 2017, 7 (5), 3622–3631. 10.1021/acscatal.6b03478. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.