

Abstract
The human and murine esophagus have some substantial differences that limit the utility of mouse as a model to study human esophagus development and disease. Due to these limitations several recent reports describe the development of methods to generate human esophageal tissues via the directed differentiation of pluripotent stem cells. Methods for differentiation are based on knowledge of years of studying embryonic development of the esophagus in vertebrate animal models. Esophageal tissues derived from human pluripotent stem cells have been used to study both development and diseases affecting the esophagus. Here, we provide a detailed protocol for the directed differentiation of human pluripotent stem cells into human esophageal organoids and organotypic raft cultures, that are highly similar, morphologically and transcriptionally, to the human esophagus epithelium. We discuss limitations of the current esophageal models and the importance of engineering more complex tissue models with muscle and enteric nerves. Moving forward, these models might be utilized for the development of personalized treatments, as well as other therapeutic solutions.
1. Introduction
The primary function of the esophagus is to transport food from the mouth to the stomach through muscle peristalsis. Its epithelium is endoderm-derived, which is enclosed in layers of mesenchyme, including fibroblasts and muscle layers, which are mesoderm-derived. The mesenchyme is innervated by the enteric nervous system, which is neural crest-derived. Correct assembly of this complex structure from the three germ layers is necessary for the proper function of the adult esophagus (Fausett & Klingensmith, 2012; Que, 2015).
Up until recently studies of esophagus development and disease were limited to vertebrate animal models. However, there are several substantial differences between murine and human esophagus highlighting the need for a human esophageal model system: The mouse esophagus epithelium consists of 4–6 layers of cells with only one layer of basal cells, while the human esophagus epithelium is 30–40 layers thick, with several layers of basal cells. The mouse epithelium lumen is lined with a keratin layer which is absent from the human epithelium. Further, the human esophagus contains papillae structures which the mouse esophagus lacks (Zhang et al., 2017). In recent years, several protocols enabling directed differentiation of human pluripotent stem cells (hPSC) into specific gastrointestinal (GI) tract organoids have been developed via the manipulation of signaling pathways in vitro that mimic stages of embryonic organogenesis (Kechele & Wells, 2019). In all cases the first step is the differentiation of hPSCs into definitive endoderm (DE), the origin of the epithelium of all GI tract organs. Next DE is patterned into either foregut, midgut or hindgut fate (D’Amour et al., 2005; McCracken et al., 2017, 2014; Múnera et al., 2017; Spence et al., 2011). The esophagus originates from the dorsal anterior foregut, which requires further manipulation of signaling pathways to generate esophageal specific tissues, either 3D organoids or organotypic rafts (Trisno et al., 2018; Zhang et al., 2018). Both esophageal organoids and raft cultures express esophagus specific epithelial markers in the correct spatial and temporal manner and are therefore excellent models for studying human esophagus epithelium development and disease (Trisno et al., 2018; Zhang et al., 2018).
All current human esophageal models only contain the epithelium and lack any stromal cell types, smooth muscle, immune cells or enteric neuroglial cell types. Given the importance of these cell types in esophageal pathophysiology it will be important to develop more complex cultures that will better mimic the human esophagus under normal and pathological conditions.
In this chapter we will provide a relatively simple and well-established step-by-step protocol for the direct differentiation of hPSC into human esophageal organoids (HEOs) and organotypic raft cultures based on Trisno et al., 2018, as well as necessary background on esophagus development and immunofluorescent analysis of final product for the verification of culture identity.
2. Significance and applications
Disorders affecting the esophagus range from rare and devastating birth defects to damage caused by gastroesophageal reflux (GERD) experienced by up to a quarter of the population in North America (Jacobs, Ku, & Que, 2012; Rosekrans, Baan, Muncan, & van den Brink, 2015; Zorn & Wells, 2009). Esophagus related congenital defects and esophagus associated diseases can dramatically affect a patient’s quality of life and survival. Identification of molecular mechanisms that are in the basis of esophagus development and disease is crucial for disease management and for development of new treatments.
The human and murine esophagus differ in several substantial ways. The human epithelium is many layers thick, whereas the mouse esophageal epithelium is only a few layers thick but has a keratinize layer facing the lumen. Furthermore, the human esophagus has sub-mucosal glands which are absent in the murine esophagus. Therefore the murine esophagus is not and ideal model for disease related studies and for treatment development in particular (Zhang et al., 2017).
Up until recently human derived esophageal models were all monolayer cultures that do not recapitulate esophageal morphology or were generated from endoscopic biopsies (Kasagi et al., 2018), which are a limited source. Development of human derived esophageal three-dimensional models that better recapitulate the human esophagus development and morphology would facilitate studies of the molecular mechanisms that drives congenital defects and diseases of the esophagus.
The protocols described below give rise to 3D esophageal epithelial cultures that can be grown as organoids or organotypic raft cultures, and these have been used to study the role of various signaling pathways on esophageal development (Trisno et al., 2018; Zhang et al., 2018), as well as progenitor cell proliferation and differentiation. Having two configurations, organoids and raft cultures, provides more flexibility for studies (Trisno et al., 2018; Zhang et al., 2018). For example, one can study the effects of gastric acid exposure on the luminal face of the esophagus epithelium to model early events triggering Barrett’s Esophagus (Iyer & Kaul, 2019) or how luminal exposure to pathogens like the human papilloma virus may cause squamous cell carcinoma (ESCC) development (Haeri, Mardany, Asadi-Amoli, & Shahsiah, 2013). Further, patient derived induced hPSCs can be used for the identification of patient specific phenotypes as well as for the development of possible personalized treatments, like in the case of ESCC tumors and other diseases that are characterized by heterogeneity between patients.
3. Morphological and transcriptional analysis of developing esophageal cultures
All of the morphological and molecular changes that we use to benchmark the differentiation of human PSCs into HEOs are based on normal embryonic development of the esophagus. The esophagus epithelium is derived from the dorsal foregut endoderm which is a simple single layer cuboidal epithelium. This epithelium expresses the markers SRY-box2 (SOX2) and tumor protein 63 (P63) and does not express respiratory markers, including NK2 homeobox 1 (NKX2-1). Correspondingly, early stage HEOs and raft cultures will have single to few layers of cells characterized by the expression of SOX2 and P63 and the lack of NKX2-1 expression (Fausett & Klingensmith, 2012; Morrisey & Rustgi, 2018; Que, 2015; Trisno et al., 2018).
Esophageal progenitors proliferate as the tissue grows and will start to differentiate, moving toward the lumen of the esophagus forming suprabasal layers. Cells populating the suprabasal layers will express differentiated epithelial markers including keratin 13 (KRT13) and involucrin (IVL) among others (Yu, Slack, & Tosh, 2005) (Fig. 1), while the basal layer of proliferating cells will express markers of proliferation, including KI67, and will continue to express SOX2 and P63. The adult human esophagus epithelium consists of 20–30 layers of cells and the HEOs and rafts cultures generated in the described protocol can give rise to similar epithelium thickness and express basal and suprabasal markers in their appropriate location. Below we describe how we use immunofluorescent analysis of markers to identify basal and suprabasal compartments in HEOs and esophageal raft cultures and to ensure the correct differentiation and assembly of the esophagus stratified squamous epithelium.
FIG. 1.
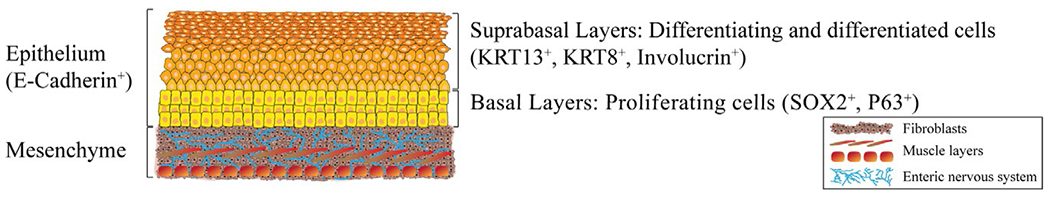
The human esophagus is consisting of mesenchyme and epithelium. The human esophagus mesenchyme is mesoderm-derived and is consists of fibroblasts and several muscle layers, which are innervated by the enteric nervous system, which is neural crest-derived. The esophagus epithelium is endoderm-derived and is a stratified squamous epithelium, expressing the epithelium marker E-Cadherin that consists of basal layers of proliferating cells, expressing SOX2 and P63 among other markers. Once the cells start to differentiate, they migrate toward the lumen and populate the suprabasal layers, expressing KRT13 and Involucrin, among other markers.
4. Overview of differentiation protocol
The protocol described below employs temporal manipulation of various signaling pathways to direct the differentiation of hPSCs into 3D esophageal cultures. We will therefore discuss the rationale behind signaling pathway manipulation at each step of the protocol. hPSC are first differentiated into the definitive endoderm (DE) (days 0–3), which gives rise to all GI tract organ epithelium, by activating the Nodal pathway with Activin A (D’Amour et al., 2005). Initial activation of the bone morphogenetic protein (BMP) pathway on first day of the culture improved the efficiency of DE differentiation. Following DE induction, inhibition of the BMP pathway by the BMP antagonist Noggin (NOG) is necessary to pattern the DE into the foregut lineages. At this step (days 3–6), addition of fibroblasts growth factor 4 (FGF4) and WNT3A promote the formation of 3D spheroids. A one-day pulse of retinoic acid (RA) is used from day 5 to day 6 of the protocol, culminating in the formation of anterior foregut (AFG) spheroids.
At this stage spheroids are collected and embedded in 3D matrix (Matrigel) and cultured in the presence of NOG for an additional 3 days. To promote epithelial growth, epithelial growth factor (EGF) and fibroblast growth factor 10 (FGF10) are added for 4 days. For the remainder of the protocol the spheroids are maintained in media with EGF and will continue to differentiate and grow in vitro for up to 2 months. We also describe a method for splitting esophageal organoids to reduce their density, which is necessary for the optimal growth and survival of the organoids in vitro (Fig. 2).
FIG. 2.

Protocol for the generation of human esophageal organoids. (A) hPSC are directly differentiated into definitive endoderm (day 3) and then into anterior foregut spheroids (day 6) which are then embedded in 3D matrix where differentiation and maturation of the spheroids continues for the generation of human esophageal organoids (HEOs). HEOs can survive up to 2 months in vitro, from day of embedding, by reducing organoids density 21 days after embedding (Day 27). Media and supplements are listed above each stage. For complete medium composition and supplements concentrations turn to Section 5.2 in the text. (B) Bright field images representing different stages of differentiation protocol. B-6 is zoom-in on dashed rectangle in B-5. Dashed circle in B-7 marks 3D matrix bubble margin. B-1, 5, 6, 7 and 8 were taken in up-right stereomicroscope (Leica, cat. # S8APO) scale bar: 500 μm. B-2, 3 and 4 were taken in inverted microscope (Nikon, TMS model), 10× magnification, scale bar—100 μm.
The HEOs that are generated in the described protocol are epithelial-only organoids and are relatively small in size, compared to gastric and intestinal organoids (Kechele & Wells, 2019). While useful for many applications, their size and the difficulty in accessing the lumen makes studying several aspects of esophagus biology challenging, like the effect of lumen acidity on the epithelium that can cause Barrett’s esophagus and esophagus adenocarcinoma (Iyer & Kaul, 2019). Therefore, we also provide a detailed protocol for the generation of organotypic raft cultures in an air-liquid interface (ALI). The rafts are generated by the dissociation of 3-weeks-old HEOs into a single cell suspension and culturing the cells as monolayer on trans-well inserts in ALI (Fig. 3).
FIG. 3.
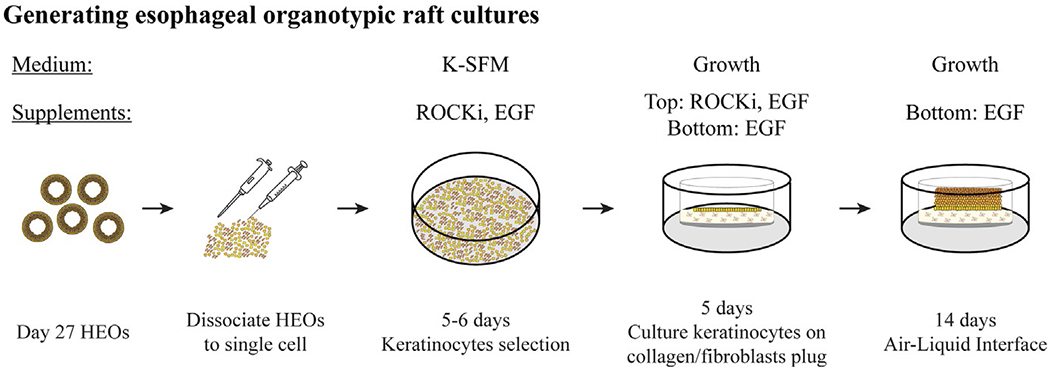
Generation of organotypic raft cultures from HEOs. Day 27 HEOs (21 days post embedding) are collected into a conical tube and dissociated to single cell using enzyme-based dissociation and serial passaging through pipette tips and syringe needles and re-cultured as monolayer for 5–6 days for keratinocyte selection. Then cells are re-dissociated and cultured on top of a collagen/mice fibroblasts plugs in trans-well inserts. After 5 days of culture, medium is removed from top compartment to generate air-liquid interface (ALI). Cells continue to proliferate and differentiate for up to 2 weeks to generate esophageal organotypic raft cultures. For complete and detailed protocol and medium composition turn to Section 5.3 in the text.
5. Step-by-step protocol
5.1. Coating plates for stem cell culture maintenance and directed differentiation
5.1.1. Materials and reagents
hESC-qualified Matrigel for coating plates (354277, Corning)
DMEM/F-12 (1132003, GIBCO)
Nunclon delta surface 6-well and 24-well tissue culture plates (6-well—140675, 24-well—142475, Nunc)
5.1.2. Protocol
Note: This protocol assumes that hPSCs are being routinely grown and passaged on 6-well plates using Matrigel and mTeSR1 and describes the protocol to generate HEOs on a 24-well plate.
Thaw hESC-qualified Matrigel in a 4 °C refrigerator.
Mix desired amount of Matrigel (according to manufacturer dilution factor) in cold DMEM/F-12. Keep Matrigel on ice at all time before dilution to prevent Matrigel polymerization.
Add the recommended coating volume to plates (500 μL per 24 well) and swirl plates to ensure the entire surface of the plate is coated.
For long-term storage (up to 2 weeks) seal coated plates with parafilm and store at 4 °C. For immediate use incubate for 1 h at room temperature or for 30min at 37 °C.
5.2. Differentiation of stem cells into esophageal organoids
The following protocol will generate three-dimensional HEOs in 24-well plate format (Fig. 2A). All procedures detailed below should be done in a sterile environment, in a biological cabinet and horizontal clean bench.
5.2.1. Materials, reagents and equipment
hPSC (~80% confluent) growing feeder-free in mTeSR1. We routinely use the H1 hESC line (NIH hESC-10-0043, WiCell research Institute). For one full 24-well plate generated from H1 cells three wells from a 6-well plate at ~80% confluency is used (Fig. 2B–1). Other embryonic and induced pluripotent stem cell lines can be used as well to generate HEOs; however, the number of hPSC wells needed for one 24-well plate of HEOs may need to be optimized.
DMEM/F-12 (1132003, GIBCO)
Accutase (A1105-01, Thermo Fisher Scientific)
15mL conical tubes (352097, BD Biosciences)
mTeSR1 media (05850, Stem Cell Technologies)
Y-27632 dihydrochloride, ROCK inhibitor—ROCKi (1254, Tocris)
RPMI medium 1640 (11875119, Invitrogen)
MEM non-essential amino acid solution (10mM, 100×, 11140-050, Invitrogen)
Defined FBS—dFBS (SH30070.02, HyClone)
ACTIVIN A (GFH6, Cell Guidance Systems)
Recombinant human BMP4 (314-BP, R&D Systems)
Recombinant human FGF4 (235-F4, R&D Systems)
Recombinant human NOGGIN (6057-NG, R&D Systems)
Recombinant human WNT3A (5036-WN, R&D Systems)
Retinoic acid (R2625, Sigma-Aldrich)
Matrigel-basement membrane matrix for embedding spheroids (354234, Corning)
Advanced DMEM/F-12 (12491-015, GIBCO)
l-Glutamine (200mM, 25030-081, Life Technologies)
Penicillin-streptomycin (100×, 15140-122, Life technologies)
HEPES buffer solution (1 M, 15630-080, Life technologies)
B27 supplement without vitamin A (50×, 12587-010, Thermo Fisher Scientific)
N2 supplement (100×, 17502-048, Invitrogen)
EGF (236-EG-01 M, R&D Systems)
Recombinant human FGF10 (345-FG, R&D Systems)
CultureOne Supplement (100×, A3320201, GIBCO)
5.2.2. Solutions preparation
| A medium: | ||
| RPMI 1640 | 500mL | |
| MEM NEAA | 5mL | |
| B medium: | ||
| RPMI 1640 | 500mL | |
| MEM NEAA | 5mL | |
| dFBS (0.2%) | 1mL | |
| C medium: | ||
| RPMI 1640 | 500mL | |
| MEM NEAA | 5mL | |
| dFBS (2%) | 10mL | |
| Growth medium: | ||
| Advanced DMEM/F-12 | 500mL | |
| HEPES Buffer solution | 7.5mL | |
| l-Glutamine | 5mL | |
| Pen-Strep | 5mL | |
| N2 | 5mL | |
| B27 | 10mL | |
5.2.3. Protocol (Fig. 2A)
5.2.3.1. hPSC dissociation and plating in 24-well plate (day-1)
To dissociate hPSCs into a single-cell suspension and re-plate on a hESC-qualified Matrigel coated 24-well plate, start with three 80% confluent hPSC H1 wells in 6-well format. From our experience there is large variability in the number of spheroids produced in each experiment due to starting cell confluency. To mitigate this it is essential to start with a consistent number of H1 cells (see step 9 below). Therefore one may need to use more wells from the initial 6-well plate of H1 cells to ensure adequate starting density in the 24-well plate.
Prepare hESC-qualified Matrigel coated 24-well plate or take out previously prepared plate from 4 °C and incubate for at least 1 h at room temperature or for 30min at 37 °C (as described in step 5.1.2).
Take out hPSC 6-well plate from incubator and manually remove any areas of differentiation under stereomicroscope, by scraping off the differentiated areas from the PSC colony using a pulled-glass Pasteur pipette or the edge of a pipette tip and aspirating off the media before Accutase treating.
Aspirate medium.
Wash each well once with 2mL of pre-warmed DMEM/F-12.
Aspirate DMEM/F-12 and add 600 μL of Accutase to each well.
Incubate 5–7 min at 37 °C until about 80% of the cells detach from the plate surface. If necessary, incubate few more minutes at 37 °C, up to 10 min in total. Tap gently on the side of the plate to detach any cells still attached.
Add 3mL of DMEM/F-12 (5× of Accutase volume) to each well, to neutralize the Accutase. Dissociate cells by pipetting up and down several times using 10mL serological pipette and transfer cells to a new 15mL conical tube, collect all three wells to one tube. Add ROCKi (5 μM final concentration) to the 15mL tube.
Centrifuge cells, 1300 RPM for 3 min at room temperature.
Carefully aspirate media and resuspend in 1mL warm mTeSR1 media and count cells using a hemocytometer or an automated cell counter. Add warm mTeSR1 media for a final concentration of 500,000–600,000 cell/mL (for 3 80% confluent 6-well wells it should be approximately 12–14mL), supplemented with Y-27632 (ROCKi) in final concentration of 10 μM. Pipette the cell solution a few times to ensure equal distribution of single cells throughout the resuspension.
Take out the 24-well hESC-qualified Matrigel coated plate from 37 °C. Aspirate DMEM-Matrigel solution from the wells.
Add 500 μL of dissociated cells to each well, approximately 250,000–300,000 cells per well.
Gently move plate back and forth, and side-to-side to ensure even distribution of the cells. This step should be done when plates are already placed in the incubator to prevent further movement after the even distribution of the cells.
Incubate in 37 °C, 5% CO2 incubator, overnight, before starting differentiation protocol below.
5.2.3.2. Differentiation protocol
Change medium daily according to the following instructions (warm medium to 37 °C before applying it to the plate):
14. Protocol Day 0: A medium supplemented with 100ng/mL ACTIVIN A and 50ng/mL BMP4 (Fig. 2B–2).
15. Day 1: B medium supplemented with 100ng/mL ACTIVIN A.
16. Day 2: C medium supplemented with 100ng/mL ACTIVIN A.
17. Day 3: C medium supplemented with 500ng/mL FGF4 and WNT3A, and 200ng/mL NOGGIN (Fig. 2B–3).
18. Day 4: C medium supplemented with 500ng/mL FGF4 and WNT3A, and 200ng/mL NOGGIN.
19. Day 5: C medium supplemented with 500ng/mL FGF4, 200ng/mL NOGGIN and 2 μM Retinoic Acid (Fig. 2B–4). After 24h, by day 6, free-floating spheroids will naturally emerge from the differentiated monolayer.
5.2.3.3. Embedding foregut spheroids in Matrigel
Spheroids are embedded in basement membrane Matrigel, which is stable at −20 °C. To prevent long-term 4 °C storage and repeated thaw-freeze cycles we recommend aliquoting Matrigel into 1.3mL aliquots (which will suffice for a one 24-well plate of embedded spheroids) and store aliquots at −20 °C (for more details regarding Matrigel storage and handling go to manufacture data sheet).
Before collecting the spheroids, thaw aliquots of embedding Matrigel (basement membrane matrix) in 4 °C refrigerator or on ice. One 24-well plate of embedded spheroids will require approximately 1.3mL of Matrigel (50 μL per well plus extra volume for pipetting error). Once thawed keep Matrigel on ice to prevent polymerization. A robust differentiation of one 24-well plate of esophagus spheroids can generate up to two 24-well plates of embedded spheroids, with 20–25 spheroids per well.
20. On day 6 free-floating spheroids will naturally emerge from the differentiated monolayer and are ready to be collected and embedded in a 3D Matrigel droplet. It is recommended to collect the spheroids 22–26 h after the last medium change, as longer exposure to RA might alter cell differentiation.
21. To collect spheroids, work under a stereomicroscope in a clean bench and gently swirl the plate until all spheroids in each well are centered (Fig. 2B–5, 6).
22. Using a 200 μL barrier pipette tip carefully collect all spheroids from each well and pool in a new 1.7mL Eppendorf tube. Be careful not to disrupt the base monolayer. You can collect spheroids from all wells in one tube or divide to several tubes, according to your experimental needs.
23. Let spheroids settle to the bottom of the tube. If needed, spin down the tube (at lowest RPM possible, at room temperature) for a few seconds to accumulate all spheroids in the pellet.
24. Aspirate medium carefully using a 200 μL tip under the stereomicroscope, without disturbing the spheroids, and wash spheroids once with 100 μL of warm DMEM/F-12.
25. Aspirate as much medium as possible without disturbing spheroids.
26. Add the appropriate amount of basement membrane Matrigel according to the number of wells and gently mix by pipetting up and down while avoiding bubble formation. For a 24-well plate add 1.25–1.3mL of Matrigel (50 μL per well). When warm, Matrigel will polymerize, so it is essential to work quickly while Matrigel is cold.
27. Carefully pipette 50 μL of spheroids-Matrigel mix into the center of a new 24-well plate well. Matrigel/spheroid mix should create a droplet in the center of the plate. Repeat for the entire 24-well plate.
28. Close the plate, let plate sit for 5 min at room temperature before carefully transferring the plate to an incubator, top side down (with the plate lid facing the shelf) to prevent spheroids from sinking to the bottom of the Matrigel droplet, for a 10–15 min incubation until Matrigel is completely solidified (Fig. 2B–7).
29. Take out the plate from the incubator and add 500 μL per well of Growth media supplemented with 100ng/mL EGF, 200ng/mL NOGGIN, 50ng/mL FGF10 and 1:100100× CultureOne supplement. In our hands, CultureOne reduces contaminating neural progenitor cells, possibly by promoting differentiation into neurons thus reducing their further proliferation. By adding CultureOne early in our HEO protocol we observed reduced numbers of neurons in the final stages of the HEO protocol. This step can be eliminated if there is little to no neural contamination.
30. Incubate plate in 37 °C, 5% CO2 incubator.
31. Change media after 3 days (day 9 of protocol) to Growth media supplemented with 100ng/mL EGF, 50ng/mL FGF10 and 1:100100× CultureOne supplement.
32. Change media after 4 additional days (day 13 of protocol) to Growth media supplemented with 100ng/mL EGF and continue changing the media every three to 4 days or earlier if medium turn yellow.
5.2.3.4. Reduction of HEOs density
Three weeks after embedding spheroids in Matrigel (day 27 of protocol), when HEOs grow in size and the phenol-red in the media changes to yellow more frequently, a reduction of culture density is required for continued organoid survival and growth for up to 2 months in vitro.
33. Cut the tip of a 1000 μL pipette tip and under stereomicroscope use the edges of the tip to remove Matrigel droplets from plate surface by carefully nudging the droplets.
34. Collect all droplets into a tube with little medium as possible.
35. Let organoids/Matrigel settle and remove any excess medium without disturbing the organoids.
36. Cut the tip of 1000 μL and 200 μL pipette tips. Starting with the 1000 μL tip, break down the Matrigel enclosing the organoids by pipetting up and down. Continue with the 200 μL tip until the organoids run smoothly through the tip.
37. Transfer all organoids into a 60mm plate with 5mL of warm DMEM/F-12. If necessary, remove any additional Matrigel remnants still surrounding the organoids sing a scalpel.
38. Swirl plate under stereomicroscope until organoids are centered, cut the tip of a 200 μL tip and collect organoids into a new tube. Collect organoids that look best: try not to collect organoids with a “fuzzy” appearance, which is possibly neuronal contamination, and avoid collecting any Matrigel remnants.
39. Let organoids settle to the bottom of the tube and aspirate any excess medium.
40. Resuspend organoids in Matrigel, 50 μL of Matrigel per well. At this stage you should culture 4–5 organoids per well. Mix by pipetting up and down with a 200 μL tip (with cut tip).
41. While working quickly collect 50 μL of organoid-Matrigel mix and pipette into the center of a new 24-well plate well. Repeat for the entire 24-well plate.
42. Close the plate, let plate sit for 5 min at room temperature before carefully transferring the plate to an incubator, top side down (as in step 28), for a short 10–15 min incubation until Matrigel is completely solidified.
43. Incubate organoids in Growth media supplemented with 100ng/mL EGF. Change medium every 3–4 days or earlier if medium turns yellow.
44. Organoids can be cultured in vitro for up to 2 months (Day 62 of protocol) from the day of Matrigel embedding (day 6 of culture) (Fig. 2B–8). At this stage dead cells will start to accumulate in the organoid lumen and will affect morphology and survival.
5.3. Esophageal raft culture protocol
HEOs generated in the protocol described above can be used to generate esophageal organotypic raft cultures in ALI, adapted from published protocols of keratinocytes and skin organotypic raft cultures (Anacker & Moody, 2012; Matrka et al., 2018; Nakahara, Peh, Doorbar, Lee, & Lambert, 2005). This can be beneficial for lumen manipulation as well as other studies that HEOs might not be suited for.
In general, 3-weeks-old HEOs are dissociated to single cells, cultured with keratinocyte specific medium, dissociated again and then cultured on trans-well inserts in ALI for up to 2 weeks (Fig. 3).
5.3.1. Materials and reagents
TrypLE Select Enzyme (1×), no phenol red (12563029, GIBCO)
1mL syringe (BD 309659, Thermo Fisher Scientific)
Syringe needles-22G× 1 ½ in (305156), 25G× 5/8 in (305122) and 27.5G×½ in (305109), BD Medical
DMEM/F-12 (1132003, GIBCO)
Keratinocyte-SFM (K-SFM) 1× (17005042, GIBCO)
Bovine Pituitary Extract, BPE (provided with K-SFM media)
EGF (236-EG-01M, R&D Systems)
Penicillin-streptomycin (100×, 15140-122, Life technologies)
Y-27632 dihydrochloride, ROCKi (1254, Tocris)
100 mm tissue culture plates (229690, CELL TREAT)
Collagen type IV from human placenta (C5533-5MG, Sigma-Aldrich)
0.1% bovine serum albumin (BSA, A3608, Sigma-Aldrich) in PBS
40μL cell strainer (22-363-547, Fisher Scientific)
RatCol® Type I Collagen (5153, Advanced BioMatrix)
10× MEM (11430030, Thermo Scientific)
Acetic Acid (A6283, Sigma-Aldrich)
Ham’s F-12 Nutrient Mix, Powder (21700075, GIBCO)
Sodium bicarbonate—NaHCO3 (S5761, Sigma-Aldrich)
Calcium chloride—CaCl2 (C4901, Sigma-Aldrich)
Mouse fibroblasts-J2-3T3 cells (EF3003, Kerafast)
10N NaOH (SX0607N-6, Sigma-Aldrich)
Transwell 24mm Polyester Membrane Inserts, 6-well plate (3450, Corning)
Trypsin-EDTA (0.05%), phenol red (25300054, Thermo Fisher)
5.3.2. Coating 100mm plates with collagen IV
Dilute Collagen IV in filtered 0.25% Acetic acid.
Coat 100 mm plates by applying 6–8mL of Collagen IV—acetic acid mix (final concentration of 1.5μg/cm2). Tilt plate back and forth to ensure coverage of the entire plate surface.
Seal plates and incubate for 1 h at room temperature.
Before use aspirate Collagen IV and wash 3 times with 10mL sterile PBS.
Aspirate PBS and leave plates open to dry inside the biological cabinet until cells are ready for culture.
5.3.3. Dissociation of HEOs and culturing in keratinocyte media
Collect 36–48 wells of 3-weeks-old HEOs (from 24-well plates, approximately 20 HEOs per well) by following Section 5.2.3, steps 34–40 (prior to reducing density), transferring the organoids to a new 15mL conical tube.
Wash organoids once with warm DMEM/F-12.
Remove as much medium as possible and add 1mL of TrypLE Select.
Incubate at 37 °C for 30–40 min. Take out every 5–7 min and dissociate by triturating them, first through 1000μL and 200μL tips, and later through series of 22.5G, 25.625G and 27.5G needles in 1mL syringe. To prevent organoids from sticking to the tips and needles can be coated with 0.1% BSA in PBS by pipetting the BSA up and down through the tip/needle few times prior to use.
Once HEOs are dissociated stop TrypLE Select reaction by adding 3–5× volume of warm DMEM/F-12.
Centrifuge at 1300 RPM for 5 min at room temperature.
Aspirate medium and resuspend cells in 1mLK-SFM medium, supplemented with 30μg/mL BPE, 10ng/mL EGF, 1:100 Pen/Strep and 10μM ROCKi (complete K-SFM medium), pass cells mix through 40μL cell strainer to remove any remaining clamps.
Count cells using a hemocytometer or an automated cell counter.
Culture 1×106 cells per 100mm collagen IV coated plate in 10mL of complete K-SFM medium plus supplements as described in step 7 of this section.
Gently swirl and move plate back and forth two to three times to ensure even distribution of the cells and incubate in 37 °C, 5% CO2 incubator.
Change medium every 2 days until cells reach confluency (5 to 6 days).
5.3.4. Preparing collagen—Mouse fibroblasts gels for raft cultures
Once the keratinocytes reach confluency, they will be dissociated to a single cell suspension and cultured on trans-well inserts coated with collagen gel containing mouse fibroblasts feeder cells. The gels need to be prepared 4 to 7 days before keratinocytes culturing.
-
Prepare working solutions:
10× F-12 media:- Dissolve 1 packet of Ham’s F-12 powder into 90mL of water.
- Add 1.176 g of sodium bicarbonate (NaHCO3).
- pH to 7.2.
- Add 23 μL of 1M CaCl2.
- Filter sterilize (long-term storage at −20 °C).
20mL Collagen solution:- 18 mL 2mg/mL rat tail collagen I (diluted according to manufacture data sheet).
- 2mL of 10× MEM.
- 400 μL 1M sodium bicarbonate (NaHCO3).
- Prepare collagen pre-mix, keep everything on ice to prevent polymerization. For 6 trans-wells inserts (6-well format):
- 2.5 mL of 10× F12 media.
- 6μL of 10N NaOH.
- 250μL penicillin—streptomycin.
- 2.5mL FBS.
- 20mL collagen solution.
Plate 1mL of collagen mix per trans-well insert, swirl plate to coat the entire surface of the trans-well insert and incubate for 5 min at room temperature.
Add 600μL single cell suspension of irradiated J2-3T3 mouse fibroblasts in concentration of 7.5×105 cells/mL to the remaining collagen mix and mix thoroughly while avoiding bubbles.
Add 2mL of collagen/fibroblasts mix on top of the 1mL solidified collagen mix in each trans-well insert. Incubate for 30 min in 37 °C incubator.
Add warm 1 mL DMEM/F-12 media supplemented with 10% FBS and 1× Pen/Strep on top of the collagen/fibroblasts plugs and incubate in a 37 °C, 5% CO2 incubator for 4–7 days. There is no need of exchanging the media during the incubation period.
5.3.5. Transferring cells from keratinocyte media to raft cultures
Take out plates with confluent cells grown in keratinocyte media from the 37 °C incubator.
Aspirate complete K-SFM medium and wash once with 5mL sterile 1× PBS.
Aspirate PBS and add 2mL of pre-warmed 0.05% Trypsin-EDTA.
Incubate for 5–7 min at 37 °C until cells detach from plate surface. Tap plate gently on the side to detach any cells left adherent.
Block the trypsin by adding 3× volume of warm DMEM/F-12, pipette up and down few times to break down cell clamps and collect the cells into a new 15mL conical tube.
Centrifuge cells at 1300 RPM, for 5 min, at room temperature.
Aspirate media and resuspend cells in 1mL warm Growth media supplemented with 100ng/mL EGF and 10μM ROCKi.
-
Count cells using a hemocytometer or an automated cell counter.
Resuspend cells in appropriate amount of Growth media supplemented with 100ng/mL EGF and 10μM ROCKi to achieve a concentration 1–1.2×106 cells per 1mL of media.
Take out trans-wells with collagen/fibroblasts gels from incubator and aspirate the media from the top compartment.
Add 2mL of warm Growth media supplemented with 100ng/mL EGF to the bottom compartment of each well.
Add 1mL per trans-well of the resuspended cells from step 8, on top of the collagen/fibroblast gels. Gently swirl and move plate back and forth two to three times to ensure even distribution of the cells and incubate plates in a 37 °C, 5% CO2 incubator.
For the first 5 days of culture change medium daily in top and bottom compartment. It is important to supplement the top compartment with ROCKi as it is crucial for cell survival.
On day five remove medium from top compartment and keep in ALI for up to 2 weeks. Change bottom compartment medium on a daily basis with Growth media supplemented with 100ng/mL EGF.
5.4. Immunofluorescence analysis of HEOs and organotypic raft cultures (Fig. 4)
FIG. 4.
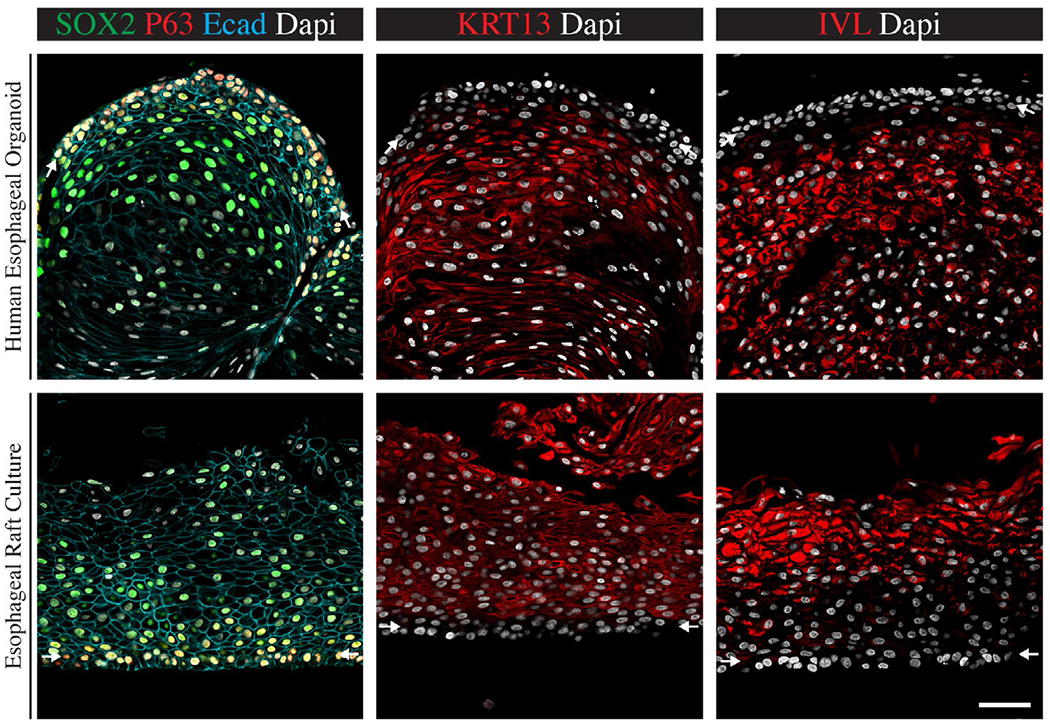
Immunofluorescent analysis of HEOs and raft cultures. HEOs (Top panel) and organotypic raft cultures (Bottom panel) express esophagus epithelium specific markers. The entire epithelium is expressing the epithelium marker E-cadherin. Basal layers cells expressing P63 and SOX2 but lack the expression of keratin 13 (KRT13) and involucrin (IVL) as marked by white arrows. Suprabasal layers of differentiated cells express KRT13 and IVL but lack P63 expression. Scale bar—50 μm.
Images where modified from Trisno, S. L., Philo, K. E. D., McCracken, K. W., Catá, E. M., Ruiz-Torres, S., Rankin, S. A., et al. (2018). Esophageal organoids from human pluripotent stem cells delineate Sox2 functions during esophageal specification. Cell Stem Cell, 23(4), 501–515.e7. doi: 10.1016/j.stem.2018.08.008 (Fig. 4), pseudo colors and brightness were altered from original images for visualization purposes.
5.4.1. Materials and reagents
4% paraformaldehyde
1× PBS
30% sucrose in 1× PBS
Tissue-Tek O.C.T Compound (25608-9030, VWR)
ImmEdge Hydrophobic Barrier PAP pen (H-400, VECTOR Laboratories)
Fisherbrand Superfrost Plus Microscope Slides (1255015, Thermo Scientific)
100×-triton (X100, Sigma-Aldrich)
Normal Donkey Serum (017000121, Jackson ImmunoResearch)
Goat anti-SOX2 (AF2018, R&D systems) (1:250)
Rabbit anti-P63 (SC-8343, Santa Cruz Biotechnology) (1:250)
Mouse anti-E-cadherin (610182, BD Bioscience) (1:500)
Rabbit anti-KRT13 (ab92551, Abcam) (1:1000)
Rabbit anti-Involucrin (HPA055211, Atlas Antibodies) (1:250)
488 conjugated donkey anti-goat secondary antibody (A11055, Thermo Fisher Scientific) (1:500)
546 conjugated donkey anti-rabbit secondary antibody (A10040, Thermo Fisher Scientific) (1:500)
647 conjugated donkey anti-mouse secondary antibody (715-605-150, Jackson ImmunoResearch laboratories) (1:500)
Fluoromount-G (0100-01, Southern Biotech)
Superslip 24× 60 #1 glass coverslips (M6045-10, Cardinal Health)
5.4.2. Tissue fixation, preparation and cryosection
Fix HEOs or raft cultures in 4% paraformaldehyde, overnight in 4 °C. For raft cultures-fix on trans-well, adding PFA to both top and bottom compartments. After fixation remove rafts from trans-well by cutting the membrane out using a scalpel and gently removing the rafts from the membrane using forceps and move to a new 6-well plate for the washes and sucrose stages before embedding in O.C.T.
Wash samples in 1× PBS, repeat 3 times.
Incubate samples in 30% sucrose/1× PBS solution overnight in 4 °C. If needed, samples can be kept in 30% sucrose for longer periods.
Take out samples from sucrose and embed in a mold immersed in O.C.T compound. Freeze O.C.T by floating the molds in 100% EtOH with dry ice.
Store molds in −80 °C freezer.
Section molds, using cryostat, into 8–12 μm thick sections and organize on Super Frost Plus Slides, marks slide borders using hydrophobic barrier PAP pen.
Store slides at −80 °C until ready for staining.
5.4.3. Immunofluorescent protocol
Take out slides from −80 °C and leave at room temperature for 5–60min.
Rehydrate slides in 1× PBS for 10min at room temperature on shaker.
Incubate 5 min at room temperature, on shaker, in 1× PBS with 0.5% triton (PBST).
Organize slides in humified chambers.
Block slides by adding 200 μL per slide of blocking solution (PBST + 5% normal donkey serum), for at least 30min at room temperature.
Incubate slides with 200 μL per slide of primary antibodies diluted in blocking solution, overnight in 4 °C (For antibodies dilutions go to Section 5.4.1).
Wash slides in PBST, 10min at room temperature, repeat three times.
Incubate slides with 200 μL per slide of secondary antibodies with DAPI diluted in blocking solution, for 1–2h at room temperatures, make sure slides are protected from light (For antibodies dilutions go to Section 5.4.1).
Wash slides in 1× PBS, 10min at room temperature, repeat 3 times.
Dry excess PBS carefully from slides (around the sections) and mount with cover slips by applying small amount of Flouromount-G and slowly covering the slide with coverslips. Make sure there are no bubbles over samples.
Let slides dry at room temperature over night before imaging them using confocal immunofluorescence microscope (Fig. 4). Make sure slides are protected from light.
Slides can be stored at 4 °C, protected from light, for longer periods.
6. Future directions
As mentioned throughout this chapter, the esophageal organoids and raft cultures, while providing an excellent model for the human esophagus epithelium, lack significant contribution from mesenchyme derivatives, as well as neurons and glia of the enteric nervous system that innervate the esophagus mesenchyme and control muscle peristalsis. The mesenchyme is involved in many aspects of the esophagus biology. During development, the mesenchyme provides molecular and physical cues to the epithelium that regulates esophagus specification from the anterior foregut, as well as other aspects of the esophagus development. In the adult esophagus, the mesenchyme provides structural support and the muscles and innervating neurons generate peristalsis that propel food down the esophagus toward the stomach.
The mesenchyme is also involved in several esophagus related disease. For example, ESCC and adenocarcinoma tumors can invade the mesenchyme and metastasize. In epidermolysis bullosa patients, where mutations in genes encoding structural proteins at the epithelium-mesenchyme junction, disease manifestations within the esophagus may result in strictures and lesions that lead to dysphagia.
The absence of mesenchyme from the current model prevents us from studying and understanding the molecular mechanisms that are the basis of mesenchyme involvement in development and disease. The lack of any cells of the enteric nervous system prevents us of studying motility related disease of the esophagus.
Therefore, a more complex model, that consists of more layers of the human esophagus, is needed to further our understanding of esophageal biology. Further, a model that better resembles the physiological conditions in the human esophagus might provide a tractable bioengineered tissue for esophageal grafts, for the treatment of esophagus injury in human, by generating cultures from patient derived induced hPSC.
There are currently several efforts to develop such models. One approach is by manipulating the monolayer that are generated at the early stages of the differentiation protocol to generate raft cultures with both epithelium and mesenchyme. Another approach uses published protocols for the directed differentiation of hPSC into mesenchyme and enteric nervous system (ENS) to generate each population of cells separately (Bajpai et al., 2010; Loh et al., 2016). Re-combining the different population of cells in 3D cultures can generate a complex model. This method was implemented successfully with ENS and human intestinal organoids to generate innervated intestinal tissue to study motility diseases (Workman et al., 2017).
Other efforts should focus on the scalability of the models and the usage of these models for the study of various esophagus disease and injury models and the therapeutic potential of esophageal raft cultures in injury models in animals.
7. Summary
In this chapter we provided a step-by-step protocol for the direct differentiation of hPSC into HEOs and esophageal organotypic raft cultures. We also provide a protocol for immunofluorescent analysis. The HEOs and rafts that are generated in this protocol provide an excellent model for human esophagus epithelium and can be used in diverse studies to study esophagus epithelium development, disease and injury. The lack of mesenchyme and ENS from the cultures prevents more advance studies. However, more complex models are currently being developed and will further advance the research in the field.
Acknowledgments
The authors are supported by the grants from the NIH, U19 AI116491 (J.M.W.), P01 HD093363 (J.M.W.), UG3 DK119982 (J.M.W.), the Shipley Foundation (J.M.W.) and the Allen Foundation (J.M.W.). We also received support from the Digestive Disease Research Center (P30 DK078392).
References
- Anacker D, & Moody C (2012). Generation of organotypic raft cultures from primary human keratinocytes. Journal of Visualized Experiments, (60), e3668. 10.3791/3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature, 463(7283), 958–962. 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, & Baetge EE (2005). Efficient differentiation of human embryonic stem cells to definitive endoderm. Nature Biotechnology, 23(12), 1534–1541. 10.1038/nbt1163. [DOI] [PubMed] [Google Scholar]
- Fausett SR, & Klingensmith J (2012). Compartmentalization of the foregut tube: Developmental origins of the trachea and esophagus. Wiley Interdisciplinary Reviews: Developmental Biology, 1(2), 184–202. 10.1002/wdev.12. [DOI] [PubMed] [Google Scholar]
- Haeri H, Mardany O, Asadi-Amoli F, & Shahsiah R (2013). Human papilloma virus and esophageal squamous cell carcinoma. Acta Medica Iranica, 51 (4), 242–245. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/23690103. [PubMed] [Google Scholar]
- Iyer PG, & Kaul V (2019). Barrett esophagus. Mayo Clinic Proceedings, 94(9), 1888–1901. 10.1016/j.mayocp.2019.01.032. [DOI] [PubMed] [Google Scholar]
- Jacobs IJ, Ku WY, & Que J (2012). Genetic and cellular mechanisms regulating anterior foregut and esophageal development. Developmental Biology, 369(1), 54–64. 10.1016/j.ydbio.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasagi Y, Chandramouleeswaran PM, Whelan KA, Tanaka K, Giroux V, Sharma M, et al. (2018). The esophageal organoid system reveals functional interplay between notch and cytokines in reactive epithelial changes. Cellular and Molecular Gastroenterology and Hepatology, 5(3), 333–352. 10.1016/j.jcmgh.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechele DO, & Wells JM (2019). Recent advances in deriving human endodermal tissues from pluripotent stem cells. Current Opinion in Cell Biology, 61, 92–100. 10.1016/j.ceb.2019.07.009. [DOI] [PubMed] [Google Scholar]
- Loh KMM, Chen A, Koh PWW, Deng TZZ, Sinha R, Tsai JMM, et al. (2016). Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell, 166(2), 451–467. 10.1016/j.cell.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrka MC, Cimperman KA, Haas SR, Guasch G, Ehrman LA, Waclaw RR, et al. (2018). Dek overexpression in murine epithelia increases overt esophageal squamous cell carcinoma incidence. PLoS Genetics, 14(3), 1–24. 10.1371/journal.pgen.1007227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken KW, Aihara E, Martin B, Crawford CM, Broda T, Treguier J, et al. (2017). Wnt/β-catenin promotes gastric fundus specification in mice and humans. Nature, 541(7636), 182–187. 10.1038/nature21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken KW, Catá EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, et al. (2014). Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature, 516(7531), 400–404. 10.1038/nature13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisey EE, & Rustgi AK (2018). The lung and esophagus: developmental and regenerative overlap. In Trends in cell biology. Elsevier Ltd. 10.1016/j.tcb.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Múnera JO, Sundaram N, Rankin SA, Hill D, Watson C, Mahe M, et al. (2017). Differentiation of human pluripotent stem cells into colonic organoids via transient activation of BMP signaling. Cell Stem Cell, 21(1), 51–64.e6. 10.1016/j.stem.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara T, Peh WL, Doorbar J, Lee D, & Lambert PF (2005). Human papillomavirus type 16 E1 E4 contributes to multiple facets of the papillomavirus life cycle. Journal of Virology, 79(20), 13150–13165. 10.1128/jvi.79.20.13150-13165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que J (2015). The initial establishment and epithelial morphogenesis of the esophagus: A new model of tracheal-esophageal separation and transition of simple columnar into stratified squamous epithelium in the developing esophagus. Wiley Interdisciplinary Reviews: Developmental Biology, 4(4), 419–430. 10.1002/wdev.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosekrans SL, Baan B, Muncan V, & van den Brink GR (2015). Esophageal development and epithelial homeostasis. American Journal of Physiology—Gastrointestinal and Liver Physiology, 309(4), G216–G228. 10.1152/ajpgi.00088.2015. [DOI] [PubMed] [Google Scholar]
- Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, et al. (2011). Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature, 470(7332), 105–110. 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trisno SL, Philo KED, McCracken KW, Catá EM, Ruiz-Torres S, Rankin SA, et al. (2018). Esophageal organoids from human pluripotent stem cells delineate Sox2 functions during esophageal specification. Cell Stem Cell, 23(4), 501–515.e7. 10.1016/j.stem.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman MJ, Mahe MM, Trisno S, Poling HM, Watson CL, Sundaram N, et al. (2017). Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nature Medicine, 23(1), 49–59. 10.1038/nm.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WY, Slack JMW, & Tosh D (2005). Conversion of columnar to stratified squamous epithelium in the developing mouse oesophagus. Developmental Biology, 284(1), 157–170. 10.1016/j.ydbio.2005.04.042. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jiang M, Kim E, Lin S, Liu K, Lan X, et al. (2017). Development and stem cells of the esophagus. Seminars in Cell & Developmental Biology, 66, 25–35. 10.1016/j.semcdb.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yang Y, Jiang M, Huang SX, Zhang W, Al Alam D, et al. (2018). 3D modeling of esophageal development using human PSC-derived basal progenitors reveals a critical role for notch signaling. Cell Stem Cell, 23(4), 516–529.e5. 10.1016/j.stem.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn AM, & Wells JM (2009). Vetebrate endoderm development. Annual Review of Cell Biology, 25, 221–251. 10.1146/annurev.cellbio.042308.113344.Vertebrate. [DOI] [PMC free article] [PubMed] [Google Scholar]