

Abstract
Chemotherapy, as one of the most commonly used treatment modalities for cancer therapy, provides limited benefits to hepatoma patients, owing to its inefficient delivery as well as the intrinsic chemo-resistance of hepatoma. Bioinformatic analysis identified the therapeutic role of a liver-specific microRNA — miR-122 for enhancing chemo-therapeutic efficacy in hepatoma. Herein, a cyclodextrin-cored star copolymer nanoparticle system (sCDP/DOX/miR-122) is constructed to co-deliver miR-122 with doxorubicin (DOX) for hepatoma therapy. In this nanosystem, miR-122 is condensed by the outer cationic poly (2-(dimethylamino) ethyl methacrylate) chains of sCDP while DOX is accommodated in the inner hydrophobic cyclodextrin cavities, endowing a sequential release manner of miR-122 and DOX. The preferentially released miR-122 not only directly induces cell apoptosis by down regulation of Bcl-w and enhanced p53 activity, but also increases DOX accumulation through inhibiting cytotoxic efflux transporter expression, which realizes synergistic performance on cell inhibition. Moreover, sCDP/DOX/miR-122 displays remarkably increased anti-tumor efficacy in vivo compared to free DOX and sCDP/DOX alone, indicating its great promising in hepatoma therapy.
Keywords: Cyclodextrin-based star copolymer nanoparticle, miR-122, Doxorubicin, Sequential release, Hepatoma chemotherapy
Graphical abstract
Highlights
-
•
Cyclodextrin-based polymeric nanoparticle was developed to co-deliver miR-122 and doxorubicin.
-
•
The nanoparticle sequentially released miR-122 and doxorubicin into HepG2 cells.
-
•
The preferentially released miR-122 induces cell apoptosis and inhibits doxorubicin efflux.
-
•
Enhanced anti-tumor effects with reduced cardiotoxicity were achieved in vivo.
1. Introduction
Hepatocellular carcinoma (HCC), also called hepatoma, is ranked as the seventh most common diagnosed cancers worldwide and represents the fourth highest cause of cancer-related mortality [1]. Surgical treatments, including radiofrequency ablation, liver resection and liver transplantation, are considered to be the first-line therapeutic strategies for HCC therapy [2,3]. However, most patients are diagnosed at intermediate advanced stage, during which they are not suitable for surgery. Recent advances in molecular target therapy [[4], [5], [6]] and immunotherapy [7,8] have provided them with alternatives, but only a limited group of patients experience clinical benefits [9]. More unfortunately, conventional chemotherapy has long been regarded as ineffective against HCC due to the insensitivity of hepatoma to chemotherapeutic agents [10]. As early as more than forty years ago, doxorubicin (DOX) was firstly explored in systemic therapy of advanced HCC, but it showed limited efficacy with a high associated mortality rate [11,12]. Despite this, DOX remains a widely used anticancer drug for multiple other cancers, and can also be administrated by transarterial chemoembolisation for intermediate advanced HCC patients [13]. Therefore, it has become extremely urgent to develop an effective strategy to overcome DOX resistance to improve the efficacy of HCC systemic chemotherapy. For DOX, one of the most important mechanisms of resistance is overexpression of ATP-binding cassette (ABC) efflux transporter such as P-glycoprotein (P-gp) and multidrug resistance-associated proteins (MRPs) that facilitate the cytotoxic efflux from cancer cells [14,15].
MicroRNAs (miRNAs) represent an abundant family of small non-coding RNAs with 18–21 bp that function as important gene regulators [16]. miRNAs play important roles in pathogenesis, development and the prognosis of cancer [17,18]. miR-122, a liver-specific miRNA accounting for 70% of the total liver miRNA population, has been reported to be vital in liver development, differentiation, homeostasis and metabolic functions [19]. miR-122 expression is frequently down-regulated in HCC and loss of miR-122 has been closely associated with HCC development and metastasis [20,21]. Several studies have revealed that miR-122 served as a tumor suppressor in HCC by inducing cell apoptosis and inhibiting cell growth [22,23]. Furthermore, restoration of miR-122 also sensitized hepatoma cells to multiple chemotherapeutics through regulating the expression of drug resistance related genes [[24], [25], [26]]. Given this, it would be of great potential to combine miR-122 with DOX to improve the overall therapeutic effect for hepatoma.
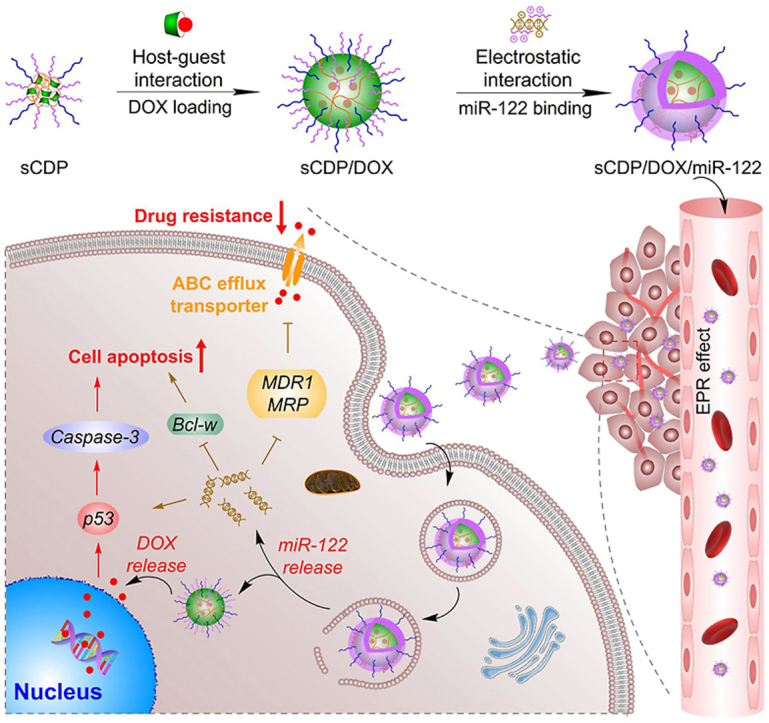
In our previous study, using a one-step atom transfer radical polymerization (ATRP), we developed a β-cyclodextrin (CD)-containing star co-polymer with a cross-linked core of CD polymer (PCD) and pH-sensitive poly (2-(dimethylamino) ethyl methacrylate) (PDMAEMA) for efficient delivery of DOX both in vitro and in vivo [27,28]. In this study, an improved structure of CD-cored star polymer with PDMAEMA and hydrophilic poly(ethylene glycol) (PEG) as the arms (denoted as sCDP) was synthesized by a continuously two-step ATRP and used as the carrier material for co-delivery of DOX and miR-122 mediating effective hepatoma treatment (Scheme 1). To be specific, the CD cavities in the inner core of sCDP provide accommodation to load hydrophobic DOX via host-guest interaction and further self-assemble into nanostructure to obtain sCDP/DOX, whereas the PDMAEMA chains are readily to condense miR-122 through electrostatic interaction to form the middle layer of the resulting nanosystem sCDP/DOX/miR-122. PEG forms the surface shell to resist serum protein adsorption and achieve enhanced tumor accumulation. With our rational design, sCDP/DOX/miR-122 accumulates at the tumor site through enhanced permeability and retention (EPR) effect and then preferentially releases miR-122 into the cytoplasm after internalization into hepatoma cells. miR-122 inhibits the expression of ABC efflux transporter, leading to an increase level of DOX concentration to enable effective cytotoxic effect. Meanwhile, miR-122 can directly induce cell apoptosis by regulating its downstream target genes to exert synergistic anti-tumor effect. In this study, the synergistic mechanism and efficacy of sCDP/DOX/miR-122 were investigated to evaluate its potential for hepatoma treatment.
Scheme 1.

Schematic illustration for (a) preparation and (b) effect mechanism of sCDP/DOX/miR-122 in HCC treatment. After intravenous administration, sCDP/DOX/miR-122 enters the blood circulation, extravasates from leaky tumor vasculature, and accumulates at the tumor site. After uptake by HCC cells (I) and endosomal escape (II), sCDP/DOX/miR-122 sequentially releases miR-122 (III) and DOX (IV) into the cytoplasm. DOX enters the cell nucleus and induces DNA damage (V), and miR-122 directly induces cell apoptosis (VI) and decreases drug resistance by inhibiting DOX efflux transporter expression (VII), thus resulting in synergistic anti-tumor effect towards hepatoma.
2. Results and discussion
2.1. Bioinformatics analyses of miR-122 expression and its correlation with DOX resistance in HCC
To valid the association of miR-122 expression and HCC tumor as well as the survival of HCC patients, we systemically analyzed the data downloaded from The Cancer Genome Atlas (TCGA). The beanplots in Fig. 1a demonstrated that miR-122 was significantly downregulated in primary HCC tumor tissues compared to the corresponding para-tumor controls (n = 48, P < 0.001). The result was also confirmed in 97 pairs of HCC and adjacent normal tissues obtained from the Gene Expression Omnibus (GEO) (Fig. S1a). In another larger cohort of HCC patients (n = 346) from TCGA, miR-122 expression in tumor tissues was negatively correlated with clinical stages (Fig. 1b). Moreover, HCC patients with lower miR-122 exhibited significantly worse prognosis including progression-free survival (PFS) (Fig. 1c, P = 0.0006) and cancer-specific survival (CSS) (Fig. S1b, P = 0.0150) than those with higher expression. We also evaluated the performances of HCC resistance to seven common chemo-therapeutic agents, and the bubble heatmap showed the landscape of their resistance performances in miR-122 low and high groups (Fig. 1d). The bubble size represented the enrichment score (ES), and the color bar indicated different FDR Q values. DOX ranked first in the drug resistance landscape, and the detail of GSEA analysis was shown in Fig. 1e. Moreover, in vitro study also confirmed that HepG2 cells transfected with Lipo2k/miR-122 were more sensitive to DOX treatment (Fig. S2). The above results implied the potential of miR-122 as a therapeutic target to enhance chemo-therapeutic efficacy in HCC.
Fig. 1.

Bioinformatics analyses of miR-122 expression and its correlation with DOX resistance in HCC. (a) miR-122 expression for 48 HCC cases of para-tumor tissues and tumor tissues in TCGA datasets. (b) miR-122 expression in tumor tissues of HCC patients at different clinical stages. RPM: reads per million microRNA reads. (c) Kaplan–Meier survival analysis of PFS of HCC patients with different miR-122 levels. (d) The relationship between miR-122 and HCC resistance to seven chemo-therapeutics. (e) The negative correlation of miR-122 expression with DOX resistance in HCC.
2.2. Preparation and characterization of sCDP/DOX/miR-122
The carrier material, sCDP, was synthesized by a continuous two-step atom transfer radical polymerization (ATRP) (Fig. 2a) of MCD and DMAEMA. MCD, a mixture of mono- and multi-methacrylate substituted β-CD (Fig. S3a) as reported in our previous study [27], was used as both the monomer and cross-linker to produce the core of PEG-[PCD] (the structure was illustrated in Fig. S3b). Initiated from PEG-[PCD], DMAEMA was further polymerized to form the arms of sCDP. The 1H NMR spectrum of sCDP was shown in Fig. 2b. The signal at 4.8 ppm (signal C1–H) was attributed to the protons on the C1 in the glucose units of CD. The signals at 4.1 (signal e) and 2.2 ppm (signal f) represented the methylene group (–CH2–OOC–) adjacent to the carboxylate group and the methylene (–CH2–N(CH3)2) group of PDMAEMA, respectively. The molar ratio of PEG: CD: DMAEMA was determined to be 1:9:34 by the peak integration ratio of signal C1–H to signal G and signal f in the 1H NMR spectrum. GPC results showed that the apparent molecular weight (Mw) of the star polymer PEG-[PCD] and sCDP were 62000 g/mol and 76000 g/mol, respectively.
Fig. 2.

Synthesis and characterization of sCDP. (a) Synthesis route of sCDP by two-step ATRP of MCD and DMAEMA. (b) The 1H NMR spectra of PEG-[PCD] and PEG-[PCD]-PDMAEMA (sCDP) in DMSO‑d6.
Using sCDP as the host carrier material, the guest molecule DOX was incorporated to form sCDP/DOX at different weight ratios of DOX to sCDP. As displayed in Table S1, the drug loading content increased with the increase of weight ratio while the drug encapsulation efficiency showed a bell-shaped dependent manner. The size of sCDP/DOX decreased as the weight ratio increased from 1/10 to 2/10, indicating DOX participation in the self-assembly process was beneficial to form sCDP/DOX with more compact structure. At higher weight ratios, the size increased as a result of more DOX loading. Positive zeta potential values of sCDP/DOX indicated that PDMEAM chains located at their outer surface. sCDP/DOX prepared at the weight ratio of 10/3 (sCDP/DOX-3) with a suitable size and relative higher surface potential was used and further characterized in the following study. The average size was measured to be 182.4 nm with a relatively narrow distribution of 0.169 by dynamic laser scattering (DLS) (Fig. 3a). According to transmission electron microscopy (TEM) image (Fig. 3b) and atom force microscopy (AFM) image (Fig. S4a and S4a′), the morphology of sCDP/DOX-3 showed irregular spherical shape with coarse surface. Host-guest competitive study further indicated that DOX was incorporated into sCDP by host-guest interaction, as it could be excluded and released from sCDP as a result of the addition of adamantane carboxylic acid (ADA), a competitive gust molecule with a higher complexation constant [29] (Fig. S5). The result demonstrated that the incorporation of CD in the star polymer was essential for DOX loading.
Fig. 3.

Characterizations of sCDP/DOX-3 and sCDP/DOX/miR-122. DLS size profile (a) and TEM image (b) of sCDP/DOX-3. (c) Agarose gel electrophoresis analysis of sCDP and sCDP/DOX-3 complexation with miR-122. DLS size profile (d) and TEM image (e) of sCDP/DOX/miR-122 prepared at the N/P ratio of 10/1. Release profiles of DOX (f) from sCDP/DOX/miR-122 and RNADy677 (g) from sCDP/DOX/RNADy677(n = 3).
Next, sCDP/DOX-3 was used to further condense miR-122 at various N/P ratios. As shown in Fig. 3c, miR-122 migration can be effectively inhibited at a N/P ratio of 10 or above, at which, the signal reduced or even disappeared because the nucleic acid dye SYBR Green could not combine miR-122 to produce any visible signal when it was completely encapsulated in the particle. Similar results were also found in our previous study [30] and other study [31]. In comparison with sCDP alone, sCDP/DOX-3 had similar miR-122 condensation ability, indicating that the inclusion of DOX had no influence on miR-122 condensation. sCDP/DOX/miR-122 prepared at all N/P ratios had smaller average sizes than sCDP/DOX-3, and the size distributions were relative narrower (Table S2). It was perhaps because PDMAEMA chains shrank as they condensed miR-122, resulting in a more compact and uniform structure. Meanwhile, the zeta potentials of all sCDP/DOX/miR-122 were less positive compared to sCDP/DOX-3, further indicating the successful complexation of miR-122 by sCDP/DOX-3. sCDP/DOX/miR-122 prepared at the N/P ratio of 10 were chosen to conduct the following experiments. The resultant sCDP/DOX/miR-122 had an average size of 160.4 nm and the size distribution was 0.177 (Fig. 3d). TEM image (Fig. 3e) and AFM image (Fig. S4b and S4b′) displayed that sCDP/DOX/miR-122 had more regular spherical shape with compact structure than sCDP/DOX-3. To investigate the stability of the assembled polymer particles, the size and size distribution of sCDP/DOX/miR-122 in PBS was monitored for a week. As shown in Fig. S6, the size of sCDP/DOX/miR-122 and polydispersity index (PDI) increased slightly within 7 days, indicating its good stability.
The release profiles of DOX and miR-122 from sCDP/DOX/miR-122 were investigated in PBS with different pHs. A double-stranded oligoRNA labeled with Dy677 (RNADy677) was used as an alternate to prepare sCDP/DOX/RNADy677 for miR-122 release study. The release rates of DOX and RNADy677 were both influenced by pH values (Fig. 3f and g). It was perhaps because the protonated PDMAEMA chains in the acid media repelled with each other and destabilized the nanoparticles. Moreover, the relatively higher solubility of DOX at pH 5.0 contributed the release of DOX. In comparison, the release rates of RNADy677 were much quicker than that of DOX. For instance, almost 80% of RNADy677 was released after 12 h at pH 5.0, while less than 20% of DOX was released under the same condition. The incorporation of CD into polymeric nanoparticles could decrease the diffusion of DOX. As DOX was incorporated into the CD cavities in the inner core of the nanoparticle, it first needed to “escape” from CD cavities and then be released from the core into the media, leading to the controlled and sustained release pattern of DOX.
2.3. Cellular uptake and increased DOX accumulation by sCDP/DOX/miR-122
In order to investigate the intracellular co-delivery of DOX and miR-122 mediated by sCDP/DOX/miR-122 in HepG2 cells, sCDP/DOX/miR-122FAM was prepared and incubated with the cells for different times. Flow cytometry analysis data showed that the cellular uptake of sCDP/DOX/miR-122FAM showed a time-dependent manner (Fig. 4a and b). At 6 h, the DOX fluorescence intensity of sCDP/DOX/miR-122FAM group was comparable to that of the free DOX group. Naked miR-122FAM was hardly internalized by the cells, since no significant fluorescence increase was observed in the naked miR-122FAM group in comparison with untreated cells.
Fig. 4.

Intracellular uptake of DOX and miR-122FAM mediated by sCDP/DOX/miR-122FAM in HepG2 cells. Intracellular levels of DOX (a) and miR-122FAM (b) determined by flow cytometry. (c) Confocal images of HepG2 cells after various treatments; Relative fluorescence intensities (RFI) of DOX (d) and miR-122FAM (e) in HepG2 cells (n = 3). RFI of DOX and miR-122FAM in HepG2 cells treated with sCDP/DOX/miR-122FAM for 4 h were designated as 100%. ***P < 0.001 was used as significant difference between these two groups.
Confocal laser scanning microscope (CLSM) was employed to further evaluate the intracellular accumulation of DOX and miR-122 after sCDP/DOX/miR-122FAM internalization. As shown in Fig. S7, DOX and miR-122FAM fluorescence could be clearly observed in HepG2 cells after treated with sCDP/DOX/miR-122FAM. The fluorescence of miR-122FAM was not observed in the naked miR-122FAM group, which was consistent as the flow cytometry result. Moreover, the DOX signal was almost distributed in cell nuclei for free DOX group, while the fluorescence signals were mainly located in the cytosol for sCDP/DOX-3 and sCDP/DOX/miR-122FAM groups. The difference was because free DOX was taken up directly by the cells through passive diffusion which could quickly enter cell nuclei, while the nanoparticles were internalized by a non-specific endocytosis pathway and then released DOX in a sustained manner. Noticeably, as incubation time prolonged, strong DOX fluorescence distributed in the whole cells with gradual accumulation of DOX in cell nuclei (Fig. 4c, d and 4e). In contrast, the DOX signal was significantly weaker in the cells treated with sCDP/DOX-3 than sCDP/DOX/miR-122 (Fig. 4d). sCDP/DOX-3-treated cells showed decreased DOX fluorescence intensity at 24 h in comparison with that at 12 h, whereas the phenomenon did not happen in sCDP/DOX/miR-122FAM group. We deduced that the increased accumulation of DOX in cells treated with sCDP/DOX/miR-122FAM was ascribed to the preferential released miR-122 from the nanoplatform for inhibiting the expression of drug-resistance proteins, which thus decreased the efflux of cytotoxic DOX from the cells.
2.4. Synergistic mechanism of miR-122 and cytotoxics by sCDP/DOX/miR-122
Prior to evaluating the gene-regulatory roles of sCDP/DOX/miR-122, cell apoptosis assay was determined by flow cytometry after staining with Annexin V-APC/7AAD. Few apoptotic cells were observed in control cells or cells treated with sCDP alone, and free DOX showed a highest cell apoptosis rate of 26.9% (Fig. 5a). As DOX was taken up directly by the cells through passive diffusion, it could quickly enter cell nuclei and showed a rapid cytotoxic effect. sCDP/DOX-3 and sCDP/miR-122 showed induction effect on cell apoptosis with apoptotic rates of 12.7% and 8.53% respectively. In contrast, an increased level of cell apoptosis rate (20.7%) was observed for cells treated with sCDP/DOX/miR-122, demonstrating that combining miR-122 with DOX exhibited enhanced effects on cell apoptosis.
Fig. 5.

sCDP/DOX/miR-122 effectively induced cell apoptosis and reduced drug-resistance protein expression. (a) Cell apoptosis of HepG2 cells qualified by flow cytometry at 48 h after various treatments. Q1: dead cells; Q2: late apoptotic cells; Q3: live cells; Q4: early apoptotic cells. (b) Quantification of miR-122 level using qRT-PCR (n = 3). (c) Relative mRNA expression of MDR1, MRP and Bcl-w determined by qRT-PCR at 48 h after various treatments (n = 3). (d) Expression levels of P-gp, MRP, Bcl-w, p53 and cleaved caspase-3 measured by western blot analysis at 48 h after various treatments. *P < 0.05, **P < 0.01 and ***P < 0.001 were used as significant difference when comparing with the control groups or between these two groups.
Next, quantitative real time polymerase chain reaction (qRT-PCR) was performed to analyze the intracellular miR-122 level in HepG2 cells after various treatments. As displayed in Fig. 5b, naked miR-122 group did not show any change in miR-122 level compared with the control group (P > 0.05). However, the miR-122 level increased substantially in sCDP/miR-122 and sCDP/DOX/miR-122 group (2299-fold and 2242-fold higher than the control, respectively), which was comparable to that of the positive control PEI/miR-122 polyplexes (2282-fold). The result demonstrated that miR-122 could be efficiently delivered into HepG2 cells by sCDP/DOX/miR-122.
According to the previous study [24,25], restoration of miR-122 promoted apoptosis and decreased drug resistance by down-regulation of Bcl-w and multi-drug resistance gene (MDR1) and up-regulation of p53. Meanwhile, miR-122 expression was negatively associated with Bcl-w expression (r = −0.2476, P < 0.001) and MRP expression (r = −0.5358, P < 0.001) from TCGA database (Fig. S8). Thus, we validated the expression of these miR-122-related downstream target genes by qRT-PCR and Western blot. As displayed in Fig. 5c, free DOX significantly enhanced MDR1 and MRP mRNA expression, while sCDP/DOX-3 showed comparable expression to the control group. As sCDP had no impact on the expression of these genes (Fig. S9), such a difference between free DOX and sCDP/DOX-3 was not attributed to sCDP. We deduced that the rapidly diffused free DOX into the HepG2 cells was easy to induce the cytotoxic efflux pump, while when it was incorporated into a sustain released nanosystem, they were internalized by endocytosis pathway and then released the drug in a sustained manner, avoiding the induction of cytotoxic efflux pump [32]. However, it was obviously that HepG2 cells treated with sCDP/miR-122 and sCDP/DOX/miR-122 showed remarkable down-regulation of MDR1, MRP and Bcl-w at mRNA level in comparison with the control group (P < 0.05). Consistently, the down-regulation of P-gp, MRP and Bcl-w and up-regulation of p53 and cleaved caspase-3 were also observed for these two groups at protein level (Fig. 5d). Considering the above results, the effect mechanism of sCDP/DOX/miR-122 could be clarified as follows. After the internalization of sCDP/DOX/miR-122, miR-122 was preferentially released and exerted its gene-regulatory role in the cytoplasm. On one hand, miR-122 reduced the expression of drug-resistance proteins (P-gp and MRP), avoiding DOX efflux from the cells. On the other hand, miR-122 directly down-regulated apoptosis-related protein Bcl-w and up-regulated p53, leading to direct cell apoptosis induction effect towards HepG2 cells. Moreover, with an increased accumulation of DOX, it entered the cell nuclei and intercalates DNA, causing DNA damage, and up-regulation of p53 [33] accompanied by activation of caspase-3. Altogether, DOX and miR-122 released from sCDP/DOX/miR-122 together exerted cell apoptosis induction effect on HepG2 cells (Scheme 1b).
2.5. Enhanced combined hepatoma chemotherapy by sCDP/DOX/miR-122
To explore the potential of sCDP/DOX/miR-122 in hematoma therapy, we first evaluated the synergistic cytotoxicity in HepG2 cells using CCK-8 assay. No significant cytotoxicity was observed after incubation of sCDP even at the concentration up to 100.0 μg/mL−1 for 48 h (Fig. S10), indicating its good biocompatibility as the carrier material. In accordance with the cell apoptosis results, free DOX exhibited significant cytotoxicity in a dose dependent manner (Fig. S11a). Naked miR-122 showed no inhibitory effect towards HepG2 cells at any concentrations since it could hardly enter the cells (Fig. S11b). Next, the cytotoxicity of sCDP/DOX/miR-122 was evaluated and compared with that of sCDP/DOX-3 and sCDP/miR-122. As shown in Fig. 6, it is noticeable that the cytotoxicity of sCDP/DOX/miR-122 was significantly higher than those of sCDP/DOX-3 and sCDP/miR-122. IC50 values of DOX in sCDP/DOX/miR-122 group were much lower than those in sCDP/DOX-3 group (Table S3). To further evaluated the synergistic effect of DOX and miR-122 co-delivered by the same nanoplatform, the combination index (CI) was calculated by Chou-Talalay equation [34]. The CI values were less than 1.0 at both 48 h and 72 h, indicating that sCDP/DOX/miR-122 exerted synergistic cytotoxic effects against HepG2 cells.
Fig. 6.

Synergisitc cytotoxicity of sCDP/DOX/miR-122 in HepG2 cells. Cell viabilities of HepG2 cells with different treatments for 48 h (a) and 72 h (b) (n = 4).
To further explore the in vivo performance of sCDP/DOX/miR-122, the pharmacokinetic and biodistribution assays were employed in healthy and HepG2 tumor-bearing BALB/c mice, respectively. The pharmacokinetic study was performed by intravenous administration of naked RNADy677 and sCDP/DOX/RNADy677. As shown in Fig. S12, naked RNADy677 was rapidly eliminated from the blood with a very short circulation time less than 30 min. In contrast, sCDP/DOX/RNADy677 showed an extended circulation time, with more than 5% of the initial sample remained in the blood after 8 h. For biodistribution study, saline, naked RNACy5.5 and sCDP/DOX/RNACy5.5 were respectively injected through tail vein, followed by harvesting the main organs and tumors 24 h post-injection for fluorescence imaging. As shown in Fig. S13a, remarkable Cy5.5 fluorescence was observed in the liver and kidney for naked RNACy5.5 group. In contrast, sCDP/DOX/RNACy5.5 group showed effectively intratumoral accumulation, with about 5-fold higher relative fluorescence intensity than naked RNACy5.5 (Fig. S13b). Meanwhile, as DOX is fluorescent itself, the DOX fluorescence was also obviously detected in the tumor tissues for sCDP/DOX/RNACy5.5 group (Fig. S13c). The result demonstrated DOX and RNA could be successfully co-delivered into the tumor by sCDP/DOX/RNA nanosystem.
We next assessed the in vivo therapeutic effect of sCDP/DOX/miR-122 on xenograft BALB/c mice model bearing HepG2 tumors. As presented in Fig. 7a, when the tumor volume reached approximately 50 mm3, free DOX, sCDP/DOX-3 and sCDP/DOX/miR-122 were administrated intravenously every three days for a total of four injections at an equivalent DOX dosage of 2.0 mg kg−1, and saline was used as a control. The saline group showed a rapid increase in average tumor volume that reached to approximately 2000 mm3 at the end of the observation period on day 42 (Fig. 7b). All of the three formulations showed inhibitory effects towards HepG2 tumors (P < 0.001). Compared with free DOX, both sCDP/DOX-3 and sCDP/DOX/miR-122 exhibited significant higher tumor suppression effect (P < 0.001) with the tumor inhibition rates of 68.3% and 83.4%, respectively, which were 1.2– and 1.5–fold superior than that of free DOX (57.2%). Notably, sCDP/DOX/miR-122 showed the most obvious suppression of tumor growth. This result was also directly observed in the image of the removed tumors and the mice at the end point (Figs. 7c and S14a). Moreover, the average weight of the isolated tumors from the mice treated with sCDP/DOX/miR-122 were 4.8–, 3.1– and 2.0–time lighter than saline, free DOX and sCDP/DOX-3, respectively (Fig. S14b). The mice treated with sCDP/DOX-3 and sCDP/DOX/miR-122 showed less weight loss compared with those treated with free DOX (Fig. S14c), indicating that the nanoparticle system effectively reduced the side effects of free DOX.
Fig. 7.

In vivo therapeutic effects of sCDP/DOX/miR-122. (a) Scheme of tumor inoculation and systemic injection (i.v. via the tail vein) of saline, free DOX, sCDP/DOX and sCDP/DOX/miR-122 in HepG2 tumor-bearing nude mice. Dose: DOX of 2.0 mg kg−1 and miR-122 of 50.0 nmol kg−1. (b) Tumor growth curves of tumor-bearing mice after various treatments (n = 6). (c) Image tumors removed from the mice at the end point of the study. (d) Immunohistochemistry images of tumor sections stained by anti-Ki67 antibody and TUNEL assay. The brown color represents staining of positive cells. (e) HE stained images of various tissues from the mice. *P < 0.05 and **P < 0.01 were used as significant difference between these two groups.
As a marker of proliferation, the expression of Ki67 was detected in tumor sections by immunohistochemical staining to evaluate the tumor cell proliferation (Figs. 7d and S15a). The control group showed the highest expression of Ki67 (79.8% positive cells), implying that the tumor cell proliferation was active without treatment. In contrast, only a few Ki67 signal was observed in sCDP/DOX/miR-122 group (5.9% positive cells), which was significantly lower than that in free DOX group (38.2% positive cells) and sCDP/DOX-3 group (17.1% positive cells). Moreover, the cell apoptosis levels in tumor sections was assessed by terminal transferase-mediated dUTP nick end-labeling (TUNEL) assays (Figs. 7d and S15b). More intensive apoptosis or necrosis signals were observed in the tumor tissues of sCDP/DOX/miR-122 group (34.7% TUNEL positive signal) than those in other groups, indicating that sCDP/DOX/miR-122 treatment showed remarkable tumor apoptosis.
Furthermore, the main organs (heart, liver, spleen, lung, and kidney) and tumor issues were processed for hematoxylin-eosin (HE) staining. Histopathological changes in the heart were clearly observed in mice treated with free DOX, while no pathological changes and injuries were detectable in the main tissues of mice treated with sCDP/DOX-3 and sCDP/DOX/miR-122 (Fig. 7e). In addition, remarkable tumor necrosis, increased nuclear atypia and lower chromatin staining were visible in sCDP/DOX/miR-122 group when compared with the other groups. The above results demonstrated that sCDP/DOX/miR-122 could efficiently inhibit tumor growth and increase tumor cell apoptosis with reduced cardiotoxicity.
3. Conclusions
In the present study, a cyclodextrin-based cationic polymeric nanoparticle system was developed to combine miR-122 and DOX for hepatoma therapy. Uniform and nanosized sCDP/DOX/miR-122 effectively co-delivered DOX and miR-122 into heptoma cells. miR-122 can be preferentially released from the nanosytem, leading to increased chemo-sensitivity and synergistic cell growth inhibition effects. Remarkably improved anti-tumor activity of sCDP/DOX/miR-122 was also demonstrated both in vitro and in vivo in comparison with sCDP/DOX-3. Our study presents an effective strategy for improving hepatoma chemotherapy.
4. Experimental section
4.1. Bioinformatics analyses
Reads per million (RPM) of miR-122 and survival information including PFS and CSS were obtained from TCGA. TCGA contains miRNA-seq data of 48 paired HCC and adjacent normal tissues, 360 HCC samples with CSS and 367 samples with PFS information. Normalized signal values of miR-122 in 97 pairs of HCC and adjacent normal tissues were obtained from GSE147892 retrieved from the GEO. The Kaplan-Meier method was used to draw survival curves, and the Log-Rank test was used to evaluate survival difference.
The gene set enrichment analysis [35] was performed to evaluate the performances of resistance to seven common agents (doxorubicin, cisplatin, mitoxantrone, vincristine, fluorouracil, alkylating agent and docetaxel) in the miR-122 low and high groups based on corresponding datasets retrieved from the Molecular Signatures Database [36], and the parameters of ES and FDR Q value were compared among different agents.
4.2. Synthesis and characterization of sCDP
The star polymer was synthesized by sequential two-step ATRP using PEG-Br as the initiator (Fig. 2a). First, CuCl (0.05 mmol, 5.0 mg) and PMDETA (0.1 mmol, 20.6 μL) were dissolved in anhydrous DMF in a two-neck flask and degassed with three freeze-pump-thaw cycles. A solution of PEG-Br (0.05 mmol, 275.3 mg) and MCD (1.0 mmol, 1.32 g) was added into the solution through a syringe under nitrogen atmosphere, and the mixture was then degassed with another two freeze-pump-thaw cycles. The reaction mixture was stirred at 90 °C for 48 h. The polymer was further purified by dialysis against deionized water for 48 h and recovered by lyophilization to obtain the star polymer PEG-[PCD]. Then, the above polymer was employed as the initiator to initiate the polymerization of DMAEMA to synthesize the star polymer PEG-[PCD]-PDMAEMA (abbreviated as sCDP). The feed ratio of [DMAEMA]: [Br]: [CuCl]: [PMDETA] was 60: 1: 1: 2. The procedure was similar to the procedure described above and the reaction mixture was stirred at 80 °C for 48 h.
The chemical structures of PEG-[PCD] and sCDP were characterized by proton nuclear magnetic resonance (1H NMR) analysis on a nuclear magnetic resonance spectrometer at 400 MHz (Avance Ш, Bruker, Germany). The apparent molecular weights of the aforementioned polymers relative to a poly(methyl methacrylate) standard were determined by gel permeation chromatography (GPC) with CoMetre 6000 LDI pump and Schambeck SFD GmbH RI2000 refractive index detector. DMF containing 0.01 M LiBr was used as mobile phase at a flow rate of 1.0 mL min−1. Polymer solution was injected through PLgel 10 μm 103 Å and 104 Å columns at 70 °C.
4.3. Preparation and characterization of sCDP/DOX/miR-122
sCDP/DOX were prepared at different weight ratios of sCDP to DOX by dialysis method. Briefly, sCDP and doxorubicin hydrochloride (DOX⋅HCl) were co-dissolved in dimethyl sulfoxide (DMSO), and triethylamine was added to remove hydrochloride. After stirring at 37 °C overnight, the mixtures were transferred into dialysis bags (Molecular weight cut-off = 7000 Da; Union Carbide Corporation, Danbury, CT, USA), dialyzed against deionized water for 24 h and finally lyophilized to yield sCDP/DOX powder. The drug loading content and encapsulation efficiency of DOX was measured by a UV spectrometer (PekinElmer Lambda 35, USA) at 480 nm according to our previous study [28].
MiR-122 was further condensed into sCDP/DOX-3 to form sCDP/DOX/miR-122 with various N/P ratios (N/P ratio was calculated as the molar ratio of N in tertiary amine group of PDMAEMA to P in phosphate group of miR-122). Briefly, sCDP/DOX-3 powder was dissolved in PBS solution (1 mM, pH 6.0) at various concentrations and miR-122 was dissolved in DNase/RNase-free water. Then, 50 μL of sCDP/DOX-3 solution was added into an equal volume of miR-122 solution (5 μg). The mixed solution was vortexed and incubated at room temperature for 30 min to ensure complete condensation. Fluorescent-labeled nanoparticles, including sCDP/DOX/miR-122FAM, sCDP/DOX/RNADy677, and sCDP/DOX/RNACy5.5 were prepared at the N/P ratio of 10/1 by the same methods with the replacement of miR-122 by the corresponding fluorescent-labeled RNA and used for in vitro and in vivo study. Polyplexes PEI/miR-122 and sCDP/miR-122 were also prepared at the N/P ratio of 10/1 and used as the controls. Characterization of the above nanoparticles and polyplexes was shown in Table S4.
The sizes, size distributions and zeta potentials of the above nanoparticles were determined by a Zetasizer Nano ZS instrument (Malvern Instruments, Worcestershire, UK) at 25 °C. The morphologies of these nanoparticles were visualized under transmission electron microscope (TEM, Hitachi HT7700, Tokyo, Japan).
4.4. DOX and RNADy677 release study
sCDP/DOX/miR-122 and sCDP/DOX/RNADy677 were respectively prepared to investigate the in vitro release of DOX and RNADy677. The fresh prepared nanoparticle solutions were diluted into 1 mL of PBS (pH 7.4) with a final DOX concentration of 0.6 mg mL−1. The solutions were then transferred to Float-A-Lyzer G2 dialysis device (Molecular weight cut-off = 100.0 kDa, Spectrum™ Spectra, Fisher Scientific, USA) and then immersed into 20.0 mL phosphate-buffered saline (PBS) solutions with pH of 7.4, 6.5 or 5.0 at 37 °C. At a predetermined interval, 5.0 μL of the solution was withdrawn and mixed with 20–fold DMSO. The UV/vis absorbance of DOX were analyzed using a microplate reader at a wavelength of 480 nm. The fluorescence intensity of RNADy677 was determined by a BioTek Synergy HT multimode microplate reader (Winooski, VT, USA) at an excitation/emission wavelength of 660/694 nm. All release tests were carried out in triplicate.
4.5. Cellular uptake and intracellular locations of sCDP/DOX/miR-122
sCDP/DOX/miR-122FAM were prepared to investigate the cellular uptake and intracellular locations by flow cytometry analysis and CLSM. For flow cytometry analysis, HepG2 cells were seeded onto 6-well plates at a density of 1.5 × 105 cells per well and cultured for 24 h sCDP/DOX/miR-122FAM were then added at DOX and miR-122 concentration of 2.0 μg mL−1 and 50.0 nM, respectively. After further incubation for 0.5 h, 2 h, 6 h or 12 h, the cells were washed with PBS, detached using 0.1% trypsine-EDTA, re-suspended in PBS, and finally analyzed on FACS caliber flow cytometer (FACSCalibur, BD Biosciences, San Jose, CA, USA).
For the CLSM study, the cells were placed into 35 mm coverglass plates at a density of 1 × 105 cells per well, incubated for 24 h and individually treated with sCDP/DOX-3 and sCDP/DOX/miR-122FAM at DOX and miR-122 concentration of 2.0 μg mL−1 and 50.0 nM. Then the cells were washed with PBS and fixed with 4% paraformaldehyde followed by treatment with 4′,6-diamidino-2-phenylindole (DAPI) for nuclei staining. After that, the cells were observed under CLSM (ZeissLSM710, Carl Zeiss, Jena, Germany). The fluorescence signals of intracellular DOX and FAM were calculated by Image J software (National Institutes of Health, Bethesda, MD, USA).
4.6. Cell apoptosis assay
HepG2 cells were seeded onto 6-well plates at a density of 1 × 105 cells per well and cultured for 24 h. Then the cells were respectively treated by fresh media containing sCDP, free DOX, sCDP/DOX-3, sCDP/miR-122, sCDP/DOX/miR-122 at sCDP concentration of 10.0 μg mL−1, DOX concentration of 1.0 μg mL−1 and miR-122 concentration of 25.0 nM. Meanwhile, untreated cells were used as the control group. After 48 h, the cells were trypsinized, harvested and separately processed using the apoptosis detection kit of Annexin V-APC/7-amino-actinomycin D (7-AAD) (Tianjin SanJian, P.R. China) according to the manufacturer's protocol. Cell apoptosis rate were detected using a flow cytometer (FACSCalibur, BD Biosciences, San Jose, CA, USA).
4.7. Quantitative real-time PCR analysis for miR-122 and mRNA expression
qRT-PCR was applied to evaluate the intracellular level of miR-122 and MDR1, MRP, Bcl-w mRNA. HepG2 cells were seeded onto 6-well plates at a density of 1 × 105 cells per well and cultured for 24 h. Then the cells were separately treated by free DOX, sCDP/DOX-3, sCDP/miR-122, sCDP/DOX/miR-122 at DOX and miR-122 concentration of 2.0 μg mL−1 and 50 nM and incubated for 48 h.
Total RNA was extracted using Trizol following the manufacturer's protocol. For miR-122 detection, reverse transcription of total RNA and qRT-PCR analysis was performed using Mir-X™ miRNA First-Strand Synthesis and SYBR® qRT-PCR kits (Takara Bio Inc., Tokyo, Japan). The expression level of miR-122 was normalized to that of U6. For mRNA detection, first strand cDNA synthesis and amplification were performed according to the protocol of GoScript™ Reverse Transcription System (Promega, Madison, WI, USA) and qRT-PCR were performed according the protocol of GoTaq® qPCR Master Mix (Promega, Madison, WI, USA). GAPDH was used as an internal control to normalize the expression of mRNA. qRT-PCR was performed on an Applied Biosystems 7500 Real-Time PCR system (ThermoFisher Scientific, Waltham, MA, USA). Data were analyzed using the 2–ΔΔCT method. The sequences of qRT-PCR primers are listed in Table S5.
4.8. Western blot assay
The cells were harvested and lysed in RIPA lysis buffer supplied with phenylmethanesulfonyl fluoride and protease inhibitor cocktail. The total protein concentration of the lysates was determined using the BCA protein assay kit (ThermoFisher Scientific, Waltham, MA, USA). An equal amount of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto poly(vinylidene fluoride) membrane (Millipore Co., Billerica, MA, USA), and blocked using 5% non-fat dried milk at RT for 1 h. The membranes with specific protein bands were separately incubated with specific primary antibodies at 4 °C overnight and followed by incubation with the corresponding second antibodies at room temperature for 1 h. The primary antibodies used were listed in Table S6. The protein bands were detected using ECL system (Cell Signaling Technology, Danvers, MA, USA) and the protein expression level was normalized against GAPDH expression.
4.9. Cytotoxicity assay
The cytotoxicity of sCDP, free DOX, naked miR-122, sCDP/DOX, sCDP/miR-122 and sCDP/DOX/miR-122 nanoparticles in HepG2 cells were evaluated by Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan). The cells were seeded into 96-well plate at a density of 5.0 × 103 per well and incubated for 24 h. Then the culture media was replaced with 100 μL of fresh media containing sample at different concentrations. The cells were further incubated for another 48 h or 72 h. After that, the cells were treated with CCK-8 reagent for 2 h and the absorbance of each well was measured by a multifunctional Varioskan Flash ELISA plate reader (ThermoFisher Scientific, Waltham, MA, USA) at 450 nm. The cell viabilities were calculated as the ratio of the absorbance values of treated cells to those of untreated cells.
4.10. In vivo anti-tumor activity
HCC xenograft mouse model was established by injecting HepG2 cells subcutaneously to four-week-old nude mice. The day on which the inoculation performed was recorded as day 0. When tumor volume reached around 50 mm3 at day 14, the mice were randomly divided into four groups (n = 6) and injected through the tail vein with saline, free DOX, sCDP/DOX-3 and sCDP/DOX/miR-122 (DOX 2.0 mg kg−1 and miR-122 50 nmol kg−1, q3d × 4). The length (L) and width (W) of tumor were measured every 3 days using a caliper, and the tumor volume were calculated as the following equation (1):
| (1) |
At the end point of therapy, the mice were sacrificed, and the major organs and tumors were collected for the subsequent histopathological and immunohistochemical analysis. The tissue samples were fixed in 4% paraformaldehyde, embedded in paraffin and cut into 5-μm tissue sections. The proliferated cells in tumor tissue were stained by rabbit polyclonal antibody against Ki67 (ab15580, Abcam, Cambrige, MA, USA). The apoptotic tumor cells were determined using the TUNEL kit (Roche, Basel, Switzerland) according to the manufacturer's instructions. Positive staining was visualized by an optical microscope (IX71, Olympus Corporation, Tokyo, Japan), and the Ki67-positive ratio and the apoptotic index were formulated as a ratio of the positive cell number to the total tumor cell number in each field. The paraffin-embedded tumors and organs were stained with HE, and then visualized using a fluorescence microscope.
4.11. Statistical analysis
All values in the figures are presented as means ± SD of at least three independent experiments. Survival curves were plotted using Kaplan-Meier's method and compared between groups by the Log-Rank test. Statistical significance among column experimental groups were analyzed using one-way analysis of variance (ANOVA) using SPSS 12.0 (SPSS Inc., Chicago, IL, USA). Statistical significance among tumor growth curves was analyzed by Repeated Measures ANOVA. All significant values shown in various figures are indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
CRediT authorship contribution statement
Qingqing Xiong: Writing – original draft, proposed the experiment plan, carried on the experiments, and wrote the manuscript draft. Yang Bai: Data curation, Formal analysis, participated in the in vitro experiments and data analysis. Run Shi: Formal analysis, performed the bioinformatics analyses. Jian Wang: conducted the Western bolt experiment. Weiguo Xu: performed the biodistribution study. Mingming Zhang: Writing – review & editing, initiated the study, and revised this manuscript. Tianqiang Song: Supervision, Writing – review & editing, supervised the study, provided financial support and revise the manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors acknowledge support from the National Natural Science Foundation of China (Grant No. 81501575 and 81802873), the National Science and Technology Major Project of China (Grant No. 2018ZX10302205), Natural Science Foundation of Tianjin (Grant No. 18JCQNJC81300), and the Key Research Project of Tianjin Health Industry (Grant No. 14KG142).
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2021.03.026.
Contributor Information
Qingqing Xiong, Email: xiongqingqing@tmu.edu.cn.
Mingming Zhang, Email: zhangmm@bme.cams.cn.
Tianqiang Song, Email: tjchi@hotmail.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA. A Cancer Journal for Clinicians. 2018;68:394. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Forner A., Reig M., Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 3.Yarchoan M., Agarwal P., Villanueva A., Rao S., Dawson L.A., Karasic T., Llovet J.M., Finn R.S., Groopman J.D., El-Serag H.B., Monga S.P., Wang X.W., Karin M., Schwartz R.E., Tanabe K.K., Roberts L.R., Gunaratne P.H., Tsung A., Brown K.A., Lawrence T.S., Salem R., Singal A.G., Kim A.K., Rabiee A., Resar L., Meyer J., Hoshida Y., He A.R., Ghoshal K., Ryan P.B., Jaffee E.M., Guha C., Mishra L., Coleman C.N., Ahmed M.M. Recent developments and therapeutic strategies against hepatocellular carcinoma. Canc. Res. 2019;79:4326. doi: 10.1158/0008-5472.CAN-19-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.-F., de Oliveira A.C., Santoro A., Raoul J.-L., Forner A., Schwartz M., Porta C., Zeuzem S., Bolondi L., Greten T.F., Galle P.R., Seitz J.-F., Borbath I., Häussinger D., Giannaris T., Shan M., Moscovici M., Voliotis D., Bruix J. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.Bruix J., Qin S., Merle P., Granito A., Huang Y.-H., Bodoky G., Pracht M., Yokosuka O., Rosmorduc O., Breder V., Gerolami R., Masi G., Ross P.J., Song T., Bronowicki J.-P., Ollivier-Hourmand I., Kudo M., Cheng A.-L., Llovet J.M., Finn R.S., LeBerre M.-A., Baumhauer A., Meinhardt G., Han G. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56. doi: 10.1016/S0140-6736(16)32453-9. [DOI] [PubMed] [Google Scholar]
- 6.Kudo M., Finn R.S., Qin S., Han K.-H., Ikeda K., Piscaglia F., Baron A., Park J.-W., Han G., Jassem J., Blanc J.F., Vogel A., Komov D., Evans T.R.J., Lopez C., Dutcus C., Guo M., Saito K., Kraljevic S., Tamai T., Ren M., Cheng A.-L. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 7.Zongyi Y., Xiaowu L. Immunotherapy for hepatocellular carcinoma. Canc. Lett. 2020;470:8. doi: 10.1016/j.canlet.2019.12.002. [DOI] [PubMed] [Google Scholar]
- 8.El-Khoueiry A.B., Sangro B., Yau T., Crocenzi T.S., Kudo M., Hsu C., Kim T.-Y., Choo S.-P., Trojan J., Welling T.H., Meyer T., Kang Y.-K., Yeo W., Chopra A., Anderson J., dela Cruz C., Lang L., Neely J., Tang H., Dastani H.B., Melero I. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kudo M. Systemic therapy for hepatocellular carcinoma: 2017 update. Oncology. 2017;93(suppl 1):135. doi: 10.1159/000481244. [DOI] [PubMed] [Google Scholar]
- 10.Asghar U., Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J. Hepatol. 2012;56:686. doi: 10.1016/j.jhep.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 11.Olweny C.L.M., Toya T., Katongole-Mbidde E., Mugerwa J., Kyalwazi S.K., Cohen H. Treatment of hepatocellular carcinoma with adriamycin. Preliminary communication, Cancer. 1975;36:1250. doi: 10.1002/1097-0142(197510)36:4<1250::aid-cncr2820360410>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 12.Lai C.-L., Lok A.S.-F., Wu P.-C., Chan G.C.-B., Lin H.-J. Doxorubicin versus no antitumor therapy in inoperable hepatocellular carcinoma. A prospective randomized trial, Cancer. 1988;62:479. doi: 10.1002/1097-0142(19880801)62:3<479::aid-cncr2820620306>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 13.Lencioni R., Llovet J.M., Han G., Tak W.Y., Yang J., Guglielmi A., Paik S.W., Reig M., Kim D.Y., Chau G.-Y., Luca A., del Arbol L.R., Leberre M.-A., Niu W., Nicholson K., Meinhardt G., Bruix J. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: the SPACE trial. J. Hepatol. 2016;64:1090. doi: 10.1016/j.jhep.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Cox J., Weinman S. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepatic Oncology. 2015;3:57. doi: 10.2217/hep.15.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonin S., Pascolo L., Crocé L.S., Stanta G., Tiribelli C. Gene expression of ABC proteins in hepatocellular carcinoma, perineoplastic tissue, and liver diseases. Mol. Med. 2002;8:318. [PMC free article] [PubMed] [Google Scholar]
- 16.Shivdasani R.A. MicroRNAs: regulators of gene expression and cell differentiation. Blood. 2006;108:3646. doi: 10.1182/blood-2006-01-030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bracken C.P., Scott H.S., Goodall G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016;17:719. doi: 10.1038/nrg.2016.134. [DOI] [PubMed] [Google Scholar]
- 18.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017;16:203. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 19.Bandiera S., Pfeffer S., Baumert T.F., Zeisel M.B. miR-122 – a key factor and therapeutic target in liver disease. J. Hepatol. 2015;62:448. doi: 10.1016/j.jhep.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Tsai W.-C., Hsu P.W.-C., Lai T.-C., Chau G.-Y., Lin C.-W., Chen C.-M., Lin C.-D., Liao Y.-L., Wang J.-L., Chau Y.-P., Hsu M.-T., Hsiao M., Huang H.-D., Tsou A.-P. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49:1571. doi: 10.1002/hep.22806. [DOI] [PubMed] [Google Scholar]
- 21.Valdmanis P.N., Kim H.K., Chu K., Zhang F., Xu J., Munding E.M., Shen J., Kay M.A. miR-122 removal in the liver activates imprinted microRNAs and enables more effective microRNA-mediated gene repression. Nat. Commun. 2018;9:5321. doi: 10.1038/s41467-018-07786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J., Zhu X., Wu L., Yang R., Yang Z., Wang Q., Wu F. MicroRNA-122 suppresses cell proliferation and induces cell apoptosis in hepatocellular carcinoma by directly targeting Wnt/β-catenin pathway. Liver Int. 2012;32:752. doi: 10.1111/j.1478-3231.2011.02750.x. [DOI] [PubMed] [Google Scholar]
- 23.Nakao K., Miyaaki H., Ichikawa T. Antitumor function of microRNA-122 against hepatocellular carcinoma. J. Gastroenterol. 2014;49:589. doi: 10.1007/s00535-014-0932-4. [DOI] [PubMed] [Google Scholar]
- 24.Fornari F., Gramantieri L., Giovannini C., Veronese A., Ferracin M., Sabbioni S., Calin G.A., Grazi G.L., Croce C.M., Tavolari S., Chieco P., Negrini M., Bolondi L. MiR-122/Cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Canc. Res. 2009;69:5761. doi: 10.1158/0008-5472.CAN-08-4797. [DOI] [PubMed] [Google Scholar]
- 25.Xu Y., Xia F., Ma L., Shan J., Shen J., Yang Z., Liu J., Cui Y., Bian X., Bie P., Qian C. MicroRNA-122 sensitizes HCC cancer cells to adriamycin and vincristine through modulating expression of MDR and inducing cell cycle arrest. Canc. Lett. 2011;310:160. doi: 10.1016/j.canlet.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 26.Cao F., Yin L.-X. miR-122 enhances sensitivity of hepatocellular carcinoma to oxaliplatin via inhibiting MDR1 by targeting Wnt/β-catenin pathway. Exp. Mol. Pathol. 2019;106:34. doi: 10.1016/j.yexmp.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Zhang M., Xiong Q., Chen J., Wang Y., Zhang Q. A novel cyclodextrin-containing pH-responsive star polymer for nanostructure fabrication and drug delivery. Polym. Chem. 2013;4:5086. [Google Scholar]
- 28.Xiong Q., Zhang M., Zhang Z., Shen W., Liu L., Zhang Q. Anti-tumor drug delivery system based on cyclodextrin-containing pH-responsive star polymer: in vitro and in vivo evaluation. Int. J. Pharm. 2014;474:232. doi: 10.1016/j.ijpharm.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J., Ma P.X. Polymeric core–shell assemblies mediated by host–guest interactions: versatile nanocarriers for drug delivery. Angew. Chem. Int. Ed. 2009;48:964. doi: 10.1002/anie.200804135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong Q., Cui M., Bai Y., Liu Y., Liu D., Song T. A supramolecular nanoparticle system based on β-cyclodextrin-conjugated poly-l-lysine and hyaluronic acid for co-delivery of gene and chemotherapy agent targeting hepatocellular carcinoma. Colloids Surf. B Biointerfaces. 2017;155:93. doi: 10.1016/j.colsurfb.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Lang J., Zhao X., Qi Y., Zhang Y., Han X., Ding Y., Guan J., Ji T., Zhao Y., Nie G. Reshaping prostate tumor microenvironment to suppress metastasis via cancer-associated fibroblast inactivation with peptide-assembly-based nanosystem. ACS Nano. 2019;13:12357. doi: 10.1021/acsnano.9b04857. [DOI] [PubMed] [Google Scholar]
- 32.Kunjachan S., Rychlik B., Storm G., Kiessling F., Lammers T. Multidrug resistance: physiological principles and nanomedical solutions. Adv. Drug Deliv. Rev. 2013;65:1852. doi: 10.1016/j.addr.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aas T., Børresen A.-L., Geisler S., Smith-Sørensen B., Johnsen H., Varhaug J.E., Akslen L.A., Lønning P.E. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996;2:811. doi: 10.1038/nm0796-811. [DOI] [PubMed] [Google Scholar]
- 34.Chou T.-C. Drug combination studies and their Synergy quantification using the chou-talalay method. Canc. Res. 2010;70:440. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 35.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. Unit. States Am. 2005;102:15545. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov Jill P., Tamayo P. The molecular Signatures database hallmark gene set collection. Cell Systems. 2015;1:417. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.