

Abstract
Mutations in the PSEN1 gene, encoding presenilin 1 (PS1), are the most common cause of familial Alzheimer’s disease(fAD). Since the first mutations in the PSEN1 gene were discovered more than 25 years ago, many postulated functions of PS1 have been investigated. The majority of earlier studies focused on its role as the catalytic component of the γ-secretase complex, which in concert with β site amyloid precursor protein cleaving enzyme 1 (BACE1), mediates the formation of Aβ from amyloid-β protein precursor (AβPP). Though mutant PS1 was originally considered to cause AD by promoting Aβ pathology through its protease function, it is now becoming clear that PS1 is a multifunctional protein involved in regulating membrane dynamics and protein trafficking. Therefore, through loss of these abilities, mutant PS1 has the potential to impair numerous cellular functions such as calcium flux, organization of proteins in different compartments, and protein turnover via vacuolar metabolism. Impaired calcium signaling, vacuolar dysfunction, mitochondrial dysfunction, and increased ER stress, among other related membrane-dependent disturbances, have been considered critical to the development and progression of AD. Given that PS1 plays a key regulatory role in all these processes, this review will describe the role of PS1 in different cellular compartments and provide an integrated view of how PS1 dysregulation (due to mutations or other causes) could result in impairment of various cellular processes and result in a “multi-hit”, integrated pathological outcome that could contribute to the etiology of AD.
Keywords: Calcium, endoplasmic reticulum, endosomal pathways, presenilin-1, vacuolar processes
INTRODUCTION
Alzheimer’s disease (AD), characterized by progressive cognitive decline, is the most common age-dependent neurodegenerative disorder. Although the disease has been studied extensively, it is not fully understood, and there are still no effective interventions that slow the disease process. Although many alterations in various cellular processes have been defined in AD, the presence of amyloid plaques, with a core of Aβ peptides, and neurofibrillary tangles (NFTs), composed primarily of abnormally phosphorylated tau, are still required for a diagnosis of AD. The vast majority of AD cases are sporadic (sAD), without a defined genetic cause, and primarily affect people who are greater than 60 years of age. However, a much smaller subset of AD cases (~5–10%) are due to autosomal dominant genetic mutations (familial AD [fAD]) and are often characterized by a more severe and rapidly progressive dementia. To date, mutations in three genes have been shown cause fAD: APP that encodes for amyloid-β protein precursor (AβPP), PSEN1 that encodes for Presenilin 1 (PS1), and PSEN2 which encodes for Presenilin 2 (PS2). Mutations in PSEN1 account for the majority of fAD cases (~70%) [1, 2].
Currently, over 180 mutations have been identified in the PSEN1 gene that span virtually all regions of this multi-pass transmembrane protein [3]. Given the variability of mutation loci in PS1, in addition to the complex and diverse medical backgrounds between patients, individuals with PS1 mutations can present with high degrees of clinical variability. Some patients with PS1 mutation present in their late 60s with a slow progression of cognitive decline, while, on the other end of the spectrum, patients with different PS1 mutations can present as early as their late 20s with rapid and severe global dysfunction involving autonomic and motor systems [4–7]. PS1 mutations also exhibit histological variability. For example, depending on the PS1 mutation, amyloid angiopathy can span the spectrum from mild to severe [8–10]. The presence of other pathological entities such as Pick’s bodies, Lewy bodies, or posterior cortical atrophy have also been differentially observed with various PS1 mutations [11–13]. Importantly, however, virtually all cases of PS1 mutation present with amyloid plaques and NFTs and are thus conferred the diagnosis of AD [14]. The neuropathological congruence of PS1-fAD and sAD, indicates that defining the various roles of PS1 in neuronal function will likely increase our understanding of the neurodegenerative processes of AD in general. Indeed, it has been shown that PS1 is necessary for synaptic function in learning and memory, and it is considered to provide a neuroprotective effect that may be attenuated as PS1 levels decrease as part of the normal aging process [15–21]. Therefore, as we consider that PS1 dysfunction is the main culprit of fAD, inadequate levels of PS1 function may also be a factor in the pathogenesis of AD in general.
Several different functions of PS1 have been described, all of which, when disrupted by mutations or diminished levels, could contribute to the pathogenesis of AD. The first described and most studied function of PS1 defines it as a multi-pass transmembrane, endo-proteolytically cleaved carboxypeptidase in the intra-membrane gamma secretase complex (PS1-γ-secretase complex). The carboxypeptidase function of PS1, also referred to as the γ-secretase function of PS1, has been elucidated by multiple studies. Wolfe et al. [22] presented clear evidence that PS1 has two transmembrane aspartates at positions D257 and D385, that are critical to this function, with D257 being essential for endoproteolysis and D385 being independently essential for γ-secretase activity/AβPP processing regardless of endoproteolysis. Taken together, PS1 mutations D257A and D385A, which are located in transmembrane domains 6 and 7, respectively flanking the endoproteolysis site, have the same dominant negative effect upon AβPP processing as PS1 deletion [22]. Of note, PS1 in this membrane bound complex associates with three other proteins, Nicastrin, APH-1 (anterior pharynx-defective 1), and PEN-2 (presenilin enhancer 2), which contribute to the folding, cleavage, and substrate processing of PS1 [23, 24]. PS1, in this complex, has been detected in multiple membranes, and cleaves type 1 transmembrane proteins. To isolate PS1 as the source of the protease active site for the γ-secretase complex, Li et al. [25] showed that potent γ-secretase inhibitors, which were chemically engineered to target an aspartyl protease active site, bound PS1. Overall, these and other studies demonstrate that PS1 is the carboxypeptidase of the γ-secretase complex, with AβPP and Notch being the most extensively documented substrates, though over 90 other substrates have been postulated [26]. However, it is important to note that not all interacting proteins may be a substrate specifically/only for cleavage, and complex-associated PS1 may have functions beyond carboxypeptidase activity. In addition to its role as a protease, PS1 can function as a calcium leak channel or a regulator of calcium channels in multiple membranes [27–33], and thus can mediate numerous cellular processes and signaling pathways that are regulated by proper calcium flux. Finally, PS1 likely plays a key role as a regulator of vacuolar metabolism, and, by proxy, general proteostasis. Here we present a brief review of the diversified functions of PS1 categorized by subcellular locations, followed by a discussion of how mutant PS1 may disrupt vacuolar processes contributing to the pathogenesis of fAD. It is noted that PS1 and PS2 are homologous in structure, and are likely similar in many functions—for example both PS1 and PS2 can function as a carboxypeptidase within the γ-secretase complex and cleave AβPP with varying kinetics; however, they also differ in substrate specificity and location. Importantly, PS2 is far less implicated in disease, so the mechanisms discussed for PS1 may not be automatically extended to PS2 [34–36].
PS1 AT THE ENDOPLASMIC RETICULUM (ER)
Although PS1 is translated at the ER along with the other components of the γ-secretase complex, the process of complex’s assembly has not yet been completely defined. Nonetheless, data indicate that the γ-secretase complex can begin assembly en-route to and/or at the cis Golgi before being packaged for delivery to the plasma membrane, internal membranes, or to compartments within the endosomal system [37]. PS1 appears to be most stable in this complex and can only undergo the endoproteolysis upon association with the other subunits of the complex, most importantly PEN2 [38]. This endoproteolysis is considered pre-requisite for PS1 to be active as a protease; to our knowledge no one has described any proteolytic roles for PS1 holoprotein monomers or putative holoprotein complexes. Monomeric γ-secretase complex subunits residual in the ER have been shown to undergo rapid degradation, so it is thought that assembly of the complex likely does not occur at this location, and that PS1 existing in any manner other than as the holoprotein in the ER does so as part of the γ-secretase complex. In conclusion, PS1 can be found at the ER, either as part of a functional γ-secretase complex or as a holoprotein, which may indicate that PS1 holoprotein is stabilized and thus regulated by other means at this location [39, 40].
Holoprotein
While enriched in the ER as part of the γ-secretase-complex, PS1 is also present as a holoprotein albeit at relatively low levels [41–43]. To our knowledge, PS1 has not been reported as a holoprotein in any other compartment except for certain regions of the nuclear envelope for which no function has yet been defined [44]. Early in the analysis of PS1 trafficking, PS1 holoprotein was found to be enriched in a digitonin-protein extraction of approximately 180 kDa and was thus presumed to likely form a specific complex in the ER [39], though these findings have neither been confirmed nor extended (e.g., putative stabilizing partners have not been identified). However, ongoing studies reveal PS1-dependent activities in the ER, some of which may be conferred by the holoprotein in certain cell models [29]. There is evidence that the general abundance and potential activities of PS1 holoprotein are physiologically involved in ER homeostasis, as overexpression of wild-type PS1 holoprotein levels was shown to alter ER calcium signaling. Although this finding is not mechanistically specific, it may prove pathogenically noteworthy given the increased abundance of ER holoprotein levels in postmortem PS1-mutant brain [45].
The mechanisms explaining altered ER calcium signaling with abnormal PS1 holoprotein levels may be multifaceted, but thus far two hypotheses have been proposed in the literature. First, PS1 holoprotein was shown to complex with ER calcium channel SERCA (sarco/endoplasmic reticulum calcium-ATPase) to regulate calcium flux in a γ-secretase independent manner [28, 46]. The other hypothesis proposes that PS1 may act directly as an ER leak channel for calcium [47–49], though this phenomenon has been debated [50, 51]. Aside from ER calcium regulation, the holoprotein may have alternative functions with regard to protein trafficking. Nixon’s research group has presented data showing that the PS1 holoprotein mediates, by direct binding, the oligosaccharyltransferase N-glycosylation of vacuolar ATPase, specifically the V0a1 subunit, as it’s translated into the ER. This glycosylation event allows for proper trafficking of the proton pump to lysosomes and, thus mutation or depletion of PS1 resulted in dysfunctional lysosomes [27, 43]. Though this work mostly focused on the altered physiology of lysosomes, the demonstrated potential of PS1 to mediate glycosylation may apply to other proteins. Therefore, mutation of PS1 holoprotein may induce states of ER stress and interrupt other pathways and systems that rely on proper protein sorting and trafficking. The fact that other groups did not observe improper glycosylation and trafficking of vacuolar ATPase in PS1 deficient neural stem cells or Drosophila melanogaster [33, 52], may indicate that diverse cell types rely on PS1 holoprotein differentially and calls for the continued study in neuronal models. Nonetheless, PS1 may modulate proteostasis within the ER in manners independent of carboxypeptidase activity and γ-secretase complex organization. By these mechanisms and probably more, mutant PS1 holoprotein likely induces ER stress through various routes which have a strong potential contribution to fAD etiology, as ER stress with calcium dysregulation is causal of synaptic dysfunction and cell death [53–55].
γ-secretase complex associated PS1
Multiple groups have detected PS1 N-terminal fragments (approximately 30kDa) and C-terminal fragments (approximately 20kDa) associated with other γ-secretase complex subunits (PEN2, Nicastrin, and APH) in ER preparations. Annaert et al. [44] demonstrated that the complex-associated form of PS1 is present in hippocampal neurons within the ER and ER sub-regions such as the rough ER. Furthermore, this group measured concentrations of post-endoproteolysis PS1 (NTF/CTF) in puncta of the ER containing sec23p and p58/ERGIC-53. Given that sec23p is a coat protein on COPII-coated vesicles, and p58/ERGIC-53 is a cargo receptor aiding the inclusion of cargo into COPII vesicles [56, 57], Annaert et al. suggested that proximity of endoproteolyzed PS1 to these regions could indicate a possible role for PS1-γ-secretase complex in ER export [44], though this supposition has yet to be confirmed. The evidence for ER localized PS1-γ-secretase complex, indicates a physiological role for endoproteolysis and other potential γ-secretase complex-mediated activities at the ER, presumably, in part, cleavage of local type 1 membrane proteins. ER-localized PS1 was present in mouse brain particularly enriched within domains of the ER called endoplasmic reticulum-mitochondria-associated membranes (ER-MAMs), which physically tether mitochondria to the ER and provide a biochemical connection, such as sharing calcium and phospholipids between the ER and mitochondria in a dynamic balance [42]. In this model, subcellular fractionation revealed that PS1 is present in the ER and ER-MAM fractions with the other components of the γ-secretase complex, which indicate that a functional γ-secretase complex is likely assembled either at this location or is trafficked to this location from the Golgi network. Furthermore, Aβ production at the ER was reduced by γ-secretase inhibition, indicating that PS1 has demonstrated catalytic activity at the ER and ER-MAM. Though the role of cleaved PS1 at the ER-MAM is not fully understood, knockdown of wild type PS1 in mouse embryonic fibroblasts altered the functional levels of ER-MAMs as measured by specific activities including upregulated synthesis of cholesterol esters, lipids, and contact sites between the ER-MAM and mitochondria. These effects, also observed in HeLa cells in which PS1 expression had been knocked down, could be rescued by expression of wild-type PS1 but not by expression of PS1 mutant D385A, the mentioned mutation that prevents AβPP processing [22]. This may indicate that the significance of the PS1-γ-secretase complex at the ER-MAM may rely in part upon AβPP processing or cleavage of other substrates. However, not all, of the alterations in ER-MAM activity noted in cells with PS1 dysregulation were directly mimicked by drug-induced γ-secretase inhibition. These results indicate that the PS1-γ-secretase complex modulates ER-MAM metabolism and activity (with a subsequent impact on mitochondria), particularly with regard to membrane composition and homeostasis, in a manner that may be independent of traditional carboxypeptidase activity [58] (see Fig. 1).
Fig. 1.
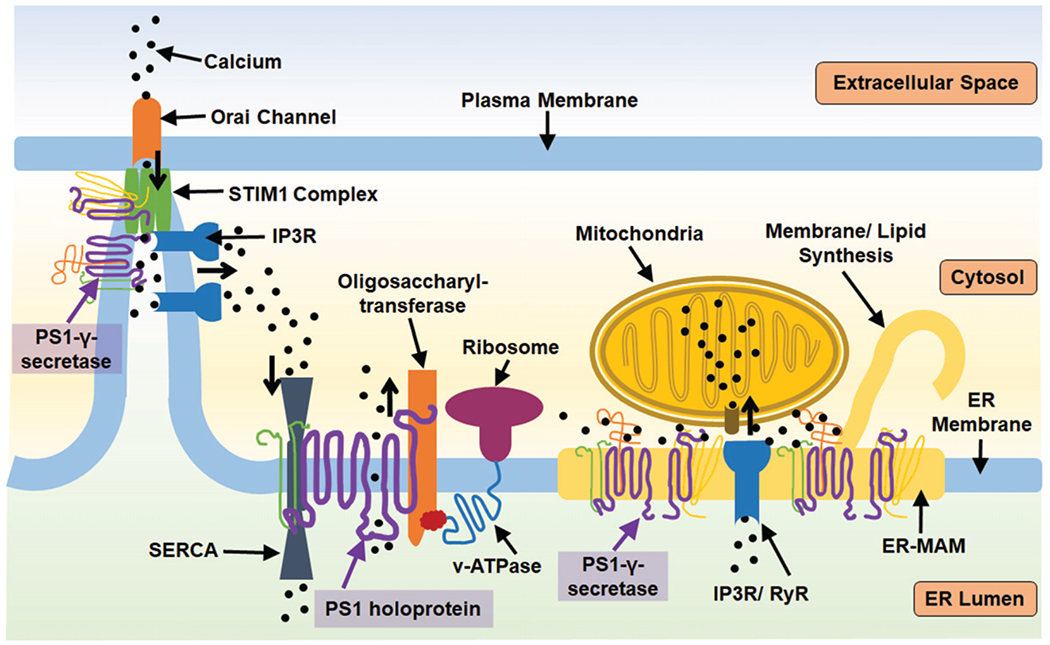
PS1 has multiple roles regulating ER activity both as a holoprotein and as a member of the γ-secretase complex. STIM1 is a calcium sensor that initiates calcium uptake via binding the Orai calcium channel at the plasma membrane-endoplasmic junction when calcium levels are transiently low. PS1 can regulate STIM1 activity by proteolysis, which is enhanced by PS1 mutation. STIM1 function is dependent upon the proper function of IP3R ([IP3 (inositol 1, 4, 5-trisphosphate) receptor] a calcium channel that releases calcium from the ER calcium when activated by IP3 ligand). IP3R is aberrantly active in the context of PS1 mutation. This IP3-STIM1 association is dependent upon ER-mitochondria association and calcium sharing, which is additionally regulated by the presence of PS1 at ER-MAMs. PS1 also regulates the expression and activity of RyR (ryanodine receptor) calcium channels which release calcium from the ER. Additionally, PS1 holoprotein can further impact calcium flux by biding SERCA, sarco/endoplasmic reticulum calcium-ATPase, which pump calcium into the ER, or PS1 may act as a leak channel itself. PS1 holoprotein has demonstrated capacity to aid oligosaccharyltransferase in glycosylation of v-ATPase, demonstrating a means through which PS1 may regulate general ER proteostasis. The role of PS1-γ-secretase at the ER-MAM (endoplasmic reticulum mitochondria associated membrane) can further impact the calcium balance of mitochondria, influencing their oxidative balance, and impact ER-MAM dependent activities such as lipid synthesis.
General PS1 expression as a modulator of ER function
The significance of PS1 at the ER has been extended by studies demonstrating physiological roles for PS1 in maintaining calcium homeostasis which are altered by PS1 deficiency or mutation. Tong et al. in 2016 [31] demonstrated that mutant PS1 M146L can increase the proteolysis of the ER calcium sensor, STIM1 (stromal interaction molecule 1). After localized ER calcium loss, STIM1 senses the depletion and oligomerizes, which initiates a fusion event between the ER and the plasma membrane as it associates with the Orai calcium channel at the plasma membrane and allows for restorative uptake of calcium. Following restoration of calcium stores, STIM1 complexes rapidly dissociate and retreat from ER-plasma membrane junctions, deactivating the Orai channel [59]. PS1’s action upon STIM1 at any point, either by the reduction of total amount of STIM1 or interruption of STIM1 at the point of activation, could inhibit store operated calcium entry. Indeed, Tong et al. did demonstrate that mutant PS1 (M146L) prevented proper restoration of ER calcium stores after ER calcium depletion [31]. They also noted that downstream of enhanced STIM1 cleavage, their neuronal model exhibited deformity of dendritic spines, characteristic of a more immature state [31], which would support findings that STIM1 dysfunction/loss of function is sufficient for improper neuronal transmission. Therefore, by this mechanism, mutant PS1 may initiate pathogenic cascades critical to the processes of neurodegeneration and AD development [60]. Furthermore, expression of mutant PS1 has been shown to increase the activation or expression of calcium channels such as ryanodine receptors (RyR) and IP3 receptors (IP3R), potentiating their calcium release [32, 61–64]. Interestingly, not only are RyR present at ER-MAMs discussed above [65], it is also postulated that in the active state, and only in the active state, IP3Rs associate with STIM1 promoting their activation [66]. Taken together, PS1 mutations seem competent to cause a nearly synergistic calcium disturbance in which they can activate the channels for calcium release and inhibit the ability of STIM1 to modulate calcium uptake.
Mitochondrial health and STIM1 function are also interrelated. STIM1 deficiency has been shown to cause an increase in the number of mitochondria with tubular morphologies. Loss of STIM1 was also shown to increase mitochondrial activity such that cells are less resilient against oxidative stress [67], and so it is plausible that these disturbances can be observed with STIM1 dysregulation secondary to the presence of mutant PS1. Though a recent study done by Korkotian et al. [68] did not focus on STIM1, they expanded upon the STIM1-mitochondria connection. They demonstrated ER calcium dysregulation and reduction of mature mushroom spines at the dendritic processes secondary to expression of PS1 mutant, M146V. Furthermore, they showed that mitochondrial uptake of calcium was directly impacted by the presence of M146V. This mitochondrial calcium uptake, in turn, mediates the signal for STIM1, and does so through the IP3/IP3R pathway [69]. As part of the improper flow of calcium into and out of the ER, cytosolic calcium levels have also been shown to increase with mutant PS1, making neurons more excitable and less stable against oxidative stress [68], which demonstrates the risk for aggressive neurodegeneration in PS1-fAD.
The impact PS1 may have on the ER and ER-associating mitochondria has been further elucidated in Caenorhabditis elegans, using mutants of the PS1 ortholog gene, sel-12. In these models, mitochondria calcium levels are elevated in the neurons of the sel-12 mutants, while ER calcium release is increased. Theses abnormalities could be rescued by reducing ER calcium release or mitochondrial calcium uptake. These data indicate that mutations in PS1 can increase ER-calcium release and reduce the ability to restore calcium levels via STIM1. Concomitantly, the increased mitochondrial calcium uptake in these sel-12 mutants correlates with changes in mitochondrial morphology and increases in mitochondrial respiration, which results in overproduction of reactive oxygen species and toxic oxidative stress [70]. Lastly, mutant PS1 may be competent to induce ER stress apart from calcium dysregulation. Cells expressing fAD-linked PS1 mutations demonstrated inhibited activation and translocation of ER stress transducers and interrupted signaling mechanisms that abate translation during the unfolded protein response (UPR) [71–73]. ER stress, mitochondrial dysfunction, and calcium dysregulation have all been proposed as central to AD. Such ER stress may specifically contribute to decreased tau degradation and increased tau phosphorylation characteristic of AD [74, 75]. Overall, there is a growing awareness that PS1 integrates protein trafficking and membrane dynamics of the ER in multiple ways and that the presence of mutant PS1 has the potential to significantly impair ER function [53, 76–79].
PS1 AT THE CELL MEMBRANE AND MEMBRANE PROXIMAL VACUOLES
PS1 localizes at the plasma membrane as part of the long-lived and stable intramembrane γ-secretase complex. The abundance of PS1 at this location seems to be dynamically linked with the endocytic activity and trafficking to and from the plasma membrane. PS1 is likely involved in proteolytic and signal-transduction activities at the plasma membrane or in traffic to the plasma membrane, most notably with regards to the production of Aβ and Notch signaling. Cleavage of AβPP to the C99 C-terminal fragment (CTF) and finally to Aβ can occur at the plasma membrane, or, perhaps more commonly, en-route to the plasma membrane in endocytic vesicles containing an abundance of BACE and AβPP [37]. PS1 is enriched in these endocytic compartments and cleaves the intramembrane C99 C-terminal fragment (CTF) of AβPP generated by BACE cleavage; these vesicles can then fuse with the plasma membrane and release their contents. For many years, it was considered that mutant PS1 had a gain in proteolytic activity and preferentially cleaved the C99 CTF of AβPP to final Aβ peptides that are longer in length with an increased frequency. It was thought that a supposed overproduction of longer, aggregate-prone Aβ species was the cause of PS1-fAD. However, studies investigating the kinetics of PS1 cleavage and Aβ production revealed that normal PS1 acts as a carboxypeptidase, which cleaves AβPP CTF sequentially to shorter and shorter products, and that most mutant PS1 disrupts this process and inappropriately releases longer Aβ peptides [80, 81]. It has been corroborated that PS1 mutations result mostly in a loss of AβPP CTF cleavage, as the increase in the Aβ42/Aβ40 ratio is often due to drastic reductions in the ability to produce Aβ40 [81–83]. Furthermore, the vast majority of the PS1 mutations result in a decreased production of Aβ peptides overall [84]. Interestingly, it has been demonstrated that the presence of a mutant PS1 gene can reduce the Aβ peptide production potential of the co-expressed wild-type PS1 [85] and that this effect is potentially mediated through direct interaction γ-secretase complexes. This mechanism gives valuable insight into how PS1 mutation can present in an autosomal dominant manner.
Loss of APP CTF cleavage due to PS1 mutation does carry through to animal models. In an AβPP transgenic mouse model, V717I, PS1 deficiency was well tolerated and actually rescued the Aβ pathology but not the cognitive defects [86], indicating that disease onset is not directly related to Aβ production. Correspondingly, there are cases of PS1 mutation in human patients where Aβ production levels are high and/or Aβ42/Aβ40 ratios are significantly increased, but these phenomena do not correlate with an early age of onset [84]. Therefore, it is improbable that the altered trends in Aβ production alone are causal in PS1-fAD. Therefore, a new framework for amyloidopathy in cases of PS1 mutation can include multiple other hypotheses. For one, PS1 mutation can alter the homeostatic variety of Aβ isoforms that are necessary for proper neuronal health. In addition, PS1 mutation may cause a build-up of AβPP and C99 CTF in the membrane compartments of neurons that could interrupt protein trafficking and overload degradative systems. Furthermore, we must consider the perspective, provided by these data, that the strongest drivers of disease could likely be independent of PS1-γ-secretase generation of Aβ, or that aberrant Aβ generation and aggregation is a consequence of improper protein regulation as a whole.
Other roles for PS1 at or in relation to the plasma membrane may be protease independent and involve protein trafficking secondary to modulation of membrane dynamics, such as organization of plasma membrane domains, protein recycling, endocytosis, endosomal trafficking, vacuolar maturation, and exocytosis. For example, Tamboli et al. [87] reported that PS1 could modulate the levels of lipid receptors at the cell surface. Specifically, it was noted that with PS1 deficiency or γ-secretase inhibition, the transmembrane receptor LRP1 (low-density lipoprotein receptor-related protein, which is a postulated γ-secretase substrate) accumulated at the cell surface and in cytoplasmic vesicles proximal to the cell surface. It should be noted that, in this study, overall LRP1 expression levels were not quantified nor was it established which stage of LRP1 trafficking was aberrant and if ligand metabolism was modified [87]. Tamboli et al. [87] also demonstrated that AβPP CTFs accumulated at the cell surface and in cytoplasmic vesicles after γ-secretase inhibition or PS1 deletion, in conjunction with reduced endocytosis of LDLR (low-density lipoprotein receptor). Although alterations in the internalization of LRP1 were not investigated in this study as downstream consequences of C99 accumulation, it is possible that PS1 deficiency could increase LRP1 cell-surface expression, in part, by inhibiting the initial endocytosis of LRP1, as was argued by Tamboli et al. [87] to be the case for LDLR. However, this possibility was not confirmed for LRP1, and, standing alone, does not account for the concomitant increase in a vesicular label for LRP1 akin to that seen for AβPP-CTF by this group. Furthermore, considering that LRP1 has a different substrate profile than LDLR, is itself a putative substrate for PS1 [88], and has a dynamic trafficking and recycling mechanism modulated by ligands and other interacting proteins [89, 90], we posit that any alterations in the location and expression levels of LRP1 secondary to PS1 dysfunction could be due to aberrant trafficking at multiple steps [87].
The dynamics of PS1-mediated LRP1 turnover are important, as LRP1 has been identified as a highly specific receptor for tau and a potential regulator of tau spread in tauopathies and AD [91]. To venture a model for how PS1’s impact on LRP1 location and trafficking may facilitate tau uptake/accumulation in cases of PS1 mutation, we propose as one possibility a mechanism described in a study of EGFR (epidermal growth factor receptor) turnover in the context of presenilin single and double knockout [92]. Using fibroblasts deficient in PS1 alone or in PS1 and PS2 (double knock-out), it was demonstrated that EGFR was endocytosed and ubiquitylated normally, but there was a reduction in the steady state sorting and trafficking of ligand-bound-EGFR to the lysosome. The majority of endocytosed EFGR is sorted out from its ligand and recycled to the cell surface, at steady state. So, with this recycling pathway remaining intact within PS1 deficient cells, while there is delayed turnover towards the lysosomal pathway, the overall EGFR levels in PS1 deficient cells increased. Proportionate to this overall increase, more EGFR was expressed at the cell surface compared to wild-type. Consequently, EGFR signaling was increased and, importantly, this phenotype, could be rescued by expression of wild-type PS1 but not by expression of mutant PS1 or wild-type PS2. This endosomal trafficking disruption that enhanced EGFR signaling, as illuminated in vitro, was evidenced in animal tissues as well [92]. Importantly, expression of the PS1 D257A mutant in a presenilin-deficient background did not rescue the steady-state level of EGFR, which the authors interpreted as an indication that catalytic γ-secretase complex activity may be involved in maintaining physiologically low EGFR levels. Although treating cells with potent γ-secretase inhibitors significantly increased accumulation of AβPP CTF, it did not increase EGFR levels by altered endocytic trafficking like in PS1-null cells. Therefore, Repetto et al. [92] concluded that regulation of EGFR by PS1 may not simply be determined by the levels of γ-secretase activity or levels of γ-secretase substrate CTF’s. Taking into account the findings of Tamboli et al. [87] and Repetto et al. [92] we propose that improper turnover of cell surface receptors in PS1 deficiency due to alterations in endocytic trafficking may be mediated by loss of PS1-γ-secretase complexes despite uncertain dependence upon a loss in protease function. These alterations may not only enhance cascade-mediated signaling, like in the case of EGFR, but alter the sorting and turnover of other AD-relevant ligands like that of tau as proposed for LRP1 [91].
It is already established that LRP1’s cysteine-rich EGF repeats and a YWTD β-propeller domain facilitate dissociation of LRP1 ligands in the sorting endocytic vesicles after uptake [90], and that LRP1 can be recycled to the cell surface thereafter [93, 94]. This pattern of rapid recycling is seen with LRP1 uptake of Apolipoprotein E [89] and is similar to the processing of EGFR, potentially making LRP1 vulnerable to PS1 dysfunction in the same fashion. If so, one could expect to see the increased cell-surface and vesicular LRP1 label that was previously reported by Tamboli et al. in the context of PS1 deficiency [87]. However, LRP1 has been noted to have a differential recycling mechanism depending on the ligand. Laatsch et al. [89] noted that in the case of LRP1 uptake of RAP (receptor associated protein), LRP1 is not rapidly recycled from the early sorting endosome, but rather, LRP1 and RAP remain associated until they reach a compartment with lower pH such as the late endosome, from which LRP1 is then recycled. This evidence of a slow recycling route for LRP1 suggests processing through endocytic recycling compartment (ERC), a network of tubular endocytic membranes that regulate recycling to the plasma membrane, [95–97]. Yet LRP1 may only recycle via this pathway when bound to certain ligands, one of which may be tau. Interestingly, PS1-γ-secretase complex function was found by Zhang et al. [98] to be critical for the protein sorting mechanisms of the ERC, which are regulated by Rab 11, Rab 4, and other proteins [98]. Similar to the dysregulation of EGFR processing in PS1 deficient cells, this research group noted that PS1 dysfunction did not inhibit endocytosis itself but noted that endocytic cargo, which in their study were transferrin and AβPP-CTFs, remained “stuck” in the ERC compartment. Potentially, like was the case with EGFR, cargo from the ERC did not correctly traffic towards lysosomes or enter a compartment that matured to the degradative endo-lysosome. Regardless of the recycling route LRP1 takes upon tau endocytosis, PS1 mutations could disturb tau-bound LRP1 trafficking towards a degradative route, making the cell vulnerable to endocytic backup and improper recycling of these proteins.
Due to increased rates of cell surface recycling secondary to interrupted traffic of cargo from sorting endosomes to lysosomes with mutant PS1 or PS1 deficiency, LRP1 may have a higher frequency or level of expression at the cell surface, such that more is available on the cell surface to enhance tau uptake. Given evidence for endosomal back-up or “traffic jams” with PS1 dysfunction, it is tempting to speculate that tau can be internalized and aggregate in endosomal or amphisomal compartments that do not properly fuse with lysosomes for cargo degradation. Vacuoles swollen with cargo in this manner are characteristic of AD brain [99, 100]. Furthermore, it was recently demonstrated that if these endosomal compartments containing tau aggregates are not properly maintained or experience any damage, they may leak their undigested contents. One could expect endosomal maintenance mechanisms to be overwhelmed without efficient lysosomal trafficking, resulting in increased leakage of aggregated tau species, which will then act as seed for endogenous tau in the cytosol [101].
The endosomal dysregulation induced by PS1 deficiency discussed above, described as “traffic jams,” are implicated in AD pathogenesis [102]. Backup of degradative cargo and/or delay in sorting may not only cause protein aggregation, but secondary increases in exocytosis. Improper retromer endosomal processing of AβPP and/or BACE1 leads to an accumulation of AβPP-CTF in endosomal membranes, for which the cell seems to compensate by increasing exosome release of AβPP CTFs [103]. Enhanced LRP1 recycling to and from the cell surface without lysosomal degradation, as expected in cases of PS1 inhibition or mutation, may likewise increase exosomal release of tau, thereby propagating its spread (illustrated in Fig. 2). Of note, LRP1 mediates uptake of Aβ but not via direct binding [104]. Taking all these data together, as illustrated in Fig. 2, we hypothesize that PS1 is necessary for balance in the endocytic system and that dysregulation caused by mutations or deficiencies may lead to tauopathy and amyloidopathy in AD as a consequence of altered membrane dynamics.
Fig. 2.
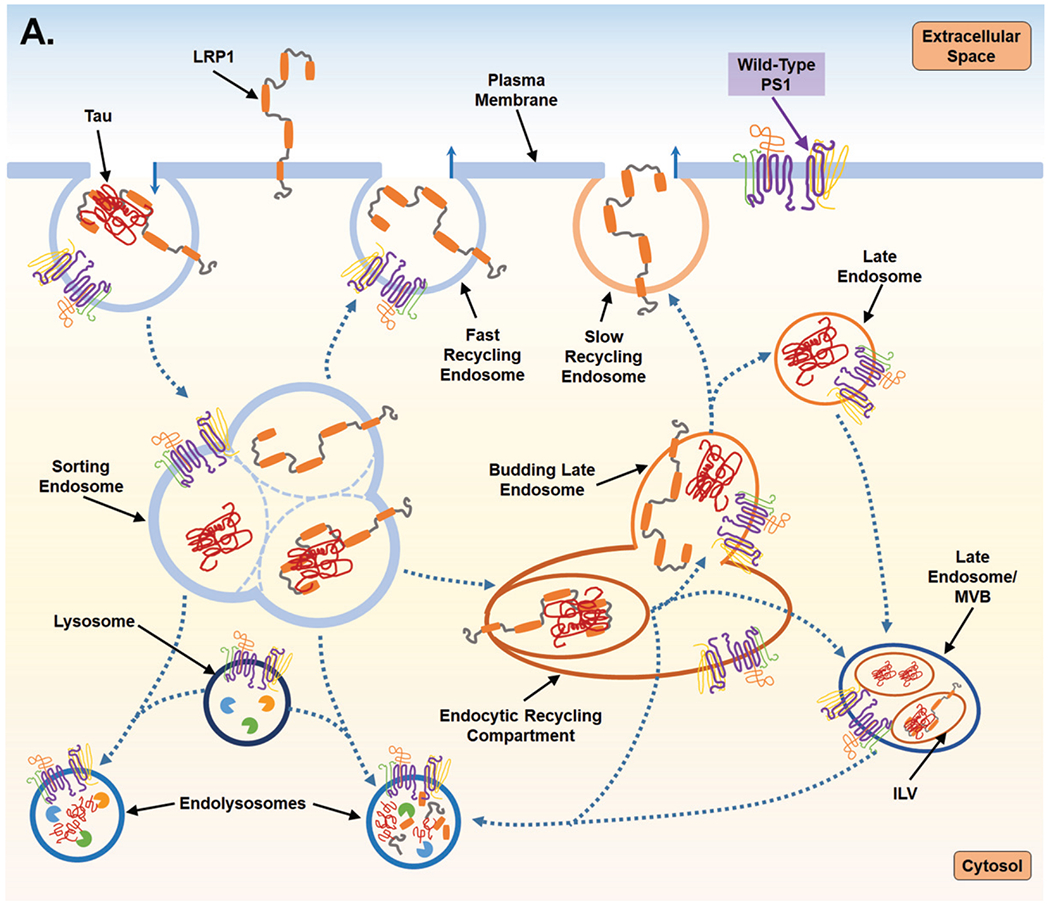
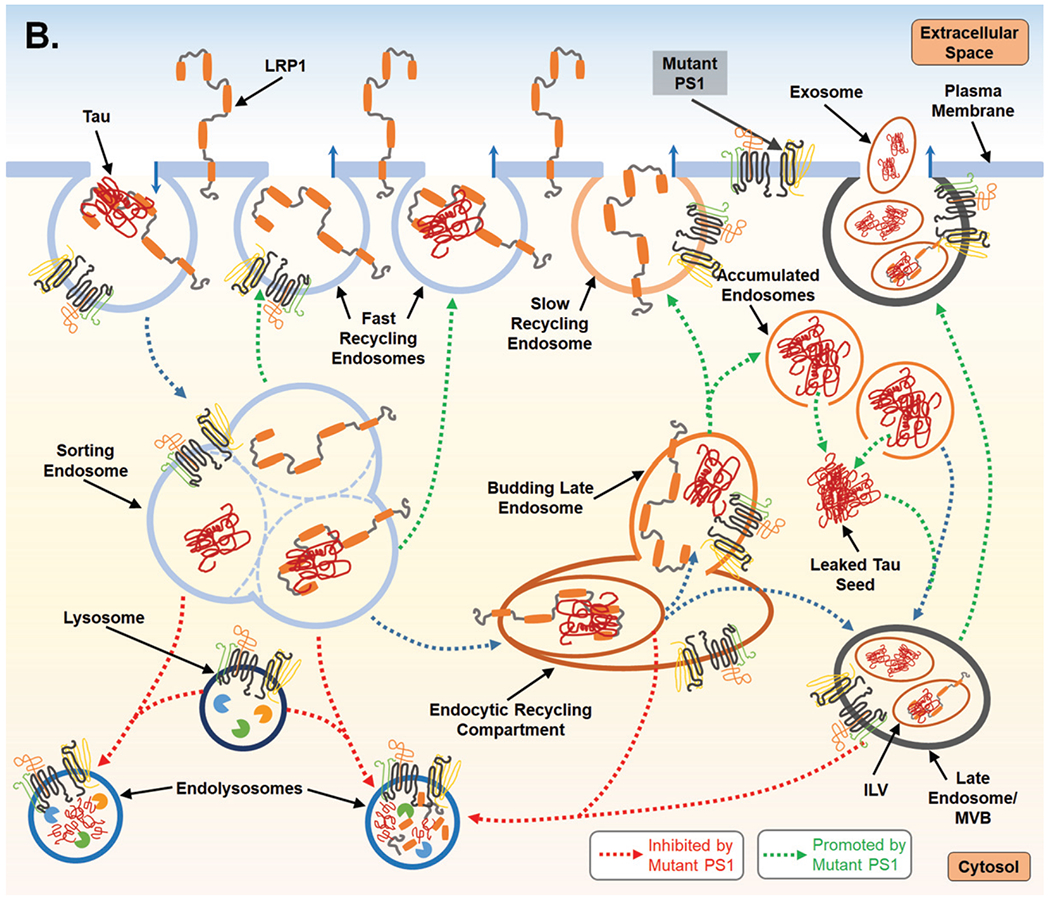
Proposed mechanism for PS1-mediated regulation of endocytosis and the subsequent impact on LPR1 turnover and tau uptake. A) PS1 is present at the cell surface and within multiple vacuolar structures. PS1 is postulated to largely localize within endocytic compartments and regulate their trafficking and fusion. PS1 is also reported to maintain lysosome function which allows for degradation of endocytic cargo after fusion event which forms the endolysosome. LPR1 (low-density lipoprotein receptor-related protein-1) binds tau at the cell surface and is internalized and trafficked to the sorting endosome. At the sorting endosome, LRP1 can be dissociated from tau and recycled to the surface; unbound tau is trafficked toward the functional degradative pathway. Alternatively, tau may remain associated with LRP1 and trafficked to the endocytic recycling compartment (ERC). From the ERC, tau and LRP1 can enter a maturing endosome and dissociate. LRP1 is recycled to the plasma membrane via the slow recycling route, whereas tau is retained in an endosomal compartment and targeted for degradation. As part of steady state LRP1 turnover, associated LRP1 and tau can also be internalized at the ERC into an intra-luminal vesicle (ILV) and trafficked together towards a multivesicular body (MVB), which is also considered a late endosome. From the MVB tau and LRP1 are degraded after fusion with a lysosome. B) Mutant PS1, however, inhibits lysosomal clearance and thus potentially return of LPR1 (with or without tau ligand) to the cell surface from the sorting endosome and from the ERC. Due to inappropriate trafficking, endosomal compartments retain cargo, increasing overall expression of both tau and LRP1. Tau can then leak from endosomal compartments and act as a seed for endogenous, cytosolic tau seen in Alzheimer’s disease. To compensate for disruption of degradative trafficking and vesicular burden, PS1 mutant cells can exocytose tau that has accumulated in MVBs, thereby propagating tau spread.
Currently no studies exist, to our knowledge, regarding the role of PS1 in mediating tau propagation, specifically, in mouse models or human cases. However, there are multiple reports of tauopathy/tau accumulation occurring secondary to PS1 mutations in mouse models and in human PS1-fAD cases [105–107]. Interestingly, it has been argued that certain genes involved with late onset AD are involved in endocytic trafficking, among other factors, may confer progression particularly through the regulation of vesicular tau secretion and uptake [108]. In mice, conditional PS1 deletion using a Cre/loxP recombinant system initiated 6 months postnatally (on a PS2 null background) results in AD-like neurodegeneration with tau hyperphosphorylation, but not plaque deposition [109,110]. Furthermore, in mice with PS1 mutation, with or without a supplemental mutation of the gene for APP, accumulate hyperphosphorylated tau [111–113]. Further investigation would be necessary to test any possible contribution of the endocytosis of LRP1 to tauopathy in cases of aberrant PS1 expression.
PS1 AT COMPARTMENTS OF THE DEGRADATIVE VACUOLAR SYSTEM
The role of PS1 in modulating membrane dynamics is evident in other subcellular compartments as well, which, aside from the trafficking and sorting discussed earlier, may further impact the function of degradative vacuoles. PS1 has been postulated to influence fusion, maturation, and possible genesis of degradative compartments including autophagosomes, late endosomes/endolysosomes, and autolysosomes (generated by fusion of cargosequestering autophagosomes and hydrolytic lysosomal compartments) [52, 114–117] (see effects of PS1 on degradative vacuolar compartments in Fig. 2). PS1-γ-secretase complex localizes to autophagosomes and lysosomes and it was even postulated to be a lysosomal hydrolase [118], though this has not been validated by subsequent studies. However, there are significant data which indicate mutant PS1 or loss of PS1 suppresses autophagic flux, though the nature of the autophagic disturbances that have been reported vary, as do the supposed mechanisms [115,119]. For example, work by Lee et al. [27, 43] demonstrated that, in the context of PS1 deficiency, lysosomal hydrolase capacity was attenuated secondary to improper acidification of lysosomes. Thus, cells became burdened with catalytically-incompetent autolysosomes or unfused autophagosomes swollen with undigested autophagic cargo. Evidence included increased numbers LC3 puncta (an autophagic membrane protein marker for autophagosomes) and increased LC3BII immunoblot signal (the lipidated form of LC3 degraded post lysosomal fusion). The increase in LC3BII on immunoblots was also observed by Neely et al. [119] in PS1 deficient cells. This increase in LC3BII was postulated to indicate a build-up of autophagosomes. Interestingly, studies looking at telencephalin (also known as ICAM-5) turnover in PS1 deficient cells also revealed accumulations of LC3-positive compartments which retained positive labeling for Apg12p, indicating that these structures were immature autophagosomes lacking proper closure of the phagophore [115]. Taken together, these data indicate that autophagosomes may not only accumulate due to improper clearance, but their formation may be aberrant and perhaps increased coincidently, or as an attempt to compensate for improper maturation and flux due to PS1 deficiency. In the work by Neely et al. [119], the increased autophagosome number in PS1 deficient cells was observed along-side an increase in Lysotracker signal that was used as measure of lysosome number. However, in the work done by Lee et al. [43] Lysotracker signal was abated in PS1 deficient cells, as expected given their proposal that loss of PS1 causes improper trafficking of v-ATPase to lysosomes. Neely et al. [119] did not propose a mechanism to explain the discrepancy in Lysotracker signals, but this difference may indicate that PS1 differentially affects degrees of acidification within vacuoles depending on the cell-type. Neely et al. [119] did confirm, however, that these changes were independent of γ-secretase inhibition with DAPT. This result might indicate that the PS1-γsecretase-complex influences autophagy and lysosomal activity in manners that are largely non-proteolytic, and that the PS1 holoprotein in the ER could significantly impact downstream vacuolar function.
Of note, the majority of the studies described above were done in non-neuronal cell lines and these exact findings were not replicated in neural stem cells in a study done by Zhang et al. [52]. However, suppression of autophagy due to PS1 deficiency has been reported in neural stem cells by Chong et al. in 2018 [116], but this finding was elucidated by a different mechanism. This work, differing from previous studies, demonstrated decreased expression of LCBI and II in the context of PS1 deficiency, indicating reduced autophagic formation, which was corroborated by a panel of changes in mRNA transcript levels for genes relevant to autophagosome and lysosome biogenesis. Lysosomal protein markers were also decreased in these PS1 deficient neural stem cells, and it was all postulated to be due to loss of normal interactions between PS1 and ERK/CREB signaling [116]. These studies indicate a role for PS1 in the function of degradative vacuoles that is conserved in multiple cell lines, but the discrepancies indicate there are nuances in how that regulation is executed depending on the cell type. Important insight into how PS1 mutations, as opposed to deletion, may alter this regulation is modeled in the work by Bustos et al. [114]. Bustos et al. demonstrated that PS1 binds Annexin A2 and other snare complexes to tether lysosomal compartments to autophagosomes to mediate fusion and degradation of cargo. It was hypothesized by this group that interaction with other adaptors could provide specificity to the Annexin A2-PS1 complex, such that they would fuse lysosomes and autophagosomes at a physiological optimum [114]. These tethering functions may not only alter fusion, but association with cytoskeletal frameworks and trafficking events necessary for fusion [120]. Furthermore, Annexin A2 has been shown to be involved in the formation of autophagosomes from the ER [114, 121] providing another route by which PS1 may play a role in the formation of fusion-competent degradative vesicles. Consideration of altered vesicular biogenesis in the context of PS1 mutation may tie the work elucidating the aberrant membrane dynamics of the ER-MAMs together with changes that have been noted in the expression of autophagosome and lysosome makers upon PS1 deficiency.
PS1 is also involved in regulating Rabs which play a major role in the regulation of vacuolar trafficking. In addition to potential interactions with Rab 11 in the aforementioned endocytic system [122], mutant PS1 was demonstrated to reduce Rab 8 expression (without changes in Rab 8 mRNA transcript levels) which reduced neurite outgrowth and vacuolar transport from the trans Golgi network to the plasma membrane [123]. Mutant PS1 also altered the location of Rab 6 and resulted in defective recycling of vesicles from the Golgi to ER [124]. These effects may be mediated by direct PS1-Rab binding or by the putative ability of PS1 to bind Rab GDP dissociation inhibitor (RabGDI) and, as demonstrated by PS1 deficiency studies, impact the association of Rab GDP with membrane fractions [125].
CONCLUDING REMARKS
It is important to emphasize that the potential mechanisms by which PS1 mutations and loss of function may alter membrane dynamics, are congruent with the observed pathological changes that occur in the AD brain. Although the presence of aggregated Aβ and NFTs are established hallmarks of AD, the mechanisms underlying their formation are not fully understood. Given what has been described above, it is likely that these protein aggregates may be signs of the underlying disturbances within the ER and vacuolar systems which are likely as substantial drivers of disease.
The vacuolar disturbances observed in models of mutant PS1 are complicated processes to study, requiring strong attention to the limitations of using a single label to distinguish membrane compartments; such caution and rigor may help to explain discrepancies in the data such as increases or decreases in Lysotracker signal with PS1 deficiency. Additionally, there remains a need to further evaluate membrane dynamics in neurons expressing various constructs of PS1 while including additional knockdown models to approximate the partial loss of function assumed by heterozygous inheritance of PS1-fAD. The catalytic roles of PS1 may be specific to the protein expression profiles of neurons, and thus mitotic cell models are limited in their use to model certain AD phenomena and protein species hallmarks such as hyperphosphorylated tau and NFTs. Additionally, the demands on membrane metabolism and trafficking in neurons are uniquely commensurate to their complex architecture and vesicular activity required for signal transduction. It is well-documented that PS1 is detectable at virtually all membrane compartments to varying degrees [37], and study of PS1 seems to demonstrate the degree to which these membranes are so tightly related, not just biochemically via calcium signaling, but physically, by docking and/or fusing to or budding from each other while exchanging lipids and proteins [126]. For example, the presence of PS1 in ER-MAMs may not only influence calcium signaling at the ER, but the changes in phospholipid synthesis at these sites that were noted in PS1 deficiency, might explain data featuring increases in autophagosome compartments in the context of PS1 dysfunction. Also, the biogenesis of lysosomal compartments and their supply with hydrolases and necessary membrane proteins are dependent upon healthy ER and Golgi networks, which in part regulate their membrane dynamics through calcium signaling [127]. Though the role of PS1 in maintaining calcium homeostasis in the ER was more mechanistically described in the literature and reviewed above, PS1 mutations have been demonstrated to significantly alter calcium levels within the Golgi, as well [128]. ER and Golgi system activities are crucial for anterograde and retrograde trafficking of endosomal compartments, and it is postulated that endosomal delivery systems may contribute to the maturation of late endosomes and thus distal generation of lysosomal compartments [129].
How PS1 may modulate these different vacuolar processes is not well delineated in neurons, but PS1 deficiency was already shown to modulate TFEB signaling [52, 116] which is in part activated by calcium signaling [79], in a manner that may inhibit lysosomal biogenesis [130, 131]. Biogenesis of an isolated membrane compartment is a different mechanism than lysosomal acidification proposed by Lee et al. [43], but both mechanisms point to loss of degradative capacity and could present very similarly with experimental read-outs such as quantification of mature hydrolases or acidic compartments. The generation, trafficking, fusion, and recycling of vacuoles in the context of PS1 mutation when examined in neuronal compartments may advance this field of study and explain AD phenomena such as tau spread and synaptic loss. Though many questions remain, there is strong evidence that PS1 mutations have many AD-related effects far beyond the Aβ generation hypothesis. Furthermore, these mutations can also disrupt the capacity for PS1 to maintain physiological membrane flux in manners that may be independent of its canonical proteolytic activities.
ACKNOWLEDGMENTS
CAD is supported by NIH F30 AG067628-01.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0598r1).
REFERENCES
- [1].Fraser PE, Yang DS, Yu G, Levesque L, Nishimura M, Arawaka S, Serpell LC, Rogaeva E, St George-Hyslop P (2000) Presenilin structure, function and role in Alzheimer disease. Biochim Biophys Acta 1502, 1–15. [DOI] [PubMed] [Google Scholar]
- [2].Janssen JC, Beck JA, Campbell TA, Dickinson A, Fox NC, Harvey RJ, Houlden H, Rossor MN, Collinge J (2003) Early onset familial Alzheimer’s disease: Mutation frequency in 31 families. Neurology 60, 235–239. [DOI] [PubMed] [Google Scholar]
- [3].Cruts M, Theuns J, Van Broeckhoven C (2012) Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33, 1340–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bagyinszky E, Youn YC, An SS, Kim S (2014) The genetics of Alzheimer’s disease. Clin Interv Aging 9, 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Robles A, Sobrido MJ, Garcia-Murias M, Prieto JM, Lema M, Santos D, Paramo M (2009) Clinical picture of a patient with a novel PSEN1 mutation (L424V). Am J Alzheimers Dis Other Demen 24, 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Portet F, Dauvilliers Y, Campion D, Raux G, Hauw JJ, Lyon-Caen O, Camu W, Touchon J (2003) Very early onset AD with a de novo mutation in the presenilin 1 gene (Met 233 Leu). Neurology 61, 1136–1137. [DOI] [PubMed] [Google Scholar]
- [7].Zekanowski C, Golan MP, Krzysko KA, Lipczynska-Lojkowska W, Filipek S, Kowalska A, Rossa G, Peplonska B, Styczynska M, Marnszak A, Religa D, Wender M, Kulczycki J, Barcikowska M, Kuznicki J (2006) Two novel presenilin 1 gene mutations connected with frontotemporal dementia-like clinical phenotype: Genetic and bioinformatic assessment. Exp Neurol 200, 82–88. [DOI] [PubMed] [Google Scholar]
- [8].Yasuda M, Maeda K, Ikejiri Y, Kawamata T, Kuroda S, Tanaka C (1997) A novel missense mutation in the presenilin-1 gene in a familial Alzheimer’s disease pedigree with abundant amyloid angiopathy. Neurosci Lett 232, 29–32. [DOI] [PubMed] [Google Scholar]
- [9].Yasuda M, Maeda S, Kawamata T, Tamaoka A, Yamamoto Y, Kuroda S, Maeda K, Tanaka C (2000) Novel presenilin-1 mutation with widespread cortical amyloid deposition but limited cerebral amyloid angiopathy. J Neurol Neurosurg Psychiatry 68, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dermaut B, Kumar-Singh S, Engelborghs S, Theuns J, Rademakers R, Saerens J, Pickut BA, Peeters K, van den Broeck M, Vennekens K, Claes S, Cruts M, Cras P, Martin JJ, Van Broeckhoven C, De Deyn PP (2004) A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann Neurol 55, 617–626. [DOI] [PubMed] [Google Scholar]
- [11].Halliday GM, Song YJ, Lepar G, Brooks WS, Kwok JB, Kersaitis C, Gregory G, Shepherd CE, Rahimi F, Schofield PR, Kril JJ (2005) Pick bodies in a family with presenilin-1 Alzheimer’s disease. Ann Neurol 57, 139–143. [DOI] [PubMed] [Google Scholar]
- [12].Yokota O, Terada S, Ishizu H, Ujike H, Ishihara T, Nakashima H, Yasuda M, Kitamura Y, Ueda K, Checler F, Kuroda S (2002) NACP/alpha-synuclein, NAC, and beta-amyloid pathology of familial Alzheimer’s disease with the E184D presenilin-1 mutation: A clinicopathological study of two autopsy cases. Acta Neuropathol 104, 637–648. [DOI] [PubMed] [Google Scholar]
- [13].Pickova T, Matej R, Bezdicek O, Keller J, van der Zee J, Van Broeckhoven C, Csefalvay Z, Rusina R (2017) Genetic Alzheimer disease and sporadic dementia with Lewy bodies: A comorbidity presenting as primary progressive aphasia. Cogn Behav Neurol 30, 23–29. [DOI] [PubMed] [Google Scholar]
- [14].Small SA, Gandy S (2006) Sorting through the cell biology of Alzheimer’s disease: Intracellular pathways to pathogenesis. Neuron 52, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jung CK, Fuhrmann M, Honarnejad K, Van Leuven F, Herms J (2011) Role of presenilin 1 in structural plasticity of cortical dendritic spines in vivo. J Neurochem 119, 1064–1073. [DOI] [PubMed] [Google Scholar]
- [16].Nikolakopoulou AM, Georgakopoulos A, Robakis NK (2016) Presenilin 1 promotes trypsin-induced neuroprotection via the PAR2/ERK signaling pathway. Effects of presenilin 1 FAD mutations. Neurobiol Aging 42, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W (1996) Alzheimer-associated presenilins 1 and 2: Neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med 2, 224–229. [DOI] [PubMed] [Google Scholar]
- [18].Suzuki T, Nishiyama K, Murayama S, Yamamoto A, Sato S, Kanazawa I, Sakaki Y (1996) Regional and cellular presenilin 1 gene expression in human and rat tissues. Biochem Biophys Res Commun 219, 708–713. [DOI] [PubMed] [Google Scholar]
- [19].Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, Seeger M, Koo E, Price DL, Sisodia SS (1996) Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci 16, 7513–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bazan NG, Lukiw WJ (2002) Cyclooxygenase-2 and presenilin-1 gene expression induced by interleukin-1beta and amyloid beta 42 peptide is potentiated by hypoxia in primary human neural cells. J Biol Chem 277, 30359–30367. [DOI] [PubMed] [Google Scholar]
- [21].Zhou Y, Zhang W, Easton R, Ray JW, Lampe P, Jiang Z, Brunkan AL, Goate A, Johnson EM, Wu JY (2002) Presenilin-1 protects against neuronal apoptosis caused by its interacting protein PAG. Neurobiol Dis 9, 126–138. [DOI] [PubMed] [Google Scholar]
- [22].Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398, 513–517. [DOI] [PubMed] [Google Scholar]
- [23].Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H (2003) PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem 278, 7850–7854. [DOI] [PubMed] [Google Scholar]
- [24].Dries DR, Yu G (2008) Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer’s disease. Curr Alzheimer Res 5, 132–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ (2000) Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694. [DOI] [PubMed] [Google Scholar]
- [26].Haapasalo A, Kovacs DM (2011) The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 25, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, Mitchell CH, Lloyd-Evans E, Nixon RA (2015) Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep 12, 1430–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM (2008) SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol 181, 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Honarnejad K, Herms J (2012) Presenilins: Role in calcium homeostasis. Int J Biochem Cell Biol 44, 1983–1986. [DOI] [PubMed] [Google Scholar]
- [30].Supnet C, Bezprozvanny I (2011) Presenilins as endoplasmic reticulum calcium leak channels and Alzheimer’s disease pathogenesis. Sci China Life Sci 54, 744–751. [DOI] [PubMed] [Google Scholar]
- [31].Tong BC, Lee CS, Cheng WH, Lai KO, Foskett JK, Cheung KH (2016) Familial Alzheimer’s disease-associated presenilin 1 mutants promote gamma-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci Signal 9, ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK (2010) Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal 3, ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, Wuytack F, Hiesinger PR, Grinstein S, Annaert W (2012) Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol 198, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lai MT, Chen E, Crouthamel MC, DiMuzio-Mower J, Xu M, Huang Q, Price E, Register RB, Shi XP, Donoviel DB, Bernstein A, Hazuda D, Gardell SJ, Li YM (2003) Presenilin-1 and presenilin-2 exhibit distinct yet overlapping gamma-secretase activities. J Biol Chem 278, 22475–22481. [DOI] [PubMed] [Google Scholar]
- [35].Yonemura Y, Futai E, Yagishita S, Suo S, Tomita T, Iwatsubo T, Ishiura S (2011) Comparison of presenilin 1 and presenilin 2 gamma-secretase activities using a yeast reconstitution system. J Biol Chem 286, 44569–44575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cai Y, An SS, Kim S (2015) Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin Interv Aging 10, 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Escamilla-Ayala A, Wouters R, Sannerud R, Annaert W (2020) Contribution of the Presenilins in the cell biology, structure and function of gamma-secretase. Semin Cell Dev Biol, doi: 10.1016/j.semcdb.2020.02.005 [DOI] [PubMed] [Google Scholar]
- [38].Prokop S, Shirotani K, Edbauer D, Haass C, Steiner H (2004) Requirement of PEN-2 for stabilization of the presenilin N-/C-terminal fragment heterodimer within the gamma-secretase complex. J Biol Chem 279, 23255–23261. [DOI] [PubMed] [Google Scholar]
- [39].Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, St George-Hyslop PH, Fraser PE (1998) The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains beta-catenin. J Biol Chem 273, 16470–16475. [DOI] [PubMed] [Google Scholar]
- [40].Rechards M, Xia W, Oorschot VM, Selkoe DJ, Klumperman J (2003) Presenilin-1 exists in both pre- and post-Golgi compartments and recycles via COPI-coated membranes. Traffic 4, 553–565. [DOI] [PubMed] [Google Scholar]
- [41].Zhang J, Kang DE, Xia W, Okochi M, Mori H, Selkoe DJ, Koo EH (1998) Subcellular distribution and turnover of presenilins in transfected cells. J Biol Chem 273, 12436–12442. [DOI] [PubMed] [Google Scholar]
- [42].Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA, Schon EA (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175, 1810–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B (1999) Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol 147, 277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Honarnejad K, Jung CK, Lammich S, Arzberger T, Kretzschmar H, Herms J (2013) Involvement of presenilin holoprotein upregulation in calcium dyshomeostasis of Alzheimer’s disease. J Cell Mol Med 17, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jin H, Sanjo N, Uchihara T, Watabe K, St George-Hyslop P, Fraser PE, Mizusawa H (2010) Presenilin-1 holoprotein is an interacting partner of sarco endoplasmic reticulum calcium-ATPase and confers resistance to endoplasmic reticulum stress. J Alzheimers Dis 20, 261–273. [DOI] [PubMed] [Google Scholar]
- [47].Nelson O, Supnet C, Tolia A, Horre K, De Strooper B, Bezprozvanny I (2011) Mutagenesis mapping of the presenilin 1 calcium leak conductance pore. J Biol Chem 286, 22339–22347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Theobald DL (2016) Presenilin adopts the ClC channel fold. Protein Sci 25, 1363–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126, 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bezprozvanny I, Supnet C, Sun S, Zhang H, De Strooper B (2012) Response to Shilling et al. ( 10.1074/jbc.M111.300491). J Biol Chem 287, 20469; author reply 20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shilling D, Mak DO, Kang DE, Foskett JK (2012) Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J Biol Chem 287, 10933–10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang X, Garbett K, Veeraraghavalu K, Wilburn B, Gilmore R, Mirnics K, Sisodia SS (2012) A role for presenilins in autophagy revisited: Normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J Neurosci 32, 8633–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Karagas NE, Venkatachalam K (2019) Roles for the endoplasmic reticulum in regulation of neuronal calcium homeostasis. Cells 8, 1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kim S, Violette CJ, Ziff EB (2015) Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 M146V mutant. Neurobiol Aging 36, 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Camandola S, Mattson MP (2011) Aberrant subcellular neuronal calcium regulation in aging and Alzheimer’s disease. Biochim Biophys Acta 1813, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jensen D, Schekman R (2011) COPII-mediated vesicle formation at a glance. J Cell Sci 124, 1–4. [DOI] [PubMed] [Google Scholar]
- [57].Fu YL, Zhang B, Mu TW (2019) LMAN1 (ERGIC-53) promotes trafficking of neuroreceptors. Biochem Biophys Res Commun 511, 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 31, 4106–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lunz V, Romanin C, Frischauf I (2019) STIM1 activation of Orai1. Cell Calcium 77, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pascual-Caro C, Espinosa-Bermejo N, Pozo-Guisado E, Martin-Romero FJ (2018) Role of STIM1 in neurodegeneration. World J Biol Chem 9, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wu B, Yamaguchi H, Lai FA, Shen J (2013) Presenilins regulate calcium homeostasis and presynaptic function via ryanodine receptors in hippocampal neurons. Proc Natl Acad Sci U S A 110, 15091–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Sudhof TC, Shen J (2009) Presenilins are essential for regulating neurotransmitter release. Nature 460, 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chakroborty S, Goussakov I, Miller MB, Stutzmann GE (2009) Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci 29, 9458–9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I (2006) Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci 26, 5180–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hajnoczky G, Csordas G, Yi M (2002) Old players in a new role: Mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32, 363–377. [DOI] [PubMed] [Google Scholar]
- [66].Sampieri A, Santoyo K, Asanov A, Vaca L (2018) Association of the IP3R to STIM1 provides a reduced intraluminal calcium microenvironment, resulting in enhanced store-operated calcium entry. Sci Rep 8, 13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Henke N, Albrecht P, Pfeiffer A, Toutzaris D, Zanger K, Methner A (2012) Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J Biol Chem 287, 42042–42052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Korkotian E, Meshcheriakova A, Segal M (2019) Presenilin 1 regulates [Ca(2+)]i and mitochondria/ER interaction in cultured rat hippocampal neurons. Oxid Med Cell Longev 2019, 7284967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Deak AT, Blass S, Khan MJ, Groschner LN, Waldeck-Weiermair M, Hallstrom S, Graier WF, Malli R (2014) IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J Cell Sci 127, 2944–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sarasija S, Laboy JT, Ashkavand Z, Bonner J, Tang Y, Norman KR (2018) Presenilin mutations deregulate mitochondrial Ca(2+) homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. Elife 7, e33052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yasuda Y, Kudo T, Katayama T, Imaizumi K, Yatera M, Okochi M, Yamamori H, Matsumoto N, Kida T, Fukumori A, Okumura M, Tohyama M, Takeda M (2002) FAD-linked presenilin-1 mutants impede translation regulation underER stress. Biochem Biophys Res Commun 296, 313–318. [DOI] [PubMed] [Google Scholar]
- [72].Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, Mori K, Rozmahel R, Fraser P, George-Hyslop PS, Tohyama M (2001) Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s disease-linked presenilin-1 mutations. J Biol Chem 276, 43446–43454. [DOI] [PubMed] [Google Scholar]
- [73].Niwa M, Sidrauski C, Kaufman RJ, Walter P (1999) A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell 99, 691–702. [DOI] [PubMed] [Google Scholar]
- [74].Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ (2012) The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol 226, 693–702. [DOI] [PubMed] [Google Scholar]
- [75].Sakagami Y, Kudo T, Tanimukai H, Kanayama D, Omi T, Horiguchi K, Okochi M, Imaizumi K, Takeda M (2013) Involvement of endoplasmic reticulum stress in tauopathy. Biochem Biophys Res Commun 430, 500–504. [DOI] [PubMed] [Google Scholar]
- [76].Hashimoto S, Saido TC (2018) Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol 8, 180024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Lee KS, Huh S, Lee S, Wu Z, Kim AK, Kang HY, Lu B (2018) Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci U S A 115, E8844–E8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mattson MP (2010) ER calcium and Alzheimer’s disease: In a state of flux. Sci Signal 3, pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tong BC, Wu AJ, Li M, Cheung KH (2018) Calcium signaling in Alzheimer’s disease & therapies. Biochim Biophys Acta Mol Cell Res 1865, 1745–1760. [DOI] [PubMed] [Google Scholar]
- [80].Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, Takeda M (2013) gamma-secretase modulators and presenilin 1 mutants act differently on presenilin/gamma-secretase function to cleave Abeta42 and Abeta43. Cell Rep 3, 42–51. [DOI] [PubMed] [Google Scholar]
- [81].Fernandez MA, Klutkowski JA, Freret T, Wolfe MS (2014) Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid beta-peptides (Abeta) by gamma-secretase to increase 42-to-40-residue Abeta. J Biol Chem 289, 31043–31052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chavez-Gutierrez L, Bammens L, Benilova l, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B (2012) The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J 31, 2261–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS (2011) Dissociation between the processivity and total activity of gamma-secretase: Implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry 50, 9023–9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sun L, Zhou R, Yang G, Shi Y (2017) Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc Natl Acad Sci U S A 114, E476–E485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhou R, Yang G, Shi Y (2017) Dominant negative effect of the loss-of-function gamma-secretase mutants on the wild-type enzyme through heterooligomerization. Proc Natl Acad Sci USA 114, 12731–12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Dewachter I, Reverse D, Caluwaerts N, Ris L, Kuiperi C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F (2002) Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci 22, 3445–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Tamboli IY, Prager K, Thal DR, Thelen KM, Dewachter I, Pietrzik CU, St George-Hyslop P, Sisodia SS, De Strooper A, Heneka MT, Filippov MA, Muller U, van Leuven F, Lutjohann D, Walter J (2008) Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J Neurosci 28, 12097–12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lleo A, Waldron E, von Arnim CA, Herl L, Tangredi MM, Peltan ID, Strickland DK, Koo EH, Hyman BT, Pietrzik CU, Berezovska O (2005)Low density lipoprotein receptor-related protein (LRP) interacts with presenilin 1 and is a competitive substrate of the amyloid precursor protein (APP) for gamma-secretase. J Biol Chem 280, 27303–27309. [DOI] [PubMed] [Google Scholar]
- [89].Laatsch A, Panteli M, Sornsakrin M, Hoffzimmer B, Grewal T, Heeren J (2012) Low density lipoprotein receptor-related protein 1 dependent endosomal trapping and recycling of apolipoprotein E. PLoS One 7, e29385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK (2008) LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev 88, 887–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Rauch JN, Luna G, Guzman E, Audouard M, Challis C, Sibih YE, Leshuk C, Hernandez I, Wegmann S, Hyman BT, Gradinaru V, Kampmann M, Kosik KS (2020) LRP1 is a master regulator of tau uptake and spread. Nature 580, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Repetto E, Yoon IS, Zheng H, Kang DE (2007) Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J Biol Chem 282, 31504–31516. [DOI] [PubMed] [Google Scholar]
- [93].Theret L, Jeanne A, Langlois B, Hachet C, David M, Khrestchatisky M, Devy J, Herve E, Almagro S, Dedieu S (2017) Identification of LRP-1 as an endocytosis and recycling receptor for beta1-integrin in thyroid cancer cells. Oncotarget 8, 78614–78632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].van Kerkhof P, Lee J, McCormick L, Tetrault E, Lu W, Schoenfish M, Oorschot V, Strous GJ, Klumperman J, Bu G (2005) Sorting nexin 17 facilitates LRP recycling in the early endosome. EMBO J 24, 2851–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Xie S, Bahl K, Reinecke JB, Hammond GR, Naslavsky N, Caplan S (2016) The endocytic recycling compartment maintains cargo segregation acquired upon exit from the sorting endosome. Mol Biol Cell 27, 108–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Grant BD, Donaldson JG (2009) Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Naslavsky N, Caplan S (2018) The enigmatic endosome - sorting the ins and outs of endocytic trafficking. J Cell Sci 131, jcs216499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zhang M, Haapasalo A, Kim DY, Ingano LA, Pettingell WH, Kovacs DM (2006) Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J 20, 1176–1178. [DOI] [PubMed] [Google Scholar]
- [99].Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J Neuropathol Exp Neurol 64, 113–122. [DOI] [PubMed] [Google Scholar]
- [100].Omata Y, Lim YM, Akao Y, Tsuda L (2014) Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer’s disease. Am J Neurodegener Dis 3, 134–142. [PMC free article] [PubMed] [Google Scholar]
- [101].Chen JJ, Nathaniel DL, Raghavan P, Nelson M, Tian R, Tse E, Hong JY, See SK, Mok SA, Hein MY, Southworth DR, Grinberg LT, Gestwicki JE, Leonetti MD, Kampmann M (2019) Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J Biol Chem 294, 18952–18966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Small SA, Simoes-Spassov S, Mayeux R, Petsko GA (2017) Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer’s disease. Trends Neurosci 40, 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sullivan CP, Jay AG, Stack EC, Pakaluk M, Wadlinger E, Fine RE, Wells JM, Morin PJ (2011) Retromer disruption promotes amyloidogenic APP processing. Neurobiol Dis 43, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Yamada K, Hashimoto T, Yabuki C, Nagae Y, Tachikawa M, Strickland DK, Liu Q, Bu G, Basak JM, Holtzman DM, Ohtsuki S, Terasaki T, Iwatsubo T (2008) The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J Biol Chem 283, 34554–34562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hendriks L, De Jonghe C, Lubke U, Woodrow S, Vanderhoeven I, Boons J, Cras P, Martin JJ, Van Broeckhoven C (1998) Immunoreactivity of presenilin-1 and tau in Alzheimer’s disease brain. Exp Neurol 149, 341–348. [DOI] [PubMed] [Google Scholar]
- [106].Dong J, Qin W, Wei C, Tang Y, Wang Q, Jia J (2017) A novel PSEN1 K311R mutation discovered in Chinese families with late-onset Alzheimer’s disease affects amyloid-beta production and tau phosphorylation. J Alzheimers Dis 57, 613–623. [DOI] [PubMed] [Google Scholar]
- [107].Sutovsky S, Smolek T, Turcani P, Petrovic R, Brandoburova P, Jadhav S, Novak P, Attems J, Zilka N (2018) Neuropathology and biochemistry of early onset familial Alzheimer’s disease caused by presenilin-1 missense mutation Thr116Asn. J Neural Transm (Vienna) 125, 965–976. [DOI] [PubMed] [Google Scholar]
- [108].Uronen RL, Huttunen HJ (2016) Genetic risk factors of Alzheimer’s disease and cell-to-cell transmission of Tau. J Neurol Neuromed 1, 17–22. [Google Scholar]
- [109].Elder GA, Gama Sosa MA, De Gasperi R, Dickstein DL, Hof PR (2010) Presenilin transgenic mice as models of Alzheimer’s disease. Brain Struct Funct 214, 127–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen Q, Nakajima A, Choi SH, Xiong X, Tang YP (2008) Loss of presenilin function causes Alzheimer’s disease-like neurodegeneration in the mouse. J Neurosci Res 86, 1615–1625. [DOI] [PubMed] [Google Scholar]
- [111].Tanemura K, Chui DH, Fukuda T, Murayama M, Park JM, Akagi T, Tatebayashi Y, Miyasaka T, Kimura T, Hashikawa T, Nakano Y, Kudo T, Takeda M, Takashima A (2006) Formation of tau inclusions in knock-in mice with familial Alzheimer disease (FAD) mutation of presenilin 1 (PS1). J Biol Chem 281, 5037–5041. [DOI] [PubMed] [Google Scholar]
- [112].Samura E, Shoji M, Kawarabayashi T, Sasaki A, Matsubara E, Murakami T, Wuhua X, Tamura S, Ikeda M, Ishiguro K, Saido TC, Westaway D, St George Hyslop P, Harigaya Y, Abe K (2006) Enhanced accumulation of tau in doubly transgenic mice expressing mutant betaAPP and presenilin-1. Brain Res 1094, 192–199. [DOI] [PubMed] [Google Scholar]
- [113].Kurt MA, Davies DC, Kidd M, Duff K, Howlett DR (2003) Hyperphosphorylated tau and paired helical filament-like structures in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Neurobiol Dis 14, 89–97. [DOI] [PubMed] [Google Scholar]
- [114].Bustos V, Pulina MV, Bispo A, Lam A, Flajolet M, Gorelick FS, Greengard P (2017) Phosphorylated Presenilin 1 decreases beta-amyloid by facilitating autophagosomelysosome fusion. Proc Natl Acad Sci U S A 114, 7148–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Esselens C, Oorschot V, Baert V, Raemaekers T, Spittaels K, Serneels L, Zheng H, Saftig P, De Strooper B, Klumperman J, Annaert W (2004) Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol 166,10411054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Chong CM, Ke M, Tan Y, Huang Z, Zhang K, Ai N, Ge W, Qin D, Lu JH, Su H (2018) Presenilin 1 deficiency suppresses autophagy in human neural stem cells through reducing gamma-secretase-independent ERK/CREB signaling. Cell Death Dis 9, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Oikawa N, Walter J (2019) Presenilins and gamma-secretase in membrane proteostasis. Cells 8, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW, Mahuran DJ (2003) Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem 278, 26687–26694. [DOI] [PubMed] [Google Scholar]
- [119].Neely KM, Green KN, LaFerla FM (2011) Presenilin is necessary for efficient proteolysis through the autophagylysosome system in a gamma-secretase-independent manner. J Neurosci 31, 2781–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Yu L, Chen Y, Tooze SA (2018) Autophagy pathway: Cellular and molecular mechanisms. Autophagy 14, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Moreau K, Ghislat G, Hochfeld W, Renna M, Zavodszky E, Runwal G, Puri C, Lee S, Siddiqi F, Menzies FM, Ravikumar B, Rubinsztein DC (2015) Transcriptional regulation of Annexin A2 promotes starvation-induced autophagy. Nat Commun 6, 8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Dumanchin C, Czech C, Campion D, Cuif MH, Poyot T, Martin C, Charbonnier F, Goud B, Pradier L, Frebourg T (1999) Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum Mol Genet 8, 1263–1269. [DOI] [PubMed] [Google Scholar]
- [123].Kametani F, Usami M, Tanaka K, Kume H, Mori H (2004) Mutant presenilin (A260V) affects Rab8 in PC12D cell. Neurochem Int 44, 313–320. [DOI] [PubMed] [Google Scholar]
- [124].Scheper W, Zwart R, Baas F (2004) Rab6 membrane association is dependent of Presenilin 1 and cellular phosphorylation events. Brain Res Mol Brain Res 122, 17–23. [DOI] [PubMed] [Google Scholar]
- [125].Scheper W, Zwart R, van der Sluijs P, Annaert W, Gool WA, Baas F (2000) Alzheimer’s presenilin 1 is a putative membrane receptor for rab GDP dissociation inhibitor. Hum Mol Genet 9, 303–310. [DOI] [PubMed] [Google Scholar]
- [126].Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77, 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Hay JC (2007) Calcium: A fundamental regulator of intracellular membrane fusion? EMBO Rep 8, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Zatti G, Burgo A, Giacomello M, Barbiero L, Ghidoni R, Sinigaglia G, Florean C, Bagnoli S, Binetti G, Sorbi S, Pizzo P, Fasolato C (2006) Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 39, 539–550. [DOI] [PubMed] [Google Scholar]
- [129].Ferguson SM (2018) Axonal transport and maturation of lysosomes. Curr Opin Neurobiol 51, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Reddy K, Cusack CL, Nnah IC, Khayati K, Saqcena C, Huynh TB, Noggle SA, Ballabio A, Dobrowolski R (2016) Dysregulation of nutrient sensing and CLEARance in presenilin deficiency. Cell Rep 14, 2166–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]