

Abstract
We developed entangled link-augmented stretchable tissue-hydrogel (ELAST), a technology that transforms tissues into elastic hydrogels to enhance macromolecular accessibility and mechanical stability simultaneously. ELASTicized tissues are highly stretchable and compressible, which enables reversible shape transformation and faster delivery of probes into intact tissue specimens via mechanical thinning. This universal platform may facilitate rapid and scalable molecular phenotyping of large-scale biological systems, such as human organs.
Engineering tissue properties with the help of hydrogels has facilitated interrogating structural and molecular organization of complex biological systems1–6. Covalent fusion of tissue and polyacrylamide (pAAm) via a chemical cross-linker allowed preservation of tissue architecture and biomolecules in a cleared tissue-gel1. This approach, termed CLARITY, has enabled fine-structural and molecular phenotyping of intact biological systems. Further engineering of various tissue-hydrogel properties has expanded the utility of the tissue-gel fusion approach. In particular, the expansion microscopy and magnified analysis of proteome techniques have enabled super-resolution imaging of biomolecules and structures by physically swelling tissue-gel hybrids2,4,7,8.
However, these approaches suffer from the fundamental limitation of hydrogels: the inverse relationship between structural stability and permeability. To enhance the molecular accessibility of tissue-gel, its permeability needs to be increased, which causes loss of mechanical stability and structural information. Structural stability of tissue-gel can be enhanced by increasing the degree of cross-linking and gel density. Increasing gel density, however, limits the penetration of molecular probes. These innate conflicting properties of hydrogels have restricted the application of the tissue-gel fusion approach to relatively small biological systems, such as zebrafish, rodent brain and small blocks of human tissues. The difficulty associated with using these methods for interrogating large-scale tissues, particularly human brain, prompted us to search for alternative approaches.
Here we introduce a tissue-hydrogel that offers structural stability while enabling fast transport of molecular probes. We hypothesized that elasticizing tissue would not only render it mechanically durable, but also allow for transient and reversible shape transformation to facilitate molecular access. Elastic hydrogel types9 include hydrogels having a unique chemical or topological cross-linking structure10,11 and double-network hydrogels12–14. These gels offer stretchability and toughness, but their syntheses are not suitable for hybridization with biological tissues. For instance, their polymer units are too large to diffuse uniformly into thick tissues before gel formation.
We discovered that high concentrations (20–60% (wt/vol)) of acrylamide (AAm) alone can polymerize to form an elastic hydrogel in a single synthesis step (Extended Data Fig. 1a). We used orders-of-magnitude-lower concentrations of thermal initiator (1 per 14,000 AAm versus 1 per 73 AAm in CLARITY) and cross-linker (1 per 220,000 AAm versus 1 per 170 AAm reported) to synthesize long polymer chains that naturally undergo entanglement with each other under a high-polymer-density environment. Compared with typical pAAm gels covalently linked by high concentrations of cross-linker (Fig. 1a), entangled pAAm gels are formed via physically slippery tangles between growing polymer chains (Fig. 1b). Such slip-links endow entangled gels with flexibility and elasticity15. Our entangled pAAm gels exhibit stability against physical stresses such as ninefold compression and tenfold stretch (Extended Data Fig. 1b,c and Supplementary Video 1).
Fig. 1 |. ELAST.

a,b, Schematic drawings describing two hydrogel-forming mechanisms: chemically cross-linked (a) and physically entangled (b) hydrogels. Physical slip links render entangled hydrogel elastic. c, Schematic illustration describing the principle of fast probe delivery in ELASTicized tissue by reversible shape transformation. Biomolecular networks in an ELASTicized tissue follow the transient shape change of the synthetic entangled gel (from left to right) in a reversible way. The timescale (t) for probe delivery into the tissue is proportional to the square of the tissue thickness (d). Transient n-fold thinning of an ELASTicized tissue can accelerate the probe delivery by n2.
Next, we tested whether in situ synthesis of such elastic pAAm gel can transmute intact tissue into an elastic platform. To prevent any covalent linking between the tissue and pAAm gel, we did not use any chemical fixatives during the gelling process. This strategy allows for the growing pAAm chains to form purely physical entanglements with endogenous biomolecular networks that are constructed by formaldehyde fixation before gelling. We found that the resulting tissue-gel possesses similar mechanical properties as entangled pAAm gels (Fig. 1c, top row).
We applied the technology to a 2-mm-thick, 6 × 8 cm2 coronal human brain hemisphere slab (Fig. 2a,b). Elastic human tissue-gels, as well as mouse tissue-gels, were stretchable and compressible (Fig. 2c, Supplementary Fig. 1 and Supplementary Videos 2 and 3), protecting large intact tissue samples from a variety of mechanical damages. Tissue elasticization was applicable to other matrix-rich organs including heart and colon as well as cerebral organoids (Extended Data Fig. 2 and Supplementary Video 4). In addition, we confirmed that biomolecules and tissue architecture are well preserved (Fig. 2d,e). We termed this technology ELAST.
Fig. 2 |. Mechanical reinforcement and reversible shape transformation of tissue specimens via ELASTicization.
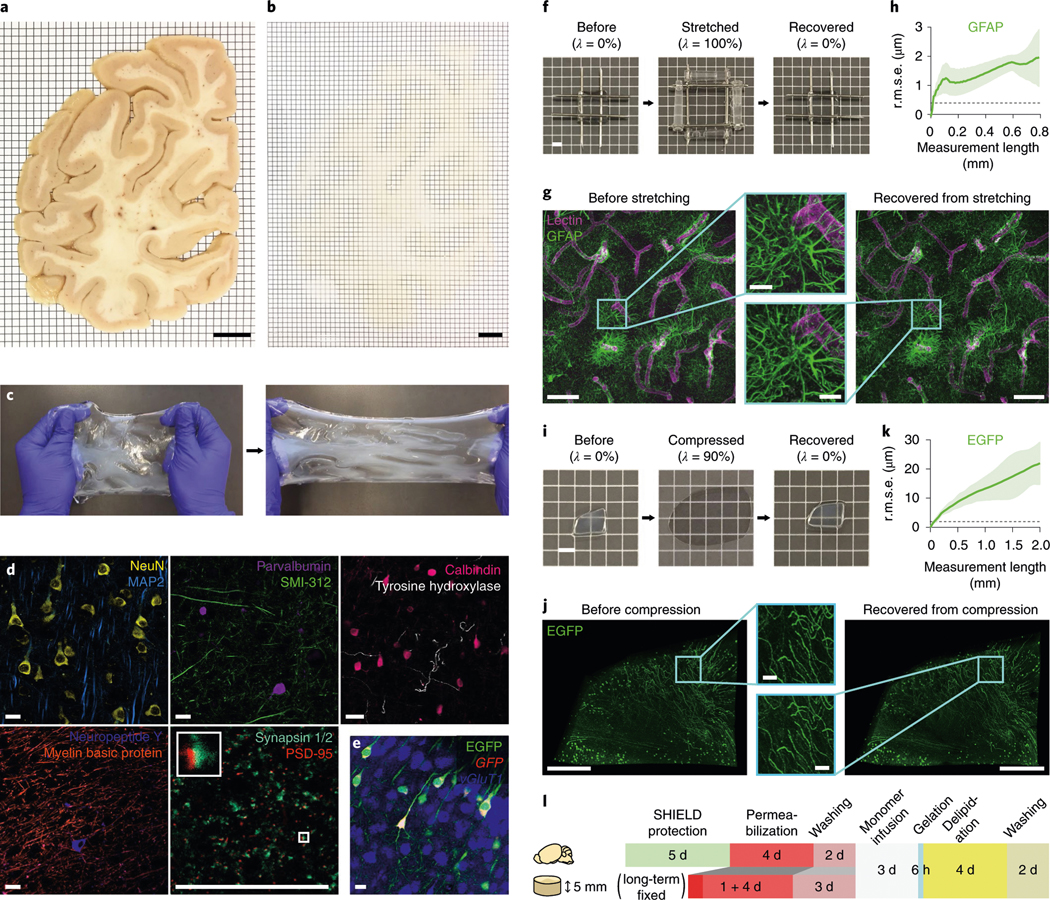
a,b, A 2-mm-thick coronal human brain hemisphere slab from the occipital pole before (a) and after (b) ELAST transformation and RI-matching. Scale bars, 1 cm. Experiment was repeated two times with similar results. c, Stretchability of an ELASTicized human brain slab. d, Immunolabeling of various proteins as indicated in ELASTicized 200-μm-thick human brain cortical samples. Scale bars, 40 μm. Experiments in c and d were repeated two times with similar results. e, In situ hybridization of GFP and vGluT1 mRNAs in an ELASTicized mouse hippocampus expressing EGFP. Scale bar, 40 μm. f, Reversible shape transformation of an ELASTicized 1-mm-thick human brain sample upon twofold stretching in both lateral dimensions. λ, tensile strain. Scale bar, 2 mm. g, Axially projected microscopic images of a prelabeled sample before stretching and after recovery (a representative image from n = 5 samples). Scale bars, 100 μm (insets, 20 μm). h, A mean ± s.d. plot of three-dimensional (3D) geometric deformation measured by the altered distances between feature correspondences in GFAP images at different length scales (n = 5 samples). Dashed line indicates the lateral pixel size of images. r.m.s.e., root mean squared error. i, Reversible shape transformation of a 1.4-mm-thick ELASTicized EGFP-expressing mouse brain sample upon ninefold compression. λ, compression strain. Scale bar, 2 mm. j, Three-dimensionally rendered microscopic images of the sample in i with RI-matching (a representative image from n = 5 samples). Scale bars, 500 μm (insets, 50 μm). k, 3D geometric deformation quantified from EGFP images (n = 5 samples), the same as in h. l, A diagram of the sample-processing procedure for ELASTicization with a suggested timeline. Upper row is for freshly perfused mouse organs, including the brain. Lower row is for about 5-mm-thick specimens from archived human brains that are typically fixed in a formalin solution for a long time. The optimal durations could vary by the degree of fixation and the sample-to-solution volume ratio.
ELAST allows the entangled biological networks to precisely follow the transient shape transformation of the hydrogel in a fully reversible way. After twofold stretching in both lateral dimensions or tenfold axial compression, ELASTicized samples showed negligible distortion, keeping subcellular architectures intact (Fig. 2f–k). These results demonstrate that ELAST enables reversible tissue shape transformation while preserving structural and molecular information of the tissue. The tissue ELASTicization is achieved through several procedural steps including biological networks formation, permeabilization, in situ polymerization of entangled hydrogels and delipidation (Fig. 2l and Supplementary Table 1). Based on the initial tissue preparation conditions, such as freshly prepared experimental animal tissues or long-term preserved archival human tissue specimens, different processing strategies can be used. The final tissue-gel size is dependent on the refractive index (RI)-matching medium used (Supplementary Fig. 2), as in other tissue transformation and clearing methods1,4,16–19.
The mechanical properties of ELASTicized tissues open new possibilities in enhancing molecular transport inside the tissue-gel. The tissue thickness, generally the shortest dimension of a tissue, determines the time required for probe penetration (t = d2/Deff, where t is dimensionless diffusion timescale, d is tissue thickness and Deff is the effective diffusivity). Therefore, we reasoned that reducing tissue thickness by transforming its shape would substantially shorten the time required for probe penetration and labeling (Fig. 1c).
To test this, we stretched elastic pAAm gels to thin them by threefold (Fig. 3a,b), and incubated them in a solution containing 150-kDa dextran-dye (Fig. 3c,d). Fitting the axial dye signal profiles to a one-dimensional (1D) transient diffusion model confirmed that the overall threefold thinning resulted in a ninefold decrease in diffusion timescale as predicted (Fig. 3e). We next tested whether thinning of ELASTicized tissues can enhance probe penetration. We stretched 1.5-mm-thick ELASTicized human brain tissues to make them two times thinner and stained them with dye-conjugated antibodies for 30 min. The thinned tissues showed a fourfold increase in antibody penetration depth compared with the unstretched samples (Fig. 3f–i).
Fig. 3 |. Thinning-mediated acceleration of probe delivery into ELASTicized tissues.
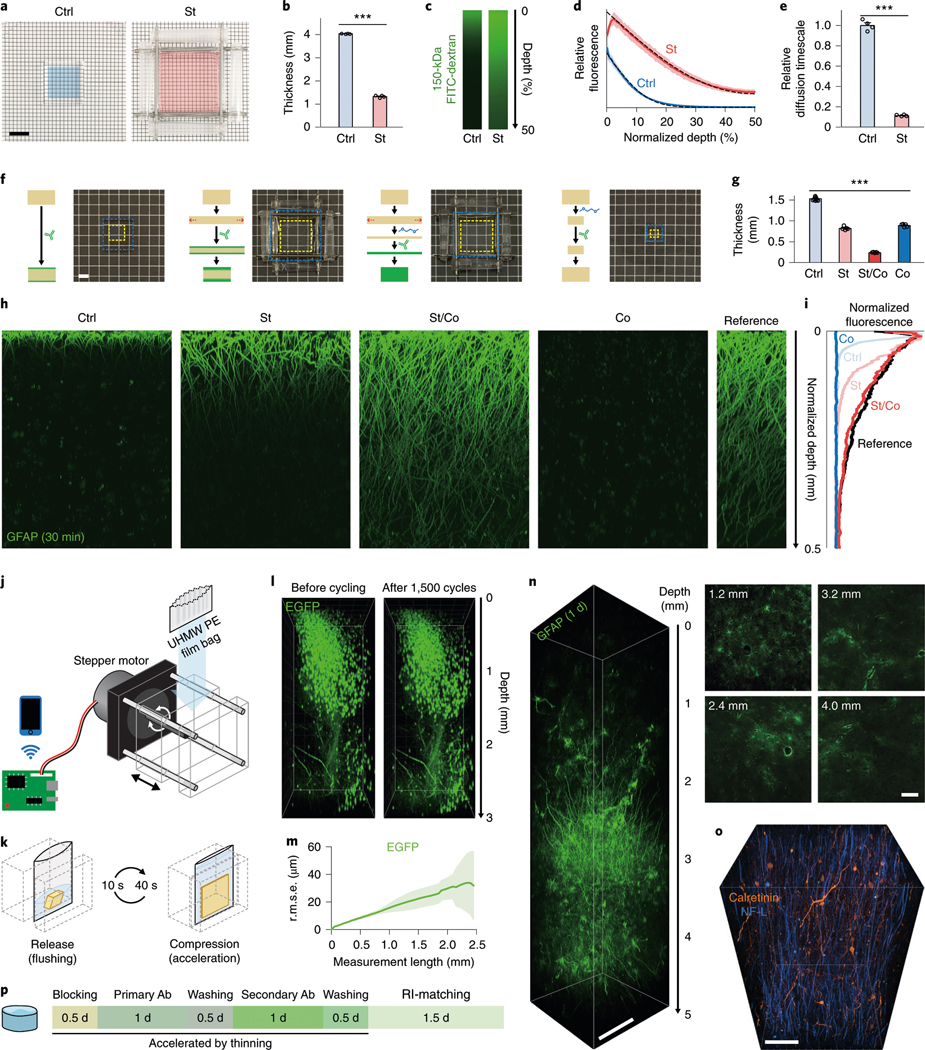
a, Control (Ctrl) and stretched (St; twofold in both lateral dimensions) entangled pAAm gels. Colored regions indicate the same areas before stretching. Scale bar, 1 cm. b, Thickness (mean with s.e.m.) change after stretching for 22.5 min (n = 4 gels; two-tailed paired t-test, ***P = 1.94 × 10−6, t = 104). c, Delivery of 150-kDa FITC-dextran into control and stretched gels for 45 min. Laterally central regions of top halves of gels were imaged and shown with lateral projections. Stretching was released before taking images. Representative images are shown from four samples in each group. d, Means ± s.d. of depth-wise relative FITC fluorescence (n = 4 gels for each group). Dashed lines show means of modeled fits (R2 (mean ± s.d.), 1.00 ± 0.0011 for Ctrl and 1.00 ± 0.0030 for St). e, A comparison of diffusion timescales (means with s.e.m.) drawn from the fitted models in d (two-tailed unpaired t-test, ***P = 3.75 × 10−5, t = 26). f, Schematic drawings of antibody delivery into tissue samples in different thinning approaches. Representative photos (10, 5, 5 and 5 tissue samples in the order shown) of ELASTicized 1-mm-thick human brain cortical samples in the conditions are shown together. Yellow and blue dashed lines indicate tissue and hydrogel areas, respectively. A 40% 1,2-dimethoxyethane solution was used for contraction (Co). Scale bar, 2 mm. g, A comparison of thicknesses (n = 10, 5, 5 and 5 samples; mean with s.e.m.; one-way analysis of variance, ***P = 4.95 × 10−24, F = 1,310; post hoc Tukey’s test yielded P < 0.05 for a comparison between St and Co, and P < 0.001 for other pairs). h, Laterally projected images of central tissue regions showing immunolabeling depths after 30 min of incubation with a fluorophore-conjugated anti-GFAP antibody. A reference image was obtained by exposing a sample’s cut surface to the antibody solution for 1 d and imaging the cut surface vertically. Experiments were repeated two times with similar results. i, A comparison of depth-wise signal intensities among images in h. j, A schematic drawing of the cyclic compression device. UHMW PE, ultrahigh molecular weight polyethylene. k, An example of a cyclic compression protocol. Each cycle consists of a compression period for acceleration and a release period for flushing with a solution. l, Three-dimensionally rendered images of an ELASTicized 3-mm-thick mouse sample expressing EGFP before and after about 1,500 times of cyclic sixfold compression over 1 d (a representative image from n = 4 samples). m, A mean ± s.d. plot of 3D geometric deformation quantified from EGFP images (n = 4 samples). n, A three-dimensionally rendered image and selected planes, showing rapid full-depth immunolabeling of an originally 5-mm-thick human brain cortical sample using cyclic compression for 1 d. The sample was compressed to 16% thickness for 40 s and released for 10 s for each cycle. A central region of the sample was imaged after RI-matching. Selected plane images were individually contrast-adjusted. Scale bar, 500 μm (selected planes, 200 μm). o, Immunolabeling of neuronal cell types and projections in a human cerebral cortex. NF-L, neurofilament light chain. Scale bar, 100 μm. Experiments in n and o were not repeated. p, A typical procedure and timeline for immunolabeling of an ELASTicized 5-mm-thick human brain sample assisted by thinning. Ab, antibody.
Additional thinning of ELASTicized tissue could further enhance probe delivery and shorten the labeling time. We discovered that 40% 1,2-dimethoxyethane in water can isotropically contract ELASTicized tissues by 1.7-fold in each dimension (Fig. 3f,g). When applied to stretched ELASTicized tissues, this solution elicited a 1D contraction that led to additional 3.4-fold thinning, making the samples thinned by 6.3-fold overall. This synergistic two-step thinning enabled complete immunostaining of the 1.5-mm-thick ELASTicized human tissue within only 30 min (Fig. 3h,i). However, when applied to unstretched tissues, isotropic contraction by 1,2-dimethoxyethane suppressed antibody penetration likely due to reduced pore size.
Although stretching combined with chemical-mediated shrinkage can shape-transform ELASTicized tissues to accelerate probe delivery, implementing this method is not straightforward. Stretching requires extra gel surrounding the tissue to interface with a stretching apparatus. In addition, 1,2-dimethoxyethane could disrupt probe–target binding. These complications together could limit the utility of this approach. We therefore sought an alternative technique for sample-thinning. We directly compressed an ELASTicized sample, instead of stretching (Supplementary Fig. 3), and periodically released the sample for flushing to avoid depletion of probes. We built a motorized device to automate this cycling (Fig. 3j,k, Supplementary Fig. 4 and Supplementary Video 5). Fivefold cyclic compression caused no noticeable tissue damage over 1 d (~1,500 cycles) and no substantial change in the quality of immunostaining (Fig. 3l,m and Supplementary Fig. 5). The increase in antibody penetration depth in cyclic compression was slightly less than that in stretching-based thinning probably due to the time required for the decompression steps (Supplementary Fig. 6).
We next applied the compression method for immunolabeling of a 5-mm-thick human brain sample within only 1 d (Fig. 3n and Supplementary Video 6), which is over 100 times faster than what we previously reported (1 d for labeling 0.5-mm-thick human brain block)1. In addition, we demonstrated the potential of this approach as a scalable platform for mapping neuronal connectivity in higher animal models and human samples by visualizing neuronal cell types and their projections in human brain tissues as well as in cerebral organoids (Fig. 3o,p, Supplementary Fig. 7, Supplementary Videos 7 and 8 and Supplementary Table 1).
In summary, we report a tissue transformation technique that simultaneously enhances mechanical properties and chemical accessibility of tissue by elasticizing it. We discovered that elastic homo-pAAm gel can be synthesized by promoting physical entanglement of long and densely packed pAAm chains. In situ synthesis of such an elastic gel transforms intact tissue into a deformable matrix that can be stretched or compressed without mechanical damage.
Transient and reversible thinning of ELASTicized tissue by cyclic compression enables fast molecular labeling. This mechanical approach has the potential to accelerate transport of a range of molecules without affecting their chemical and functional properties. As a step toward such extension, we demonstrated the compatibility of ELAST with a variety of antibodies, nuclear and lipophilic dyes, and messenger RNA probes (Fig. 2d,e, Supplementary Fig. 8 and Supplementary Tables 2 and 3).
The current ELASTicization protocol takes about 20 d to pro cess fresh mouse organs or archived human tissues with similar tissue thickness, but once ELASTicization is completed, half- centimeter-thick tissues can be stained within 1 d. Such rapid staining could be particularly useful for biological applications requiring multiple rounds of staining and imaging20. In addition, we found that the mechanical stability of ELAST enables cryo-sectioning of intact ELASTicized tissues into 3–10-μm-thick sections after immunofluorescence observation, allowing downstream histology such as hematoxylin and eosin staining (Extended Data Fig. 3).
We envision that ELAST will expedite scaling of tissue phenotyping approaches to larger systems by simultaneously overcoming two major bottlenecks: slow molecular labeling and low tissue integrity21,22. For instance, the lateral dimension of human organ slices is orders of magnitude larger than that of rodent organ sections, rendering human specimens more prone to mechanical damage than animal tissues with the same thickness. ELAST not only transforms tissue into a virtually indestructible platform, it also enables labeling thicker tissues at a faster rate. Together with the versatility and simplicity of the method, ELAST will facilitate investigation of higher animal models and clinical human samples.
online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41592–020-0823-y.
Methods
Reagents and reagent preparation.
Solutions for making pAAm hydrogels were prepared using AAm (A9099; MilliporeSigma), 2% N,N′-methylenebisacrylamide (MBAA) solution (161–0142; Bio-Rad Laboratories) and VA-044 (Wako Chemicals). A 10% (wt/vol) VA-044 stock solution was prepared in deionized (DI) water, aliquoted and kept at −20 °C. Frozen VA-044 stock aliquots were thawed and used only during the same day. For hydrogel experiments, the gel monomers and the VA-044 stock solution were directly dissolved in DI water as needed. For ELAST tissue preparation, an ELAST stock solution without VA-044 was prepared in DI water (0.1% volume was left to add the VA-044 stock solution later) with 30% (wt/vol) AAm, 0.0003% MBAA (using 2% solution), 0.2% (wt/vol) Triton X-100 (0694; VWR Scientific) using a 10% (wt/vol) stock solution in DI water and 1× PBS using Gibco 10× PBS (70011–044; Life Technologies). The ELAST stock solution was kept at 4 °C for up to 6 months and added by 0.1% volume of the VA-044 stock solution for each use to make a final ELAST solution with 0.01% (wt/vol) VA-044. Perfusion and Off solutions used for the stabilization underharsh conditions via intramolecular epoxide linkages to prevent degradataion (SHIELD) were prepared using polyglycerol 3-polyglycidyl ether (GE38) provided by CVC Thermoset Specialties of Emerald Performance Materials23. SHIELD perfusion solution was prepared with 2.5% (wt/vol) GE38 and 4% paraformaldehyde (PFA) using a 32% solution (15714-S; Electron Microscopy Sciences) in 0.1 M sodium phosphate (diluting 1 M sodium phosphate buffer (P2072; Teknova)) and 50 mM sodium chloride (diluting 5 M sodium chloride (S5845; Teknova)). A supernatant was collected and used by vortexing the solution for 1 min and repeating (1) centrifugation for 10 min at 4 °C and 7,200 relative centrifugal force (r.c.f.) and (2) collecting a supernatant until the supernatant was clear. SHIELD-Off solution was prepared by omitting PFA from the SHIELD perfusion solution. SHIELD-On solution was prepared by combining 2 ml of 1 M sodium carbonate (S7795; MilliporeSigma) with 0.02% (wt/vol) sodium azide, 2 ml of 1 M sodium bicarbonate (S5761; MilliporeSigma) with 0.02% (wt/vol) sodium azide and 36 ml of DI water. A clearing solution was prepared by dissolving 6% (wt/vol) SDS (75746; MilliporeSigma), 50 mM sodium sulfite (S0505; MilliporeSigma) and 0.02% (wt/vol) sodium azide (S2002; MilliporeSigma) in 0.1 M phosphate buffer, and the pH was adjusted to 7.4. Washing solutions were prepared as 0.1% (wt/vol) Triton X-100 in PBS (diluting 10× PBS (P0496; Teknova)) either with (PBST) or without (PBST without azide) 0.02% (wt/vol) sodium azide. PBS with 0.02% (wt/vol) sodium azide (PBS with azide) was also used as a washing solution. A blocking solution was prepared by adding 5% volume of normal goat serum (ab7481; Abcam) to PBST. See Supplementary Tables 2 and 3 for antibody and probe purchase and usage for mouse and human brain tissues. A 1,2-dimethoxyethane solution was prepared by mixing 1,2-dimethoxyethane (307432; MilliporeSigma) and PBST in a volume-to-volume ratio of 4:6. Antibody fixation solution was prepared in PBS with 4% PFA using a 32% solution and Gibco 10× PBS. Two types of RI-matching immersion media were used: (1) an immersion medium of a lower contraction power, named exPROTOS, and (2) an immersion medium of a higher contraction power, named dPROTOS. exPROTOS was prepared by dissolving 125 g iohexol, 3 g diatrizoic acid and 5 g N-methyl-d-glucamine in 100 ml of DI water, followed by RI adjustment to 1.458 by adding DI water, and then mixing 90% volume of the medium with 10% volume of 2,2′-thiodiethanol (16782; MilliporeSigma), which yielded a final RI of 1.465. dPROTOS (RI = 1.52) was prepared by mixing 150 ml of 2,2′-thioodiethanol, 150 ml of dimethyl sulfoxide and 200 ml of DI water, and dissolving 530 g of iohexol in the mixture. AAm, MBAA, 1,2-dimethoxyethane and PFA are toxic chemicals requiring caution and should be handled in a chemical hood.
Entangled hydrogel experiments.
All hydrogels were polymerized in 50-ml conical tubes (referred to as polymerization tubes). The common preparation sequence is as follows: (1) a polymerization tube was purged with a nitrogen gas (NI UHP300; Airgas East) at 80 kPa (at a regulator level) for 1 min; (2) a gel solution was put in the tube; (3) the tube was additionally purged with a nitrogen gas for 15 s and sealed using a screw cap connected to 12-psi nitrogen gas; and (4) the tube was vertically placed in a custom-built heating device. Gel solutions to confirm dense gel formation without MBAA were prepared in a fixed ratio between monomer and initiator concentrations. Gel solutions of 2 ml were prepared in 15-ml conical tubes and bubbled using a nitrogen gas-connected glass capillary (1B100F-4; World Precision Instruments) at a low gas pressure for 1 min. The solutions (1 ml) were transferred to polymerization tubes. Low-concentration (4% (wt/vol) AAm with and without 0.05% (wt/vol) MBAA) solutions with 0.04% (wt/vol) VA-044 were polymerized at room temperature (RT), and high-concentration (40% (wt/vol) AAm with and without 0.5% (wt/vol) MBAA) solutions with 0.4% (wt/vol) VA-044 were polymerized on ice to avoid runaway heat generation. The tubes were left for 1 d and photos were taken. To test compression, the top of each 15-ml conical tube was cut into a 2-cm-long cylinder, and the original top was superglued to a squared no. 1 coverslip (48393070; VWR Scientific). Gel solutions (40% (wt/vol) AAm and 0.005% (wt/vol) VA-044 with and without 0.5% (wt/vol) MBAA) of 2 ml were prepared in 15-ml conical tubes and nitrogen gas-bubbled for 1 min. The solutions (1.5 ml) were transferred to the cut cylinders. The cylinders were put in polymerization tubes. Polymerization was completed at 37 °C after about 15 min, and the tubes were kept overnight for stabilization. Gels of 9-mm thickness were collected, and each was placed on a vise jaw. Two fragments of 1-mm-thick slide glasses were left on the jaw as a thickness guide. The vice was tightened and then released as a compression demonstration. To test stretching, 2-ml gel solutions (5 M AAm and 0.005% (wt/vol) MBAA) were prepared, transferred to 1-ml syringes (SS-01T; Fisher Scientific) and nitrogen gas-bubbled for 1 min. Each syringe was sealed with Parafilm and covered with a Blu-Tack adhesive (Bostik) ball. The syringes were put in polymerization tubes filled with DI water and polymerized and stabilized at 35 °C for 1 d. The syringes were cut, and the gels were taken out and used for a stretching demonstration.
Diffusion experiment and modeling using hydrogels.
Two-millimeter-thick cartridges were made of two 1-mm-thick slide glasses spaced by supergluing four stacks of two slide glass fragments near the four edges. The lateral and bottom edges were taped, and further secured with superglue. A gel solution of 30% (wt/vol) AAm, 0.0003% (wt/vol) MBAA and 0.01% (wt/vol) VA-044 in DI water was poured into the cartridges and polymerized in polymerization tubes at 33 °C for 6 h. After swelling extracted gels in excess DI water at 37 °C for 3 d with daily exchange for fresh water, swollen 4-mm-thick gel slabs were cut into squares (2 × 2 cm2). Any top regions having less-swollen sizes were excluded. A stretching apparatus was made with four 56-mm-long, 1.6-mm-diameter stainless steel rods (cut from 1263K24; McMaster-Carr) and four 38-mm-long tube pieces cut from polypropylene 1,250-μl pipette tips having two holes in a 32-mm-long interval. Each of the squared gels was first pierced using 25-gauge syringe-needles (305125; BD) along the four lateral edges in 16-mm-long intervals, then the rods were inserted into these piercings and finally the gels were stretched with the aid of the pipette tip pieces. A micrometer (395–371-30; Mitutoyo America) was used to measure the thicknesses of unstretched gels and stretched gels after immersing in PBST for 22.5 min. Each thickness was measured three times, and the median was reported.
A 150-kDa fluorescein isothiocyanate (FITC)-dextran (FD150S; MilliporeSigma) stock solution was prepared at 10 mg ml−1 in DI water, aliquoted and frozen at −20 °C. An FITC-dextran solution in PBST was freshly prepared at 0.2 mg ml−1 using the stock solution. Control and stretched gels were incubated in 40-ml FITC-dextran solutions on a gentle shaker for 45 min at RT. The stretched gels were recovered by removing the apparatus immediately after the incubation. Each of the stained gels was mounted in an imaging chamber made of a slide glass, a WillCo dish (HBSB-5030; WillCo Wells) and two spacers using four slide glasses. The topmost 3-mm thickness of a central region (0.3 × 0.3 mm2) of the mounted gel was imaged using the Olympus FV1200MPE laser-scanning confocal microscope with a 10×, 0.3-numerical aperture (NA) water-immersion objective (UMPLFLN 10XW) and a 488-nm argon laser. Mounting and imaging were completed within 3 min. The fluorescence signals were subtracted by depth-wise background signals obtained from an unstained gel imaged in the same way.
A 1D transient diffusion model was used to fit the FITC-dextran distribution profiles:
where C(x, t) is the nondimensional relative FITC-dextran concentration at depth x at time t and Deff is the effective diffusivity. Because of symmetry, the lower half of the gel was assumed to have a mirror distribution of the upper half. The initial and boundary conditions for the partial differential equation are:
where T is the half-thickness of a gel. The equation was numerically solved using custom MATLAB (R2016a; MathWorks) scripts to derive Deff using least-squares curve fitting. The surface data points showing reduced concentration possibly due to regurgitation were excluded from the fitting and then reconstructed from the fitted solution for display. Obtained Deff values were converted to relative dimensionless diffusion timescale for a comparison.
Preparation of human brain samples.
Six coronal human brain hemisphere slabs (about 1-cm thickness) in a formalin solution were obtained from three autopsy brains at the Neuropathology Core of the Massachusetts Alzheimer Research Center. The brains were collected and banked in accordance with approval from the local Institutional Review Board. Subjects with no evidence of neurological disease on clinical grounds at the time of death and no evidence of notable disease processes upon full neuropathologic examination were selected. The formalin solution was replaced with PBS with azide upon arrival, and the slabs were stored at 4 °C until use. Whole-area coronal hemisphere slabs of 2-mm thickness were obtained by microtome-sectioning of the original thick slabs using a custom-built flexure-based vibrating-blade microtome. Each of the thick slabs was embedded in a 1.5% (wt/vol) agarose gel in PBS, where any gel on the bottom surface was removed, and superglued on a glass plate. The glass plate was superglued on a microtome chamber, and the chamber was filled with PBS. A frequency of 60 Hz and a blade propagation speed of 0.3 mm s−1 were used. For large tissue blocks used for compression testing and thick-tissue labeling, cortical regions were manually cut using a razor blade to include a thickness of up to 5 mm from the pial surface. For measurement of tissue size changes, 2–3-mm-thick cortical blocks were obtained by manual cutting. Horizontal tissue samples of 1-mm thickness used for antibody delivery comparison and stretching-related deformation analysis were obtained by slicing with a vibrating-blade microtome (VT1200; Leica Biosystems) of several-millimeters-thick horizontal cortical blocks that were manually cut. The pial surface region (200–300-μm thickness) of each sample was trimmed off. The obtained 1-mm-thick samples were manually cut into square pieces (2 × 2 mm2). A coronal slice having an original thickness of about 1.5 mm was used for the antibody compatibility test.
ELASTicization of archived human brain specimens.
The long-term formalin-fixed human tissue samples were first permeabilized using clearing solution, for which a 70 °C overnight step was followed by a 56 °C step, if any. See Supplementary Table 1 for details of each experiment. Permeabilized samples were washed with PBST without azide. At this point, it is important to ensure the removal of most of the azide and sulfite from the tissues, because these chemicals quench radicals during AAm polymerization and impede gel formation. Samples were then incubated in an ELAST solution at 4 °C overnight (1-mm-thick samples), for 5 d (5-mm-thick samples) or for 8–9 d (2-mm-thick slabs). Other than for large-area slabs, polymerization cartridges were prepared using two 1-mm-thick slide glasses spaced at four edges by superglued stacks of slide glass and coverslip fragments that matched the thickness of each sample in the ELAST solution. After taping with transparent plastic tape along lateral and bottom edges and then tailoring and supergluing the tape, samples were inserted into the cartridges. The space in each cartridge was filled carefully with the same ELAST solution used to incubate the tissues, ensuring that there were no apparent bubbles. The cartridges were put in 50-ml conical tubes that were prepurged with a nitrogen gas at 12 psi for 1 min. The tubes were then additionally purged for 15 s and sealed with screw caps connected to a nitrogen gas at 12 psi. Samples were polymerized at 20–37 °C for 6 h. In a cold environment, each 2-mm-thick hemisphere slab incubated in ELAST solution was placed on a glass plate (260232; Ted Pella) having superglued 2-mm-thick spacers (stacks of two slide glass fragments) at four corners of the plate. Another glass plate was first placed on top of the sample at a height of 3 mm, guided by two stacks of three slide glasses. Then, the space above the sample was filled with the same solution, and the cover plate was lowered to 2-mm height. The glass plates were taped, leaving a small opening at one corner, and sealed using a superglue, which was cured at 4 °C. The cartridge space was filled with the same solution using a 20-ml needle-syringe while avoiding bubble-trapping. The cartridge was placed in a plastic bag with a zipper lock. The bag was purged with nitrogen gas at 12 psi for 10 min, closed, placed in a water bath where the temperature was maintained at 33 °C, and polymerized and stabilized overnight. Embedded samples were cut from the gels using a razor blade to have adequate gel margins and cleared in clearing solution at 37 °C overnight (1-mm-thick samples) or at 56 °C for 5 d (5-mm-thick samples) or 2 d (2-mm-thick slabs, in a 500-ml glass bottle) with exchange of the solution every 1 or 2 d. Fully cleared samples were washed with PBST at 37 °C for 6 h (1-mm-thick samples) or 2–3 d (other samples) with exchange of the solution two or three times. Gels containing samples for the antibody delivery speed comparison were cut into squares (6 × 6 mm2) using a razor blade so as to have one tissue sample for each square at the center. Thick samples used for compression testing and thick-tissue labeling were carefully isolated from the surrounding gels.
Preparation and ELASTicization of mouse tissue samples.
All experimental protocols were approved by the Massachusetts Institute of Technology (MIT) Institutional Animal Care and Use Committee and the Division of Comparative Medicine and were in accordance with guidelines from the National Institutes of Health. All experiments using mice were conducted in strict adherence to the ethical regulations of MIT Institutional Animal Care and Use Committee and the Division of Comparative Medicine. Thy1-GFP-M transgenic mice (2–4 months old, male and female) were housed in a 12-h light/dark cycle with unrestricted access to food and water. Mice were transcardially perfused with 20 ml of ice-cold PBS followed by 20 ml of ice-cold SHIELD perfusion solution at a flow rate of 5 ml min−1. Organs were extracted and incubated in 15–20 ml of the same perfusion solution at 4 °C for 48 h with shaking. The organs were transferred to 20 ml of prechilled SHIELD-Off solution and incubated at 4 °C for 24 h with shaking. The organs were placed in 40 ml of SHIELD-On solution and incubated at 37 °C for 24 h. SHIELD-fixed organs were washed with PBS with azide at RT overnight. Brains were split into hemispheres using a razor blade. The brain hemispheres and other organs were delipidated passively in clearing solution at 37 °C for 12–14 d, 45 °C for 10 d, or 45 °C for 2 d and 56 °C for 3 d until the opaque core disappeared. Cleared tissues were washed with 50 ml of PBST without azide or with 0.02% (wt/vol) thimerosal (for in situ hybridization; T5125; MilliporeSigma) at 37 °C for 2–3 d with three to four solution exchanges. The tissues were incubated in ELAST solution at 4 °C for 3–5 d with daily solution exchange. One or two brain hemispheres or other organs were inserted into a polymerization cartridge, and polymerization was performed at 20–37 °C for 6 h. Embedded samples were cleared in clearing solution at 37 °C for 5–9 d or 56 °C for 4 d with solution exchange every 2 d, and then washed with PBST at 37 °C for 2–4 d with daily solution exchange. Surrounding gels were carefully removed from the samples during or after washing. The 1-mm-thick mouse coronal brain hemisphere slices used for demonstrations of stretching, measurement of size changes and antibody screening were prepared by embedding microtome-sectioned 1-mm-thick slices. See Supplementary Table 1 for the detailed protocol for each of the mouse samples in the current study.
Thinning of ELASTicized tissue samples.
A 4.5-mm-thick ELASTicized human sample used for the compression demonstration was placed on a 15 cm (width) × 15 cm (height) × 1.3 cm (thickness) glass plate (CG-1904–18; Chemglass Life Sciences) together with two spacers of approximately 0.42-mm thickness using stacks of three no. 1 coverslips at two lateral edges of the plate. The sample was covered with another plate and photos were taken before and after compression. The thickness of the coverslip stacks was the target final compressive thickness of the sample. Mouse organs and cerebral organoids used for the compression demonstration were tested similarly, but spacers were not used. ELASTicized mouse brain samples for the quantification of compression-related deformation were obtained from an ELASTicized mouse brain hemisphere: one hemisphere was manually cut using a razor blade to obtain a 3-mm-thick block, and this block was microtome-sliced to get a 1.4-mm-thick slice; the slice was chopped into several pieces using a razor blade. Obtained pieces were compressed between two glass plates with dimensions of 75 mm (width) × 51 mm (height) × 1.2 mm (thickness) (71862–01, Electron Microscopy Sciences). A no. 1 coverslip was placed on each of the lateral edges of the bottom glass plate to guide a tenfold compression. Samples were compressed ten times for at least 1 min, and photos were taken before and after each compression. The mouse brain samples to measure the compressed area were obtained by chopping an ELASTicized 1-mm-thick brain slice. The samples having a thickness greater than 2 mm were first compressed to 2 mm between two 1.2-mm-thick glass plates with two stacks of two slide glasses as spacers to ensure all top and bottom surface regions were in contact with the plates. After taking photos, the samples were further compressed to 1 mm by reducing the heights of the spacers. After taking photos again, compressed areas were measured using ImageJ (National Institutes of Health) and compared between 2-mm and 1-mm compressions.
A stretching apparatus was made with four 14.5-mm-long needles cut from 25-gauge syringe-needles and 10.5-mm-long tube pieces cut from polypropylene 10-μl pipette tips having two holes in an 8-mm-long interval. ELASTicized 1-mm-thick human samples were pierced by the needles along four lateral edges in an 8-mm-long interval and stretched with the aid of the pipette tip pieces. Samples for the quantification of stretching-related deformation were stretched overnight at 37 °C. Photos were taken before stretching, after overnight stretching and after >2-h recovery. The thicknesses of ELASTicized 1-mm-thick human samples were measured by a micrometer three times, and the median was chosen for each sample. The thicknesses of stretched samples were measured after stretching followed by incubation in 2 ml of PBST at RT for 3 h. The thicknesses of contracted samples were measured after incubation in a 10-ml 1,2-dimethoxyethane solution at 37 °C overnight and then at RT for 6 h for a full incubation to dismiss a transient opaqueness. After incubating stretched samples in a 5-ml 1,2-dimethoxyethane solution at RT for 1 h, exchanging the solution, and incubating at 37 °C overnight and then at RT for 1 h, the thicknesses of the stretched and contracted samples were measured.
Labeling of thin ELASTicized brain samples.
For the antibody compatibility test with human brain tissue, an approximately 1.5-mm-thick human brain coronal slice was ELASTicized and then microtome-sliced to obtain 200-μm-thick sections. A slow blade propagation speed (0.1 mm s−1) and a high amplitude (1 mm) were used to slice the elastic sample. The sections were cut into small pieces (about 2 × 2 mm2) and put in a 24-well plate. Samples were incubated in PBST with primary antibodies overnight, washed with PBST three times for 2 h each, incubated in PBST with secondary antibodies overnight and washed with PBST twice for 2 h each. The well plate was placed on a gentle shaker at 37 °C during the procedure.
For the antibody compatibility test with mouse brain tissue, 1-mm-thick SHIELD-protected mouse brain hemisphere slices were used. Control samples were first vibratome-sliced into 100-μm-thick sections at this step. Both 100-μm-thick control samples and 1-mm-thick samples to be ELASTicized were delipidated in clearing solution (200 mM SDS in pH 9 borate buffer) for the same amount of time (1.5 d at 45 °C). One-millimeter-thick samples were ELASTicized, vibratome-sliced into originally 100-μm-thick sections, additionally delipidated for 8 h at 37 °C, washed at 37 °C overnight and incubated in blocking solution at 37 °C for 3 h. Small pieces were obtained from both groups of samples, and each pair was immunostained separately at 37 °C: primary antibody (1:300) overnight, washing with PBST three times for 8 h, secondary antibody (1:300) overnight and washing with PBST three times for 6 h. Lectin (1:300) was added at the primary antibody staining step. DAPI (D1306; Life Technologies) was dissolved in DI water at a concentration of 5 mg ml−1, and this stock solution was used at a 1:10,000 dilution during the second time of washing after secondary antibody staining. Dil (D282; Life Technologies) was prepared at a concentration of 2.5 mg ml−1, and the solution was used at a 1:1,000 dilution at the secondary antibody staining step.
For the antibody delivery speed comparison, ELASTicized 1-mm-thick human samples were stained with a 10-μl anti-glial fibrillary acidic protein (GFAP) antibody conjugated with Alexa Fluor 594 in a 50-ml conical tube with 1 ml of PBST on a gentle shaker at RT for 30 min. Stretched samples were put in the antibody solution immediately after stretching. Stretched and contracted samples were prepared by incubating stretched samples in a 3-ml 1,2-dimethoxyethane solution at RT for 30 min (exchanging the solution once after 10 min). After exchanging again with fresh 1-ml 1,2-dimethoxyethane solution, antibody was added. Contracted samples were prepared by incubating samples in a 3-ml 1,2-dimethoxyethane solution at 37 °C overnight (exchanging once) and then at RT for 3 h. Antibody-stained samples were washed with PBST at RT for 30 min. All stretched samples were recovered from stretching and left in PBST for 30 min before imaging.
To compare cyclic compression with other methods in regard to antibody delivery speed in the same setting, a manual cyclic compression setting was used. A plastic film bag of about 6 cm (height) by 2 cm (width) was made using an impulse heat sealer. After putting a sample into the bottom of the bag, a horizontal midline was sealed leaving a small hole at one side. PBST (1 ml) was added to the bottom chamber, and 10 μl of anti-GFAP antibody was added to the top chamber. The bag was then completely sealed, and the lower chamber area was placed between two slide glasses with spacers at two sides that matched a whole thickness of the bag with a sample compressed by sixfold. A manual cyclic compression was applied for 30 min by repeating a compression for 50 s that pushed the solution out to the upper chamber and a release for 10 s by compressing the upper chamber. After staining, the bag was cut with a razor blade, and the sample was taken out and washed with PBST for 2 h before imaging.
One ELASTicized 1-mm-thick sample was used to test the compatibility of thinning methods with cell-type and projection markers. The sample was stretched twofold laterally and then antibody-stained. A 1,2-dimethoxyethane solution (35:65 volume ratio) was used for anti-neurofilament light chain antibody, and other primary and secondary antibodies were stained in PBST.
Thick-tissue immunolabeling with cyclic compression.
An ELASTicized 5-mm-thick human sample (lateral dimensions of 12 and 7 mm) was photobleached to remove autofluorescence from lipofuscin pigments. A photobleaching device was built by wrapping a 1-l glass beaker with a strip of white light-emitting diodes (Ledmo SMD5630; Shenzhen Xiangliang Lighting). The sample in PBST was placed in the center of the horizontally positioned device at 4 °C for 3 d, then preblocked in a blocking solution at 37 °C for 1 d on a gentle shaker and then kept in the same solution until antibody-staining. A cyclic compression device was custom-built using a Nema 23 bipolar stepper motor (23HS16–0084S; OSM Technology) connected to a Raspberry Pi 3 B+ motherboard (Raspberry Pi Foundation) with a dedicated stepper motor controller (2348; Adafruit). The axial movement part of the device consisted of a 60-mm cage system (two LCP02 and one LCP03; Thorlabs) and custom-made polycarbonate plates. One plate was fixed in place while the other was driven with a leadscrew cut from an M6-threaded rod (1078N11; McMaster-Carr), which was coupled to the shaft of the stepper motor. A sample container bag with a width of 5.5 cm and height of 15 cm was made of an ultrahigh-molecular-weight polyethylene film (85655K12; McMaster-Carr) and shaped using an impulse heat sealer. To minimize sticking of samples to the walls during a flushing cycle, the bag was vertically grooved in a 5-mm interval by alternative folding. Two films of 5.5-cm height were fused with the bag vertically along the midline using the heat sealer so as to provide wings for attaching the bag to the chamber plates. The bag was inserted into the movement chamber of the device in a 37 °C environment, and the four wings of the bag were fastened and then attached to the chamber plates using tape. The sample was put into the bag with 5 ml of blocking solution, and 50 μl of antibody was added. Compression and relaxation movements operated by the stepper motor and leadscrew were remotely controlled using a Python script. For an 8-mm-thick swollen sample, the chamber was operated with a travel distance of 10 mm (starting at an 11.25-mm height and compressing to a 1.25-mm height). The travel speed was set to 2 mm s−1, and the cycling time intervals were set to 40 s for compression and acceleration and 10 s for relaxation and flushing (by means of upward and downward movements). After antibody incubation for 1 d, the sample was washed with PBST at 37 °C overnight on a gentle shaker. Before RI-matching, the antibody in the sample was fixed by incubating in an antibody fixation solution at 37 °C for 6 h and washing with PBS with azide twice for 2 h each at 37 °C.
RI-matching.
The whole-area human brain hemisphere slabs of 2-mm thickness, the immunostained 5-mm-thick human sample, the 1.4-mm-thick mouse samples for the compression test and the originally 3-mm-thick mouse samples for the cyclic compression test were optically cleared by RI-matching. The 2-mm-thick slabs were incubated in 100–150 ml of exPROTOS in a 250-ml glass bottle at 37 °C for 1 d, exchanging the medium twice. Other samples, including the immunostained thick sample, the 1.4-mm-thick ELASTicized mouse brain samples and the ELASTicized 3-mm-thick mouse brain samples, were RI-matched using dPROTOS. Samples were incubated at 37 °C according to the following sequences: for the 5-mm-thick human sample, 10 ml of 50% medium (mixed with DI water) for 6 h, 10 ml of 100% medium overnight and fresh 10 ml of medium for 6 h; and for the 1.4-mm-thick samples, 1.5 ml of 50% medium for 2 h and 1.5 ml of 100% medium overnight. Other samples that were microscopically imaged were not RI-matched due to sufficient transparency and/or unnecessity of imaging deep regions. Mouse and human brain tissue samples to measure their size changes were first incubated in dPROTOS at 37 °C for 1 d, and photos were taken. The same procedure was repeated with exPROTOS.
Microscopic imaging of ELASTicized tissues.
For the antibody delivery speed comparison, two strips of Blu-Tack adhesive were placed on a slide glass, and each of the 1-mm-thick samples was put between the strips. A squared no. 1 coverslip was placed and gently compressed to fit the sample thickness. About 100 μl of PBST was added to prevent the sample from drying. The samples were imaged using the Olympus FV1200MPE laser-scanning confocal microscope with a 20×, 1.0-NA water-immersion objective (XLUMPLFLN 20XW) and a 559-nm diode laser. Central tissue locations were chosen to avoid misinterpretation by lateral diffusion, and longitudinal regions (8:1) were imaged along the whole depth with a constant laser power and gain parameter and then projected to xz planes using Imaris software (Bitplane). For labeling and imaging of the 5-mm-thick human sample, a Blu-Tack adhesive strip was placed on a 60-mm-diameter petri dish (351007; Corning) along the sample boundary, and three Blu-Tack adhesive balls were put along the dish boundary as spacers. The sample incubated in the RI-matching medium was placed on the dish and covered by a WillCo dish with no bubble-trapping. The WillCo dish was gently compressed to match the sample thickness, and the space between the two dishes was filled with the same medium. The gap between the two dishes exposed to the air was sealed with a long Blu-Tack strip. The mounted sample was left at 37 °C for 2 h for RI stabilization. The sample was imaged using the same Olympus microscope with a 10×, 0.6-NA CLARITY-optimized objective (XLPLN10XSVMP, 8.0-mm working distance) and a 559-nm diode laser. The objective was immersed in the same RI-matching medium poured in the WillCo dish, and imaging was started 30 min after installation to stabilize the RI around the objective. A central tissue region was imaged along the whole depth with a constant laser power and gain parameter. The imaged volume was three-dimensionally rendered using the Imaris software, and a video showing axial navigation was generated using ImageJ. The thin sections used for the antibody compatibility test were mounted on slide glasses with Blu-Tack adhesive balls at four corners and covered with no. 1 coverslips. The space near the samples was filled with PBST. Human tissue sections were imaged using the Leica TCS SP8 laser-scanning confocal microscope with a white-light laser source for excitations at 488 and 647 nm. A 63×, 1.3-NA glycerol-immersion objective (HC PL APO CS2) was used for the samples stained with anti-synapsin 1/2 and anti-PSD-95 antibodies, and a 25×, 0.95-NA water-immersion objective (HCX IRAPO L) was used for other samples. Mouse tissue sections were imaged using the Olympus microscope with a 20×, 1.0-NA water-immersion objective.
Fluorescence in situ hybridization (FISH).
Thy1-GFP-M mouse SHIELD-ELAST brain hemisphere was used. All clearing steps were performed at 37 °C instead of 45 °C. A SHIELD-ELAST coronal hemisphere tissue with 3-mm thickness was prepared by vibratome-slicing of the hemisphere. The slice was split into 2 mm × 1 cm × 3 mm blocks, and A block was incubated in the prehybridization buffer containing 2× SSC, 30% formamide, 10% dextran sulfate (average molecular weight, 500 kDa; D8906; MilliporeSigma) and 1 mg ml−1 denatured/snap-cooled salmon sperm DNA (D7656; MilliporeSigma) at 37 °C for 1 h. Then the FISH probes with 3′ hybridization chain reaction (HCR) initiator overhangs (B1-YFP, 20 nucleotides; B5-vGluT1, 35 nucleotides) were added at final concentration of 1 nM and further incubated at 37 °C overnight23. The tissue block was washed by 2× SSCT (0.1% triton X-100), 30% formamide buffer at 37 °C for 1 h, three times. Additional 2× SSCT washing for 1 h, two times at RT was performed to remove any residual formamide. The tissue block was then immersed in the amplification buffer containing 2× SSC, 10% dextran sulfate buffer for 1 h. While pre-incubating, aliquots of HCR hairpins (B1-Alexa Fluor 647, B5-Alexa Fluor 568) were heat-denatured at 95 °C for 90 s, and snap-cooled in ice at least for 15 min. Cooled HCR hairpins were added to the amplification buffer at the final concentration of 120 nM and the tissue was incubated at RT overnight. After the HCR process was finished, the tissue was washed by 2× SSCT for 1 h, three times at RT and prepared for imaging by immersing in 2× SSCT. The sample was imaged using the Leica microscope with a 25×, 0.95-NA water-immersion objective.
ELASTicization and labeling of cerebral organoids.
Organoids were grown according to the protocol by Lancaster and Knoblich24, with the addition of dual SMAD inhibition between day 6 and day 9 to drive neural differentiation, as previously described25. Organoids were fixed after 35 d of culture with 4% PFA. Organoids were rinsed in PBS three times, and transferred into ice-cold 2% SHIELD solution with 100 mM phosphate buffer (pH 7.2) for 2 d. Organoids were subsequently transferred into 100 mM sodium carbonate buffer (pH 10) and incubated at 37 °C for 1 d. Organoids were extensively washed with PBS then cleared in 0.2 M SDS in 50 mM phosphate buffer (pH 7.3) for 48 h at 55 °C. Organoids were washed with PBST without azide at 37 °C for 1 d, and were incubated in ELAST solution at 4 °C for 1 d. Four organoids were inserted into a polymerization cartridge, and polymerization was performed at 35 °C for 6 h. The organoids were delipidated passively at 37 °C for 3 d, and then washed in PBST at 37 °C for 1 d.
An ELASTicized organoid was incubated at 37 °C in blocking solution for 3 h using cyclic compression (36-s wait time, 9-s flushing time), then stained using cyclic fourfold compression with 15 μg of DAPI, 10 μg of anti-MAP2 antibody conjugated with Alexa Fluor 594 (677807; BioLegend) and 4 μg of anti-vimentin antibody conjugated with Alexa Fluor 647 (801803; BioLegend) in 1 ml of blocking solution for 12 h at 37 °C, and finally washed with cyclic compression in PBST for 12 h at 37 °C. A ~200-μm-thick depth-wise cross-section was cut from the organoid using two parallel, adjacent steel razor blades, making sure to cut through a laterally central section of the organoid to interrogate the most inaccessible core of the organoid. Depth images of the labeled organoid were acquired using the Leica confocal microscope with ultraviolet (405 nm) and white-light lasers (594 and 647 nm) with a 25×, 0.95-NA water-immersion objective.
Quantification of thinning-related deformation.
ELASTicized human samples of original 1-mm thickness were stained with anti-GFAP antibody conjugated with Alexa Fluor 594 and tomato lectin conjugated with DyLight 488. The samples were mounted in the same manner as the samples used for the antibody delivery speed comparison and left under the same microscope setting for 0.5–2 h until all noticeable sample movement had ceased. A 100-μm surface depth of each sample was imaged with a 2-μm z-step size using the Olympus microscope with a 20×, 1.0-NA water-immersion objective, a 488-nm argon laser and a 559-nm diode laser. The imaged samples were stretched using the stretching apparatus and left in PBST at 37 °C overnight. The samples were then released from stretching and recovered in PBST at RT for 1–2 h. The samples were mounted, stabilized and imaged in the same way. The GFAP channel was used for quantitative analysis using a method described previously4. In brief, GFAP filament features of each sample were detected in three dimensions and corresponded between two images obtained before and after stretching. The correspondence information was used to generate a regularly spaced three-dimensional deformation mesh. The deformation-related distance change between each pair of mesh points was measured as an error. For each of the measurement distances, the square root of the average of squared errors was collected from each tissue sample.
ELASTicized mouse samples in 1.4-mm thickness used for the compression test were imaged before compression and after recovery. Before compression, samples were RI-matched and enhanced green fluorescent protein (EGFP) signals were imaged using the Olympus microscope with a 20×, 1.0-NA water-immersion objective and a 488-nm argon laser. Images were three-dimensionally rendered by the Imaris software for display. Imaged samples were then recovered in PBST and subjected to the compression test while photos were taken. After the test, the samples were RI-matched and imaged again. Obtained EGFP images before and after compression were quantitatively analyzed using the same method for the stretching test with human samples and GFAP images. ELASTicized 3-mm-thick mouse samples were obtained from an ELASTicized brain hemisphere by manual slicing and chopping with a razor blade and used for the cyclic compression test in the same manner. The samples were RI-matched and imaged before and after cycles of 42-s 5–6-fold compression and 12-s release at RT over approximately 1 d. The Olympus microscope was used with a 10×, 0.6-NA CLARITY-optimized objective.
Comparisons of depth-wise immunoreactivity.
To compare compression versus passive GFAP staining, three 1.5-mm-thick ELASTicized human tissue blocks were pre-incubated passively at 37 °C for 6 h in blocking solution, and then stained using cyclic fivefold compression (40-s wait time, 10-s flushing time) with 10 μg of anti-GFAP antibody conjugated with Alexa Fluor 488 (clone 2E1; 644704; BioLegend) in 2 ml of blocking solution for 6 h at RT. The tissues were then washed passively using PBST for 3 h at 37 °C, after which they were cut vertically in the middle, exposing the cut surfaces. The cut surfaces were passively stained using 1.5 μg of anti-GFAP antibody conjugated with Alexa Fluor 594 (clone GA5; 8152; Cell Signaling Technologies) in 300 μl of PBST for 3 h at 37 °C, and then washed passively in PBST for 1 h at 37 °C. To compare passive versus passive GFAP staining, three 1.5-mm-thick ELASTicized human tissue blocks were cut vertically in the middle, and then pre-incubated passively at 37 °C for 6 h in blocking solution. The cut surfaces were passively stained with 1.5 μg of anti-GFAP antibody conjugated with Alexa Fluor 488 (clone 2E1) and 1.5 μg of anti-GFAP antibody conjugated with Alexa Fluor 594 (clone GA5) in 300 μl of PBST for 3 h at 37 °C, and then washed in PBST for 1 h at 37 °C. ‘Noise’ group images (used to compare signal intensity agreement between noise and the passive 594 channel) were generated by drawing pixel values from a uniform distribution from 0 to 65,535. To compare mouse anti-GFP versus EGFP, an ELASTicized 1-mm-thick Thy1-eGFP mouse coronal hemisphere tissue block was pre-incubated passively at 37 °C for 6 h in blocking solution, and then stained using cyclic fivefold compression (40-s wait time, 10-s flushing time) with 10 μl of chicken anti-GFP antibody (A10262; Invitrogen) in 3 ml of blocking solution for 3 h at 37 °C. After 3 h of cyclic compression washing in blocking solution (with two solution changes) at 37 °C, the sample was secondary-stained using cyclic compression for 3 h at 37 °C with 10 μl of goat anti-chicken IgY antibody conjugated with Alexa Fluor 647 (ab150175; Abcam) in 3 ml of blocking solution, followed by cyclic compression washing in PBST for 1 h at 37 °C. All samples’ cut surfaces were then imaged using a 20×, 0.5-NA water-immersion objective with the Leica TCS SP8 confocal microscope with a white-light laser source for 488-nm and 594-nm wavelengths.
Each z-stack image was post-processed with histogram equalization (using the stack histogram, 0.3% pixels saturated) using ImageJ. The depth-wise signal intensity agreement (Ak) of each yz-plane image optical section at a given axial position k was computed with:
where N and M are the number of pixels in the two lateral dimensions of each image plane, and Iλ,ijk represents the signal intensity of the λ wavelength image channel at lateral pixel position (i,j) and axial pixel position k. The depth-wise coefficient of variation (CoV) was computed with:
where the mean and standard deviation are computed by:
where Z is the number of pixels in the depth-wise direction. The upper and lower bounds of the 95% confidence intervals were computed with ±2 s.d. from the depth-wise mean signal agreement between the three samples in each group. To compute the statistical significance between each group, two-tailed Student’s t-tests were run using the depth-wise mean signal intensity agreements of each group as sample observations. All analyses were performed using custom scripts in numpy, scipy and matplotlib packages in Python 3.6 (https://www.python.org/).
Downstream histology of ELASTicized tissue.
A block (approximate lateral dimensions of 2 × 2 mm2) from an ELASTicized 1-mm-thick mouse brain hemisphere slice was first immunostained as follows: blocking at 37 °C for 6 h, 6 μg of rabbit anti-NeuN antibody (ab104225; Abcam) in 500 μl of blocking solution at 37 °C for 2 d, washing with PBST three times at 37 °C overnight, 12 μg of donkey anti-rabbit IgG antibody conjugated with Alexa Fluor Plus 594 (A32754; Life Technologies) in 500 μl of PBST at 37 °C overnight and washing with PBST three times at 37 °C for 8 h. A 200-μm-thickness volume of the stained sample was imaged with the Zeiss LSM 780 confocal microscope with a 10×, 0.45-NA dry objective and a 561-nm diode laser.
After confocal fluorescent microscopic imaging, the sample was cryopreserved in 15% sucrose (S0389; MilliporeSigma) in PBS until sinking followed by 30% sucrose in PBS overnight. Sections of varying thicknesses from 3 to 30 μm (original thickness) were obtained by cryosectioning with the Leica CM1850. Originally 10-μm-thick sections were chosen for the following histological staining, placed on slide glasses, spread with a paint brush and air-dried. The sections were stained as follows: Harris hematoxylin solution (20022; Muto Pure Chemicals) for 10 min, washing carefully with DI water by pipetting, a drop of 1% hydrochloric acid (7647–01-0; Junsei Chemical), washing with DI water and 0.5% eosin Y solution (102439; Merck) for 2 min. The sections were then dehydrated with a series of ethanol solutions, dipped in xylene and mounted on slide glasses with a Dako mounting medium (CS70330; Agilent Technologies). The samples were imaged using the Leica DM6000 B bright-field microscope with a 2.5×, 0.07-NA dry objective.
Statistics and reproducibility.
Sample numbers and replication types are described in figure legends, where all replicates are either different hydrogels or different tissue samples. For the thickness measurement of hydrogels, the thicknesses of four gels were measured before and after stretching and analyzed using a two-tailed paired t-test. For the comparison of diffusions in hydrogels, eight gels were randomly assigned to either control or stretching group. An effective diffusion coefficient was computed from each gel, and the resulting diffusion timescales were compared using a two-tailed unpaired t-test. For the thickness measurement of ELASTicized human brain samples, ten samples were collected and their thicknesses were measured. The samples were then split into two groups of stretching and contraction-only so as to have average thicknesses as close as possible between the two groups. Thicknesses of stretched samples were measured after stretching and then after contraction, and thicknesses of after contraction. One-way analysis of variance was used to compare the thicknesses among the four groups. A post hoc Tukey’s test was performed to compare thicknesses between each pair of groups. Five, five and four samples were prepared for deformation analysis with stretching, manual compression and cyclic compression, respectively, and images obtained before and after stretching or compression were used for error quantification. Tissue size changes by ELASTicization and RI-matching with different media were measured with six mouse samples and four human samples. A tissue size of each sample was estimated as a square root of the product of two crossing lines connecting representative points. The margins of surrounding gels were instead measured for transparent human samples after RI-matching. For the compressed area measurement experiment, four samples were tested. Most of the other experiments, such as the antibody validation, the antibody delivery speed comparison, stretching and RI-matching of large-area slabs, compression of thick samples, depth-wise comparison of EGFP signal and anti-GFP immunoreactivity by cyclic compression, and downstream histology, were carried out at least twice with the same results. The thick-tissue immunolabeling with a 5-mm-thick human brain sample, compression of various mouse organs and immunolabeling of a cerebral organoid were performed once.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Dense entangled pAAm hydrogels that are elastic, tough, and stretchable.

a, High-concentration acrylamide (AAm) solution forms an entangled hydrogel in the absence of cross-linker, N, N′-methylenebisacrylamide (MBAA). b, A compression test on 40% (wt/vol) pAAm hydrogels with (upper panels) and without (lower panels) 0.5% (wt/vol) MBAA. The gels were prepared with 0.005% (wt/vol) VA-044 and nitrogen gas-bubbling. Gels with 9-mm thickness were compressed to 1 mm. λ, compression strain. c, A demonstration of stretching a 36% (wt/vol) entangled pAAm gel (no MBAA and 0.005% (wt/vol) VA-044 with nitrogen gas-bubbling). Representative images are shown from experiments repeated two times (a and b) and three times (c) with similar results.
Extended Data Fig. 2 |. ELASTicization and compression of various mouse organs and a cerebral organoid.

a, Compression of ELASTicized mouse organs. Samples were manually compressed between two glass plates. The experiment was not repeated. b, Compression of an ELASTicized cerebral organoid. Scale bars, 5 mm. Experiment was repeated two times with similar results.
Extended Data Fig. 3 |. Downstream histology of ELASTicized tissue.

a, An ELASTicized 1-mm-thick mouse brain slice block was immunolabeled with an anti-NeuN antibody and imaged by confocal fluorescence microscopy. b, The same sample in a was then cryo-sectioned into 10-μm-thick sections. One section was histologically stained with hematoxylin and eosin with following a series of dehydration steps using ethanol solutions and xylene. The image was obtained by a bright-field microscope. Note that the image planes were not intended to match. Scale bar (adjusted to match the original tissue dimensions), 200 μm. The hematoxylin and eosin staining experiment in b was repeated six times with similar results using two NeuN-stained samples as in a.
Supplementary Material
Acknowledgements
We thank the entire Chung Laboratory for support and discussions. We thank S.-C. Chen and J. Wang for the development of the vibrating-blade microtome, and D. H. Yun for its operation. We also thank K. Xie for mouse sample preparation, Y. Tian for the initial diffusion modeling work and R. Thorndike-Breeze for comments on improving the manuscript. We thank H. Jung and H. Kim for providing the histology facility and helping the H&E experiment. K.C. was supported by the Burroughs Wellcome Fund Career Awards at the Scientific Interface, the Searle Scholars Program, the Packard Award in Science and Engineering, the NARSAD Young Investigator Award, the Institute for Basic Science grant no. IBS-R026-D1 and the McKnight Foundation Technology Award. W.G. was supported by the National Science Foundation Graduate Research Fellowship under grant no. 1122374. A.A. was supported by the JPB Foundation (Picower Fellowship). M.P.F. was partially supported by NIA grant no. P50 AG005134. This work was supported by the JPB Foundation (grant nos. PIIF and PNDRF), the NCSOFT Cultural Foundation, and the NIH (grant nos. 1-DP2-ES027992, U01MH117072).
Footnotes
Competing interests
The ELAST concepts and applications are covered in a pending patent application owned by MIT (K.C. and T.K.). K.C. is a cofounder of LifeCanvas Technologies, a startup that provides solutions for 3D tissue processing.
Data availability
All data supporting the findings of this study are included in figures and videos as representative images or data points in the plots. Additional images other than the representative images are available from the corresponding author upon reasonable request. Resources that may help enable general users to establish the methodology are freely available online (http://www.chunglabresources.org).
Code availability
The custom code used in this study is available from the corresponding author upon reasonable request.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41592-020-0823-y.
Supplementary information is available for this paper at https://doi.org/10.1038/s41592-020-0823-y.
Peer review information Nina Vogt was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
References
- 1.Chung K. et al. Structural and molecular interrogation of intact biological systems. Nature 497, 332–337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen F, Tillberg PW & Boyden ES Expansion microscopy. Science 347, 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sylwestrak EL, Rajasethupathy P, Wright MA, Jaffe A & Deisseroth K. Multiplexed intact-tissue transcriptional analysis at cellular resolution. Cell 164, 792–804 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ku T. et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat. Biotechnol 34, 973–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X. et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ueda HR et al. Tissue clearing and its applications in neuroscience. Nat. Rev. Neurosci 21, 61–79 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Chen F. et al. Nanoscale imaging of RNA with expansion microscopy. Nat. Methods 13, 679–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park H-E et al. Scalable and isotropic expansion of tissues with simply tunable expansion ratio. Adv. Sci 6, 1901673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang YS & Khademhosseini A. Advances in engineering hydrogels. Science 356, eaaf3627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okumura Y & Ito K. The polyrotaxane gel: a topological gel by figure-of-eight cross-links. Adv. Mater 13, 485–487 (2001). [Google Scholar]
- 11.Zhong MJ, Wang R, Kawamoto K, Olsen BD & Johnson JA Quantifying the impact of molecular defects on polymer network elasticity. Science 353, 1264–1268 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Gong JP, Katsuyama Y, Kurokawa T & Osada Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater 15, 1155–1158 (2003). [Google Scholar]
- 13.Sun J-Y et al. Highly stretchable and tough hydrogels. Nature 489, 133–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong JP Materials both tough and soft. Science 344, 161–162 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Ball RC, Doi M, Edwards SF & Warner M. Elasticity of entangled networks. Polymer 22, 1010–1018 (1981). [Google Scholar]
- 16.Hama H. et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat. Neurosci 14, 1481–1488 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Ke M-T, Fujimoto S & Imai T. SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat. Neurosci 16, 1154–1161 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Pan C. et al. Shrinkage-mediated imaging of entire organs and organisms using uDISCO. Nat. Methods 13, 859–867 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Murakami TC et al. A three-dimensional single-cell-resolution whole-brain atlas using CUBIC-X expansion microscopy and tissue clearing. Nat. Neurosci 21, 625–637 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Murray E. et al. Simple, scalable proteomic imaging for high-dimensional profiling of intact systems. Cell 163, 1500–1514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao S. et al. Cellular and molecular probing of intact human organs. Cell 180, 796–812 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubota SI et al. Whole-body profiling of cancer metastasis with single-cell resolution. Cell Rep. 20, 236–250 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Park Y-G et al. Protection of tissue physicochemical properties using polyfunctional crosslinkers. Nat. Biotechnol 37, 73–83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lancaster MA & Knoblich JA Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc 9, 2329–2340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellios N. et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatr 23, 1051–1065 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.