

Abstract
Hepatocytes are the primary functional cells of the liver that perform essential roles in homeostasis, regeneration and injury. Most mammalian somatic cells are diploid and contain pairs of each chromosome, but there are also polyploid cells containing additional sets of chromosomes. Hepatocytes are among the best described polyploid cells, with polyploids comprising more than 25% and 90% of the hepatocyte population in humans and mice, respectively. Cellular and molecular mechanisms that regulate hepatic polyploidy have been uncovered, and in recent years, diploid and polyploid hepatocytes have been shown to perform specialized functions. Diploid hepatocytes accelerate liver regeneration induced by resection and may accelerate compensatory regeneration after acute injury. Polyploid hepatocytes protect the liver from tumor initiation in hepatocellular carcinoma (HCC) and promote adaptation to tyrosinemia-induced chronic injury. This review describes how ploidy variations influence cellular activity and presents a model for context-specific functions for diploid and polyploid hepatocytes.
Keywords: polyploidy, aneuploidy, liver injury, liver regeneration, hepatocellular carcinoma
A. Introduction
Cellular identity is the product of numerous characteristics influenced by developmental stage, organ specificity, differentiation status, injury, etc. An important and frequently overlooked aspect of cellular identity is chromosome number (“n”). Most somatic cells contain homologous pairs of each chromosome. For example, humans have 23 chromosomes, and diploid cells (2n) have 23 chromosome pairs for a total of 46 chromosomes. There are also somatic cells with higher chromosome content: polyploid cells. Polyploidy refers to an increase in the sets of chromosomes beyond the diploid state. Human polyploid cells can have 92 (tetraploid, 4n), 184 (octaploid, 8n), 368 (hexadecaploid, 16n) or even greater numbers of chromosomes. Polyploidy is frequently associated with aberrant cell cycling and chromosomal instability associated with tumorigenesis, but polyploidy is also found in healthy tissues, including cardiac myocytes, megakaryocytes, giant trophoblasts, muscle and hepatocytes (reviewed in1). In fact, recent studies identified polyploidy in cardiac interstitial cells – cells that were never associated with polyploidy – which indicates that ploidy diversity may be more common than previously appreciated2. Here, we focus on liver and discuss the effect of chromosome variations on hepatocyte identity and function.
B. Chromosome variations in the liver
Diploid hepatocytes form polyploid daughters during postnatal development, a process that has been called physiological polyploidy3. Polyploid hepatocytes, in turn, can generate diploid daughter cells that can subsequently re-polyploidize. The ploidy state is dynamic as hepatocytes undergo periods of polyploidization and ploidy reduction.
B1. Mechanisms of physiological polyploidization
In hepatocytes, ploidy is regulated by the number of nuclei per cell (typically one or two) and DNA content of each nucleus: 2n, 4n, 8n, etc. (Fig. 1A)4,5. Mononucleate hepatocytes are either diploid or polyploid, depending on the DNA content of the single nucleus: diploid (one 2n nucleus), tetraploid (one 4n nucleus), etc. Binucleate hepatocytes are always polyploid but can be tetraploid (two 2n nuclei), octaploid (two 4n nuclei) or higher. Multiple cellular mechanisms have been shown to induce polyploidy, including endoreplication, mitotic slippage and cell fusion, but acytokinetic mitosis (also called cytokinesis failure) is the dominant mechanism in healthy hepatocytes (reviewed in6). At birth, hepatocytes are exclusively diploid and during postnatal development the first binucleate tetraploid daughters are generated by a subset of proliferating diploid hepatocytes that undergo acytokinetic mitosis7. These binucleate tetraploid daughter cells can then produce a pair of mononucleate tetraploid daughters with a round of cell cycling that includes mitosis followed by successful cytokinesis. The cycle of acytokinetic mitosis followed by traditional mitosis continues such that in adult mice ≥90% of hepatocytes are polyploid and ≥25% of adult human hepatocytes are polyploid (Fig. 1B)8–10. Diploid and polyploid hepatocytes are distributed throughout the liver lobule, although transcriptomic11,12 and mapping13 studies demonstrate modest enrichment of polyploids in periportal regions.
Figure 1.
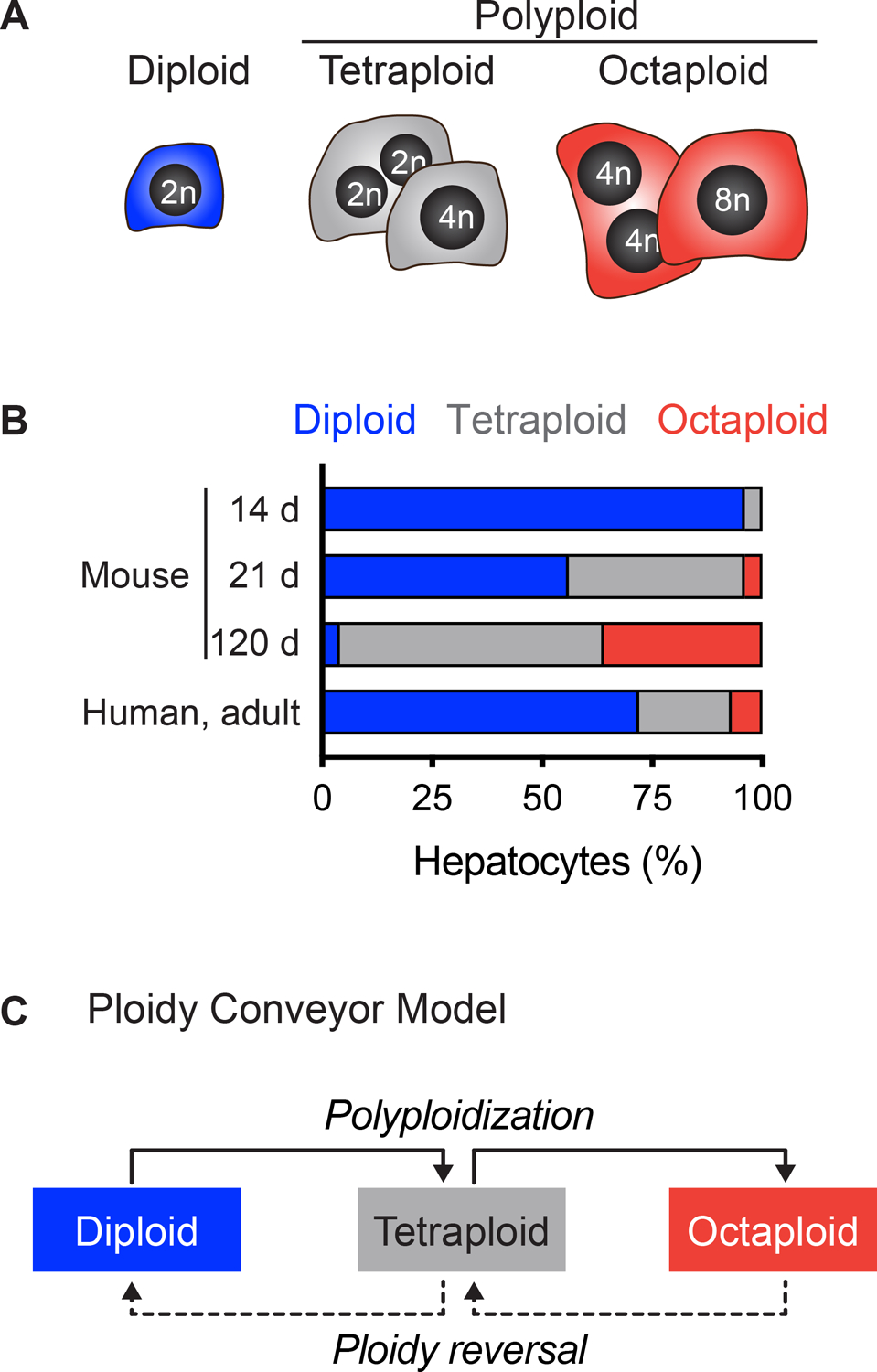
Diploidy and polyploidy in the liver. (A) Diploid hepatocytes have a single nucleus with 2n chromosome content. Polyploid hepatocytes are mono- or binucleate and can have tetraploid, octaploid or higher overall chromosome content. (B) The degree of polyploidy increases with age. The percentage of diploid, tetraploid and octaploid hepatocytes is shown for 14-, 21- and 120-day-old mice8,18,29 and adult humans10. (C) The ploidy conveyor model incorporates hepatic polyploidization, ploidy reversal and re-polyploidization.
Numerous signals have been implicated in hepatic polyploidy development and are described in comprehensive reviews elsewhere5,6,14. Here, we highlight a few of the most important pathways. First, insulin signaling changes dramatically at the suckling/weaning transition, and this promotes AKT-dependent acytokinetic mitosis and emergence of binucleate hepatocytes in rats7. Second, E2F7 and E2F8 are expressed maximally during weeks one through seven of postnatal development15. Mice with liver-specific deletion of E2f7 and E2f8 develop and function normally, but their livers are depleted of polyploid hepatocytes15,16. E2F7 and E2F8 influence ploidy by antagonizing E2f1 expression and inhibiting pro-cytokinesis genes15. Third, miR-122 is the most abundantly expressed microRNA in the adult livers17. Similar to E2f7/E2f8 knockouts, Mir122−/− mice are depleted of polyploid hepatocytes, indicating that miR-122 is required for normal polyploidization18. In wild-type mice (WT), miR-122 expression spikes during weeks two to three, the period when binucleate hepatocytes expand 10-fold. miR-122 binds to seed sequences of pro-cytokinesis genes, which antagonizes centralspindlin complex formation and promotes cytokinesis failure. Finally, it is also important to understand how hepatocytes can tolerate polyploid chromosome content since polyploidy in most tissues leads to cell cycle arrest or apoptosis. For example, a p53-regulated “tetraploid checkpoint” has been hypothesized to restrict polyploid cells19,20. New work from Sladky and colleagues has shown that supernumerary centrosomes in polyploid cells activate the PIDDosome complex, which regulates cell cycle arrest in a p53/p21-dependent manner21,22. Intriguingly, PIDDosome members Casp2 and Pidd1 are direct transcriptional targets of E2F1, further implicating the E2F family in hepatic ploidy regulation. Collectively, hepatic polyploidy is regulated by a complex network of signals where insulin, E2F family members and miR-122 regulate polyploidization in a temporal manner, and miR-122 acts as liver-specific driver of polyploidy.
B2. Ploidy reversal and aneuploidy
Hepatic polyploidy is reversible. In 2009 and 2010 our group showed that polyploid hepatocytes could fully repopulate Fah−/− mouse livers over 2–3 months9,23. Briefly, in this model system, recipient mice lack FAH, a critical enzyme in the tyrosine catabolic pathway24,25. Fah−/− mice typically die from tyrosinemia shortly after birth but are maintained and kept healthy with the drug 2-(2-nitro-4-trifluoro- methylbenzoyl)-1,3-cyclo-hexanedione (NTBC), which prevents accumulation of toxic intermediates in tyrosine catabolism26. FAH+ hepatocytes are transplanted intrasplenically or through the portal vein, and a subset of donor cells engraft in the liver. When NTBC is removed, the donor hepatocytes have a selective advantage and can proliferate extensively to fully repopulate the liver with FAH+ healthy hepatocytes. In our experiment, purified wild-type (WT) octaploid hepatocytes were isolated by fluorescence activated cell sorting (FACS) and transplanted into Fah−/− recipients9. During 500-1,000-fold expansion, proliferating octaploid hepatocytes produced octaploid and hexadecaploid daughters, as expected, but surprisingly also produced lower ploidy daughters with tetraploid and diploid chromosome content. In 2020, utilizing an elegant lineage tracing approach, Matsumoto and colleagues demonstrated ploidy reversal by single polyploid hepatocytes in vivo followed by re-polyploidization during liver repopulation27. Moreover, they demonstrated ploidy reversal after liver injury induced by carbon tetrachloride, thioacetamide and 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC), suggesting that ploidy reversal occurs during compensatory hepatocyte proliferation. The cycle of polyploidization, ploidy reversal and re-polyploidization is called the ploidy conveyor (Fig. 1C)9,14,27.
The mechanism by which polyploid hepatocytes undergo ploidy reversal involves multipolar cell division. Polyploid hepatocytes have multiple centrosomes during mitosis (e.g., four centrosomes per tetraploid cell, eight centrosomes per octaploid cell). Proliferating hepatocytes in vitro and in vivo form multipolar spindles that in some cases lead to three- or four-way nuclear segregation, permitting a single tetraploid hepatocyte, for example, to produce up to four diploid daughters8,9. One of the consequences of having multiple spindles during mitosis is chromosome segregation errors leading to partial or whole chromosome gains and/or losses and emergence of aneuploid hepatocytes. The frequency of aneuploid hepatocytes in healthy livers has been the subject of intense debate. We originally described chromosome segregation errors and aneuploidy in healthy mouse and human hepatocytes in 2009–2012 using metaphase cytogenetic G-banding analysis where isolated donor hepatocytes are cultured briefly, arrested in metaphase and the first dividing hepatocytes are karyotyped.8,9,20,23. Karyotypes of >600 mouse hepatocytes revealed that half of adult hepatocytes are aneuploid, with all chromosomes equally affected18,28,29. In 2014, Knouse et al. performed single-cell DNA sequencing on hepatic nuclei and found 5% of the sequenced hepatocytes were aneuploid30, an observation that was recently confirmed by a separate group22. A study in 2018 observed low aneuploidy in hepatocytes with normal tissue organization and cell polarity, but chromosome segregation errors were elevated in hepatocytes dividing in vitro31. It was hypothesized that disrupted tissue architecture increased chromosome segregation errors by dividing polyploid hepatocytes, leading to aneuploid daughter cells31. This concept is further supported by Matsumoto and colleagues where lineage tracing showed that multipolar divisions and marker loss by polyploid hepatocytes occurred rarely in vivo27. The idea that normal tissue architecture ensures chromosome segregation fidelity in dividing hepatocytes could explain why metaphase cytogenetic analysis, which involves brief in vitro culturing, reveals a higher percentage of aneuploid hepatocytes. Thus, the degree of hepatic aneuploidy is much lower than 50% and is likely in the range of 2–5%. Considering the large number of hepatocytes per liver (e.g., hundreds of millions in humans), it stands to reason that there are millions of hepatocytes with random chromosome gains and/or losses.
C. Chromosome variations in liver regeneration, cancer and chronic injury
Polyploidy was recognized in regenerating liver in the early 1900s, most likely because of the ease in detecting binucleate hepatocytes in sections of liver tissue32. Over the last two decades, the liver field has finally begun to delineate specialized functions of diploid and polyploid hepatocytes during liver regeneration and injury.
C1. Polyploidy in Liver Regeneration
The liver has an extraordinary capacity to regenerate, which is best illustrated by the response to partial hepatectomy (PH) involving 2/3 surgical removal of liver33,34. In mice, mature hepatocytes in the remaining liver tissue undergo one to two cell divisions to restore liver mass after one week34. Polyploid hepatocytes were originally considered to be terminally differentiated with low proliferative potential35,36, but more recent studies have shown that polyploid hepatocytes contribute to liver regeneration induced by PH37. Liver repopulation studies in the Fah−/− model have shown extensive in vivo proliferation by polyploid hepatocytes9,26,29,38. Only recently, head-to-head comparisons of regenerative capacity have revealed differences between ploidy subsets.
First, our group investigated repopulation capacity using competitive transplantation in the Fah−/− model system. Genetically marked diploid and octaploid hepatocytes from WT donors were mixed and transplanted into Fah−/− recipients9. Complete liver repopulation involves up to eight rounds of cell division by each donor cell, therefore a cell population with proliferative advantage would out-compete a disadvantaged cell population. Surprisingly, after complete liver repopulation, the ratio of donor-derived hepatocytes matched the ratio of input cells. For example, in Fah−/− mice transplanted with a hepatocyte mixture (10% diploid and 90% octaploid), donor-derived cells remained in the same ratio after repopulation (10% donor-derived diploids and 90% donor-derived octaploids). These results suggested that diploid and octaploid hepatocytes proliferate equivalently. Consistent with this finding, genetically marked diploid and polyploid hepatocytes formed clones of equivalent size after chemical-induced liver injury, indicating similar repopulation potential27. However, direct comparison of long-term proliferation by each population is problematic because of the ploidy conveyor where hepatocytes can change their ploidy during cell division (with diploid hepatocytes becoming polyploid and polyploid hepatocytes becoming diploid).
Secondly, to avoid the dynamic changes in ploidy associated with the ploidy conveyor, competitive repopulation experiments were performed with E2f7/E2f8-deficient hepatocytes29. As discussed earlier, liver-specific deletion of E2f7 and E2f8 largely blocks polyploidization such that approximately 80% of hepatocytes are diploid and 20% are polyploid, ratios that are stable during aging and liver regeneration. E2f7/E2f8-deficient (mostly diploid) and WT hepatocytes (mostly polyploid) were co-transplanted into Fah−/− mice. In every case, the E2f7/E2f8-deficient hepatocytes massively out-competed WT hepatocytes, suggesting that stably diploid hepatocytes maintain a robust proliferative advantage. Finally, to avoid potential off-target effects associated with E2f7/E2f8 deletion, ploidy subsets were tracked during PH-induced liver regeneration in WT mice29. Hepatocytes were harvested at defined intervals after PH and their ploidy and cell cycle status measured. Diploid hepatocytes were found to enter the cell cycle earlier and complete the cell cycle faster than polyploids, while polyploid hepatocytes progressed through the cell cycle after a delay. These results have been confirmed by others39, and it has been suggested that elevated p21 expression by polyploid hepatocytes restricts or delays their proliferation22. Taken together, the extent by which ploidy affects hepatocyte proliferation varies depending on the regenerative demand. During long-term proliferation (e.g., liver repopulation by cell therapy, chronic liver damage), diploid-derived and polyploid-derived hepatocytes have equivalent proliferative capacity, and this is likely due to the ploidy conveyor. In contrast, during short-term proliferation (e.g., early liver regeneration) diploid hepatocytes have an initial proliferative advantage.
C2. Polyploidy in Liver Cancer
Hepatocellular carcinoma (HCC) is the most common form of liver cancer in the world40. Considering the wealth of evidence that polyploidy is associated with increased disease severity in cancers, it is tempting to hypothesize that hepatic polyploidy contributes to HCC formation41. However, it has been well documented that HCCs in patients and rodent models are enriched with diploid hepatocytes, suggesting that diploid hepatocytes are dominant drivers of HCC and that polyploidy may actually protect the liver from tumorigenesis42–44.
Recent work has identified polyploidy-specific mechanisms for HCC protection. Zhang and colleagues used a “tunable” system to modulate ploidy45. They generated livers with high-degree hepatic polyploidy by deletion of AnIn and livers with minimal polyploidy by E2f8 deletion. Mice were treated with the tumor initiator diethylnitrosamine (DEN) and HCC formation was monitored. Consistent with another report46, E2f8-deficient mice with mostly diploid livers were highly susceptible to HCC formation and formed abundant tumors. In contrast, AnIn-deficient mice with highly polyploid livers developed few, if any, tumors. To elucidate a mechanism for tumor resistance by polyploid hepatocytes, the authors focused on ploidy-specific effects associated with oncogenes and tumor suppressor genes (TSG). Mice with mostly diploid or polyploid livers were equally sensitive to overexpression of the oncogene MYC. However, TSG loss revealed striking differences in each mouse model. Inactivation of one copy of a TSG, such as Pten or Apc, in a diploid cell led to loss-of-heterozygosity (LOH) and increased transformation potential. In polyploid hepatocytes, additional chromosome sets effectively provided backup copies of TSG to compensate for LOH. Thus, polyploidy acts as a buffer that protects hepatocytes from transformation and tumorigenesis, specifically in the context of TSG loss. Our group also compared HCC formation in WT and E2f7/E2f8-deficient mice using the DEN and phenobarbital model for HCC induction and expansion, respectively29. Compared to WT mice with mostly polyploid livers, E2f7/E2f8-deficient mice with mostly diploid livers formed tumors as early as three months and developed extensive tumor burden by nine months, suggesting that diploid hepatocytes are immediate drivers of tumor growth. Although never proven, this concept is supported by liver regeneration studies (described in section C1) where proliferating diploid hepatocytes outperform slower cycling polyploids. Collectively, recent studies indicate that the polyploid state could provide protection from tumorigenesis through multiple mechanisms – by providing extra TSG copies during LOH and restricting hepatocyte proliferation. It is important to emphasize that these studies were performed with genetic models involving highly diploid and highly polyploid livers, which makes it difficult to distinguish between ploidy-specific effects and gene-specific effects (e.g., altered hepatocyte function resulting from AnIn or E2f7/E2f8 deletion). Future work should directly address the oncogenic potential of WT diploid and polyploid hepatocytes.
Despite observations that HCCs are comprised of diploid hepatocytes, Desdouets and colleagues recently found the opposite in a cohort of 75 HCC patients10. Careful analysis revealed the HCCs were enriched with mononuclear polyploid hepatocytes, which correlated with poor differentiation, increased inflammation, p53 mutation and increased expression of proliferation genes. Hence, the role of polyploid hepatocytes in HCC progression is complex and likely depends on other genetic drivers.
While acytokinetic mitosis is the dominant mechanism leading to polyploid hepatocyte formation in healthy liver, polyploid hepatocytes are also formed by heterotypic cell fusion of hepatocytes and myelomonocytic blood cells23,47,48. Studies in mice have shown fusion events affect approximately 0.001% of hepatocytes49, and in humans fusion-derived hepatocytes have also been observed, albeit rarely50. Transplantation of WT bone marrow into lethally irradiated Fah−/− mice leads to formation of fusion-derived hepatocytes that acquire FAH and proliferate extensively when tyrosinemia is induced by NTBC withdrawal23,47,48. Intriguingly, most fusion-derived hepatocytes are aneuploid, as detected by metaphase cytogenetic analysis23. Fusion-derived cells have been implicated in a variety of diseased tissues, including the intestine51 and breast cancer cells52, and fusion hybrids were recently shown to correlate with patient survival53. The role of fusion-derived hepatocytes in HCC remains to be examined. Thus, future studies are necessary to build a comprehensive picture of hepatic ploidy populations and fusion-derived hepatocytes in HCC pathogenesis.
C3. Polyploidy during chronic liver injury
Chronic liver injury induced by various insults has been associated with alterations in hepatic ploidy in humans and rodents. One study found that polyploid hepatocytes exhibited higher infection rates of the malaria parasite Plasmodium than diploids, indicating that Plasmodium parasites preferentially infected polyploid hepatocytes54. Other studies have looked at the relationship between liver polyploidy and Hepatitis B (HBV) and Hepatitis C viral infections (HCV). Reductions and increases in hepatic polyploidy have been observed in patients with hepatitis infections, making it unclear if polyploidy contributes to disease progression or resistance. Moreover, many of these patients have other liver complications, including cirrhosis and HCC, making it difficult to disentangle the ploidy phenotypes strictly related to hepatitis infection42,55,56. Hepatic polyploidy has also been shown to be higher in livers affected by metabolic overload. Madra et al. observed increased levels of polyploidy in mice administered high doses of iron57. Additionally, it was seen that Long Evans Cinnamon rats, which accumulate copper in the liver due to a genetic defect in a copper transporter gene, had increased hepatic polyploidy58. As certain diseases and disorders are characterized by hepatic accumulation of iron (thalassemias, hereditary hemochromatosis and chronic blood transfusion)59 and copper (Wilson’s disease)60, it is important that studies investigate the relationship between polyploidy and metabolic overload.
Polyploidy levels have also been shown to increase in response to metabolic and oxidative stress. Gentric et al. observed increases of hepatic polyploidy in mouse models of nonalcoholic fatty liver disease (NAFLD) and patients with nonalcoholic steatohepatitis (NASH)3. Cell cycle analysis demonstrated a significant increase in polyploid hepatocytes and tissue microscopy showed dramatic enrichment for mononucleate cells with high nuclear content. They termed this unique ploidy distribution pathological polyploidy, which contrasts with physiological polyploidy that increases throughout postnatal development and is defined predominantly by binucleate hepatocytes. The authors identified oxidative damage as the primary driver of these ploidy changes. NAFLD induces oxidative damage in hepatocytes, which leads to DNA damage response and cell cycle arrest. As a result, hepatocytes undergo atypical cell cycles where they replicate DNA, transiently arrest in G2, skip mitosis and re-enter the cell cycle. This pattern repeats with mononucleate polyploid hepatocytes becoming even more highly polyploid. The function of these highly polyploid hepatocytes in fatty liver disease is currently unknown, and it remains to be determined whether they contribute to disease progression or resistance61.
C4. Aneuploidy and resistance to chronic liver injury
The frequency of aneuploid hepatocytes with random chromosome gains and/or losses has been the subject of much debate (section B2). The newest estimates using single-cell DNA sequencing indicate low-level aneuploidy is common, affecting ≤5% of hepatocytes22,30. A major unanswered question is how this diverse “background” aneuploidy affects liver function8,14,18,28,30,62. We previously demonstrated that aneuploidy can be protective in chronic liver injury28. During chronic tyrosinemia in Hgd+/− Fah−/− mice, livers were spontaneously repopulated by injury-resistant endogenous hepatocytes that clonally expanded. Chromosome analysis revealed that aneuploid hepatocytes with whole or partial loss of Chromosome 16 accounted for half of the regenerating nodules. Thus, in this model system, pre-existing aneuploid hepatocytes lacking Chromosome 16 had a profound selective advantage during chronic tyrosinemia injury. These disease-resistant hepatocytes proliferated extensively over several months to repopulate the diseased liver while simultaneously maintaining normal liver function and preventing liver failure. Consistent with these observations, very recently, we observed E2f7/E2f8-deficient mice lacked aneuploidy and were susceptible to liver failure and death when bred onto the Hgd+/− Fah−/− background, which is likely caused by an inability to adapt via this aneuploidy mechanism63.
Aneuploidy and liver adaptation may also play a role in human liver disease, notably HCV infection. HCV is a major health issue, leading to fibrosis and cirrhosis and at times progressing to liver cancer, liver failure and death64–67. Studies have shown HCV often disrupts the expression of genes, including tight junction proteins Claudin-1 and Occludin, that regulate hepatocyte cell polarity and liver tissue architecture68–70. Considering that disrupted tissue architecture promotes chromosome segregation errors and aneuploidy31, it is likely that HCV-infected livers have elevated numbers of aneuploid hepatocytes. Notably, several groups observed 25–50% of pre-neoplastic macroscopic nodules from HCV-infected cirrhotic livers were monoclonal71–73, suggesting they derived from a single cell with a survival advantage. This raises the question of whether aneuploid karyotypes can help hepatocytes resist disease, such as HCV infection, and proliferate in a clonal manner to aid liver regeneration. In the future, it will be important to determine if unique aneuploid karyotypes, involving particular chromosome gains/losses, are associated with specific liver diseases and injuries.
D. A model for context-specific functions for diploid and polyploid hepatocytes
Findings from many laboratories have clearly demonstrated that diploid and polyploid hepatocytes perform specialized roles. We propose a model wherein ploidy populations differentially affect disease progression in three areas: cancer, acute injury and chronic injury (Fig. 2). First, in HCC, diploid hepatocytes are major drivers of HCC progression, which is supported by the observation that most HCC in rodents and patients are diploid enriched42–44, although there are important exceptions10. Diploid hepatocytes are sensitive to TSG LOH, and once transformed, can proliferate quickly29,45. In contrast, polyploid hepatocytes with double (or more) gene copies are buffered from TSG loss and limit HCC initiation. The protective effect of polyploid hepatocytes may be most impactful in mice where >90% of hepatocytes are polyploid. At least 25% of hepatocytes are polyploid in humans, so their protective effect in patient HCC may be reduced (Fig. 1B). Secondly, diploid hepatocytes play a dominant role after acute injury such as surgical removal of liver tissue29,39. In response to proliferative cues, diploid hepatocytes proliferate earlier and faster than polyploids, indicating that diploids are the immediate drivers of liver regeneration. We hypothesize that diploids also drive compensatory proliferation after acute insults, such as drug-induced liver injury. For example, in the case of acetaminophen overdose involving extensive centrilobular hepatocyte death, the liver undergoes compensatory proliferation and recovers when the ratio of regenerating cells to necrotic cells is high74. Proliferation by diploid hepatocytes in early stages of compensatory proliferation likely tips the ratio toward “regeneration” and facilitates liver healing. The role of hepatic ploidy populations in drug-induced injury requires investigation. Finally, diploid and polyploid hepatocytes both perform critical functions during chronic liver injury. Proliferating polyploid hepatocytes undergo nuclear segregation errors to generate aneuploid daughters at low frequency, indicating that polyploid hepatocytes serve as cellular intermediates to promote aneuploidy8,9,22,30,63. After long-term tyrosinemia injury in mice, a subset of injury-resistant aneuploid hepatocytes (likely diploid hepatocytes derived by ploidy reversal) undergo clonal expansion to repopulate the liver and maintain function28. A similar process may also occur in HCV patients with clonal, healthy hepatic nodules71–73. However, it is also possible that clonal expansion by aneuploid hepatocytes can exacerbate chronic disease, such as cirrhosis and HCC (reviewed in34), although aneuploid hepatocytes were rarely found in cirrhotic liver nodules from five HCC patients75.
Figure 2.
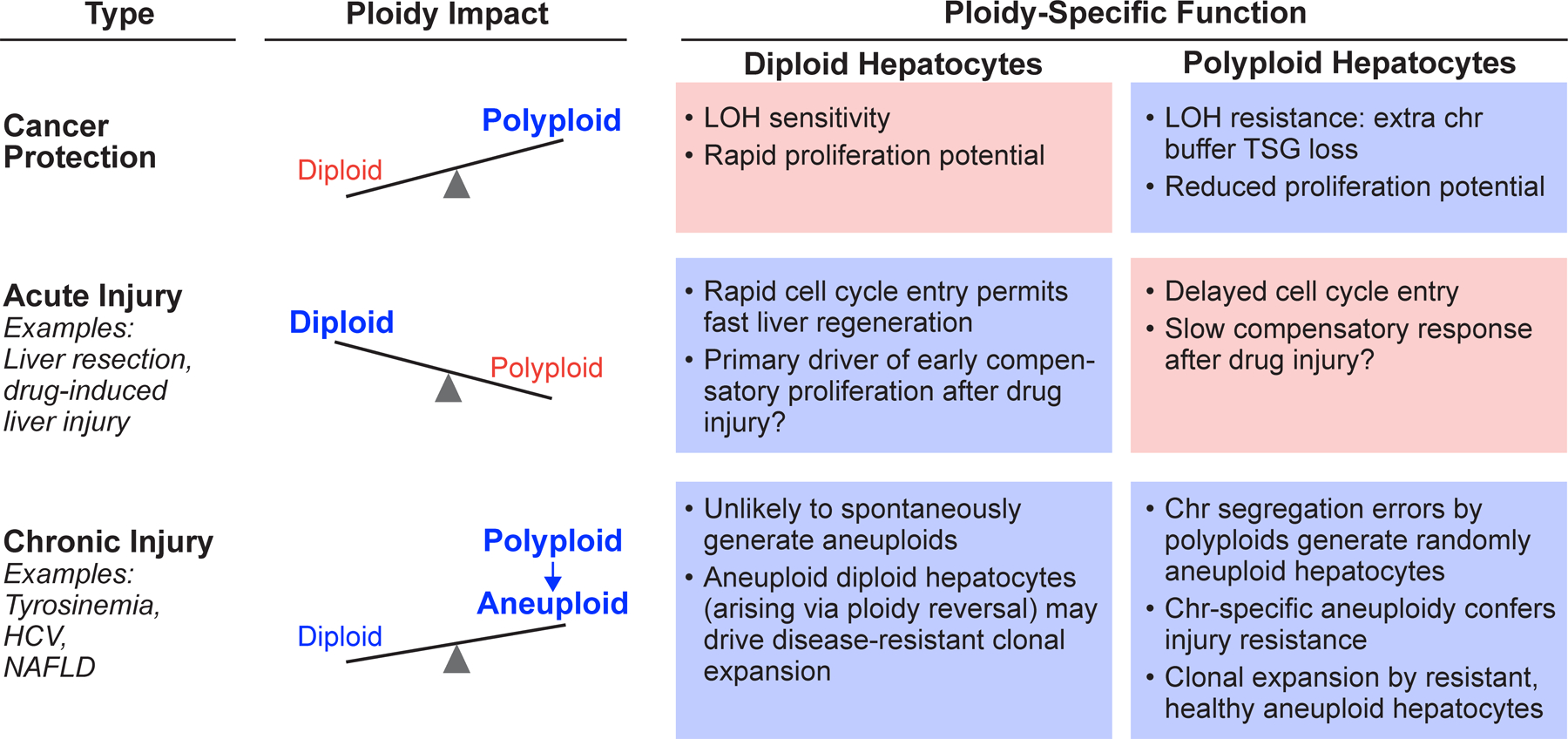
Context-specific roles for diploid and polyploid hepatocytes in acute injury, chronic injury and cancer. “Ploidy Impact,” represented by scales, indicates the relative importance of each ploidy population. “Ploidy-Specific Function” for diploid and polyploid hepatocytes is highlighted blue (major contribution) or pink (minor contribution). Abbreviations: loss-of-heterozygosity, LOH; chromosome, chr; tumor suppressor gene, TSG; hepatitis c virus, HCV; non-alcoholic fatty liver disease, NAFLD.
In summary, the development of novel therapies to improve liver health and treat disease requires a better understanding of liver function and regeneration. Chromosomal variations play a role in liver pathophysiology where diploid hepatocytes drive immediate liver regeneration through rapid proliferation and polyploids guard against liver tumorigenesis while also facilitating adaptation to chronic liver injury via aneuploid intermediates. Progress in liver therapy innovation and design hinges on our ability to elucidate the mechanisms driving liver homeostasis and regeneration, incorporating all facets of liver function and physiology, and ultimately deriving treatment stratagems that improve patient health and well-being.
Main Concepts and Learning Points.
Polyploid hepatocytes are common in human and rodent livers.
Polyploidy is reversible – proliferating polyploid hepatocytes can generate diploid daughter cells.
Diploid and polyploid hepatocytes perform specialized roles in cancer, regeneration and injury response.
A comprehensive understanding of hepatic polyploidy is lacking, and additional studies will determine how chromosome variations contribute to liver homeostasis and disease.
Acknowledgements
This work was supported by grants to AWD from the NIH (R01 DK103645) and the Commonwealth of Pennsylvania. Thanks to Evan Delgado, Nairita Roy and Frances Alencastro for insightful discussions. PDW and AWD apologize to authors whose work was not cited because of limited space.
Footnotes
Conflict of Interest
None
References
- 1.Patterson M, Swift SK. Residual Diploidy in Polyploid Tissues: A Cellular State with Enhanced Proliferative Capacity for Tissue Regeneration? Stem Cells Dev. 2019;28(23):1527–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broughton KM, Khieu T, Nguyen N, et al. Cardiac interstitial tetraploid cells can escape replicative senescence in rodents but not large mammals. Commun Biol. 2019;2:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gentric G, Maillet V, Paradis V, et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. The Journal of clinical investigation. 2015;125(3):981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gentric G, Desdouets C. Polyploidization in liver tissue. The American journal of pathology. 2014;184(2):322–331. [DOI] [PubMed] [Google Scholar]
- 5.Pandit SK, Westendorp B, de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends in cell biology. 2013;23(11):556–566. [DOI] [PubMed] [Google Scholar]
- 6.Donne R, Saroul-Ainama M, Cordier P, Celton-Morizur S, Desdouets C. Polyploidy in liver development, homeostasis and disease. Nat Rev Gastroenterol Hepatol. 2020. [DOI] [PubMed] [Google Scholar]
- 7.Celton-Morizur S, Merlen G, Couton D, Margall-Ducos G, Desdouets C. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. The Journal of clinical investigation. 2009;119(7):1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duncan AW, Hanlon Newell AE, Smith L, et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology. 2012;142(1):25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duncan AW, Taylor MH, Hickey RD, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467(7316):707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bou-Nader M, Caruso S, Donne R, et al. Polyploidy spectrum: a new marker in HCC classification. Gut. 2020;69(2):355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katsuda T, Hosaka K, Matsuzaki J, et al. Transcriptomic Dissection of Hepatocyte Heterogeneity: Linking Ploidy, Zonation, and Stem/Progenitor Cell Characteristics. Cell Mol Gastroenterol Hepatol. 2020;9(1):161–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duncan AW. Single-Cell and Bulk Transcriptome Profiling Reveals Unique Features of Diploid and Polyploid Hepatocytes. Cell Mol Gastroenterol Hepatol. 2020;9(1):193–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanami S, Ben-Moshe S, Elkayam A, Mayo A, Bahar Halpern K, Itzkovitz S. Dynamic zonation of liver polyploidy. Cell Tissue Res. 2017;368(2):405–410. [DOI] [PubMed] [Google Scholar]
- 14.Duncan AW. Aneuploidy, polyploidy and ploidy reversal in the liver. Seminars in cell & developmental biology. 2013;24(4):347–356. [DOI] [PubMed] [Google Scholar]
- 15.Pandit SK, Westendorp B, Nantasanti S, et al. E2F8 is essential for polyploidization in mammalian cells. Nature cell biology. 2012;14(11):1181–1191. [DOI] [PubMed] [Google Scholar]
- 16.Chen HZ, Ouseph MM, Li J, et al. Canonical and atypical E2Fs regulate the mammalian endocycle. Nature cell biology. 2012;14(11):1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu SH, Ghoshal K. MicroRNAs in Liver Health and Disease. Current pathobiology reports. 2013;1(1):53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu SH, Delgado ER, Otero PA, et al. MicroRNA-122 regulates polyploidization in the murine liver. Hepatology. 2016;64(2):599–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minn AJ, Boise LH, Thompson CB. Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage. Genes & development. 1996;10(20):2621–2631. [DOI] [PubMed] [Google Scholar]
- 20.Kurinna S, Stratton SA, Coban Z, et al. p53 regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology. 2013;57(5):2004–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fava LL, Schuler F, Sladky V, et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes & development. 2017;31(1):34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sladky VC, Knapp K, Soratroi C, et al. E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Dev Cell. 2020;52(3):335–349 e337. [DOI] [PubMed] [Google Scholar]
- 23.Duncan AW, Hickey RD, Paulk NK, et al. Ploidy reductions in murine fusion-derived hepatocytes. PLoS genetics. 2009;5(2):e1000385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grompe M The pathophysiology and treatment of hereditary tyrosinemia type 1. Seminars in liver disease. 2001;21(4):563–571. [DOI] [PubMed] [Google Scholar]
- 25.Grompe M, Lindstedt S, al-Dhalimy M, et al. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nature genetics. 1995;10(4):453–460. [DOI] [PubMed] [Google Scholar]
- 26.Overturf K, Al-Dhalimy M, Finegold M, Grompe M. The repopulation potential of hepatocyte populations differing in size and prior mitotic expansion. The American journal of pathology. 1999;155(6):2135–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumoto T, Wakefield L, Tarlow BD, Grompe M. In Vivo Lineage Tracing of Polyploid Hepatocytes Reveals Extensive Proliferation during Liver Regeneration. Cell Stem Cell. 2020;26(1):34–47 e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duncan AW, Hanlon Newell AE, Bi W, et al. Aneuploidy as a mechanism for stress-induced liver adaptation. The Journal of clinical investigation. 2012;122(9):3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilkinson PD, Delgado ER, Alencastro F, et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatology. 2019;69(3):1242–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knouse KA, Wu J, Whittaker CA, Amon A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(37):13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knouse KA, Lopez KE, Bachofner M, Amon A. Chromosome Segregation Fidelity in Epithelia Requires Tissue Architecture. Cell. 2018;175(1):200–211 e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milne LS. The histology of liver tissue regeneration. J Path and Bact. 1909;13:127–158. [Google Scholar]
- 33.Higgins G, Anderson GM. Experimental pathology of the liver. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol. 1931;12:186–202. [Google Scholar]
- 34.Michalopoulos GK. Hepatostat: Liver regeneration and normal liver tissue maintenance. Hepatology. 2017;65(4):1384–1392. [DOI] [PubMed] [Google Scholar]
- 35.Fausto N, Campbell JS. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev. 2003;120(1):117–130. [DOI] [PubMed] [Google Scholar]
- 36.Gupta S Hepatic polyploidy and liver growth control. Semin Cancer Biol. 2000;10(3):161–171. [DOI] [PubMed] [Google Scholar]
- 37.Miyaoka Y, Ebato K, Kato H, Arakawa S, Shimizu S, Miyajima A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Current biology : CB. 2012;22(13):1166–1175. [DOI] [PubMed] [Google Scholar]
- 38.Weglarz TC, Degen JL, Sandgren EP. Hepatocyte transplantation into diseased mouse liver. Kinetics of parenchymal repopulation and identification of the proliferative capacity of tetraploid and octaploid hepatocytes. The American journal of pathology. 2000;157(6):1963–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen F, Jimenez RJ, Sharma K, et al. Broad Distribution of Hepatocyte Proliferation in Liver Homeostasis and Regeneration. Cell Stem Cell. 2020;26(1):27–33 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. [DOI] [PubMed] [Google Scholar]
- 41.Merlo LM, Wang LS, Pepper JW, Rabinovitch PS, Maley CC. Polyploidy, aneuploidy and the evolution of cancer. Advances in experimental medicine and biology. 2010;676:1–13. [DOI] [PubMed] [Google Scholar]
- 42.Anti M, Marra G, Rapaccini GL, et al. DNA ploidy pattern in human chronic liver diseases and hepatic nodular lesions. Flow cytometric analysis on echo-guided needle liver biopsy. Cancer. 1994;73(2):281–288. [DOI] [PubMed] [Google Scholar]
- 43.Rua S, Comino A, Fruttero A, et al. Flow cytometric DNA analysis of cirrhotic liver cells in patients with hepatocellular carcinoma can provide a new prognostic factor. Cancer. 1996;78(6):1195–1202. [DOI] [PubMed] [Google Scholar]
- 44.Saeter G, Schwarze PE, Nesland JM, Seglen PO. Diploid nature of hepatocellular tumours developing from transplanted preneoplastic liver cells. Br J Cancer. 1989;59(2):198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang S, Zhou K, Luo X, et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev Cell. 2018;44(4):447–459 e445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kent LN, Rakijas JB, Pandit SK, et al. E2f8 mediates tumor suppression in postnatal liver development. The Journal of clinical investigation. 2016;126(8):2955–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Willenbring H, Akkari Y, et al. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422(6934):897–901. [DOI] [PubMed] [Google Scholar]
- 48.Willenbring H, Bailey AS, Foster M, et al. Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nat Med. 2004;10(7):744–748. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Montini E, Al-Dhalimy M, Lagasse E, Finegold M, Grompe M. Kinetics of liver repopulation after bone marrow transplantation. The American journal of pathology. 2002;161(2):565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Myerson D, Parkin RK. Donor-derived hepatocytes in human hematopoietic cell transplant recipients: evidence of fusion. Virchows Arch. 2019;474(3):365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rizvi AZ, Swain JR, Davies PS, et al. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(16):6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang LN, Huang YH, Zhao L. Fusion of macrophages promotes breast cancer cell proliferation, migration and invasion through activating epithelial-mesenchymal transition and Wnt/beta-catenin signaling pathway. Arch Biochem Biophys. 2019;676:108137. [DOI] [PubMed] [Google Scholar]
- 53.Gast CE, Silk AD, Zarour L, et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci Adv. 2018;4(9):eaat7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Austin LS, Kaushansky A, Kappe SH. Susceptibility to Plasmodium liver stage infection is altered by hepatocyte polyploidy. Cell Microbiol. 2014;16(5):784–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melchiorri C, Bolondi L, Chieco P, Pagnoni M, Gramantieri L, Barbara L. Diagnostic and prognostic value of DNA ploidy and cell nuclearity in ultrasound-guided liver biopsies. Cancer. 1994;74(6):1713–1719. [DOI] [PubMed] [Google Scholar]
- 56.Toyoda H, Bregerie O, Vallet A, et al. Changes to hepatocyte ploidy and binuclearity profiles during human chronic viral hepatitis. Gut. 2005;54(2):297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Madra S, Styles J, Smith AG. Perturbation of hepatocyte nuclear populations induced by iron and polychlorinated biphenyls in C57BL/10ScSn mice during carcinogenesis. Carcinogenesis. 1995;16(4):719–727. [DOI] [PubMed] [Google Scholar]
- 58.Yamada T, Sogawa K, Kim JK, et al. Increased polyploidy, delayed mitosis and reduced protein phosphatase-1 activity associated with excess copper in the Long Evans Cinnamon rat. Res Commun Mol Pathol Pharmacol. 1998;99(3):283–304. [PubMed] [Google Scholar]
- 59.Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366(4):348–359. [DOI] [PubMed] [Google Scholar]
- 60.Gaetke LM, Chow-Johnson HS, Chow CK. Copper: toxicological relevance and mechanisms. Arch Toxicol. 2014;88(11):1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsu SH, Duncan AW. Pathological polyploidy in liver disease. Hepatology. 2015;62(3):968–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faggioli F, Vezzoni P, Montagna C. Single-cell analysis of ploidy and centrosomes underscores the peculiarity of normal hepatocytes. PloS one. 2011;6(10):e26080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilkinson PD, Alencastro F, Delgado ER, et al. Polyploid Hepatocytes Facilitate Adaptation and Regeneration to Chronic Liver Injury. The American journal of pathology. 2019;189(6):1241–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alter MJ. Epidemiology of hepatitis C virus infection. World journal of gastroenterology : WJG. 2007;13(17):2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Martel C, Maucort-Boulch D, Plummer M, Franceschi S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology. 2015;62(4):1190–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264–1273 e1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pawlotsky JM. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology. 2014;146(5):1176–1192. [DOI] [PubMed] [Google Scholar]
- 68.Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. Journal of virology. 2009;83(4):2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mee CJ, Farquhar MJ, Harris HJ, et al. Hepatitis C virus infection reduces hepatocellular polarity in a vascular endothelial growth factor-dependent manner. Gastroenterology. 2010;138(3):1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benedicto I, Molina-Jimenez F, Barreiro O, et al. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology. 2008;48(4):1044–1053. [DOI] [PubMed] [Google Scholar]
- 71.Paradis V, Dargere D, Bonvoust F, et al. Clonal analysis of micronodules in virus C-induced liver cirrhosis using laser capture microdissection (LCM) and HUMARA assay. Lab Invest. 2000;80(10):1553–1559. [DOI] [PubMed] [Google Scholar]
- 72.Paradis V, Laurendeau I, Vidaud M, Bedossa P. Clonal analysis of macronodules in cirrhosis. Hepatology. 1998;28(4):953–958. [DOI] [PubMed] [Google Scholar]
- 73.Ochiai T, Urata Y, Yamano T, Yamagishi H, Ashihara T. Clonal expansion in evolution of chronic hepatitis to hepatocellular carcinoma as seen at an X-chromosome locus. Hepatology. 2000;31(3):615–621. [DOI] [PubMed] [Google Scholar]
- 74.Mazaleuskaya LL, Sangkuhl K, Thorn CF, FitzGerald GA, Altman RB, Klein TE. PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet Genomics. 2015;25(8):416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin YH, Zhang S, Zhu M, et al. Mice With Increased Numbers of Polyploid Hepatocytes Maintain Regenerative Capacity But Develop Fewer Tumors Following Chronic Liver Injury. Gastroenterology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]