

Abstract
Glycoside hydrolases require carboxyl groups as catalysts for their activity. A retaining xylanase from Streptomyces olivaceoviridis E-86 belonging to glycoside hydrolase family 10 possesses Glu128 and Glu236 that respectively function as acid/base and nucleophile. We previously developed a unique mutant of the retaining xylanase, N127S/E128H, whose deglycosylation is triggered by azide. A crystallographic study reported that the transient formation of a Ser–His catalytic dyad in the reaction cycle possibly reduced the azidolysis reaction. In the present study, we engineered a catalytic dyad with enhanced stability by site-directed mutagenesis and crystallographic study of N127S/E128H. Comparison of the Michaelis complexes of N127S/E128H with pNP-X2 and with xylopentaose showed that Ser127 could form an alternative hydrogen bond with Thr82, which disrupts the formation of the Ser–His catalytic dyad. The introduction of T82A mutation in N127S/E128H produces an enhanced first-order rate constant (6 times that of N127S/E128H). We confirmed the presence of a stable Ser–His hydrogen bond in the Michaelis complex of the triple mutant, which forms the productive tautomer of His128 that acts as an acid catalyst. Because the glycosyl azide is applicable in the bioconjugation of glycans by using click chemistry, the enzyme-assisted production of the glycosyl azide may contribute to the field of glycobiology.
Keywords: retaining GH10 β-xylanase, chemical rescue, switching enzyme with azide, histidine catalyzed glycosidase, rational protein design
Abbreviations
BsNagZ, β-N-acetylglucosaminidase from Bacillus subtilis; BSA, bovine serum albumin; GH, glycoside hydrolase; SoXyn10A, GH family 10 β-1,4-xylanase from Streptomyces olivaceoviridis E-86; HAD, L-3-hydroxyacyl-CoA dehydrogenase; pNP, p-nitrophenol; pNP-X2, p-nitrophenyl-β-D-xylobioside; SEA, switching enzyme with azide; CfXyn10A, β-1,4-glycanase from Cellulomonas fimi; X5, xylopentaose; WT, wild-type.
INTRODUCTION
Endo-β-1,4-xylanase (EC 3.2.1.8) hydrolyzes β-1,4-glycosidic linkages within the xylan backbone to produce xylooligosaccharides of various lengths. A retaining xylanase from Streptomyces olivaceoviridis E-86 (SoXyn10A, formerly known as FXYN) consists of N-terminal catalytic and C-terminal xylan-binding domains, which are classified into glycoside hydrolase family 10 (GH10) and carbohydrate-binding module family 13, respectively, based on their primary structures.1),2) SoXyn10A is one of the most widely investigated glycoside hydrolases for studies on reaction mechanism and substrate specificity.3),4),5),6),7),8),9) In most cases, the retaining enzymes possess glutamic or aspartic acids as acid/base catalyst and nucleophile.10),11) The catalytic domain of SoXyn10A forms a (β/α)8 TIM-barrel structure belonging to clan GH-A, and contains an acid/base catalyst (Glu128) and nucleophile (Glu236) at the active site cleft.6),8) The histidine residue (His) generally serves as a good catalyst of glycosidases. For example, His234 works as an acid/base catalyst in a β-N-acetylglucosaminidase from Bacillus subtilis (BsNagZ) that has a unique Asp–His catalytic dyad (Fig. 1A).11),12) To investigate whether His works as a catalytic residue in SoXyn10A, we previously constructed mutant enzymes, E128H (the acid/base catalyst replaced by His) and E236H (the nucleophile replaced by His). However, the activity of E236H was completely abolished and that of E128H was drastically decreased.6) In particular, E128H was reactivated several folds by the addition of sodium azide.6) This is known as chemical rescue, which is used for the identification of catalytic residues.13),14)
Fig. 1. Proposed reaction mechanisms of the glycosylation of BsNagZ (A), SEA (B), and T82A-SEA (C).

T82A-SEA and SEA15) contain a Ser–His pair, and BsNagZ11) contains an Asp–His catalytic dyad at their active sites. In both cases, the histidine residue serves as an acid catalyst during the glycosylation step. E•S complex, Michaelis complex; E–I complex, covalent glycosyl–enzyme intermediate; E•P complex, enzyme–product complex.
In our subsequent study, we successfully isolated a N127S/E128H double mutant from a parent enzyme (E128H) by random mutagenesis.15) The activity of N127S/E128H can be regulated by the use of sodium azide, and therefore, we named it switching enzyme with azide (SEA).15) SEA is active against p-nitrophenyl-β-D-xylobioside (pNP-X2) and appears to contain a Ser–His catalytic dyad similar to the Asp–His catalytic dyad of BsNagZ (Fig. 1).11),12),15) In SEA and BsNagZ, His serves as a general acid catalyst during glycosylation step. The activity of SEA for pNP-X2 was low and that for a natural substrate was lost.
To further explore the low activity of SEA, the Michaelis complex (E•S complex) structures of SEA with pNP-X2 and with xylopentaose (X5) were previously studied.15) These structures showed that an essential hydrogen bond between Ser127 and His128 was eliminated only in the E•S complex with X5, and Ser127 formed an alternative hydrogen bond with Thr82 (Fig. 1B). The hydrogen bond between Asp and His of the Asp–His catalytic dyad of BsNagZ is essential for catalysis.11) Therefore, we predicted that a disrupted hydrogen bond between Ser127 and His128 would affect the velocity of the glycosylation step. To overcome this problem, we attempted to stabilize the interaction by replacing Thr82 with Val and Ala, which cannot form hydrogen bonds.
Here, we report an enhancement of the glycosylation step of SEA based on its crystal structures. The designed mutants, T82A-SEA and T82V-SEA, exhibited higher activity against pNP-X2 than the parent SEA enzyme. Crystal structures of T82A-SEA have been determined to confirm the enhanced activity of the mutants. The E•S complex and covalent glycosyl–enzyme intermediate (E–I complex) structures of T82A-SEA revealed that the Ser–His catalytic dyad of T82A-SEA was stably formed.
MATERIALS AND METHODS
Construction of mutant xylanases.
pET28b/SEA plasmid DNA harboring the sea gene was prepared as described previously.15) In addition, an improved megaprimer PCR mutagenesis strategy was used to construct the mutant xylanases.16) For substituting Thr82 with Val, the sea fragment was amplified using the following PCR primers: T7 lac promoter primer 5′-TAATACGACTCACTATAGGGGAATTGTG-3′ and T82V reverse primer 5′-TGCCAGGCCAGGACGTGGCCGCGCACCT-3′ (substituted nucleotides are underlined). For substituting Thr82 with Ala, T7 lac promoter primer and T82A reverse primer 5′-TGCCAGGCCAGGGCGTGGCCGCGCACCT-3′ (substituted nucleotides are underlined) were used. The amplified megaprimers were purified by phenol/chloroform extraction and ethanol precipitation. Intact sea fragment was amplified using the megaprimers and T7 terminator primer 5′-ATGCTAGTTATTGCTCAGCGG-3′. The PCR products were also purified using the phenol/chloroform extraction and ethanol precipitation. The purified DNA fragments were cleaved with NcoI and HindIII and ligated into the pET28b(+) vector (Novagen Inc., Madison, WI, USA) in order to produce pET28b/T82V-SEA and pET28b/T82A-SEA. The sequences were checked with nucleotide sequencing with an automated DNA sequencing system (model 3100, Applied BioSystems, Inc. Foster City, CA, USA).
Enzyme production and purification.
Production of wild-type (WT) and mutant xylanases in Escherichia coli BL21 codon plus (DE3)-RIL (Stratagene Inc., La Jolla, CA, USA) was performed as described previously.6) For characterization, a construct, CD (303), consisting of only the catalytic module (residues 1–303) was used, while the full-length enzyme (residues 1–436) was used for crystallization.4) To facilitate purification, His6-tag was added to the C-terminus of the expressed CD (303). The His6-tagged enzymes were purified by Ni-nitrilotriacetic acid agarose (Qiagen GmbH, Hilden, Germany) affinity chromatography as described previously.6) Purification of the full-length mutant enzymes was performed using lactose-Sepharose affinity chromatography as previously reported.17) Concentration of mutant enzymes was measured using a BCA protein assay kit (Sigma, St. Louis, MO, USA) using bovine serum albumin (BSA) as a standard.
Enzyme assay.
Enzyme activity was determined colorimetrically under the conditions reported previously.15) Steady-state kinetic parameters were determined with pNP-X26) and soluble oat-spelt xylan7) as substrates. For pNP-X2, 450 μL of substrate solution containing 25 % McIlvaine buffer (a mixture of 0.1 M citric acid and 0.2 M Na2HPO4, pH 6.0), 0.5 mg/mL BSA, and 300 mM sodium azide was preincubated at 30 °C for 5 min. Then, 50 μL of the enzyme solution was added to start the azidolysis reaction. The amount of p-nitrophenol (pNP) released was monitored continuously by measuring the absorbance at 400 nm with a spectrophotometer (DU 630: Beckman Coulter, Inc., Brea, CA, USA). For the soluble oat-spelt xylan, the 200 μL of the substrate solution (same reaction composition as for pNP-X2) was used and the reaction conditions were same as that for pNP-X2. The reaction was stopped by heating the mixture at 100 °C for 10 min. The amount of reducing sugar was determined using the copper-bicinchoninic acid method by measuring the absorbance at 560 nm with xylobiose as a standard.18) The kinetic parameters were determined by Eadie-Hofstee plot in at least three independent measurements. The concentration of substrates was set to the appropriate values as described previously.6),15)
Monitoring of the reaction cycles.
Reaction cycles of SEA was monitored using a 1-mL syringe according to the following procedures:
Step I: To immobilize the SEA onto a resin, the C-terminal His6-tagged purified SEA (10 mg; 0.30 μmol) was subjected to a 1-mL HisTrap™ HP column (GE Healthcare UK Ltd., Buckinghamshire, England) equilibrated with 25 % McIlvaine buffer (pH 7.0) at room temperature.
Step II: One milliliter of pNP-X2 solution (1.2 μmol) was loaded onto the SEA-immobilized column to produce a covalent glycosyl–enzyme intermediate, and then the column was washed with 1.1 mL of 25 % McIlvaine buffer (pH 7.0) to remove cleaved pNP and unreacted pNP-X2. The eluates were harvested into the same 15-ml disposable centrifuge tube (pNP fraction).
Step III: To elute the trapped sugars, 1 mL of 25 % McIlvaine buffer (pH 7.0) supplemented with 300 mM sodium azide was applied to the column. The column was washed two times with 2 mL of 25 % McIlvaine buffer (pH 7.0) to remove sodium azide form the column. The pNP fraction was reloaded onto the same column and steps II and III were repeated four times. A 50-μL aliquot of the pNPfraction was mixed with an equal volume of 0.2 M Na2CO3, and absorbance was measured at 400 nm to determine the amount of pNP released.
Crystallography.
Crystals of T82A-SEA in complex with X5 and pNP-X2 were prepared separately using a soaking technique as described previously.15) The X-ray diffraction experiments of T82A-SEA in complex with pNP-X2 (T82A-SEA/pNP-X2) were performed using charge-coupled device cameras of the NW12A station at the Photon Factory Advanced Ring for Pulse X-rays (PF-AR, Tsukuba, Japan) (λ = 1.0000 Å). The X-ray diffraction data set of T82A-SEA in complex with X5 (T82A-SEA/X5) was collected using an in-house R-AXIS VII imaging plate detector with a copper rotating-anode generator (λ= 1.5418 Å; Rigaku Corporation, Tokyo, Japan). The crystals were flash-cooled in a nitrogen stream at 100 K. The diffraction data were collected in 1.0° oscillation steps for the range of 100° (T82A-SEA/X5) and 180° (T82A-SEA/pNP-X2). The collected images were processed using the HKL2000 program suite.19) The structures were solved by a molecular replacement method by using MOLREP20) in the CCP4 package,21) with the coordinates of SEA (PDB code 2D1Z) as a search model. Manual model rebuilding and refinement were performed using XtalView22) and CNS.23) The clear electron densities corresponding to the sugar molecules were found in the Fobs–Fcalc maps around the active site. The crystallographic R-factor (Rfree) for T82A-SEA/pNP-X2 and T82A-SEA/X5 converged to 17.5 % (19.7 %) and 17.9 % (24.9 %), respectively. Stereochemistry of the final models was analyzed using PROCHECK.24) A summary of the data collection and refinement statistics is provided in Table 1. The atomic coordinates and structure factors of T82A-SEA/pNP-X2 (accession code 5GQD) and T82A-SEA/X5 (accession code 5GQE) have been deposited in the Protein Data Bank (http://wwpdb.org/). All figures were prepared using PyMol (DeLano Scientific LLC, Palo Alto, CA, USA).
Table 1.
Statistics for data collection and refinement.
| Data set | T82A-SEA/X5 | T82A-SEA/pNP-X2 |
|---|---|---|
| Data collection statistics | ||
| Space group | P212121 | P212121 |
| Unit cell (Å) | a = 75.1 | a = 78.2 |
| b = 94.0 | b = 94.0 | |
| c = 137.6 | c = 140.1 | |
| X-ray source | R-AXIS VII | AR-NW12A |
| Wavelength (Å) | 1.5418 | 1.0000 |
| Resolution (Å)* | 50–2.50 (2.59–2.50) | 50–1.80 (1.86–1.80) |
| Total reflections | 551,614 | 1,305,054 |
| Unique reflections | 34,484 (3,268) | 96,353 (9,237) |
| Completeness (%)* | 96.3 (97.2) | 98.4 (97.0) |
| Rmerge (%)* | 7.8 (29.9) | 6.3 (28.6) |
| I/σI * | 21.4 (5.8) | 32.6 (6.1) |
| Redundancy* | 4.1 (4.0) | 7.3 (6.9) |
| Refinement statistics | ||
| Resolution range (Å) | 39.16–2.50 | 40.09–1.80 |
| No. of reflections | 33,153 | 94,683 |
| R-factor/Rfree (%) | 17.9/24.9 | 17.5/19.7 |
| r.m.s.d. from ideal | ||
| Bond lengths (Å) | 0.005 | 0.005 |
| Bond angles (°) | 1.3 | 1.3 |
| Average B-factor (Å2) | ||
| All atoms | 30.2 | 18.5 |
| Protein (chains A/B) | 27.9/29.9 | 16.6/15.0 |
| Water atoms | 36.6 | 35.3 |
| Sugars (chain A/B) | 42.4/51.5 | 15.7/10.8 |
| Ramachandran plot (%) | ||
| Favored (chain A/B) | 87.5/87.7 | 89.9/89.9 |
| Allowed (chain A/B) | 12.5/12.3 | 10.1/10.1 |
| Disallowed (chain A/B) | 0.0/0.0 | 0.0/0.0 |
| PDB code | 5GQE | 5GQD |
*Values in parentheses are for the highest resolution shell.
RESULTS AND DISCUSSION
Interaction between Ser127 and Thr82 destabilizes that between Ser127 and His128.
SEA contains two mutations, N127S and E128H, in its active site. SEA was active against pNP-X2, whereas no activity was detected when a natural substrate such as oat-spelt xylan was used.15) We previously published the structures of E•S complex of SEA in complex with pNP-X2 [SEA/pNP-X2(E•S), PDB code 2D20] and with X5 (SEA/X5, 2D24).15) The SEA/pNP-X2(E•S) structure shows that an Nδ1 atom of His128 forms a hydrogen bond with the Oγ atom of Ser127 (distance of 2.5 Å) and appears to form a Ser–His catalytic dyad. A similar catalytic dyad (Asp–His catalytic dyad) is present in BsNagZ.11) In SEA and BsNagZ, His serves as a general acid catalyst during the glycosylation step (Fig. 1). As shown in Fig. 2, the SEA/pNP-X2(E•S) and SEA/X5 structures showed that an imidazole ring of His128 in SEA/X5 was rotated by 25° in comparison with SEA/pNP-X2(E•S). Rotation of χ1 angle (95°) on the Ser127 side chain in SEA/X5 resulted in the formation of an alternative hydrogen bond between the Oγ atom of Ser127 and the Oγ1 atom of Thr82 (2.9 Å) (Fig. 2). This indicates that the hydrogen bond between Ser127 and His128 plays an important role in maintaining the appropriate orientation of the imidazole ring as described in a previous report.25) We therefore assume that the low velocity of the glycosylation step of SEA is attributable to the disturbance of the hydrogen bond between Ser127 and His128.
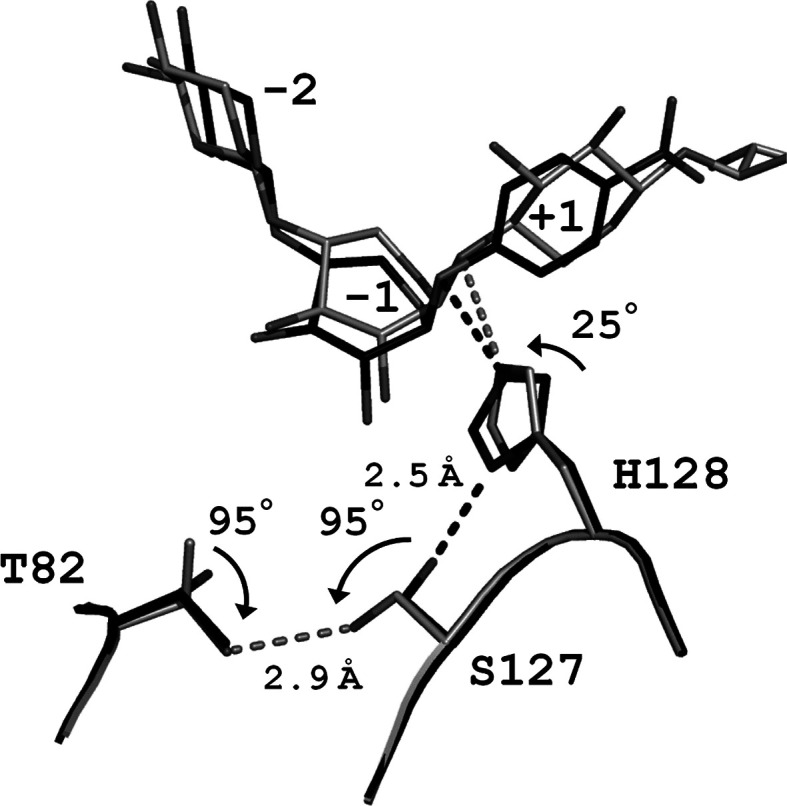
Fig. 2. Close-up view of the superposition of the two active sites of E•S complexes of SEA with pNP-X2 and with X5.
SEA/pNP-X2(E•S) (PDB code 2D20) and SEA/X5 (2D24) are indicated by black and light-gray stick models, respectively. For clarity, binding sugars of SEA/X5 at subsites +2 and +3 are not shown.
Construction and characterization of site-directed mutants.
To stabilize the hydrogen bond between Ser127 and His128, Thr82 was replaced with Ala (T82A-SEA) and Val (T82V-SEA); both do not form a hydrogen bond with Ser127 and do not induce any steric hindrance. Activity of the mutant enzymes was low in the absence of general nucleophiles, but addition of sodium azide significantly reactivated the velocity. Therefore, the kinetic parameters of the mutant enzymes in the presence of sodium azide were estimated (Table 2). The kcat (2.14 min−1) and Km (0.0362 mM) values of T82V-SEA against pNP-X2 were lower than those of SEA, resulting in an approximately 1.5-fold increase in the kcat/Km value (59.1 min−1 mM−1). A significant increment in the kcat value of T82A-SEA (18.9 min−1) results in the kcat/Km value of T82A-SEA (139 min−1 mM−1) reaching one-tenth of the WT level (1.08 × 103 min−1 mM−1) (Table 2). The azidolysis activity against pNP-X2 indicated that the glycosylation step of T82A-SEA was enhanced.
Table 2.
Steady-state kinetic parameters of pNP-X2 in the presence of sodium azide.
|
kcat (min−1) |
Km (mM) |
kcat/Km (min−1 mM−1) |
|
|---|---|---|---|
| T82V-SEA | 2.14 ± 0.14 | 0.0362 ± 0.0031 | 59.1 |
| T82A-SEA | 18.9 ± 0.5 | 0.136 ± 0.003 | 139 |
| SEA* | 3.14 ± 0.32 | 0.0789 ± 0.0033 | 39.8 |
| WT-SoXyn10A* | 2,530 ± 34** | 2.37 ± 0.05 | 1,080 |
Next, the activity of SEA, T82V-SEA, and T82A-SEA against oat-spelt xylan was measured in both the presence and absence of sodium azide. T82A-SEA was active only when sodium azide was present, but the others showed no activity irrespective of high enzyme and high substrate concentrations. We therefore determined the kinetic parameters of WT-SoXyn10A and T82A-SEA using oat-spelt xylan as a substrate (Table 3). The azidolysis velocity of T82A-SEA was very low, which took several days for measurement of the time course. The half-life activity of T82A-SEA at 30 °C was found to be five days, and it retained 96 % of its activity 2 days later (data not shown), therefore, the parameters of WT-SoXyn10A and T82A-SEA were assayed at 30 °C within two days. The kcat value of T82A-SEA was 0.05 min−1, which was about 104-fold lower than that of WT-SoXyn10A (838 min−1) (Table 3).
Table 3.
Steady-state kinetic parameters of soluble oat-spelt xylan in the presence of sodium azide.
|
kcat (min−1) |
Km (mg mL−1) |
|
|---|---|---|
| SEA | ND* | ND* |
| T82A-SEA | 0.05 ± 0.01 | 2.20 ± 0.40 |
| WT-SoXyn10A | 838 ± 61 | 1.02 ± 0.17 |
* Activity not detected.
Substrate-binding structures of T82A-SEA.
Because T82A-SEA showed considerably higher activity than T82V-SEA and was active against both synthetic and natural substrates, we determined the crystal structure of E•S complex of T82A-SEA with X5 (T82A-SEA/X5, PDB code 5GQE) by using a soaking technique according to the method described previously.15) The E–I complex of T82A-SEA with X5 could not be observed owing to its very low activity, and uncleaved X5 was bound to the subsites (Fig. 3A). Instead, the E–I complex structure of T82A-SEA with pNP-X2 (T82A-SEA/pNP-X2, PDB code 5GQD) was determined. The crystal structures of T82A-SEA/X5 and T82A-SEA/pNP-X2 contained two noncrystallographic symmetry-related molecules, A and B. The root-mean-square (rms) deviation of all Cα atom pairs between T82A-SEA/X5 and SEA/X5 was 0.31 Å, and that between T82A-SEA/pNP-X2 and E–I complex of SEA with pNP-X2 [SEA/pNP-X2(E–I), PDB code 2D22] was 0.20 Å; this indicated that their overall structures were almost identical except for the mutated residue (T82A). The soaked sugars were trapped in the active sites of these structures. An uncleaved X5 was bound to the subsites −2 to +3 of the T82A-SEA/X5 (molecule B). A xylose unit at the subsite −1 of molecule B adopted a 1S3 skew-boat conformation like the Michaelis complex in SEA/X5 (Fig. 3A).15) A xylobiose moiety of pNP-X2 was accumulated as a covalent adduct at the active sites in T82A-SEA/pNP-X2 (molecules A and B). The subsite −1 of T82A-SEA/pNP-X2 was occupied by the 4C1 chair conformation of the xylose unit whose anomeric carbon was inverted to form a covalent bond with the Oε2 atom of Glu236 (Fig. 3B). Our results demonstrate that the reaction mechanism of T82A-SEA was double displacement mechanism and employed conformations 4C1 and 1S3 during the reaction process, as observed in SEA.15) This conformational itinerary is typical of an anti-protonators that act on xylo-configured substrates.26),27)
Fig. 3. Crystal structures of E·S and E–I complexes of T82A-SEA.

Close-up views around the active site of T82A-SEA/X5 (5GQE) (A) and T82A-SEA/pNP-X2 (5GQD) (B) are shown with the Fobs–Fcalc omit electron density maps contoured at 3.0σ The key residues and hydrogen bonds are shown as stick models and dashed lines, respectively. The sugar units occupying subsite –1 in the E·S and the E–I complexes adopted the 1S3 and 4C1 conformations, respectively.
Insight into the glycosylation step of T82A-SEA.
To elucidate the mechanism of enhancement of the azidolysis activity of T82A-SEA, we compared the structures around the active site of SEA and T82A-SEA during the glycosylation step. Because there were no residues around Ser127 within the hydrogen-bonding fragments, the Oγ atom of Ser127 in T82A-SEA structures exclusively interacted with the Nδ1 atom of His128 to form the Ser–His catalytic dyad (Figs. 4A and 4B). The catalytically important role of the interaction between Asp and His in the Asp–His–Ser catalytic triad and Asp–His catalytic dyad is to orient the proper tautomer of His.25),28),29) Our previous saturation mutagenesis study of SEA at position 127 revealed that only three mutants having the Ser127, Cys127, and Thr127 mutations showed detectable activity.15) These N127X mutants containing hydroxyl or thiol groups at the γ-position of their side chain are able to form a hydrogen bond with Nδ1 atom of His128 and are thought not to cause severe steric hindrance. Therefore, we believe that the Ser–His catalytic dyad was present in T82A-SEA, and that Ser127 played a role in maintaining appropriate orientation of His128, where it acted as an acid catalyst. However, there is a significant difference between Ser–His catalytic dyad of the present study and the Asp–His catalytic dyad reported previously, which is the negative charge of Asp (Fig. 1). Mutational studies on the Asp–His catalytic dyad of ribonuclease A,28) glucose 6-phosphate dehydrogenase,29) and Glu–His catalytic dyad of L-3-hydroxyacyl-CoA dehydrogenase (HAD)30) suggested that the negative charge of Asp or Glu modulates pKa of the His and is required for efficient catalysis. Indeed, the activity of E170Q mutant of HAD, which has no negative charge, is 100-fold lower than that of WT HAD.30) We therefore assumed that the absence of the negatively charged residue in Ser–His catalytic dyad of T82A-SEA perturbs modulation of the pKa of His128.
Fig. 4. Active site structures of enzyme–substrate complexes.

T82A-SEA/X5 (A), T82A-SEA/pNP-X2 (B), SEA/X5 (C), SEA/pNP-X2(E-I) (2D22) (D), and Cex in complex with 2-deoxy-2-fluoro-xylobiose (2XYL) (E). The residues essential for catalysis and bound substrates are represented by stick drawings. Hydrogen bonds are shown in broken lines. Important hydrogen-bonding distances are indicated and labeled.
The azidolysis activity of T82A-SEA against the natural substrate was restored to five orders of magnitude lower level compared with hydrolysis activity of WT-SoXyn10A (Table 3). To account for the extremely low azidolysis activity of T82A-SEA, the region around the active sites of T82A-SEA and β-1,4-glycanase CfXyn10A from Cellulomonas fimi (formerly known as Cex) was studied. CfXyn10A, a homologue of SoXyn10A, is also classified as GH10 and shows high sequence identity with SoXyn10A (49 %). A highly conserved hydrogen bond between the Nδ2 atom of Asn126 in Cex (corresponding to Asn127 of SoXyn10A) and the C-2 hydroxyl group of the sugar at subsite −1 (distance of 3.3 Å) is considered to stabilize the transition state (Fig. 4E).31),32),33) Our SEA and T82A-SEA structures lacked this catalytically important hydrogen bond interaction between Asn127 and the substrate (Figs. 4A, 4B, 4C, and 4D). Moreover, the N127S mutation created a space around subsite −1 and likely increased the flexibility of the bound substrate at the active site. Recovery of the missing hydrogen bond and rigidity of the substrate may presumably lead to further enhancement of the glycosylation step of T82A-SEA against natural substrates (e.g., oat-spelt xylan).
One-pot enzymatic synthesis of glycosyl azide.
The reaction catalyzed by SEA stopped at the deglycosylation step in the absence of general nucleophiles, resulting in the accumulation of stable covalent glycosyl–enzyme intermediates. When general nucleophiles such as sodium azide are added to the intermediate, the deglycosylation step immediately proceeds to form β-xylobiosyl azide as the sole product.15) This indicates that the reaction cycles can be regulated by sodium azide. We therefore monitored the reaction using a His6-tagged SEA-immobilized Ni-chelating column by measuring the amount of liberated pNP using pNP-X2 as the substrate. The amount of released pNP increased with an increase in the reaction cycles (Fig. 5A). This showed that a rapid and convenient method to isolate the reaction product (β-xylobiosyl azide) and unreacted substrate pNP-X2 could be established (Fig. 5B). The resulting β-xylobiosyl azide may be used for bioconjugation such as glycan synthesis and fluorescent labeling by using click chemistry.34),35),36),37) Thus, production of glycosyl azide may contribute to the field of glycobiology. The positions of the mutations (Asn127 and Glu128) in SEA are highly conserved across the clan GH-A enzymes with varied substrate preferences.1),15) Therefore, introduction of these mutations into other clan GH-A enzymes may produce switching enzymes that spawn various types of glycosyl azides. Our rapid and convenient one-pot enzymatic synthesis method is expected to serve as a model system for the production of such glycosyl azides (Fig. 5B).
Fig. 5. Monitoring of reaction cycles of SEA (A) and schematic drawing of a rapid and convenient method for glycosyl azide preparation using SEA-immobilized column (B).

(A) Reaction cycles of SEA were monitored by using pNP-X2 as a substrate (see text for details). The amount of released pNP (open circles) was measured. (B) The C-terminal His6-tagged SEA was immobilized onto a Ni column (I). pNP-X2 was loaded on the SEA-immobilized column (II). Glycosyl azide was eluted by adding sodium azide (III). The procedures (II) and (III) were repeated. Detailed procedures are described in the text.
To summarize, we have created T82A-SEA with an enhanced glycosylation step based on the three-dimensional structures. In addition, structural analyses of E•S and E–I complexes for T82A-SEA revealed that the hydrogen-bonding interaction between the Nε2 atom of His128 and the Oγ atom of Ser127 plays an important role in catalysis by forming the Ser–His catalytic dyad. Because the enzymatic reaction of SEA can be regulated by the presence or absence of sodium azide, it is easy to produce β-xylobiosyl azide from pNP-X2 by using the SEA-immobilized column. However, preparation of pNP-X2 requires considerable time and money, and therefore, we hope that the azidolysis should be reactivated against natural substrates. Alternative strategies using artificial substrates such as 4,6-dimethoxy-1,3,5-triazine sugars, which are easily synthesized by one-step procedure, are highly attractive.38)
ACKNOWLEDGMENTS
We thank Dr. Keitarou Kimura of National Agriculture and Food Research Organization for careful reading of our manuscript and his fruitful comments. We also thank the staff of the Photon Factory for the X-ray data collection. This work was supported in part by the Program for the Promotion of Basic Research Activities for Innovative Bioscience (BRAIN) and New Energy and Industrial Technology Development Organization (NEDO) in Japan.
REFERENCES
- 1).Henrissat B.: A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J., 280, 309-316 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Cantarel B.L., Coutinho P.M., Rancurel C., Bernard T., Lombard V., and Henrissat B.: The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res., 37, D233-D238 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Kaneko S., Ichinose H., Fujimoto Z., Kuno A., Yura K., Go M., Mizuno H., Kusakabe I., and Kobayashi H.: Structure and function of a family 10 beta-xylanase chimera of Streptomyces olivaceoviridis E-86 FXYN and Cellulomonas fimi Cex. J. Biol. Chem., 279, 26619-26626 (2004). [DOI] [PubMed] [Google Scholar]
- 4).Kaneko S., Ito S., Fujimoto Z., Kuno A., Ichinose H., Iwamatsu S., and Hasegawa T.: Importance of Interactions of the α-Helices in the Catalytic Domain N- and C- terminals of the Family 10 Xylanase from Streptomyces olivaceoviridis E-86 to the Stability of the Enzyme. J. Appl. Glycosci., 56, 165-171 (2009). [Google Scholar]
- 5).Kaneko S., Ichinose H., Fujimoto Z., Iwamatsu S., Kuno A., and Hasegawa T.: Substrate recognition of a family 10 Xylanase from Streptomyces olivaceoviridis E-86: A study by site-directed mutagenesis to make an hindrance around the entrance toward the substrate-binding cleft. J. Appl. Glycosci., 56, 173-179 (2009). [Google Scholar]
- 6).Kuno A., Shimizu D., Kaneko S., Hasegawa T., Gama Y., Hayashi K., Kusakabe I., and Taira K.: Significant enhancement in the binding of p-nitrophenyl-β-D-xylobioside by the E128H mutant F/10 xylanase from Streptomyces olivaceoviridis E-86. FEBS Lett., 450, 299-305 (1999). [DOI] [PubMed] [Google Scholar]
- 7).Kuno A., Kaneko S., Ohtsuki H., Ito S., Fujimoto Z., Mizuno H., Hasegawa T., Taira K., Kusakabe I., and Hayashi K.: Novel sugar-binding specificity of the type XIII xylan-binding domain of a family F/10 xylanase from Streptomyces olivaceoviridis E-86. FEBS Lett., 482, 231-236 (2000). [DOI] [PubMed] [Google Scholar]
- 8).Fujimoto Z., Kuno A., Kaneko S., Yoshida S., Kobayashi H., Kusakabe I., and Mizuno H.: Crystal structure of Streptomyces olivaceoviridis E-86 β-xylanase containing xylan-binding domain. J. Mol. Biol., 300, 575-585 (2000). [DOI] [PubMed] [Google Scholar]
- 9).Fujimoto Z., Kaneko S., Kuno A., Kobayashi H., Kusakabe I., and Mizuno H.: Crystal structures of decorated xylooligosaccharides bound to a family 10 xylanase from Streptomyces olivaceoviridis E-86. J. Biol. Chem., 279, 9606-9614 (2004). [DOI] [PubMed] [Google Scholar]
- 10).Vocadlo D.J., and Davies G.J.: Mechanistic insights into glycosidase chemistry. Curr. Opin. Chem. Biol., 12, 539-555 (2008). [DOI] [PubMed] [Google Scholar]
- 11).Litzinger S., Fischer S., Polzer P., Diederichs K., Welte W., and Mayer C.: Structural and kinetic analysis of Bacillus subtilis N-acetylglucosaminidase reveals a unique Asp-His dyad mechanism. J. Biol. Chem., 285, 35675-35684 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Fushinobu S., Alves V.D., and Coutinho P.M.: Multiple rewards from a treasure trove of novel glycoside hydrolase and polysaccharide lyase structures: New folds, mechanistic details, and evolutionary relationships. Curr. Opin. Struct. Biol., 23, 652-659 (2013). [DOI] [PubMed] [Google Scholar]
- 13).Ly H.D., and Withers S.G.: Mutagenesis of glycosidases. Annu. Rev. Biochem, 68, 487-522 (1999). [DOI] [PubMed] [Google Scholar]
- 14).Viladot J.L., de Ramon E., Durany O., and Planas A.: Probing the mechanism of Bacillus 1,3-1,4-β-D-glucan 4-glucanohydrolases by chemical rescue of inactive mutants at catalytically essential residues. Biochemistry, 37, 11332-11342 (1998). [DOI] [PubMed] [Google Scholar]
- 15).Suzuki R., Fujimoto Z., Ito S., Kawahara S., Kaneko S., Taira K., Hasegawa T., and Kuno A.: Crystallographic snapshots of an entire reaction cycle for a retaining xylanase from Streptomyces olivaceoviridis E-86. J. Biochem., 146, 61-70 (2009). [DOI] [PubMed] [Google Scholar]
- 16).Séraphin B., and Kandels-Lewis S.: An efficient PCR mutagenesis strategy without gel purification [correction of "purificiation"] step that is amenable to automation. Nucleic Acids Res., 24, 3276-3277 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Ito S., Kuno A., Suzuki R., Kaneko S., Kawabata Y., Kusakabe I., and Hasegawa T.: Rational affinity purification of native Streptomyces family 10 xylanase. J. Biotechnol., 110, 137-142 (2004). [DOI] [PubMed] [Google Scholar]
- 18).Waffenschmidt S., and Jaenicke L.: Assay of reducing sugars in the nanomole range with 2,2´-bicinchoninate. Anal. Biochem., 165, 337-340 (1987). [DOI] [PubMed] [Google Scholar]
- 19).Otwinowski Z. and Minor W.: Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307-326 (1997). [DOI] [PubMed] [Google Scholar]
- 20).Vagin A., and Teplyakov A.: Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr., 66, 22-25 (2010). [DOI] [PubMed] [Google Scholar]
- 21).Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A., McNicholas S.J., Murshudov G.N., Pannu N.S., Potterton E.A., Powell H.R., Read R.J., Vagin A., and Wilson K.S.: Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr., 67, 235-242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).McRee D.E.: XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol., 125, 156-165 (1999). [DOI] [PubMed] [Google Scholar]
- 23).Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., and Warren G.L.: Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr., 54, 905-921 (1998). [DOI] [PubMed] [Google Scholar]
- 24).Laskowski R.A., MacArthur M.W., D.S. Moss, and Thornton J.M.: PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst., 26, 283-291 (1993). [Google Scholar]
- 25).Sprang S., Standing T., R.J. Fletterick, Stroud R.M., Finer-Moore J., Xuong N.H., Hamlin R., Rutter W.J., and Craik C.S.: The three-dimensional structure of Asn102 mutant of trypsin: Role of Asp102 in serine protease catalysis. Science, 237, 905-909 (1987). [DOI] [PubMed] [Google Scholar]
- 26).Davies G.J., Ducros V.M., Varrot A., and Zechel D.L.: Mapping the conformational itinerary of β-glycosidases by X-ray crystallography. Biochem. Soc. Trans., 31, 523-527 (2003). [DOI] [PubMed] [Google Scholar]
- 27).Vasella A., Davies G.J., and Böhm M.: Glycosidase mechanisms. Curr. Opin. Chem. Biol., 6, 619-629 (2002). [DOI] [PubMed] [Google Scholar]
- 28).Schultz L.W., Quirk D.J., and Raines R.T.: His⋯Asp catalytic dyad of ribonuclease A: Structure and function of the wild-type, D121N, and D121A enzymes. Biochemistry, 37, 8886-8898 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Cosgrove M.S., Gover S., Naylor C.E., Vandeputte-Rutten L., Adams M.J., and Levy H.R.: An examination of the role of Asp-177 in the His-Asp catalytic dyad of Leuconostoc mesenteroides Glucose 6-Phosphate Dehydrogenase: X-ray structure and pH dependence of kinetic parameters of the D177N mutant enzyme. Biochemistry, 39, 15002-15011 (2000). [DOI] [PubMed] [Google Scholar]
- 30).Barycki J.J., O'Brien L.K., Strauss A.W., and Banaszak L.J.: Glutamate 170 of human L-3-Hydroxyacyl-CoA dehydrogenase is required for proper orientation of the catalytic histidine and structural integrity of the enzyme. J. Biol. Chem., 276, 36718-36726 (2001). [DOI] [PubMed] [Google Scholar]
- 31).White A., Tull D., Johns K., Withers S.G., and Rose D.R.: Crystallographic observation of a covalent catalytic intermediate in a β-glycosidase. Nat. Struct. Biol., 3, 149-154 (1996). [DOI] [PubMed] [Google Scholar]
- 32).Williams S.J., Notenboom V., Wicki J., Rose D.R., and Withers S.G.: A new, simple, high-affinity glycosidase inhibitor: Analysis of binding through x-ray crystallography, mutagenesis, and kinetic analysis. J. Am. Chem. Soc., 122, 4229-4230 (2000). [Google Scholar]
- 33).Notenboom, V., Williams S.J., Hoos R., Withers S.G., and Rose D.R.: Detailed structural analysis of glycosidase/inhibitor interactions: Complexes of Cex from Cellulomonas fimi with xylobiose-derived aza-sugars. Biochemistry, 39, 11553-11563 (2000). [DOI] [PubMed] [Google Scholar]
- 34).Kolb H.C., Finn M.G., and Sharpless K.B.: Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed Engl., 40, 2004-2021 (2001). [DOI] [PubMed] [Google Scholar]
- 35).Nierengarten J.F., Iehl J., Oerthel V., Holler M., Illescas B.M., Muñoz A., Martín N., Rojo J., Sánchez-Navarro M., Cecioni S., Vidal S., Buffet K., Durka M., and Vincent S.P.: Fullerene sugar balls. Chem. Commun. , 46, 3860-3862 (2010). [DOI] [PubMed] [Google Scholar]
- 36).Sánchez-Navarro M., Muñoz A., Illescas B.M., Rojo J., and Martín N.: Fullerene as multivalent scaffold: Efficient molecular recognition of globular glycofullerenes by concanavalin A. Chem. Eur. J., 17, 766-769 (2011). [DOI] [PubMed] [Google Scholar]
- 37).Kolb H.C., and Sharpless K.B.: The growing impact of click chemistry on drug discovery. Drug Discov. Today, 8, 1128-1137 (2003). [DOI] [PubMed] [Google Scholar]
- 38).Kobayashi A., Tanaka T., Watanabe K., Ishihara M., Noguchi M., Okada H., Morikawa Y., and Shoda S.: 4,6-Dimethoxy-1,3,5-triazine oligoxyloglucans: Novel one-step preparable substrates for studying action of endo-β-1,4-glucanase III from Trichoderma reesei. Bioorg. Med. Chem. Lett., 20, 3588-3591 (2010). [DOI] [PubMed] [Google Scholar]