

Abstract
Much remains unknown about the roles of CD4+ T helper cells in shaping localized memory B cell and CD8+ T cell immunity in the mucosal tissues. Here, we report that lung T helper cells provide local assistance for the optimal development of tissue-resident memory B and CD8+ T cells after the resolution of primary influenza virus infection. We have identified a population of T cells in the lung that exhibit characteristics of both follicular T helper and TRM cells, and we have termed these cells as resident helper T (TRH) cells. Optimal TRH cell formation was dependent on transcription factors involved in T follicular helper and resident memory T cell development including BCL6 and Bhlhe40. We show that TRH cells deliver local help to CD8+ T cells through IL-21–dependent mechanisms. Our data have uncovered the presence of a tissue-resident helper T cell population in the lung that plays a critical role in promoting the development of protective B cell and CD8+ T cell responses.
INTRODUCTION
Tissue-resident memory B (BRM) and T (TRM) cells that reside in the mucosal sites have recently been identified and characterized (1–5). BRM and TRM cells are able to mount rapid recall responses in situ against invading pathogens before pathogen dissemination and, therefore, are thought to provide immediate and superior protection against secondary infections (1, 6–8). However, the mechanisms underlying the development and persistence of robust BRM and TRM cell responses in the respiratory tract are largely undefined. Furthermore, whether there are cellular and molecular pathways that can be targeted to simultaneously promote both BRM and TRM cell responses for maximal protection against pathogen reinfections remains unknown.
Influenza virus causes up to 56,000 deaths annually in the United States since 2010 (9). Influenza virus infection induces potent development of protective BRM and CD8+ TRM cell responses in the respiratory tract (5, 10, 11). Compared with memory B cells in the secondary lymphoid organs, lung BRM cells have enhanced percentages of cells poised for cross-reactive memory repertoires and contribute to early plasmablast responses after influenza reinfection (1, 5, 12). Similarly, influenza-specific lung CD8+ TRM cells can rapidly respond to heterologous influenza virus reinfection before the virus can replicate to high titers (13, 14). However, lung-protective CD8+ TRM cell responses are short-lived in nature (15, 16), which poses a potential roadblock for developing TRM-centered vaccination strategies. Therefore, better understanding the cellular and molecular mechanisms regulating the development and/or maintenance of lung-protective BRM and/or CD8+ TRM cell responses may aid the design of future influenza vaccines.
CD4+ T helper (TH) cells are important in antiviral immune responses by providing essential “help” for the development of effector and memory CD8+ T and germinal center B (BGC) cell responses (17–19). CD4+ T cell help, particularly during the priming stage, is vital for the development of circulating memory T (TMEM) and CD103+ TRM cells after primary influenza infection (20, 21). Similarly, CD4+ T cell help, mediated mainly through follicular helper T (TFH) cells in the secondary lymphoid organs, is required for B cells to form GC and to produce high-affinity antibodies (Abs) during influenza infection (22–24). However, whether CD4+ T cells can assist local B and CD8+ T cell memory responses at the mucosal tissue after the resolution of primary infection is unknown. Several recent studies have also identified a population of PD-1Hi “TFH-like” cells in the peripheral tissues during autoimmunity (25–27). However, the developmental cues and the precise physiological function of these cells remain largely elusive. During influenza infection, the existence of a lung TFH-like cell population that can potentially sustain lung BGC cell responses has been recently demonstrated (26, 28, 29) but is still controversial (5, 27).
Here, we have identified a population of lung CD4+ TH cells that are developed after influenza viral clearance and coexhibit TFH and TRM features. On the basis of their gene expression, sessile features, and functional properties, we termed these cells tissue-resident T helper (TRH) cells. TRH cells provide local help for the generation of optimal BGC and BRM cell responses, as well as a CD8+ TRM population that is critical for the protection against heterologous influenza virus infection (30, 31). Our findings reveal a respiratory tissue-resident TH cell population that is important in assisting local development of protective memory B and CD8+ T cell responses.
RESULTS
Lung CD4+ T cells provide “late” help for the formation of BGC, BRM, and CD8+ TRM cell responses in situ
We sought to determine whether CD4+ T cell help can assist local B and CD8+ T cell memory responses at the mucosal tissue after the clearance of primary infection. We used a mouse model of influenza A virus PR8/34 (PR8) infection, in which viral clearance occurs within 10 days postinfection (d.p.i.) (32–34). We infected wild-type (WT) mice with PR8 virus and depleted CD4+ T cells with α-CD4 (GK1.5) injections starting at 14 d.p.i. (Fig. 1A). CD4+ T cells were largely depleted in the spleen and the lung (fig. S1A). At 42 d.p.i., we analyzed lung tissue B and T cell responses through intravenous (i.v.) injection of α-CD45 for 5 min before mouse sacrifice as reported (31, 35, 36). In this setting, CD45i.v.− cells were within lung tissue, whereas CD45i.v.+ cells were in lung blood vessels. CD4+ T cell depletion disrupted the formation of inducible bronchus-associated lymphoid tissue (iBALT), which contained B cell and CD4+ T cell aggregates (28, 37, 38) (Fig. 1B and fig. S1B). CD4+ T cell depletion also abrogated lung BGC responses (fig. S1C). We then examined influenza hemagglutinin-specific B (HA-B) cell responses in the lung and spleen after CD4+ T cell depletion using influenza HA-tetramer. Influenza PR8 HA tetramer mainly stained lung B cells of mice with prior PR8 but not X31 infection, which indicated the specificity of the HA-tetramer (fig. S1D). CD4+ T cell depletion did not affect lung circulating (CD45i.v.+) nor splenic HA-B cell responses but drastically diminished total and HA-B cells in the lung tissue (CD45i.v.−) at the memory stage (Fig. 1C and fig. S1, E to G).
Fig. 1. Lung CD4+ T cells deliver localized help to B and CD8+ T cells.

(A to D) WT mice were infected with PR8 strain of influenza virus and treated with control (Ctl) IgG or α-CD4 starting at 14 d.p.i. Mice were injected with α-CD45 intravenously 5 min before sacrifice at 42 d.p.i. (A) Experimental scheme. (B) Representative confocal images of iBALT in the lung. Lung sections were stained with α-CD4 (red), α-B220 (green), and DAPI (blue). (C) Frequencies and cell number of influenza HA-B cells in the lung tissue or blood vessels. (D) Lung tissue CD8+, CD8+CD69+, or CD8+CD69+CD103+ NP366–374 or PA224–233 TRM cells were enumerated. (E to I) WT mice were infected with PR8 and treated with Ctl IgG or α-CD4 (starting at 14 d.p.i.) in the presence of daily injection of FTY720 (starting at 13 d.p.i.). (E) Schematic of experimental design. (F) BGC, (G) total HA-B (CD45i.v.−HA-B), or (H) HA-BRM cells were enumerated by flow cytometry. (I) Lung tissue CD8+, CD8+CD69+, or CD8+CD69+CD103+ NP366–374 TRM cells were enumerated. (J to L) WT mice were infected with PR8 and received Ctl IgG and high or low dose of α-CD4. (J) Experimental scheme. (K) Cell number of BGC, HA-BRM, or NP-BRM cells in the lung tissue. (L) Lung parenchymal CD8+, CD8+CD69+, or CD8+CD69+CD103+ NP366–374 TRM cells were enumerated. In (B) to (D) were the representative data from at least two independent experiments (four to five mice per group). In (F), (G), and (I) to (L), data were pooled from two (F to G and I) or three (K and L) independent experiments (two to five mice per group). P values were calculated by unpaired two-tailed Student’s t test in (C), (D), and (F) to (I). P values in (J) and (K) were analyzed by one-way ANOVA. Data are means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. n.s., not significant.
We next examined influenza-specific CD8+ memory T cell responses in the lung and spleen after CD4+ T cell depletion. To do so, we checked lung CD8+ memory T cell responses against two dominant influenza major histocompatibility complex class I (MHC-I) H-2Db–restricted epitopes, nucleoprotein (NP) peptide 366 to 374 (NP366–374) and polymerase peptide 224 to 233 (PA224–233) at 42 d.p.i. (31). It has been shown before that CD8+ memory T cells against NP366–374 or PA224–233 epitope exhibit distinct phenotypic and recall properties (31, 39–41). Specifically, lung CD8+ NP366–374 memory T cells are highly protective and dominate over the CD8+ PA224–233 memory T cells in the secondary recall expansion upon rechallenge with heterotypic influenza virus (31, 39–41). Late CD4+ T cell depletion did not affect lung circulating or parenchymal total CD8+ or PA224–233 memory T cell population (Fig. 1D and fig. S1, H to J) but caused a significant decrease of the magnitude of CD8+ NP366–374 memory T cells in the lung tissue compartment (Fig. 1D and fig. S1, H to I). The magnitudes of parenchymal CD8+ CD69+ NP366–374 TRM or CD8+ CD69+ CD103+ NP366–374 TRM cells were also diminished after late CD4+ T cell depletion (Fig. 1D and fig. S1K). Late CD4+ T cell depletion did not affect the percentages of CD103+ cells within the CD8+ CD69+ NP366–374 TRM population (fig. S1L). This finding is in contrast with the results observed after CD4+ T cell depletion before influenza infection (21). Contrary to the diminished magnitude of lung CD8+ NP366–374 TRM cells, late CD4+ T cell depletion did not decrease splenic CD8+ NP366–374 or CD8+ PA224–233 TMEM cells (fig. S1M). These data suggest that continuous CD4+ T cell help after viral clearance is required for the persistence of optimal B and CD8+ T cell responses against a dominant protective epitope in the lung at the memory stage.
CD4+ T cells could possibly provide help to B or CD8+ T cells either in the circulation or in the lung. To determine whether lung tissue CD4+ T cells can provide local help, we infected WT mice with PR8 virus and depleted CD4+ T cells at 14 d.p.i. in the presence of FTY720 (Fig. 1E), a compound that blocks lymphocyte migration (42). FTY720 treatment drastically diminished T and B cell circulation in the blood (fig. S2A). The depletion of CD4+ T cells abolished BGC cell development, total HA-B cells, and HA-specific BRM (HA-BRM) cells (identified as CD45i.v.− B220+ GL7− IgD− IgM− CD38+ HA+) in the lung (Fig. 1, F to H, and fig. S2B) even in the presence of FTY720. Influenza-specific immunoglobulin G (IgG) in bronchoalveolar lavage fluid was also significantly decreased after CD4+ T cell depletion (fig. S2C). Further, CD4+ T cell depletion diminished lung tissue CD8+ NP366–374-specific TMEM, CD8+ CD69+ TRM, and CD8+ CD69+ CD103+ TRM cells but did not affect the number of total CD8+ TRM cells (Fig. 1I and fig. S2D). Similar data were observed after CD4+ T cell depletion in the presence of FTY720 during H3N2 X31 influenza virus infection (fig. S2, E and F). Thus, lung CD4+ T cells can provide in situ help to local B and CD8+ T cells for the optimal development of BGC, BRM, and CD8+ NP366–374-specific TRM cells after viral clearance.
We next sought to examine whether lung tissue CD4+ T cells are sufficient for the generation of BGC, BRM, and CD8+ NP366–374-specific TRM cell responses after the clearance of the primary infection, in the absence of circulating CD4+ T cells. We injected low or high doses of α-CD4 into PR8-infected WT mice at 14 d.p.i. Low dose of α-CD4 treatment largely ablated CD4+ T cells in the blood but not in the lung parenchyma, whereas the high-dose α-CD4 injection depleted both circulating and lung parenchymal CD4+ T cells (Fig. 1J and fig. S2, G and H). High dose, but not low dose, of α-CD4 treatment diminished the magnitude of lung BGC, influenza HA, or NP-specific BRM (NP-BRM) and CD8+ NP366–374 TRM (including CD69+ TRM or CD69+CD103+ TRM) cells (Fig. 1, K and L). Together, our data suggest that lung tissue CD4+ T cells, rather than CD4+ T cells in the circulation, provide late “local help” for the optimal generation of BGC, BRM, and CD8+ TRM responses after influenza infection.
Identification of a population of TFH-like cells in the lung
We next sought to identify the lung CD4+ T cell populations that may mediate the local help to B and/or CD8+ T cells. To do so, we performed single-cell RNA sequencing (scRNA-seq) on CD45i.v.− lung parenchymal CD4+ T cells at 28 d.p.i. Hierarchical clustering analysis identified five separated CD4+ T cell populations within the lung parenchyma (Fig. 2A and fig. S3, A and B). These cell populations included TH1 effector/memory-like cells [expressing high levels of chemokine receptor Cxcr6 and TH1 transcription factor Tbx21 (T-bet); cluster 0], T cells recently entering the lung or circulating effector memory T cells [expressing the transcription factor Klf2 and sphingosine 1-phosphate receptor 1 (S1pr1), which mediate lymphocyte migration; cluster 1], regulatory T (Treg) cells (expressing Treg lineage–specific transcription factor Foxp3; cluster 2), TH17-like (expressing TH17 signature genes Il17/Ccr6/Rora; cluster 4), and a cluster of T cells exhibiting features of TFH cells (expressing TFH transcription factor Bcl6 and signature cytokine Il21; cluster 3) (Fig. 2B and fig. S3C). Cluster 3 CD4+ T cells also expressed higher levels of TFH-associated surface molecules Pdcd1 (PD-1), Cxcr5, and Izumo1r (FR4) (Fig. 2B).
Fig. 2. Identification of a population of TFH-like cells in the lung tissue.

WT (A to E) or IL-21-VFP reporter (F) mice were infected with PR8. (A) t-Distributed stochastic neighbor embedding (tSNE) plot of scRNA-seq analysis of sorted lung CD45i.v.−CD4+CD44Hi cells (pooled from five mice) at 28 d.p.i. (B) Heat map of indicated genes in each cluster from scRNA-seq data. (C) Kinetics of the percentages of PD-1HiFR4Hi population in lung tissue, total CD4+, or influenza NP-specific (NP311–325) CD4+ T cells. (D) Expression of TFH cell–associated markers in lung total or influenza-specific PD-1HiFR4Hi, PD-1LowFR4Low, or splenic TFH (CD4+CD44HiPD-1+CXCR5+) cells at 28 d.p.i. (E) Frequency positive for IFN-γ, IL-17, or IL-4 by lung PD-1HiFR4Hi, PD-1LowFR4Low, or spleen TFH cells were identified by intracellular staining at 28 d.p.i. (F) IL-21-VFP expression in lung CD4+ PD-1HiFR4Hi, PD-1LowFR4Low, or spleen TFH at 28 d.p.i. In (C) to (F), representative plots, histograms, and graphs were collected from at least two independent experiments (two to four mice per group). P values in (E) and (F) were analyzed by one-way ANOVA. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Flow cytometry analysis identified a population of total CD4+ or influenza-specific lung CD4+ NP311–325 PD-1HiFR4Hi T cells that expressed TFH-associated markers including B-cell lymphoma 6 (BCL6), inducible T-cell costimulator (ICOS), and P2X purinoceptor 7 (P2RX7) but relatively lower levels of CXCR5 compared with splenic TFH cells (Fig. 2, C and D, and fig. S3, D and E). Similar to splenic TFH cells, lung CD4+ PD-1HiFR4Hi T cells expressed low levels of interferon-γ (IFN-γ) and interleukin-17 (IL-17; Fig. 2E and fig. S3F). However, splenic TFH cells expressed significantly higher IL-4 than lung CD4+ PD-1HiFR4Hi T cells (Fig. 2E and fig. S3F). Using IL-21-vivid Verde fluorescent protein (VFP) reporter mice (43), we found that lung CD4+ PD-1HiFR4Hi T cells expressed high levels of IL-21, almost comparable with those of splenic TFH cells (Fig. 2F). These data suggest that there is a lung tissue CD4+ T cell population transcriptionally and phenotypically resembling TFH cells in the secondary lymphoid organs.
Transcriptional profiling reveals lung TFH-like cells exhibit TRM gene signature
To gain more insights into the phenotype and identity of lung TFH-like cells, we sought to compare the transcriptional signatures of lung TFH-like cells to splenic TFH, non-TFH, and lung non–TFH-like cell signatures. Foxp3+ Treg cells expressed folate receptor 4 (FR4), glucocorticoid-induced TNFR-related protein (GITR), and programmed cell death protein 1 (PD-1) (albeit their PD-1 levels were lower than TFH-like cells) (Figs. 2B and 3A and fig. S3C). To minimize the potential contribution of Treg cells on the transcriptional profiles of lung TFH-like cells, we excluded splenic or lung GITR+ CD4+ T cells, which were mostly Foxp3+ cells, in our sorting (fig. S4A). We then sorted splenic non-TFH (CD4+CD44HiGITR−CXCR5LowPD-1Low), splenic TFH (CD4+CD44Hi GITR−CXCR5HiPD-1Hi), lung non–TFH-like (CD45i.v.− CD4+CD44Hi GITR−PD-1LowFR4Low), and lung TFH-like cells (CD45i.v.− CD4+ CD44HiGITR−PD-1HiFR4Hi) and performed RNA-seq analysis.
Fig. 3. Transcriptional profiling reveals PD-1HiFR4Hi cells exhibit both TFH and TRM gene signatures.
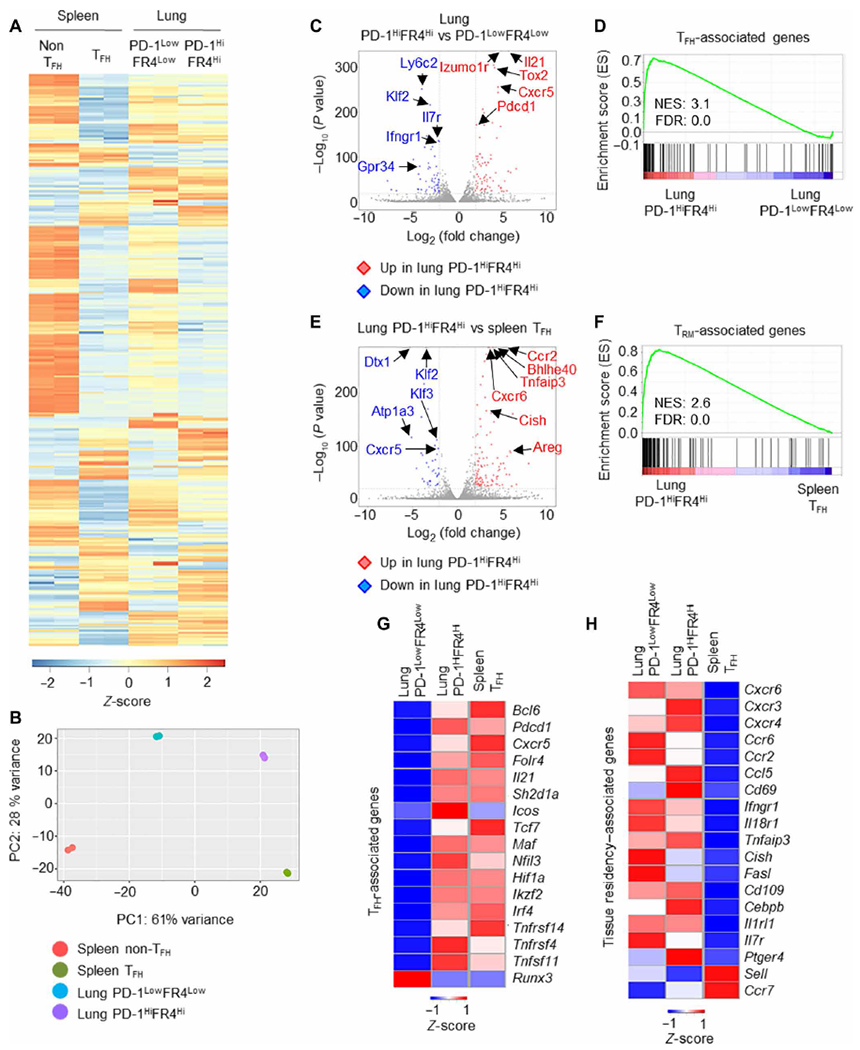
(A to F) WT mice were infected with PR8. Lung PD-1HiFR4Hi or PD-1LowFR4Low CD4+ T cells and splenic TFH or non-TFH cells were sorted after exclusion of GITRHi Treg cells, and RNA-seq analysis was performed at 28 d.p.i. (A) Heatmap of DEGs among lung PD-1HiFR4Hi, PD-1LowFR4Low CD4+ T cells, and splenic TFH or non-TFH cells. (B) Principal component analysis (PCA) of RNA-seq data of lung PD-1HiFR4Hi, PD-1LowFR4Low CD4+ T cells, and splenic TFH or non-TFH cells. (C) Volcano plot of RNA-seq analysis of lung PD-1HiFR4Hi or PD-1 LowFR4Low CD4+ T cells. (D) GSEA of the core TFH signature genes in lung CD4+ PD-1HiFR4Hi and CD4+ PD-1LowFR4Low cells. (E) Volcano plot of RNA-seq analysis on lung PD-1HiFR4Hi CD4+ T and splenic TFH cells. (F) GSEA of the core tissue residency signature genes of TRM cells in lung PD-1HiFR4Hi and splenic TFH cells. (G and H) WT mice were infected with PR8. Lung PD-1HiFR4Hi or PD-1LowFR4Low CD4+ T cells and splenic TFH cells were sorted at 28 d.p.i. Nanostring analysis on 560 immune-associated genes was performed. The expression of TFH-associated genes (G) or tissue residency–associated genes (H) in the three cell populations was depicted. For RNA-seq, data were from duplicates of pooled samples (n = 15). For nanostring analysis, data were from pooled samples (n = 10). ES, enrichment score; NES, normalized enrichment score; FDR, false discovery rate.
Differential gene expression and principal components analysis revealed that those four different CD4+ T cell populations have distinct gene expression patterns, although lung CD4+ PD-1HiFR4Hi cells were more distinct from splenic non-TFH cells relative to splenic TFH or lung CD4+ PD-1LowFR4Low cells (Fig. 3, A and B, and fig. S4B). When directly compared with lung CD4+ PD-1LowFR4Low cells, lung CD4+ PD-1HiFR4Hi cells highly expressed TFH-associated genes including Il21, Tox2, and Pdcd1 (Fig. 3C). Gene Set Enrichment Analysis (GSEA) showed that lung CD4+ PD-1HiFR4Hi cells had an enrichment of TFH-associated genes (44) relative to CD4+ PD-1LowFR4Low cells (Fig. 3D). Conversely, lung CD4+ PD-1LowFR4Low cells expressed higher levels of Ly6c and Il7r and showed enhanced enrichment of genes in transforming growth factor–β, hypoxia, and Notch signaling relative to PD-1HiFR4Hi cells (Fig. 3C and fig. S4C). When compared with splenic TFH cells, lung CD4+ PD-1HiFR4Hi cells showed increased expression of genes associated with tissue migration, retention, and function including Ccr2, Bhlhe40, and Cxcr6 (Fig. 3E) (45, 46). GSEA revealed that lung CD4+ PD-1HiFR4Hi cells had significant enrichment of TRM-associated genes relative to splenic TFH cells (Fig. 3F) (47). Lung CD4+ PD-1HiFR4Hi cells also had higher expression of genes associated with IL-2/signal transducers and activators of transcription 5 (STAT5), nuclear factor κB (NF-κB), and interferons (IFN) signaling, whereas splenic TFH cells had higher enrichment of Myc and phosphatidylinositol 3-kinase (PI3K)–mammalian target of rapamycin (mTOR) signaling (fig. S4D). Thus, these RNA-seq analyses indicate that lung CD4+ PD-1HiFR4Hi cells exhibit transcriptional signatures of both TFH cells and TRM cells.
To confirm these observations, we sorted splenic TFH, lung CD4+ PD-1HiFR4Hi, or lung CD4+ PD-1LowFR4Low cells and performed Nanostring analysis of 560 immune-associated genes (31). Compared with lung CD4+ PD-1LowFR4Low cells, splenic TFH and lung CD4+ PD-1HiFR4Hi cells expressed higher levels of TFH-associated genes including Bcl6, Sh2d1a, and Tcf7 (Fig. 3G). Compared with splenic TFH cells, both PD-1Hi FR4Hi and PD-1Low FR4Low lung CD4+ T cell populations had enhanced expression of genes associated with tissue migration and residency but diminished expression of lymphoid migration or retention molecules Sell (CD62L) and Ccr7 (Fig. 3H). These data suggest that lung CD4+ PD-1HiFR4Hi cells exhibit a “hybrid” gene signature of both conventional TFH cells and TRM cells.
Lung CD4+ PD-1HiFR4Hi T cells are tissue resident
PD-1HiFR4Hi cells expressed higher levels of CD69, CXCR6, and Bhlhe40 molecules associated with T cell migration, retention, and function in the respiratory mucosal tissue, compared with splenic TFH cells (Fig. 4A). To confirm their tissue residency, we performed parabiosis experiments and joined the circulation of PR8-infected CD45.1+ and CD45.1+ CD45.2+ congenic mice at 21 d.p.i. (Fig. 4B). We examined CD4+ T cell exchange between the two parabionts after 2 weeks of parabiosis. Close to 40 to 60% of splenic CD4+ T cells in parabiont hosts were derived from their parabiont pair (Fig. 4C). Within lung intravenous CD45 Ab protected tissue CD4+ T cell compartment, PD-1HiFR4Hi total CD4+ or antigen (Ag)–specific CD4+ NP311–325 T cells exhibited limited exchange between the two parabionts (Fig. 4, D and E), suggesting that these cells are mostly tissue resident. Most of Ag-specific CD4+ NP311–325 T cells are tissue resident (Fig. 4E), whereas total lung CD4+ PD-1LowFR4Low T cells showed a higher circulating rate than those of CD4+ PD-1HiFR4Hi cells (Fig. 4D). Thus, lung CD4+ PD-1HiFR4Hi cells are lung tissue–resident T cells exhibiting both TFH and TRM features. On the basis of their gene signature, protein expression, cytokine production, and tissue residency property, we termed these cells as TRH cells.
Fig. 4. Lung PD1HiFR4Hi CD4+ T cells are tissue resident.
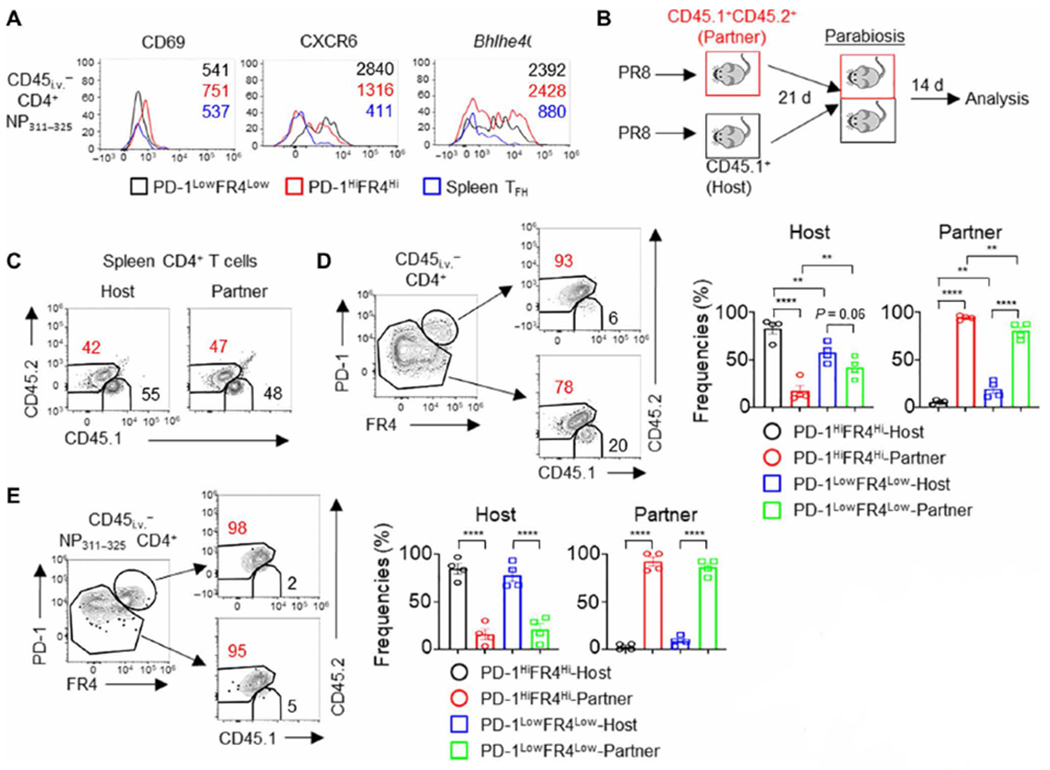
(A) WT mice were infected with PR8. The expression of CD69, CXCR6, and Bhlhe40 in lung CD4+ PD-1HiFR4Hi, CD4+ PD-1LowFR4Low NP311–325 T cells, or splenic TFH cells at 28 d.p.i. (B to E) CD45.1+ (host) or CD45.1+ CD45.2+ (partner) WT mice were infected with PR8. Parabiosis surgery was performed at 21 d.p.i. Mice were euthanized 14 days later for analysis. (B) Schematic of parabiosis experiments. (C) Composition of Host-derived or Partner-derived CD4+ T cells in the spleen of the parabionts. (D) Frequencies of Host-derived or Partner-derived cells in the lung PD-1HiFR4Hi or PD-1LowFR4Low total CD4+ T cell compartment. (E) Frequencies of Host-derived or Partner-derived cells in influenza-specific lung CD4+ PD-1HiFR4Hi or CD4+ PD-1LowFR4Low NP311–325 T cell compartment. In (A), the representative histograms were from at least two independent experiments (three to four mice per group). In (C) to (E), parabiosis data were pooled from two different experiments. P values were calculated by one-way ANOVA in (D) and (E). Data are means ± SEM. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Optimal TRH responses depend on both TFH and TRM transcription factors
We next sought to investigate the molecular mechanisms regulating lung TRH cell development. We first examined whether lung TRH cell development is dependent on the master transcription factor of TFH cells, BCL6 (48, 49). We infected WT (Bcl6fl/fl) or Bcl6ΔT mice with PR8 virus and examined total and influenza-specific TRH or non-TRH cells in the lung tissue at 28 d.p.i. T cell–specific BCL6 deficiency greatly diminished both the frequencies and the magnitude of lung TRH responses but not those of non-TRH responses (Fig. 5, A to C). Consistent with prior work (50, 51), T cell–specific BCL6 deficiency also diminished splenic TFH responses (Fig. 5D).
Fig. 5. Both BCL6 and Bhlhe40 are required for optimal lung TRH cell responses.

(A to D) Bcl6fl/fl or Bcl6ΔT mice were infected with PR8. (A) Representative dot plot, (B) frequencies, and (C) cell numbers of TRH or non-TRH in lung CD45i.v.− total CD4+ or CD45i.v.− influenza-specific CD4+ NP311–325 T cells at 28 d.p.i. (D) Frequencies (top) or cell numbers (bottom) of splenic total TFH or NP311–325 TFH at 28 d.p.i. (E) GSEA of the Bhlhe40-associated genes in lung TRH (PD-1HiFR4Hi) and spleen TFH cells. (F to H) Bhlhe40fl/fl or Bhlhe40ΔT mice were infected with PR8. (F) Representative dot plot, (G) frequencies (top), or cell numbers (bottom) of lung NP311–325 TRH or non-TRH cells at 28 d.p.i. (H) Active caspase 3/7–fluorochrome-labeled inhibitors of caspases (FLICA)+ cells in lung NP311–325 TRH or non-TRH cells at 28 d.p.i. In (A) to (D) and (F) to (G), data were pooled from two independent experiments (three to four mice per group). In (H), representative data were from at least two independent experiments (four mice per group). P values were calculated by unpaired two-tailed Student’s t test. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Bhlhe40 is critical for the development of tissue-resident CD8+ T cell responses (46). Because lung TRH and non-TRH cells expressed high levels of Bhlhe40 (Fig 4A), we investigated the roles of Bhlhe40 in lung TRH responses relative to splenic TFH cells. Lung TRH cells were enriched with Bhlhe40-associated genes (46) compared with splenic TFH cells (Fig. 5E). T cell–specific Bhlhe40 deficiency (Bhlhe40ΔT) modestly increased the frequencies of TRH cells relative to non-TRH cells within the influenza-specific NP311–325 CD4+ T cell population but not in the total lung CD4+ T cell population (Fig. 5, F and G, and fig. S5, A and B). Bhlhe40 deficiency in T cells significantly diminished total and influenza-specific CD4+ T cells in the lung tissue but not in the spleen (fig. S5, C and D). These data suggest that Bhlhe40 is required for the establishment of the overall lung-resident CD4+ T cell population after influenza infection, which was also observed with the lung-resident CD8+ T cells (46). As a result, the magnitude of both TRH and non-TRH cells in the lung was significantly decreased (Fig. 5G and fig. S5B). In contrast, Bhlhe40 deficiency did not alter either the frequencies or the magnitude of the splenic TFH response (fig. S5, A, B, E, and F). Previously, we have reported that Bhlhe40 is required for the survival of CD8+ T cells in nonlymphoid tissues (46). Consistent with that observation, Bhlhe40 deficiency resulted in enhanced cellular apoptosis in both lung TRH and non-TRH cells (Fig. 5H and fig. S5G). These data indicate that Bhlhe40 is likely not important for the acquisition of TFH-like features in TRH cells but is vital in sustaining TRH cell survival in respiratory mucosal tissues. Thus, the optimal formation of lung TRH cells requires transcription factors involved in both TFH (BCL6) and TRM (Bhlhe40) development. Conversely, the formation of splenic TFH cells is dependent on BCL6 but not Bhlhe40, and the development of lung PD-1LowFR4Low cells (probably consist of conventional TRM cells) is dependent on Bhlhe40 but not BCL6.
TRH cells assist the formation of protective BGC, BRM, and CD8+ TRM cell responses
We hypothesized that TRH cells are responsible for mediating the effects of CD4+ T cell help on lung local B and CD8+ T cells. Consistent with that hypothesis, T cell–specific BCL6 deficiency led to diminished BGC, BRM, and CD8+ NP366–374 TRM cell responses (fig. S6, A to E). Furthermore, T cell–specific Bhlhe40 deficiency resulted in diminished lung tissue CD8+ T, B, BRM, and CD8+ TRM cell responses, although it is possible that Bhlhe40 deficiency in CD8+ T cells directly contributes to the diminished CD8+ TRM cell phenotype in these mice as previously shown (fig. S7, A to C) (46).
To specifically determine whether TRH cells are required for the development of memory B and CD8+ T cells in the lung, we generated CD4+ T cell–specific inducible BCL6-deficient mice (Bcl6ΔCD4 ERT2). Tamoxifen treatment efficiently caused gene recombination in CD4+ T cells but only minimally in other lymphocytes in Bcl6ΔCD4 ERT2 mice (fig. S8, A to C). We then infected WT (Bcl6fl/fl) or Bcl6ΔCD4 ERT2 mice with PR8 and administered tamoxifen daily from 12 to 16 d.p.i. (five times) to specifically ablate BCL6 in CD4+ T cells after influenza infection (Fig. 6A). To exclude the contribution of lymphoid organ TFH cells in providing the “late” help to lung B (52) and CD8+ T cells, we treated the mice with FTY720 daily to block T and B cell migration starting at 11 d.p.i. At the lung, the magnitude of CD45i.v.− CD4+ T and B cells was decreased in the inducible BCL6-ablated group (fig. S8D). As with the constitutive BCL6 deficiency in T cells, inducible CD4+ T cell–specific BCL6 ablation resulted in diminished TRH but not non-TRH responses in the lung (fig. S8E). The ablation of lung TRH responses significantly diminished BGC, influenza HA-BRM, and NP-BRM responses in the lung (Fig. 6, B to D). CD8+ NP366–374 TRM responses were also significantly impaired after TRH ablation (Fig. 6E and fig. S8F). These data suggest that lung TRH cells are important in assisting the development of local B and CD8+ T cell memory responses in situ.
Fig. 6. TRH cells are required for the development of lung protective CD8+ TRM and B cell immunity.

(A to J) Bcl6fl/fl or Bcl6ΔCD4ERT2 mice were infected with PR8. (A to E) Tamoxifen was administrated daily (12 to 16 d.p.i.) in the presence of daily FTY720 administration (11 to 34 d.p.i.). (A) Schematic of experimental design. Cell numbers of (B) BGC, (C) HA-BRM, (D) NP-BRM, (E) CD8+CD69+ NP366–374 TRM, or CD8+CD69+ PA224–233 TRM at 35 d.p.i. (F to J) Tamoxifen was administrated daily from 21 to 25 d.p.i. in the presence of daily FTY720 administration (20 to 41 d.p.i.). (F) Schematic of experimental design. Cell numbers of (G) BGC, (H) HA-BRM, (I) NP-BRM, (J) CD8+CD69+ NP366–374 TRM, or CD8+CD69+ PA224–233 TRM at 42 d.p.i. (K) Bcl6fl/fl or Bcl6ΔCD4ERT2 mice were infected with X31 strain (H3N2) of influenza. Tamoxifen was administrated daily (12 to 16 d.p.i.) in the presence of daily FTY720 administration (11 to 34 d.p.i.). Representative dot plot (left) and cell numbers (right) of X31 strain-specific BRM or cross-reactive HA-BRM (to H3N2 A/Uruguay/716/07 strain) at 35 d.p.i. (L and M) Bcl6fl/fl (n = 11) or Bcl6ΔCD4ERT2 (n = 15) mice were infected with X31 and administered tamoxifen from 12 to 16 d.p.i. Mice were rechallenged with PR8 at 42 d.p.i. in the presence of FTY720 (starting from 41 days). (L) Schematic of experimental design. (M) Host mortality following PR8 challenge was monitored. (N and O) Bcl6fl/fl or Bcl6ΔCD4ERT2 mice were infected with X31 and administered tamoxifen. Mice were challenged with PR8 at 40 d.p.i. in the presence of FTY720. Lung (N) infectious virus titers and (O) viral gene expression were measured at day 2 post rechallenge. In (A) to (H) and (J) to (K), all data were pooled from two (C, D, G, H, J, and K) or three (B and E) independent experiments (two to five mice per group). In (A) to (O), P values were calculated by unpaired two-tailed Student’s t test. P value of survival study in (M) was calculated by log-rank test. Data are means ± SEM. *P < 0.05, **P < 0.01, and ****P < 0.0001.
To examine the roles of TRH cells in the maintenance of lung tissue B and CD8+ T cell responses, we treated WT or Bcl6ΔCD4 ER2 mice with tamoxifen starting from 21 to 25 d.p.i. to ablate TRH cell responses at the memory stage (Fig. 6F). We then examined BGC, BRM, and CD8+ TRM cell responses at 42 d.p.i. (Fig. 6, G to J). TRH cell ablation at the memory stage did not lead to significant decrease of BRM cell magnitude but significantly impaired BGC cell responses at 6 weeks postinfection (Fig. 6, G to I). These data suggest that TRH cells help program lung BRM cell development but may not be directly required for their maintenance at the memory stage. However, TRH cells are continuously needed for sustaining lung BGC cell responses (Fig. 6G). These data are consistent with the data showing that BGC cell responses are important for BRM cell development in the first 3 weeks but may not notably contribute to lung BRM cells after 20 d.p.i. (5). TRH cell ablation at the memory stage also diminished CD8+ NP366–374 TRM cell responses, suggesting that lung TRH cells continuously provide local help for the maintenance of CD8+ NP366–374 TRM cells (Fig. 6J). Together, our data indicate that TRH cells are vital for programming lung BRM cell development before 20 d.p.i. and are necessary for both the optimal development and maintenance of BGC and CD8+ NP366–374 TRM cell responses at the memory stage.
Lung memory B cells generated from local BGC cells harbored high portions of cross-reactive B cells to H3N2 A/Uruguay/716/07 strain (Urg) after influenza X31 (H3N2) virus infection (12). To examine whether TRH cells help to generate those cross-reactive BRM cells, we infected WT or Bcl6ΔCD ERT2 mice with X31 virus and injected the mice with tamoxifen in the presence of FTY720 as shown in fig. S8A. We then checked X31 strain–specific HA-BRM and cross-reactive HA-BRM cells against H3N2 Urg (12) using flow cytometry. TRH ablation resulted in diminished strain-specific (X31 HA+ and Urg HA−) and cross-reactive (both X31 HA+ and Urg HA+) BRM cell responses (Fig. 6K). These data indicate potential roles of TRH cells in the development of both strain-specific and cross-reactive B cell immunity, although the function of those cross-reactive B cells in the protective immunity remains to be determined.
Because CD8+ TRM [particularly NP366–374 TRM (31)] and possibly BRM cells are important in mediating host heterologous protection (5), we next sought to determine whether the ablation of TRH cells impairs host protective immunity against heterologous virus infection. To do so, we used a heterologous infection and challenge model in which X31 virus was used as the primary infection and lethal PR8 virus was used as the secondary challenge (46). PR8 and X31 viruses differ in the viral surface proteins but share internal viral proteins such as NP (53). Hence, CD8+ TRM and possibly BRM cells against internal viral epitopes (mainly against viral NP protein) can provide heterologous protection (5, 31). We infected WT or Bcl6ΔCD4ERT2 mice with X31 virus and treated the mice with tamoxifen. We confirmed that TRH ablation affects CD8+ NP366–374 TRM, BGC, and BRM cell development in the X31 model at 35 d.p.i. (fig. S8G). We then rechallenged the mice with a lethal dose of PR8 in the presence of FTY720 to block the contribution of circulating memory CD8+ T and B cells at 42 d.p.i. (Fig. 6L). Close to 40% of Bcl6ΔCD4ERT2 mice succumbed, whereas WT mice were fully protected with lethal PR8 infection (Fig. 6M). Consistent with the increased mortality, lung from Bcl6ΔCD4ERT2 mice exhibited increased infectious virus titer and elevated viral gene (NP and M2) expression compared with those of control mice (Fig. 6, N and O). Thus, TRH cells are required for optimal protection against secondary heterologous infection, most likely through their help for the development of protective CD8+ TRM and BRM response.
Identification of the factors mediating TRH-derived “local” help
CD40-CD40L interaction is critical for T cell help of BGC cell responses (5, 51). Lung influenza–specific TRH cells express modestly higher levels of CD40L than non-TRH cells (fig. S9, A and B). CD40L blockade diminished lung BGC formation and the magnitude of HA-BRM responses in the lung tissue (fig. S9, C and D). These data indicate that CD40L may be necessary for TRH cells to facilitate BGC and BRM cell responses, but further studies are required to firmly establish this idea.
The vast majority of lung IL-21-VFP+ cells were CD4+ T cells after influenza infection at 28 d.p.i. (fig. S10A). IL-21-VFPHi cells, which expressed the highest levels of IL-21 than those IL-21-VFPLow cells (fig. S10B), were mainly TRH cells (Fig. 7A). In addition, T cell–specific BCL6 deficiency impaired Il21 expression in the lung (fig. S10C). These data suggest that TRH cells are the main IL-21 producers in the lung after influenza virus infection. Because IL-21 is an important cytokine that has been implicated in facilitating BGC, memory B and CD8+ effector and memory T cell responses (54), we blocked IL-21 signaling starting at 14 d.p.i. through intraperitoneal injection of α-IL-21R in the presence of FTY720 (Fig. 7B). IL-21 signaling blockade did not impair BGC or influenza HA-BRM cell responses in the lung tissue (Fig. 7, C and D). However, IL-21 signaling blockade significantly diminished total CD8+ and NP366–374 TRM cell responses, suggesting that TRH cells help CD8+ NP366–374 TRM cells establishment and/or maintenance through IL-21 (Fig. 7E and fig. S10D). IL-21R blockade did not significantly diminish PA224–233- or PB1703–711-specific TRM cell responses (fig. S10E), indicating that the roles of IL-21 in TRM cell maintenance may be epitope specific. Lung local blockade of IL-21 signaling (intranasal delivery of α-IL-21R) also diminished lung CD8+ NP366–374 TRM magnitude (Fig. 7F) but not splenic memory T cells (Fig. 7, G and H, and fig. S10F) nor lung parenchymal BGC or HA-B cell responses (fig. S10G).
Fig. 7. TRH help to CD8+ T cells is IL-21 dependent.

(A) Representative dot plot of IL-21Hi or IL-21Low CD4+ T cells from the IL-21-VFP reporter mice infected with PR8 (28 d.p.i.). (B to E) WT mice were infected PR8 with or without IL-21R blockade through intraperitoneal (i.p.) route starting at 14 d.p.i. in the presence of FTY720 administration (13 to 34 d.p.i.). (B) Experimental scheme. Cell numbers of lung parenchymal (C) BGC, (D) HA-BRM, and (E) CD8+CD69+ NP366–374, or CD8+CD69+ PA224–233 TRM cells. (F to J) WT mice were infected with PR8 with or without IL-21R blockade through intranasal (i.n.) route at 14 d.p.i. (F) Experimental scheme. (G) Frequencies or (H) cell numbers of lung tissue CD8+CD69+ NP366–374 or CD8+CD69+ PA224–233 TRM. (I) Percentage of apoptotic cells were identified by active caspase 3/7-FLICA staining within lung CD8+ NP366–374 TRM or splenic CD8+ NP366–374 TMEM at 28 d.p.i. (J) Representative histogram of BATF expression in lung CD8+ NP366–374 or CD8+ PA224–233 TRM of mice received with Ctl IgG or α-IL21R at 35 d.p.i. FMO, fluorescence minus one control. (K to M) WT mice were infected with PR8 with or without IL-21R blockade in the presence of FTY720. Mice were challenged with X31 at 40 d.p.i. (K) Experimental scheme. (L) Lung infectious virus titer or (M) viral gene expression were measured at day 2 after rechallenge. In (A) and (I) to (M), representative data were from at least two independent experiments (four to five mice per group). In (C), (E), and (G) to (H), data were pooled from two independent experiments (three to five mice per group). P values of all experiments were calculated by unpaired two-tailed Student’s t test. Data are means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
IL-21R signaling blockade led to enhanced cellular apoptosis but not proliferation, specifically in CD8+ NP366–374 TRM but not those of splenic memory T cells (Fig. 7I and fig. S10H). Compared with CD8+ PA224–233 TRM cells, CD8+ NP366–374 TRM cells exhibited higher expression of genes associated with IL-21 signaling including Batf (fig. S11, A and B) (55). Furthermore, there was a higher expression of IL-21R in the CD8+ NP366–374 TRM cells than CD8+ PA224–233 TRM cells (fig. S11C). These data suggest that CD8+ NP366–374 TRM cells, but not CD8+ PA224–233 TRM cells, potentially receive IL-21 signaling in the lung at the memory stage after influenza virus infection. Consistent with the data, IL-21R blockade diminished basic leucine zipper transcription factor, ATF-like (BATF) expression in CD8+ NP366–374 TRM but not CD8+ PA224–233 TRM cells at 35 d.p.i. (Fig. 7J), indicating that TRH cells provide local help to CD8+ NP366–374 TRM cells in an IL-21–dependent manner. We then infected WT mice with PR8, treated the mice with α-IL-21R or control IgG in the presence of FTY720, and then challenged the mice with X31 at 6 weeks postinfection (Fig. 7K). Mice that received α-IL-21R treatment had enhanced infectious virus titers and elevated viral gene expression after X31 rechallenge (Fig. 7, L and M). Similar findings were observed in the model of primary X31 infection followed with PR8 virus rechallenge (fig. S11, D and E). Thus, IL-21–mediated TRH cell help to CD8+ T cells promotes optimal heterologous immunity against influenza reinfection.
DISCUSSION
Here, we have identified a requirement for “in situ” CD4+ T cell help in the respiratory mucosa, which is mediated by what we have termed TRH cells, in the development of localized protective memory responses after influenza virus infection. TRH cells comanifest phenotypic and transcriptional hallmarks of both TFH and TRM cells. Tissue PD-1HiCXCR5LowBCL6Int TFH-like cells have been previously observed (25–29). However, the cellular identity and developmental cues regulating their development have previously remained largely elusive. Furthermore, the physiological function of these cells beyond their help on the generation of BGC cells was unknown.
Using multiple lines of approaches including high or low doses of α-CD4 depletion, inducible CD4+ T cell–specific BCL6 ablation, and IL-21 blockade combined with long-term FTY720 treatment, we have provided comprehensive evidence that localized CD4+ T cell help, mediated by TRH cells and IL-21, is required for the optimal generation of a population of protective TRM cells (H-2Db-restricted NP366–374 TRM cells) after the resolution of primary infection. It was previously shown that continuous CD4+ T cell help beyond T cell priming (7 d.p.i.) was not required for the differentiation of CD103+ CD8+ TRM cells, as measured by the levels of CD103 expression in GP33–41 TRM cells after infection with recombinant influenza X31 virus expressing lymphocytic choriomeningitis virus GP33–41 peptide (21). Here, we found that persistent late CD4+ T cell help after viral clearance was required for the maintenance of the overall NP366–374 TRM population (both CD103− CD69+ and CD103+ CD69+ TRM cells). As a result, consistent with prior work (21), CD4+ T cell depletion after the resolution of primary infection did not affect the percentages of CD103+ TRM cells nor CD103 expression level per cell within the total NP366–374 CD8+ CD69+ TRM cell population. In addition, late CD4+ T cell help to Ag-specific TRM cells appears to be epitope specific as the generation and/or the maintenance of PA224–233 or PB1703–711 TRM cells are largely CD4+ T cell help or IL-21 independent.
An interesting question is why lung TRH cell help, in the form of IL-21 provision, is specifically required for CD8+ NP366–374 TRM cell responses. We have shown previously that CD8+ NP366–374 TRM cells receive constant Ag engagement in the lung at the memory stage due to the chronic deposition of high levels of influenza NP Ag (31). Compared with conventional CD8+ PA224–233 TRM cells, CD8+ NP366–374 TRM cells express high levels of PD-1 and exhibit “exhausted-like” phenotypes similar to those of T cells receiving chronic Ag exposure during chronic viral infections (31, 56, 57). Consequently, the maintenance of these TRM cells requires persistent MHC-I-dependent stimulation at the memory stage (31) similar to those of exhausted CD8+ T cells (56). CD4+ T cell help in the form of IL-21 has been recently demonstrated to sustain those PD-1–expressing CD8+ T cells during chronic viral infection and tumor growth (58). Thus, CD4+ T cell help and IL-21 signaling may be specifically required for maintaining the survival of lung CD8+ TRM cells receiving persistent low levels of in situ antigenic stimulation. Consistent with this idea, NP366–374 TRM cells express higher levels of molecules associated with IL-21 signaling, including IL-21R and BATF.
Influenza virus is able to undergo antigenic shift, drift, and reassortment to escape previously established host immunity. Current influenza vaccines require yearly update and only provide high levels of protection when influenza vaccine strains match exactly with the circulating strains. Much of the attention has been given to the development of potential universal influenza vaccines recently. The common goals of various universal influenza vaccine candidates are to induce broadly neutralizing influenza Ab, strong CD8+ memory T cell responses against conserved epitopes and/or high levels of localized lung-resident mucosal immunity that can restrict viral spreading early (59–62). Because of the high mutation rates of influenza viruses, it is conceivable that the induction of “all inclusive” immune responses, i.e., induction of strong both B and CD8+ T cell immunity in the mucosal tissue, may be required for the ultimate success of a universal influenza vaccine (62). Because of the distinct ability of TRH cells in assisting local BRM and TRM cell development, it is tempting to speculate that the specific promotion of TRH cells may simultaneously enhance both memory B and CD8+ T cell immunity in the respiratory tract during vaccination, thereby providing rapid cross-reactive protection against broad-spectrum viruses.
In summary, our data have revealed a complex T-B cell interaction network that is programmed by lung TRH cells for the maintenance of protective local respiratory immunity following acute influenza virus infection. Moving forward, it is of substantial interest to dissect the mechanisms modulating the development of TRH cells and the precise function of TRH cells in assisting the development of local B and CD8+ T cell immunity during infection. The role of TRH cells in vaccination and in other contexts remains to be examined.
METHODS
Study design
The goal of the study was to determine the roles of tissue-resident CD4+ TTH cells in coordinating protective tissue-resident CD8+ and B cell responses after respiratory viral infection. We first examined lung CD8+ TRM, BGC, and BRM cell responses after the depletion of CD4+ T cells after influenza viral clearance. We performed scRNA-seq and bulk RNA-seq, identified a tissue TFH-like cell population coexhibiting both TFH and TRM gene features, and termed as TRH cells. We used T cell–specific BCL6- or Bhlhe40-deficient mice to examine whether optimal TRH cell development and/or maintenance were dependent on both TFH and TRM transcription factors. Using T cell–specific BCL6-deficient mice, we specifically ablated TRH cells to measure lung CD8+ TRM and B cell responses after primary influenza virus infection, as well as protection against a heterotypic influenza virus reinfection. Last, we used IL-21 reporter mice and an IL-21R blocking Ab to determine whether TRH-dependent CD8+ T cell help is mediated by IL-21. Experiments were conducted in replicates as indicated in figure legends. No outliers were removed.
Mice and influenza viral infection
WT C57BL/6, CD45.1, and IL-21 VFP reporter mice were purchased from the Jackson Laboratory (JAX) and bred in-house. To generate CD45.1+ and CD45.2+ (CD45.1+/CD45.2+) mice, CD45.1 mice were crossed with C57BL/6 mice. Bcl6fl/fl were generated as previously reported (50). Bcl6ΔT were generated by crossing with CD4-Cre transgenic. Bcl6ΔCD4ERT2 were generated by crossing with CD4-ERT2 transgenic mice. Bcl6fl/fl or Bcl6ΔCD4ERT2 mice were additionally crossed with Rosa26 LSL–yellow fluorescent protein (JAX) mice for the determination of the efficiency of tamoxifen-induced gene recombination. Bhlhe40fl/fl or Bhlhe40ΔT mice were generated as previously reported (46). All animal protocols were approved by the Institutional Animal Care and Use Committees (IACUC) of the Mayo Clinic (Rochester, MN). Sex- and age-matched 8- to 10-week- old mice of both sexes were used in the experiments. Influenza A/PR8/34 [~200 plaque-forming units (PFU) per mouse in the primary infection and ~1 × 104 PFU/mouse in the secondary infection] and influenza A X31 (~800 PFU/mouse in the primary infection and ~1 × 105 PFU/mouse in the secondary infection) were infected into the mice by intranasal under anesthesia as reported before (33). Both real death and humane sacrifice were counted as mouse mortality. According to the institute IACUC policy, we humanely euthanized infected mice when their weight was below 70% of original weight.
Ab administration in vivo
Influenza-infected WT mice were administrated with control IgG or various neutralizing or depleting Ab as described in the related Results sections. For CD4+ T cell depletion with high dose of α-CD4, mice were injected with 250 μg of α-CD4 weekly (clone: GK1.5, BioXCell) starting at 14 d.p.i. For circulating CD4+ T cell depletion, mice were intraperitoneally injected with 40 μg of α-CD4 for the first dose followed with 10 μg of α-CD4 weekly. CD40L blockade was achieved by the injection of 250 μg of α-CD40L (clone: MR-1, BioXCell) weekly, respectively. For systemic IL-21R blockade, 500 μg of α-IL21R (clone: 4A9, BioXCell) was injected intraperitoneally weekly starting at 14 d.p.i. For lung local IL-21R blockade, 50 μg of α-IL21R was injected through intranasal route weekly starting at 14 d.p.i. In some experiments, FTY720 (1 mg/kg; Cayman) was administrated via intraperitoneal injection daily from 13 d.p.i. to block lymphocyte migration until mouse euthanasia.
Tamoxifen treatment
To induce gene recombination in Bcl6ΔCD4ERT2 mice, tamoxifen (Sigma-Aldrich) was diluted in warm sunflower oil (Sigma-Aldrich) and daily treated via intraperitoneal route for five consecutive times. Each application was 2 mg per mouse.
Immunohistochemistry and immunofluorescence
Left lobe of the whole lung was harvested and fixed in 10% formaldehyde (Thermo Fisher Scientific) until embedding. Fixed lung tissues were embedded in paraffin, sectioned at 10-μm thickness. To identify tertial lymphoid structure, the lung tissue slide was stained with hematoxylin and eosin by the Mayo Clinic Histology Core Laboratory (Scottsdale, AZ). To measured iBALT structure, and lung tissue sections were deparaffinized in CitriSolv (Thermo Fisher Scientific) for 30 min and then immersed in ethanol series from 100, 95, 85, and 75% to distill H2O for 5 min each for tissue hydration. For Ag retrieval, hydrated sections were steamed for 20 min in 1 mM EDTA (pH 8.0). Lung sections were blocked with Super Block Blocking buffer (Thermo Fisher Scientific) for 1 hour at room temperature. Anti–B220-eflour660 (clone: 4SM95, Invitrogen), anti–CD4-eflour570 (clone: RA3-6B2, Invitrogen), and/or anti–GL7-Alexa488 (clone: GL7, BioLegend) were stained on the lung tissue sections overnight at 4°C. After washing in 0.1% PBST [phosphate-buffered saline (PBS) with Tween 20], the slides were counterstained with 4′, 6-diaminodino-2-phenylindole (DAPI) and mounted. Tissue staining was reviewed, and representative images were acquired on a Zeiss LSM 780 confocal system (Carl Zeiss).
Parabiosis surgery
To examine tissue residency of lung TRH or non-TRH cells, parabiotic surgery was performed. CD45.1+ or CD45.1+/CD45.2+ mice were infected with influenza PR8. Three weeks later, mice were anesthetized with ketamine and xylazine. The lateral skin area was shaved and disinfected, and then a matching incision was made from the olecranon to the knee joint of each mouse. Matching area was opposed with continuous sutures. Parabionts were then allowed to rest for 14 days before analysis. Equilibration of parabionts was confirmed in the peripheral blood before tissue analysis.
Plaque assay
Monolayers of Madin-Darby canine kidney cells in a 12-well plate were washed twice with 1× PBS. The cells were then inoculated with serial 10-fold dilutions of supernatants of the lung homogenate in serum-free medium. After incubation at 37°C for 1 hour, cells were washed twice with PBS and overlaid with minimum essential medium containing 0.5% agarose (Lonza) and TPCK-trypsin (0.5 gm/ml). After 3 days in culture, the cells were fixed with 4% of formaldehyde and stained with 0.1% crystal violet solution.
Single-cell RNA sequencing
Sorted CD45i.v.−CD4+CD44Hi T cells from pooled lung cells of mice (five mice) infected with influenza virus (28 d.p.i.) were loaded on the Chromium Controller (10× Genomics). Single-cell libraries were prepared using the Chromium Single Cell 3′ Reagent kit (10× Genomics) following the manufacturer’s instruction. Paired-end sequencing was performed using an Illumina HiSeq 2500 in rapid-run mode. The CellRanger software package (10× Genomics) was used to align and quantify sequencing data from 10× Genomics. All scRNA-seq analyses were performed in R using the package Seurat (version 2.0) (63). The same number of cells from each group was used for log normalization, followed by identifying differentially expressed genes (DEGs) using variance-stabilizing transformation. DEGs in Fig. 2B were selected on the basis of the signature genes expressed by circulating memory T cells, TH1, TH17, Treg, or TFH cells.
Bulk RNA sequencing
Lung CD45i.v.−CD4+CD44HiGITR−PD-1HiFR4Hi, CD45i.v.−CD4+ CD44HiGITR−PD-1LowFR4Low, splenic CD4+CD44HiGITR−PD-1Hi CXCR5+ TFH, or splenic CD4+CD44HiGITR−PD-1low CXCR5− non-TFH cells were sorted from a total of 15 mice that were infected with influenza virus (28 d.p.i.). RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) following the manufacture’s recommendation. After quality control, high-quality [Agilent Bioanalyzer RNA Integrity Number (RIN) >7.0] total RNA was used to generate the RNA sequencing library. Complementary DNA synthesis, end repair, A-base addition, and ligation of the Illumina indexed adapters were performed according to the TruSeq RNA Sample Prep Kit v2 (Illumina, San Diego, CA). Paired-end libraries were sequenced on an Illumina HiSeq 4000 following Illumina’s standard protocol using the Illumina cBot and HiSeq 3000/4000 PE Cluster Kit. Base calling was performed using Illumina’s RTA software (version 2.5.2). Paired-end RNA-seq reads were aligned to the mouse reference genome (GRCm38/mm10) using RNA-seq spliced read mapper Tophat2 (v2.1.1). Pre- and postalignment quality controls, gene level raw read count, and normalized read count (i.e., fragments per kilobase million, FPKM) were performed using RSeQC package (v2.3.6) with National Center for Biotechnology Information (NCBI) mouse RefSeq gene model. DEGs were identified on the basis of the results of DESeq2 Wald tests. DEGs were presented in the form of volcano plots. For visualization, data were logarithmic transformed, and genes that exhibited log2 fold change values > 2 and log10 P > 25 between compared groups were highlighted. For functional analysis, GSEA was used to identify enriched gene sets, from the hallmark collection of MSigDB, having up- and down-regulated genes, using a weighted enrichment statistic and a log2 ratio metric for ranking genes.
Intravascular labeling with α-CD45 and preparation of lung cell suspension
Mice were intravenously injected with 2 μg of α-CD45 (clone: 30-F11; Tonbo Biosciences), which was diluted in 300 μl of sterile PBS 5 min before sacrifice. To prepare single cells from lung tissue, lung was cut into small pieces, digested with type 2 collagenase (Worthington Biochemical), and dissociated in 37°C for 30 min with Gentle-MACS (Miltenyi). Cells were further ground through 70-μm cell strainer (Falcon) and washed with plain Iscove’s Modified Dulbecco’s Medium (Gibco). After red blood cell lysis, cells were centrifuged and resuspended in cold fluorescence-activated cell sorting buffer (PBS, 2 mM EDTA, 2% fetal bovine serum, and 0.09% sodium azide) for flow cytometry analysis. Lung circulating immune cells are i.v. Ab+, and lung tissue immune cells are defined by i.v. Ab−. Lung TRM cells were defined as CD45i.v.−CD8+ tetramer+CD69+. Lung BRM cells were defined as CD45i.v.− B220+ GL7− IgD− IgM− CD38+ influenza B cell Ag (HA or NP)+.
B cell antigens
Influenza PR8-HA protein was a gift from M. C. Crank (National Institutes of Health). PR8-NP protein was purchased from Sino Biological. Purified Ags were biotinylated using an EZ-Link Sulfo-NHS-LCBiotinylation kit (Thermo Fisher Scientific) using a 1:1.3 M ratio of biotin to Ag. To make tetramers, biotinylated Ags were mixed with streptavidin–phycoerythrin (PE; PJ27S; ProZyme) at the ratio determined or at a 5:1 ratio using the biotin concentration provided by the manufacturer as described before (64). After a 30-min incubation on ice, unconjugated biotinylated Ag was often removed by several rounds of dilution and concentration using a 100-kDa Amicon Ultra (MilliporeSigma) or 300-kDa Nanosep centrifugal devices (Pall). Tetramers were stored at 1 ຼM in 1× Dulbecco’s phosphate-buffered saline (DPBS) at 4°C before use. H3N2 X31-HA conjugated with allophycocyanin and H3N2 Urg (Uruguay)–HA conjugated with PE were previously reported (12).
Quantification and statistical analysis
Statistical analysis was done using GraphPad Prism 7.0 (GraphPad Software) and presented as means ± SEM. Unpaired or paired Student t tests (two group comparisons) and one-way or two-way analysis of variance (ANOVA) analysis (multiple group comparisons) were used in data analysis. Log-rank test was used for analysis of survival study.
Supplementary Material
Fig. S1. “Late” CD4+ T cell help shapes respiratory mucosal memory CD8+ T and B cell immunity.
Fig. S2. Parenchymal tissue CD4+ T cells deliver local help to CD8+ T and B cells.
Fig. S3. scRNA-seq identifies a population of lung TH cells that exhibit TFH features.
Fig. S4. Total RNA-seq identifies PD-1HiFR4Hi cells that exhibit features of both TFH and TRM.
Fig. S5. Bhlhe40 deficiency impairs tissue CD4+ T cell responses.
Fig. S6. T cell–specific BCL6 deficiency leads to impaired lung CD8+ memory and B cell immunity.
Fig. S7. Bhlhe40 is required for lung memory CD8+ T and B cell responses.
Fig. S8. TRH cells help local development of memory CD8+ T and B cells.
Fig. S9. CD40L signal is likely involved in TRH cell help to tissue B cells.
Fig. S10. TRH cells help CD8+ TRM cells through IL-21–dependent mechanisms.
Fig. S11. Epitope-specific IL-21 signaling in CD8+ T cells.
Fig. S12. Graphical summary.
Acknowledgments:
We thank NIH tetramer core for tetramers and Mayo flow cytometry core for technical assistance. We thank B. Graham and M. Crank for PR8 HA protein used in this study.
Funding:
This study was funded by the U.S. NIH R01s AI112844, AI147394, AI154598, and AG047156 (to J.S.); R01 AI125741 and RO1 AI148403 (to W.C.); and R01 AI057459 (to M.H.K.).
Footnotes
REFERENCES AND NOTES
- 1.Onodera T, Takahashi Y, Yokoi Y, Ato M, Kodama Y, Hachimura S, Kurosaki T, Kobayashi K, Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc. Natl. Acad. Sci. U.S.A 109, 2485–2490 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schenkel JM, Masopust D, Tissue-resident memory T cells. Immunity 41, 886–897 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mueller SN, Mackay LK, Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 16, 79–89 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Milner JJ, Goldrath AW, Transcriptional programming of tissue-resident memory CD8(+) T cells. Curr. Opin. Immunol. 51, 162–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, Randall TD, The establishment of resident memory B cells in the lung requires local antigen encounter. Nat. Immunol. 20, 97–108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, Carbone FR, Tscharke DC, Heath WR, Mueller SN, Mackay LK, Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 19, 183–191 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, Fonseca R, Burbach BJ, Hickman HD, Vezys V, Fife BT, Masopust D, Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol. 19, 173–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jameson SC, Masopust D, Understanding subset diversity in T cell memory. Immunity 48, 214–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rolfes MA, Foppa IM, Garg S, Flannery B, Brammer L, Singleton JA, Burns E, Jernigan D, Olsen SJ, Bresee J, Reed C, Annual estimates of the burden of seasonal influenza in the United States: A tool for strengthening influenza surveillance and preparedness. Influenza Other Respir. Viruses 12, 132–137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, Farber DL, Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 7, 501–510 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takamura S, Kohlmeier JE, Establishment and maintenance of conventional and circulation-driven lung-resident memory CD8(+) T cells following respiratory virus infections. Front. Immunol. 10, 733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adachi Y, Onodera T, Yamada Y, Daio R, Tsuiji M, Inoue T, Kobayashi K, Kurosaki T, Ato M, Takahashi Y, Distinct germinal center selection at local sites shapes memory B cell response to viral escape. J. Exp. Med. 212, 1709–1723 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMaster SR, Wilson JJ, Wang H, Kohlmeier JE, Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-γ poduction. J. Immunol. 195, 203–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pizzolla A, Wakim LM, Memory T cell dynamics in the lung during influenza virus infection. J. Immunol. 202, 374–381 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Slutter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, Harty JT, Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol. 2, eaag2031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cauley LS, Environmental cues orchestrate regional immune surveillance and protection by pulmonary CTLs. J. Leukoc. Biol. 100, 905–912 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J, Braciale TJ, Role of T cell immunity in recovery from influenza virus infection. Curr. Opin. Virol. 3, 425–429 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakanishi Y, Lu B, Gerard C, Iwasaki A, CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 462, 510–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.lijima N, Iwasaki A, Access of protective antiviral antibody to neuronal tissues requires CD4 T-cell help. Nature 533, 552–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belz GT, Wodarz D, Diaz G, Nowak MA, Doherty PC, Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J. Virol. 76, 12388–12393 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Guan T, Hu Y, Cauley LS, Craft J, Kaech SM, CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity 41, 633–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crotty S, A brief history of T cell help to B cells. Nat. Rev. Immunol. 15, 185–189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papillion A, Powell MD, Chisolm DA, Bachus H, Fuller MJ, Weinmann AS, Villarino A, O’Shea JJ, Leon B, Oestreich KJ, Ballesteros-Tato A, Inhibition of IL-2 responsiveness by IL-6 is required for the generation of GC-TFHcells. Sci. Immunol. 4, eaaw7636 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allie SR, Randall TD, Pulmonary immunity to viruses. Clin. Sci. (Lond.) 131, 1737–1762 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, Donlin LT, Henderson LA, Wei K, Mizoguchi F, Teslovich NC, Weinblatt ME, Massarotti EM, Coblyn JS, Helfgott SM, Lee YC, Todd DJ, Bykerk VP, Goodman SM, Pernis AB, Ivashkiv LB, Karlson EW, Nigrovic PA, Filer A, Buckley CD, Lederer JA, Raychaudhuri S, Brenner MB, Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rao DA, T cells that help B cells in chronically inflamed tissues. Front. Immunol. 9, 1924 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van DV, Beier KC, Pietzke LJ, Al Baz MS, Feist RK, Gurka S, Hamelmann E, Kroczek RA, Hutloff A, Local T/B cooperation in inflamed tissues is supported by T follicular helper-like cells. Nat. Commun. 7, 10875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan HX, Esterbauer R, Vanderven HA, Juno JA, Kent SJ, Wheatley AK, Inducible bronchus-associated lymphoid tissues (iBALT) serve as sites of B cell selection and maturation following influenza infection in mice. Front. Immunol. 10, 611 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hornick EE, Zacharias ZR, Legge KL, Kinetics and phenotype of the CD4 T cell response to influenza virus infections. Front. Immunol. 10, 2351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodland DL, Kohlmeier JE, Migration, maintenance and recall of memory T cells in peripheral tissues. Nat. Rev. Immunol. 9, 153–161 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, Huang S, Shan J, Ma C, Ye Z, Xiang M, Limper AH, Porquera EC, Kohlmeier JE, Kaplan MH, Zhang N, Johnson AJ, Vassallo R, Sun J, PD-1hiCD8+resident memory T cells balance immunity and fibrotic sequelae. Sci. Immunol. 4, eaaw1217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hufford MM, Kim TS, Sun J, Braciale TJ, Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J. Exp. Med. 208, 167–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun J, Madan R, Karp CL, Braciale TJ, Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 15, 277–284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, Sun J, Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity 39, 833–845 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D, Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goplen NP, Huang S, Zhu B, Cheon IS, Son YM, Wang Z, Li C, Dai Q, Jiang L, Sun J, Tissue-resident macrophages limit pulmonary CD8 resident memory T cell establishment. Front. Immunol. 10, 2332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, Woodland DL, Lund FE, Randall TD, Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat. Med. 10, 927–934 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Hwang JY, Randall TD, Silva-Sanchez A, Inducible bronchus-associated lymphoid tissue: Taming inflammation in the lung. Front. Immunol. 7, 258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas PG, Keating R, Hulse-Post DJ, Doherty PC, Cell-mediated protection in influenza infection. Emerg. Infect. Dis. 12, 48–54 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.La Gruta NL, Kedzierska K, Pang K, Webby R, Davenport M, Chen W, Turner SJ, Doherty PC, A virus-specific CD8+ T cell immunodominance hierarchy determined by antigen dose and precursor frequencies. Proc. Natl. Acad. Sci. U.S.A. 103, 994–999 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ballesteros-Tato A, Leon B, Lee BO, Lund FE, Randall TD, Epitope-specific regulation of memory programming by differential duration of antigen presentation to influenza-specific CD8(+) T cells. Immunity 41, 127–140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baeyens A, Fang V, Chen C, Schwab SR, Exit strategies: S1P signaling and T cell migration. Trends Immunol. 36, 778–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marnik EA, Wang X, Sproule TJ, Park G, Christianson GJ, Lane-Reticker SK, Jain S, Duffy T, Wang H, Carter GW, Morse HC 3rd, Roopenian DC, Precocious interleukin 21 expression in naive mice identifies a natural helper cell population in autoimmune disease. Cell Rep. 21, 208–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, Sen JM, Berg LJ, Gattinoni L, McGavern DB, Schwartzberg PL, TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep. 12, 2099–2110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, Friedman AL, Shen Y, Farber DL, Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z, Beckermann KE, Wu Y, Jenkins JW, Siska PJ, Vincent BG, Prakash YS, Peikert T, Edelson BT, Taneja R, Kaplan MH, Rathmell JC, Dong H, Hitosugi T, Sun J, The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8(+) T cell fitness and functionality. Immunity 51 , 491–507.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beura LK, Fares-Frederickson NJ, Steinert EM, Scott MC, Thompson EA, Fraser KA, Schenkel JM, Vezys V, Masopust D, CD4(+) resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J. Exp. Med. 216, 1214–1229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S, Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, Dong C, Bcl6 mediates the development of T follicular helper cells. Science 325, 1001–1005 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, Dent AL, Insights into the role of Bcl6 in follicular Th cells using a new conditional mutant mouse model. J. Immunol. 191, 3705–3711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crotty S, T follicular helper cell biology: A decade of discovery and diseases. Immunity 50, 1132–1148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunzli M, Schreiner D, Pereboom TC, Swarnalekha N, Litzler LC, Lotscher J, Ertuna YI, Roux J, Geier F, Jakob RP, Maier T, Hess C, Taylor JJ, King CG, Long-lived T follicular helper cells retain plasticity and help sustain humoral immunity. Sci. Immunol. 5, eaay5552 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Rutigliano JA, Sharma S, Morris MY, Oguin TH III, McClaren JL, Doherty PC, Thomas PG, Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells. J. Virol. 88, 1636–1651 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian Y, Zajac AJ, IL-21 and T cell differentiation: Consider the context. Trends Immunol. 37, 557–568 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xin G, Schauder DM, Lainez B, Weinstein JS, Dai Z, Chen Y, Esplugues E, Wen R, Wang D, Parish IA, Zajac AJ, Craft J, Cui W, A critical role of IL-21-induced BATF in sustaining CD8-T-cell-mediated chronic viral control. Cell Rep. 13, 1118–1124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wherry EJ, Kurachi M, Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R, Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, Cui W, CD4(+) T cell help is required for the formation of a cytolytic CD8(+) T cell subset that protects against chronic infection and cancer. Immunity 51, 1028–1042.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zens KD, Chen JK, Farber DL, Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 1, e85832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clemens EB, van de Sandt C, Wong SS, Wakim LM, Valkenburg SA, Harnessing the power of T cells: The promising hope for a universal influenza vaccine. Vaccines 6, 18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angeletti D, Yewdell JW, Is it possible to develop a “universal” influenza virus vaccine? Outflanking antibody immunodominance on the road to universal influenza vaccination. Cold Spring Harb. Perspect. Biol. 10, a028852 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Estrada LD, Schultz-Cherry S, Development of a universal influenza vaccine. J. Immunol. 202, 392–398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steach HR, DeBuysscher BL, Schwartz A, Boonyaratanakornkit J, Baker ML, Tooley MR, Pease NA, Taylor JJ, Cross-reactivity with self-antigen tunes the functional potential of naive B cells specific for foreign antigens. J. Immunol. 204, 498–509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheon IS, Son YM, Jiang L, Goplen NP, Kaplan MH, Limper AH, Kita H, Paczesny S, Prakash YS, Tepper R, Ahlfeld SK, Sun J, Neonatal hyperoxia promotes asthma-like features through IL-33-dependent ILC2 responses. J. Allergy Clin. Immunol. 142, 1100–1112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goplen NP, Wu Y, Son YM, Li C, Wang Z, Cheon IS, Jiang L, Zhu B, Ayasoufi K, Chini EN, Johnson AJ, Vassallo R, Limper AH, Zhang N, Sun J, Tissue-resident CD8+ T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci. Immunol.5, eabc4557 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. “Late” CD4+ T cell help shapes respiratory mucosal memory CD8+ T and B cell immunity.
Fig. S2. Parenchymal tissue CD4+ T cells deliver local help to CD8+ T and B cells.
Fig. S3. scRNA-seq identifies a population of lung TH cells that exhibit TFH features.
Fig. S4. Total RNA-seq identifies PD-1HiFR4Hi cells that exhibit features of both TFH and TRM.
Fig. S5. Bhlhe40 deficiency impairs tissue CD4+ T cell responses.
Fig. S6. T cell–specific BCL6 deficiency leads to impaired lung CD8+ memory and B cell immunity.
Fig. S7. Bhlhe40 is required for lung memory CD8+ T and B cell responses.
Fig. S8. TRH cells help local development of memory CD8+ T and B cells.
Fig. S9. CD40L signal is likely involved in TRH cell help to tissue B cells.
Fig. S10. TRH cells help CD8+ TRM cells through IL-21–dependent mechanisms.
Fig. S11. Epitope-specific IL-21 signaling in CD8+ T cells.
Fig. S12. Graphical summary.